
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dual orthogonal metal-complexes and their utilization for the versatile fabrication of smart interpenetrating polymer networks†
Thomas
Bätz
ab,
Michael F.
Agyemang
cde,
Josefine
Meurer
ab,
Julian
Hniopek
 cde,
Stefan
Zechel
ab,
Michael
Schmitt
cd,
Jürgen
Popp
cde,
Martin D.
Hager
abf and
Ulrich S.
Schubert
*abdf
cde,
Stefan
Zechel
ab,
Michael
Schmitt
cd,
Jürgen
Popp
cde,
Martin D.
Hager
abf and
Ulrich S.
Schubert
*abdf
aLaboratory of Organic and Macromolecular Chemistry (IOMC), Friedrich Schiller University Jena, Humboldtstr. 10, 07743 Jena, Germany. E-mail: ulrich.schubert@uni-jena.de
bJena Center of Soft Matter (JCSM), Friedrich Schiller University Jena, Philosophenweg 7, 07743 Jena, Germany
cInstitute of Physical Chemistry (IPC), Friedrich Schiller University Jena, Helmholzweg 4, 07743 Jena, Germany
dAbbe Center of Photonics (ACP), Friedrich Schiller University Jena, Albert-Einstein-Straße 6, 07745 Jena, Germany
eLeibniz Institute of Photonic Technology, e. V. Jena, Albert-Einstein-Straße 9, 07745 Jena, Germany
fHelmholtz Institute for Polymers in Energy Applications Jena (HIPOLE Jena), Lessingstraße 12-14, 07743 Jena, Germany
First published on 6th December 2024
Abstract
This article presents a versatile method for the preparation of smart interpenetrating polymer networks (IPNs). Based on two different orthogonal binding motives, IPNs are generated by simply mixing ligand containing polymers and corresponding metal salts leading to a polymeric material featuring shape-memory abilities. Due to the utilization of heteroleptic complexes as crosslinks in one of the two networks, simple tuning of the crosslinking density, as well as the generation of IPNs made out of three different main polymers is possible. The structure–property-relationships of the resulting IPNs are investigated in a detailed fashion applying FT-Raman spectroscopy, thermogravimetric analysis (TGA), dynamic-mechanical-thermal analyses (DMTA) as well as thermo-mechanical analyses (TMA). In the scope of thermo-mechanical analysis (TMA) excellent shape-memory properties, with strain fixity rates near 100% and strain recovery rates up to 93%, could be observed.
Introduction
Over many decades, polymers have become an integral part of our daily life. Based on their unique and tunable properties they are used in nearly every application area.1–3 However, the typically applied polymers, which are produced to fulfil a single task over their lifetime, are no longer sufficient. Moreover, we are searching for materials, which are able to adapt their function(s) and properties as a result of experiencing an environmental change.4 This triggered adaption is beneficial for certain applications and can open completely new fields. For this reason, the research topic dealing with so-called stimuli-responsive polymers has emerged.5–7 One very promising way to obtain such switchable properties in polymeric materials is the implementation of reversible covalent8 or supramolecular crosslinks.9 A variety of such reversible moieties have already been successfully used to synthesize switchable polymers, featuring abilities such as self-healing10 or shape-memory.11Shape-memory polymers (SMPs) have the unique ability to “remember” a predetermined shape.11 After the fabrication of those polymers in their original, so-called permanent shape, it is possible to convert them, under the respective conditions, into a completely new temporary shape. Thereafter, they keep this new shape until an external trigger leads to the induction of the recovery step, in which the “memorized” permanent shape is restored.11,12 The design of SMPs requires, in minimum, two different building units.11,13 Firstly, a triggerable part, called switching-unit, is important. For this building unit, in general any interaction with a certain degree of reversibility can be utilized. Here for example, thermal transitions (e.g., glass transition14 or transition between crystallization and melting15,16) but of course also reversible covalent17,18 or supramolecular bonds19,20 can be mentioned. The other required part is the stable phase. This unit is important for the structural integrity of SMPs and further provides the driving force for the recovery step. It usually consists of covalent21 or physical crosslinks.22 Furthermore, there are also some recent research approaches, in which this part could be generated via an interpenetrating polymer network (IPN) structure.23,24
Metallopolymers are a special class in the field of polymers, which combine unique properties.25 On the one hand, they have all the advantages of polymers, such as cheap and easy production, low weight, corrosion-resistance and many more.1 But at the same time, the integrated metal-complex, whose binding strength and properties can be easily adjusted by the choice of ligand and/or metal salt(s), provides an ideal opportunity for the design of switchable and intelligent materials.26 There are numerous reports in literature that deal with metallopolymers for a wide variety of stimuli responsive materials for several application fields, e.g., as sensors,27 for drug-delivery28 or self-healing.29,30
In order to fabricate smart materials, which are addressable with multiple stimuli and/or show multiple responses, different dynamic binding motifs, which are orthogonal, need to be integrated.31 This orthogonality can be obtained by the utilization of completely different binding concepts, like, e.g., hydrogen bonds in combination with metal–ligand interactions.32 Even though the benefits of metallopolymers, metal–ligand interactions are more likely to interfere with each other, resulting in a lack of orthogonality, which is why such systems solely based on metal complexes are rather limited in literature. Nevertheless, our group already presented a few examples in which SMPs could be generated entirely based on metallopolymers featuring excellent shape-memory abilities.33–35 In this context, two metal complexes of different stability formed the basis for both required structural units. In one of those studies the two complexes, namely a bis-terpyridine and a tris- or bis-trityl-histidine complex, could be utilized to achieve an IPN structure in a very simple manner.35 Thus, this study will be the starting point for the current investigation in which a better tunability of the polymeric structure of the IPN should be obtained by applying heteroleptic metal complexes for the first time. This introduction leads to the possibility to tune one phase of the IPN individually without influence the other part of the polymer.
The first network is generated via the formation of a homoleptic complexes (bis-terpyridine). On the other hand, the second network is crosslinked via the formation of heteroleptic complexes (formed between zinc-tetraphenyl porphyrin (ZnTPP) and pyridine (Py)). In this way it is also possible to selectively influence the crosslinking density in the second network. Furthermore, this novel synthetic approach opens the way to include multiple polymers in a single subnetwork (via heteroleptic complexes) of the resulting IPNs broadening the chemistry of IPNs in general.
Results and discussion
Supramolecular binding motifs
The orthogonality of the two utilized metal complexes is the key parameter for the formation of the desired interpenetrating polymer network structure. Furthermore, the utilized metal complexes should be addressable to obtain smart properties in the resulting materials and feature a different stability in order to represent both required building units for shape-memory polymers. In one of our former studies we utilized a stable bis-terpyridine complex to crosslink one, and a much weaker trityl-histidine based complex, for the formation of the second polymer network.35 These two binding motifs were found to be fully orthogonal in the solid state, in particular for the here used combination of iron and zinc ions.The stable bis-terpyridine complex was also selected in this study to obtain a network acting as stable phase during the shape-memory process.
