
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Converting high modulus water-based elastomeric core–shell nanoparticle films from viscoelastic to predominantly elastic using di-epoxide crosslinking†
Mollie
Osborne-Richards
 *a,
David
Ring
b,
Xuelian
Wang
a,
Sarah
Wall
a,
Steve
Edmondson
a and
Brian R.
Saunders
*a
*a,
David
Ring
b,
Xuelian
Wang
a,
Sarah
Wall
a,
Steve
Edmondson
a and
Brian R.
Saunders
*a
aDepartment of Materials, University of Manchester, MECD(A), Manchester, M1 7HL, UK. E-mail: brian.saunders@manchester.ac.uk; mollie.osborne-richards@postgrad.manchester.ac.uk
bSynthomer (UK) Ltd, Temple Fields, Harlow, Essex, CM20 2BH, UK
First published on 29th November 2024
Abstract
Most elastomers are formed using solvent-based processes which result in an environmental burden. Consequently, elastomers formed using water-based nanoparticle dispersions are highly desirable. Here, we investigate elastomer-like films based on water-dispersible carboxylic acid-containing core–shell (CS) nanoparticles. The nanoparticles contain a poly(n-butylacrylate) (PBA) core and a poly(BA-co-acrylonitrile-co-methacrylic acid) shell. We react the –COOH groups of the shell with a di-epoxide (1,4-butanediol diglycidyl ether, BDDE) which replaces dissipative hydrogen bonds in the nanoparticle elastomer films with covalent bonds. The reaction with BDDE enables the transformation of a stretchable dissipative film (shear modulus of 9.0 MPa with 20% strain energy recovery) into a predominantly elastic film (shear modulus of 0.20 MPa with almost 100% energy recovery). Our optimum system, CS-0.5, has a shear modulus of 0.40 MPa, an impressive strain-at-break of greater than 300% and an energy recovery of 80%. The strain-at-break is increased to more than 450% using a monofunctional epoxide. We further explore the inter- and intra-nanoparticle nature of the di-epoxide reaction and how the mechanical properties can be tuned by varying the method of film formation. The facile approach introduced here enables the tuning of the mechanical properties of elastomeric core–shell nanoparticle films from dissipative to predominantly elastic on demand.
Introduction
Elastomer materials are crosslinked polymers that can be stretched by significant elongation and then return to their original size with little to no permanent deformation.1–3 The term elastomer usually refers to a fully crosslinked network. If there is incomplete crosslinking then, strictly, the material will be viscoelastic. Elastomer materials are used for a wide range of applications as they can be adhesive,4–6 strong,7 stretchable8–10 and moldable.11,12 Therefore, the characteristics of elastomers need to be easily tuneable for the desired application.13,14 A common method for preparing elastomers is by solution processing; however, this process often uses harmful solvents8,15 which are released as volatile organic compounds into the atmosphere and raise environmental concerns.16–18 Consequently, there is an urgent need to develop more environmentally friendly elastomer syntheses.19 The present study demonstrates a new water-based synthesis for preparing high modulus, core–shell (CS) nanoparticle elastomer-like films with highly tuneable mechanical properties that are shown to be controllable via nanoparticle composition, nanoparticle crosslinking and film preparation methods.Elastomers are crosslinked by either physical crosslinking or chemical crosslinking.20 Common crosslinking strategies employ covalent bonding21,22 and hydrogen bonding,20,23,24 with dynamic bonding (including disulfide bonding25,26) coming into the spotlight in recent years.21,27,28 To create more environmentally friendly elastomers, solvents are being replaced with water-based dispersions that form elastomeric films.29–35 For such systems, the elastomeric film is formed as the water evaporates and the particles deform. Interpenetration then occurs at the particle–particle interface, causing coalescence.15,31 To improve the mechanical properties of such films, inter-particle, physical crosslinking can be introduced by the addition of hydrogen bonding or ionic interactions.16,31,36
Additionally, CS nanoparticles are advantageous for film property control because the core and the shell can be separately modified to tune elastomer mechanical properties.37–39 They have been studied extensively for a range of applications from semiconductors40,41 to drug delivery.42,43 Crucially, CS nanoparticles have been investigated as sustainable alternatives to solvent-based polymer films.31,35,44,45 The core of the CS nanoparticles is usually synthesized first followed by addition of the shell which forms around the core in situ43 as a uniform layer.46 The properties of both the core and the shell can be separately tailored, resulting in versatile CS nanoparticles.46,47
Previously we investigated the nanostructure of elastomeric films based on CS nanoparticles with a poly(n-butyl acrylate) (PBA) core and a shell containing a copolymer of BA, acrylonitrile (AN), methacrylic acid (MAA) and 1,4-butanediol diacrylate (BDDA).37 The inter-particle crosslinking in those PBA/(PBA–AN–MAA–BDDA) nanoparticle films was from hydrogen bonds and free-radical coupling of vinyl groups that occurred when the films were heated. The film's mechanical properties were tunable via the MAA content and/or the extent of vinyl-based inter-particle crosslinking.48,49 Unfortunately, the versatility of that approach was hindered by the oxygen-triggered radical crosslinking reaction that inevitably occurred at the high temperatures required for vinyl crosslinking.37 In contrast, here we introduced a new CS dispersion preparation method coupled with a low-temperature crosslinking method that uses a non-free-radical di-epoxide reaction. The di-epoxide will crosslink across polymer chains within the shell of the particles as the ring opens and reacts with the –COOH groups. We hypothesized that the CS elastomer-like films would have profound mechanical property tuneability whilst maintaining high elongation-at-break.
In this study, we sought to establish a facile method for covalently interlinking CS nanoparticle films using 1,4-butanediol diglycidyl ether (BDDE) added to the nanoparticle dispersion (Scheme 1). The di-epoxide is shown to act as a crosslinker and change the properties of the CS-x films via two-stage intra- and inter-particle reactions (x is the mass of BDDE added in g). In the first stage (Stage I, pre-film casting) BDDE is reacted with the –COOH groups. Excess BDDE remains after Stage I and subsequently reacts during elastomer film formation (Stage II, post-film casting). This method dramatically changes the mechanical properties of the CS nanoparticle films, transforming them from dissipative to predominantly elastic films. All the films in the study are viscoelastic materials. We show that the extent of elastic behaviour is highly tunable via the BDDE concentration used. We separately investigate the contributions from Stage I and Stage II reactions on the final film mechanical properties by isolating the contributions from each stage. We demonstrate that the greatest change in mechanical and elastic properties of the films occurs when both Stage I and II reactions occur. This study provides a new and scalable water-based method to tune the mechanical properties of nanoparticle-based elastomer films.
