
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoregular cyclic poly(3-hydroxybutyrate) enabled by catalyst-controlled tacticity and topology†
Celine R.
Parker‡
 ,
Zhen
Zhang‡
,
Ethan C.
Quinn‡
,
Liam T.
Reilly
and
Eugene Y.-X.
Chen
*
,
Zhen
Zhang‡
,
Ethan C.
Quinn‡
,
Liam T.
Reilly
and
Eugene Y.-X.
Chen
*
Department of Chemistry, Colorado State University, Fort Collins, CO 80523-1872, USA. E-mail: eugene.chen@colostate.edu
First published on 14th March 2025
Abstract
Chemocatalytic routes to poly(3-hydroxybutyrate) (P3HB), a natural biodegradable polyester that has long been considered as a more sustainable alternative to non-(bio)degradable polyolefins, can deliver P3HB materials with catalyst-controlled diverse stereomicrostructures for tunable properties. P3HB topology brings in another way of modulating its properties, but selective synthesis of cyclic P3HB is challenging; yet it is harder to simultaneously control both tacticity and topology. Here we introduce an effective strategy to selectively synthesize cyclic isotactic or syndio-rich P3HB by using bulky C2-symmetric salen-ligated metal catalysts bearing an initiating ligand that is also a good leaving group, and a relatively long and ionic metal–polymer bond, both facilitating chain end-to-end cyclization. The resulting cyclic P3HB topology has been extensively characterized and differentiated from its linear counterpart, including direct visualization. When compared to its linear counterpart, cyclic P3HB of both stereomicrostructures shows noticeably superior properties, including higher melting and decomposition temperatures, better processability, and higher overall mechanical toughness.
Introduction
High-volume commodity plastics, which are typically hard-to-recycle and/or non-(bio)degradable, pose acute environmental concerns due to their growing environmental accumulation and reliance on non-renewable petrochemical feedstocks.1–3 In our current global recycling climate, ∼22% of plastics escape collection, become pollution, and cause serious concerns towards human, animal, and environmental health and safety.4,5 Multifaceted approaches and innovations are required to resolve this issue including, but not limited to, increased participation in the current recycling infrastructure, development of a circular polymer economy, and further consideration of the end-of-life options of new plastics prior to widespread adoption.6–11 When contemplating this last facet, the utility of biodegradable plastics becomes apparent.Poly(3-hydroxyalkanoate)s (PHAs) are a class of bio-sourced and biodegradable polyesters that can breakdown into CO2, water, and biomass under managed or unmanaged environments, making them a more sustainable alternative to non-(bio)degradable plastics.12–16 Isotactic poly(3-hydroxybutyrate) (it-P3HB) is the most widely studied PHA and is a strong material (ultimate tensile stress, σB ∼35 MPa) with excellent barriers to gas (water vapor and oxygen) permeation.17,18 Biological fermentation routes from biorenewable feedstocks have enabled the commercialization of a few selected PHAs, but large-scale implementation of these PHAs has been limited due to their high production cost and inability to tune PHA properties by varying their stereomicrostructures without changing chemical composition (e.g., by copolymerization).19–21 Efforts to reduce the mechanical brittleness of it-P3HB by these routes further complicate its synthesis or processing,22 which are better resolved using stereocontrolled chemocatalysis to produce commercially viable P3HB. Examples include the ring-opening polymerization (ROP) of four-membered β-butyrolactone (BBL)21,23–25 and stereoselective ROP of the eight-membered racemic dimethyl diolide (rac-8DLMe), the latter of which has enabled access to stereoperfect it-P3HB with quantitative isotacticity (>99% meso triads [mm]) and melting transition temperature (Tm) being essentially identical to microbial it-P3HB.26 In seeking to further enhance PHA material properties, the 8DL platform has been extended to produce PHAs of varying tacticities and side-chain substituents, some of which show polyolefin-like properties.27–30
Seeking to diversify the topology of P3HB available via the ROP of 8DL monomers, cyclic P3HB became an attractive synthetic target due to its potential for enhanced material properties such as thermal stability and melt-processability. Although early rheological studies had inconclusive results due to linear contaminants in the cyclic polymers of interest,31–33 techniques such as high-performance liquid chromatography in the critical condition resolved these issues and enabled the study of the pure cyclic polymers for revealing their distinct material properties.34–36 Without chain ends, cyclic polymers exhibit lowered shear viscosities as a result of their reduced entanglement.35,37,38 Having no chain ends also reduces their degrees of conformational freedom, resulting in a lower hydrodynamic volumes and radii of gyration relative to linear counterparts.39 Some cyclic polymers have also been shown to have increased degradation temperatures (Td) over their linear counterparts.40–43 Recent synthetic advancements have resulted in production of cyclic polymers able to be topologically characterized without exhaustive refinement.43–47 Based on these performance-advantaged properties, cyclic polymers show promise for applications ranging from medicine48,49 to electronics50–52 and materials science.53
Cyclic polymers were first shown to be a side product formed via ring-chain equilibration during polycondensation, the discovery of which laid the foundation of the Jacobsen–Stockmeyer theory and has since sparked a prolific amount of research on the topic of cyclic polymers.54–60 Kricheldorf and co-workers first synthesized cyclic polyesters by utilizing cyclic distannoxane initiators through ring-expansion polymerization (REP), producing a series of polyesters from lactone monomers.61 Utilizing BBL, cyclic, syndio-rich P3HB (c/sr-P3HB) with a probability of racemic linkages between two consecutive monomer units (Pr) up to 0.62 was produced. Although this strategy required heating (50–75 °C) for up to 6 days and the polymer retained the hydrolytically unstable distannoxane initiator linkages, making it an unsuitable method for broad implementation. Zwitterionic REP of BBL using an N-heterocyclic carbene, 1,3-dimesitylimidazolin-2-ylidene (SIMes), was then utilized to produce cyclic atactic P3HB (c/at-P3HB).45 Through omitting the use of a protic initiator, c/at-P3HB was also obtained using a zinc oxazinolate catalyst through the proposed back-biting reactions to cleave a labile chain-end.62 The resulting cyclic polymer had a nominally higher degradation temperature (temperature at 5% weight loss, Td,5%) than its linear counterpart. The most recent report of c-P3HB was synthesized from BBL using an [OSSO]-type ligated iron catalyst.63 However, stereocontrolled c-P3HB materials with high molar mass and narrow dispersity have been challenging to obtain. Accordingly, this work addresses this challenge via the metal-catalyzed ROP of 8DLMe with bulky C2-symmetric salen-ligated metal catalysts that can control not only the stereochemistry of the polymerization but also the chain end-to-end cyclization reaction. The catalysts that were discovered from this work to exhibit such catalytic properties are the lanthanum (La)-salen catalysts that carry an initiating ligand that is also a good leaving group, and relatively long and ionic La-OR (polymer) bond, both of which facilitate chain end-to-end cyclization.