The weaker complex, crosslinking the second network and acting as switching unit in the SMPs, was exchanged this time by a heteroleptic binding motif, enabling a better and easier tunability of the IPN composition. In this context, the ZnTPP-Py interaction represents an excellent candidate due to its labile nature and resistance against competing terpyridine (Tpy) ligands. This orthogonality is well-known in literature and derived from steric constraints.36,37 While the ZnTPP provides only one free axial binding site, the multidentate Tpy is not able to coordinate the zinc core. In a previous study we investigated this behavior in a detailed fashion using isothermal titration calorimetry (ITC) and were able to show that the intrinsic orthogonality remains if the binding motifs are implemented in a polymeric environment.38,39
Ligand containing polymers
Initially ligand containing monomers, namely 6-(2,2′:6′2′′-terpyridin-4′-yloxy)-hexyl methacrylate (Tpy-MA) as well as N-(4-(10,15,20-triphenylporphyrin-5-yl)phenyl) methacrylamide (TPP-MA), were synthesized. While Tpy-MA was prepared according to a literature procedure,29TPP-MA was obtained by the treatment of 4-(10,15,20-triphenylporphyrin-5-yl)aniline with methacrylic anhydride. A detailed description can be found in the ESI.†Afterwards, the reversible-addition–fragmentation-chain-transfer (RAFT) polymerization technique was applied to obtain the polymers P1 to P3 which bear different ligands in their side chains. The synthesis of all polymers is presented in Scheme 1. 2,2′-Azo-bis(2-methylpropionitrile) (AIBN) was utilized as initiator for all polymerizations. As chain transfer agent (CTA) either 2-cyano-2-propyl dodecyl trithiocarbonate (CPDT) (P2) or 2-cyano-2-propyl benzodithioate (CPDB) (P1 and P3) was selected. For the synthesis of P1, Tpy-MA was copolymerized with butyl methacrylate (BMA) as main monomer. The polymer P2 was prepared by copolymerization of the formerly synthesized TPP-MA with 2-ethylhexyl methacrylate (2-EHMA). Metalation of the porphyrin moieties with zinc(II) acetate lead to the polymer P2-Zn. The quantitative metalation of all porphyrin side groups was verified by the comparison of the respective NMR spectra (Fig. S3†).38 Due to the loss of the NH-protons, the respective signal at negative ppm-values completely disappeared in the spectrum of P2-Zn. 2-EHMA was also utilized as main monomer for the synthesis of P3a to P3c, together with 4-vinyl pyridine (4-VPy) as commercially available ligand monomer. Furthermore, in contrast to P1 and P2, the ligand monomer content was varied for the three different polymerizations (increasing 4-VPy content from P3a to P3c), to enable a simple tuning of the crosslinking density in the network two later on. Additionally, the polymer P3d was synthesized via the copolymerization of styrene and 4-VPy with the same ligand content obtained in P3c. A detailed description of all polymerizations and the metalation of P2 is presented in the ESI (Table S1†). The molar masses as well as dispersities of the synthesized polymers were determined via size exclusion chromatography (SEC). All polymers revealed a unimodal molar mass distribution with low dispersities, which is characteristic for controlled radical polymerization techniques like RAFT.40 The determined molar masses of the linear polymers are rather comparable, ranging from Mn-values of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 g mol−1 (P1) to 15400 g mol−1 (P3b). The only exception was found for P3d which only revealed a Mn of 8100 g mol−1. For this polymerization styrene was utilized as comonomer, which is known to yield in lower monomer conversions when utilizing RAFT.41 A decrease in molar mass could be observed for P2-Zn when compared to the non-metalated polymer P2, which indicates a decrease of the hydrodynamic radius. This might result from intramolecular interactions between the metalated side-chain moieties leading to a varied hydrodynamic behavior after complexation. The 1H NMR spectra were used to calculate the exact composition of the final polymers. In particular for P3a to P3d the ligand content played an important role, since one goal of this study was to investigate the influence of the crosslinking density of the switchable polymer network on the shape-memory abilities of the resulting IPNs. It was found that the resulting polymer compositions nearly match the ratio of main monomer to ligand monomer which was utilized for the synthesis. The results of the SEC investigations as well as the determined compositions are listed in Table 1. Further information is given in the ESI (Fig. S2–S7†).
500 g mol−1 (P1) to 15400 g mol−1 (P3b). The only exception was found for P3d which only revealed a Mn of 8100 g mol−1. For this polymerization styrene was utilized as comonomer, which is known to yield in lower monomer conversions when utilizing RAFT.41 A decrease in molar mass could be observed for P2-Zn when compared to the non-metalated polymer P2, which indicates a decrease of the hydrodynamic radius. This might result from intramolecular interactions between the metalated side-chain moieties leading to a varied hydrodynamic behavior after complexation. The 1H NMR spectra were used to calculate the exact composition of the final polymers. In particular for P3a to P3d the ligand content played an important role, since one goal of this study was to investigate the influence of the crosslinking density of the switchable polymer network on the shape-memory abilities of the resulting IPNs. It was found that the resulting polymer compositions nearly match the ratio of main monomer to ligand monomer which was utilized for the synthesis. The results of the SEC investigations as well as the determined compositions are listed in Table 1. Further information is given in the ESI (Fig. S2–S7†).
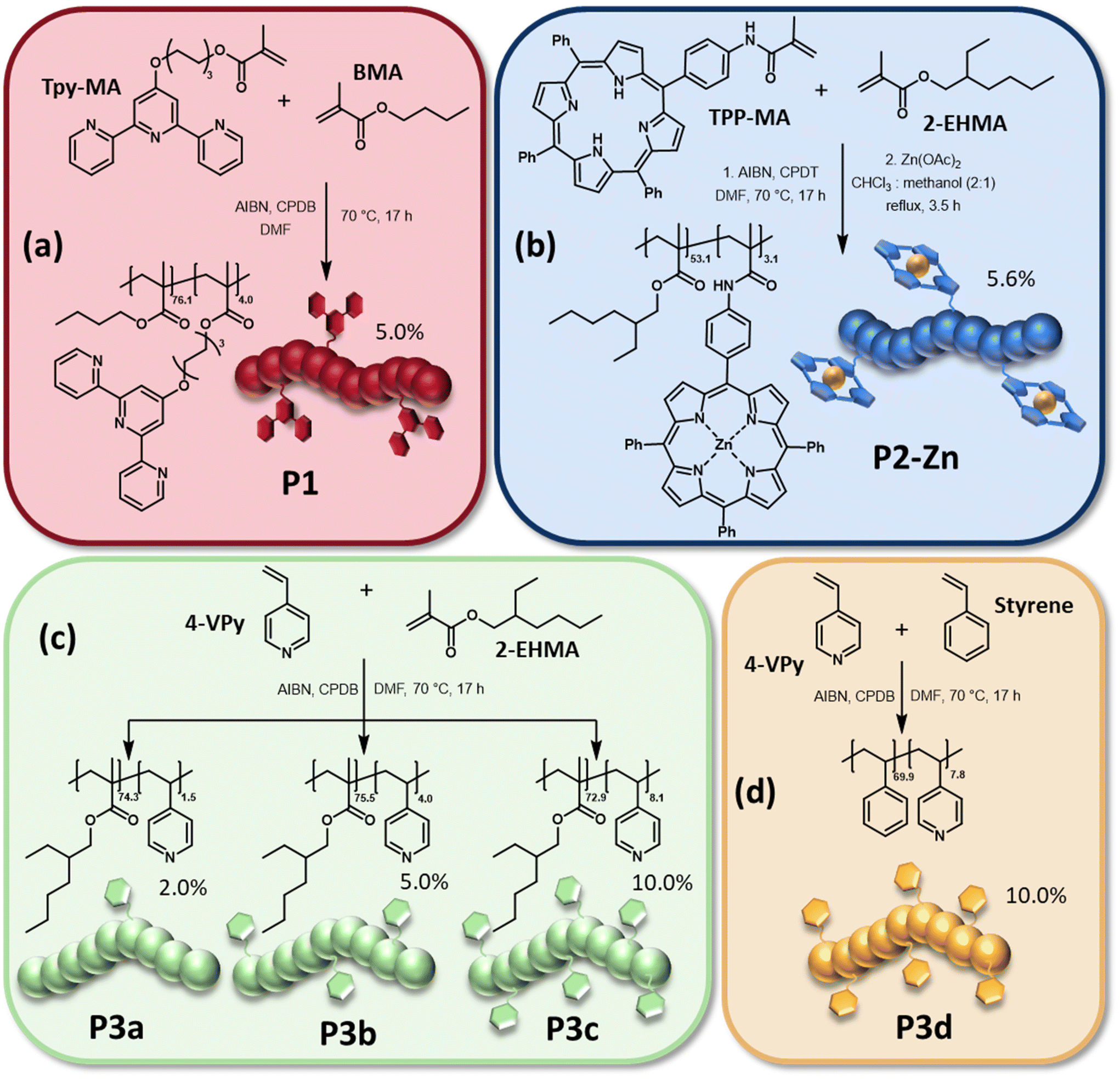 | ||
| Scheme 1 Schematic representation of the synthesis of (a) the terpyridine ligand containing polymer P1, (b) the zinc-tetraphenyl porphyrin containing polymer P2-Zn as well as the pyridine containing polymers (c) based on 2-EHMA (P3a to P3c) and (d) based on styrene (P3d). | ||
| Size exclusion chromatographya | 1H NMR spectroscopyb | |||||
|---|---|---|---|---|---|---|
| M n [g mol−1] | M w [g mol−1] | Đ | Monomer | Calculated ratio | Ligand content [%] | |
|
a Eluent: CHCl3/NEt3/i-PrOH [94:4:2], PMMA standard (P1, P2, P2-Zn and P3a to P3c) or PS standard (P3d)
b 300 MHz, CD2Cl2.
