
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and cell-induced luminescence of post-functionalisable ionisable polyesters from the Passerini 3-component polymerisation†
Lewis
O'Shaughnessy
 a,
Akosua
Anane-Adjei
b,
Mariarosa
Mazza
b,
Naoto
Hori
a,
Pratik
Gurnani
*c and
Cameron
Alexander
*a
a,
Akosua
Anane-Adjei
b,
Mariarosa
Mazza
b,
Naoto
Hori
a,
Pratik
Gurnani
*c and
Cameron
Alexander
*a
aDivision of Molecular Therapeutics and Formulation, Boots Science Building, School of Pharmacy, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: cameron.alexander@nottingham.ac.uk
bAdvanced Drug Delivery, Pharmaceutical Sciences, R&D, AstraZeneca, UK
cUCL School of Pharmacy, University College London, 29-39 Brunswick Square, Bloomsbury, London WC1N 1AX, UK. E-mail: p.gurnani@ucl.ac.uk
First published on 13th December 2024
Abstract
Recent developments in polymer synthesis methods allow the preparation of new materials across a wide chemical space and with high atom efficiency. In this work, we developed a strategic approach to the synthesis of a diverse array of novel ionisable polyesters via a modular and iterative synthetic approach. We initially designed a range of diacid monomers and subsequently used the Passerini 3-component reaction as a simple, one-pot step growth polymerisation to build a library of novel, highly functionalised polymers. This library was expanded further by the application of thiol/ene and copper-initiated azide/alkyne click chemistries to introduce further structural diversity into the materials. Selected polymers were found to internalise readily in a model cell line (HEK-293T), and for a subset of these polymers, bright luminescence was observed in the cells upon internalisation, even though neither the cells, nor the polymers, were luminescent on their own at these wavelengths. Our synthetic approach offers a versatile platform for the systematic development and modification of novel macromolecules and for cell-labelling components.
Introduction
Multicomponent reactions (MCRs), which combine 3 or more components in a single, one-pot reaction, have revolutionised our ability to access complex products at high atom economy, high yield and in mild solvents whilst also providing orthogonality with multiple functional groups.1–3 There now exists a wide range of MCRs, each of which combines a unique combination of substrates into a different product. For example, the Passerini 3-component reaction (P3CR) combines an aldehyde with a carboxylic acid and an isocyanide to form an α-acyloxy amide.4 Meanwhile, the A3 reaction involves the reaction of aldehyde, an alkyne and an amine to form a propargyl-amine,5 while further possibilities are offered by the Mannich reaction, which reacts a non-enolizable aldehyde with a primary or secondary amine and an enolizable carbonyl group to form β-amino-carbonyl compounds.6 The enormous diversity in products derived from MCRs has enabled the development of high throughput combinatorial libraries to screen for putative drug products7 and for the total synthesis of natural products.8MCRs also represent an ideal strategy to impart complex chemical functionality to synthetic macromolecules, with the potential to encode previously inaccessible chemistries. There is now a growing body of literature reporting the use of MCRs in polymer chemistry. This includes P3CR chemistries to synthesise novel, multifunctional acrylate9 or diene10 monomers that can be subsequently polymerised to form networks and cross-linked materials. Subsequent work has also seen the P3CR utilised as a technique to functionalise polymers, with pendant carboxylic acid groups.11 The Ugi 4-component reaction has similarly been used to generate diene monomers that were later polymerised by both acyclic diene metathesis and by thiol–ene addition.12 The reaction has been used more broadly in polymer science as a method to link different components into block copolymers.13 MCRs have also played a role in polymer post-modification, including the use of the Kabachnik–Fields reaction to generate α-amino phosphonate motifs on polymer side chains,14 whilst the Debus–Radziszewski reaction has been used to crosslink polymers by converting pendant amine groups into imidazolium groups.15
More recently, MCRs have been directly employed to produce polymers in multicomponent polymerisations (MCPs) where several units are incorporated as single monomers in the repeating polymer chain. Most commonly this has been exploited using an AB + C type step growth MCP including the P3CR,16–18 Ugi 4-component reaction19 and the Debus–Radziszewski reaction,20 but has also been conducted via an A2 + B2 route.10 One of the key advantages of MCPs is the ability to produce a chemically diverse library of synthetic macromolecules often imparting more than one pendant functional group per repeating unit in a single polymerisation step.
Of these MCP techniques, the Passerini-3 component polymerisation (P3CP) remains among the most popular due to its mild conditions with high yields, 100% atom economy, minimal by-products and use of simple acids, aldehydes and isocyanides.17,21–25 P3CP is a relatively recent development, first reported in 2011 via so-called A2 B2 monomers, when simple, unfunctionalized diacid and dialdehyde monomers were combined with four different isocyanides.10 AB type P3CP was later established in 2014, when novel aldehyde/acid monomers were prepared, which were then successfully reacted with a range of isocyanides.16 Subsequently, more functionalised monomers have been successfully integrated into the reaction, resulting in polymers that display a large variety of addressable chemistries.18,22,24,26 Recent advances have seen a number of novel macromaterials synthesised via the P3CR and P3CP, including light27 and heat28 responsive polymers, self-healing materials29 and modified nanocellulose materials for peptide delivery.30
Accordingly, P3CP derived materials offer considerable promise for the development of novel biomaterials, as the diversity in functionality in many ways mimics those of natural polymers. Many current biomaterials are derived from proteins and polysaccharides because of this high functionality, and also due to their intrinsic biodegradability and established biological properties.31 The most explored applications are in wound healing, implants, tissue regeneration scaffolds and drug delivery.32 Synthetic polymers have the potential to replace some of these natural macromolecules in biomaterials applications, as the additional chemical versatility they offer allows the tailoring of properties specifically towards the desired application.31 Passerini derived polymers have a great advantage as biomaterials because they are derived from ester and amide linkages, so are likely to be biodegradable.17,22,26,33 In addition, the range of functional groups tolerated by the reaction allows for the synthesis of materials that can be used as prepared or modified further according to the intended use.
Polycationic biomaterials have been used to enhance drug delivery into cells and as carriers for nucleic acid therapeutics and vaccines.34–39 In addition, a number of polycationic materials have shown intrinsic antibacterial and antifungal properties, owing to their ability to bind to or disrupt cellular membranes.40,41 Accordingly, there is considerable interest in expanding the toolkit for synthesising ionisable polymers and in making libraries of these materials for biological screening.33,37–39 However, the original P3CP reaction is incompatible with primary amine groups, as this leads to competition with the Ugi 4-component reaction, instead forming a bis-amide.42,43 Therefore, although ionisable RAFT polymers have been synthesised from P3CR-derived monomers,44 the preparation of ionisable polymers by direct MCPs remains relatively unexplored. For this polymerisation approach to be translated towards materials or biological applications, there is a need for the development of new monomer families. In this work, we have generated a set of modular diacid monomers and exploited the A2 B2 approach to P3CP to develop a library of novel ionisable polymers. To the best of our knowledge, this is the first reported use of P3CP to directly synthesise protonatable polymers. The range of monomers studied allowed for the integration of a wide range of functionality into our materials, which could then be further enhanced by polymer post-modification via click-chemistries.
Experimental
Materials
The following chemicals were used as received: allylamine (Aldrich), propargylamine (Acros Organics), 1-(3-aminopropyl)imidazole (Aldrich), piperazine (Aldrich), N,N-dimethylethylenediamine (Aldrich), N,N′-dimethylethylenediamine (Aldrich), N,N′,N′′-trimethyldiethylenetriamine (ABCR), tert-butyl acrylate (Acros Organics), magnesium sulphate (Aldrich), acetyl chloride (Aldrich), tert-butyl isocyanide (Aldrich), cyclohexyl isocyanide (Aldrich), n-butyl isocyanide (Aldrich), methyl isocyanoacetate (Alfa Aesar), azobisisobutyronitrile (Aldrich), benzyl mercaptan (Aldrich), thioglycerol (Aldrich), 6-mercaptohexanol (Aldrich), 2-(dimethylamino)ethanethiol hydrochloride (Aldrich), pyridine (Aldrich), 3-mercaptopropionic acid (Aldrich), benzyl bromide (Aldrich), sodium azide (Aldrich), 3-bromo-propanol (Aldrich), 4-bromomethyl benzoic acid (Aldrich), sodium ascorbate (Acros Organics), copper(II) sulphate anhydrous (Aldrich), and ethylenediaminetetraacetic acid disodium salt dihydrate (Aldrich).Glutaraldehyde was purchased as a 50 wt% solution in water (Aldrich) and was lyophilised before use.
