
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of dendronized polymers through Pd-initiated C1 polymerization of diazoacetates with different generation ester-type dendron groups†
Hiroaki
Shimomoto
 *,
Takumi
Kubo
,
Kazunari
Nishigawa
,
Hazuki
Okuda
,
Makoto
Ishimoto
,
Tomomichi
Itoh
and
Eiji
Ihara
*
*,
Takumi
Kubo
,
Kazunari
Nishigawa
,
Hazuki
Okuda
,
Makoto
Ishimoto
,
Tomomichi
Itoh
and
Eiji
Ihara
*
Department of Applied Chemistry, Graduate School of Science and Engineering, Ehime University, 3 Bunkyo-cho, Matsuyama 790-8577, Japan. E-mail: shimomoto.hiroaki.mx@ehime-u.ac.jp; ihara@ehime-u.ac.jp; Fax: +81-89-927-9949; Fax: +81-89-927-8547; Tel: +81-89-927-9949, +81-89-927-8547
First published on 6th January 2025
Abstract
A series of diazoacetates with an ester-type dendron unit of 1st to 3rd generation as an ester substituent were newly prepared and their Pd-initiated polymerization was conducted to afford dendronized C1 polymers with the SEC-estimated number-average molecular weight (Mn,SEC) of up to 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 and a relatively narrow molecular weight distribution (Đ = 1.12–1.37). The glass transition temperature (Tg) of these dendronized polymers increased with the increasing generation of the dendron unit, where the degree of increase (ΔTg) was much larger than that observed with their vinyl polymer counterparts (polymethacrylates with the same dendron substituents). The diazoacetate with the 3rd generation dendron unit was successfully copolymerized with benzyl diazoacetate to afford a copolymer, whose dendron-derived units were converted to those with multiple hydroxy groups via deprotection of acetonide-protecting groups at the peripheral positions.
700 and a relatively narrow molecular weight distribution (Đ = 1.12–1.37). The glass transition temperature (Tg) of these dendronized polymers increased with the increasing generation of the dendron unit, where the degree of increase (ΔTg) was much larger than that observed with their vinyl polymer counterparts (polymethacrylates with the same dendron substituents). The diazoacetate with the 3rd generation dendron unit was successfully copolymerized with benzyl diazoacetate to afford a copolymer, whose dendron-derived units were converted to those with multiple hydroxy groups via deprotection of acetonide-protecting groups at the peripheral positions.
Introduction
Transition-metal initiated polymerization of diazoacetate is one of the representative examples of C1 polymerization, where the backbone of carbon–carbon (C–C) main chain polymers is constructed from “one carbon unit”.1–8 All the main chain carbon atoms of the polymers obtained by the C1 polymerization of diazoacetate have an alkoxycarbonyl group (ester) as a substituent, which is the most striking characteristic of the polymers, in comparison to their vinyl polymer counterpart, poly(alkyl acrylate), whose alkoxycarbonyl substituents are located on every other carbon atom on the main chain. The structural feature of C1 polymers is expected to bring about enhanced physical properties or functionalities because of the dense packing of the ester substituents around the polymer main chain, and we and other research groups have indeed observed enhanced properties in some C1 polymers in comparison to their vinyl polymer counterparts with the same ester substituents.9–28As initiators for the polymerization, some Rh and Pd complexes have been reported to be effective, yielding high number-average molecular weight (Mn) polymers in good yield. Our group has been focusing on the development of Pd-based initiating systems for the diazoacetate polymerization, reporting some effective systems such as (NHC)Pd(nq)/NaBPh4 (NHC = N-heterocyclic carbene, nq = naphthoquinone),29 π-allylPdCl/NaBPh4,30,31 and (nq)2Pd/NaBPh4.32 Although these initiating systems can afford high Mn polymers in good yield, it is difficult to control Mn of the product, and the controlled polymerization of diazoacetate with Pd-based initiating systems is one of our current important objectives. (Some Pd-based initiators have been reported to be effective for the controlled polymerization of diazoacetate by other research groups.33–35)
Meanwhile, in the course of our research, we have found and reported that, when the monomer has a sterically bulky ester substituent such as cyclotriphosphazene13,36 and benzyl ether-type dendrons,26 the π-allylPdCl/NaBPh4-initiated polymerization of such diazoacetates proceeds in a controlled manner to give polymers with a narrow molecular weight distribution (MWD). In particular, for the latter monomer, we have revealed that the use of the dendron unit with a higher generation leads to a narrower MWD of the C1 polymer products.26 In addition, we introduced protected carboxyl groups at the peripheral positions of the benzyl ether-type dendron unit of the monomer, and after its polymerization followed by deprotection we observed enhanced acidity of the resulting C1 polymer in comparison to its vinyl polymer counterpart.
Herein, we attempted to introduce ester-type dendron units37–40 in place of the above-described benzyl ether-type dendron units as an ester substituent of the monomers. Whereas the ester-type dendron has been utilized for preparation of dendronized polymers via ring-opening metathesis polymerization (ROMP) and radical polymerization so far,41–45 the results obtained here will be the first example of a dendronized C1 polymer with an ester-type dendron. In addition, because the C1 polymer with a benzyl-ether type dendron described above is the only example of a dendronized C1 polymer, this attempt to prepare new diazoacetates with dendron units and to investigate their polymerization behavior should be worthwhile for extending the scope of dendronized polymers46–51 as well as developing the promising chemistry of C1 polymerization. Diazoacetate with an ester-type dendron substituent with a higher generation is expected to undergo controlled polymerization in a similar manner to that with a benzyl ether-type dendron26 because of the steric bulkiness of the substituent as well. In addition, because the acetonide-protecting groups at the peripheral positions of the dendron unit can be removed to liberate multiple hydroxy groups, the incorporation of the ester-type dendron unit via copolymerization with other diazoacetates can be an effective general method for introducing highly hydrophilic substituents into the C1 polymer backbone. We report the synthesis of new diazoacetates with a series of ester-type dendron substituents and their polymerization behavior with π-allylPdCl/NaBPh4 as an initiator. In addition, copolymerization of a dendron-containing monomer with other less bulky diazoacetates and deprotection of the acetonide-protecting group to yield C1 copolymers with multiple hydroxy groups are described.