|
||||||
| P1 | 12500 |
15700 |
1.25 | Tpy-MA | 1 | 5.0 |
| BMA | 19 | |||||
| P2 | 15000 |
18900 |
1.26 | TPP-MA | 1 | 5.6 |
| P2-Zn | 12900 |
17300 |
1.34 | 2-EHMA | 17 | |
| P3a | 14900 |
20500 |
1.37 | 4-VPy | 1 | 2.0 |
| 2-EHMA | 48 | |||||
| P3b | 15400 |
19200 |
1.25 | 4-VPy | 1 | 5.0 |
| 2-EHMA | 19 | |||||
| P3c | 15300 |
18800 |
1.23 | 4-VPy | 1 | 10.0 |
| 2-EHMA | 9 | |||||
| P3d | 8100 | 9100 | 1.11 | 4-VPy | 1 | 10.0 |
| styrene | 9 | |||||
Interpenetrating metallopolymer networks
After the synthesis of the linear ligand containing polymers, the preparation of the interpenetrating metallopolymer networks was the next step. As mentioned before, those IPNs should be based on two different networks. Network one, which should be formed from P1 in the presence of iron sulfate, is thereby crosslinked via stable bis-terpyridine-iron(II)-complexes, and network two is formed through the formation of the heteroleptic complexes between P2-Zn and the pyridine side chains of P3a to P3d. A schematic representation of the synthesis of the different IPNs is presented in Fig. 1. A detailed description is provided in the ESI (Table S2†).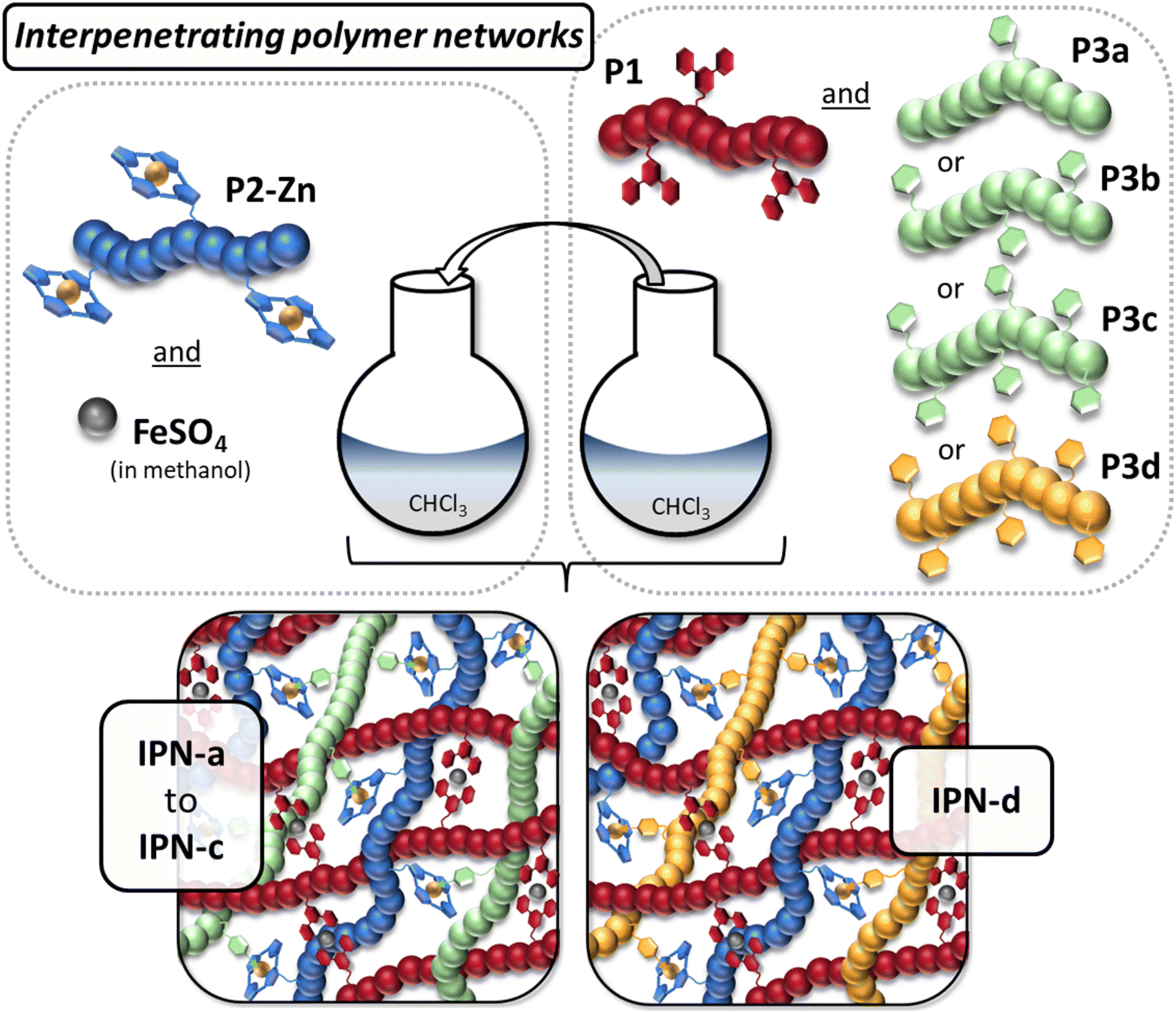 | ||
| Fig. 1 Schematic representation of the preparation of the interpenetrating metallopolymer networks IPN-a to IPN-d. | ||
For all IPNs, an equimolar ratio of [Tpy-ligand] to [TPP-ligand] to [Py-ligand] was utilized, which was calculated based on the composition, determined in the 1H NMR spectra. Firstly, two separate mixtures were prepared. Mixture one contained P2-Zn (dissolved in chloroform) and the required amount of iron(II) sulfate (dissolved in a small amount of methanol). For the other mixture, the required amounts of P1 and P3 were dissolved in chloroform. Subsequently, mixture two was given to mixture one leading to the formation of the interpenetrating networks based on the orthogonality of both supramolecular binding motifs. This procedure was performed analogously for all different pyridine containing polymers (P3a to P3d), resulting in four different IPNs (IPN-a to IPN-d). While the IPNs-a–c should feature a different crosslinking density, regarding network two, IPN-c and -d only differ in the main monomer of P3. Furthermore, four different model metallopolymer networks were prepared. MP-1 was synthesized as reference sample for the stable phase based on P1 by the addition of FeSO4.
Furthermore, three different metallopolymers were synthesized as reference sample for the labile networks, crosslinked via ZnTPP-Py interactions. Those networks (MP-2a, MP-2c and MP-2d) were prepared by mixing P2-Zn with P3a (EHMA-based with the lowest pyridine content), P3c (EHMA-based with the highest pyridine content), as well as P3d (styrene-based pyridine polymer). The general procedure and the exact amounts are provided in the ESI (Table S3†). In general, the model metallopolymers enable a detailed interpretation of the investigations of the structurally complex IPNs (Raman spectra or mechanical analyses) later on.
Raman spectroscopic investigations
To validate the formation of the interpenetrating networks based on the orthogonality of both supramolecular binding motifs, Raman spectroscopy measurements were performed on the interpenetrating networks IPN-a to IPN-d, the polymers P1 to P3, as well as the model samples MP-1 to MP-2. Raman spectroscopy offers insights into ligand binding, complexation, and structural changes, consequently representing an effective tool for studying the selective formation of orthogonal metal complexes.33,35Characteristic vibrational bands for the binding of vinyl pyridine to the porphyrin complex were first identified by investigating the model complexes ZnTPP and ZnTPP-Py (Fig. S36†). Here, characteristic shifts of bands in three regions of interest could be identified, which can be used to confirm the binding of Py to the ZnTPP complex. Namely, the band doublet at 390/410 cm−1, which can be assigned to νZn–N vibrations,42,43 is further split with the band at 390 cm−1 being redshifted by ca. 8 cm−1 and the band at 410 cm−1 being blueshifted by ca. 5 cm−1. This is caused by the additional electron density donated by the pyridine ligand into the metal orbitals. A further characteristic shift can be identified for the band at 1083 cm−1, which can be assigned to νC–H vibrations of the pyrrole subunits in the porphyrin ligand.42 Here, a redshift of ca. 5 cm−1 is observable. Further redshifts, also attributed to the porphyrin centered modes, can be observed for the bands at 1365 (in-plane skeletal mode of pyrrole unit; −3 cm−1), 1462 (νC–C, bridge; −8 cm−1) and 1496 cm−1 (νC–C, pyrrole; −2 cm−1).42 All of these shifts are caused by partial population of π* orbitals due to the coordination of the axial pyridine, which is an effect well known from literature and weakens the associated bonds, thus redshifting the vibrational frequencies.44,45 To confirm that, these changes are also visible in a polymeric environment, model complexes MP2-a to MP2-c were also measured (Fig. S36,† top lines). A polymeric environment is known to cause band broadenings, to influence the positions of some bands slightly and of course introduces additional, potentially overlapping bands.33 Due to this, only the band shifts for the νZn–N vibration at 390 cm−1 and the νC–C vibration at 1462 cm−1 are visible here. Nevertheless, these shifts are still characteristic enough to allow a clear identification of the formed ZnTPP-Py complex.
