
Polymerization-induced self-assembly nanomaterials based on dynamic covalent bonds
Zhenyu
Wan
 ,
Nankai
An
,
Chang
Xu
,
Mingxin
Zheng
and
Jinying
Yuan
*
,
Nankai
An
,
Chang
Xu
,
Mingxin
Zheng
and
Jinying
Yuan
*
Department of Chemistry, Key Laboratory of Organic Optoelectronics & Molecular Engineering of Ministry of Education, Tsinghua University, Beijing, China. E-mail: yuanjy@mail.tsinghua.edu.cn
First published on 26th December 2024
Abstract
Polymerization-induced self-assembly (PISA) has emerged as a versatile and powerful methodology for the in situ generation of polymeric nanostructures with diverse morphologies and functionalities. Currently, dynamic covalent bonds (DCBs), known for their reversible and stimulus-responsive nature, offer a sophisticated tool for the precise modulation of polymer assemblies. The incorporation of DCBs into PISA facilitates the disaggregation, morphological transition, surface modification, controlled release, and intra- and inter-micellar crosslinking of assemblies, thereby expanding the applications of PISA assemblies in drug delivery, targeted controlled release, molecular recognition, sensing, and modifiable micelle-crosslinked gels. The combination of PISA with DCBs offers a promising approach for designing adaptive and tunable block copolymer nano-object systems, providing new insights and opportunities in the field of polymer chemistry. This review discusses the integration of dynamic covalent chemistry, including disulfide bonds, boronate ester bonds, imine bonds, and [2 + 2] cycloaddition, within the PISA framework and provides guidelines for future research on the development of dynamically responsive and multifunctional PISA nanomaterials.
 Zhenyu Wan | Zhenyu Wan received her B.S. degree in 2022 from the Department of Chemistry, Tsinghua University. She is pursuing her Ph.D. under the supervision of Prof. Jinying Yuan in the Department of Chemistry, Tsinghua University. Her current research mainly focuses on polymerization-induced self-assembly. |
 Nankai An | Nankai An received his B.S. degree in 2020 from the Department of Chemistry, Tsinghua University. He is pursuing his Ph.D. under the supervision of Prof. Jinying Yuan in the Department of Chemistry, Tsinghua University. His current research mainly focuses on polymerization-induced self-assembly. |
 Jinying Yuan | Prof. Jinying Yuan received her Bachelor's degree (1987) from the Department of Applied Chemistry, University of Science and Technology of China (USTC) and received her Ph.D. (2000) from the Department of Polymer Science and Engineering, USTC. After two years of postdoctoral research at the College of Chemistry and Molecular Engineering, Peking University, she joined the Department of Chemistry, Tsinghua University in 2002, where she became a full professor in 2011. Her current research is mainly focused on the methodology of polymer synthesis and functional polymer materials, especially on controlled polymerization, stimuli-responsive polymers and polymerization-induced self-assembly. |
1. Introduction
Over the past few decades, the self-assembly of block copolymers has attracted significant academic interest due to its ability to generate ordered nanostructures with diverse morphologies, including spheres, rods, lamellae, vesicles, and more complex assemblies through microphase separation of chemically distinct blocks.1,2 The conventional solution self-assembly techniques typically generate copolymer nano-assemblies in rather low concentrations with limited suitability for subsequent commercial applications. Since the discovery and development of polymerization-induced self-assembly (PISA) by Pan's group, a novel one-pot method for the preparation of stable, uniform and shape-controllable polymeric nanomaterials based on the phase separation and re-organization of block copolymers has attracted significant attention in polymer chemistry. As an innovative and particularly noteworthy method with significant advantages in terms of efficiency and convenience, PISA enables the straightforward preparation of block copolymer nanoparticles (NPs) with high solid content and various morphologies.3,4 In this process, solvophilic polymer chains act as stabilizers and the specific monomers polymerize into new insoluble blocks. As the degree of polymerization (DP) of the solvophobic segments increases, the growing copolymers undergo in situ self-assembly, yielding NPs with specific morphologies.5–11 The copolymer nanomaterials prepared by PISA have found potential applications in controlled delivery,12–19 bioimaging,20–22 nanoactuators,23–25 catalysis,26–30 functional coatings,31–33 sensing,34,35 and other fields.36–41Dynamic covalent bonds (DCBs) have emerged as a powerful concept in materials science, garnering extensive research attention across multiple areas. While generally exhibiting relatively high chemical stability similar to classical irreversible covalent bonds, DCBs can also uniquely undergo reversible formation, cleavage, or exchange reactions in response to external stimuli (temperature, pH, light, chemical reagents, etc.).42–44 The past few decades have witnessed remarkable advances in dynamic covalent chemistry, from typically utilized types (such as disulfides, esters, imines, boroxine/boronic esters, Diels–Alder reactions, etc.) to developing novel reversible linkages (such as X-yne adducts,45,46 reversible isocyanate chemistry,47–50etc.), and the further establishment of orthogonal systems capable of independent multi-stimuli responsiveness.51–53 The dynamic covalent strategy has enabled the rational design of stable and multifunctional materials such as adaptive polymeric architectures, self-healing materials, recyclable and reproducible networks, and stimuli-responsive sensing systems, with applications from biomedical engineering to soft robotics.54–60
In the field of PISA, the incorporation of DCBs represents a significant advancement. This strategy not only broadens the morphological diversity of self-assembled nanostructures, but also introduces stimuli-responsive features through covalent bond formation and cleavage. The unique characteristics of DCBs enable precise control over the crosslinking density, facilitate the post-modification, and allow for the fabrication of adaptive nanomaterials capable of structural reconfiguration in response to environmental stimuli. Beyond such microscopic nanomaterials, DCB-based PISA has facilitated the development of smart micelle-crosslinked gel networks exhibiting tunable chemical and mechanical properties along with self-healing behaviors.69,71,83 This review summarizes the current research on dynamic covalent chemistry applied in PISA, including disulfide bonds, boronate ester bonds, imine bonds, and [2 + 2] cycloaddition (Table 1). In part of these works, dynamic covalent bonding has been used as a practical chemical synthesis method without utilization of the dynamic properties. And in the other studies, DCBs are used to achieve specific stimulus-responsive disaggregation,61–64 morphological transition,65–67,72–74 surface functionalization,35,68,75,76 controlled release,77,78 and crosslinking and discrosslinking69–71,79–83 of PISA NPs, which are of greater interest to this review. Additionally, the review discusses the application potential of these stimulus-responsive changes in drug delivery, targeted drug release, sensing, fabrication of functional micelle-crosslinked gels with diverse mechanical properties, etc. In general, this review summarizes the significant progress in PISA research based on dynamic covalent chemistry, highlights the broad applications and proposes potential future research directions.
| Bond type | PISA systema | Stimulib | Function and application | Ref. |
|---|---|---|---|---|
| a EG: ethylene glycol; DIPEMA: 2-(diisopropylamino)ethyl methacrylate; CBMA: N,N-cystamine bismethacrylamide; OEGMA: oligo(ethylene glycol) methyl ether methacrylate; GlyMA: glycidyl methacrylate; SKM: solketal methacrylate; BMOD: bis(2-methacryloyl)oxyethyl disulfide; CouMA: coumarin-methacrylate; BMA: benzyl methacrylate; GMA: glycerol methacrylate; HPMA: 2-hydroxypropyl methacrylate; MTC: 3-methylene-1,9-dioxo-5,12,13-trithiocyclopentadecane-2,8-dione; HPMAAm: 2-hydroxypropyl methacrylamide; CysMA: cystamine methacrylamide hydrochloride; AMPS: 2-acrylamido-2methylpropanesulfonic acid; DSDMA: disulfide-based dimethacrylate; Bu3P: tributylphosphine; APBA: 3-aminophenylboronic acid; CPBA: 4-carboxyphenylboronic acid; DMA: N,N-dimethylacrylamide; BAPhA: 3-acrylamidophenylboronic acid; PBBMA: 4-pinacolboronylbenzyl methacrylate; VPBA: 4-vinylphenylboronic acid; St: styrene; AEAM: NH3+-based N-2-aminoethyl acrylamide hydrochloride; DAAM: diacetone acrylamide; PDCA: pyridine-2,6-dicarboxaldehyde; VBA: vinyl benzaldehyde; DOX: doxorubicin; DMAEMA: (N,N-dimethylamino)ethyl methacrylate; MAEBA: p-(methacryloxyethoxy)benzaldehyde; DEMA: 2-((3-(4-(diethylamino)phenyl) acryloyl)oxy)ethyl methacrylate; NBMA: 2-nitrobenzyl methacrylate; CMA: 7-(2-methacryloyloxy-ethoxy)-4-methyl-coumarin; TCMAm: 7-(4-(trifluoromethyl)coumarin) methacrylamide; DMAc: N,N-dimethylacrylamide; BA: n-butyl acrylate; AEMC: 7-(2-acryloyloxyethoxy)-4-methylcoumarin. b DTT: dithiothreitol; GSH: glutathione; TCEP: tris(2-carboxyethyl)phosphine. | ||||