Results and discussion
Monomer syntheses
For the synthesis of ester-type dendron-containing diazoacetates required for this study, we should prepare ester-type dendrons having a hydroxy group at their core, which will be transformed to a diazoacetyl group with the standard diazoacetylation using bromoacetyl bromide and N,N′-ditosylhydrazine.52 In addition, for efficient progress of C1 polymerization of the sterically bulky diazoacetates, we considered that the presence of a CH2CH2 linker between the dendron and diazoacetyl group would be suitable. The syntheses of the desired precursor compounds meeting these conditions, G1-OH in Scheme 1 and its G2,3-derivatives G2-OH and G3-OH in Scheme 2, have been reported by Hedrick and coworkers,39 who used esterification of acetonide-protected bis-MPA 1 [bis-MPA: 2,2-bis(hydroxymethyl) propionic acid] with 2-benzyloxyethanol, followed by deprotection with hydrogenation with Pd/C for the introduction of a 2-hydroxyethyl group as shown in Scheme 1(a) for the synthesis of G1-OH as a representative example. However, in order to avoid the use of hydrogenation for benzyl-ether deprotection, we attempted to use partially silyl-protected ethylene glycol 2 for the introduction of the linker unit. The non-protected hydroxy group in 2 was used for the esterification of 1 to yield G1-Si, followed by TBAF-mediated deprotection of a silyl-group to afford G1-OH as shown in Scheme 1(b). | ||
| Scheme 1 Methods for preparation of hydroxyethyl ester of acetonide protected bis-MPA. | ||
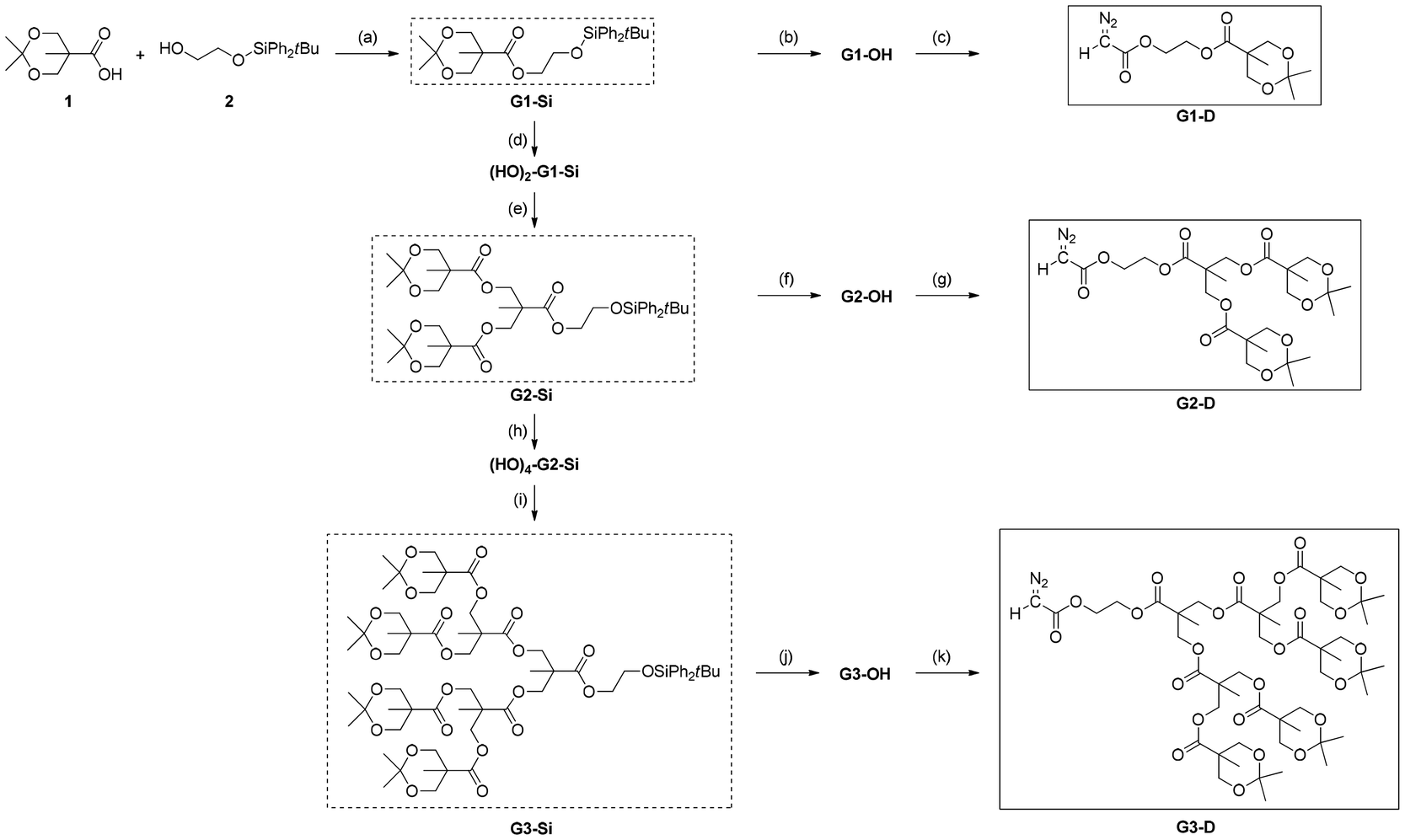 | ||
| Scheme 2 Synthetic routes for diazoacetates with an ester-type dendron G(1–3)-D. Reagents and conditions: (a) diisopropyl azodicarboxylate (DIAD), PPh3, THF, RT, 12 h (88%); (b) tetrabutylammonium fluoride (TBAF), THF, RT, 3 min (71%); (c) bromoacetyl bromide, pyridine, THF, 0 °C, 3 h; N,N′-ditosylhydrazine, 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), THF, 0 °C, 3 h (66%); (d) CuCl2·2H2O, −10 to 0 °C, 20 h (81%); (e) 1, DIAD, PPh3, THF, RT, 46 h (72%); (f) TBAF, THF, RT, 15 h (66%); (g) bromoacetyl bromide, pyridine, THF, 0 °C, 5 h; N,N′-ditosylhydrazine, DBU, THF, 0 °C, 5 h (64%); (h) CuCl2·2H2O, −10 to 0 °C, 20 h (70%); (i) 1, DIAD, PPh3, THF, 50 °C, 72 h (63%); (j) TBAF, THF, RT, 30 min (77%); (k) bromoacetyl bromide, pyridine, THF, 0 °C, 7 h; N,N′-ditosylhydrazine, DBU, THF, 0 °C, 7 h (55%). | ||
Then, as shown in Scheme 2, the 1st generation diazoacetate monomer G1-D was successfully prepared by diazoacetylation of the precursor alcohol G1-OH with the standard procedure reported by Fukuyama and coworkers.52 In addition, as summarized in Scheme 2, starting from G1-Si, the systematic divergent syntheses of G2-D and G3-D were also realized by repeating (1) the acetonide-deprotection with CuCl2·2H2O, (2) esterification of the hydroxy groups with 1, (3) silyl deprotection, and (4) diazoacetylation. Experimental procedures and characterization data for the isolated new compounds are described in the ESI.†
Homopolymerization of diazoacetates with an ester-type dendron G(1–3)-D
Following the results of the polymerization of diazoacetates containing a benzyl ether-type dendron in our previous publication,26 we tried to polymerize diazoacetates with a series of ester-type dendrons, G(1–3)-D, using π-allylPdCl/NaBPh4 as an initiator (Scheme 3 and Table 1). As shown in runs 1 and 2, when polymerization of G1-D with a feed ratio of [G1-D]/[Pd] = 50 was conducted at room temperature and −20 °C, the results at −20 °C were better with respect to Mn and polymer yield. Then, at −20 °C, the polymerization was conducted with increasing feed ratios of 100 and 200 (runs 3 and 4), resulting in the gradual increase of Mn with the feed ratio of up to Mn,SEC = 16700, with a moderate polymer yield of 61% and 64%. A further increase of the feed ratio to [G1-D]/[Pd] = 300 did not lead to a higher Mn, but lower polymer yield (run 5). MWDs of products in runs 1–5 in the range of Đ = 1.29–1.37 were relatively narrow for products obtained with this initiator.