With these characteristic bands in mind, in a next step IPN-a to IPN-d were studied in the same manner. Due to the additional presence of Tpy ligands, introduced by P1, the band at 1462 cm−1 could not be used for the identification of the pyridine complex, as a strong Tpy band overlaps here. Notwithstanding this, the νZn–N band, which is nicely separated from overlapping bands can be used to clearly prove the formation of the desired ZnTPP-Py complexes within the IPNs.
Fig. 2 depicts the Raman spectra of P2-Zn (as a comparison) and the IPNs in the region of interest. For all IPNs clear redshifts of 2.7 to 4.5 cm−1 can be identified, which, keeping in mind the spectral precision of <0.5 cm−1 of the used Raman device, clearly proves the presence of the pyridine complexes.
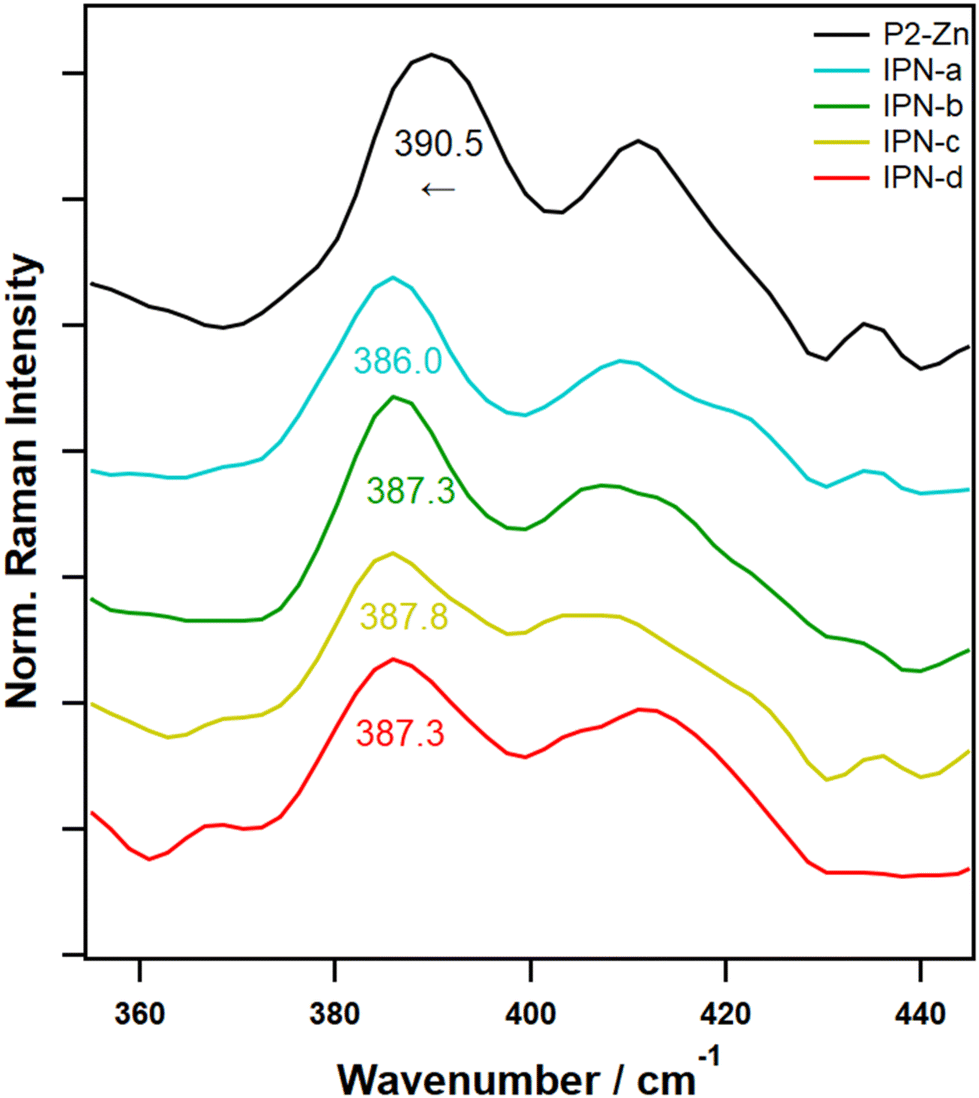 | ||
| Fig. 2 FT-Raman spectra of P2-Zn compared with IPN-a to IPN-d in the wavenumber region of interest (360 to 440 cm−1) to confirm vinyl pyridine bonding to the porphyrin complex via the observable characteristic band-shift. | ||
Furthermore, utilizing the same FT-Raman study, the formation of the bis-terpyridine-iron(II)-complexes inside the IPNs could be confirmed. The effect of the formation of this complex on the Tpy vibrations has been extensively described in previous studies. Thereby in general, a shift of the strong band located at 996 cm−1 (δC–C) to higher wavenumbers can by observed and cis–trans transition and shortening of the C–C bonds induces more complex changes of the region between 1540 and 1620 cm−1 (νC–C/N).33–35 While the changes in the higher wavenumber region cannot be easily observed for these IPNs, as there is a large amount of overlap with signals from P2-Zn and P3, the characteristic blueshift of the band at 996 cm−1 could be observed for all IPNs (Fig. S37†) proving the formation of both the stabile and labile complex required for their function. Furthermore, no significant uncoordinated ligand fractions could be detected. Therefore, the complex formation can be regarded as virtually quantitative.
Regarding the implementation of several dynamic metal-complexes inside the prepared IPNs, a potential interference or metal exchange upon thermal cycling needs to be excluded. Raman spectroscopy represents a powerful method in this context, since it provides insights on the molecular level, i.e. the nature of the reversible binding motifs.
To examine the long-term stability of the binding motifs under thermal stress, a long-term Raman spectroscopic experiment was performed. For this purpose, IPN-c was treated at a reasonable operating temperature for the shape-memory process of 80 °C for in total 19 days. The sample was allowed to cool to room temperature every day and a Raman spectrum was recorded (Fig. S55†). In this context, any changes in complex formation during thermal cycling should be visible in significant band shifts over time. The band location of the νZn–N vibration at approx. 390 cm−1, which is characteristic for the ZnTPP-Py complex, was determined for every measurement (Fig. S56†). It can be seen from Table S9† that the band location continuously variates between 386.0 and 387.9 cm−1 and does not follow an observable trend over time. Thus, it can be concluded that the ZnTPP-Py binding motif is present after every thermal cycle as intended and no thermal induced cross-interactions with the present tpy complex, such as potential transmetalation, occurs.
Thermal properties
A very important parameter for thermally triggered polymers is their thermal behavior. For this reason, firstly the degradation temperature (Td) of all building block polymers and IPNs was determined applying thermo gravimetric analysis (TGA). The investigation of the thermal stability is important to ensure that no degradation of the samples occurs during the manufacturing steps or the thermally triggered switching. The TGA measurements revealed for all polymers and IPNs a Td above 200 °C. Additionally, within temperature dependent dynamic-mechanical analysis (DMTA), the mechanical properties of the four IPNs as well as those of the reference metallopolymers (MP-2a, MP-2c and MP-2d) were investigated.A plot of those measurements, showing the corresponding loss factors (tan(δ)) depending on the temperature, is presented in Fig. 3. Interestingly, the resulting curve of tan(δ) revealed two maxima for the model polymers MP-2a and MP-2c. One is located at moderate temperatures of 50 to 70 °C and one at higher temperatures around 100 °C.