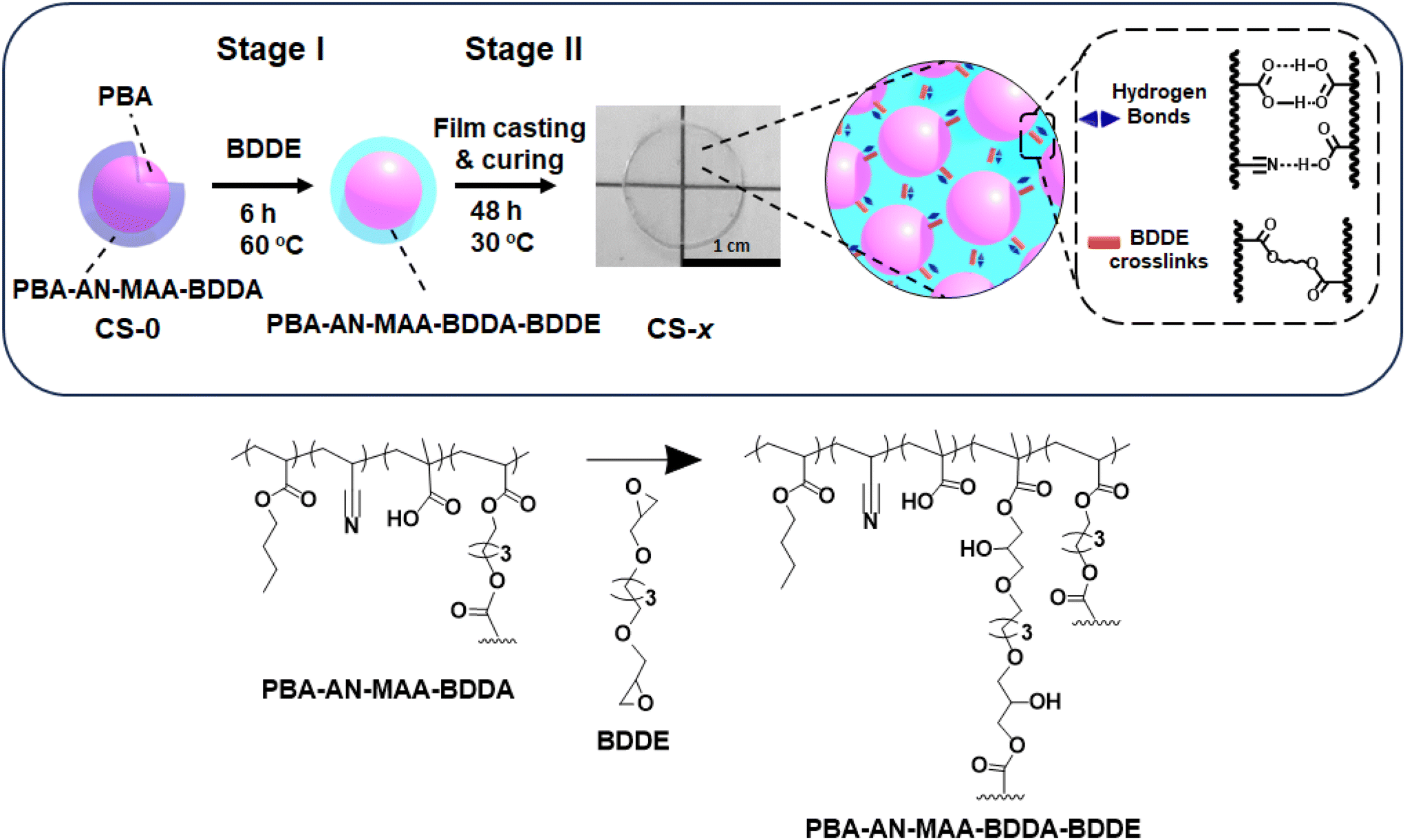 | ||
| Scheme 1 Depiction of the formation of CS-x films (x is the mass of BDDE added in g) after the first pre-film casting reaction with BDDE (Stage I) followed by the post-casting reaction during film formation (Stage II). Stages I and II indicate the two stages of reaction of BDDE with the –COOH functional groups of the particles. BA, AN, MAA and BDDA are n-butylacrylate, acrylonitrile, methacrylic acid and 1,4-butanediol diacrylate, respectively. BDDE is 1,4-butanediol diglycidyl ether. The reaction of some of the –COOH groups in the PBA–AN–MAA–BDDA shell with BDDE gives additional covalent crosslinking (depicted). | ||
Results and discussion
Core–shell nanoparticle reaction with BDDE during the pre-film casting stage (Stage I)
The as-made parent CS nanoparticles (denoted as CS-0) were prepared by semi-batch emulsion polymerization (Scheme S1†) as described in the Experimental section using a method inspired by a microgel study.50 The growth of the CS-0 nanoparticles was monitored by dynamic light scattering (Fig. S1a†) and gravimetric measurements. The constant number of particles (NP, Fig. S1b†) showed that there was no secondary nucleation (see Additional Note 1†). We reacted the CS-0 nanoparticles with increasing masses of BDDE in the dispersion as depicted in Scheme 1 (Stage I) to prepare CS-x nanoparticles. The reaction of the –COOH groups with BDDE is necessarily intra-particle in nature at this pre-film casting stage (i.e., Stage I, Scheme 1). The number-average diameters of the nanoparticles (106–109 nm) are not affected by addition of BDDE (Fig. 1a–d) and the nanoparticles are spherical with a standard deviation of <15 nm (Fig. S2 and Table S1†). The nanoparticle dispersions were analyzed by potentiometric titration (Fig. S3†) which gave the MAA concentration (wt%) and pKa value of each sample (Fig. 1e and Table S1†). As the mass of BDDE (x) added during the reaction increases, more epoxide reacted with the –COOH groups, reducing the MAA concentration in the nanoparticles. The pKa of the nanoparticles increased as the MAA concentration decreased in line with earlier reports for related nanoparticles.51 From the titration data, the percentage of –COOH groups that reacted with BDDE during Stage I was calculated (Fig. S4†). This value increased from 8.5% for the CS-0.25 system to 21.3% for the CS-1 system. Consequently, there was unreacted “free” BDDE present after Stage I. We show below that this free BDDE subsequently reacted during film formation (Stage II in Scheme 1). Because there are two stages of BDDE reaction, we term films prepared using both the pre-film casting and post-film casting reactions as Stage I + II films. The CS-x nanoparticles are pH responsive as shown in Fig. 1f. Whilst the dz value for the initial CS-0 sample increased from 100 to 125 nm as the pH increased from 4 to 10, the reaction of BDDE with the nanoparticles decreased the pH response. Indeed, the nanoparticles with the highest proportion of –COOH reacted (i.e., CS-1) only swelled to 107 nm at pH 10. The decrease in swelling with increasing x at pH 10 is shown in Fig. 1g. The data for pH 10 and pH 4 approach convergence as x increases. This decrease in swelling is due to the BDDE reacting with the RCOOH and is a key indicator of the crosslinking reaction, depicted in Scheme 1. Furthermore, the nanoparticles become more negatively charged with the increase in the pH due to –COO− formation, with the zeta potential increasing to ∼ −40 mV (Fig. S5†) at pH 10.0 compared to ∼ −20 mV at pH 4.0. Taken together, these data confirm that –COOH groups remain for all the CS-x systems after the Stage I reaction.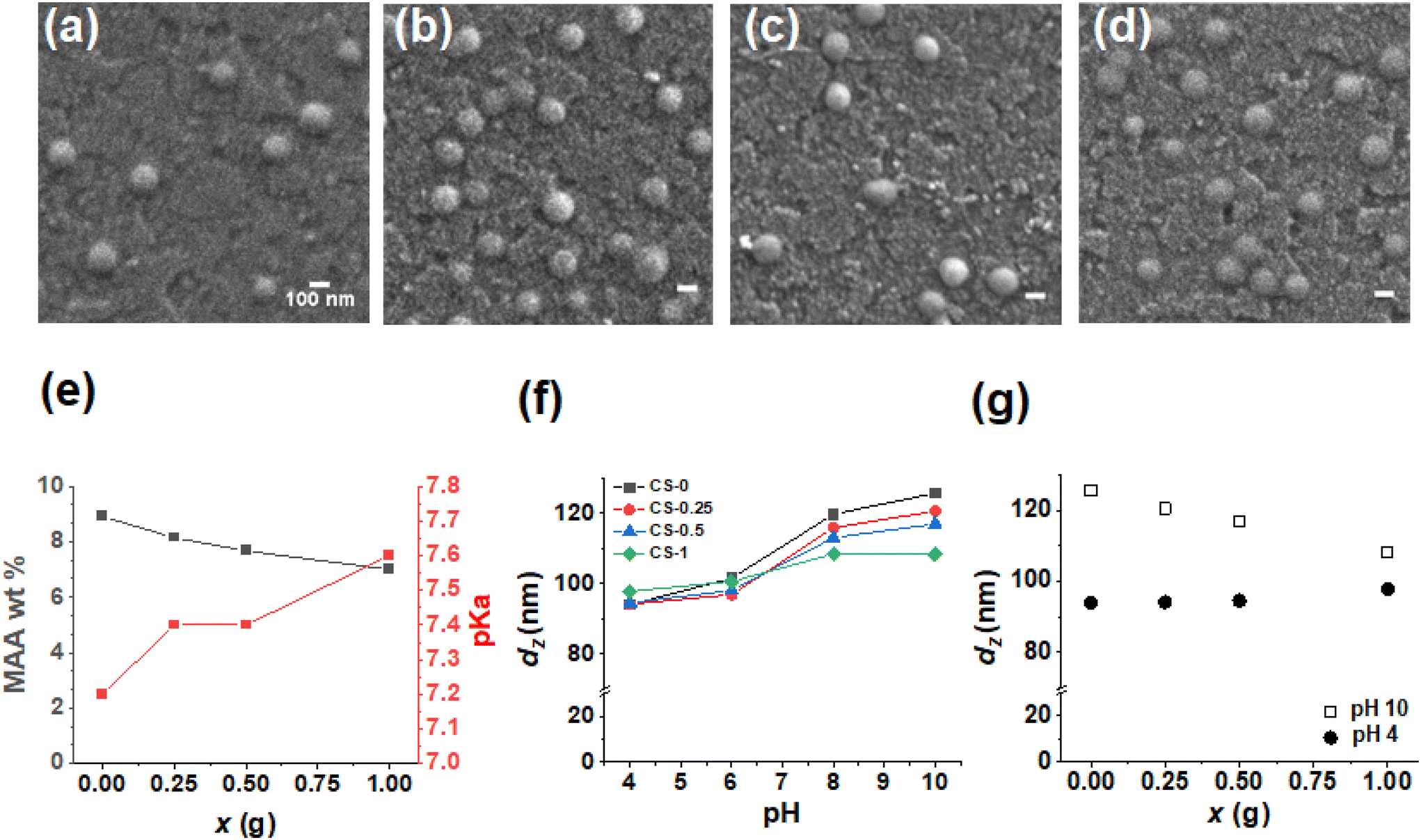 | ||
| Fig. 1 SEM images of (a) CS-0, (b) CS-0.25, (c) CS-0.5 and (d) CS-1 nanoparticles deposited from water. Scale bars: 100 nm. (e) Concentration of MAA wt% in the CS-x nanoparticles and their pKa values. (f) Measured z-average diameters of the nanoparticles at different pH values. (g) Data from (f) at pH 4 and pH 10 plotted as a function of x. | ||
Analysis of Stage I + II films cast from dispersions containing reacted and free BDDE
Stage I + II films were prepared by casting CS-x dispersions at 30 °C for 48 h. The dispersions used contained CS-x nanoparticles that had partially reacted with BDDE, as well as free BDDE in the continuous phase (Scheme 1). This method resulted in thin transparent films (Fig. 2a). The films prepared in this way contained less than 1 wt% residual water as judged by thermogravimetric analysis for a CS-0.5 film (Fig. S6†). To investigate the effect the crosslinking had on the film surface topology, the films were studied using AFM (Fig. S7†). Individual nanoparticles can be discerned for CS-0. However, irregularly shaped features are seen on the surface of the CS-x films which indicates that extensive coalescence of nanoparticles occurred as the films formed, which is due to their lower glass transition temperature (Tg) values (discussed below).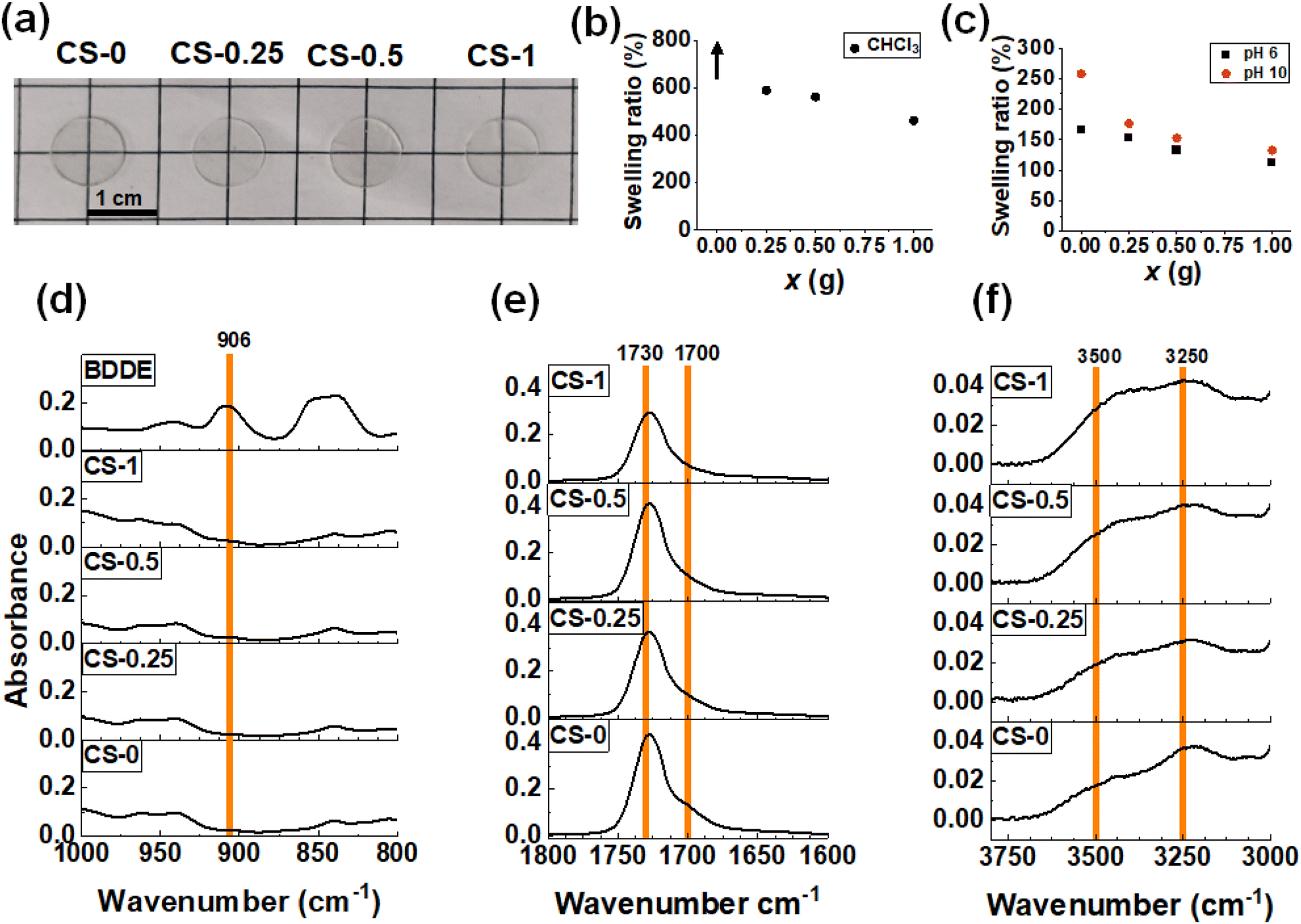 | ||