| Disulfide bond | PEG-b-P(DIPEMA-co-CBMA) | DTT as the reductant | Disaggregation | 61 |
| Disulfide bond | POEGMA-b-P(GlyMA-co-CBMA) | DTT as the reductant | Disaggregation | 62 |
| Disulfide bond | POEGMA-b-P(SKM-co-BMOD) | GSH as the reductant | Disaggregation | 63 |
| Disulfide bond, [2 + 2] cycloaddition | POEGMA-b-P(GlyMA-co-CBMA-co-CouMA), POEGMA-b-P(BMA-co-CBMA-co-CouMA) | DTT as the reductant, UV | Disaggregation | 64 |
| Disulfide bond | PGMA-b-P(HPMA-co-MTC) | TCEP as the reductant | Worm-to-sphere transition | 65 |
| Disulfide bond | PHPMAAm-b-PCysMA/PAMPS | UV | Vesicle-to-lamella and vesicle-to-sphere transition | 66 |
| Disulfide bond | PEG-b-P(DIPEMA-co-CBMA) | GSH as the reductant | Worm-to-sphere, lamella-to-sphere and vesicle-to-worm/sphere transition | 67 |
| Disulfide bond | P(GMA-st-DSDMA)-b-PHPMA | Bu3P as the reductant | Thiol functionalization | 68 |
| Disulfide bond | P(GMA-co-DSDMA)-b-PHPMA, PHPMA-b-PGMA-S-S-PGMA-b-PHPMA | TCEP as the reductant | Crosslinked worm gels | 69 |
| Disulfide bond | P(GMA-co-GlyMA)-b-PHPMA, cystamine | — | Crosslinked worm gels | 70 |
| Disulfide bond | PBMA-b-POEGMA-b-PBMA, PGlyMA-b-POEGMA-b-PGlyMA | UV | Crosslinked worm gels | 71 |
| Boronate ester bond | PGMA-b-PHPMA, APBA | pH | Vesicle-to-worm transition | 72 |
| Boronate ester bond | PGMA-b-PHPMA, CPBA | pH | Worm-to-sphere and vesicle-to-worm transition | 73 |
| Boronate ester bond | PDMA-b-PBAPhA | Hydrolysis | Sphere-to-rod transition | 74 |
| Boronate ester bond | POEGMA-b-PPBBMA | — | Boronic functionalization | 75 |
| Boronate ester bond | PVPBA-b-PSt | Fructose | Boronic functionalization and fructose sensing | 35 |
| Imine bond | PHPMA-b-P(AEAM-co-DAAM), PDCA | — | Zn(II)-coordination | 76 |
| Imine bond | POEGMA-b-P(St-co-VBA), DOX | pH | Controlled release of DOX | 77 |
| Imine bond | PDMAEMA-b-PMAEBA, DOX | pH | Controlled release of DOX | 78 |
| [2 + 2] cycloaddition | PHPMA-b-PDEMA | UV | Crosslinking | 79 |
| [2 + 2] cycloaddition | PHPMAAm-b-P(NBMA-co-CMA) | UV | Crosslinking | 80 |
| [2 + 2] cycloaddition | PHPMAAm-b-P(DIPEMA-co-CMA) | UV | Crosslinking | 81 |
| [2 + 2] cycloaddition | PEG-b-P(HPMA-co-TCMAm) | UV | Crosslinking | 82 |
| [2 + 2] cycloaddition | PDMAc-b-P(BA-co-AEMC) | UV | Reversible crosslinking | 83 |
2. PISA based on disulfide bonds
Disulfides are typically obtained through the oxidation of thiols in an alkaline environment, while reducing conditions can facilitate the cleavage of disulfide bonds (Scheme 1a). Furthermore, exchange reactions of disulfide bonds can occur via anionic or radical pathways (Scheme 1b and c).84,85 In living organisms, disulfide bonds formed between cysteine residues play an important role in ensuring the correct folding and conformation of proteins, maintaining the stability of protein structures and preserving their physiological activity.86,87 The incorporation of disulfide bonds into polymers has become increasingly prevalent, allowing materials to achieve self-healing and stimulus responsiveness. The utilization of disulfide bonds in the PISA method enables controlled delivery, morphological transition and redox responsiveness of the NPs, being conducive to constructing crosslinked gels.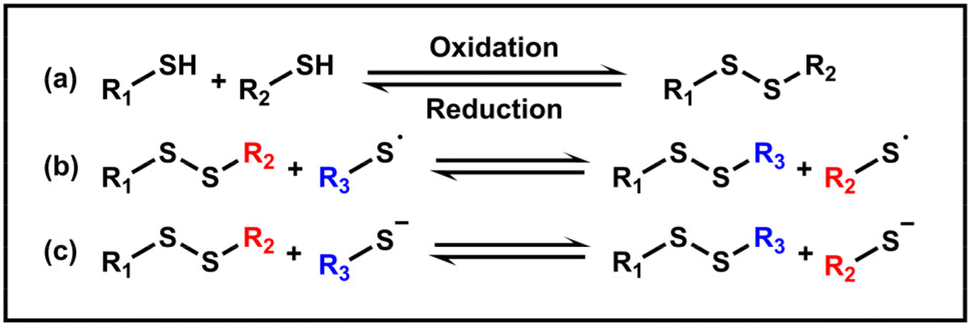 | ||
| Scheme 1 Reversible formation and exchange mechanism of disulfide bonds. | ||
2.1. Disulfide bonds leading to disassembly
The cleavage and rearrangement of disulfide bonds under various stimuli lead to a common strategy for regulating the disassembly behavior of PISA assemblies. PISA is a one-pot process where chain extension of a solvophilic macro-initiator with solvophobic monomers leads to in situ self-assembly of block copolymer nano-objects. The growing solvophobic blocks drive the self-assembly process, resulting in various morphologies such as spheres, worms, vesicles or others determined by the packing parameter, solvent type, solid content, etc.88 Therefore, to realize the disassembly triggered by disulfide cleavage, a common method is to incorporate a difunctional monomer as a crosslinker during the polymerization of the solvophobic segment. Typically, such crosslinking monomers usually have lower reactivity and are added in less than 15% of the solvophobic segment monomers to prevent excessive crosslinking that could restrict chain mobility and hinder the evolution of morphologies during PISA.61 Although disulfide cleavage represents a feasible approach for triggering disassembly, considering the facts that determine PISA morphology, the cleavage of side chains alone may not provide complete disassembly, as the intrinsic solvophobicity of the core-forming blocks remains largely unchanged. Therefore, effective disassembly strategies often combine disulfide bond cleavage with additional mechanisms that alter the solvophobicity of the core blocks, such as protonation or hydrolysis of the solvophobic segments. This dual-responsive approach ensures more efficient disassembly of the PISA nano-objects based on disulfide bonds under specific stimuli.Currently, the disassembly of PISA assemblies based on disulfide bonding is typically realized using a solvophobic variable second block and a small amount of disulfide crosslinking units. The assemblies become solvophilic and reducing agents such as dithiothreitol (DTT), glutathione (GSH), etc. are then used to break the disulfide bonds which maintain the morphology, thus causing disassembly. Pan's group reported in situ disulfide crosslinking in PISA through reversible addition–fragmentation chain transfer polymerization (RAFT) dispersion copolymerization of 2-(diisopropylamino)ethyl methacrylate (DIPEMA) and the disulfide crosslinker N,N-cystamine bismethacrylamide (CBMA) with a macromolecular chain transfer agent (macro-CTA) polyethylene glycol-4-(4-cyanopentanoic acid) dithiobenzoate (PEG-CPADB) and prepared vesicles exhibiting dual pH and reductant responsiveness.61 The disaggregation occurs through the cleavage of disulfide bonds of CMBA in the presence of DL-DTT in an acidic aqueous solution, enabling the controlled release of encapsulated cargoes (Fig. 1a). Without DTT, the vesicles only exhibit pH-dependent reversible swelling and shrinking due to the protonation of PDIPEMA. Using a similar strategy, Reuther and coworkers implemented the photo-controlled atom transfer radical polymerization-induced self-assembly (PhotoATR-PISA) process using poly(oligo(ethylene glycol) methyl ether methacrylate) (POEGMA) as the solvophilic block, poly(glycidyl methacrylate) (PGlyMA) as the solvophobic block and CBMA as the crosslinking agent.62 Under acidic conditions with DTT, disulfide crosslinking was cleaved along with the ring-opening hydrolysis of PGlyMA epoxide groups, followed by the disassembly and release of encapsulated cargoes.
 | ||
| Fig. 1 Schematic illustration of disulfide-based stimulus-responsive disassembly of PISA NPs: (a) reductant-responsive vesicles based on PEG-b-P(DIPEMA-co-CBMA). (b) Reductant-responsive vesicles based on POEGMA-b-P(SKM-co-BMOD). Adapted with permission from ref. 63. Copyright 2021 The Royal Society of Chemistry. | ||
Using a similar PISA method, a reduction-responsive double hydrophilic block copolymer (DHBC) vesicle was reported. Goto and coworkers reported a reversible complexation mediated polymerization PISA formulation using poly(oligo(ethylene glycol) methyl ether methacrylate)-iodide (POEGMA-I) as the macroinitiator and solketal methacrylate (SKM) as the monomer.63 As the polymerization proceeded, a small amount of bis(2-methacryloyl)oxyethyl disulfide (BMOD) crosslinker was added to provide the vesicles with stable skeletons against acidic environments and dynamic properties under specific stimuli. The DHBC vesicles were then generated by the hydrolysis with HCl solution, through which the hydrophobic PSKM was converted into hydrophilic poly(glycerol methacrylate) (PGMA) segments (Fig. 1b). The reductant-responsiveness of DHBC vesicles was demonstrated by the encapsulation and release study that showed a 75% release of dye molecule Fast Green FCF from vesicles treated with GSH, compared to a 13% release without GSH. These biocompatible nano-capsules, consisting of neutral and biocompatible POEGMA and PGMA segments and without the use of heavy metals or sulfur compounds, hold the potential for extensive applications in biomedicine, cosmetics and household products.
Briefly, the utilization of disulfide-containing difunctional monomers in PISA can stabilize the nano-assemblies and provide reduction-responsiveness. Therefore, the resulting systems can serve as cargo carriers, thereby showing good potential for drug delivery and controlled release. In terms of practical applications, reduction conditions, potential toxicity and stability in different environments require comprehensive consideration.