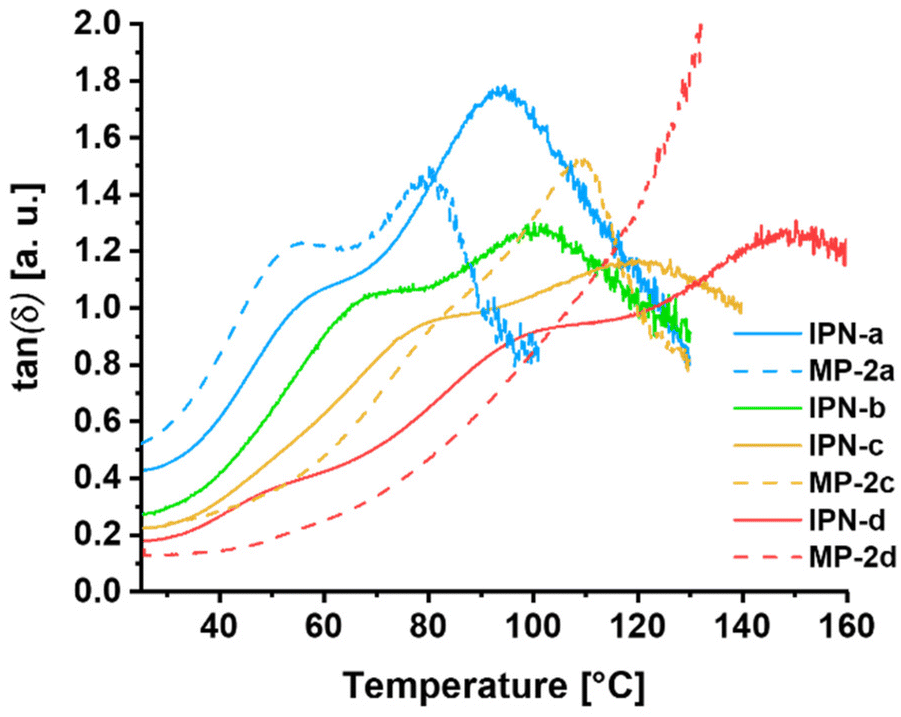 | ||
| Fig. 3 Comparison of the DMTA measurements (temperature dependent tan(δ)-curves) of the model polymers MP-2a, MP-2c and MP-2d as well as the interpenetrating metallopolymer networks IPN-a to IPN-d. | ||
Our assumption is that the lower one results from the glass transition temperature of the polymer chains, while the second one indicates the activation (induced mobility due to partial decomplexation and/or ligand exchange) of the ZnTPP-Py complex. By comparing MP-2a with MP-2c, which only differ regarding their crosslinking density, different intensities for the two signals are visible. For MP-2c, the model polymer with the highest crosslinking density, the signal at higher temperature is dominant, while the other one is nearly invisible and looks more like a shoulder. In the case of MP-2a, there is a clear peak at around 50 °C and a similarly large elevation later in the curve at around 100 °C. In our opinion, the different trend of the curves can be explained very well by the composition of MP-2a.
By combining the results of SEC and the calculated monomer ratio in P3a, it becomes clear that on average the polymer chains carry only 1 to 2 pyridine moieties per chain. However, these polymer chains are unable to form a network structure with P2-Zn, but only cause a supramolecular chain extension at the ZnTPP moieties of P2-Zn, leading to a graft polymer-like architecture. This different structure could explain the strong visibility of the glass transition related signal at moderate temperatures in the case of MP-2a and its “missing” for the sample MP-2c, featuring a relatively high supramolecular crosslinking density. Furthermore, the maxima are shifted towards higher temperature with increasing crosslinking density, which is expected for the glass transition of higher crosslinked networks.
The fact that also the second maximum is shifted in the same direction indicates that not only the nature of the reversible bond but also the overall content influences the tan(δ) maximum. This behavior can be explained by the thermal activation mechanism of the supramolecular crosslinks in the networks. With a temperature dependent shift of the equilibrium between stable and activated complexes, a progressive decrease in crosslinking density is expected upon heating. The system will show a maximum in tan(δ) when the loss modulus dominates the storage modulus at a certain degree of crosslinking. Samples with a higher initial crosslinking density will reach this point at higher temperatures, assuming the same relative activation state and temperature dependent equilibrium.
The two signals are also observable for the corresponding IPNs with the same observable trends. However, there is a clear temperature shift of those signals compared to the model systems, which is probably caused by the additional network in these samples, as it has also been shown for other IPNs in the literature.46 Interestingly, in the case of MP-2d no maximum of tan(δ) could be identified, while for the corresponding IPN three maxima are observable.
In addition to the two processes above, the third at higher temperature (150 °C) might be induced by the additional polystyrene backbone and/or the more stable bis-Tpy crosslinks.
Differential scanning calorimetry (DSC) represents another method to measure thermal transitions within the polymers and IPNs. For P1 a Tg of 18 °C could be determined, while P3d revealed Tg of 104 °C. These glass transition temperatures are within the expected range based on the utilized main monomers BMA and styrene.47,48 In contrast no Tg was observable for all linear polymers containing EHMA. For the methacrylate-based IPNs (IPN-a and IPN-b) an increase of the glass transition (from 30 to 44 °C) could be found within an increase of the crosslinking density of the switching phase. Simultaneously, an increase in crosslinking density seems to fade out any visible glass transition within the DSC curves. While the curves are in general difficult to evaluate (Tg of IPNa needed to be evaluated from the second heating cycle) IPN-c and IPN-d showed no observable Tg, at all. In contrast, DMTA revealed tan(δ) maxima in all cases. The respective DSC traces may indicate a thermal transition, which however feature a too low intensity to be evaluated properly. In this context the tan(δ) method can be regarded as a more sensitive method and provides better insight in temperature dependent processes.
All TGA, DSC and DMTA plots as well as a summary of the detected temperatures during those measurements are presented in the ESI (see Fig. S8–S29 and Tables S4–S6†).
Shape-memory ability
The investigation of shape-memory abilities requires an initial processing step to predetermine the permanent shape of the IPNs. This step was performed analogously to literature procedures,33–35 leading to rectangular polymer specimen (see photo in Fig. 4a) which represent a suitable form for manual tests as well as thermo-mechanical analyses. Subsequently, within manual tests the general ability of the IPNs to reveal a shape-memory effect was investigated. During those manual tests (Fig. S35†) it was already observable that the shape-memory abilities of the IPNs strongly depend on the pyridine content of P3. IPN-a, which was prepared using P3a (2% Py) stayed in the temporary shape even though the recovery step was triggered, while IPN-c, which contained P3c (10% Py), showed a full restorage of the original form within a few seconds. Exemplarily, a photo-series of the manual test for the sample IPN-c is displayed in Fig. 4a.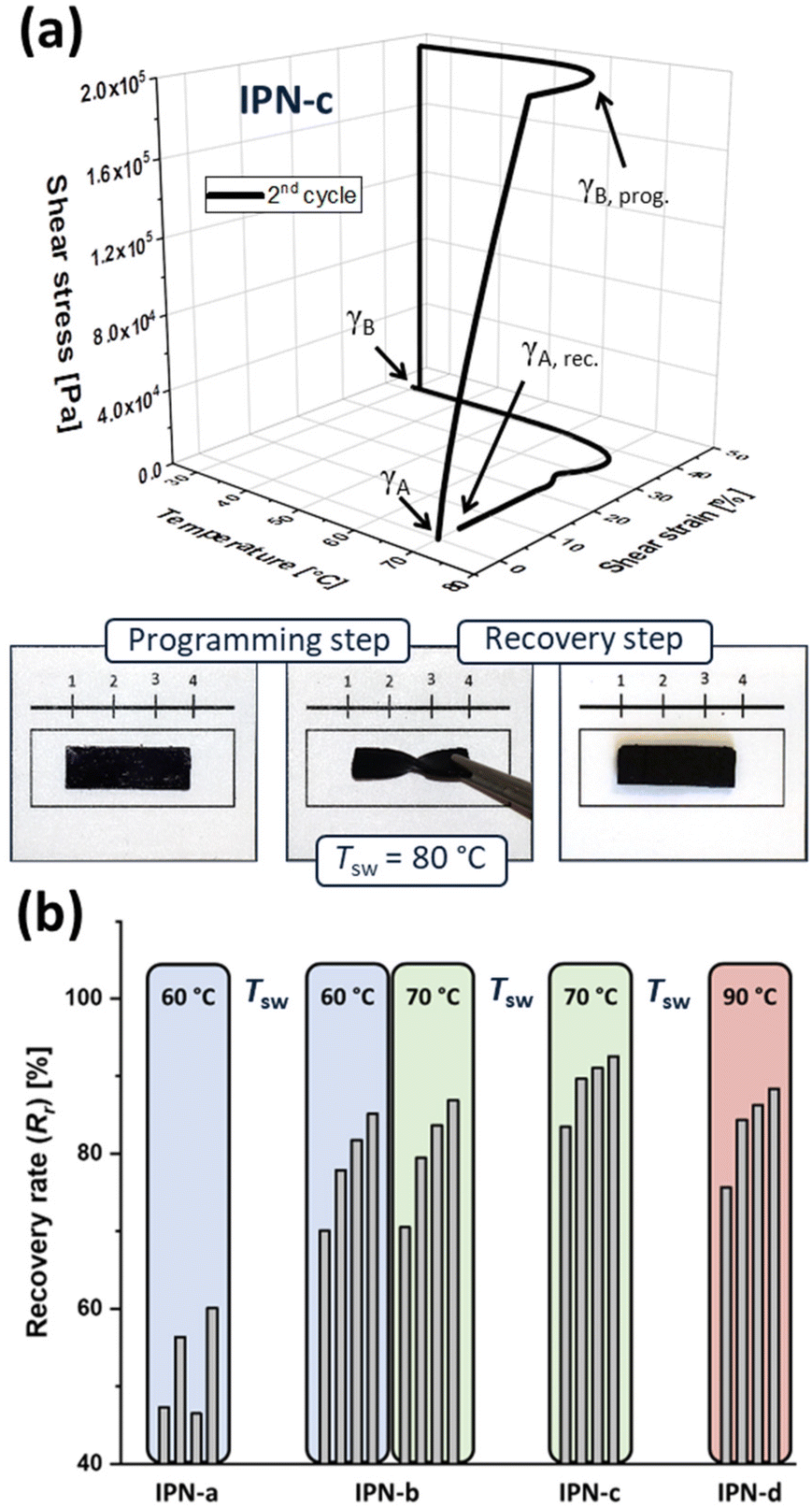 | ||
| Fig. 4 (a) 3D-TMA plot (2nd cycle) and photo-series of the manual shape-memory test for IPN-c and (b) comparison of the strain fixity and recovery rates of the IPNs determined via TMA over four cycles. | ||
In order to quantify the shape-memory abilities of all IPNs, thermo-mechanical analysis (TMA) followed the manual test. Within these measurements, it is possible to calculate the strain fixity (Rf) and strain recovery rates (Rr) according to eqn (1) and (2), which indicate the quality of the fixation of the temporary form as well as the recovery step to the original form. The calculations are based on shear strain percentages (γ), which are recorded during the TMA. The permanent shape (γA) serves as reference point for the measurement and is consequently 0%.