Fig. 2 (a) Photographs of each of the Stage I + II films (thickness = 0.5 mm). (b) The swelling ratios of the films in CHCl3 after 5 days. The arrow shows that the CS-0 film, which swelled considerably, became too brittle to measure. (c) Swelling ratio of the films in water at pH 6 and 10. Expanded views of the FTIR spectra for the (d) epoxide region, (e) C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O region and (f) –OH region (see the text). O region and (f) –OH region (see the text). | ||
Based on the DLS data (Fig. S1†), the core and shell of the CS-0 nanoparticles occupy ∼34% and 66% of the particle volume, respectively. We envisage the morphology depicted in the top right of Scheme 1 wherein soft PBA cores are dispersed within a continuous phase of inter-meshed (coalesced) nanoparticle shells. Accordingly, the mechanical properties of the CS-x films result directly from those of the constituent nanoparticles. We have shown previously48 that the mechanical properties of this general type of core–shell film can be described by the isostrain model52 for biphasic gels. In the present case, a soft PBA core is dispersed within a relatively hard PBA–AN–MAA–BDDA–BDDE (shell) matrix. In such a model, the shear modulus (G) for the CS-x film is the volume fraction weighted sum of that from the core and shell parts of the constituent nanoparticles. Because of the dominance of the shell volume fraction and their likely relatively high G values, we assume that the G values for the present systems are dominated by that of the nanoparticle shells.
We conducted swelling tests in CHCl3 (Fig. S8a†) as well as in water at pH 6 and pH 10 to probe crosslinking changes in the CS-x films.53 Unlike the CS-0 film which swelled to the point of dissolution in CHCl3, the CS-x films exhibited good tolerance to CHCl3, as evidenced by the finite mass swelling ratios (Fig. 2b). As x increased, the swelling ratio of the films decreased, indicating that crosslinking occurred between the polymer chains due to the reaction with BDDE. The films were also immersed in water at pH 6 (Fig. S8b†) and pH 10 (Fig. S8c†). The swelling ratios for the CS-x films (Fig. 2c) decreased as x increased, again indicating that higher crosslinking occurred at higher x values. Lower –COOH contents in the higher x films likely also contributed to less swelling. The swelling ratios are higher in pH 10 water than in pH 6 water which shows that unreacted –COO− groups drive the swelling of these films at pH 10. These data are consistent with the DLS data (Fig. 1f) and support our view that the Stage I + II CS-x films contained both –COOH groups and covalent crosslinks due to the reaction of some of the original –COOH groups with BDDE.
FTIR spectra for each film were measured (Fig. S9a†). The BDDE spectrum has a peak at 906 cm−1 due to the epoxide54–56 which is not present in the spectra for the CS-x films (Fig. 2d). Scrutiny of these signals in the epoxide region (Fig. 2d) confirms that the epoxy has reacted after CS-x film formation. Thus, by the end of Stage II there is no detectable unreacted BDDE. As x is increased in the CS-x films and –COOH reacts with the epoxide, changes are seen in the CO region of the FTIR spectra (Fig. 2e). As the amount of –COOH that reacts increases with x, using up hydrogen bonding sites, the shoulder peak at 1700 cm−1 (which is due to –COOH) decreases (Fig. 2e). This is shown in Fig. S9b† where the absorbance ratio of the 1700 cm−1 shoulder to the sum of the 1700 cm−1 shoulder and the 1730 cm−1 ester peak57 is plotted. Additionally, as the –COOH group reacts and opens the epoxy ring (Scheme 1), a shifting peak is identified in the –OH spectral region (Fig. S9a†) which is expanded in Fig. 2f. The CS-0 film has a broad –OH peak at 3250 cm−1, whereas upon reaction of –COOH with the epoxide, a new –OH bond58 is formed at 3500 cm−1, which grows as x increases and is plotted in Fig. S9c† as a function of x. Consequently, the FTIR spectra show that reaction between the –COOH groups and epoxide groups occurred. Hence, less hydrogen bonding occurred in the films as x increased due to the ring-opening reaction of BDDE with –COOH and covalent bond formation (Scheme 1).