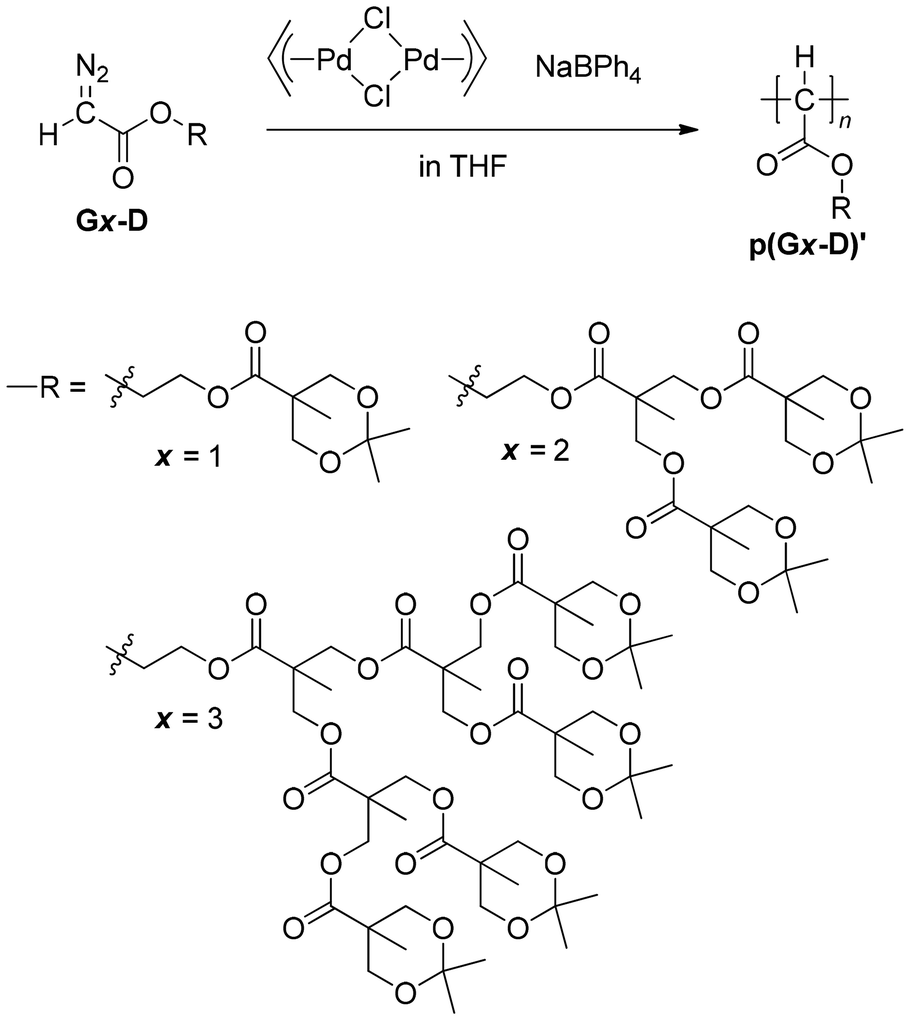 | ||
| Scheme 3 Homopolymerization of diazoacetates with an ester-type dendron G(1–3)-D. | ||
| Run | Monomer (M) | Temp. | [M]/[Pd] | Yieldb (%) | M n | Đ | M n,MALS | Đ MALS |
T
g
(°C) |
|---|---|---|---|---|---|---|---|---|---|
| a In THF for 15 h, [monomer] = ca. 0.1 M, [NaBPh4]/[Pd] = ca. 1.2. b Determined by gravimetry after purification with preparative SEC. c Determined by SEC calibrated using PMMA standards. d M n,MALS values were determined by SEC-MALS. The average degree of polymerization (DP) values were calculated from Mn,MALS. Apparent initiator efficiency (IE) values were calculated from Mn,MALS and polymer yield. e Determined by DSC measurements. | |||||||||
| 1 | G1-D | RT | 50 | 48 | 4900 | 1.29 | |||
| 2 | G1-D | −20 °C | 50 | 56 | 6500 | 1.29 | 10900 (DP = 42, IE = 0.66) |
1.34 | 5 |
| 3 | G1-D | −20 °C | 100 | 61 | 10900 |
1.32 | |||
| 4 | G1-D | −20 °C | 200 | 64 | 16700 |
1.37 | |||
| 5 | G1-D | −20 °C | 300 | 51 | 13800 |
1.30 | |||
| 6 | G2-D | RT | 50 | 35 | 4200 | 1.12 | |||
| 7 | G2-D | −20 °C | 50 | 49 | 8700 | 1.19 | 12700 (DP = 24, IE = 1.0) |
1.21 | 10 |
| 8 | G2-D | −20 °C | 100 | 50 | 10300 |
1.14 | |||
| 9 | G2-D | −20 °C | 200 | 42 | 14200 |
1.31 | |||
| 10 | G3-D | −20 °C | 50 | 30 | 7000 | 1.16 | 39 | ||
As for the polymerization of G2-D, the results of polymerization at −20 °C are again better than those at room temperature (runs 6 and 7), and with the increase of the [G2-D]/[Pd] feed ratio to 100 and 200 at −20 °C (runs 8 and 9), the Mn,SEC increased up to 14200. Compared to the results of G1-D, MWDs of p(G2-D)s obtained at −20 °C were narrower in the range of Đ = 1.14–1.31; these low Đ values cannot be considered as higher controllability of G2-D than that of G1-D but indicated the same level of controllability, on the basis of the comparison with the data reported for their vinyl polymer counterparts as discussed later in this paper. Meanwhile, the polymer yield became slightly lower (42–50%), suggesting that the sterically larger ester substituent in G2-D brought about lower reactivity because of its steric demand during the polymerization. Fig. 1 shows 1H NMR spectra of G2-D and p(G2-D)′ of a sample obtained in run 6, where we can observe broad signals in the spectrum of p(G2-D)′ corresponding to each signal in the spectrum of the monomer except for the signal for the H on the diazo-bearing carbon atom, which was converted to a main chain CH exhibiting a broad signal at 3.0–3.8 ppm in the spectrum of p(G2-D)′.