| Rf = γB/γB, prog. × 100% | (1) |
| Rr = (γB, prog. – γA, rec.)/(γB, prog. – γA) × 100% | (2) |
For the temporary shape two different values are defined. γB, prog. represents the shear strain after applying torsional stress at the switching temperature, while γB stands for the shear strain after fixation (cooling) and release of the shear stress. Finally, γA, rec. reveals the remaining shear strain in the specimen after the recovery step. For each IPN four consecutive cycles were recorded without reprocessing the samples in between. In each case a significantly lower recovery rate could be determined in the first cycle, while Rr is slightly increasing through the cycles.
This behavior is well-known in literature for SMPs and can be ascribed to remaining residual stress as well as thermal history of the specimen.49 Furthermore, this observation provides strong evidence for the stability and absence of cross-interactions between the utilized binding motifs in addition to the spectroscopic findings. A potential metal-exchange would result in shifted binding affinities, and, consequently, decreased shape-memory performance due to unintended activation of the stable phase. Such behavior cannot be observed during thermal cycling. This obtained result is also in line with previous measurements in solution, which revealed a selective formation of ZnTPP-Py and bis-terpyridine systems simultaneously.38
Exemplarily, the resulting 3D-TMA plot of the second cycle for IPN-c is presented in Fig. 4a. Furthermore, all calculated recovery rates are presented and compared in Fig. 4b. Further information as well as all TMA plots are presented in the ESI (Table S7 and Fig. S29–S33†). While every tested sample showed excellent fixity rates (near 100%), the TMA measurements confirmed the assumption, already made in the manual test. A clear structure–property-relationship, relating recovery and crosslinking density, is visible. The poor shape-memory abilities of IPN-a can easily be explained by its overall composition.
As mentioned before, P3a is not able to really form the second network with P2-Zn, due to the low Py-content. For this reason, IPN-a should not be able to form an interpenetrating polymer network structure. Instead, a semi-IPN in which a network of P1 and the iron(II)-ions is formed, which is interspersed with supramolecular connected graft polymer chains out of P3a and P2-Zn. The fact that this semi-IPN has extremely poor shape-memory properties provides evidence that the IPN structure, based on the supramolecular bonding, is essential for the shape-memory effect. Besides that, an increasing crosslinking density of the switching phase (ZnTPP-Py network) appears to be beneficial for the shape-memory process. IPN-b (5% Py) still reveals good recovery rates up to 87% while excellent values could be obtained for IPN-c (10% Py, Rr = 93%). Presumably a higher crosslinking density is causative for a larger difference in flexibility of the switching unit between the fixed and activated (dynamic) state, which influences the shape-memory behavior.
Furthermore, the crosslinking density also affects the optimal switching temperature, since more thermal energy is required to obtain flexibility in the specimen. Since in all of the upper IPNs the switching phase is solely build up by EHMA-based copolymers (EHMA is known to induce a relatively low glass transition),47 the required switching temperatures may be predominantly effected by the nature and content of the labile ZnTPP-Py complexes.
In the case of IPN-d, the additional styrene backbone of P3-d provides a higher Tg. Hence, the thermal properties of the switching phase are presumably determined by an interplay between this glass transition and the metal–ligand crosslinks, resulting in an increased optimal switching temperature of 90 °C, even though the crosslinking density is identical to IPN-c. Thus, the implementation of a mixed network as switching phase (enabled by the utilization of a heteroleptic binding motif) offers the possibility to further adjust the usable switching temperatures.
Rewriting potential
Since the presented IPNs are based on two orthogonal metal ligand interactions, the materials feature additional smart properties beside single one-way shape-memory behavior. The bis-terpyridine-iron(II)-complexes (crosslinks of the stable network/phase) remain stable during the upper TMA cycles, but can still be activated at even higher temperatures. Under these circumstances the material should exhibit a vitrimer-like behavior enabling a complete reprocessing of the samples as well as the possibility to rewrite the permanent shape.In order to investigate this, the sample of IPN-c (utilized for TMA) was grinded manually into granules. Subsequently, the material was hot-pressed again (see ESI†) into a rectangular specimen under identical conditions, which was further investigated by TMA (sample IPN-c*). An initial TMA cycle was performed to erase remaining residual stress and thermal history resulting from the reprocessing step. The following second cycle is displayed in Fig. 5. IPN-c* revealed still a very good Rf close to 100% as well as a Rr of 86%, which represents only a slight decrease compared to the value obtained from the second TMA cycle of IPN-c (90%). These findings clearly demonstrate a reprocessability of the sample without a significant loss of shape-memory properties.
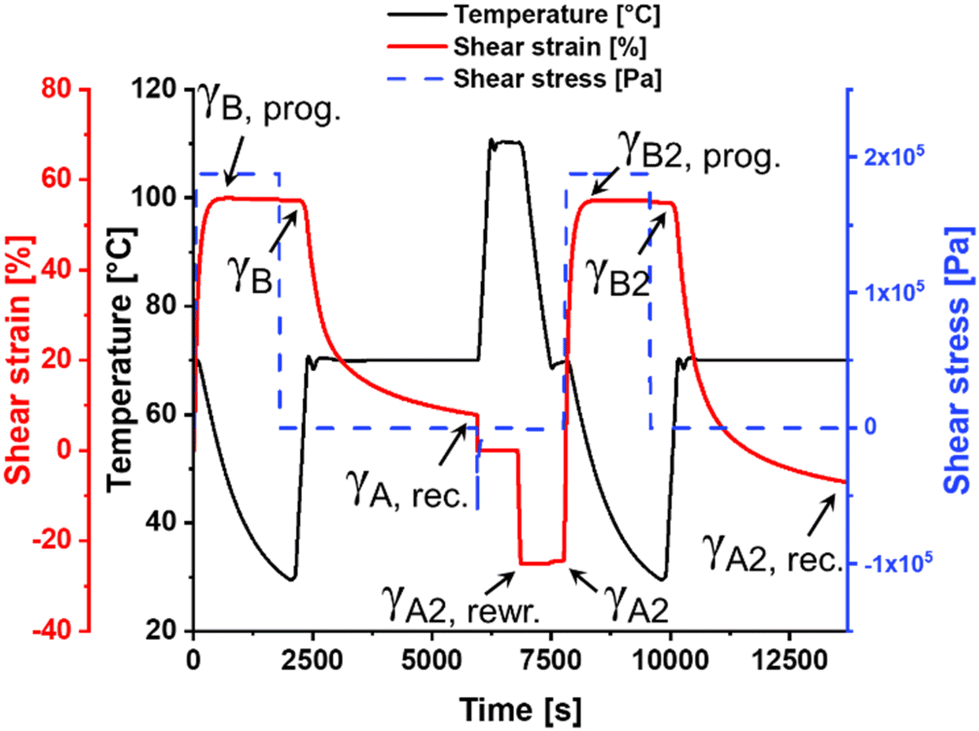 | ||
| Fig. 5 2D-TMA plot (rewriting experiment) of IPN-c*. | ||
After thermo-mechanical characterization, the specimen was heated to 110 °C, which leads again to an activation (induced mobility due to partial decomplexation and/or ligand exchange) of the more stable Tpy complexes, enabling a permanent deformation and, thus, a rewriting of the permanent shape. This was realized by twisting in opposite direction until a torsional shear strain of −25% was reached (Fig. 5, γA, rewr.).