Chloroform-d (99.8% D atom), methanol-d4 (99% D atom), dimethyl-sulfoxide-d6 (>99.5% D atom) and deuterium oxide (99.9% D atom) were obtained from Aldrich and used as received.
All solvents were obtained from Fisher Scientific and were of HPLC grade and used without purification.
HEK-293T cells were obtained from ATCC and were cultured in Dulbecco's Modified Eagle's Medium (DMEM) for cell culture (Gibco, ThermoFisher) supplemented with foetal bovine serum, L-glutamine and penicillin streptomycin from ThermoFisher. Prior to fluorescent imaging, cells were incubated in Opti-MEM reduced serum medium (Gibco, ThermoFisher). PrestoBlue reagent was purchased from Promega, UK. Phosphate buffered saline (PBS) was obtained from Gibco, ThermoFisher.
Methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500–550 g mol-1. Analyte samples were filtered through a membrane with 0.22 μm pore size before injection. Experimental molar mass (Mn, SEC) and dispersity (Đ) values of synthesised polymers were determined by conventional calibration using Cirrus Size Exclusion Chromatography (SEC) software.
500–550 g mol-1. Analyte samples were filtered through a membrane with 0.22 μm pore size before injection. Experimental molar mass (Mn, SEC) and dispersity (Đ) values of synthesised polymers were determined by conventional calibration using Cirrus Size Exclusion Chromatography (SEC) software.
Monomer D7 was synthesised by mixing 1-(3-aminopropyl)imidazole with 2.4 equivalents of tert-butyl acrylate and heating the resultant mixture at 70 °C for 48 hours before removing excess acrylate under vacuum.46 The tert-butyl groups were then removed by the same method as described for monomers D1 to D6, but after removing solvents and reagents under vacuum, the product was redissolved in methanol and precipitated from ice cold diethyl ether, repeated a total of three times to improve the purity. The resultant impure mix containing monomer D7 was used crude in the polymerisation reaction.
:1 DCM/N,N-dimethylformamide (DMF), dimethyl sulphoxide (DMSO) or 9:1 water:DMF) and the reaction mix was stirred at room temperature for 30 minutes. At this point 2 equivalents of isocyanide was added and the reaction mix was stirred at room temperature for a further 2 days. Polymers were purified by precipitation twice in diethyl ether, or by dialysis against ultra-pure water as appropriate.
For reactions with 2-(dimethylamino)ethanethiol hydrochloride, additional purification was required before precipitation. An excess of pyridine (2 ml per 100 mg of starting material polymer) was added and the reaction mix was stirred for 10 minutes and then filtered before precipitation from ice cold diethyl ether was conducted as before.
:1 mix of DMF/MeOH along with 1 equivalent of azide, 0.45 equivalents of sodium ascorbate and 0.1 equivalents of copper(II) sulphate (CuSO4). The reaction mix was heated to 45 °C for 24 hours, then diluted with water and dialysed against a 5% w/v solution of ethylenediaminetetraacetic acid disodium salt (EDTA-Na2) in a 1 kDa cut-off membrane for 24 hours and then dialysed for a further 24 hours against ultra-pure water.
Desired azide compounds were prepared by dissolving an appropriate halide in DMF with 1.3 equivalents of sodium azide and stirring at room temperature for 24 hours. The reaction mix was then poured over ice and extracted three times with DCM. The combined organic phase was dried over magnesium sulphate and concentrated under vacuum.
000 cells per well in 200 μL DMEM and allowed to attach for 24 h at 37 °C, 5% CO2 in a humidified incubator to reach 90% confluency. After that, growth media was removed. Polymers were dissolved at 640 μg mL−1 in 25 mM sodium acetate buffer and then diluted 1 in 10 in Opti-MEM to a final concentration of 64 μg mL−1 and 200 μL of polymer/Opti-MEM solution was applied to each well. Cells were incubated for a further 24 hours at 37 °C, 5% CO2 in a humidified incubator, then all media was removed, and the cells were gently washed with 100 μL of PBS before imaging.
Results and discussion
Monomer design and synthesis
In order to produce a chemically diverse library of synthetic macromolecules, we planned to utilise an A2 + B2 + 2C type P3CP with dicarboxylic acid (A2), dialdehyde (B2) and mono-isocyanide (C) monomers to yield a polyester chain. This combination of reagents results in a polymer chain of alternating units formed from the diacid and the dialdehyde, with pendant amide groups primarily formed from the isocyanide component. To incorporate structural diversity into the resultant polymers, we decided to synthesise a range of novel diacid monomers, which could then be combined with the commercially available dialydehyde glutaraldehyde and a range of isocyanides. We sought to incorporate easily functionalisable handles into several of the monomers to allow for post-modification of the resultant polymers. We hypothesised that we could adapt established Aza-Michael addition chemistries to produce a range of beta-amino diacid monomers, which could be further polymerised with the dialdehyde and isocyanide to produce multicomponent polymers with an ionisable polyester backbone and amide pendant chains.45For the initial monomer synthesis, we produced diacid monomer D1 by an Aza-Michael addition between allylamine with tert-butyl acrylate, followed by an acid-mediated deprotection of the tert-butyl groups conducted by generating hydrochloric acid in situ via the reaction of acetyl chloride (AcCl) with water, which served to unveil the desired diacid moiety. Reaction progress could be followed by 1H NMR spectroscopy, showing in the first step the complete conversion of acrylate groups and in the second stage, the complete elimination of the distinctive tertiary-butyl group.
The result was an alkene functionalised pendant group across a β-amino diacid. Following optimisation, this route proceeded under mild conditions, with high yield, and required minimal work-up or purification; the product of each step could be isolated by washing and separation procedures only, with no requirement for intensive procedures like column chromatography or distillation, which makes this chemistry amenable for scale up.
Encouraged by the initial monomer synthesis, we then sought to generate a broader library of starting materials into the monomer synthesis method to adapt the monomer structure and consequently introduce further functionality into the final polymer. The synthesis pathway proved robust enough that the Aza-Michael addition with tert-butyl acrylate proceeded successfully with propargylamine as well as piperazine, N,N-dimethylethylenediamine, N,N′-dimethylethylenediamine and N,N′,N′′-trimethyldiethylenetriamine. Likewise, the deprotection procedure could also be applied to these new substrates without requiring any modifications, to yield diacid monomers D2–D6. In each case, the combined yield over the two steps ranged from 75–90%. Monomers were characterised by NMR, MS and FTIR; in the same manner as for monomer D1, the formation of each monomer could be tracked by 1H NMR spectroscopy, showing the conversion of the acrylate groups following Aza-Michael addition, and elimination of the tertiary-butyl group during deprotection. Following deprotection it was also possible to identify the carboxylic acid moiety by the presence of a broad peak in the FTIR spectrum at approximately 2900–2600 cm−1. Data can be found in full in the ESI.†
However, Michael addition with 1-(3-aminopropyl)imidazole following this protocol proved unsuccessful; with acrylate 1H NMR signals remaining. Instead, it proved necessary to proceed via a high-temperature, solvent-free Michael addition as described by Zhao et al.46 The product was then deprotected as described above, but was impure. Partial purification was achieved by precipitating the reaction mix dropwise in ice-cold diethyl ether. The precipitate containing monomer D7 was then dried and used without further purification in subsequent polymerisation reactions.