2.2. Disulfide bonds leading to morphological transition
Based on the regulation of the chemical composition and the topological structure of solvophobic blocks triggered by disulfide bond breaking, it is possible to control the morphological transition of assemblies into a lower-order morphology instead of undergoing a complete disassembly as in the above examples discussed in section 2.1. The first thing that comes to mind is that by decreasing the solvophobic blocks, the reduced packing parameter induces a degradation of the assembly morphology. For example, Paulusse et al. used a cyclic disulfide monomer, 3-methylene-1,9-dioxo-5,12,13-trithiocyclopentadecane-2,8-dione (MTC), to introduce disulfide bonds into the copolymer backbone when copolymerizing GMA and 2-hydroxypropyl methacrylate (HPMA) in the aqueous PISA, forming worm-like diblock copolymer nano-objects.65 Then they found that the irreversible morphological change from worm to spherical shape could be achieved by cutting disulfide bonds inside the hydrophobic blocks using excessive tris(2-carboxyethyl)phosphine (TCEP) (Fig. 2a). Such transformation could help the discharge of nano-objects from living organisms and has potential research value in the biomedical field.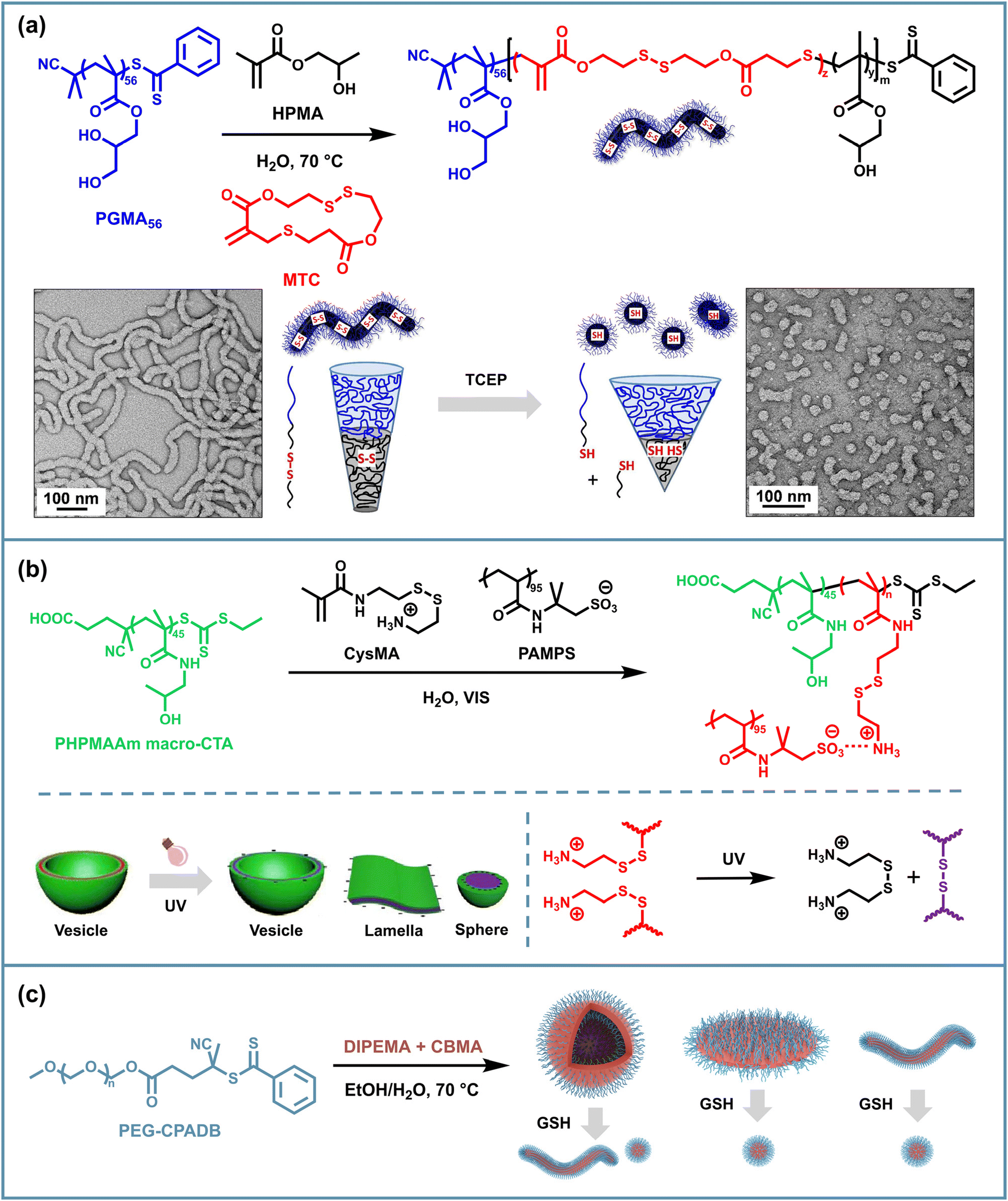 | ||
| Fig. 2 Schematic illustration of disulfide-based stimulus-responsive morphological transformation of PISA NPs: (a) reductant-responsive worms based on PGMA-b-P(HPMA-co-MTC). Adapted with permission from ref. 65. Copyright 2016 American Chemical Society. (b) UV-responsive vesicles based on PHPMAAm-b-PCysMA/PAMPS. Adapted with permission from ref. 66. Copyright 2019 American Chemical Society. (c) Reductant-responsive worms, lamellae and vesicles based on PEG-b-P(DIPEMA-co-CBMA). | ||
In addition to introducing cleavable disulfide sites into the polymer backbone, disulfide bonds on the side chains can likewise be molecularly designed to achieve morphological transformation of the micelles. Cai et al. presented a method of visible light-initiated polymerization-induced electrostatic self-assembly (PIESA) to synthesize polyion complex nano-vesicles using 2-hydroxypropyl methacrylamide (HPMAAm) and cystamine methacrylamide hydrochloride (CysMA) monomers.66 They found that UV irradiation of such vesicles resulted in a rapid exchange reaction of locally enriched disulfide motifs and the rapid release of cystamine salts, generating crosslinked spheres, lamellae and vesicles in one pot from one single vesicle precursor, depending on the exposure time (Fig. 2b). Such a transition could be attributed to the charge imbalance, the variation and crosslinking of core-forming segments.
Apart from the use of special disulfide-containing monomers described above, difunctional disulfide monomers can also be added to provide side-chain crosslinking along with the PISA process, but this usually limits the chain mobility of the solvophobic blocks, thereby preventing the morphology transition of spherical micelles to higher-order forms. Hong et al. found that the less reactive disulfide crosslinking monomer CBMA incorporated at the late stage of RAFT PISA of PEG90-CPADB and DIPEMA allowed higher-order morphologies to be generated before the crosslinking and gave solvophobic branched structures, which further promoted the transition to more advanced morphologies.67 Accordingly, this provides a feasible solution for morphological degradation triggered by side-chain disulfide breakage and loss of crosslinked structures. After the incubation in GSH, worm-like and lamellar micelles degraded into spherical micelles, while vesicles transformed into worms and spheres (Fig. 2c). However, in the case of DPPDIPEMA = 150, the assemblies maintained their vesicular morphology, since these structures were also formed at the same DPPDIPEMA without adding CBMA.
In summary, studies using disulfide bonds to trigger morphology transformations of assemblies have mainly focused on morphology degradation. Current methods include decreasing solvophobic segments by breaking or exchanging disulfide bonds, as well as altering the topology of the solvophobic segments. Additionally, disulfide-mediated transitions to higher-order morphologies of PISA assemblies as well as the reversible transformations between different morphologies can also be explored to expand their potential in the development of novel smart-responsive materials.
2.3. Disulfide bonds leading to surface modification
Due to the reductant-responsive cleavage of disulfide bonds, thiol-functionalized nano-objects can be easily prepared from the PISA NPs with a solvophilic corona containing disulfide bonds. Armes et al. used HPMA to chain-extend a macro-CTA copolymerized with GMA and a small amount of disulfide-based dimethacrylate (DSDMA). The grown water-insoluble PHPMA chain drove in situ self-assembly, producing diblock copolymer vesicles in a concentrated aqueous solution.68 The disulfide bonds in the PGMA stabilizer chains can be cleaved using excess tributylphosphine (Bu3P) to generate thiol groups on the vesicle surface. The reactivity of thiol groups facilitates further modifications such as cationization and the introduction of fluorescent tags (Fig. 3). Disulfide bonds offer a versatile approach for the surface modification of PISA nano-objects, enabling the creation of thiol-functionalized NPs that can undergo further modifications. This fabrication concept can be further extended to other types of dynamic covalent bonds to prepare diverse surface-functionalized polymer vesicles, which requires the extension of solvophilic segment crosslinking monomers for PISA. | ||
| Fig. 3 Schematic illustration of surface-functionalized PISA vesicles based on disulfide cleavage of P(GMA-st-DSDMA)-b-PHPMA. Adapted with permission from ref. 68. Copyright 2012 American Chemical Society. | ||
2.4. Disulfide bonds to construct crosslinked gels
In addition to introducing disulfide bonds inside NPs, disulfide bonds can be used to link different nano-objects to create macroscopic micelle crosslinked materials which could combine the advantages of physical crosslinking of efficiently prepared PISA assemblies with the responsive reversible properties of dynamic covalent bonding and may exhibit unique mechanical characteristics. To achieve inter-micellar crosslinking, which requires the crosslinking of the solvophilic shells of different NPs, disulfide-containing solvophilic segment crosslinking monomers, chain transfer agents and initiators can be used during polymerization, and besides that, additional disulfide-containing crosslinking agents can also be added after assembly. For the former approach, Armes et al. designed two strategies for introducing disulfide linkages into the solvophilic stabilizer block of RAFT-synthesized PGMA-PHPMA worm-shaped micelles.69 The first approach involved statistically copolymerizing GMA and the difunctional disulfide monomer DSDMA as the hydrophilic segments to form disulfide units on side chains, followed by the chain extension of the hydrophobic PHPMA block. The second approach involved using a difunctional RAFT agent with a disulfide bond on the backbone to copolymerize GMA and HPMA (Fig. 4a). Both routes were capable of yielding freestanding worm gels. Oscillatory rheology studies confirmed that both routes exhibited an increase in gel strength with the increasing disulfide contents, especially for gels (Route 2) as they had more inter-worm crosslinks. Furthermore, this enhanced gel modulus was reversible for both types of gels through reductive disulfide cleavage with excess TCEP, resulting in thiol-functionalized worm gels with a modulus similar to that of non-functionalized gels. These results demonstrated the formation of inter-worm covalent bonds via disulfide exchange. They then devised a new scheme for preparing disulfide crosslinked PISA gels, namely synthesizing P(GMA-co-GlyMA)-b-PHPMA block copolymer worms via RAFT aqueous dispersion polymerization, followed by nucleophilic substitution between the water-soluble reagent cystamine and impermeable epoxy groups located within the P(GMA-co-GlyMA) stabilizer chain.70 This derivatization introduced disulfide functional groups, and two types of worm gels can be obtained depending on the cystamine/epoxy molar ratio. The use of a large excess of cystamine produces essentially linear, primary amine-functionalized worms, which form a soft gel that preserves the thermal response characteristics of the precursor worm gel. Conversely, the adoption of stoichiometric amounts of cystamine results in stronger covalent crosslinked worm gels that no longer exhibit thermo-responsive behavior. | ||
| Fig. 4 Schematic illustration of disulfide-crosslinked PISA worm gels: (a) reductant-responsive worm gels based on PGMA-PHPMA using the disulfide-based methacrylic comonomer DSDMA or a GMA45-S-S-GMA45 disulfide-based difunctional macro-CTA. Adapted with permission from ref. 69. Copyright 2015 American Chemical Society. (b) UV-responsive worm gels based on PBMA-b-POEGMA-b-PBMA and PGlyMA-b-POEGMA-b-PGlyMA, and the overall recovery of the adsorption capacity (qt) for both PISA24 (PBMA-b-POEGMA-S-S-POEGMA-b-PBMA) and Ctrl9 (PBMA-b-POEGMA-b-PBMA) calculated based on the obtained kinetic data for three UV-regeneration cycles. Adapted with permission from ref. 71. Copyright 2024 American Chemical Society. | ||
Similar to Route 2 with disulfide bonds on polymer backbones proposed by Armes et al., Reuther's group implemented PhotoATR-PISA via the chain extension from a POEGMA-based disulfide-functionalized macroinitiator with benzyl methacrylate (BMA) to in situ form BAB-type triblock copolymer PISA gels.71 They discovered that due to the π–π interaction between aromatic rings, the resulting gels exhibited an adsorption capacity for phenanthrene (PNT, a potential carcinogen) which could be UV-regenerated based on the UV-exchangeability of disulfide bonds through multiple cycles of UV exposure in pure water and adsorption tests in PNT solution. This study showed that such worm-gels exhibited superior capabilities in adsorption capacity recovery over three cycles compared to one nondynamic control sample, providing research directions for the design of dynamic disulfide-crosslinked PISA materials (Fig. 4b). However, the authors found that the calculated adsorption capacities for PISA gels were significantly lower than those for conventional adsorbents, suggesting the need for further morphology modification of PISA assemblies, the introduction of different functional monomers into the NP cores or coronas and improvement of the disulfide bond crosslinking density.