The efficiency of this reprogramming step (Effrewr.) can be determined as the fixity of the rewritten permanent shape, calculated according to eqn (3), where γA, rec. represents the remaining shear strain of the preceding TMA cycle. γA2, rewr. displays the defined negative torsion (−25%) and γA2 the remaining shear strain after fixation (cooling).
| Effrewr. = (γA, rec. – γA2)/(γA, rec. – γA2, rewr.) × 100% | (3) |
The rewriting process of IPN-c* was found to be highly efficient (Effrewr. = 98%). In order to examine the ability of the sample to recover the new permanent shape, an additional TMA cycle was recorded (Fig. 5, t > 7500 s). The procedure of this measurement is identical to the previous TMA, just utilizing the rewritten shape (γA2) as reference point. It is clearly visible that the observed recovery step exceeds the initial permanent form (γA = 0%) and the rewritten permanent shape is restored. A Rr of 78% could be determined, which appears to be low at first glance. Keeping in mind that the performed measurement represents an initial TMA cycle after processing, the value needs to be compared with Rr of IPN-c, which was obtained in the first TMA cycle (84%). In this context, also only a slight decline in shape-memory ability is observable.
The results indicate that rewriting of the permanent shape based on the more stable Tpy complexes is feasible and does not negatively influence the shape-memory properties in a drastic manner.
Experimental part
Materials and methods
All chemicals were used as received from TCI (Eschborn, Germany), Sigma Aldrich (Darmstadt, Germany), Alfa Aesar (Kandel, Germany), Thermo Fisher Scientific (Geel, Belgium) and Acros Organics (Geel, Belgium) if not otherwise stated. All solvents were dried over molecular sieve under nitrogen atmosphere. The stabilizer in the used liquid monomers was removed over a short aluminium oxide (AlOx) column (neutral AlOx, obtained from Molecula, Darlington, UK).Nuclear magnetic resonance (NMR) spectra were measured using a Bruker AC 300 (300 MHz) spectrometer at 298 K. The chemical shift is given in parts per million (ppm on δ scale) related to deuterated solvent.
Elemental analysis (EA) was performed utilizing a Vario El III instrument (Elementar, Langenselbold, Germany).
Size exclusion chromatography (SEC) measurements were performed on the following setup: Shimadzu with CBM-20A (system controller), DGU-14A (degasser), LC-20AD (pump), SIL-20AHT (auto sampler), CTO-10AC vp (oven), RID-10A (RI detector), PSS SDV guard/1000 Å/1000000 Å column (5 μm particle size), chloroform/isopropanol/triethylamine [94/2/4] (eluent) with 1 mL min−1 at 40 °C, poly(methyl methacrylate) or polystyrene (standard).
Thermo gravimetric analyses (TGA) were carried out under nitrogen atmosphere with a heating rate of 10 K min−1 using a Netzsch TG 209 F1 Iris (Selb, Germany).
Differential scanning calorimetry (DSC) was measured on a Netzsch DSC 204 F1 Phoenix instrument (Selb, Germany) under a nitrogen atmosphere with a heating rate of 20 K min−1 (first and second heating cycle) and 10 K min−1 (third heating cycle).
Thermo mechanical analyses (TMA) were performed according to a literature procedure33–35 utilizing a MCR 301 rheometer from Anton Paar (Graz, Austria) using the convection oven device CTD 450. The samples were measured with a solid rectangular fixture setup (SRF12-SN13529), Anton Paar (Graz, Austria) in dimensions of approximately 30 × 10 mm (length, width) and a thickness of approx. 1.5 to 2.5 mm. The resulting sample gap was set to 20 to 21 mm. The software RheoCompassTM V1.24.549-Release 64-bit (Anton Paar, Graz, Austria) was applied for operating the rheometer as well as for analysis. The data was exported as txt-files and evaluated and processed with OriginPro 2019 (OriginLab Corporation, Northampton, MA, USA). A detailed description for the TMA utilized for the investigation of the shape-memory and the rewriting abilities is presented in the ESI.†
Raman-spectroscopic measurements were performed using a Multispec Fourier-transform Raman-Spectrometer (Bruker Corporation, Billerica, Massachusetts, United States of America) in the range 0 and 4000 cm−1 with a spectral resolution of 4 cm−1. The Raman excitation light at 1064 nm was provided by a Nd:YAG laser (Klastech DeniCAFC-LC-3/40, Dortmund, Germany). Due to the photothermal stability of the samples, the power of the Laser was varied between 10 and 1000 mW to ensure no burning occurred and a total of 500 scans were accumulated for each of the samples to boost the signal-to-noise ratio.
To investigate the long-term temperature stability, IPN-c was heated to 80 °C in a Nabertherm S27 oven (Nabertherm GmbH, Lilienthal, Lower Saxony, Germany). The temperature was maintained for 19 days. Every day, the sample was cooled to room temperature and a Raman spectrum was recorded.
Synthesis of ligand monomers
6-(2,2′:6′2′′-Terpyridin-4′-yloxy)-hexyl methacrylate (Tpy-MA) and 4-(10,15,20-triphenylporphyrin-5-yl)aniline (TPP-NH2) were synthesized according to literature procedures.29,50N-(4-(10,15,20-Triphenylporphyrin-5-yl)phenyl)methacryl-amide (TTP-MA) was prepared by treatment of TPP-NH2 with methacrylic anhydride. The detailed procedure is presented in the ESI.†
Synthesis of ligand containing polymers
The linear ligand containing polymers P1, P2 and P3-a to P3-d were synthesized applying the RAFT-polymerization technique according to a literature procedure.34 A detailed description of the polymerization conditions as well as of the metalation of P2 (preparation of P2-Zn) is presented in the ESI (see Table S1†).Synthesis of interpenetrating metallopolymer networks and reference metallopolymer networks
Detailed information for the synthesis of the interpenetrating metallopolymer networks IPN-a to IPN-d and the reference metallopolymer networks MP-1 and MP-2a to MP-2d is given in the ESI (see Tables S2 and S3†).Conclusion
Within this article, we presented a simple method for the preparation of interpenetrating metallopolymer networks featuring shape-memory abilities. Firstly, different linear polymers were synthesized bearing terpyridine-, zinc-tetraphenyl porphyrin- or pyridine side-groups. By simply mixing those polymers together with iron sulfate, interpenetrating metallopolymer networks were obtained due to the orthogonality of the two different complexes. The desired complex formation was proven via FT-Raman spectroscopy. The structure–property-relationships of all samples were investigated in a detailed fashion by thermogravimetric analysis as well as thermo-mechanical investigations. In particular, the thermo-mechanical analysis revealed a strong correlation of the shape-memory abilities and the overall composition of the IPNs. These material properties, in combination with spectroscopic data and previous investigations in solution,38 strongly indicate the remaining orthogonality of the applied binding motifs during the shape-memory process.The results also clearly exhibit that the crosslinking density of the switchable polymer networks is crucial for shape memory capabilities and can easily be tuned by the incorporation of a heteroleptic metal-complex. Moreover, this structural feature enables the fabrication of IPNs, incorporating three different polymers at the same time, in our case: BMA, 2-EHMA and styrene. In this context it was possible to include multiple polymers in a single phase of the resulting shape-memory materials. Furthermore, the incorporation of orthogonal addressable metal-complexes as a structural feature enables the fabrication of IPNs which are fully recyclable and rewritable.