Polymerisation
Having produced a library of diacids (D1–7), we then sought to evaluate their ability to participate in an A2 + B2 + 2C P3CP step-growth polymerisation. In the first instance, this was achieved by combining D1 as a model diacid monomer along with a dialdehyde, glutaraldehyde, and a monofunctional isocyanide, tertiary-butyl isocyanide. The isocyanide and dialdehyde monomers were chosen because they are commercially available and functionally simple, so would avoid adding additional synthetic complexity at this stage. In addition, the use of tertiary-butyl isocyanide has previously been well established in Passerini polymerisations.18 D1 was combined with an equivalent amount of glutaraldehyde, before the addition of two equivalents of tertiary butyl isocyanide (I1), and allowed to react at room temperature for 48 hours, as outlined in Fig. 1. Purification was achieved by repeated precipitation from ice-cold diethyl ether, with no additional work-up required. Following this methodology, P3CP proceeded successfully under ambient conditions and delivered the desired polymer D1–I1 as a brittle, pale yellow solid, in good yield (>90%).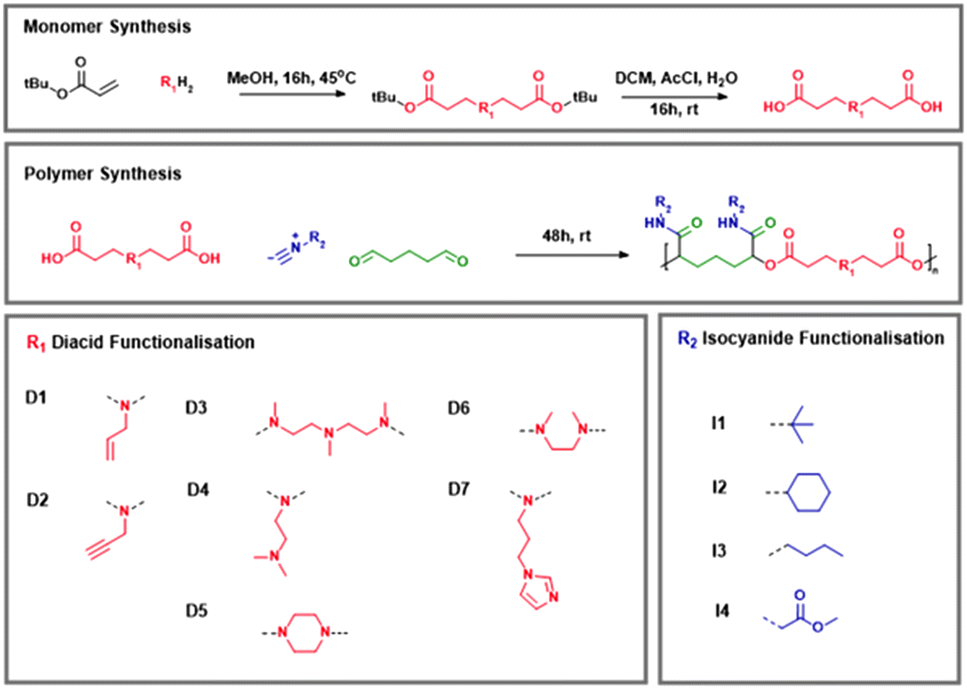 | ||
| Fig. 1 Strategies for the synthesis of novel diacid monomers and Passerini 3-component reaction derived polymers. | ||
Polymerisation was confirmed by both 1H NMR spectroscopy and SEC. The 1H NMR spectrum of D1–I1 is shown in Fig. 2. The distinctive multiplet at 4.95 ppm is highly characteristic of the CH group between the ester and amide groups that form via the P3CR – in this instance, this correlates with the atom labelled C in Fig. 2A. The progress of the reaction was followed by SEC analysis of reaction samples extracted at intervals throughout the course of the reaction (Fig. 2B).
 | ||
| Fig. 2 (A) 1H NMR spectrum of Passerini derived polymer D1–I1; (B) DMF-SEC chromatograms of polymer D1–I1 conducted at intervals throughout the synthesis. | ||
As anticipated for a step-growth polymerisation procedure, initially, we observed the presence of a large number of small oligomers, which appear to be the most prominent peaks in SEC chromatograms up to the 2 hour time point. Over time, these oligomers were replaced with larger polymers, which can be seen by the steady increase in the size of peaks corresponding to higher molecular weights over the next few time points. The molar mass of the polymer increased until it reached a maximum of 5300 g mol−1 at 48 hours, after which, no further increase in molar mass was detected. As a result, all subsequent reactions were stopped after 48 hours.
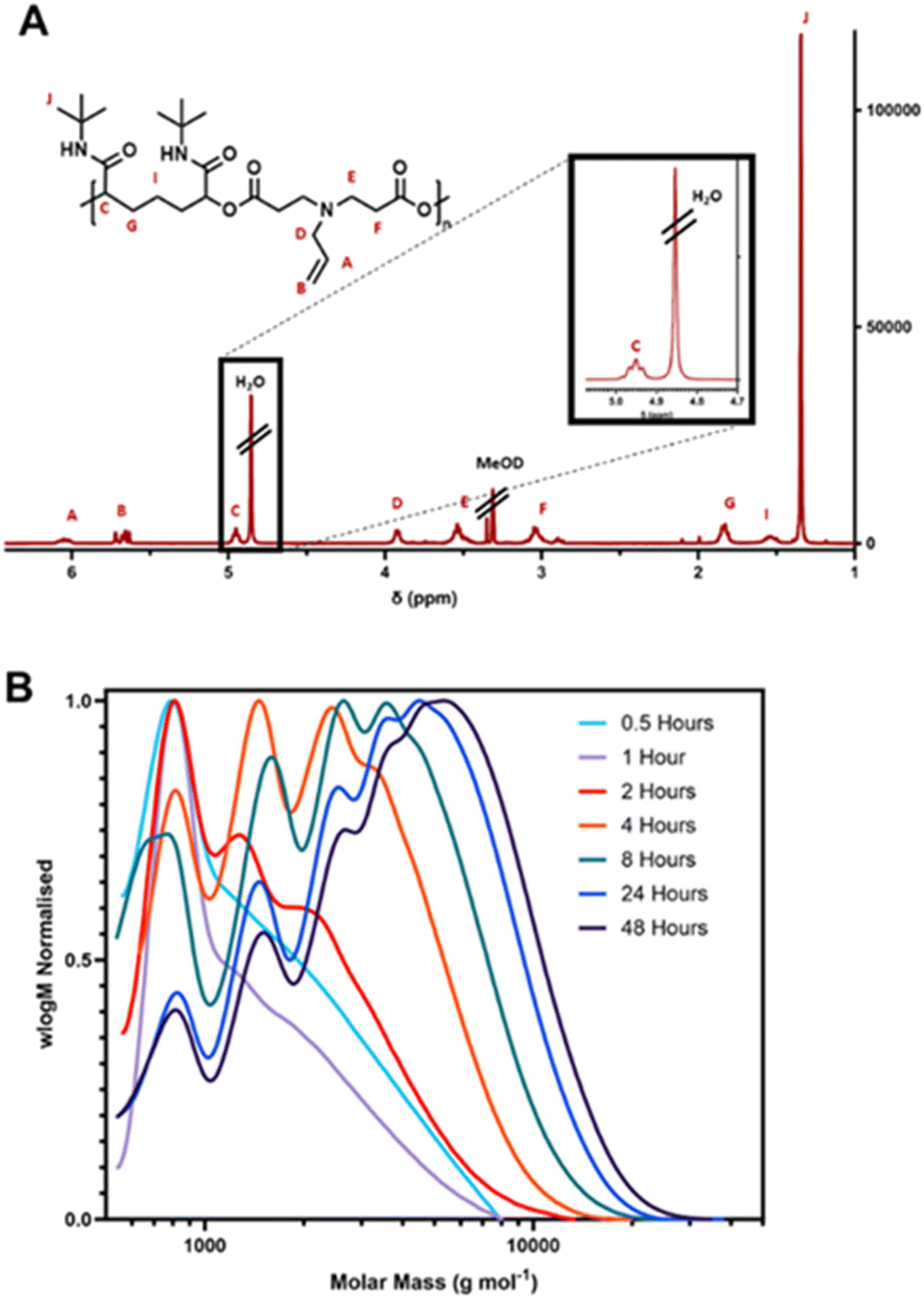
Whilst the initial polymerisation of D1–I1 was conducted in a 9:1 mixture of DCM/DMF, to investigate an alternative solvent system, the polymerisation was also tested in THF. In addition, the polymerisation was conducted at both 20 °C and 35 °C in the DCM/DMF solvent system and at 20 °C, 35 °C and 50 °C in THF. All other elements of the methodology were unchanged. The reaction proceeded successfully under all five conditions, with the target polymer isolated and characterised by NMR. The length of the resultant polymers was studied via SEC, with the results shown below in Fig. 3 and Table 1. The longest polymer chains were obtained from the reactions conducted at 20 °C in DCM/DMF and at 50 °C in THF. However, the THF reaction led to a significantly lower yield (less than half that of the DCM/DMF reaction) and additionally the sample was significantly discoloured; a dark orange colour relative to the pale yellow observed for the product obtained under all the other conditions. For these reasons, the DCM/DMF solvent mix was used for remaining polymerisations unless noted otherwise.