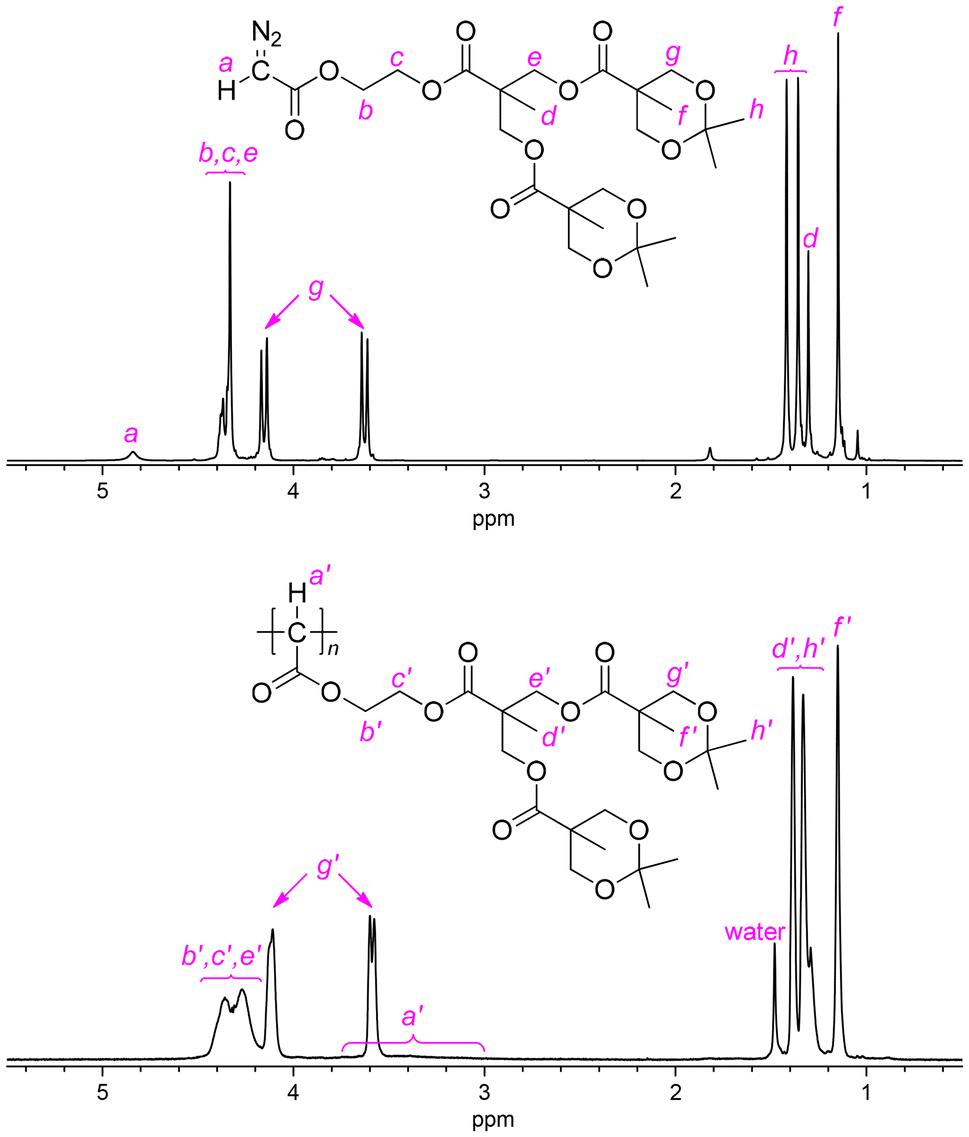 | ||
| Fig. 1 1H NMR spectra of G2-D (upper) and p(G2-D)′ (lower) recorded in CDCl3. | ||
In contrast to G1-D and G2-D affording polymers in moderate yield (ca. 50%), the polymerization of G3-D with a [G3-D]/[Pd] feed ratio of 50 afforded a polymer in a low yield of 30% (run 10), suggesting that the ester substituent of G3-D is too bulky for the polymerization to proceed effectively. Also, the narrow MWD of the p(G3-D)′ (Đ = 1.16) suggests that the polymerization of G3-D would proceed with the same level of controllability as those of G1-D and G2-D as well (see discussion below).
SEC-MALS measurements were carried out for the samples in runs 2 and 7 in Table 1. As reported in our previous publication for C1 polymers obtained from diazoacetates, the Mn,MALS values were 1.5–1.7 times higher than those estimated by standard PMMA-calibrated SEC. In addition, on the basis of the Mn,MALS and polymer yield, the apparent initiator efficiency ([Pd acting as an initiator]/[Pd employed]) was calculated for these samples and is listed in Table 1. These data indicate that while 66% of π-allylPdCl employed for the polymerization of G1-D acted as an initiator, the Pd complex almost quantitatively participated in the initiation in the case of polymerization of G2-D. These results suggest more efficient initiation of G2-D with bulkier ester substituents than G1-D.