Such micelle-crosslinked gels can be easily obtained by crosslinking the solvophilic shells of PISA NPs using the methods described above. However, current crosslinked gels with disulfide bonds are typically worm gels, which correlates with the fact that the multiple contacts and entanglements of worm-like micelles facilitate the formation of physical gels89 and also promote inter-micelle crosslinking. Since their interactions mainly originate from non-covalent physical crosslinking, the structures of such worm micelle entangled networks tend to have notable deformability and low modulus, which restricts their application value. Considering this limitation, expanding micelle-crosslinked gels with disulfide bonds to include spherical or vesicular micelles is expected to improve the mechanical properties.83,90–93 Taking advantage of the PISA method to prepare highly concentrated nano-assemblies and modifiable polymer chains, and combining the dynamic characteristics of disulfide bonds, it is possible to prepare hydrogels exhibiting both high toughness and strength with specific drug-carrying nanodomains, adjustable functions and tunable viscoelasticity, which can be used for biomedical scaffolds and other materials.
3. PISA based on boronate ester bonds
Boronic acids can react with diols through an esterification reaction to reversibly form boronate esters (Scheme 2). In aqueous solution, the equilibrium favors boronate esters when the pH is above the pKa of boronic acids, while below this pKa, the equilibrium shifts toward boronic acids and diols.43 Therefore, this dynamic equilibrium allows boronate ester bonds to be utilized not only for their reversible nature, but also for creating materials that respond to specific conditions through tailored structures. Recent studies have employed boronate ester bonds in the design of PISA processes, where these bonds enable the reversible alteration of nano-object morphology, pH responsiveness, and molecular recognition. The integration of boronate ester bonds into PISA thus offers a versatile approach for developing functional materials with tunable properties.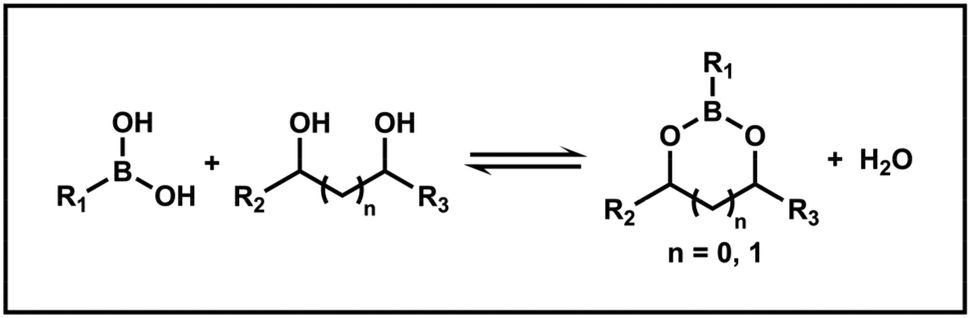 | ||
| Scheme 2 Reversible formation mechanism of boronate ester bonds. | ||
3.1. Boronate ester bonds leading to morphological transition
Boronate ester bonds can be used to regulate the morphological transitions of PISA NPs, and phenylboronic acid and its derivatives are commonly used as additive reagents in studies. Boronic acid undergoes condensation reactions with diols in the stabilizer chains of NPs under alkaline conditions, leading to a decrease in the packing parameter and subsequent morphological degradation. Armes et al. prepared PGMA-b-PHPMA vesicles via aqueous PISA and then added 3-aminophenylboronic acid (APBA) to form phenylboronate ester bonds with the cis-diol groups of the PGMA block at a pH of around 10.72 Consequently, a morphological transformation was induced from vesicles to worms or spheres (Fig. 5a), which depended on different concentrations of APBA. This was ascribed to the increase in effective mass and the introduction of anionic charge of the PGMA stabilizer block, thereby reducing the packing parameter. Using an increasing concentration of APBA, such a transition can also occur in the presence of salt or buffer, or with thicker-walled vesicles prepared by relatively long PHPMA blocks. They also discovered the controlled release of silica NPs encapsulated within the vesicles by varying the solution pH, the APBA concentration and the membrane thickness.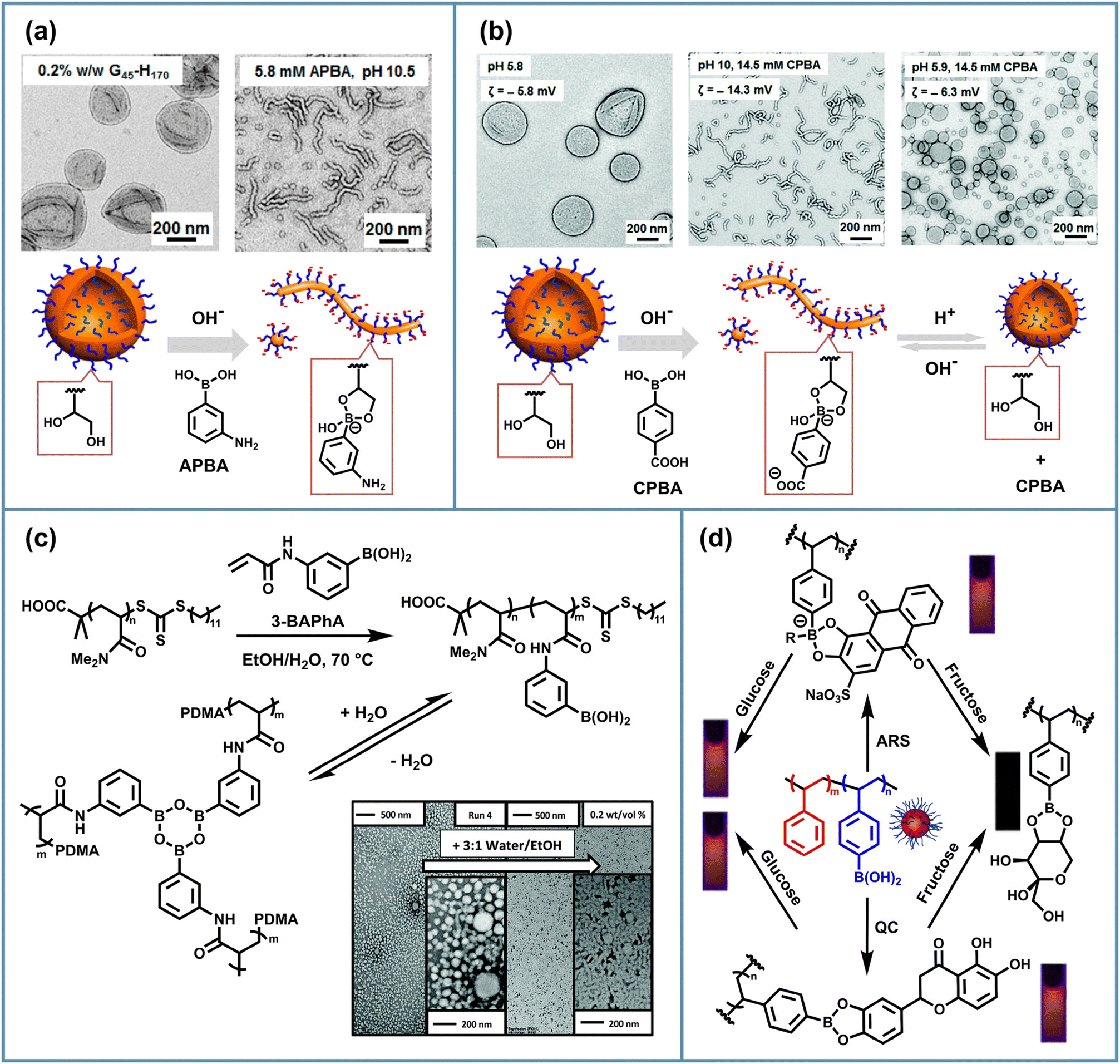 | ||
Fig. 5 Schematic illustration of borate-based stimulus-responsive morphological transformation and functionalization of PISA NPs: (a) vesicle-to-worm transition of PGMA-b-PHPMA vesicles after APBA addition at pH = 10.5. Adapted with permission from ref. 72. Copyright 2017 American Chemical Society. (b) Reversible worm-to-sphere and vesicle-to-worm transition of PGMA-b-PHPMA NPs driven by switching pH in the presence of CPBA. Adapted with permission from ref. 73. Copyright 2017 The Royal Society of Chemistry. (c) Sphere-to-rod transition of PDMA-b-PBAPhA after a 100-fold dilution with 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/EtOH. Adapted with permission from ref. 74. Copyright 2022, The Royal Society of Chemistry. (d) Detection performance for fructose and glucose of PVPBA-b-PSt nanospheres. Adapted with permission from ref. 35. Copyright 2022 Elsevier Ltd. :1 water/EtOH. Adapted with permission from ref. 74. Copyright 2022, The Royal Society of Chemistry. (d) Detection performance for fructose and glucose of PVPBA-b-PSt nanospheres. Adapted with permission from ref. 35. Copyright 2022 Elsevier Ltd. | ||
In addition, they achieved reversible morphological transitions of PGMA-b-PHPMA copolymer nano-objects through the generation of phenylboronic ester bonds between 4-carboxyphenylboronic acid (CPBA) and the 1,2-diol groups on the PGMA chains.73 Due to the pH-dependent nature of boronate ester bonds, the reversible vesicle-to-worm or worm-to-sphere transitions can be realized by adjusting the pH value. They observed that the transition from vesicles to worms was reversible at relatively low copolymer concentrations but became irreversible at higher concentrations. Conversely, the transformation of worms to spheres is reversible over a wide range of copolymer concentrations (Fig. 5b) and can lead to in situ gelation and de-gelation at higher concentrations. These works using additive phenylboronic acid to react with diols in stabilizer chains to drive morphological transitions of PISA NPs have provided insights into the design of novel PISA carriers for drugs or enzymes. Considering the different pH conditions for various target applications in the field of pharmaceuticals and daily chemicals, other phenylboronic acid derivatives that incorporate electron-donating or electron-withdrawing groups thus exhibiting increased or decreased pKa values can be used to modulate the response conditions of similar phenylboronic acid/PISA vesicle systems, providing design strategies for targeted delivery and controlled release.