 | ||
| Fig. 3 DMF-SEC chromatograms of polymer D1–I1 synthesised from two different solvents at different temperatures. | ||
Having established that novel diacid monomer D1 was compatible with Passerini polymerisation, we then expanded the range of materials under investigation. Diacid D2 was introduced first and behaved similarly to D1, and when polymerised with glutaraldehyde and tert-butyl isocyanide (I1) led to the synthesis of D2–I1. We then explored the introduction of a wider variety of isocyanide compounds, cyclohexyl isocyanide, n-butyl isocyanide and methyl isocyanoacetate, leading to the successful synthesis of polymers D1–I2, D2–I2, D2–I3 and D2–I4. With cyclohexyl isocyanide and n-butyl isocyanide, these reactions proceeded similarly to the tertiary-butyl isocyanide polymerisations and delivered the desired polymers in yields of over 60%, with full purification achieved by precipitation from ice cold diethyl ether, with no changes to the methodology required. However, while polymerisation with methyl isocyanoacetate led mainly to the desired product, the polymer could not be fully isolated either by precipitation or dialysis and remained impure.
Polymers were subsequently synthesised from the remaining diacid monomers with tertiary-butyl isocyanide. Diacids D3–D6 were insufficiently soluble in DCM/DMF to proceed with the reaction as originally developed, necessitating a change in solvent. Monomers D3, D5 and D6 proved wholly soluble in deionised water, and polymerisation was conducted in 9:1 water/DMF; the addition of DMF assured complete solubilisation of the other monomer compounds and resulted in the greatest polymer chain length relative to the other solvent systems tested. Diacid D4 proved completely soluble in DMSO, and polymerisation was conducted in 100% DMSO, which again resulted in the greatest chain length from the solvent systems tested. Purification of the resultant polymers was conducted not by precipitation, but instead by dialysis against ultrapure water for 24 hours.
Diacid D7 was suitably soluble in DCM/DMF to proceed with polymerisation via the original procedure, but to completely purify the polymer, precipitation proved insufficient, and it was again necessary to dialyse the polymer against ultrapure water for 24 hours.
In total, successful reactions with different combinations of diacid and isocyanide monomers led to the synthesis of 11 unique Passerini derived polymers. The range of structures incorporated shows the versatility of the reaction, and the products are anticipated to have a range of different properties resulting from their structural diversity.
The resultant polymers were all characterised by NMR and SEC, and the yields, masses and dispersities (Đ) are shown in Table 2 below. Full characterisation data can be found within the ESI.†
| Polymer |
M
na (Da) |
Đ | Yield |
|---|---|---|---|
| a Calculated by SEC using 0.05 M LiBr in DMF at 50 °C as the eluent. | |||
| D1–I1 | 5000 | 1.38 | 92% |
| D2–I1 | 4000 | 1.72 | 93% |
| D3–I1 | 4200 | 1.22 | 7% |
| D4–I1 | 1900 | 1.41 | 5% |
| D5–I1 | 1100 | 1.25 | 13% |
| D6–I1 | 4400 | 1.22 | 10% |
| D7–I1 | 4500 | 1.26 | 7% |
| D1–I2 | 2900 | 1.23 | 78% |
| D2–I2 | 2900 | 1.41 | 66% |
| D2–I3 | 3500 | 1.20 | 69% |
| D2–I4 | 3700 | 1.70 | 4% (impure) |
| D1–I1–T1 | 2400 | 1.49 | 41% |
| D1–I1–T2 | 2000 | 1.30 | 68% |
| D1–I1–T3 | 2800 | 1.41 | 42% |
| D1–I1–T4 | 2400 | 1.24 | 17% |
| D1–I1–T5 | 4700 | 1.31 | 72% |
| D1–I2–T2 | 2600 | 1.51 | 18% |
| D1–I2–T4 | 2900 | 2.03 | 61% |
| D2–I1–A1 | 4300 | 1.22 | 8% |
| D2–I1–A2 | 6700 | 1.31 | 50% |
| D2–I1–A3 | 2800 | 1.36 | 39% |
| D2–I2–A1 | 3500 | 1.24 | 51% |
| D2–I2–A2 | 3200 | 1.34 | 40% |
| D2–I2–A3 | 3700 | 1.25 | 62% |
| D2–I3–A2 | 2000 | 1.63 | 31% |
Polymer post-functionalisation
Post-modification of polymers offers an opportunity to incorporate additional functional groups onto the polymer chain, providing a means to modify the properties of the polymers, potentially tailoring them towards desired applications. Post-functionalisation has been applied across polymer science to achieve a wide range of goals, from cross-linking polymers into hydrogels,49 incorporation of sugar units for interaction with carbohydrate binding sites50 and the addition of fluorescent labels for polymer imaging.48 Such modification requires the polymers to possess functional groups that are compatible with further reactions for modification, so monomers D1 and D2 were designed to incorporate alkene or alkyne functional group side chains. These motifs are amenable to post-functionalisation via thiol/ene and azide/alkyne click chemistries, and are well tolerated by the P3CP process, so are preserved in the resultant polymer chains. Click chemistries offer simple yet efficient means to modify these handles with a wide variety of different substrates. They offer further benefits as they are typically highly selective and can proceed under mild conditions.47 Developing click methodologies compatible with our novel polymer system would therefore open up a large amount of chemical space for future expansion of the polymer library. | ||
| Fig. 4 (A) Synthesis of post-modified Passerini-derived polymers via AIBN-initiated thiol/ene reaction. (B) 1H NMR spectra of polymer D1–I1 (top) and the product D1–I1–T1 (bottom) following functionalisation via thiol/ene reaction. | ||
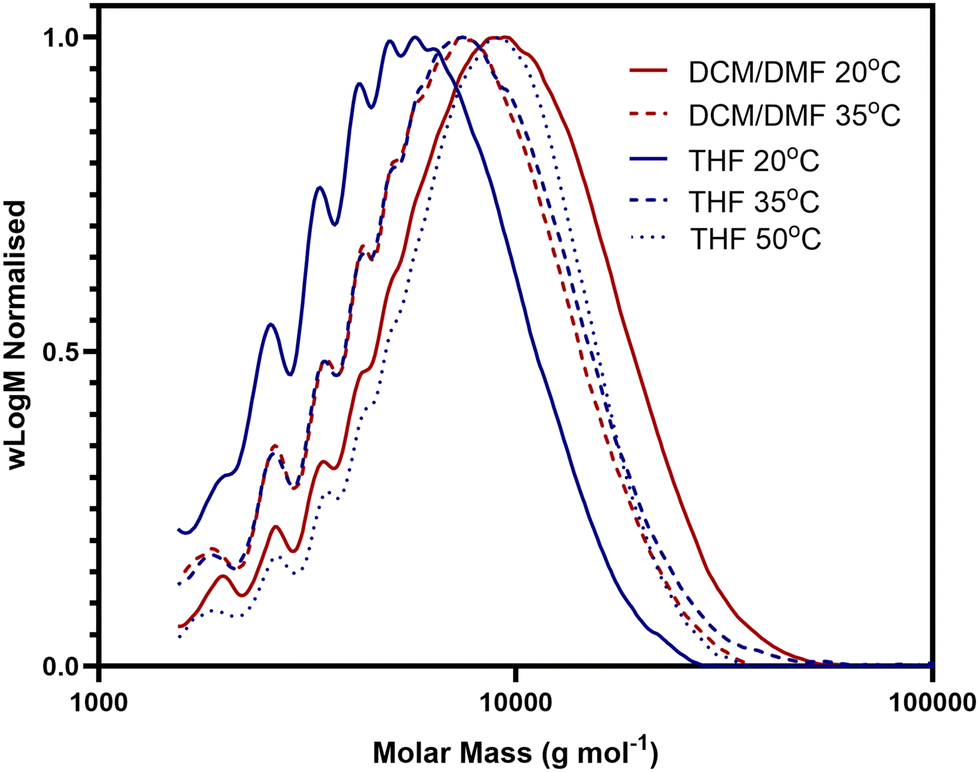