Experimental details
Materials
All monomers and catalysts employed in this study were synthesized according to literature procedures.26,29,64 Benzyl alcohol (BnOH) and HPLC-grade dichloromethane were stirred for 12 h on activated calcium hydride and distilled before use. All syntheses and manipulations of air- and moisture-sensitive materials were carried out in flamed Schlenk-type glassware on a dual-manifold Schlenk line, on a high-vacuum line, or in an N2-filled glovebox. Sodium iodide, dithranol, α-cyano-4-hydroxycinnamic acid, trifluoroacetic acid (TFA), sodium trifluoroacetate (NaTFA), 9 kg mol−1 polyethylene glycol (PEG), and trans-2-[3-(4-tert-butylphenyl)-2-propenylidene]malononitrile (DCTB) were obtained from TCI America and used as received. ACS grade tetrahydrofuran, ACS grade chloroform, and ACS grade acetonitrile were obtained from Sigma Aldrich and used as received.General polymerization procedure
Polymerizations were performed in glass reactors inside an inert N2 glovebox at ambient temperature (∼23 °C). The reactor was first charged with a predetermined amount of monomer and solvent (typically dichloromethane (DCM)), and a mixture of catalyst and/or initiator in solvent was stirred at ambient temperature for 10 minutes in another reactor. The polymerization was initiated by rapid addition of the catalyst solution to the monomer solution. After a desired time, a 0.01 mL of aliquot was taken from the reaction mixture and quenched with a 5% solution of benzoic acid in CDCl3 for 1H NMR analysis to obtain the percent monomer conversion data. The polymerization was later quenched by addition of HCl/methanol (5% solution, 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of polymerization solution to quenching solvent). The quenched mixture was then filtered, redissolved in a minimal amount of dichloromethane, and precipitated into an excess amount of cold methanol while stirring. This process was repeated a total of three times and the polymer was then dried in a vacuum oven at 60 °C for 12 h to a constant weight.
:1 ratio of polymerization solution to quenching solvent). The quenched mixture was then filtered, redissolved in a minimal amount of dichloromethane, and precipitated into an excess amount of cold methanol while stirring. This process was repeated a total of three times and the polymer was then dried in a vacuum oven at 60 °C for 12 h to a constant weight.
Instrumentation
MALDI-TOF-MS was also carried out on a MALDI TOF/TOF UltrafleXtreme high resolution mass spectrometer with a 355 nm modified Nd:YAG laser. Solutions were prepared as 20:1:1 mixtures of dithranol:analyte:NaTFA and then 1 μL was added to a ground well on a stainless-steel MicroScout plate and allowed to air dry. Similarly a 20:1:1 mixture of DCTB:PEG:NaTFA solutions was prepared and added to adjacent wells on the plate and allowed to air dry, serving as the external calibration. The raw data was processed in the FlexAnalysis software (version 2.4, Bruker Daltonics).
Results and discussion
Catalyst-controlled synthesis of stereoregular c-P3HB
To selectively synthesize stereoregular c-P3HB from rac-8DLMe, it is of necessity to identify a stereoselective catalyst capable of promoting faster polymerization relative to selective backbiting at the chain end (Fig. 1A). Trityl salen-ligated yttrium silylamide complex 1 (Fig. 1B), when combined with a stoichiometric amount of BnOH co-initiator, undergoes spontaneous protonolysis to cleave the [Y]–N bond and generate in situ the [Y]-OBn active propagating species, and is known to produce linear, stereoperfect it-P3HB from rac-8DLMe.26 The strategy here for the synthesis of cyclic, it-P3HB (c/it-P3HB) is to avoid using the BnOH co-initiator, as the bulky silylamide ligand already present on the precatalyst, [Y]-[N(SiHMe2)2], has been shown to be effective to trigger backbiting by a lanthanum catalyst chain end resulting in end-to-end cyclization.40 The challenge is to achieve high selectivity for the cyclic topology and suppress or prevent the formation of mixed linear and cyclic topologies that can result from indiscriminate transesterification along polyester chains.65,66 Utilizing this catalyst-controlled cyclization strategy would also enable observing the catalyst's stereoselectivity, thereby achieving the control of both P3HB stereomicrostructures and topologies, which had not been previously reported. | ||
| Fig. 1 (A) Sketched synthetic pathways from dimethyl diolide (8DLMe) to linear (l) and cyclic (c) P3HB. (B) Selected catalysts, monomers, and stereomicrostructures of P3HB discussed herein. All it-P3HB discussed is stereo-perfect, with [mm] > 99%. | ||
At the outset, rac-8DLMe (0.2 M, DCM, ∼25 °C) was polymerized using 1 (5 mol%). After achieving full monomer conversion within 1 min, the reaction was left to cyclize for 7 h, and it was then quenched and analyzed by MALDI-TOF-MS. The resulting MS spectrum (Fig. S1†) of P3HB indicated two series of molecular ion peaks. The lower molar mass series, A, was assigned to c-P3HB with no chain ends [Mend = 0 + 23 Na+ g mol−1] and the higher-molar mass series, B, was assigned to l-P3HB with hydroxy/H chain ends [Mend = 18 + 23 Na+ g mol−1]. Further characterization by 13C NMR (Fig. 2C) revealed that the polymer maintained the desired perfect isotacticity ([mm] > 99%), providing evidence that the stereoselectivity of this catalyst was not impacted by the initiator identity (silylamide vs. alkoxide) (Table 1).