Similar to disulfide-based PISA, the design of attaching boronic acid groups in polymer chains is an important direction to be investigated, eliminating the need for external boronic acid reagents to drive morphology transformation. Hsu and Delaittre et al. proposed a boronate-containing monomer 4-pinacolboronylbenzyl methacrylate (PBBMA) and achieved the in situ introduction of boronate ester fragments in PISA nanospheres.75 An acrylate analogue 4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)benzyl acrylate (TBA) was reported by Thang's group to chain extent poly-N,N-dimethylacrylamide (PDMA) macro-CTA in RAFT-mediated PISA and achieved a range of higher order PISA nano-objects, including cubic and hexagonal mesophases.94 Sumerlin and Wooley et al. conducted the RAFT polymerization using the monomer 3-acrylamidophenylboronic acid (3-BAPhA) to construct diblock copolymers and performed post-assembly procedures.95,96 Zetterlund and Aldabbagh et al. further expanded the application of phenylboronic acid-substituted acrylamide monomers to PISA and also studied the effect of pinacol-protected and unprotected monomers on polymerization.74 Using a PDMA96-derived macro RAFT agent to carry out dispersion polymerization of 3-BAPhA, boroxine-crosslinked nanospheres were obtained, which can undergo a water-responsive morphological transition from spheres to rods and worms while diluting (Fig. 5c). This was assumed to be a result of the hydrolysis of boroxine moieties in hydrophobic cores, which changes the interfacial energy and polymer–solvent interactions. Additionally, to circumvent the formation of boroxine crosslinking, pinacol-protected 3-BAPhA was then used as the hydrophobic segment monomer, and unsurprisingly, higher-order nano-objects including worms and vesicles were observed. Based on the preliminary findings of this work on the in situ crosslinking of phenylboronic acid moieties during PISA and their water-responsiveness, it is possible to further expand the application of boroxine crosslinked structures in PISA, leading to the possible construction of water-responsive PISA assemblies.
3.2. Boronate ester bonds leading to boronic functionalization
In addition to driving NP morphology transformation, boronate ester bonds can be used to functionalize PISA nano-objects by incorporating monomers containing boronic acid units, which could have potential applications in boron neutron capture therapy, recognition and sensing of saccharide molecules. Hsu and Delaittre et al. synthesized the boronic-ester-functionalized monomer PBBMA and copolymerized POEGMA-b-PPBBMA to form NPs with a boron-rich core and a biocompatible shell by RAFT-meditated PISA, which has the potential for anticancer therapy.75 In order to take advantage of the dynamic properties of boronate ester bonds, nano-assemblies have been prepared by immobilizing boron-containing groups in a solvophilic shell to readily serve as fructose sensors based on the reversible reactions of boronic acids and diols. Qian et al. synthesized poly (4-vinylphenylboronic acid)-b-polystyrene (PVPBA-b-PSt) NPs via PISA in methanol and constructed a fluorescence sensor system based on the displacement assay of PVPBA-b-PSt nanosphere/PBA-Alizarin Red S (ARS) and PVPBA-b-PSt nanosphere/quercetin (QC) complexes to detect fructose.35 The authors found that the fluorescence was quenched with the addition of fructose but remained emitting with glucose (Fig. 5d). This was attributed to the fact that the binding force of fructose to boronic acid was greater than that of ARS and QC, while the binding force of glucose was even lower. In summary, there remains significant research space in exploring the applications of boronate-functionalized PISA NPs. For example, the study of dynamic boronate ester bonding for glucose detection is apparently of greater importance in the biomedical field.4. PISA based on imine bonds
The imine bond is a widely used dynamic covalent bond, usually formed by condensation of a carbonyl group and a primary amine, which is also referred to as the Schiff base bond. The condensation starts with the nucleophilic attack of the amine on the carbonyl carbon, followed by dehydration. Under acidic conditions, the hydrolysis process tends to occur (Scheme 3a). Additionally, a new imine bond can also be generated through an imine exchange reaction with another amine or via an imine metathesis with another imine (Schemes 3b and c).43 Due to its intrinsic dynamic connectivity and unique pH-responsiveness, the imine bond now holds promising applications in the field of polymer science and nanomaterials.97,98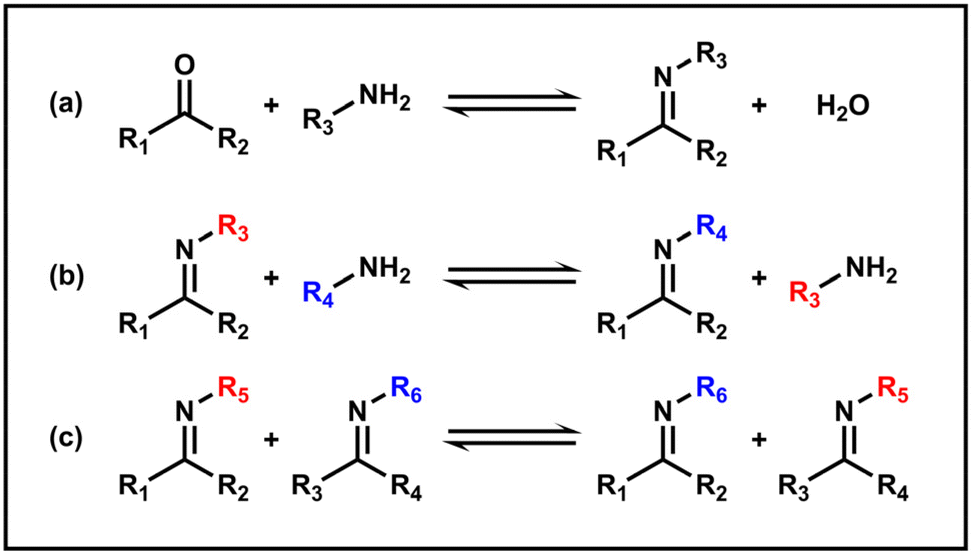 | ||
| Scheme 3 Reversible formation and exchange mechanism of imine bonds. | ||
The imine bond is an efficient coupling method due to the mild reaction conditions and high reaction rates and could be used as a practical synthetic approach in PISA. Cai et al. reported an aqueous dispersion RAFT PISA of diacetone acrylamide (DAAM) and NH3+-based N-2-aminoethyl acrylamide hydrochloride (AEAM) using a PHPMA-derived macro-CTA.76 Then the conjugation with pyridine-2,6-dicarboxaldehyde (PDCA) via imine bonds was carried out to form ligand motifs that can enable coordination with zinc ions. Such PISA NPs containing metal centers with a large accessible specific surface area show potential as precursors for metalloenzyme-inspired aqueous catalysts. Furthermore, imine bonds can be transformed into secondary amines with more stable C–N single bonds generated by reductive amination reactions, by using reducing agents such as sodium borohydride (NaBH4) and sodium cyanoborohydride (NaBH3CN). Armes et al. synthesized a hydrophilic aldehyde-functional OEGMA monomer to form copolymer micelles with aldehyde-containing shells which can further undergo reductive amination for stable bonding with amino acids and model proteins to achieve potential bio-applications.99,100
Moreover, the studies of PISA based on imine bonds manifesting their dynamic properties can provide specific stimulus responsiveness to NPs and have attracted greater interest. To maintain the dynamic nature of imine bonds and their pH-responsiveness in PISA micelles, several relevant studies of the controlled loading and release of doxorubicin (DOX), a commonly used chemotherapy drug with an amine group, have been reported. Boyer and Davis et al. synthesized a poly(oligo(ethylene glycol) methacrylate)-b-(poly(styrene)-co-poly(vinyl benzaldehyde)) (POEGMA-b-P(St-co-VBA)) copolymer PISA NPs with a range of morphologies through one-pot RAFT dispersion polymerization.77 DOX could be conjugated with the aldehyde groups in the NP cores, based on the pH-sensitive dynamic imine bonds. Therefore, a pH-controlled drug release was achieved with more than 80% of DOX released from the NPs at pH 5, while a very slow release (around 10% DOX) was observed at neutral pH. Similarly, Pan's group carried out RAFT PISA of p-(methacryloxyethoxy)benzaldehyde (MAEBA) using a poly((N,N-dimethylamino)ethyl methacrylate) (PDMAEMA) based macro-CTA, and DOX was then conjugated into the nano-object cores.78 They also observed the stable binding (less than 1% release) of DOX in the neutral environment and rapid release behavior in an acidic environment (Fig. 6). Then the intracellular experiments of DOX release showed that the main localization of free DOX occurred in acidic organelles, further demonstrating the pH-responsiveness of imine bonds in PISA-DOX systems. Such preparation of drug-carrying PISA NPs with pH-regulated release behaviors provides new insights for the development of highly concentrated nano drug carriers with controlled release properties.
 | ||
| Fig. 6 Schematic illustration of imine-containing PISA NPs based on PDMAEMA-b-PMAEBA and DOX, and the DOX release profiles of vesicles–DOX (a1, a2, a3), nanorods–DOX (b1, b2, b3), nanowires–DOX (c1, c2, c3), and spheres–DOX (d1, d2, d3) in the aqueous buffer solutions at pH = 7.4 (a3, b3, c3, and d3), at pH = 6.0 (a2, b2, c2, and d2) and at pH = 5.0 (a1, b1, c1, and d1). Adapted with permission from ref. 78. Copyright 2016 American Chemical Society. | ||
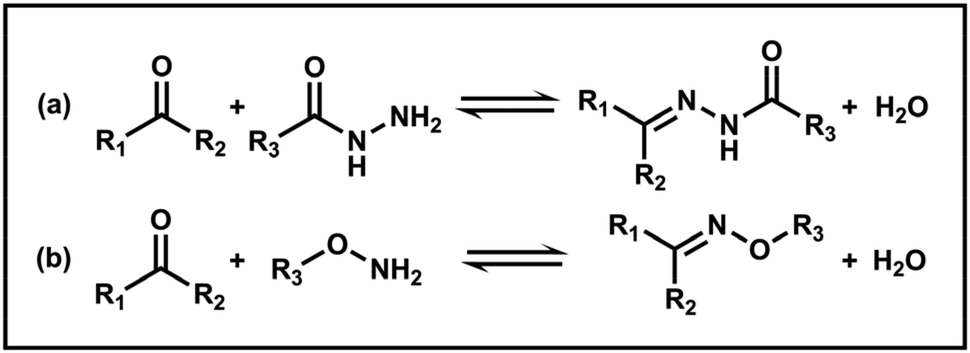
However, imine chemistry generally suffers from hydrolytic instability, which limits its range of applications. The susceptibility of imines to hydrolysis can be averted by using more stable acylhydrazones or oximes. Acylhydrazones and oximes are prepared by reacting aldehydes or ketones with acylhydrazides and hydroxylamines, respectively (Scheme 4). They are more stable than imines due to the lone pair of electrons on the electronegative atoms adjacent to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds.42 Additionally, reversibility can be achieved under mild conditions and controlled by pH. For example, Armes and coworkers reported a series of poly(N,N-dimethylacrylamide)-b-polydiacetone acrylamide (PDMAC-b-PDAAM) diblock copolymer nano-objects via PISA and used adipic acid dihydrazide (ADH) to react selectively with the pendent ketone groups on the hydrophobic PDAAM chains to form hydrazone moieties at pH 4 in aqueous solution.101 Nevertheless, further studies on the dynamic response of acylhydrazone bonds have yet to be carried out.