Passerini derived polymers have previously been shown to be compatible with thiol/ene chemistries and so we investigated a range of techniques to develop a suitable protocol for the modification of our novel polymers derived from diacid monomer D1.47,55 In reactions between polymer D1–I1 and benzyl mercaptan, thermally initiated coupling using azobisisobutyronitrile (AIBN) proved superior to thermal initiation via 4,4′-azobis(4-cyanopentanoic acid) (ACVA), or UV initiation reactions using 2,2-dimethoxy-2-phenylacetophenone (DMPA). Following reaction progression by NMR, we were able to establish reaction completion by the loss of alkene peaks at approximately 5.5–6.0 ppm. We observed that the AIBN reaction reached completion overnight in just one step, whereas the ACVA and DMPA reaction required multiple days along with repeated dosing of both catalyst and thiol for complete conversion of the alkene group. This difference may result from the higher temperatures used for the AIBN reaction, as the polymers synthesised here are significantly sterically hindered and lack torsional flexibility owing to their structure. This rigidity and hindrance may restrict access to the reaction site at lower temperatures. Ultimately, we established an AIBN catalysed procedure that involved combining the alkene functionalised polymer with 10 equivalents of thiol and 0.55 equivalents of AIBN under an inert atmosphere. Reaction progress was followed by NMR (Fig. 4B) and SEC (Fig. S1†), allowing us to observe the complete loss of the alkene peaks (highlighted in blue) from the polymer, and the presence of the new side chain peaks (highlighted in green). Following the successful thiol/ene reaction, polymers were purified by repeated precipitation from ice-cold diethyl ether.
The initial procedure was developed with benzyl mercaptan, due to the distinctive NMR signature of the benzene ring, which was beneficial for early analysis during the method development. Following success with this model substrate, the same methodology was successfully applied to both polymers D1–I1 and D1–I2 and a range of different thiol substrates, chosen to incorporate a broad range of different functionalities and chemical properties, with each functional group anticipated to have different impacts on polymer properties. This allowed us to develop a small library of thiol/ene functionalised polymers (Fig. 4A), detailed in Table 2. The molecular weight of the polymers as measured by SEC was in many cases observed to decrease following the thiol/ene reaction, this was generally considered to be a result of the changing identity of the polymer side chain impacting behaviour of the polymer within the column, which in turn could impact elution times, although it is also possible that the high temperatures used could have led to some degree of degradation of the polymer chain during the reaction.
We therefore sought to modify the alkyne functionalised polymers derived from monomer D2 by reaction with azide functionalised substrates. Polymer functionalisation was initially tested between polymer D2–I1 and benzyl azide as a test substrate. We utilised a synthetic route that used sub-stoichiometric amounts of copper and regenerated the active species throughout the reaction.48 In this instance, the polymers were combined with 1 equivalent of azide along with 0.45 equivalents of sodium ascorbate and just 0.1 equivalents of copper sulphate. Polymers were then purified by dialysis against a solution of EDTA disodium salt, and then subsequent dialysis against deionised water, followed by lyophilisation.
Once this protocol was successfully established with D2–I1 and benzyl azide it was then expanded to the remaining D2-derived polymers and other azide substrates (Fig. 5A), once more, these substrates were chosen to test diverse functional groups both for compatibility with the reaction protocol and for impacts on polymer properties. Reaction progress was followed by NMR (Fig. 5B) and SEC (Fig. S2†), allowing us to track completion by observing the complete loss of the alkyne peak (highlighted in blue, Fig. 5B) from the polymer D2–I1. Additionally, the successful formation of the triazole ring could be observed by the presence of the distinctive signal for the triazole proton at approximately 7.3 ppm, which can be seen in Fig. 5B (highlighted in green) for the product D2–I1–A2. FTIR spectra from before and after the reaction show the loss of the alkyne C–H stretch at 3300 cm−1 and CΞC stretch at 2140 cm−1 showing conversion of the alkyne group; (Fig. S3†). As before, some of the functionalised polymers were found to differ in apparent molar mass compared to the unfunctionalised starting materials. This could again be the result of the functionalised polymers interacting differently with the SEC column, leading to different elution times.
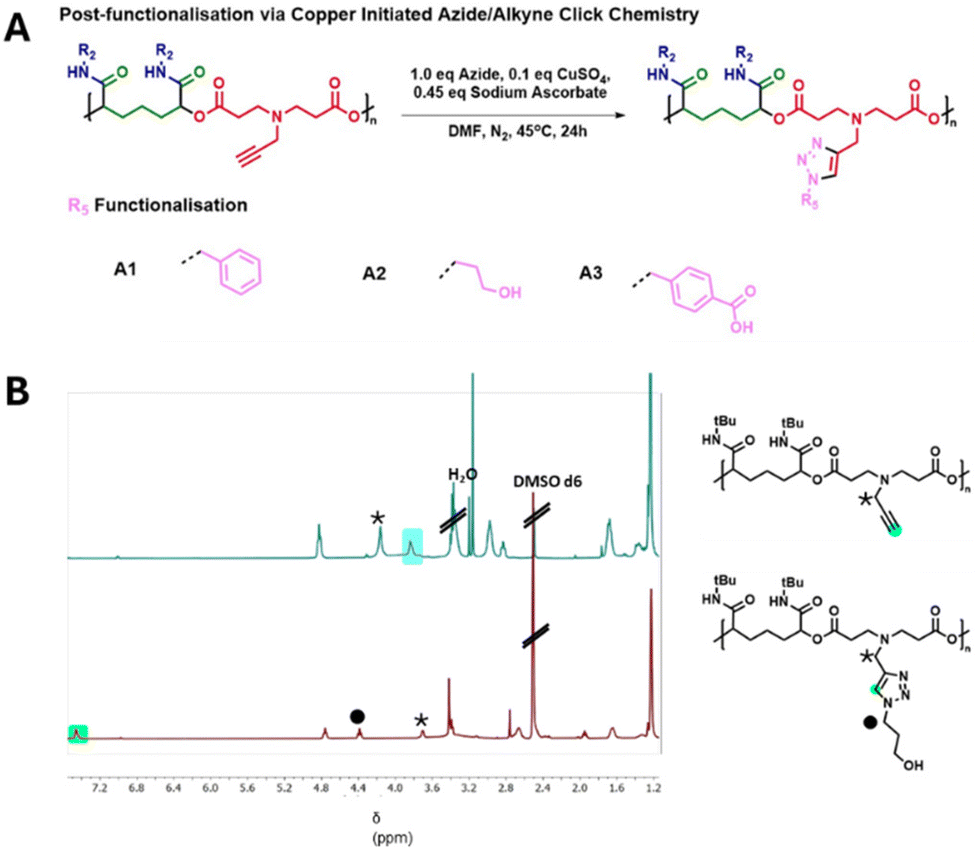 | ||
| Fig. 5 (A) Synthesis of post-modified Passerini derived polymers via copper-initiated azide/alkyne click. (B) 1H NMR spectra of polymer D2–I1 (top) and the product D2–I1–A2 (bottom) following functionalisation via copper/click chemistry. | ||
Luminescence properties of polymers in intracellular environments