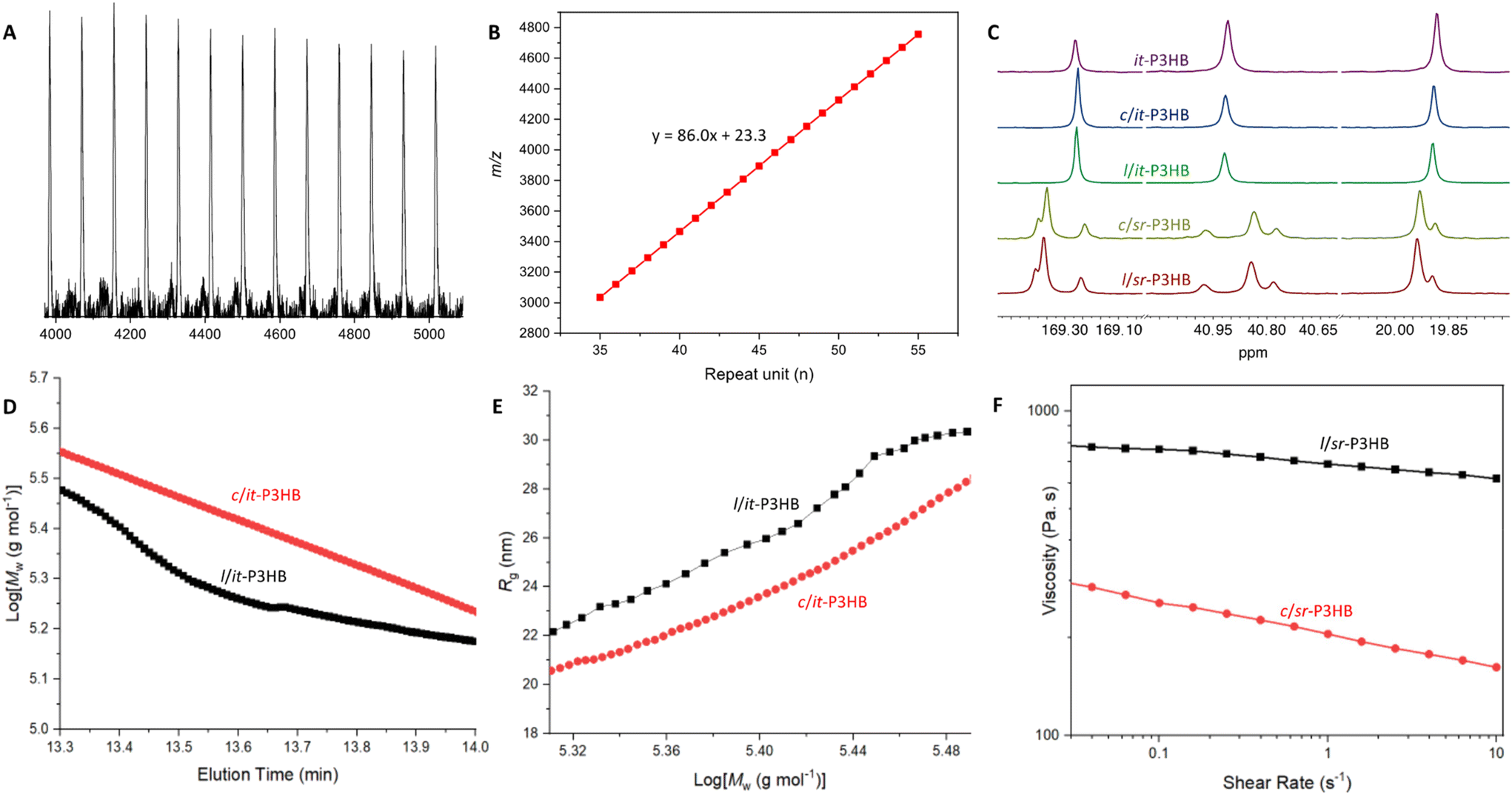 | ||
| Fig. 2 (A) MALDI-TOF-MS spectrum of c/it-P3HB produced using 2. (B) Plot of the mass-to-charge ratio (m/z) vs. the theoretical number of repeat units for c/it-P3HB produced using 2. (C) 13C NMR overlay of P3HB produced in this work: it-P3HB (top, purple) produced using 1, c/it-P3HB (2nd from top, blue) produced using 2, l/it-P3HB (center, green) produced using 2 and BnOH, c/sr-P3HB (2nd from bottom, yellow) produced using 3, l/sr-P3HB (bottom, red) produced using 3 and BnOH. (D) Log of weight-average molar mass (Mw, g mol−1) per elution time (as a function of their hydrodynamic radius) of c/it-P3HB (red, 164 kg mol−1, Đ = 1.21) and l/it-P3HB (black, 141 kg mol−1, Đ = 1.07). (E) Radius of gyration (Rg) per log[Mw] of c/it- and l/it-P3HB. (F) Shear sweeps of l/sr-P3HB (black, 152 kg mol−1, Đ = 1.17) and c/sr-P3HB (red, 143 kg mol−1, Đ = 1.22). | ||
| P3HB | [M] | Catalyst |
M
nb (kg mol−1) |
Đ |
P
rc |
P
mc |
[mm]c (%) | [rr]c (%) | [mr] + [rm]c (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Reaction conditions unless otherwise stated: no alcohol co-initiator, [M] = 0.2 M, dichloromethane (DCM), 7 h, 25 °C. Other details can be found in Table 2. b Absolute weight-average molar mass (Mw), number-average molar mass (Mn), and dispersity (Đ = Mw/Mn) determined by size exclusion chromatography (SEC) coupled with a Wyatt DAWN HELEOS II multi (18) angle light scattering detector and a Wyatt Optilab TrEX dRI detector and performed at 40 °C in chloroform. c Calculated from semiquantitative 13C NMR (CDCl3, 25 °C). d Determined as a mixture of cyclic and linear topologies by MALDI-TOF-MS. Molar mass of the oligomers synthesized for MS analysis were not determined (n.d.) by SEC. e [M] = 0.5 M, 12 h. f BnOH as initiator, [M] = 0.01 M, 0.5 h. g [M] = 0.1 M, 12 h. h BnOH as initiator, [M] = 1.0 M, 7 h. | |||||||||
| c + l/it-P3HBd | rac-8DLMe | 1 | n.d. | n.d. | 0 | >0.99 | >99 | 0 | 0 |
| c/it-P3HBe | rac-8DLMe | 2 | 93.3 | 1.28 | 0 | >0.99 | >99 | 0 | 0 |
| l/it-P3HBf | rac-8DLMe | 2 | 141 | 1.07 | 0 | >0.99 | >99 | 0 | 0 |
| c/sr-P3HBg | meso-8DLMe | 3 | 143 | 1.22 | 0.80 | 0.20 | 0 | 60 | 40 |
| l/sr-P3HBh | meso-8DLMe | 3 | 152 | 1.17 | 0.80 | 0.20 | 0 | 60 | 40 |
| Sample | Polymer | Catalyst | Concentration (mol L−1) | [M]/[Cat] equiv. | Time (h) | Conv. (%, 1H NMR) | M n (kg mol−1, SEC) | Đ |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: dichloromethane (DCM), ∼25° C, inert (N2) atmosphere. b Benzyl alcohol (BnOH) used as an initiator (1 eq.). c Benzyl alcohol (BnOH) used as an initiator (0.5 eq.). | ||||||||