Mechanical properties of the Stage I + II films
To examine the mechanical properties of the CS-x films as a function of temperature, DMA data were measured to obtain the storage (E′) and loss (E′′) modulus and tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ = (E′′/E′) values. The E′ values show strong temperature dependencies (Fig. 3a) and decrease by several orders of magnitude as the temperature increases from −70 °C. This is due to the glass transition temperature (Tg) of the PBA core and, subsequently, the PBA–AN–MAA–BDDA shells of the nanoparticles being exceeded as the temperature increases. Moreover, there is a strong decrease in E′ between 0 and 25 °C with increasing x. Hence, the reaction with BDDE strongly decreases film stiffness at room temperature. Furthermore, as the temperature increases above 100 °C, the E′ values show increasingly plateau-like behaviour which is most pronounced for the Cs-x films with x ≥ 0.5. Such higher temperature E′ plateaus are indicative of elastic behaviour.
δ = (E′′/E′) values. The E′ values show strong temperature dependencies (Fig. 3a) and decrease by several orders of magnitude as the temperature increases from −70 °C. This is due to the glass transition temperature (Tg) of the PBA core and, subsequently, the PBA–AN–MAA–BDDA shells of the nanoparticles being exceeded as the temperature increases. Moreover, there is a strong decrease in E′ between 0 and 25 °C with increasing x. Hence, the reaction with BDDE strongly decreases film stiffness at room temperature. Furthermore, as the temperature increases above 100 °C, the E′ values show increasingly plateau-like behaviour which is most pronounced for the Cs-x films with x ≥ 0.5. Such higher temperature E′ plateaus are indicative of elastic behaviour.
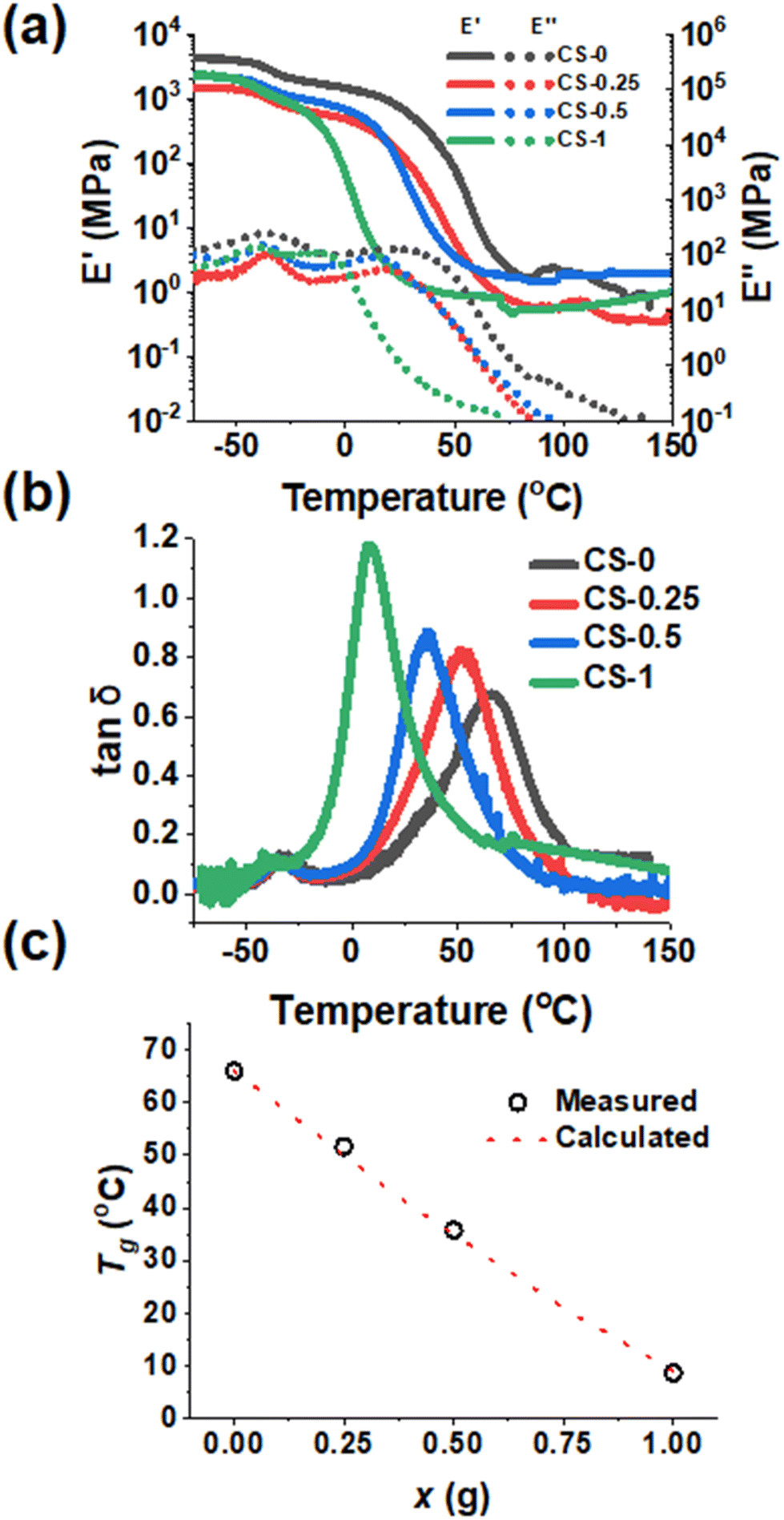 | ||
| Fig. 3 (a) Storage modulus (E′) and loss modulus (E′′) from representative DMA measurements versus temperature. (b) Variation of representative tanδ (=E′′/E′) as a function of temperature for the films. (c) Glass transition temperature (Tg) of each film sample measured using the main peak of the tanδ versus temperature data shown in (b) compared to the calculated Tg using the Fox equation. | ||
The peak of the tanδ versus temperature data (Fig. 3b) is indicative of the Tg.59,60Fig. 3b shows two peaks that correspond to Tg values. The lower temperature peak at ∼ −40 °C is from the PBA core of the CS-x nanoparticles.37,60 This peak is consistent with soft PBA nanoparticle cores dispersed within a harder inter-meshed shell matrix as depicted in Scheme 1. The core Tg did not change as x increased, showing that shell crosslinking does not affect chain mobility in the core. The tanδ peak which occurs in a temperature range from 0 °C to 70 °C is attributed to the shell of the nanoparticles.37 An increase in x caused a decrease in these Tg values (Fig. 3c). We applied the Fox equation for predicting poly(A-co-B) copolymer glass transition temperature values (equation (S2), Additional Note 2†) to our present systems.61 The results from the Fox equation are shown in Fig. 3c. The agreement between the measured and calculated Tg values for the x = 0.25 and 0.5 systems is remarkably good. It follows that the reaction with BDDE (which governs the Tg) occurs uniformly throughout the shells of these core–shell nanoparticles and that such copolymer shells form a well-mixed continuous phase within the final nanoparticle films.
The addition of a crosslinker would normally be expected to increase the Tg of an elastomer62 and so the present trend from Fig. 3c appears counterintuitive at first sight. However, as BDDE reacted, the amount of –COOH available for inter-chain hydrogen bonding in the film decreased. This decrease of inter-chain hydrogen bonding (and dynamic crosslinks) increased chain mobility. Whilst covalent crosslinks were produced by the BDDE reaction, their effect on the Tg appears to have been overwhelmed by the decrease in hydrogen bonding between –COOH groups, decreasing the Tg.
The CS-x films underwent tensile testing and show distinctive changes as x is increased (Fig. 4a). (Tensile data for all systems appear in Table S2.†) When x = 0, there was initial elastic deformation followed by plastic deformation as the modulus drops beyond the first 20% strain. As x increased, the effect of plastic deformation dramatically decreased, leaving elastic deformation as the dominating effect. We assume that hydrogen bonds involving –COOH are responsible for the plastic deformation. The shear modulus of the films (Fig. 4b) falls from 9.0 MPa when x = 0 to 1.1, 0.40 and 0.20 MPa for the CS-0.25, CS-0.5 and CS-1 films, respectively. The stress-at-break decreased strongly with the increase in x (Fig. 4c), while the strain-at-break (Fig. 4d) remained high (>300%) for the CS-x films when x = 0, 0.25 and 0.5, before decreasing to a respectable value of 142% for the CS-1 film. For these CS-x nanoparticle elastomer-like films, the modulus is considered to reflect intra-particle crosslinking, and the strain-at-break reflects inter-particle crosslinking.48 The decrease in strain-at-break for the CS-1 system (Fig. 4d) is attributed to a major change from dissipative hydrogen bonding to non-dissipative covalent inter-particle crosslinking which can accommodate less fracture energy.63–65 In contrast, the CS-x systems with x ≤ 0.5 had more –COOH groups that could dissipate strain via dynamic hydrogen bonds and, therefore, retained higher strain-at-break values.