Many synthetic polymers have found applications in biology, from carriers for enhanced delivery of therapeutics,22 to synthetic extra-cellular scaffolds.63 In our previous work,22 we found that amphiphilic Passerini block co-polymers with no charged or reactive groups in their backbone or side chains were well-tolerated in specific cell lines at concentrations up to 0.5 mg mL−1. However, for the materials in this study, the presence of ionisable amines in the polymer main chains was a potential concern for membrane interaction or cytotoxicity. Consequently, we thought it prudent to test how well human cells, in this case, the widely used HEK-293T cell line, would tolerate the amine-containing Passerini polymers. Initial assays showed that the polymers were not toxic in this cell line (Fig. S4†). However, microscopy investigations revealed that, despite lacking typical chromophores, a number of these polymers induced luminescence in the GFP channel when incubated with the HEK-293T cells. The results of this can be seen in Fig. 6.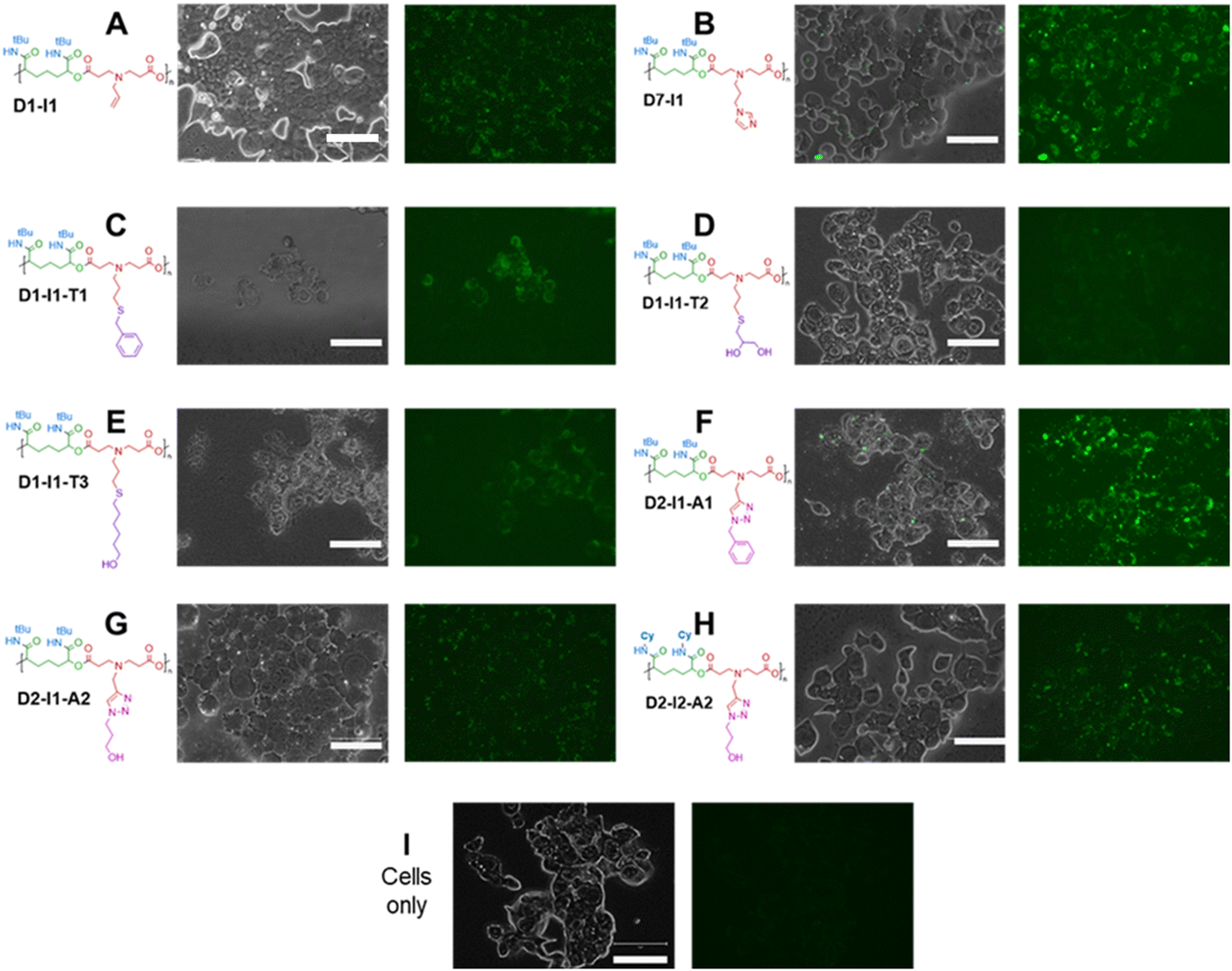 | ||
| Fig. 6 Combined brightfield and fluorescence microscopy images (left in each case) and fluorescence channel images (right in each pair), depicting HEK-293T cells incubated for 24 hours with polymers at 64 μg mL−1 in OptiMEM reduced serum medium. Fluorescence imaging captured with excitation at 482 nm, emission at 524 nm. (A) D1–I1; (B) D7–I1; (C) D1–I1–T1; (D) D1–I1–T2; (E) D1–I1–T3; (F) D2–I1–A1; (G) D2–I1–A2; (H) D2–I2–A2; (I) cells only (polymer free), media only control. Fluorescence images all enhanced by 10% for contrast to facilitate visualisation. Scale bar = 75 μm. | ||
Polymeric luminescence without conventional fluorophores is not unprecedented, with studies of PAMAM dendrimers dating back to 2001 which reported unexpected optical properties, and the topic was reviewed in 2019.64 More recent papers include observation of green fluorescence in poly(ester amides) by Zia et al.65 and an observation of fluorescence arising from peptides without ‘conventional’ fluorophores from Monti et al.66 In most instances, the occurrence of what has been termed non-traditional intrinsic luminescence (NTIL) has been ascribed to chain-association and confinement phenomena, in a manner similar to aggregation-induced emission (AIE). One common feature is the presence of tertiary amine functionality, and it has also been noted that increasing the rigidity of a potential light-emitting (or lumogenic) unit within a polymeric structure can increase the emission intensity of the material.67–69 All the polymers in this study contained tertiary amines, and the most emissive, i.e. D7–I1 and D2–I1–A1, contained aromatic units, but these were not highly conjugated. Rigidity may have been a factor, as the pendant t-butyl groups at the side-chain amides would likely reduce mobility adjacent to these sites, with the effect of ‘locking’ parts of the polymer main chain in set conformations. Thus, some of the criteria for NTIL are met by these Passerini systems. However, the luminescence phenomena were only observed when the polymers were incubated with cells, suggesting that other components were important in the induction of light emission. To eliminate the possibility of cellular autofluorescence, cells from the same cell line were incubated in the absence of polymer and imaged under the same conditions (Fig. 6I). No light emission was detected under these conditions and so we conclude that the luminescence was due to a combination of the polymers with cellular components rather than the cells or polymers alone. Solutions of a range of polymers were prepared in different aqueous solvents and components of cell media, and the resulting emission profiles were measured. We also tested polymer luminescence in the presence and absence of copper ions, in 25 mM sodium acetate buffer, ultra-pure water, phosphate buffered saline and in OptiMEM reduced serum medium (Fig. S5–S12). In all these cases, no significant luminescence was apparent unless the polymers were internalised in the HEK-293T cells. We also carried out experiments in a different cell line, the triple negative breast cancer MDA-MB-231 line, and again luminescence was observed with the brightest emission from polymer D2–I1–A1 (ESI Fig. S13†). It seems likely that aggregation occurred intracellularly, as evidenced by punctate luminescence in discrete regions in the cellular interiors (ESI Fig. S14†). However, further dye-labelling experiments to determine specific intracellular compartment co-localisation were not able to give unequivocal data owing to the broad band light emission of the polymers. We suggest that macromolecular crowding in regions of the cellular interior may have caused aggregation-induced emission from the polymers. It is well-established that the cytosolic interior is a tightly regulated environment, with high concentrations of biomacromolecules and a fine balance of water and electrolytes. It is also known that under stress conditions, cells can alter processes to reduce damage, including the aggregation of macromolecules in tight networks to mitigate dehydration and by formation of ‘stress granules’ with phase-separated regions of nucleic acids and their associated binding proteins.70 Although we observed no acute toxicities of the polymers in this study, it is possible that the cells in culture did experience temporary osmotic shock on the ingress of the Passerini polymers, and any resulting increase in macromolecular crowding would result in aggregation, and alignment of acylamino, carbonyl and amine groups on adjacent polymer chains. These functional groups have all been reported to generate non-traditional intrinsic luminescence,71,72 and experiments are now ongoing to establish a detailed mechanism underlying this cell-induced luminescence with these Passerini-type polymers.