| 2 | c/it-P3HB | 2 | 0.5 | 200/1 | 12 | >99 | 93.3 | 1.28 |
| 3 | l/it-P3HBb | 2 | 0.01 | 600/1 | 0.5 | >99 | 141 | 1.07 |
| 4 | c/sr-P3HB | 3 | 0.1 | 150/1 | 12 | >99 | 143 | 1.22 |
| 5 | l/sr-P3HBb | 3 | 1 | 1000/1 | 7 | 59 | 152 | 1.17 |
| 6 | c/it-P3HB | 2 | 0.005 | 200/1 | 12 | >99 | 164 | 1.21 |
| 7 | l/sr-P3HBc | 3 | 1 | 500/1 | 36 | >99 | 283 | 1.26 |
| 8 | c/sr-P3HB | 3 | 1 | 150/1 | 20 | >99 | 162 | 1.28 |
We hypothesized that the effectiveness of the cyclization involving the metal catalyst chain-end and silylamide (initiating) chain end is affected by the [M]-OR bond length and ionic character. Thus, increasing this bond length and ionic character by switching from a group 3 metal (Y) to a lanthanide metal (Ln) possessing a larger ionic radius would allow for more facile backbiting/cyclization reactions leading to the more selective formation of c-P3HB. Lanthanum (La) fits these criteria as it has the largest ionic radius of the Ln series66,67 and La-based catalysts have previously been shown to be highly active towards the ROP of rac- and meso-8DLMe producing stereoperfect it-P3HB (Mn = 190 kg mol−1, Đ = 1.03, Pm > 0.99) and syndiotactic (st-) P3HB (Mn = 16.9 kg mol−1, Đ = 1.09, Pr = 0.92).29 Additionally, La([NSiMe3]2)3 has previously been utilized to produce cyclic poly(γ-butyrolactone) and cyclic 3,4-trans-cyclohexyl-fused poly(γ-butyrolactone) under neat conditions due to the favorable leaving group ability of the silylamine chain end that facilitates backbiting from the ionic La-OR (polymer) chain end, inducing cyclization.40,68
Based on the above reasoning, we subsequently employed La-based catalyst 2 (Fig. 1B) for the same ROP ([rac-8DLMe] = 0.2 M, 5 mol% 2, DCM, ∼25 °C). Gratifyingly, MALDI-TOF-MS analysis of the polymer sample showed the presence of only a single series of molecular ion peaks attributed to c-P3HB with no chain ends [Mend = 0 + 23 Na+ g mol−1] (Fig. 2A and B). This result supported the above hypothesis that the longer [M]-OR bond length and higher ionic character facilitates chain end-to-end cyclization, thus leading to selective c-P3HB formation, although other alternative cyclization pathways could not be ruled out without further detailed studies to probe all possible cyclization mechanisms. Higher molar mass c/it-P3HB can be obtained by simply adjusting the monomer-to-catalyst/initiator ratio. For example, employing the ratio of rac-8DLMe:2 = 400:1, the ROP accordingly yielded c/it-P3HB with higher molar mass (Mn = 93.3 kg mol−1, Đ = 1.28).