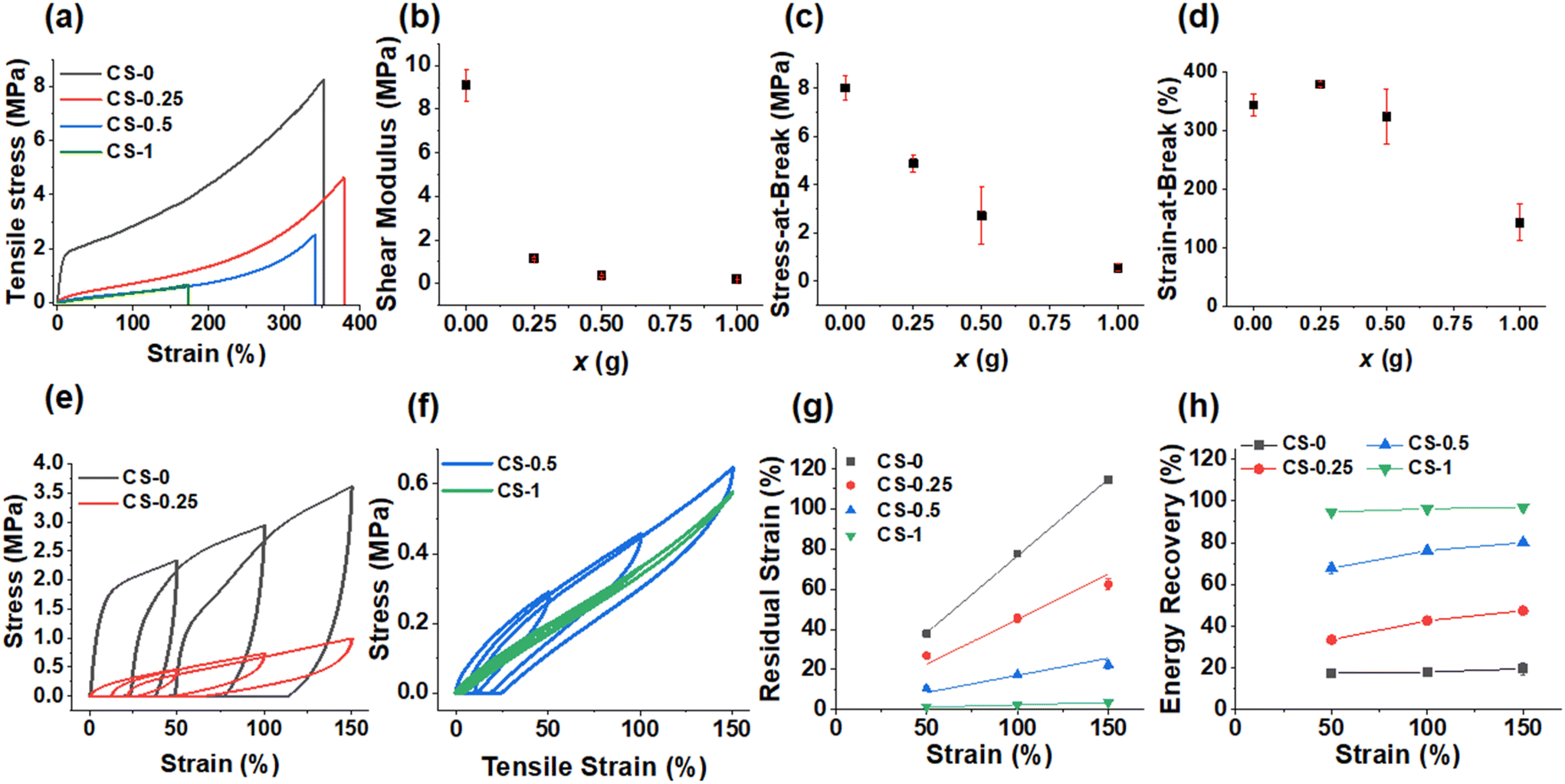 | ||
| Fig. 4 (a) Representative tensile stress/strain data for the Stage I + II films. The (b) shear modulus, (c) stress-at-break and (d) strain-at-break are shown for each system. Representative cyclic tensile data are shown for (e) CS-0 and CS-0.25 as well as (f) CS-0.5 and CS-1.0. The (g) residual strain values and (h) energy recovery values measured at each final tensile strain from (e) and (f) are shown. | ||
Cyclic tensile measurements were performed to provide greater insight into the CS-x film mechanical properties (Fig. 4e and f). For CS-0, the steep increase in tensile stress corresponds to the initial elastic deformation which then yields with plastic deformation. This yielding caused the material to deform irreversibly, and thus, not return to 0% strain when the stress was removed. When x > 0, the amount of irreversible deformation for the CS-x films decreased. The residual strain values measured from Fig. 4e and f are plotted in Fig. 4g and increase linearly with tensile strain. The residual strain values at 150% dramatically decrease from ∼115% to less than 5% as x increases from 0 to 1.0 revealing greatly increased elasticity.
When comparing the energy recovery data (calculated from the area ratio of the reverse to forward stress vs. strain sweeps in Fig. 4e and f), the energy recovery at all applied strains (Fig. 4h) increased from ∼20% for CS-0 to 97% for CS-1. The trends shown in Fig. 4g and h demonstrate a major change from sacrificial –COOH-based crosslinking to covalent (permanent) crosslinking for the CS-x films when x increases from 0 to 1.0. Hence, the reactions of the nanoparticles with BDDE transform the dissipative predominantly viscoelastic CS-0 film into a predominantly elastic CS-1 film. Whilst these films are all viscoelastic materials, the contribution of the viscous component to the overall mechanical properties decreases with increasing x. The CS-0.25 and CS-0.5 films contain mixtures of dynamic crosslinks (from –COOH) and covalent crosslinks (from –COOH plus di-epoxide reaction). In particular, the CS-0.5 system is noteworthy because it has a high modulus (0.40 MPa), high strain energy recovery (80%) and high strain-at-break (323%).
We inferred above from the titration data (Fig. S3†) and the formulations used to prepare these films that (a) free BDDE was present after the Stage I reaction (Scheme 1) and (b) from the FTIR data (Fig. 2b) that free BDDE was consumed during Stage II. A key question concerns the importance of the BDDE reactions during Stage I and Stage II for the dramatic (and tuneable) mechanical property changes observed in Fig. 3 and 4. We address this question in the next section.
The Stage I reaction occurred in the dispersed state and was conducted at 60 °C for 6 h. The Stage II reaction was conducted at 30 °C for 48 h and also began in the dispersed state. We propose that the reactions that occurred in Stages I and II were broadly similar at the molecular level. If the Arrhenius rule of thumb that the reaction rate doubles when the temperature increases by 10 °C is applied to the present systems, it follows that the Stage I and Stage II reactions used are equivalent in terms of the extent of reaction. (Stage II may have occurred at a rate that was eight times slower but the duration was eight times longer than Stage I.) However, the transformation of a dispersion into a dry film of coalesced particles that is unique to Stage II would likely affect the reaction kinetics in that stage. It is plausible that a significant part of the BDDE reaction occurred after extensive nanoparticle coalescence had taken place.