N bonds.42 Additionally, reversibility can be achieved under mild conditions and controlled by pH. For example, Armes and coworkers reported a series of poly(N,N-dimethylacrylamide)-b-polydiacetone acrylamide (PDMAC-b-PDAAM) diblock copolymer nano-objects via PISA and used adipic acid dihydrazide (ADH) to react selectively with the pendent ketone groups on the hydrophobic PDAAM chains to form hydrazone moieties at pH 4 in aqueous solution.101 Nevertheless, further studies on the dynamic response of acylhydrazone bonds have yet to be carried out.
 | ||
| Scheme 4 Reversible formation mechanism of acylhydrazone bonds and oxime bonds. | ||
5. PISA based on [2 + 2] cycloaddition
Cycloaddition is now commonly recognized as a method to access dynamic covalent C–C bonds due to its ability to allow reversible formation and breakage of normally stable C–C single bonds under certain light or heat conditions. Photoinduced [2 + 2] cycloaddition is a well-studied method for introducing dynamic C–C bonds during the PISA process, usually involving cinnamate and coumarin moieties (Scheme 5).102 Due to the photosensitive nature of the [2 + 2] reaction, the relevant monomers can be easily incorporated during the polymerization and are not affected by the heating conditions. Consequently, it becomes a useful method for crosslinking PISA NPs because of its simple reaction conditions, lack of byproducts, no need for additives, and no impact on the assembly process as well as morphologies. | ||
| Scheme 5 Reversible dimerization mechanism of cinnamate and coumarin. | ||
First, the [2 + 2] cycloaddition method has primarily been used as a convenient photo-crosslinking strategy for PISA assemblies. For example, RAFT polymerization of a cinnamate-containing monomer, 2-((3-(4-(diethylamino)phenyl) acryloyl)oxy)ethyl methacrylate (DEMA) from a PHPMA derived macro-CTA was reported to yield sphere, worm and vesicle micelles which can be easily stabilized using 365 nm UV irradiation.79 Pan and Hong et al. copolymerized 2-nitrobenzyl methacrylate (NBMA) and the photosensitive monomer 7-(2-methacryloyloxy-ethoxy)-4-methyl-coumarin (CMA) with PHPMAAm macro-CTA via RAFT PISA, providing the resulting NPs with the ability to form carboxyl groups and core-crosslinking.80 Under UV irradiation, over 60% of the coumarin moieties participated in dimerization within 2 h for sphere, worm, lamella and vesicle NPs. They then changed the solvophobic segment monomer to DIPEMA and CMA to copolymerize from PHPMAAm and constructed permeability-tunable vesicles.81 By varying the irradiation time, the crosslinking density can be adjusted, thus modulating the pore size and selectivity of the membrane. Considering the wavelength-selective features, Boyer and Yoew et al. exploited the wavelength orthogonality of the UV-induced [2 + 2] coumarin cycloaddition reaction and the red light-mediated RAFT PISA.82 The photo-responsive monomer 7-(4-(trifluoromethyl)coumarin) methacrylamide (TCMAm) was copolymerized with HPMA and PEG-macro-CTA under low energy red light (λmax = 595 nm), with no dimerization occurring. Instead, crosslinked NPs with a stabilized morphology were obtained after UV (λmax = 365 nm) illumination.
Predictably, the aforementioned coumarin-dimerization-based core-crosslinked NPs could undergo a dissociation reaction under 254 nm UV illumination.103 Considering different dynamic covalent bonds with orthogonal reactivity, Reuther et al. combined dynamic S–S bonds and C–C bonds to construct a series of tunable orthogonal reversible covalent (TORC) core-crosslinked polymeric nanogels with nanosphere, worm-like and tubesome morphologies via PhotoATR-PISA. They utilized the solvophilic POEGMA macroinitiator to chain-extend PGlyMA or PBMA and added CBMA and coumarin-methacrylate (CouMA) as the dynamic covalent core crosslinkers.64 The pH-responsiveness of epoxy groups, the reductant-responsiveness of disulfide bonds, and the UV-responsiveness of coumarin dimers provided the copolymer assemblies with an AND-gate release behavior via the sequential addition of stimuli (Fig. 7a). After incubation in HCl solution, irradiation with 254 nm UV light and the addition of DTT, the worm-like sample exhibited sustained release with more than 80% total cargo discharged, and nanospheres showed an over 90% release under the same stimuli after 7 days of incubation with DTT. It is noteworthy that after the sequence of stimuli, nearly complete disassembly of the spherical structures took place, whereas worm-like NPs underwent a similar swelling process to the nanospheres but remained structurally intact. Such a combination of different types of dynamic covalent chemistry provides a feasible direction for achieving controlled multiple release of PISA NPs. Additionally, Huo et al. utilized the photoswitched reversible dimerization/cleavage reaction of coumarin motifs inside PISA NPs, achieving the decoupled regulation of fracture stress and modulus of micellar crosslinked hydrogels. These hydrogels consisted of assembled spheres via RAFT PISA of N,N-dimethylacrylamide (DMAc), n-butyl acrylate (BA) and 7-(2-acryloyloxyethoxy)-4-methylcoumarin (AEMC), followed by inter-micellar crosslinking with acrylamide.83 Then they implemented iterative 365/254 nm UV irradiation to trigger the reversible dimerization and cleavage of coumarin moieties, discovering the reversible enhancement and recovery of the hydrogel fracture stress (Fig. 7b). This research provides a new understanding relevant to the design of PISA hydrogels with tunable mechanical properties and improved toughness.
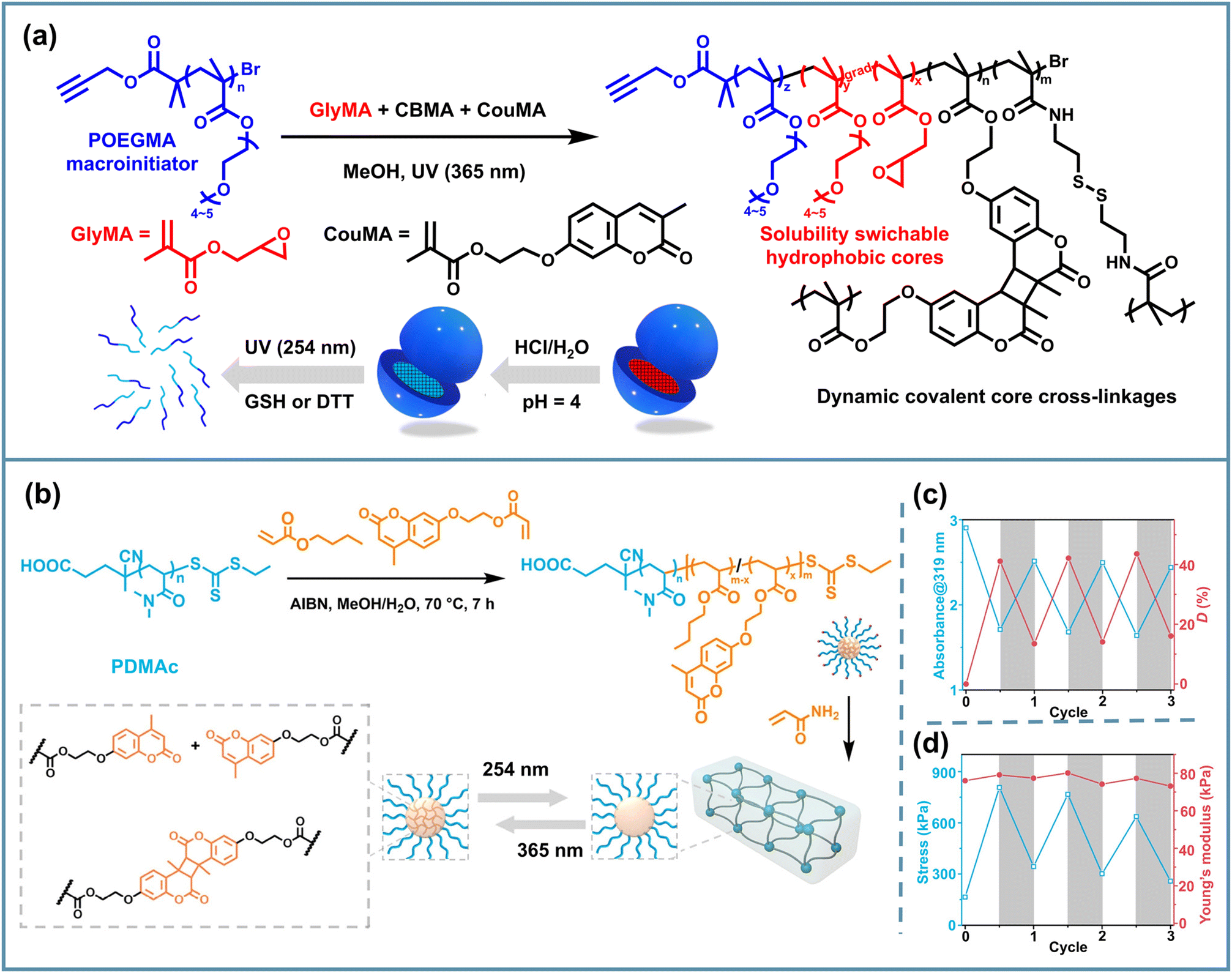 | ||
| Fig. 7 (a) Schematic illustration of UV- and reductant-responsive PISA NPs based on POEGMA-b-P(GlyMA-co-CBMA-co-CouMA). Adapted with permission from ref. 64. Copyright 2023 The Royal Society of Chemistry. (b) Schematic illustration of a UV-responsive PISA gel containing a dynamic C–C bond based on PDMAc-b-P(BA-co-AEMC); (c) the absorbance at 319 nm, dimerization degree, (d) fracture stress and Young's modulus of the hydrogel iteratively irradiated with 365/254 nm lamps. Adapted with permission from ref. 83. Copyright 2024 American Chemical Society. | ||
So far, research on [2 + 2] cycloaddition in PISA mainly focuses on UV-induced crosslinking to generate more stable assemblies. A limited number of studies have explored the light-controlled reversible reaction in PISA NPs, which expands the application for controlled release and modulation of mechanical properties of hydrogels. There remains significant potential in other areas such as light-responsive drug delivery systems, UV self-healing materials and coatings. In addition, broad wavelength compatibility can be achieved through molecular design and tuning of light-responsive units. Furthermore, as a widely studied dynamic reversible cycloaddition reaction, the Diels–Alder reaction possesses the advantages of catalyst-free, high stability and thermal reversibility, making it attractive for a wide range of studies such as thermally reparable materials104–108 and thermo-control fluorescent platforms,109–111 and yet there is still a need for studies incorporating DA reactions in PISA studies, which would be a direction to proceed in the future.