After accomplishing the synthetic target of c/it-P3HB, we tested the hypothesis that this method could be generalized to c-P3HB materials with other stereomicrostructures, thanks to the catalyst-site-controlled stereoselectivity. Thus, salen-ligated La catalyst 3 (Fig. 1B) with tert-butyl substituents in the ortho and para positions was selected to polymerize meso-8DLMe to c/- and l/sr-P3HB materials, using 3 alone and 3 + BnOH, respectively, otherwise employing the same conditions. The cyclic topology of c/sr-P3HB was again observed by MALDI-TOF-MS (Fig. S2†), confirming the generalizability of the cyclic polymer synthesis strategy. Next, higher-molar mass sr-P3HB samples were prepared for further materials testing: l/sr-P3HB (Mn = 152 kg mol−1, Đ = 1.17) and c/sr-P3HB (Mn = 143 kg mol−1, Đ = 1.22). Their tacticities were measured by 13C NMR, showing [mm] = 0%, [mr + rm] = 40%, [rr] = 60% and Pr = 0.80 for both the cyclic and linear polymers (Fig. 2C), again validating that the identity of the initiating species did not impact the stereoselectivity of this catalyst-site-controlled ROP stereoselectivity.
Characterization of synthesized P3HB topologies
MALDI-TOF-MS is an effective and rapid method to screen the topology of P3HB produced by catalysts 2 and 3, but this method is limited to the low molar-mass range, up to ∼5000 m/z for the P3HB analyzed under the current conditions (Fig. 2A). Upon obtaining higher molar mass samples for cyclic and linear it- and sr-P3HBs, further characterization was performed to differentiate between the linear and cyclic topologies. First, size-exclusion chromatography (SEC) experiments comparing their weight-average molar mass (Mw) per elution volume showed that c/it-P3HB (164 kg mol−1, Đ = 1.21) had higher Mw than l/it-P3HB at identical elution times (141 kg mol−1, Đ = 1.07) (Fig. 2D), which aligns with previously observed trends when comparing cyclic and linear polymers.33,46,47 This observed trend is due to the fact that cyclic polymers have fewer degrees of conformational freedom compared to their linear counterparts and thus have reduced hydrodynamic volume.39 Next, the radii of gyration (Rg) of c/it- and l/it-P3HB were measured through multi-angle light scattering and then compared (Fig. 2E). As expected, the Rg values of c/it-P3HB were shown to be lower than their linear counterparts.In an attempt to directly visualize the cyclic topology of c/it-P3HB compared with l/it-P3HB, atomic force microscopy (AFM) was employed to differentiate these two topologies. Thin slices of highly ordered pyrolytic graphite (HOPG) were spin coated with dilute solutions of dissolved P3HB and the resulting substrate depositions were subsequently imaged by AFM. Consistently, images of the c/it-P3HB sample (170 kg mol−1, Đ = 1.26) comprised of large distinct circular aggregates (Fig. S3†). In sharp contrast, images of l/it-P3HB (141 kg mol−1, Đ = 1.07) possessed a strikingly different structural pattern (Fig. S3†). Despite unsuccessfully obtaining images at the single chain level to provide direct topological characterization, the linear and cyclic samples consistently produced contrasting images which allowed for qualitative differentiation between cyclic and linear samples of P3HB with high molar masses. The origin and nature of the apparent differences in substrate deposition/aggregation behavior were not investigated in detail but remain topics of interest.
Thermal, rheological, and mechanical properties of c-P3HB
After validating the topology of the respective stereoregular P3HB samples, we subsequently performed comparative studies of their material properties to uncover their performance differences. Initial characterizations by 13C NMR revealed that c/it-P3HB produced by catalyst 2 also had perfect isotacticity (Pm > 0.99, [mm] > 99%, Fig. 2C). Consequently, through thermal analysis this c/it-P3HB (Mn = 93.3 kg mol−1, Đ = 1.28) was shown to be a semi-crystalline polymer with a Tm of 174 °C and an enthalpy of fusion ΔHf = 106 J g−1 (Fig. 3A, Table 3). This Tm is 4 °C higher than its linear analogue (Mn = 141 kg mol−1, Đ = 1.07) with a Tm = 170 °C (ΔHf = 124 J g−1), which may indicate tighter packing in the crystal lattice of the cyclic polymer than its linear counterpart. Likewise, the crystallization temperature (Tc) of 107 °C of c/it-P3HB was found to be considerably higher than that (87.8 °C) of l/it-P3HB (Fig. 3B). The Td,5% of c/it-P3HB was measured to be 245 °C which was 10 °C higher than l/it-P3HB with a Td,5% of 235 °C (Fig. 3C), which can be attributed to the enhanced stability of c/it-P3HB conferred by the lack of chain ends. Following the same trend, c/sr-P3HB (Mn = 143 kg mol−1, Đ = 1.22) exhibited a higher Tm of 132 °C (ΔHf = 43.0 J g−1) compared to its linear counterpart (Mn = 152 kg mol−1, Đ = 1.17) with a Tm of 129 °C (ΔHf = 35.2 J g−1). Similar to c/it-P3HB, c/sr-P3HB also exhibited a higher Td,5% of 252 °C than l/sr-P3HB which had a Td,5% of 245 °C. A combination of increased Tm values due to better packing and enhanced Td,5% values due to the lack of chain ends demonstrate superior thermal properties of c-P3HB to its counterpart l-P3HB in both it and sr-stereomicrostructures.| P3HB |
T
ma (°C) |
ΔHfa (J g−1) |
T
ca (°C) |
ΔHca (J g−1) |
T
d,5%b (°C) |
T
maxb (°C) |
X
cc (%) |
|---|---|---|---|---|---|---|---|
| a Values obtained using differential scanning calorimetry (DSC) at a heating and cooling rate of 10 °C min−1. b Values obtained using thermogravimetric analysis (TGA) at a heating rate of 10 °C min−1. c Degree of crystallinity. Calculated according to ESI Note 1.† | |||||||