Fortunately for us, A. Hult and coworkers reported the preparation and physical properties of polymethacrylates with the same ester-type dendron as ours with 1st to 4th generation via atom transfer radical polymerization (ATRP).41,42 Coincidentally, as shown in Chart 1, the oxyethylene spacer unit between the dendron and the main chain of the polymethacrylates is exactly the same as ours, making the comparison here more relevant. In their report, they prepared polymethacrylate with a 1st generation dendron via ATRP and polymers with higher generations via divergent transformation (post-polymerization modification) therefrom. Importantly to us, the SEC-determined Đ value gradually decreased with the increase of the dendron generation, from 1.24 to 1.11 (Chart 1), even though the polymethacrylate backbone is identical for these dendronized polymers. These results indicated that probably because of the higher rigidity and bulkiness of the polymer chain with the increase of the dendron generation, the SEC-determined Đ value became lower. Accordingly, the results of the apparent narrower MWD with the increase of dendron generation in the C1 polymerization of G(1–3)-D cannot be considered as improved controllability of the polymerization as well. However, these results at least indicate that the relatively high controllability for p(G1-D)′ is retained for p(G2-D)′ and p(G3-D)′.
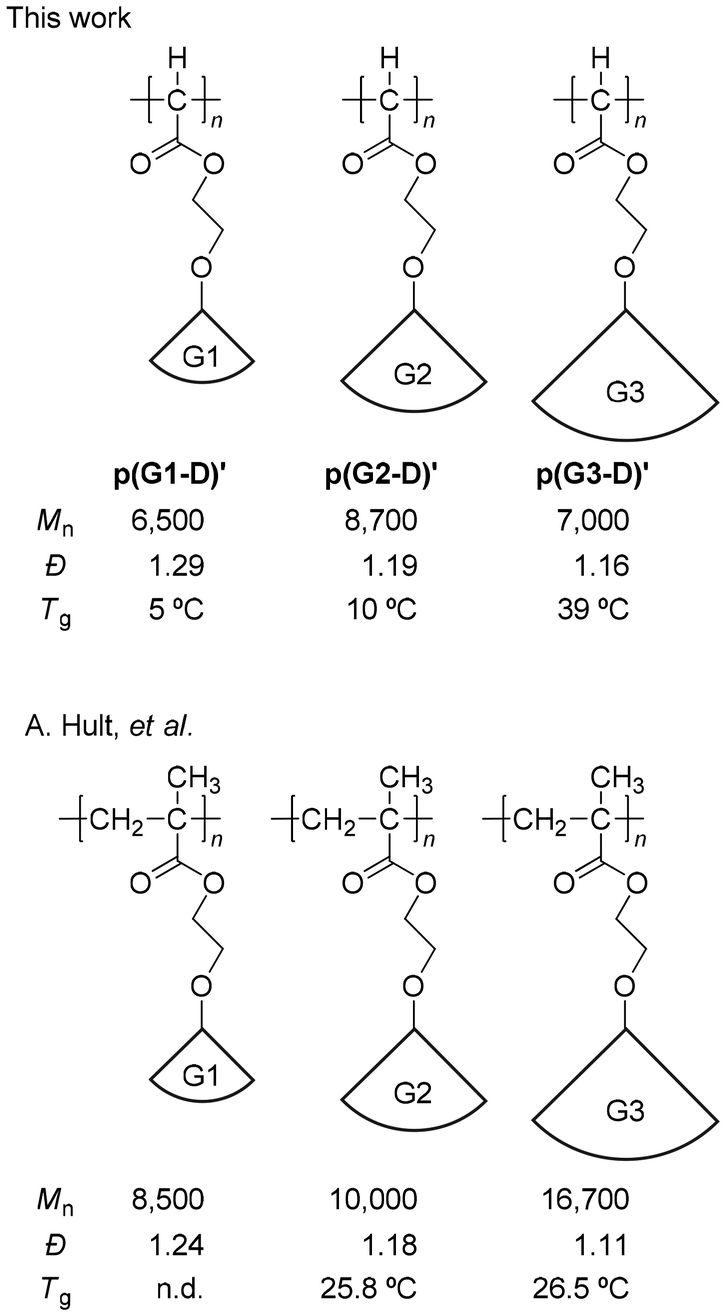 | ||
| Chart 1 Comparison of Mn, Đ, and Tg values between p[G(1–3)-D]′ and the corresponding polymethacrylates. | ||
Fig. 2 shows the charts of DSC measurements of p[G(1–3)-D]′ samples obtained in runs 2, 7, and 10. It is clearly demonstrated that the glass transition temperature (Tg) increased with the increase of the dendron generation, indicating that the increased side chain bulkiness would restrict the main chain mobility. In particular, the higher Tg (39 °C) of p(G3-D)′ despite its lower Mn indicates that the steric demand caused by the ester-type dendron G3 unit led to severe restriction for the main chain mobility.
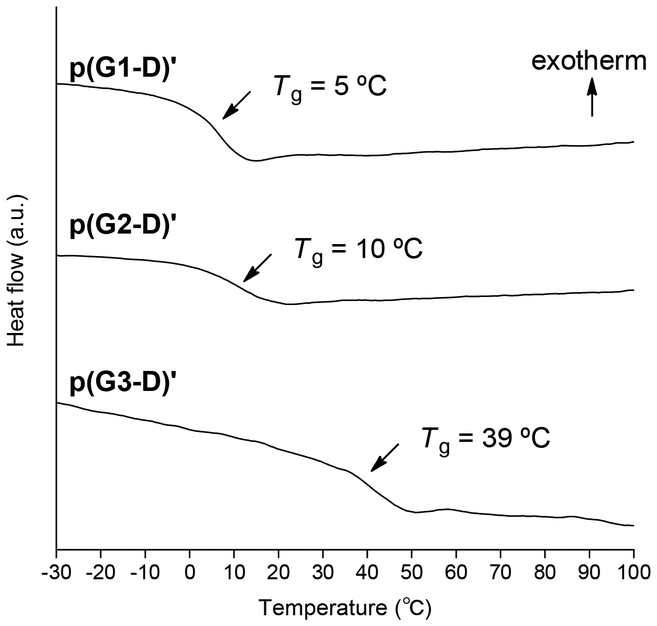 | ||
| Fig. 2 DSC thermograms of p[G(1–3)-D]′ samples (second heating, heat rate = 10 °C min−1). | ||
With respect to the observed Tgs, the results reported by A. Hult and coworkers41,42 are again quite helpful for us to conduct reasonable comparison of the C1 polymers with their vinyl polymer counterparts. They reported that the Tg increase was 0.7 °C with the dendron generation increase from G2 to G3 as listed in Chart 1. In contrast, the Tgs of our polymers increased from 10 to 39 °C, whose ΔTg is a much larger value of 29 °C. These results clearly demonstrate that because of the denser packing of the ester-type dendron in our dendronized C1 polymers without any CH2 unit in the main chain, the restriction of the main chain mobility caused by the generation increase from G2 to G3 is enhanced to a much greater extent compared to their vinyl polymer counterparts.