Conclusions
The methods developed here have led to the successful development of a range of novel, modular diacid monomers, which can be polymerised by exploiting Passerini 3-component polymerisation to synthesise a library of novel polymers via a robust, efficient procedure. This led to a broad polymer library, which was further enhanced by utilising thiol/ene and azide/alkyne click chemistries to achieve a wide degree of post-functionalisation. Post-functionalisation is anticipated to impact the properties of the polymers, and could be exploited further for a process of rapid, iterative polymer design so that the resultant polymers can be adapted for a wide range of applications. A large area of chemical space remains untapped in the modification of these polymers and many more substrates could be integrated into the monomer synthesis, polymer synthesis and the post-functionalisation process. Future studies will explore further the use of these materials to label cells without conventional fluorophores.Author contributions
Lewis O'Shaughnessy conceptualisation, methodology, validation, formal analysis, investigation, visualisation, writing – original draft. Mariarosa Mazza: conceptualisation, supervision; writing – review& editing; funding acquisition, resources Akosua Anane-Adjei: conceptualisation, supervision, writing – review & editing, funding acquisition, resources Naoto Hori: conceptualisation, supervision, writing – review & editing. Pratik Gurnani: conceptualisation, supervision, writing – review & editing. Cameron Alexander: conceptualisation, supervision; writing – review & editing; funding acquisition, resources.Data availability
All data in this paper are included in the ESI.† Other relevant data are available on request via University College London, the University of Nottingham, p.gurnani@ucl.ac.uk or cameron.alexander@nottingham.ac.uk.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank UKRI/EPSRC for funding [grant EP/S023054/1] and the Royal Society [Wolfson Research Merit Award WM150086 to CA]. We also thank Tom Hyde, Esme Ireson and Paul Cooling for expert technical support. The Nanoscale & Macroscale Research Centre (NMRC) is acknowledged for providing the facilities for TEM and related analysis, and we thank the School of Life Sciences imaging facility (SLIM) and staff for their contribution to this publication.References
- H. Liu, P. Kanjilal and S. Thayumanavan, Polym. Int., 2022, 71, 562–568 CrossRef CAS.
- B. Wei, W. Li, Z. Zhao, A. Qin, R. Hu and B. Z. Tang, J. Am. Chem. Soc., 2017, 139, 5075–5084 CrossRef CAS PubMed.
- R. Kakuchi, Angew. Chem., Int. Ed., 2014, 53, 46–48 CrossRef CAS PubMed.
- S. Kumar, A. Arora, S. Kumar, R. Kumar, J. Maity and B. K. Singh, Eur. Polym. J., 2023, 190, 112004 CrossRef CAS.
- I. Jesin and G. C. Nandi, Eur. J. Org. Chem., 2019, 2019, 2704–2720 CrossRef CAS.
- M. Arend, B. Westermann and N. Risch, Angew. Chem., Int. Ed., 1998, 37, 1044–1070 CrossRef PubMed.
- S. Oelmann, A. Travanut, D. Barther, M. Romero, S. M. Howdle, C. Alexander and M. A. R. Meier, Biomacromolecules, 2019, 20, 90–101 CrossRef CAS PubMed.
- M. Konstantinidou, K. Kurpiewska, J. Kalinowska-Tłuscik and A. Dömling, Eur. J. Org. Chem., 2018, 2018, 6714–6719 CrossRef CAS.
- A. Sehlinger, O. Kreye and M. A. R. Meier, Macromolecules, 2013, 46, 6031–6037 CrossRef CAS.
- O. Kreye, T. Tóth and M. A. R. Meier, J. Am. Chem. Soc., 2011, 133, 1790–1792 CrossRef CAS PubMed.
- B. Alkan, O. Daglar, S. Luleburgaz, B. Gungor, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Polym. Chem., 2022, 13, 258–266 RSC.
- O. Kreye, O. Türünç, A. Sehlinger, J. Rackwitz and M. A. Meier, Chemistry, 2012, 18, 5767–5776 CrossRef CAS PubMed.
- B. Yang, Y. Zhao, C. Fu, C. Zhu, Y. Zhang, S. Wang, Y. Wei and L. Tao, Polym. Chem., 2014, 5, 2704–2708 RSC.
- R. Kakuchi and P. Theato, ACS Macro Lett., 2014, 3, 329–332 CrossRef CAS PubMed.
- K.-S. Krannig, D. Esposito and M. Antonietti, Macromolecules, 2014, 47, 2350–2353 CrossRef CAS.
- A. Sehlinger, R. Schneider and M. A. R. Meier, Eur. Polym. J., 2014, 50, 150–157 CrossRef CAS.
- X.-X. Deng, L. Li, Z.-L. Li, A. Lv, F.-S. Du and Z.-C. Li, ACS Macro Lett., 2012, 1, 1300–1303 CrossRef CAS PubMed.
- M. Zhao, N. Liu, R.-H. Zhao, P.-F. Zhang, S.-N. Li, Y. Yue and K.-L. Deng, ACS Appl. Bio Mater., 2019, 2, 1714–1723 CrossRef CAS PubMed.
- A. Sehlinger, P.-K. Dannecker, O. Kreye and M. A. R. Meier, Macromolecules, 2014, 47, 2774–2783 CrossRef CAS.
- S. Saxer, C. Marestin, R. Mercier and J. Dupuy, Polym. Chem., 2018, 9, 1927–1933 RSC.
- A. K. Pearce, A. Travanut, B. Couturaud, V. Taresco, S. M. Howdle, M. R. Alexander and C. Alexander, ACS Macro Lett., 2017, 6, 781–785 CrossRef CAS PubMed.
- A. Travanut, P. F. Monteiro, S. Smith, S. M. Howdle, A. M. Grabowska, B. Kellam, M. A. R. Meier and C. Alexander, J. Mater. Chem. B, 2022, 10, 3895–3905 RSC.
- J. Zhang, X.-X. Deng, F.-S. Du and Z.-C. Li, in Sequence-Controlled Polymers: Synthesis, Self-Assembly, and Properties, American Chemical Society, 2014, vol. 1170, ch. 15, pp. 223–234 Search PubMed.
- X.-W. Kan, X.-X. Deng, F.-S. Du and Z.-C. Li, Macromol. Chem. Phys., 2014, 215, 2221–2228 CrossRef CAS.
- Y. Cui, M. Zhang, F.-S. Du and Z.-C. Li, ACS Macro Lett., 2017, 6, 11–15 CrossRef CAS PubMed.
- A. Travanut, P. F. Monteiro, S. Oelmann, S. M. Howdle, A. M. Grabowska, P. A. Clarke, A. A. Ritchie, M. A. R. Meier and C. Alexander, Macromol. Rapid Commun., 2021, 42, 2000321 CrossRef CAS PubMed.
- X.-F. Sun, X. Zhang, S.-P. Song, Y.-Q. Yao, Y. Zhang, C.-L. Wang, J.-J. Sun and Q.-F. Wang, Chin. J. Polym. Sci., 2024, 42, 570–578 CrossRef CAS.
- Y. Zhang, S. Liu and C. Xu, Polym. Chem., 2024, 15, 2677–2686 RSC.