Mechanical properties of Stage I films cast from dispersions without free BDDE
To investigate the effect of the Stage I reaction, a CS-1 dispersion was dialyzed against water for 5 days prior to film formation (depicted in Fig. 5a) to remove unreacted (free) BDDE. These Stage I films are denoted CS-1-D. Notably, the DMA data (Fig. 5b) show that the peak of CS-1-D has a much higher Tg (57 °C) than measured for the (Stage I + II) CS-1 film (9 °C, Fig. 3c). The Tg is also 10 °C lower than that for the CS-0 film (67 °C, Fig. 3c). The tensile data (Fig. 5c) show that the CS-1-D film has less plastic deformation than the CS-0 film but more than the CS-1 film. The modulus of CS-1-D (5.0 MPa) is much lower than that of the CS-0 system (9.0 MPa) but much higher than that of CS-1 (0.20 MPa) (Fig. 5d). The stress-at-break values (Fig. 5e) for the CS-1-D film is in between those of the CS-0 and CS-1 films. As it was established above that the BDDE reaction decreases the Tg, modulus and stress-at-break, the data for CS-1-D reveal that a significant reaction of BDDE occurred during Stage I of the CS-1 film, prepared using the Stage I + II method. Furthermore, if the Stage I reaction (Fig. 5a) was exclusively intra-particle in nature, then one would expect that the strain-at-break, which is dominated by inter-particle crosslinks, would be unaffected. This suggestion is confirmed by the strain-at-break value for CS-1-D of 318% which is very close to that for CS-0 (330%, Fig. 5f).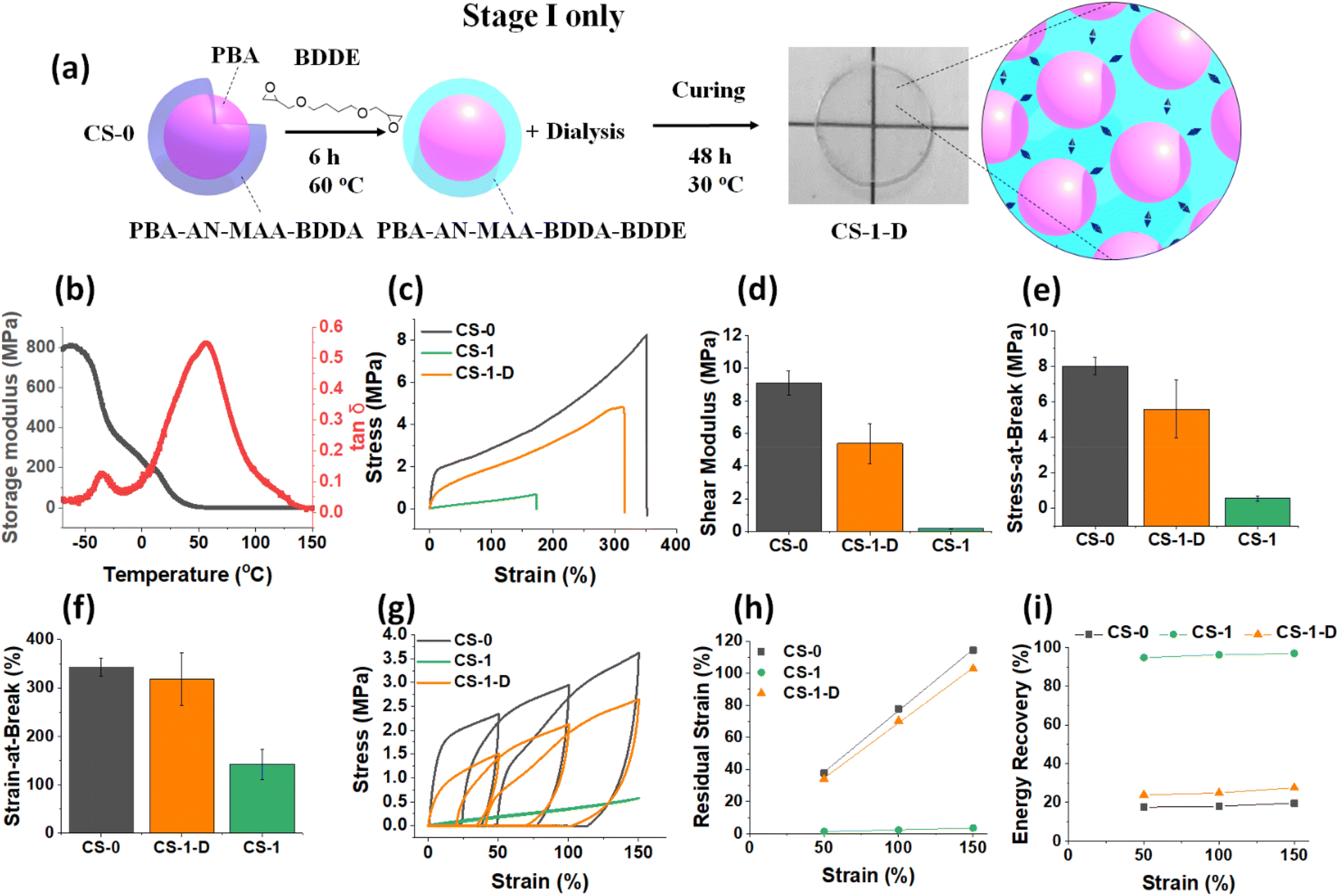 | ||
| Fig. 5 (a) Depiction of the formation of CS-1-D films where the CS-1 dispersion is dialyzed to remove residual BDDE prior to film formation. The CS-1-D films have only the Stage I reaction with BDDE. (b) Variation of E′ and tanδ with temperature measured for the CS-1-D film. (c) Representative stress–strain curve of CS-1-D compared to CS-0 and (Stage I + II) CS-1 data. The (d) shear modulus, (e) stress-at-break and (f) strain-at-break values are shown. (g) Representative cyclic tensile data are shown for the CS-1-D film and compared to those from the CS-0 and CS-1 films. The (h) residual strain values and (i) energy recovery values measured at each final tensile strain from (f) are shown. | ||
Cyclic tensile data for the CS-1-D film also show a plastic yield point and the data appear closer to CS-0 than to CS-1 in behavior (Fig. 5g). Notably, the residual strain (Fig. 5h) and energy recovery (Fig. 5i) at each strain measured are close to the values observed for the CS-0 film. These data confirm the dissipative nature of the CS-1-D films which is due to the dominance of reversible –COOH crosslinks in the nanoparticle shells. It follows from the Stage I CS-1-D film data discussed here that for the CS-1 film prepared using the Stage I + II method, free BDDE and the Stage II reaction are vital to fully develop the dissipative-to-elastic transition achieved for the CS-x films.
Mechanical properties of Stage II films cast from dispersions containing added BDDE
An interesting question arising from the Stage I CS-1-D film data discussed above is whether simply using a Stage II reaction only can achieve the pronounced mechanical property changes observed for the CS-x (x = 0.25, 0.5, 1.0) films shown in Fig. 3 and 4. Accordingly, we studied films prepared using the Stage II process only. These films were formed by addition of BDDE to a CS-0 dispersion without a Stage I reaction (Fig. 6a). This system is denoted as CS-0 + BDDE. The masses of BDDE and CS-0 nanoparticles used were the same as those used to prepare the CS-1 film using the Stage I + II process (Scheme 1). From the DMA data for the CS-0 + BDDE film (Fig. 6b), the Tg of the film is 41 °C. This is much higher than that for the CS-1 film (9 °C). The Tg is also considerably lower than that for CS-0 (67 °C, Fig. 3c). These data show that a substantial proportion of the –COOH groups reacted with BDDE during the Stage II process.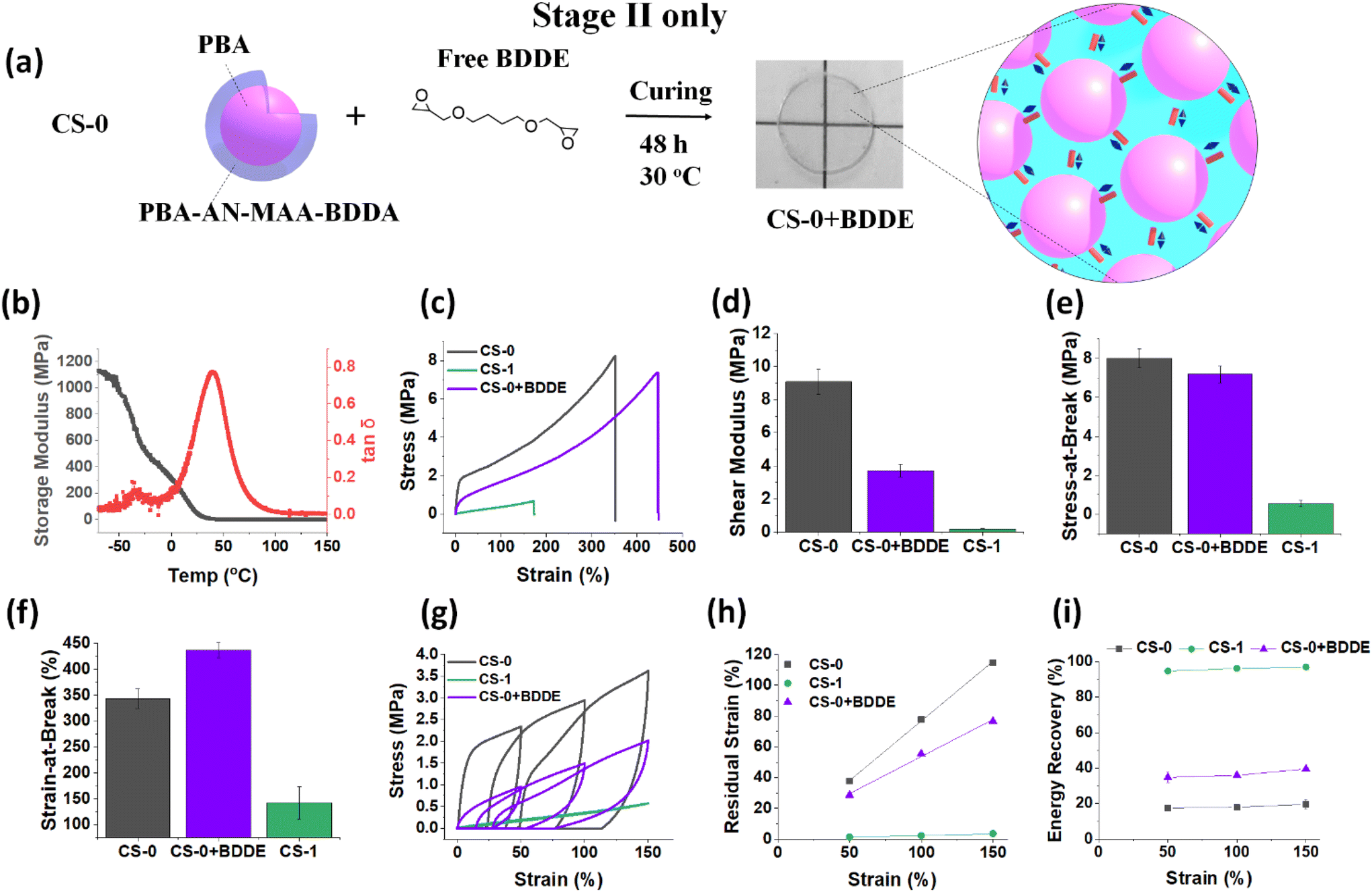 | ||
| Fig. 6 (a) Depiction of the formation of CS-0 + BDDE films where the CS-0 dispersion is mixed with free BDDE prior to film formation. The CS-0 + BDDE films were prepared using only the Stage II reaction with BDDE. (b) Variation of E′ and tanδ with temperature measured for the Stage I CS-0 + BDDE film. (c) Representative stress–strain curve of CS-0 + BDDE compared to CS-0 and CS-1 data (Stage I + II). The (d) shear modulus, (e) stress-at-break and (f) strain-at-break values are shown. (g) Representative cyclic tensile data are shown for the CS-0 + BDDE film and compared to those from the CS-0 and CS-1 films. The (h) residual strain and (i) energy recovery measured at each strain are also shown. | ||
Tensile data comparing CS-0 + BDDE with CS-0 and CS-1 are shown in Fig. 6c. The plastic deformation seen for the CS-0 film is present for the CS-0 + BDDE film. Furthermore, the CS-0 + BDDE modulus (Fig. 6d) is in between those of CS-1 and CS-0 while the stress-at-break (Fig. 6e) value is closer to that of CS-0 than that of CS-1. Interestingly, the strain-at-break (Fig. 6f) value has increased greatly. The latter result implies that the Stage II reaction enables improved energy dissipation. Hence, the Stage II only process results in permanent crosslinks (from BDDE) as well as H-bonding crosslinks (from residual COOH) at an optimum ratio to provide increased stretchability.