Copolymerization of G3-D with benzyl diazoacetate (BDA)
Although homopolymerization of G3-D did not proceed effectively (run 10, Table 1), we can expect that the monomer with the bulky ester substituent can be used as a comonomer with other diazoacetates with a non-sterically demanding ester substituent. Considering facile determination of the copolymer composition using signal intensities in the 1H NMR spectra of the resulting copolymers, we chose benzyl diazoacetate (BDA) as a less bulky comonomer for the copolymerization with G3-D and the results are summarized in Table 2. The copolymerization with a feed ratio of [G3-D]/[BDA] = 1:5 ([monomers]/[Pd] = 100:1) resulted in the formation of a copolymer with Mn,SEC = 12900 and a composition of [G3-D′]/[BDA′] = 1:5.4 (run 1 in Table 2), which was determined from the signal intensity ratio in the 1H NMR spectrum of the copolymer; in the spectrum of Fig. 3(a), the composition was calculated by using the intensity of OCH2 of BDA′ at 4.3–5.1 ppm and that of all Hs of G3-D′ except for Me–Hs and main chain CH of BDA′ at 2.7–4.3 ppm. Whereas the composition of the product in the above example was close to the monomer feed ratio, when the relative feed ratio of G3-D was increased to [G3-D]/[BDA] = 1:2 in run 2, the composition of the product was [G3-D′]/[BDA′] = 1:4.2, whose G3-D′ composition was much lower than that in the feed ratio, suggesting that the bulkiness of G3-D restricts its efficient incorporation into the product in the copolymerization with a relatively high G3-D content in the feed ratio. The Mn,MALS of the copolymer sample obtained in run 2 was 27600, which was again much higher than that estimated by SEC Mn,SEC (= 13000) with calibration using standard PMMAs.
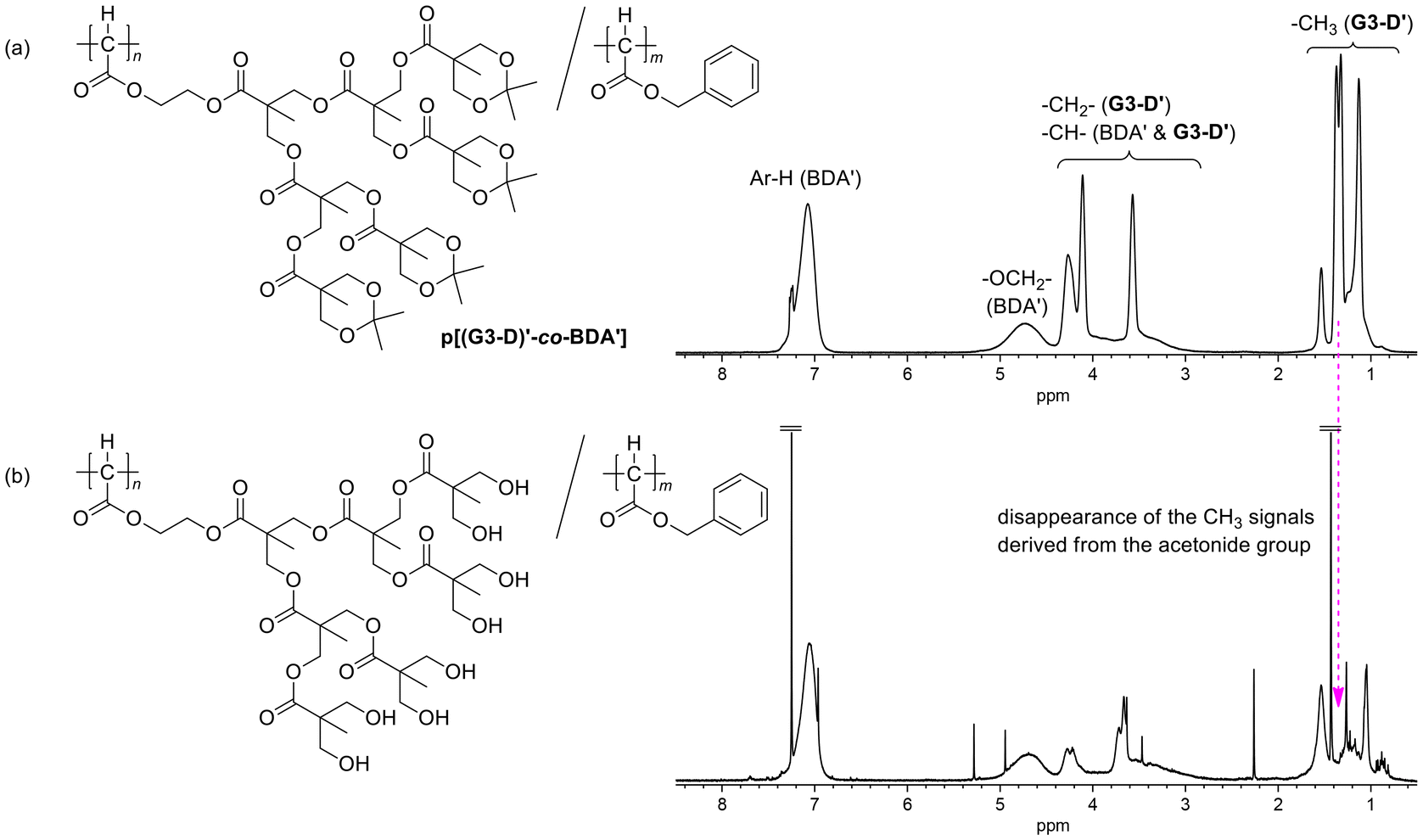 | ||
| Fig. 3 1H NMR spectra of (a) p[(G3-D)′-co-BDA′] (run 1 in Table 2) and (b) its deprotected product. | ||
| Run | Feed ratio ([G3-D]/[BDA]) | Yieldb (%) | Compositionc ([G3-D′]/[BDA′]) | M n | Đ | M n,MALS | Đ MALS | T g (°C) |
|---|---|---|---|---|---|---|---|---|
| a In THF for 15 h; [NaBPh4]/[Pd] = ca. 1.2. b Determined by gravimetry after purification with preparative SEC. c Determined by 1H NMR measurements. d Determined by SEC calibrated using PMMA standards. e Determined by SEC-MALS. f Determined by DSC measurements. | ||||||||
| 1 | 1:5 |
52 | 1:5.4 |
12900 |
1.44 | |||
| 2 | 1:2 |
41 | 1:4.2 |
13000 |
1.42 | 27600 |
1.23 | 16 |
| 3 | 0:1 |
67 | 11000 |
1.48 | 18400 |
1.33 | 26 | |
Run 3 in Table 2 shows the results of homopolymerization of BDA under the same conditions as those in runs 1 and 2. Although the comparison is not straightforward because the molecular weight of G3-D is much higher than that of BDA, it is at least confirmed that the use of G3-D as a comonomer did not prevent the progress of the polymerization of BDA.