- Y. Nakamura, Y.-S. Huang, C.-F. Huang and S. Samitsu, Chem. Commun., 2024, 60, 5270–5273 RSC.
- Y. Khine, Y. Li, W. Ge and M. H. Stenzel, Colloid Polym. Sci., 2024 DOI:10.1007/s00396-024-05265-8.
- A. Girotti, S. Escalera-Anzola, I. Alonso-Sampedro, J. González-Valdivieso and F. J. Arias, Pharmaceutics, 2020, 12(11), 1115 CrossRef CAS PubMed.
- E. Troy, M. A. Tilbury, A. M. Power and J. G. Wall, Polymers, 2021, 13(19), 3321 CrossRef CAS PubMed.
- A. Elsherbeny, H. Bayraktutan, U. C. Oz, C. Moloney, J. C. Ashworth, A. M. Grabowska and C. Alexander, Adv. Ther., 2024, 7, 2300330 CrossRef CAS.
- B. Ballarín-González and K. A. Howard, Adv. Drug Delivery Rev., 2012, 64, 1717–1729 CrossRef PubMed.
- A. Bernkop-Schnürch, Adv. Drug Delivery Rev., 2018, 136–137, 62–72 CrossRef PubMed.
- A. E. Neitzel, G. X. De Hoe and M. V. Tirrell, Curr. Opin. Solid State Mater. Sci., 2021, 25, 100897 CrossRef CAS.
- H. Bayraktutan, R. J. Kopiasz, A. Elsherbeny, M. Martinez Espuga, N. Gumus, U. C. Oz, K. Polra, P. F. McKay, R. J. Shattock, P. Ordóñez-Morán, A. Mata, C. Alexander and P. Gurnani, Polym. Chem., 2024, 15, 1862–1876 RSC.
- H. Bayraktutan, P. Symonds, V. A. Brentville, C. Moloney, C. Galley, C. L. Bennett, A. Mata, L. Durrant, C. Alexander and P. Gurnani, Biomaterials, 2024, 311, 122647 CrossRef CAS PubMed.
- N. K. Dastgerdi, N. Gumus, H. Bayraktutan, D. Jackson, K. Polra, P. F. McKay, F. Atyabi, R. Dinarvand, R. J. Shattock, L. Martinez-Pomares, P. Gurnani and C. Alexander, Nanoscale Adv., 2024, 6, 1409–1422 RSC.
- X. Y. Cai, J. Z. Li, N. N. Li, J. C. Chen, E.-T. Kang and L. Q. Xu, Biomater. Sci., 2016, 4, 1663–1672 RSC.
- K. Kasza, B. Richards, S. Jones, M. Romero, S. N. Robertson, K. R. Hardie, P. Gurnani, M. Cámara and C. Alexander, ACS Appl. Mater. Interfaces, 2024, 16, 5412–5425 CrossRef CAS PubMed.
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef.
- R. M. I. Ugi, U. Fetzer and C. Steinbrückner, Angew. Chem., 1959, 71, 386 Search PubMed.
- Z. Zha, J. Li and Z. Ge, ACS Macro Lett., 2015, 4, 1123–1127 CrossRef CAS PubMed.
- N. Suleman, R. S. Kalhapure, C. Mocktar, S. Rambharose, M. Singh and T. Govender, RSC Adv., 2015, 5, 34967–34978 RSC.
- X. Zhao, J. Chen, M. Qiu, Y. Li, Z. Glass and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 20083–20089 CrossRef CAS PubMed.
- Y.-Z. Wang, X.-X. Deng, L. Li, Z.-L. Li, F.-S. Du and Z.-C. Li, Polym. Chem., 2013, 4, 444–448 RSC.
- A. B. Anane-Adjei, N. L. Fletcher, R. J. Cavanagh, Z. H. Houston, T. Crawford, A. K. Pearce, V. Taresco, A. A. Ritchie, P. Clarke, A. M. Grabowska, P. R. Gellert, M. B. Ashford, B. Kellam, K. J. Thurecht and C. Alexander, Biomater. Sci., 2022, 10, 2328–2344 RSC.
- A. E. Rydholm, C. N. Bowman and K. S. Anseth, Biomaterials, 2005, 26, 4495–4506 CrossRef CAS PubMed.
- L. Nurmi, J. Lindqvist, R. Randev, J. Syrett and D. M. Haddleton, Chem. Commun., 2009, 2727–2729, 10.1039/B904249K.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- R. L. A. David and J. A. Kornfield, Macromolecules, 2008, 41, 1151–1161 CrossRef CAS.
- A. Gress, A. Völkel and H. Schlaad, Macromolecules, 2007, 40, 7928–7933 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS PubMed.
- M. Uygun, M. A. Tasdelen and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 103–110 CrossRef CAS.
- J.-F. Lutz, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS PubMed.
- J. A. Opsteen and J. C. M. van Hest, Chem. Commun., 2005, 57–59, 10.1039/B412930J.
- N. Vinson, Y. Gou, C. R. Becer, D. M. Haddleton and M. I. Gibson, Polym. Chem., 2011, 2, 107–113 RSC.
- G. Acik, C. E. Cansoy and M. A. Tasdelen, J. Appl. Polym. Sci., 2019, 136, 47072 CrossRef.
- B. Pektas, G. Sagdic, O. Daglar, S. Luleburgaz, U. S. Gunay, G. Hizal, U. Tunca and H. Durmaz, Polymer, 2022, 253, 124989 CrossRef CAS.
- N. V. Tsarevsky, K. V. Bernaerts, B. Dufour, F. E. Du Prez and K. Matyjaszewski, Macromolecules, 2004, 37, 9308–9313 CrossRef CAS.
- D. D. Díaz, S. Punna, P. Holzer, A. K. McPherson, K. B. Sharpless, V. V. Fokin and M. G. Finn, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 4392–4403 CrossRef.
- H.-Y. Mi, X. Jing and L.-S. Turng, J. Cell. Plast., 2014, 51, 165–196 CrossRef.
- D. A. Tomalia, B. Klajnert-Maculewicz, K. A. M. Johnson, H. F. Brinkman, A. Janaszewska and D. M. Hedstrand, Prog. Polym. Sci., 2019, 90, 35–117 CrossRef CAS.
- A. Zia, J. R. Finnegan, J. P. Morrow, W. Yin, J. J. Jasieniak, E. Pentzer, S. Thickett, T. P. Davis and K. Kempe, Biomacromolecules, 2021, 22, 4794–4804 CrossRef CAS PubMed.
- A. Monti, C. Bruckmann, F. Blasi, M. Ruvo, L. Vitagliano and N. Doti, Chem. Commun., 2021, 57, 3720–3723 RSC.
- P. K. S. Antharjanam, M. Jaseer, K. N. Ragi and E. Prasad, J. Photochem. Photobiol., A, 2009, 203, 50–55 CrossRef CAS.
- L. Pastor-Pérez, Y. Chen, Z. Shen, A. Lahoz and S. E. Stiriba, Macromol. Rapid Commun., 2007, 28, 1404–1409 CrossRef.
- X. Liu, Y. Zeng, J. Liu, P. Li, D. Zhang, X. Zhang, T. Yu, J. Chen, G. Yang and Y. Li, Langmuir, 2015, 31, 4386–4393 CrossRef CAS PubMed.
- A. Kitamura, S. Oasa, H. Kawaguchi, M. Osaka, V. Vukojević and M. Kinjo, Sci. Rep., 2023, 13, 11834 CrossRef CAS PubMed.
- N. Jiang, Y.-J. Meng, X. Pu, C.-Y. Zhu, S.-H. Tan, Y.-H. Xu, Y.-L. Zhu, J.-W. Xu and M. R. Bryce, ACS Mater. Lett., 2024, 24–31, DOI:10.1021/acsmaterialslett.4c02100.
- N. Jiang, D. Zhu, Z. Su and M. R. Bryce, Mater. Chem. Front., 2021, 5, 60–75 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01165a |
| This journal is © The Royal Society of Chemistry 2025 |