Author contributions
Synthesis: T. B., J. M.; characterization: T. B, J. M.; Raman spectroscopy: M. F. A.; DMTA/TMA: J. M.; writing – original draft: T. B., M. F. A., J. M.; supervision: S. Z., M. S., J. P., M. D. H., U. S. S.; conceptualization: S. Z., M. S., J. P., M. D. H., U. S. S.; writing – review & editing: J. H., S. Z., M. S., J. P., M. D. H., U. S. S.; funding acquisition: S. Z., J. P., M. D. H., U. S. S.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Deutsche Forschungsgemeinschaft (CRC/TRR 234 “Catalight”, subproject C2, project number: 364549901). Furthermore, the project was funded by the Carl-Zeiss Foundation (Perspektiven 2019, project number: P2019-02-001).References
-
M. A. A. AlMaadeed, D. Ponnamma and A. A. El-Samak, in Polymer Science and Innovative Applications, ed. M. A. A. AlMaadeed, D. Ponnamma and M. A. Carignano, Elsevier, 2020, pp. 1–19 Search PubMed
.
-
N. S. K. Gowthaman, H. N. Lim, T. R. Sreeraj, A. Amalraj and S. Gopi, in Biopolymers and their Industrial Applications, ed. S. Thomas, S. Gopi and A. Amalraj, Elsevier, 2021, pp. 351–372 Search PubMed
- H. Namazi, Bioimpacts, 2017, 7, 73–74 Search PubMed
- H. Meng and H. Jinlian, J. Intell. Mater. Syst. Struct., 2010, 21, 859–885 Search PubMed
- F. Liu and M. W. Urban, Prog. Polym. Sci., 2010, 35, 3–23 Search PubMed
- C. d. l. H. Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 Search PubMed
- X. Liu, F. Liu, T. Abdiryim and R. Jamal, Polym. Rev., 2023, 64, 306–370 Search PubMed
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93 Search PubMed
- X. Yan, F. Wang, B. Zheng and F. Huang, Chem. Soc. Rev., 2012, 41, 6042–6065 Search PubMed
- S. Wang and M. W. Urban, Nat. Rev. Mater., 2020, 5, 562–583 Search PubMed
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034–2057 Search PubMed
- C. Ni, D. Chen, Y. Yin, X. Wen, X. Chen, C. Yang, G. Chen, Z. Sun, J. Wen, Y. Jiao, C. Wang, N. Wang, X. Kong, S. Deng, Y. Shen, R. Xiao, X. Jin, J. Li, X. Kong, Q. Zhao and T. Xie, Nature, 2023, 622, 748–753 Search PubMed
- M. D. Hager, S. Bode, C. Weber and U. S. Schubert, Prog. Polym. Sci., 2015, 49–50, 3–33 Search PubMed
- M. Ahmad, J. Luo, B. Xu, H. Purnawali, P. J. King, P. R. Chalker, Y. Fu, W. Huang and M. Miraftab, Macromol. Chem. Phys., 2011, 212, 592–602 Search PubMed
- A. Lendlein, A. M. Schmidt, M. Schroeter and R. Langer, J. Polym. Sci., Part A:Polym. Chem., 2005, 43, 1369–1381 Search PubMed
- K. Uto, Y. Matsushita and M. Ebara, Polym. Chem., 2023, 14, 1478–1487 Search PubMed
- P. Yan, W. Zhao, B. Zhang, L. Jiang, S. Petcher, J. A. Smith, D. J. Parker, A. I. Cooper, J. Lei and T. Hasell, Angew. Chem., Int. Ed., 2020, 59, 13371–13378 Search PubMed
- G. Zhang, W. Peng, J. Wu, Q. Zhao and T. Xie, Nat. Commun., 2018, 9, 4002 Search PubMed
- J. Li, J. A. Viveros, M. H. Wrue and M. Anthamatten, Adv. Mater., 2007, 19, 2851–2855 Search PubMed
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 Search PubMed
- N. Y. Choi, S. Kelch and A. Lendlein, Adv. Eng. Mater., 2006, 8, 439–445 Search PubMed
- B. K. Kim, S. Y. Lee and M. Xu, Polymer, 1996, 37, 5781–5793 Search PubMed
- S. Zhang, Y. Feng, L. Zhang, J. Sun, X. Xu and Y. Xu, J. Polym. Sci., Part A:Polym. Chem., 2007, 45, 768–775 Search PubMed
- L. Wang, F. Zhang, S. Du and J. Leng, ACS Appl. Mater. Interfaces, 2023, 15, 21496–21506 Search PubMed
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 Search PubMed
- K. Y. Zhang, S. Liu, Q. Zhao and W. Huang, Coord. Chem. Rev., 2016, 319, 180–195 Search PubMed
- B. J. Holliday, T. B. Stanford and T. M. Swager, Chem. Mater., 2006, 18, 5649–5651 Search PubMed
- G.-M. Bao, L. Wang, H.-Q. Yuan, X.-Y. Wang, T.-X. Mei and M.-R. Qu, RSC Adv., 2016, 6, 109453–109459 Search PubMed
- S. Bode, L. Zedler, F. H. Schacher, B. Dietzek, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, Adv. Mater., 2013, 25, 1634–1638 Search PubMed
- M. Enke, F. Jehle, S. Bode, J. Vitz, M. J. Harrington, M. D. Hager and U. S. Schubert, Macromol. Chem. Phys., 2017, 218, 1600458 Search PubMed
- K. M. Herbert, S. Schrettl, S. J. Rowan and C. Weder, Macromolecules, 2017, 50, 8845–8870 Search PubMed
- X. Wang, J. Xu, Y. Zhang, T. Wang, Q. Wang, S. Li, Z. Yang and X. Zhang, Nat. Commun., 2023, 14, 4712 Search PubMed
- J. Meurer, J. Hniopek, T. Bätz, S. Zechel, M. Enke, J. Vitz, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, Adv. Mater., 2021, 33, 2006655 Search PubMed
- J. Meurer, T. Bätz, J. Hniopek, S. Zechel, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, J. Mater. Chem. A, 2021, 9, 15051–15058 Search PubMed
- J. Meurer, T. Bätz, J. Hniopek, C. Bernt, S. Zechel, M. Schmitt, J. Popp, M. D. Hager and U. S. Schubert, J. Mater. Chem. A, 2022, 10, 25106–25117 Search PubMed
- K. Mahata, M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2010, 132, 15933–15935 Search PubMed
- M. Schmittel, Isr. J. Chem., 2019, 59, 197–208 Search PubMed
- T. Bätz, M. Enke, S. Zechel, M. D. Hager and U. S. Schubert, Macromol. Chem. Phys., 2021, 222, 2100295 Search PubMed
- T. Bätz, I. Anufriev, C.-H. Pai, T. Schuett, I. Nischang, S. Zechel, M. D. Hager, Y.-T. Chan and U. S. Schubert, ACS Appl. Polym. Mater., 2023, 5, 1354–1363 Search PubMed
- G. Moad, E. Rizzardo and S. H. Thang, Chem. – Asian J., 2013, 8, 1634–1644 Search PubMed
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 Search PubMed
- T. S. Rush, P. M. Kozlowski, C. A. Piffat, R. Kumble, M. Z. Zgierski and T. G. Spiro, J. Phys. Chem. B, 2000, 104, 5020–5034 Search PubMed
- A. A. JarzÈ©cki, P. M. Kozlowski, P. Pulay, B.-H. Ye and X.-Y. Li, Spectrochim. Acta, Part A, 1997, 53, 1195–1209 Search PubMed
- M. Nappa and J. S. Valentine, J. Am. Chem. Soc., 1978, 100, 5075–5080 Search PubMed
- F. D'Souza, Y.-Y. Hsieh and G. R. Deviprasad, Inorg. Chem., 1996, 35, 5747–5749 Search PubMed
- M. Sangermano, G. Malucelli, A. Priola and M. Manea, Prog. Org. Coat., 2006, 55, 225–230 Search PubMed
- F. Fleischhaker, A. P. Haehnel, A. M. Misske, M. Blanchot, S. Haremza and C. Barner-Kowollik, Macromol. Chem. Phys., 2014, 215, 1192–1200 Search PubMed
- J. Rieger, J. Therm. Anal., 1996, 46, 965–972 Search PubMed
- R. Dolog and R. A. Weiss, Macromolecules, 2013, 46, 7845–7852 Search PubMed
- N. S. Lebedeva, Y. A. Gubarev, E. S. Yurina, E. N. Smirnova and S. A. Syrbu, Colloid Polym. Sci., 2017, 295, 2173–2182 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01079e |
| This journal is © The Royal Society of Chemistry 2025 |