Next, we attempted to remove the acetonide-protecting groups of the copolymer sample obtained in run 1 in Table 2 with aqueous HCl-treatment in THF. As shown in Fig. 3(b), disappearance of the Me–H signals derived from the acetonide group after the HCl-treatment was clearly observed in comparison to the precursor polymer in Fig. 3(a). Although the presence of many hydrophilic hydroxy groups at the peripheral positions of the dendron unit could change the polymer solubility,53,54 the product after the deprotection was still soluble in organic solvents such as CHCl3, probably because of the relatively low content of the G3-D unit in the copolymer. These results indicate that the copolymerization of G3-D with other diazoacetates can be an effective method for introducing the highly hydrophilic repeating unit in the C1 polymer after the deprotection.
Conclusions
We have demonstrated that Pd-initiated polymerization of diazoacetates with a series of ester-type dendrons, G(1–3)-D, proceeded to afford a new type of dendronized C1 polymer, p[G(1–3)-D]′. Although MWDs of p(G2-D)′ and p(G3-D)′ with higher generation dendrons were narrower than that of p(G1−D)′, these results did not indicate that steric bulkiness of the ester substituent led to higher controllability of the polymerization, but showed that the polymerization of G(1–3)-D proceeded with a similar level of controllability, on the basis of the comparison with the reported data of their vinyl polymer counterparts. Tgs of the dendronized polymers clearly increased with the increase of the dendron generation, indicating that the steric demand of the ester substituent restricts the mobility of the polymer main chain. In particular, the degree of Tg increase (ΔTg) is much larger than that observed in the reported data of their vinyl polymer counterparts, clearly demonstrating the much higher level of rigidity of the C1 polymer main chain because of the denser packing of the substituents along the main chain. Copolymerization of G3-D with BDA proceeded to afford copolymers, and their acetonide-protecting groups were successfully removed with HCl treatment, demonstrating that the use of G(1–3)-D as a comonomer with other diazoacetates is an effective method for incorporating multiple hydroxy groups in the C1 polymer backbone. These results indicate that the new dendron-containing diazoacetates can be utilized for developing new functional C1 polymers, which is now underway in our laboratory.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by JSPS KAKENHI (Grant Numbers JP18H02021, JP19K05586, JP19K22219, JP21H01988, JP22K05219, and JP24K01540). The authors thank the Advanced Research Support Center (ADRES) in Ehime University for its assistance in NMR measurements and elemental analyses.References
- E. Ihara, Adv. Polym. Sci., 2010, 231, 191–231 CrossRef CAS.
- E. Jellema, A. L. Jongerius, J. N. H. Reek and B. de Bruin, Chem. Soc. Rev., 2010, 39, 1706–1723 RSC.
- N. M. G. Franssen, A. J. C. Walters, J. N. H. Reek and B. de Bruin, Catal. Sci. Technol., 2011, 1, 153–165 RSC.
- C. R. Cahoon and C. W. Bielawski, Coord. Chem. Rev., 2018, 374, 261–278 CrossRef CAS.
- E. Ihara and H. Shimomoto, Polymer, 2019, 174, 234–258 CrossRef CAS.
- H. Shimomoto, Polym. J., 2020, 52, 269–277 CrossRef CAS.
- F. Li, L. Xiao, B. Li, X. Hu and L. Liu, Coord. Chem. Rev., 2022, 473, 214806 CrossRef CAS.
- E. Ihara, Polym. J., 2025, 57, 1–23 CrossRef CAS.
- E. Ihara, R. Okada, T. Sogai, T. Asano, M. Kida, K. Inoue, T. Itoh, H. Shimomoto, Y. Ishibashi and T. Asahi, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1020–1023 CrossRef CAS.
- M. Tokita, K. Shikinaka, T. Hoshino, K. Fujii, J. Mikami, N. Koshimizu, K. Sakajiri, S. Kang, J. Watanabe and K. Shigehara, Polymer, 2013, 54, 995–998 CrossRef CAS.
- N. M. G. Franssen, B. Ensing, M. Hegde, T. J. Dingemans, B. Norder, S. J. Picken, G. O. R. Alberda van Ekenstein, E. R. H. van Eck, J. A. A. W. Elemans, M. Vis, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2013, 19, 11577–11589 CrossRef CAS PubMed.
- H. Shimomoto, E. Itoh, T. Itoh, E. Ihara, N. Hoshikawa and N. Hasegawa, Macromolecules, 2014, 47, 4169–4177 CrossRef CAS.
- H. Shimomoto, H. Asano, T. Itoh and E. Ihara, Polym. Chem., 2015, 6, 4709–4714 RSC.
- H. Shimomoto, K. Shimizu, C. Takeda, M. Kikuchi, T. Kudo, H. Mukai, T. Itoh, E. Ihara, N. Hoshikawa, A. Koiwai and N. Hasegawa, Polym. Chem., 2015, 6, 8124–8131 RSC.
- N. Koshimizu, Y. Aizawa, K. Sakajiri, K. Shikinaka, K. Shigehara, S. Kang and M. Tokita, Macromolecules, 2015, 48, 3653–3661 CrossRef CAS.
- H. Shimomoto, A. Oda, M. Kanayama, T. Sako, T. Itoh, E. Ihara, N. Hsoshikawa, A. Koiwai and N. Hasegawa, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1742–1751 CrossRef CAS.
- H. Shimomoto, T. Uegaito, S. Yabuki, S. Teratani, T. Itoh, E. Ihara, N. Hoshikawa, A. Koiwai and N. Hasegawa, Solid State Ionics, 2016, 292, 1–7 CrossRef CAS.
- H. Shimomoto, M. Kikuchi, J. Aoyama, D. Sakayoshi, T. Itoh and E. Ihara, Macromolecules, 2016, 49, 8459–8465 CrossRef CAS.