Cyclic tensile data were also obtained (Fig. 6g). The residual strain (Fig. 6h) for CS-0 + BDDE is significantly higher than that for the CS-1 film at all the applied strains, while the energy recovery (∼35%) is much lower than that of CS-1 (Fig. 6i). Importantly, these data show that using only the Stage II reaction does not provide the complete change from dissipative to predominantly elastic mechanical properties observed for the Stage I + II CS-1 film as evidenced by the comparison in Fig. 6h and i. Hence, it is only by using the complete Stage I + II process (two-stage BDDE reaction, Scheme 1) that the nanoparticle films can fully transition from a dissipative CS-0 film to a predominantly elastic CS-1 film with almost 100% energy recovery. There appears to be a synergistic process requiring both Stage I and II processes for complete mechanical property transformation.
We note that the discussion above assumed that BDDE was bifunctional and formed crosslinks between –COOH groups. This assumption was tested by replacing BDDE with the monoepoxide species butyl glycidyl ether (BGE), as discussed in Additional Note 3 and depicted in Scheme S2.† BGE was reacted with the CS-0 dispersion using the Stage I + II method. This system is denoted as CS-0-BGE. Variable pH DLS data and titration data are presented in Fig. S10† whilst SEM data are shown in Fig. S11.† The CS-0-BGE films became brittle and fragmented when placed in CHCl3 (Fig. S12†), implying little, if any, permanent crosslinking. The Tg determined from DMA data (Fig. S13a†) was 44 °C. Tensile data are shown in Fig. S13.† The modulus and stress-at-break values were both lower than those for CS-0 (Table S2†). The cyclic tensile data (Fig. S13f†) showed strong dissipative behaviour with relatively high residual strain (Fig. S13g†), poor energy recovery (28%, Fig. S13h†) and no evidence of significant elasticity. These dissipative behaviours are expected for a ring-opening reaction of CS-0 with a mono-functional epoxide (BGE) because no covalent crosslinking should have been added. The data contrasted with CS-0.5 and CS-1 which were both much more elastic (80% and 97% energy recovery respectively). This comparison confirms that BDDE reacted as a bifunctional crosslinker. It is noted that the modulus, strain-at-break and stress-at-break values for the CS-0-BGE system are all higher than the values for the CS-0.5 and CS-1 films. As discussed in Additional Note 3,† we speculate that these differences originate from the spatial constraints of a difunctional monomer reaction. Briefly, the two epoxy groups of BDDE must react in the same location, whereas BGE is not constrained in this way. Accordingly, we propose that more inter-chain hydrogen bond pairs can occur for the BGE films compared to the BDDE films.
Conclusions
We have introduced a facile new water-based method for the more environmentally friendly synthesis of CS nanoparticle films with highly tuneable mechanical properties. The films can be changed from a dissipative, high modulus system (CS-0, 9.0 MPa, 20% energy recovery) to a lower-modulus, elastic system (CS-1, 0.20 MPa, 97% energy recovery) simply using low-temperature di-epoxide crosslinking. Intermediate compositions contain a tuneable mixture of dynamic and covalent bonding. The CS-0.5 film suggests to be the most improved elastic material with a modulus of 0.40 MPa and energy recovery of 80% while it retains an impressive strain-at-break of more than 300%. This value can be increased to greater than 450% using a monofunctional epoxide (BGE). This tuneability comes about from the decrease of dissipative –COOH crosslinks, decrease in the Tg, and introduction of covalent crosslinks. The CS-0.5 film is a softer and more elastic material with decreased water and CHCl3 swelling compared to the CS-0 films. CS-0.5 may have potential utility where moderate stiffness, high stretchability, energy recovery and/or solvent resistance are required. These may include washers, gaskets, sporting equipment, mats, protective clothing, soft actuators and surface coatings.The reaction between the –COOH groups and BDDE is shown to occur by crosslinking in two stages which is tuneable by the mass of BDDE used or when the BDDE is reacted (in Stage I or Stage II or both). While higher modulus, high strain-at-break films with some elastic behaviours are achievable by just Stage II, it was revealed that the combination of both Stage I + II provides greater mechanical property changes than Stage I or Stage II individually with greater elasticity. Not only does this novel approach provide highly tuneable mechanical properties, but it is also water-based which may reduce the negative environmental impact of elastomers. The simple approach used to covalently crosslink –COOH-containing nanoparticle films demonstrated here in the case of nanoparticles made of a PBA core and a poly(BA-co-AN-co-MAA) shell could, in principle, be applied to other water-based nanoparticle systems with accessible carboxylated functional groups, such as carboxylated NBR latex for example.66
Author contributions
M. O-R.: Investigation; visualization; writing – original draft. D. R.: Conceptualization; supervision. X. W.: Investigation. S. W.: Resources. S. E.: Validation; writing – review & editing. B. R. S.: Conceptualization; data curation; project administration; supervision; writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. O-R. and B. R. S. gratefully acknowledge the financial support from Synthomer for this research.References
- R. J. Young and P. A. Lovell, Introduction to Polymers, Francis Group, 2011 Search PubMed.
- L. McKeen, in Effect of Temperature and other Factors on Plastics and Elastomers, Elsevier, 2008, ch. 5 Search PubMed.
- J. R. Halladay and R. J. D. Vecchio, Bonding of Elastomers, De Gruyter, Berlin, 2020 Search PubMed.
- S. Song, H. Hou, J. Wang, P. Rao and Y. Zhang, J. Mater. Chem. A, 2021, 9, 3931 CAS.
- C. Lou, E. Liu, T. Cheng, J. Li, H. Song, G. Fan, L. Huang, B. Dong and X. Liu, ACS Omega, 2022, 7, 5825 CrossRef CAS PubMed.
- Z. Zhang, N. Ghezawi, B. Li, S. Ge, S. Zhao, T. Saito, D. Hun and P. Cao, Adv. Funct. Mater., 2021, 31, 2006298 CrossRef CAS.
- Y. Zhu, Q. Shen, L. Wei, X. Fu, C. Huang, Y. Zhu, L. Zhao, G. Huang and J. Wu, ACS Appl. Mater. Interfaces, 2019, 11, 29373 CrossRef CAS.
- X. Zhu, K. Han, C. Li, J. Wang, J. Yuan, Z. Pan and M. Pan, ACS Appl. Mater. Interfaces, 2023, 15, 19414 CrossRef CAS.
- Y. Xu, S. Zhou, Z. Wu, X. Yang, N. Li, Z. Qin and T. Jiao, J. Chem. Eng., 2023, 446, 143179 Search PubMed.