6. Conclusion
In summary, the exploration of PISA nanomaterials utilizing DCBs represents a new frontier in polymer science. Currently, DCBs used in PISA nanomaterials can be categorized into two main approaches: (1) introducing functional groups into the core or shell of individual assemblies for internal crosslinking or functionalization, and (2) achieving crosslinking among different NPs to create new macroscopic materials. The incorporation of DCBs, such as disulfide bonds, boronate ester bonds, imine bonds, and the [2 + 2] cycloaddition structure, in PISA processes endows the PISA NPs with unique advantages, including enhanced structural stability, dynamic properties, stimulus responsiveness, a tunable morphology and controllable functionality.Despite the significant advances in PISA with DCBs, several challenges demand further attention. First, the introduction of dynamic covalent units in polymerization, such as crosslinking monomers containing disulfide bonds, impacts PISA processes’ morphology evolution and raises concern about reproducibility. The precise control of polymerization kinetics for these monomers requires systematic investigation, particularly in scaling up production from laboratory to industrial scales. Second, PISA based on DCBs commonly focuses on classic disulfide bonds, boronate ester bonds, etc., and has relatively few combinations of multiple DCBs. Newly developed bond types in the past two decades will facilitate the discovery of DCB-based PISA patterns with unique responsiveness and properties.45–47,50 Besides, while the combination of multiple DCBs in PISA shows promise,64 further explorations are needed for more reliable PISA systems with orthogonal responses to different external environments, aiming at expanding the prospects for practical applications under different operating conditions. Third, further development of macroscopic materials based on PISA NPs, such as nanocomposite hydrogels, elastomers, and shape memory polymers containing DCBs, is also required, along with a better understanding of their long-term stability, mechanical properties, and cost-effective manufacturing processes. Additionally, standardized protocols for industrial preparation and characterization are needed to ensure consistent product performance.
Looking forward, diverse applications of PISA nanomaterials with DCBs are anticipated in a wide range of fields, including drug delivery, molecular recognition, sensing, tissue engineering, intelligent materials and other broader areas. Future research may focus not only on expanding the fundamental chemical design of these systems, but also on developing reliable, reproducible, and environmentally/economically friendly preparation methods, advancing the versatility and practicability of DCB-based PISA nanomaterials.
Author contributions
Zhenyu Wan was responsible for writing the manuscript and creating the figures and schemes. Jinying Yuan contributed to the conceptualization and provided essential resources. Nankai An, Chang Xu, Mingxin Zheng and Jinying Yuan all contributed suggestions. All authors participated in the manuscript revision and approved the final version.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22071131, 52273106 and 52473114). We thank Dr Yinyin Bao in the Department of Chemistry and Applied Biosciences at ETH Zurich for the support on manuscript revision.References
- J. Rodriguezhernandez, F. Checot, Y. Gnanou and S. Lecommandoux, Prog. Polym. Sci., 2005, 30, 691–724 CrossRef CAS.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- W.-M. Wan, C.-Y. Hong and C.-Y. Pan, Chem. Commun., 2009, 5883 RSC.
- W.-M. Wan and C.-Y. Pan, Polym. Chem., 2010, 1, 1475 RSC.
- S. L. Canning, G. N. Smith and S. P. Armes, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS.
- X. Chen, L. Liu, M. Huo, M. Zeng, L. Peng, A. Feng, X. Wang and J. Yuan, Angew. Chem., Int. Ed., 2017, 56, 16541–16545 CrossRef CAS PubMed.
- W. Zhang, C. Hong and C. Pan, Macromol. Rapid Commun., 2019, 40, 1800279 CrossRef PubMed.
- C. Liu, C.-Y. Hong and C.-Y. Pan, Polym. Chem., 2020, 11, 3673–3689 RSC.
- F. D'Agosto, J. Rieger and M. Lansalot, Angew. Chem., Int. Ed., 2020, 59, 8368–8392 CrossRef.
- N. An, X. Chen and J. Yuan, Polym. Chem., 2021, 12, 3220–3232 RSC.
- Z. Zhao, S. Lei, M. Zeng and M. Huo, Aggregate, 2023, e418 Search PubMed.
- V. Ladmiral, M. Semsarilar, I. Canton and S. P. Armes, J. Am. Chem. Soc., 2013, 135, 13574–13581 CrossRef CAS PubMed.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- C. J. Mable, R. R. Gibson, S. Prevost, B. E. McKenzie, O. O. Mykhaylyk and S. P. Armes, J. Am. Chem. Soc., 2015, 137, 16098–16108 CrossRef CAS PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Biomacromolecules, 2016, 17, 2992–2999 CrossRef CAS PubMed.
- Q. Ye, M. Huo, M. Zeng, L. Liu, L. Peng, X. Wang and J. Yuan, Macromolecules, 2018, 51, 3308–3314 CrossRef CAS.
- L. D. Blackman, S. Varlas, M. C. Arno, Z. H. Houston, N. L. Fletcher, K. J. Thurecht, M. Hasan, M. I. Gibson and R. K. O'Reilly, ACS Cent. Sci., 2018, 4, 718–723 CrossRef CAS.
- W. Zhang, C. Hong and C. Pan, Macromol. Rapid Commun., 2019, 40, 1800279 CrossRef.
- H. Phan, M. Cossutta, C. Houppe, C. Le Cœur, S. Prevost, I. Cascone, J. Courty, J. Penelle and B. Couturaud, J. Colloid Interface Sci., 2022, 618, 173–184 CrossRef CAS PubMed.
- L. Esser, N. P. Truong, B. Karagoz, B. A. Moffat, C. Boyer, J. F. Quinn, M. R. Whittaker and T. P. Davis, Polym. Chem., 2016, 7, 7325–7337 RSC.
- W. Zhao, H. T. Ta, C. Zhang and A. K. Whittaker, Biomacromolecules, 2017, 18, 1145–1156 CrossRef CAS PubMed.
- P. Li, M. Sun, Z. Xu, X. Liu, W. Zhao and W. Gao, Biomacromolecules, 2018, 19, 4472–4479 CrossRef CAS.
- M. Huo, Z. Wan, M. Zeng, Y. Wei and J. Yuan, Polym. Chem., 2018, 9, 3944–3951 RSC.
- M. Zheng, Q. Ye, X. Chen, M. Zeng, G. Song, J. Zhang and J. Yuan, Chem. Commun., 2022, 58, 6922–6925 RSC.
- C. Xu, M. Zheng, Y. Wei and J. Yuan, Chem. – Eur. J., 2024, 30, e202303586 CrossRef CAS.
- X. Zhang, A. F. Cardozo, S. Chen, W. Zhang, C. Julcour, M. Lansalot, J. Blanco, F. Gayet, H. Delmas, B. Charleux, E. Manoury, F. D'Agosto and R. Poli, Chem. – Eur. J., 2014, 20, 15505–15517 CrossRef CAS.
- S. Chen, A. F. Cardozo, C. Julcour, J.-F. Blanco, L. Barthe, F. Gayet, M. Lansalot, F. D'Agosto, H. Delmas, E. Manoury and R. Poli, Polymer, 2015, 72, 327–335 CrossRef CAS.
- S. Chen, A. F. Cardozo, C. Julcour, J.-F. Blanco, L. Barthe, F. Gayet, M. Lansalot, F. D'Agosto, H. Delmas, E. Manoury and R. Poli, Polymer, 2015, 72, 327–335 CrossRef CAS.
- E. Lobry, A. F. Cardozo, L. Barthe, J.-F. Blanco, H. Delmas, S. Chen, F. Gayet, X. Zhang, M. Lansalot, F. D'Agosto, R. Poli, E. Manoury and C. Julcour, J. Catal., 2016, 342, 164–172 CrossRef CAS.
- J. Wan, B. Fan and S. H. Thang, Nanoscale Adv., 2021, 3, 3306–3315 RSC.
- B. T. T. Pham, D. Nguyen, V. T. Huynh, E. H. Pan, B. Shirodkar-Robinson, M. Carey, A. K. Serelis, G. G. Warr, T. Davey, C. H. Such and B. S. Hawkett, Langmuir, 2018, 34, 4255–4263 CrossRef CAS.
- L. Luppi, T. Babut, E. Petit, M. Rolland, D. Quemener, L. Soussan, M. A. Moradi and M. Semsarilar, Polym. Chem., 2019, 10, 336–344 RSC.
- N. An, X. Chen, M. Zheng and J. Yuan, Chem. Commun., 2023, 59, 7595–7598 RSC.
- Q. Ye, M. Zheng, X. Chen, D. Li, W. Tian, J. Zhang and J. Yuan, Acta Polym. Sin., 2019, 50, 344–351 CAS.
- T. Li, J. Liu, X.-L. Sun, W.-M. Wan, L. Xiao and Q. Qian, Polymer, 2022, 253, 125005 CrossRef CAS.
- M. J. Derry, T. Smith, P. S. O'Hora and S. P. Armes, ACS Appl. Mater. Interfaces, 2019, 11, 33364–33369 CrossRef CAS.
- S. J. Hunter and S. P. Armes, Langmuir, 2020, 36, 15463–15484 CrossRef CAS PubMed.
- Y. Du, S. Jia, Y. Chen, L. Zhang and J. Tan, ACS Macro Lett., 2021, 10, 297–306 CrossRef CAS.
- M. Zeng, S. Zhou, X. Sui and J. Yuan, Chin. J. Chem., 2021, 39, 3448–3454 CrossRef CAS.
- S. Zhou, M. Zeng, Y. Liu, X. Sui and J. Yuan, Macromol. Rapid Commun., 2022, 43, 2200010 CrossRef CAS.
- D. Li, N. Liu, M. Zeng, J. Ji, X. Chen and J. Yuan, Polym. Chem., 2022, 13, 3529–3538 RSC.
- F. García and M. M. J. Smulders, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3551–3577 CrossRef.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- M. M. Perera and N. Ayres, Polym. Chem., 2020, 11, 1410–1423 RSC.
- X. Fu, A. Qin and B. Z. Tang, J. Polym. Sci., 2024, 62, 787–798 CrossRef CAS.