- H. Shimomoto, T. Kudo, S. Tsunematsu, T. Itoh and E. Ihara, Macromolecules, 2018, 51, 328–335 CrossRef CAS.
- K. Shikinaka, K. Suzuki, H. Masunaga, E. Ihara and K. Shigehara, Polym. Int., 2018, 67, 495–499 CrossRef CAS.
- T. Takaya, T. Oda, Y. Shibazaki, Y. Hayashi, H. Shimomoto, E. Ihara, Y. Ishibashi, T. Asahi and K. Iwata, Macromolecules, 2018, 51, 5430–5439 CrossRef CAS.
- D. S. Tromp, M. Lankelma, H. de Valk, E. de Josselin de Jong and B. de Bruin, Macromolecules, 2018, 51, 7248–7256 CrossRef CAS PubMed.
- X. Li, Y. Sun, J. Chen, Z. Wu, P. Cheng, Q. Li, J. Fang and D. Chen, Polym. Chem., 2019, 10, 1575–1584 RSC.
- X. Li, B. Mu, C. Chen, J. Chen, J. Liu, F. Liu and D. Chen, Macromolecules, 2019, 52, 6913–6926 CrossRef CAS.
- H. Shimomoto, T. Yamada, T. Itoh and E. Ihara, Polym. J., 2020, 52, 51–56 CrossRef CAS.
- H. Shimomoto, R. Hohsaki, D. Hiramatsu, T. Itoh and E. Ihara, Macromolecules, 2020, 53, 6369–6379 CrossRef CAS.
- H. Shimomoto, S. Tsunematsu, T. Itoh and E. Ihara, Polym. Chem., 2021, 12, 689–701 RSC.
- H. Shimomoto, I. Katashima, H. Murakami, T. Itoh and E. Ihara, Macromolecules, 2023, 56, 4639–4648 CrossRef CAS.
- E. Ihara, Y. Ishiguro, N. Yoshida, T. Hiraren, T. Itoh and K. Inoue, Macromolecules, 2009, 42, 8608–8610 CrossRef CAS.
- E. Ihara, M. Akazawa, T. Itoh, M. Fujii, K. Yamashita, K. Inoue, T. Itoh and H. Shimomoto, Macromolecules, 2012, 45, 6869–6877 CrossRef CAS.
- H. Shimomoto, M. Nakajima, A. Watanabe, H. Murakami, T. Itoh and E. Ihara, Polym. Chem., 2020, 11, 1774–1784 RSC.
- H. Shimomoto, S. Ichihara, H. Hayashi, T. Itoh and E. Ihara, Macromolecules, 2019, 52, 6976–6987 CrossRef CAS.
- J.-H. Chu, X.-H. Xu, S.-M. Kang, N. Liu and Z.-Q. Wu, J. Am. Chem. Soc., 2018, 140, 17773–17781 CrossRef CAS PubMed.
- A. V. Zhukhovitskiy, I. J. Kobylianskii, A. A. Thomas, A. M. Evans, C. P. Delaney, N. C. Flanders, S. E. Denmark, W. R. Dichtel and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 6473–6478 CrossRef CAS PubMed.
- X.-Q. Yao, Y.-S. Wang and J. Wang, Macromolecules, 2021, 54, 10914–10922 CrossRef CAS.
- F. Kato, A. Chandra, M. Tokita, H. Asano, H. Shimomoto, E. Ihara and T. Hayakawa, ACS Macro Lett., 2018, 7, 37–41 CrossRef CAS PubMed.
- H. Ihre, A. Hult and E. Söderlind, J. Am. Chem. Soc., 1996, 118, 6388–6395 CrossRef CAS.
- H. Ihre, A. Hult, J. M. J. Fréchet and I. Gitsov, Macromolecules, 1998, 31, 4061–4068 CrossRef CAS.
- A. Würsch, M. Möller, T. Glauser, L. S. Lim, S. B. Voytek, J. L. Hedrick, C. W. Frank and J. Hilborn, Macromolecules, 2001, 34, 6601–6615 CrossRef.
- M. Malkoch, E. Malmström and A. Hult, Macromolecules, 2002, 35, 8307–8314 CrossRef CAS.
- A. Nyström and A. Hult, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 3852–3867 CrossRef.
- A. M. Nyström, I. Furó, E. Malmström and A. Hult, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4496–4504 CrossRef.
- M. Malkoch, A. Carlmark, A. Woldegiorgis, A. Hult and E. E. Malmström, Macromolecules, 2004, 37, 322–329 CrossRef CAS.
- K. O. Kim and T.-L. Choi, Macromolecules, 2013, 46, 5905–5914 CrossRef CAS.
- J. Noh, G. I. Peterson and T.-L. Choi, Angew. Chem., Int. Ed., 2021, 60, 18651–18659 CrossRef CAS PubMed.
- A. D. Schlüter and J. P. Rabe, Angew. Chem., Int. Ed., 2000, 39, 864–883 CrossRef.
- A. Zhang, L. Shu, Z. Bo and A. D. Schlüter, Macromol. Chem. Phys., 2003, 204, 328–339 CrossRef CAS.
- Y. Chen and X. Xiong, Chem. Commun., 2010, 46, 5049–5060 RSC.
- K. A. Andreopoulou, M. Peterca, D. A. Wilson, B. E. Partridge, P. A. Heiney and V. Percec, Macromolecules, 2017, 50, 5271–5284 CrossRef CAS.
- Y. Zhu, H. Jiang, W. Wu, X.-Q. Xu, X.-Q. Wang, W.-J. Li, W.-T. Xu, G. Liu, Y. Ke, W. Wang and H.-B. Yang, Nat. Commun., 2023, 14, 5307 CrossRef CAS PubMed.
- L. Ren, X. Lu, W. Li, J. Yan, A. K. Whittaker and A. Zhang, J. Am. Chem. Soc., 2023, 145, 24906–24921 CAS.
- T. Toma, J. Shimokawa and T. Fukuyama, Org. Lett., 2007, 9, 3195–3197 CrossRef CAS PubMed.
- W. Li, A. Zhang and A. D. Schlüter, Macromolecules, 2008, 41, 43–49 CrossRef CAS.
- W. Li, A. Zhang, K. Feldman, P. Walde and A. D. Schlüter, Macromolecules, 2008, 41, 3659–3667 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01324g |
| This journal is © The Royal Society of Chemistry 2025 |