- A. Hanif, T. Q. Trung, S. Siddiqui, P. T. Toi and N. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 27297 CAS.
- M. Bobnar, N. Derets, S. Umerova, V. Domenici, N. Novak, M. Lavric, G. Cordoyiannis, B. Zalar and A. Resetic, Nat. Commun., 2023, 14, 764 CAS.
- Z. Pei, Y. Yang, Q. Chen, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Mater., 2013, 13, 36 Search PubMed.
- Z. Xie, B. Hu, R. Li and Q. Zhang, ACS Omega, 2021, 6, 9319 CAS.
- S. Zhang, K. Liu, T. Wu, M. Xu and S. Shen, J. Phys. Chem. C, 2020, 124, 24429–24434 CAS.
- M. J. Gonzalez-Alvarez, J. Paternoga, K. Breul, H. Cho, M. Z. Roshandel, M. Soleimani and M. A. Winnik, Polym. Chem., 2017, 8, 2931 RSC.
- M. Argaiz, F. Ruiperez, M. Aguirre and R. Tomovska, Polymers, 2021, 13, 3098 CrossRef CAS.
- R. S. Gurney, A. Morse, E. Siband, D. Dupin, S. P. Armes and J. L. Keddie, J. Colloid Interface Sci., 2015, 448, 8 CAS.
- C. Guizard, B. Boutevin, F. Guida, A. Ratsimihety, P. Amblard, J. C. Lasserre and S. Naiglin, Sep. Purif. Technol., 2001, 22–23, 23 Search PubMed.
- S. Debnath, S. Kaushal and U. Ojha, ACS Appl. Polym. Mater., 2020, 2, 1006 CAS.
- H. Ye, K. Zhang, D. Kai, Z. Li and X. Loh, Chem. Soc. Rev., 2018, 47, 4545 CAS.
- S. Kim, H. Jeon, S. Shin, S. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 Search PubMed.
- Y. Cai, L. Yan, Y. Wang, Y. Ge, M. Liang, Y. Chen, H. Zou and S. Zhou, J. Chem. Eng., 2022, 436, 135156 CrossRef CAS.
- R. Yang, Y. Yao, Z. Duan, H. Tai, Y. Jiang, Y. Zheng and D. Wang, Langmuir, 2020, 36, 3029 CrossRef CAS.
- H. Xu, J. Ji, H. Li, J. Tu, Z. Fan, X. Zhang and X. Guo, Chem. Eng. J., 2023, 475, 146018 CrossRef CAS.
- Y. Luo, J. Chen, G. Situ, C. Li, C. Zhang, F. Li, C. Li, Z. Luo and X. Zhang, J. Chem. Eng., 2023, 469, 143958 CrossRef CAS.
- K. Mantala and D. Crespy, Macromolecules, 2023, 56, 7332 CAS.
- M. Guerre, C. Taplan, R. Nicolay, J. M. Winne and F. E. D. Prez, J. Am. Chem. Soc., 2018, 140, 13272–13284 CAS.
- S. K. Raut, S. Sarkar, P. Mondal, A. Meldrum and N. K. Singha, J. Chem. Eng., 2023, 453, 139641 Search PubMed.
- Q. Shi, W. Wu, B. Yu, M. Ren, L. Wu and C. Zhang, RSC Adv., 2022, 12, 35396 CAS.
- H. Abdeldaim, B. Reck, K. J. Roschmann and J. M. Asua, Macromolecules, 2023, 56, 3304 CAS.
- N. Jimenez, N. Ballard and J. M. Asua, Macromolecules, 2019, 52, 9724 CrossRef CAS.
- I. Tiggelman and P. C. Hartmann, Prog. Org. Coat., 2009, 67, 76 CrossRef.
- P. A. Steward, J. Hearn and M. C. Wilkinson, Adv. Colloid Interface Sci., 2000, 86, 195 CrossRef CAS PubMed.
- X. Jiang, J. Li, M. Ding, H. Tan, Q. Ling, Y. Zhong and Q. Fu, Eur. Polym. J., 2007, 43, 1838 CrossRef CAS.
- F. Lossada, D. Jiao, X. Yao and A. Walther, ACS Macro Lett., 2020, 9, 70 CAS.
- Y. Liu, Z. Tang, S. Wu and B. Guo, ACS Macro Lett., 2019, 8, 193 CAS.
- J. Turton, S. Worral, M. S. Musa, A. H. Milani, Y. Yao, P. Shaw, D. Ring and B. R. Saunders, Polym. Chem., 2021, 12, 466 CAS.
- O. Pinprayoon, R. Groves, P. A. Lovell, S. Tungchaiwattana and B. R. Saunders, Soft Matter, 2011, 7, 247 CAS.
- M. D. Besteti, D. M. G. Freire and J. C. Pinto, Macromol. React. Eng., 2011, 5, 518 CrossRef CAS.
- S. Brovellie, R. D. Schaller, S. A. Crooker, F. Garcia-Santamaria, Y. Chen, R. Viswanatha, J. A. Hollingsworth, H. Htoon and V. I. Klimov, Nat. Commun., 2011, 2, 280 CrossRef.
- P. Reiss, M. Protiere and L. Li, Small, 2009, 5, 154–168 CrossRef CAS.
- J. M. Chan, L. Zhang, K. P. Yuet, G. Liao, J. Rhee, R. Langer and O. C. Farokhzad, Biomaterials, 2009, 30, 1627–1634 CrossRef CAS.
- R. G. Chaudhuri and S. Paria, Chem. Rev., 2011, 112, 2373–2433 Search PubMed.
- D. Juhue and J. Lang, Macromolecules, 1995, 28, 1306 CAS.
- E. Limousin, N. Ballard and J. Asua, J. Appl. Polym. Sci., 2019, 136, 47608 Search PubMed.
- R. A. Ramli, W. A. Laftah and S. Hashim, RSC Adv., 2013, 3, 15543 CAS.
- J. Rosenfeld, F. Ganachaud and D. Lee, J. Colloid Interface Sci., 2024, 653, 1753 CAS.
- M. S. Musa, A. H. Milani, P. Shaw, G. Simpson, P. A. Lovell, E. Eaves, N. Hodson and B. R. Saunders, Soft Matter, 2016, 12, 8112 RSC.
- S. Tungchaiwattana, R. Groves, P. A. Lovell, O. Pinprayoon and B. R. Saunders, J. Mater. Chem., 2012, 22, 5840 RSC.
- B. E. Rodriguez, M. S. Wolfe and M. Fryd, Macromolecules, 1994, 27, 6642 CAS.
- O. Pinprayoon, A. Saiani, R. Groves and B. R. Saunders, J. Colloid Interface Sci., 2009, 336, 73 CAS.
- E. R. Morris, Carbohydr. Polym., 1992, 17, 65 Search PubMed.
- G. Lee, H. Y. Song, S. Choi, C. B. Kim, K. Hyun and S. Ahn, Macromolecules, 2022, 55, 10366 CAS.
- Y. Zhang, L. Yuan, G. Liang and A. Gu, ACS Sustainable Chem. Eng., 2023, 11, 9264 CAS.
- Z. Yang, H. Li, Y. Zhong, X. Lai, J. Ding, Z. Chen and X. Zeng, ACS Appl. Mater. Interfaces, 2022, 14, 44878 CAS.
- T. Kimura and M. Hayashi, J. Mater. Chem. A, 2022, 10, 17406 RSC.
- L. Xiao, W. Li, Z. Liu, K. Zhang, S. Li, Y. Wang, J. Chen, J. Huang and X. Nie, ACS Sustainable Chem. Eng., 2022, 10, 9829 CrossRef CAS.
- M. Hayashi and R. Yano, Macromolecules, 2020, 53, 182 CrossRef CAS.
- M. K. Singh and A. Singh, Characterization of Polymers and Fibres, Woodhead Publishing, USA, 2022 Search PubMed.
- J. Brandrup, E. H. Immergut and E. A. Grulke, Polymer Handbook, John Wiley & Son, New York, 4 edn, 1999 Search PubMed.
- W. Brostow, R. Chiu, I. M. Kalogeras and A. Vassilikou-Dova, Mater. Lett., 2008, 62, 3152 CrossRef CAS.
- K. Bandzierz, L. Reuvekamp, J. Dryzek, W. Dierkes, A. Blume and D. Bielinski, Materials, 2016, 9, 607 Search PubMed.
- X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899 CAS.
- J. Slootman, V. Waltz, J. C. Yeh, C. Baumann, R. Göstl, J. Comtet and C. Creton, Phys. Rev. X, 2020, 10, 041045 CAS.
- Y. Yao, B. Liu, Z. Xu, J. Yang and W. Liu, Mater. Horiz., 2021, 8, 2742 CAS.
- G. Zhang, H. Feng, K. Liang, Z. Wang, X. Li, X. Zhou, B. Guo and L. Zhang, Sci. Bull., 2020, 65, 889 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01073f |
| This journal is © The Royal Society of Chemistry 2025 |