| c/it-P3HB | 174 | 106 | 107 | 93.6 | 245 | 276 | 73 |
| l/it-P3HB | 170 | 124 | 87.8 | 74.1 | 235 | 259 | 84 |
| c/sr-P3HB | 132 | 43.0 | 92.9 | 56.4 | 252 | 285 | 29 |
| l/sr-P3HB | 129 | 35.2 | 83.7 | 50.0 | 245 | 279 | 24 |
 | ||
| Fig. 3 (A) Overlay of DSC trace, 2nd heating scan (10 °C min−1). (B) Overlay of DSC trace cooling scan (10 °C min−1). (C) Representative stress–strain curves of l/sr-P3HB (black, 283 kg mol−1, Đ = 1.26) and c/sr-P3HB (red, 163 kg mol−1, Đ = 1.28). Strain rate = 5 mm min−1. (D) Decomposition temperature overlay of P3HB produced in this work. | ||
Based on the enhanced thermal properties associated with c-P3HB, further assessment of its melt-processability was conducted by observing its shear viscosity through rheology. It was commonly observed that a cyclic polymer exhibits a lower viscosity than its linear counterpart due to reduced chain entanglement.33,35–38,69 Shear sweeps were conducted on c/sr-P3HB (Mn = 143 kg mol−1, Đ = 1.22) and l/sr-P3HB (Mn = 152 kg mol−1, Đ = 1.17). Consistently, the viscosity of c/sr-P3HB was found to be considerably lower than its linear counterpart at the same shear rates (Fig. 2F). Reduced viscosity can enhance processability, therefore c-P3HB shows this additional property advantage.
Lastly, mechanical performance was tested by uniaxial tensile testing and compared between l/sr-P3HB (283 kg mol−1, Đ = 1.26) and c/sr-P3HB (163 kg mol−1, Đ = 1.28). Shown to be a tough, ductile thermoplastic, the l/sr-P3HB exhibited a Young's modulus (E) = 230 ± 22 MPa, tensile strength (σ) = 38.1 ± 2.2 MPa, and elongation at break (εB) = 299 ± 27%. Similarly, the c/sr-P3HB was also shown to be a tough, ductile thermoplastic, with noticeably enhanced tensile values (even with a more than 100 kg mol−1 lower molar mass): E = 298 ± 65 MPa, σ = 40 ± 0.8 MPa, and εB = 375 ± 11% (Fig. 3D). Overall, the above results showed enhanced thermal, mechanical, and rheological properties of c-P3HB over its linear counterpart.
Conclusions
This study has revealed that, in the chemocatalytic synthesis of stereoregular P3HB by the metal-catalyzed ROP of eight-membered diolide monomers, rac-8DLMe and meso-8DLMe, the catalyst site can impart control over not only the stereoregularity (e.g., stereoperfect it-P3HB and sr-P3HB) but also the topology of the resulting P3HB materials. In particular, while the use of the typical [Y]-[N(SiHMe2)2] + ROH initiating system leads to formation of l-P3HB, removal of the ROH co-initiator triggers generation of a significant portion of c-P3HB in the product mixture, due to backbiting cyclization and the silylamide being a better leaving group than the typical alkoxide group. More interestingly, simply switching the salen-ligated catalyst's metal center from [Y] to [La], the latter of which exhibits a longer [M]-OR (polymer) length and ionic character, renders selective formation of c-P3HB, attributed to enhanced chain end-to-end cyclization upon completion of polymerization.The linear and cyclic P3HB topologies were characterized and compared extensively, through a suite of analytical techniques, including NMR, MALDI-TOF-MS, SEC, DSC, TGA, rheological and tensile tests, as well as direct visualization by AFM imaging. Most notably, for both types of P3HB stereomicrostructures, it-P3HB and sr-P3HB, when compared to its linear counterpart, cyclic P3HB demonstrates noticeably superior properties, including higher thermal transitions (Tm and Tc) and decomposition temperature (Td); better processability (lower shear viscosity); and somewhat but noticeable higher Young's modulus, ultimate tensile strength, fracture stain, and thus overall toughness. While further investigation into the underlying cyclization mechanism is warranted, these results highlight the important role of the [M]-X (X = initiating nucleophile, leaving group, or polymer chain) bond properties (length, ionic character, metal size) in controlling the selectivity of the resulting polymer topology and should stimulate further research on selective synthesis of cyclic polymers with specific property enhancements.
Author contributions
C. R. P., Z. Z., & E. C. Q.: investigation, data curation, writing – original draft, writing – review & editing, visualization, validation, and methodology. L. T. R.: imaging, visualization, writing – review & editing. E. Y.-X. C.: resources, writing – review & editing, supervision, project administration, and funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Advanced Materials and Manufacturing Technologies Office (AMMTO), and Bioenergy Technologies Office (BETO), performed as part of the BOTTLE Consortium, which includes the members from Colorado State University, and funded under contract no. DE-AC36-08GO28308 with the National Renewable Energy Laboratory, operated by the Alliance for Sustainable Energy. The authors also thank the Analytical Resources Core (RRID:SCR_021758) and Dr Alyssa Winter May at Colorado State University for instrument access, training, and assistance with sample analysis using the Bruker Ultraflex, which was obtained using NSF grant no. 2117934.References
- S. B. Borrelle, J. Ringma, K. L. Law, C. C. Monnahan, L. Lebreton, A. McGivern, E. Murphy, J. Jambeck, G. H. Leonard, M. A. Hilleary, M. Eriksen, H. P. Possingham, H. De Frond, L. R. Gerber, B. Polidoro, A. Tahir, M. Bernard, N. Mallos, M. Barnes and C. M. Rochman, Science, 2020, 369, 1515–1518 CAS.