- T. Jadhav, B. Dhokale, Z. M. Saeed, N. Hadjichristidis and S. Mohamed, ChemSusChem, 2024, 17, e202400356 Search PubMed.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- W. Liu, S. Yang, L. Huang, J. Xu and N. Zhao, Chem. Commun., 2022, 58, 12399–12417 RSC.
- J. Lai, X. Xing, H. Feng, Z. Wang and H. Xia, Polym. Chem., 2023, 14, 4381–4406 RSC.
- B. Spitzbarth and R. Eelkema, Cell Rep. Phys. Sci., 2024, 5, 102010 Search PubMed.
- A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC.
- J. F. Reuther, S. D. Dahlhauser and E. V. Anslyn, Angew. Chem., Int. Ed., 2019, 58, 74–85 CrossRef CAS PubMed.
- L. You, Chem. Commun., 2023, 59, 12943–12958 RSC.
- M. Podgórski, B. D. Fairbanks, B. E. Kirkpatrick, M. McBride, A. Martinez, A. Dobson, N. J. Bongiardina and C. N. Bowman, Adv. Mater., 2020, 32, 1906876 CrossRef PubMed.
- N. J. Van Zee and R. Nicolaÿ, Prog. Polym. Sci., 2020, 104, 101233 CrossRef CAS.
- N. Zheng, Y. Xu, Q. Zhao and T. Xie, Chem. Rev., 2021, 121, 1716–1745 CrossRef CAS PubMed.
- Q. Zhang, D.-H. Qu, B. L. Feringa and H. Tian, J. Am. Chem. Soc., 2022, 144, 2022–2033 CrossRef CAS PubMed.
- A. Tageldin, C. A. Omolo, V. O. Nyandoro, E. Elhassan, S. Z. F. Kassam, X. Q. Peters and T. Govender, J. Controlled Release, 2024, 371, 237–257 CrossRef CAS PubMed.
- M. Zheng, Y. Wang, D. Hu, M. Tian, Y. Wei and J. Yuan, Aggregate, 2024, e624 Search PubMed.
- Z. Liu, Y. Tang, Y. Chen, Z. Lu and Z. Rui, Chem. Eng. J., 2024, 497, 154710 CrossRef CAS.
- M. Chen, J.-W. Li, W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Macromolecules, 2019, 52, 1140–1149 CrossRef CAS.
- A. Shahrokhinia, R. A. Scanga, P. Biswas and J. F. Reuther, Macromolecules, 2021, 54, 1441–1451 CrossRef CAS.
- J. Sarkar, K. B. J. Chan and A. Goto, Polym. Chem., 2021, 12, 1060–1067 RSC.
- S. Tafazoli, A. Shahrokhinia, S. Rijal, J. Garay, R. A. Scanga and J. F. Reuther, Polym. Chem., 2023, 14, 4957–4969 RSC.
- L. P. D. Ratcliffe, C. Couchon, S. P. Armes and J. M. J. Paulusse, Biomacromolecules, 2016, 17, 2277–2283 CrossRef CAS PubMed.
- L. Huang, Y. Ding, Y. Ma, L. Wang, Q. Liu, X. Lu and Y. Cai, Macromolecules, 2019, 52, 4703–4712 CrossRef CAS.
- J. Kadirkhanov, C.-L. Yang, Z.-X. Chang, R.-M. Zhu, C.-Y. Pan, Y.-Z. You, W.-J. Zhang and C.-Y. Hong, Polym. Chem., 2021, 12, 1768–1775 RSC.
- J. Rosselgong, A. Blanazs, P. Chambon, M. Williams, M. Semsarilar, J. Madsen, G. Battaglia and S. P. Armes, ACS Macro Lett., 2012, 1, 1041–1045 CrossRef CAS PubMed.
- N. J. Warren, J. Rosselgong, J. Madsen and S. P. Armes, Biomacromolecules, 2015, 16, 2514–2521 CrossRef CAS PubMed.
- L. P. D. Ratcliffe, K. J. Bentley, R. Wehr, N. J. Warren, B. R. Saunders and S. P. Armes, Polym. Chem., 2017, 8, 5962–5971 RSC.
- A. Shahrokhinia, S. Tafazoli, S. Rijal, D. B. Shuster, R. A. Scanga, D. J. Morefield, J. Garay, R. A. Rocheleau, M. Bagheri Kashani, R. Nagarajan, O. G. Apul and J. F. Reuther, Macromolecules, 2024, 57, 628–639 CrossRef CAS.
- R. Deng, M. J. Derry, C. J. Mable, Y. Ning and S. P. Armes, J. Am. Chem. Soc., 2017, 139, 7616–7623 CrossRef CAS PubMed.
- R. Deng, Y. Ning, E. R. Jones, V. J. Cunningham, N. J. W. Penfold and S. P. Armes, Polym. Chem., 2017, 8, 5374–5380 RSC.
- H. S. Dhiraj, F. Ishizuka, A. Elshaer, P. B. Zetterlund and F. Aldabbagh, Polym. Chem., 2022, 13, 3750–3755 RSC.
- L.-C. S. Huang, D. Le, I.-L. Hsiao, S. Fritsch-Decker, C. Hald, S.-C. Huang, J.-K. Chen, J. R. Hwu, C. Weiss, M.-H. Hsu and G. Delaittre, Polym. Chem., 2021, 12, 50–56 RSC.
- Y. Jiang, N. Xu, J. Han, Q. Yu, L. Guo, P. Gao, X. Lu and Y. Cai, Polym. Chem., 2015, 6, 4955–4965 RSC.
- B. Karagoz, L. Esser, H. T. Duong, J. S. Basuki, C. Boyer and T. P. Davis, Polym. Chem., 2014, 5, 350–355 RSC.
- L. Qiu, C.-R. Xu, F. Zhong, C.-Y. Hong and C.-Y. Pan, ACS Appl. Mater. Interfaces, 2016, 8, 18347–18359 CrossRef CAS PubMed.
- J. Huang, D. Li, H. Liang and J. Lu, Macromol. Rapid Commun., 2017, 38, 1700202 CrossRef PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, Biomacromolecules, 2017, 18, 1210–1217 CrossRef CAS PubMed.
- W.-J. Zhang, C.-Y. Hong and C.-Y. Pan, ACS Appl. Mater. Interfaces, 2017, 9, 15086–15095 CrossRef CAS PubMed.
- S. Xu, J. Yeow and C. Boyer, ACS Macro Lett., 2018, 7, 1376–1382 CrossRef CAS PubMed.
- Z. Zhao, L. Fan, G. Song and M. Huo, Chem. Mater., 2024, 36, 1436–1448 CrossRef CAS.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- A. G. Orrillo and R. L. E. Furlan, Angew. Chem., Int. Ed., 2022, 61, e202201168 CrossRef CAS PubMed.
- C. S. Sevier and C. A. Kaiser, Nat. Rev. Mol. Cell Biol., 2002, 3, 836–847 CrossRef CAS PubMed.
- P. J. Hogg, Trends Biochem. Sci., 2003, 28, 210–214 CrossRef CAS PubMed.
- J. Rieger, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- J. R. Lovett, M. J. Derry, P. Yang, F. L. Hatton, N. J. Warren, P. W. Fowler and S. P. Armes, Chem. Sci., 2018, 9, 7138–7144 RSC.
- J.-H. Lee, J. P. Gustin, T. Chen, G. F. Payne and S. R. Raghavan, Langmuir, 2005, 21, 26–33 CrossRef CAS PubMed.
- X. Hao, H. Liu, Y. Xie, C. Fang and H. Yang, Colloid Polym. Sci., 2013, 291, 1749–1758 CrossRef CAS.
- P. Wang, G. Deng, L. Zhou, Z. Li and Y. Chen, ACS Macro Lett., 2017, 6, 881–886 CrossRef CAS.
- X. Li, H. Mutlu, C. Fengler, M. Wilhelm and P. Theato, Polym. Chem., 2021, 12, 361–369 RSC.
- B. Fan, J. Wan, J. Zhai, X. Chen and S. H. Thang, ACS Nano, 2021, 15, 4688–4698 CrossRef CAS PubMed.
- J. Zou, S. Zhang, R. Shrestha, K. Seetho, C. L. Donley and K. L. Wooley, Polym. Chem., 2012, 3, 3146–3156 RSC.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Chem. Commun., 2009, 2106 RSC.
- Y. Xin and J. Yuan, Polym. Chem., 2012, 3, 3045 RSC.
- L. Liu, C. Kong, M. Huo, C. Liu, L. Peng, T. Zhao, Y. Wei, F. Qian and J. Yuan, Chem. Commun., 2018, 54, 9190–9193 RSC.
- E. E. Brotherton, M. J. Smallridge and S. P. Armes, Biomacromolecules, 2021, 22, 5382–5389 CrossRef CAS PubMed.
- H. Buksa, E. C. Johnson, D. H. H. Chan, R. J. McBride, G. Sanderson, R. M. Corrigan and S. P. Armes, Biomacromolecules, 2024, 25, 2990–3000 Search PubMed.
- S. J. Byard, M. Williams, B. E. McKenzie, A. Blanazs and S. P. Armes, Macromolecules, 2017, 50, 1482–1493 CrossRef CAS PubMed.
- H. Frisch, D. E. Marschner, A. S. Goldmann and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2018, 57, 2036–2045 CrossRef CAS PubMed.
- S.-A. Safavi-Mirmahalleh, M. Golshan, B. Gheitarani, M. Salami Hosseini and M. Salami-Kalajahi, Eur. Polym. J., 2023, 198, 112430 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- J. Zhang, Y. Niu, C. Huang, L. Xiao, Z. Chen, K. Yang and Y. Wang, Polym. Chem., 2012, 3, 1390 RSC.
- G. A. Appuhamillage, J. C. Reagan, S. Khorsandi, J. R. Davidson, W. Voit and R. A. Smaldone, Polym. Chem., 2017, 8, 2087–2092 RSC.
- K. Yang, J. C. Grant, P. Lamey, A. Joshi-Imre, B. R. Lund, R. A. Smaldone and W. Voit, Adv. Funct. Mater., 2017, 27, 1700318 CrossRef.
- L. Yang, X. Lu, Z. Wang and H. Xia, Polym. Chem., 2018, 9, 2166–2172 RSC.
- J. Ji, D. Hu, J. Yuan and Y. Wei, Adv. Mater., 2020, 32, 2004616 CrossRef CAS PubMed.
- D. Hu, L. Mao, M. Wang, H. Huang, R. Hu, H. Ma, J. Yuan and Y. Wei, ACS Appl. Mater. Interfaces, 2022, 14, 3485–3495 CrossRef CAS PubMed.
- D. Hu, H. Huang, R. Li, J. Yuan and Y. Wei, Sci. China: Chem., 2022, 65, 1532–1537 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2025 |