- A. Chamas, H. Moon, J. Zheng, Y. Qiu, T. Tabassum, J. H. Jang, M. Abu-Omar, S. L. Scott and S. Suh, ACS Sustainable Chem. Eng., 2020, 8, 3494–3511 CAS.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 Search PubMed.
- P. G. C. Nayanathara Thathsarani Pilapitiya and A. S. Ratnayake, Cleaner Mater., 2024, 11, 100220 CrossRef CAS.
- OECD, Global Plastics Outlook, 2022 Search PubMed.
- R. W. Clarke, G. Rosetto, T. Uekert, J. B. Curley, H. Moon, B. C. Knott, J. E. McGeehan and K. M. Knauer, Mater. Adv., 2024, 5, 6690–6701 RSC.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS.
- R. Meys, A. Kätelhön, M. Bachmann, B. Winter, C. Zibunas, S. Suh and A. Bardow, Science, 2021, 374, 71–76 CrossRef CAS.
- F. Vidal, E. R. van der Marel, R. W. F. Kerr, C. McElroy, N. Schroeder, C. Mitchell, G. Rosetto, T. T. D. Chen, R. M. Bailey, C. Hepburn, C. Redgwell and C. K. Williams, Nature, 2024, 626, 45–57 CrossRef CAS.
- A. Dhaini, V. Hardouin-Duparc, A. Alaaeddine, J.-F. Carpentier and S. M. Guillaume, Prog. Polym. Sci., 2024, 149, 101781 CrossRef CAS.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Trends Chem., 2019, 1, 148–151 CrossRef CAS.
- K. W. Meereboer, M. Misra and A. K. Mohanty, Green Chem., 2020, 22, 5519–5558 RSC.
- H.-M. Müller and D. Seebach, Angew. Chem., Int. Ed. Engl., 1993, 32, 477–502 CrossRef.
- B. Laycock, P. Halley, S. Pratt, A. Werker and P. Lant, Prog. Polym. Sci., 2013, 38, 536–583 CrossRef CAS.
- A. Sangroniz, J.-B. Zhu, X. Tang, A. Etxeberria, E. Y.-X. Chen and H. Sardon, Nat. Commun., 2019, 10, 3559 CrossRef PubMed.
- D. K. Bedade, C. B. Edson and R. A. Gross, Molecules, 2021, 26, 3463 CrossRef CAS PubMed.
- H. Park, H. He, X. Yan, X. Liu, N. S. Scrutton and G.-Q. Chen, Biotechnol. Adv., 2024, 71, 108320 CrossRef CAS.
- A. H. Westlie, E. C. Quinn, C. R. Parker and E. Y.-X. Chen, Prog. Polym. Sci., 2022, 134, 101608 CrossRef CAS.
- G. J. M. de Koning and P. J. Lemstra, Polymer, 1993, 34, 4089–4094 CrossRef CAS.
- J. Bruckmoser, S. Pongratz, L. Stieglitz and B. Rieger, J. Am. Chem. Soc., 2023, 145, 11494–11498 CrossRef CAS PubMed.
- H.-Y. Huang, W. Xiong, Y.-T. Huang, K. Li, Z. Cai and J.-B. Zhu, Nat. Catal., 2023, 6, 720–728 CrossRef CAS.
- M. S. Young, A. M. LaPointe, S. N. MacMillan and G. W. Coates, J. Am. Chem. Soc., 2024, 146, 18032–18040 CrossRef CAS PubMed.
- X. Tang and E. Y.-X. Chen, Nat. Commun., 2018, 9, 2345 CrossRef PubMed.
- E. C. Quinn, A. H. Westlie, A. Sangroniz, M. R. Caputo, S. Xu, Z. Zhang, M. Urgun-Demirtas, A. J. Müller and E. Y.-X. Chen, J. Am. Chem. Soc., 2023, 145, 5795–5802 CrossRef CAS PubMed.
- X. Tang, A. H. Westlie, L. Caporaso, L. Cavallo, L. Falivene and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2020, 59, 7881–7890 CrossRef CAS PubMed.
- X. Tang, A. H. Westlie, E. M. Watson and E. Y.-X. Chen, Science, 2019, 366, 754–758 CrossRef CAS PubMed.
- Z. Zhang, E. C. Quinn, J. L. Olmedo-Martínez, M. R. Caputo, K. A. Franklin, A. J. Müller and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2023, 62, e202311264 CrossRef CAS PubMed.
- G. B. McKenna, G. Hadziioannou, P. Lutz, G. Hild, C. Strazielle, C. Straupe, P. Rempp and A. J. Kovacs, Macromolecules, 1987, 20, 498–512 CrossRef CAS.
- J. Roovers, Macromolecules, 1985, 18, 1359–1361 CrossRef CAS.
- J. Roovers, Macromolecules, 1988, 21, 1517–1521 CrossRef CAS.
- P. G. Santangelo, C. M. Roland, T. Chang, D. Cho and J. Roovers, Macromolecules, 2001, 34, 9002–9005 CrossRef CAS.
- M. Kapnistos, M. Lang, D. Vlassopoulos, W. Pyckhout-Hintzen, D. Richter, D. Cho, T. Chang and M. Rubinstein, Nat. Mater., 2008, 7, 997–1002 CrossRef CAS PubMed.
- R. Pasquino, T. C. Vasilakopoulos, Y. C. Jeong, H. Lee, S. Rogers, G. Sakellariou, J. Allgaier, A. Takano, A. R. Brás, T. Chang, S. Gooßen, W. Pyckhout-Hintzen, A. Wischnewski, N. Hadjichristidis, D. Richter, M. Rubinstein and D. Vlassopoulos, ACS Macro Lett., 2013, 2, 874–878 CrossRef CAS PubMed.
- Y. Doi, K. Matsubara, Y. Ohta, T. Nakano, D. Kawaguchi, Y. Takahashi, A. Takano and Y. Matsushita, Macromolecules, 2015, 48, 3140–3147 CrossRef CAS.
- D. Richter, S. Gooßen and A. Wischnewski, Soft Matter, 2015, 11, 8535–8549 RSC.
- J. Roovers, in Cyclic Polymers, ed. J. A. Semlyen, Springer Netherlands, Dordrecht, 2002, pp. 347–384, DOI:10.1007/0-306-47117-5_10.
- M. Hong and E. Y.-X. Chen, Nat. Chem., 2016, 8, 42–49 CrossRef CAS PubMed.
- J. A. Kaitz, C. E. Diesendruck and J. S. Moore, J. Am. Chem. Soc., 2013, 135, 12755–12761 CrossRef CAS PubMed.
- H. Li, J. Ollivier, S. M. Guillaume and J.-F. Carpentier, Angew. Chem., Int. Ed., 2022, 61, e202202386 CrossRef CAS PubMed.
- J.-B. Zhu, E. M. Watson, J. Tang and E. Y.-X. Chen, Science, 2018, 360, 398–403 CrossRef CAS PubMed.
- F. M. Haque and S. M. Grayson, Nat. Chem., 2020, 12, 433–444 CrossRef CAS PubMed.
- W. Jeong, J. L. Hedrick and R. M. Waymouth, J. Am. Chem. Soc., 2007, 129, 8414–8415 CrossRef CAS.
- M. L. McGraw, R. W. Clarke and E. Y.-X. Chen, J. Am. Chem. Soc., 2021, 143, 3318–3322 CrossRef CAS PubMed.
- L. Zhou, L. T. Reilly, C. Shi, E. C. Quinn and E. Y.-X. Chen, Nat. Chem., 2024, 16, 1357–1365 CrossRef CAS PubMed.
- N. Nasongkla, B. Chen, N. Macaraeg, M. E. Fox, J. M. J. Fréchet and F. C. Szoka, J. Am. Chem. Soc., 2009, 131, 3842–3843 CrossRef CAS PubMed.
- B. Zhang, H. Zhang, Y. Li, J. N. Hoskins and S. M. Grayson, ACS Macro Lett., 2013, 2, 845–848 CAS.
- M. Ball, Y. Zhong, B. Fowler, B. Zhang, P. Li, G. Etkin, D. W. Paley, J. Decatur, A. K. Dalsania, H. Li, S. Xiao, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2016, 138, 12861–12867 CrossRef CAS PubMed.
- Z. Miao, A. M. Esper, S. S. Nadif, S. A. Gonsales, B. S. Sumerlin and A. S. Veige, React. Funct. Polym., 2021, 169, 105088 CAS.
- J. E. Poelma, K. Ono, D. Miyajima, T. Aida, K. Satoh and C. J. Hawker, ACS Nano, 2012, 6, 10845–10854 CAS.
- M. Romio, L. Trachsel, G. Morgese, S. N. Ramakrishna, N. D. Spencer and E. M. Benetti, ACS Macro Lett., 2020, 9, 1024–1033 CAS.
- C. Hu, E. Louisy, G. Fontaine and F. Bonnet, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 3175–3179 CrossRef CAS.
- H. Jacobson, C. Beckmann and W. Stockmayer, J. Chem. Phys., 1950, 18, 1607–1612 CAS.
- H. Jacobson and W. H. Stockmayer, J. Chem. Phys., 1950, 18, 1600–1606 CAS.
- H. R. Kricheldorf, S. M. Weidner and F. Scheliga, Polym. Chem., 2020, 11, 2595–2604 CAS.
- J. Ochs, C. A. Pagnacco and F. Barroso-Bujans, Prog. Polym. Sci., 2022, 134, 101606 CAS.
- J. A. Semlyen, Cyclic polymers, Springer, 2000 Search PubMed.
- Y. Tezuka, Topological Polymer Chemistry: Progress of Cyclic Polymers in Syntheses, Properties and Functions, 2012 Search PubMed.
- H. R. Kricheldorf and S.-R. Lee, Macromolecules, 1995, 28, 6718–6725 CrossRef CAS.
- M. Shaik, J. Peterson and G. Du, Macromolecules, 2019, 52, 157–166 CrossRef CAS.
- S. Impemba, F. Della Monica, A. Grassi, C. Capacchione and S. Milione, ChemSusChem, 2020, 13, 141–145 CrossRef CAS PubMed.
- Q. Liu, C. Meermann, H. W. Görlitzer, O. Runte, E. Herdtweck, P. Sirsch, K. W. Törnroos and R. Anwander, Dalton Trans., 2008, 6170–6178 RSC.
- A. V. Prasad, L. P. Stubbs, Z. Ma and Z. Yinghuai, J. Appl. Polym. Sci., 2012, 123, 1568–1575 CrossRef CAS.
- A. Duda and A. Kowalski, in Handbook of Ring–Opening Polymerization, 2009, pp. 1–51 Search PubMed.
- R. Anwander, in Lanthanides: Chemistry and Use in Organic Synthesis, ed. S. Kobayashi, Springer Berlin Heidelberg, Berlin, Heidelberg, 1999, pp. 1–61 Search PubMed.
- J.-B. Zhu and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2018, 57, 12558–12562 CrossRef CAS PubMed.
- M. Rubinstein, Phys. Rev. Lett., 1986, 57, 3023–3026 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01304b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |