DOI:
10.1039/D4PY01369G
(Paper)
Polym. Chem., 2025,
16, 1478-1485
Organocatalyzed photoredox radical cyclopolymerization of methacrylate- and acrylamide-crotonate hybrid monomers†
Received
30th November 2024
, Accepted 21st February 2025
First published on 21st February 2025
Abstract
Cascade bond-forming polymerizations have emerged as a powerful strategy for the synthesis of polymeric materials with advanced structures and properties. Among these, radical cyclopolymerization is one of the most extensively studied methodologies, although it still faces several challenges, including low cyclization rates, unwanted crosslinking, and lack of spatiotemporal control. Herein, we report the design and synthesis of methacrylate- and acrylamide-crotonate hybrid monomers, which undergo a visible light-driven, organocatalyzed photoredox ATRP-type cyclopolymerization. This approach eliminates crosslinking and proceeds through a sophisticated sequence of intramolecular cyclization and intermolecular propagation, yielding diverse homopolymers and block copolymers with cyclic repeating units. Additionally, factors influencing the control over polymer chain growth, such as light intensity and additives, are explored.
Introduction
The merger of reversible-deactivation radical polymerization (RDRP) with visible-light photocatalysis has been recognized as a mild and versatile strategy for synthesizing polymers with diverse structures and functionalities.1–7 This approach not only imparts excellent control over polymer chain growth, tailoring the molecular weights (MWs) and narrowing the molecular weight distributions,8 but also provides unique spatiotemporal control which enables a broad scope of applications ranging from photolithography to 3D manufacturing.9–12 Among the numerous mechanisms employed in photocontrolled RDRP, atom transfer radical polymerization (ATRP) is one of the most extensively studied controlled radical polymerization methodologies.6,13 In contrast to the early ATRP examples mediated by Cu(I)14 or Ru(II)15 complexes under thermal conditions, the development of photocontrolled ATRP has focused on two dimensions: sustainable photoredox processes and an expanded monomer scope. To address concerns about transition metal contaminations, considerable efforts have been directed toward the development of efficient photocatalysts (PCs), shifting from metal complexes16–22 to metal-free systems. As such, a variety of purely organic molecules, including perylene,23,24 phenothiazines,25–28 dihydrophenazines,29,30 phenoxazines,31,32 carbazoles,33 thienothiophenes,34 dihydroacridines,35 and oxygen-/sulfur-doped polycyclic arenes,36,37 have proven effective and practical for organocatalyzed ATRP (O-ATRP), yielding well-defined polymers with high chain-end fidelity and a controlled topology. On the other hand, methacrylates have dominated the research of O-ATRP, while progress involving other vinyl monomers (e.g., acrylates,27,28,35,38 acrylamides,39,40 acrylonitrile,26,41 and styrene23–33) remains limited. Expanding the scope of polymerizable monomers, particularly those with intriguing polymerizability profiles, is highly desirable for further advancing this field.
In contrast to conventional single-reactivity monomers, multi-functionalized molecules hold the potential for cascade reactions in polymerizations, leading to advanced polymers with unique structures and properties.42–45 A premier example is the radical cyclopolymerization of diene monomers, where sequential intramolecular cyclization and propagation ideally proceed to afford linear polymers with cyclic repeating units (Fig. 1a).46 In the realm of free-radical cyclopolymerization, various divinyl monomers have been designed to (a) enable the direct preparation of functional polymeric materials, such as poly(diallyldimethylammoniumchloride)47 and its copolymers with acrylamide for water treatment, and (b) address the alternation challenge in copolymerization of two distinct vinyl monomers through an alternating intra-intermolecular chain mechanism. Additionally, conventional nitroxide-mediated radical polymerization (NMP),48 reversible addition-fragmentation chain transfer (RAFT),49–57 and ATRP58–61,62–64 techniques have been applied to radical cyclopolymerization, offering precise control over MWs, dispersities, and chain-end livingness, which enables sequence-defined copolymerization (Fig. 1b). Despite these advancements, a photocontrolled radical cyclopolymerization has yet to be realized, likely due to long-standing problems, such as low cyclization rates and inevitable crosslinking, which complicate the process and hinder the effective control by a photocatalyst.
 |
| | Fig. 1 (a) Concept of radical cyclopolymerization. (b) Cyclopolymerization using transition-metal-based ATRP technology. (c) Photocontrolled radical cyclopolymerization in this work. | |
We hypothesized that a vinyl monomer bearing a pendent internal alkene functionality would help minimize these unwanted side reactions in radical cyclopolymerizaiton. As depicted in Fig. 1c, a highly reducing PC in its excited state (PC*) can donate an electron to the dormant chain-end bromide A, thus generating a propagating carbon radical B and the [PC˙+][Br−] complex. Upon vinyl addition of species B to the monomer, intermediate C is formed, which afterwards can undergo multiple pathways: (a) radical species may be recaptured by the [PC˙+][Br−] complex (C to F); (b) C may cyclize in a 5-exo-trig manner (C to D) or (c) a 6-endo-trig manner (C to G). Notably, the radical addition of B to the internal C–C double bond should be kinetically unfavorable due to its higher steric hindrance than the adjacent vinyl group. Finally, deactivation of radical species D and G by [PC˙+][Br−] results in the formation of dormant polymers E and H, respectively. Given the limited redox window of commonly used PCs, the resulting inactive alkyl chain-end bromide H is unlikely to be re-activated for propagation,65 representing an unwanted pseudo-termination pathway. In this work, we report the synthesis of malonate-linked methacrylate-crotonate and p-toluenesulfonamide-linked acrylamide-crotonate monomers, and their highly selective radical cyclopolymerization by using a visible-light-driven photoredox ATRP approach.
Results and discussion
Initial radical cyclopolymerization of M1
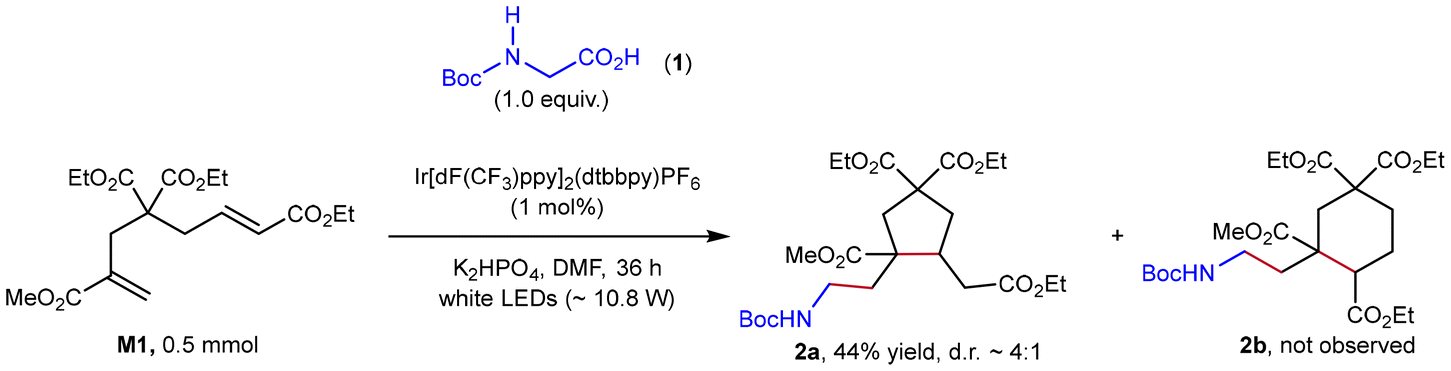
To valid our hypothesis, M1, diethyl-2-bromo-2-methylmalonate (DBMM), and N,N′-di(2-naphthyl)dihydrophenazine (1)29 were selected as the monomer, initiator and photocatalyst, respectively. Gratifyingly, a 0.5 mmol-scale polymerization of M1 under conditions of [M1]/[DBMM]/[1] = 500/10/1 in 1.0 mL of N,N-dimethylacetamide (DMAc), with white LED irradiation (∼10.8 W) and a fan to maintain the temperature at ∼30 °C, proceeded smoothly and reached 81% monomer conversion to give highly soluble PM1 with a number-average molecular weight (Mn) of 8.7 kDa and a moderate dispersity (Đ) of 1.60 (Table 1, entry 1). The 1H NMR spectrum of the obtained PM1 (Fig. S44†) showed no signal of vinyl or alkenyl protons, suggesting an exclusive cyclopolymerizaton rather than the vinyl polymerization. As mentioned in Fig. 1c, cyclopentyl and cyclohexyl units are likely formed via 5-exo-trig- and 6-endo-trig cyclization, respectively. However, due to their high structural similarity, the ratios of these two units cannot be determined based on the 1H NMR spectrum of PM1. Inspired by Niu's56 and Huang's66 strategy that utilizes a small-molecule model reaction to illustrate the structure of complex polymers, we performed a photoredox-catalyzed stoichiometric reaction of M1 with a glycine derivative67 as the radical precursor (Fig. 2). This reaction afforded the 5-membered product 2a as a ∼4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers in 44% yield, while its six-membered analogous product 2b was not observed (Fig. S10 and 11†), indicating a favorable 5-exo-trig reaction pathway. Accordingly, the core structure of repeating units of the obtained PM1 was ascertained as cyclopentane.
:1 mixture of diastereomers in 44% yield, while its six-membered analogous product 2b was not observed (Fig. S10 and 11†), indicating a favorable 5-exo-trig reaction pathway. Accordingly, the core structure of repeating units of the obtained PM1 was ascertained as cyclopentane.
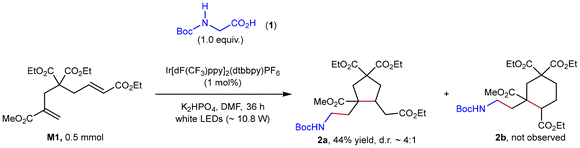 |
| | Fig. 2 Model reaction of M1 with glycine derivative 2 under photoredox conditions. | |
Table 1 Photocontrolled radical cyclopolymerization of M1a
Effect of solvents, photocatalysts, and initiators
A brief screening of solvents (Table 1, entries 1–3) suggested that chlorobenzene (PhCl) was superior to others in achieving higher monomer conversion (92%) and producing PM1 with a Mn (15.1 kDa) close to its theoretical value and a lower Đ of 1.59 (entry 3). Replacing PC1 with N,N′-di(4-CF3-phenyl)dihydrophenazine (PC2) or core-modified N-(1-naphthyl)phenoxazine (PC3) led to slightly decreased monomer conversions and higher dispersity (Đ = 1.64–1.70, entries 4 and 5). Examination of other commonly used O-ATRP alkyl bromide initiators, including ethyl α-bromophenylacetate (EBP) and methyl 2-bromopropionate (M2BP), revealed that DBMM outperformed these initiators, affording polymers with the best monomer conversion and dispersity (entries 6 and 7 vs. entry 3). Importantly, the radical cyclopolymerization did not proceed in the absence of light (entry 8), a PC (entry 8), or an initiator (entry 10), providing strong evidence for the photoredox ATRP mechanism. Additionally, polymerization performed with [M1]/[DBMM]/[1] = 500/25/1 produced PM1 with predictable Mn = 7.2 kDa and moderate Đ = 1.59 (entry 11), while attempts to target higher degree of polymerization (DP) were unsuccessful (entries 12 and 13), probably due to increased termination events.
Investigation of the role of light intensity
Compared to typical O-ATRP, which produces well-defined poly(methacrylates) with Đ < 1.2, this photoredox radical cyclopolymerization of M1 generally afforded PM1 with Đ ∼ 1.6. This higher dispersity is likely due to the incorporation of the crotonate moiety, which introduces a secondary radical species that is structurally similar to the propagating radical in the ATRP of acrylates. Since the propagating rate constant (kp) of acrylates is an order of magnitude higher than that for methacrylates,68 the chain growth through crotonate-to-methacrylate cross propagation is challenging to control. In addition, the occasional formation of six-membered cyclic repeating units (C to G to H, Fig. 1c) likely contributes to chain termination, further complicating the control of polymer chain growth.
To gain deeper insights into the overall control of the polymerization, several experiments were performed. First, the role of light intensity in the polymerization of [M1]/[DBMM]/[1] = 500/10/1 in PhCl at ∼30 °C was investigated. In fact, first-order kinetics were revealed for polymerizations conducted under white LED irradiation ranging from 1.4 to 10.8 W (Fig. 3a). The rates of polymerization roughly correlated to the intensity of irradiation and decreased with lower light intensity, as it actually influences the concentration of the excited-state PC,69 which in turn regulates the radical cyclopolymerization. Importantly, two PM1 samples, synthesized under 10.8 W (Fig. 3b) and 1.4 W irradiation (Fig. 3c), were analyzed by matrix-assisted laser desorption ionization time of flight mass (MALDI-TOF MS) spectrometry. The analyses revealed that increased light intensity can accelerate side reactions, such as radical coupling and hydrogen atom abstraction to form a C–H chain end.
 |
| | Fig. 3 (a) Plots of natural log of monomer conversion as a function of time for polymerization of [M1]/[DBMM]/[PC1] = 500/10/1 performed in PhCl (0.5 M), at ∼30 °C, with 1.4–10.8 W white LED irradiation. MALDI-TOF MS spectra of low-MW PM1 synthesized with (b) 10.5 W and (c) 1.4 W white LED irradiation. | |
Kinetic study and chain-end fidelity
We then monitored the polymerization of [M1]/[DBMM]/[PC1] = 500/10/1 in PhCl (0.5 M) at ∼30 °C, with 1.4 W white LED irradiation. First, a linear increase in polymer MW as a function of monomer conversion was observed (Fig. 4a), demonstrating the robustness of the O-ATRP process. Second, a pulsed irradiation experiment was performed, where the cyclopolymerization proceeded with light, paused in the dark, and revived upon re-irradiation (Fig. 4b and c). Third, the excellent chain-end fidelity allowed chain extension with methyl methacrylate (MMA) using the isolated PM1 (Mn = 4.9 kDa, Đ = 1.60) as the macroinitiator (Fig. 4d). The gel-permeation chromatography (GPC) traces before and after the chain extension confirmed the successful preparation of the block copolymer PM1-b-PMMA (Fig. 4e).
 |
| | Fig. 4 (a) Plots of measured Mn and Đ as a function of monomer conversion for polymerization of [M1]/[DBMM]/[PC1] = 500/10/1 performed in PhCl (0.5 M), at ∼30 °C, with 1.4 W white LED irradiation. (b) Plots of natural log of monomer conversion as a function of time and (c) overlaid GPC traces for the pulsed irradiation experiment. (d) Chain-extension polymerization from a PM1 macroinitiator with MMA. (e) Overlayed GPC traces for chain-extension polymerization. | |
Effect of bromide additives
From the perspective of the reversible-deactivation mechanism in ATRP, the addition of bromide salts primarily shifts the equilibrium to the dormant Br chain ends, thereby enhancing control over the polymer chain growth.35 We investigated the effects of LiBr on cyclopolymerization of [M1]/[DBMM]/[1] = 500/10/1 in PhCl at ∼30 °C under ∼1.4 W white LED irradiation (Table 2, entries 1–3). The inclusion of LiBr, regardless of its concentration, unexpectedly led to an increase in dispersity, probably due to its poor solubility in PhCl. In polymerizations of M1 performed in EtOAc, the polymerization rates were negatively correlated to the concentration of LiBr, although the dispersities of the obtained PM1 remained high (entries 4–6). We then turned to tetrabutylammonium bromide (TBAB) as an additive. Encouragingly, despite a reduction in monomer conversion, the addition of 3.0 equiv. of TBAB to DBMM resulted in the formation of PM1 with a Mn of 11.8 kDa and a low Đ of 1.38 (entry 9). More importantly, the MALDI-TOF MS analysis of low molecular-weight PM1 synthesized under such conditions (entry 9) indicated high chain-end fidelity (Fig. S3†), underscoring the importance of the bromide anion in the photoredox deactivation process.
Table 2 Effect of bromide additives on photoredox radical cyclopolymerization of M1a
| Entry |
Additive |
Solvent |
[M1]/[DBMM]/[1][Br] |
Conv.b (%) |
M
n,theoc (kDa) |
M
nd (kDa) |
Đ
|
|
Polymerizations performed using 0.5 mmol of M1, 0.01 mmol of DBMM, 0.001 mmol of 1, 0.01–0.03 mmol of bromide additive, 1.0 mL of PhCl, and irradiated with white LEDs (∼1.4 W) for 12 h. A cooling fan was used to maintain the temperature at ∼30 °C.
Measured by crude 1H-NMR.
M
n,theo = MW(initiator) + MW(M1) × conversion × ([M1]/[initiator]).
Determined by gel permeation chromatography (GPC) in THF (1.0 mL min−1, 40 °C) and calibrated with polystyrene standards.
|
| 1 |
LiBr |
PhCl |
500/10/1/10 |
79 |
14.9 |
13.4 |
1.91 |
| 2 |
LiBr |
PhCl |
500/10/1/20 |
77 |
14.5 |
11.9 |
1.90 |
| 3 |
LiBr |
PhCl |
500/10/1/30 |
76 |
14.3 |
12.1 |
1.97 |
| 4 |
LiBr |
EtOAc |
500/10/1/10 |
77 |
14.5 |
11.8 |
1.79 |
| 5 |
LiBr |
EtOAc |
500/10/1/20 |
65 |
12.3 |
7.5 |
1.95 |
| 6 |
LiBr |
EtOAc |
500/10/1/30 |
55 |
10.4 |
7.4 |
1.96 |
| 7 |
TBAB |
PhCl |
500/10/1/10 |
79 |
14.9 |
15.1 |
1.59 |
| 8 |
TBAB |
PhCl |
500/10/1/20 |
85 |
16.0 |
13.6 |
1.48 |
| 9 |
TBAB |
PhCl |
500/10/1/30 |
68 |
12.8 |
11.8 |
1.38 |
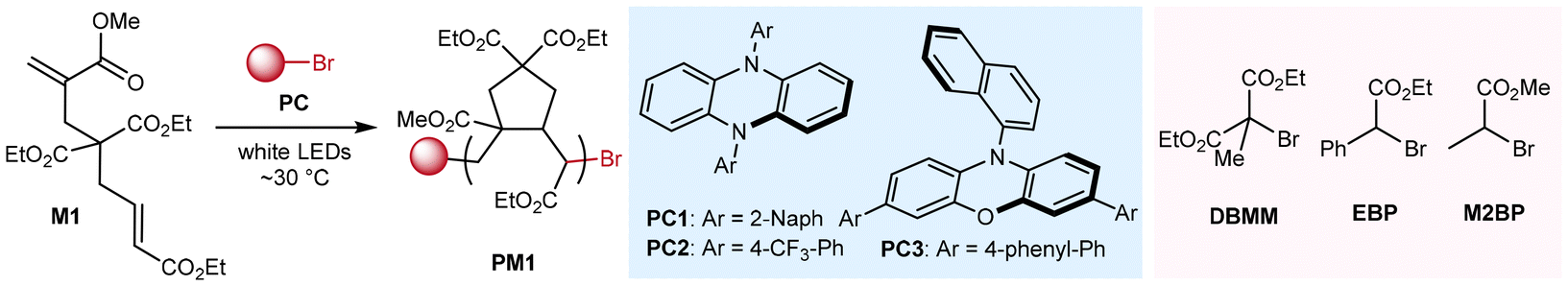
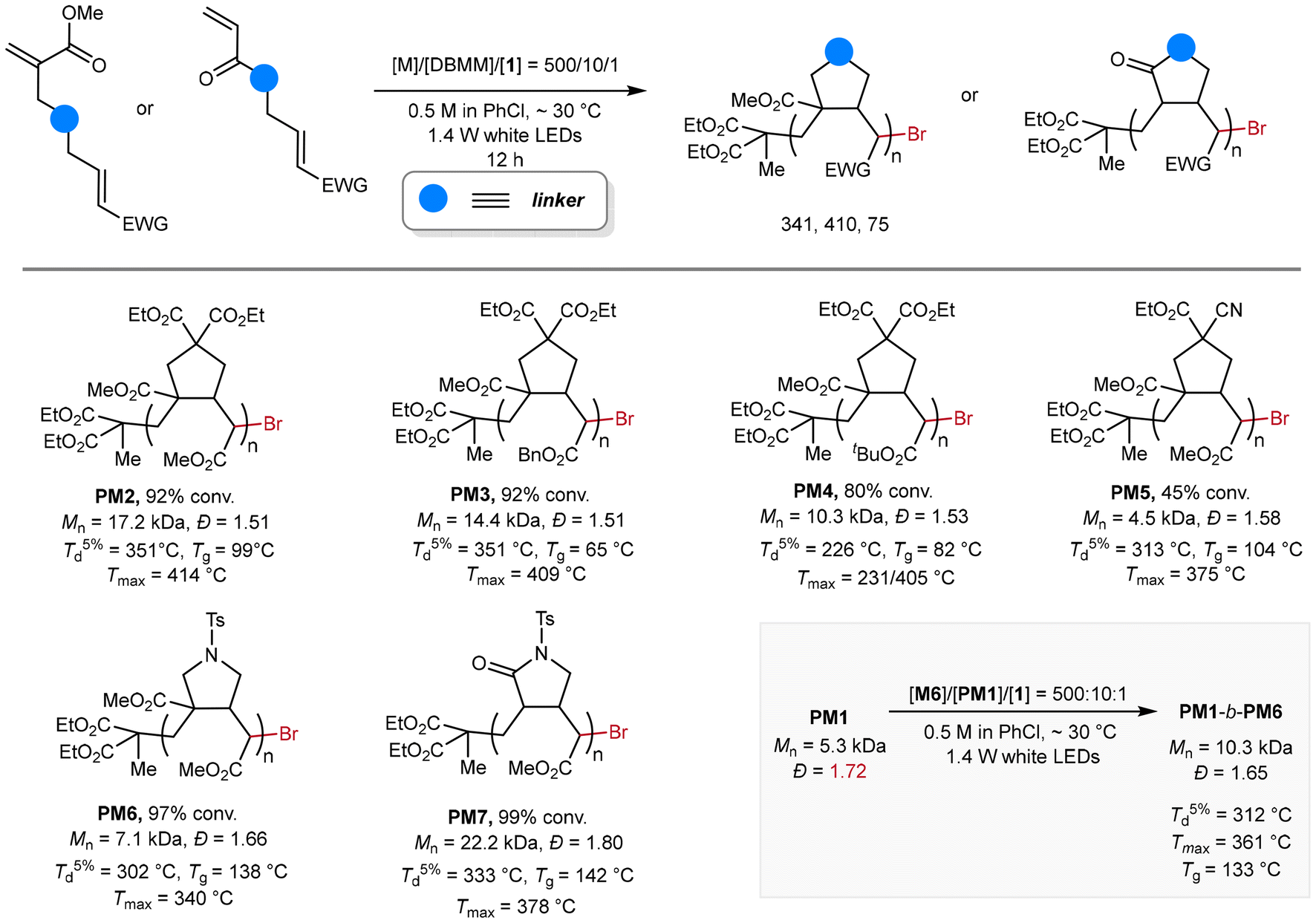
Scope of methacrylate-crotonate diene monomers
With the optimized conditions in hand, we next explored the generality of this radical cyclopolymerization method (Fig. 5). Diene monomers containing methyl- (M2), benzyl- (M3), and tert-butyl crotonate (M4) motifs were successfully polymerized by using this organocatalyzed photoredox protocol, affording the corresponding PM2–PM4 of 10.3–17.2 kDa and with moderate dispersities of ∼1.5. It is noteworthy that the monomer structure significantly impacts the polymerizability. First, the substituent effect of the crotonate motif was observed. Compared to M2 and M3, M4 bearing a sterically bulky tert-butyl group exhibited lower reactivity under identical polymerization conditions, as evidenced by lower monomer conversion (80%). Second, the linker's chemical structure principally dominates the monomer's cyclopolymerizability. For instance, polymerization of M5, which contains a cyanoacetate linkage, proceeded more slowly, reaching only 45% conversion to produce PM5 with Mn = 4.5 kDa and Đ = 1.58. In contrast, polymerizations of p-toluenesulfonamide-linked M6 and acrylamide-crotonate monomer M7 achieved near quantitative conversions, yielding PM6 (Mn = 7.1 kDa and Đ = 1.66) and PM7 (Mn = 22.2 kDa and Đ = 1.80), respectively. Finally, the block copolymer PM1-b-PM6 (Mn = 12.0 kDa and Đ = 1.57) was successfully synthesized using PM1 (Mn = 5.3 kDa and Đ = 1.71) as the macroinitiator and M6 as the comonomer, as evidenced by the shift of retention time in the GPC traces before and after the copolymerization (Fig. S9†).
 |
| | Fig. 5 Monomer scope. | |
The thermal properties of the obtained polymers were then investigated. First, the thermal stability of PM1–PM7 was examined by thermogravimetric analysis (TGA) in terms of decomposition temperature at 5% weight loss (Td5%) and maximum decomposition temperature (Tmax), which is defined as the peak value in relative derivative thermogravimetry (DTG). PM1–PM3 generally exhibited high Td5% of 314–351 °C and Tmax of 409–410 °C, while those of PM4 significantly decreased to 226 °C and 231 °C, respectively, probably due to the fast decomposition of the pendent tert-butyl group. Nitrogen-containing polymers, PM5–PM7, seem to be less stable in terms of lower Td5% (302–333 °C) and Tmax (340–378 °C). Second, the glass transition temperatures (Tgs) of PM1–PM4 were measured by differential scanning calorimetry (DSC), ranging from 65 to 99 °C. The Tgs of nitrogen-containing PM5–PM7 (104 to 142 °C) are higher than those for polymers bearing cyclopentane units, indicating the presence of more rigid cyclic structures.
Experimental
Materials and methods
All chemicals were purchased from TCI, J&K, Energy Chemical, and Adamas-beta, and were used as received without further purification. All syntheses and handling of air- and moisture-sensitive chemicals were performed in a N2-filled glove box.
1H NMR and 13C NMR spectroscopy
1H NMR and 13C NMR spectra were recorded on a Bruker 400 Hz (100 Hz for 13C) spectrometer at ambient temperature. Chemical shifts (δ) for both 1H and 13C NMR spectra are given in ppm relative to tetramethylsilane. All NMR spectra were referenced to the residual solvent (CHCl3) signal (δ = 7.26 ppm for 1H NMR and δ = 77.00 ppm for 13H NMR).
Gel permeation chromatography (GPC)
Analysis of polymers’ number-average molecular weight (Mn) and dispersity (Đ) was performed using a Waters e2695 system (with one guard column and two Styragel columns) coupled with a Waters 2414 refractive index detector (calibrated with 10 polystyrene standards). The analysis was performed at 40 °C using THF as the eluent at a flow rate of 1.0 mL per minute.
Thermo-gravimetric analysis (TGA)
Decomposition temperatures (Td5%) at 5% weight loss and maximum rate decomposition temperatures (Tmax) of the obtained polymers were measured by thermal gravimetric analysis (TGA) on a TA Q50 analyzer, TA instruments. Polymer samples were measured by heating the polymer samples from 25 °C to 700 °C at the rate of 10 °C min−1.
Differential scanning calorimetry (DSC)
Glass transition temperatures (Tg) of the obtained polymers were measured by differential scanning calorimetry (DSC) on a TA Q20 analyzer, at a rate of 10 °C min−1. All Tg values were obtained from a second scan.
Conclusions
In summary, we have designed and synthesized a series of diene monomers that integrate methacrylates or acrylamide with crotonates. By using N,N′-di(2-naphthyl)dihydrophenazine 1 as the photocatalyst and DBMM as the initiator, radical polymerization of these hybrid monomers proceeded exclusively through a sequence of intramolecular cyclization and intermolecular propagation, resulting in polymers with unique cyclic repeating units. Kinetic studies, along with the effects of light intensity and bromide additives, combined with MALDI-TOF mass spectrometry analysis, have provided valuable insights into this radical cyclopolymerization driven by the photoredox ATRP mechanism. This approach enables straightforward synthesis of a range of novel homopolymers and block copolymers with low to moderate dispersities. With these findings presented in this work, we believe that photocontrolled radical cyclopolymerization is a promising and practical strategy for accessing new polymeric materials with distinct properties.
Author contributions
G.-Y. and Y.-L. T. performed the experiments. G.-Y., Y.-L. T. and K. L. analyzed the data. D.-F. C. directed the project and wrote the manuscript with assistance from all authors. G.-Y. and Y.-L. T. contributed equally to this work.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
The authors declare no competing financial interest.
Acknowledgements
This work was financially supported by the NSFC (grant no. 22101273) and the Fundamental Research Fund for the Central Universities (WK9990000111). This work was partially carried out at the Instruments Center for Physical Science, University of Science and Technology of China (USTC).
References
- F. A. Leibfarth, K. M. Mattson, B. P. Fors, H. A. Collins and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 199–210 CrossRef CAS PubMed.
- X. Pan, M. A. Tasdelen, J. Laun, T. Junkers, Y. Yagc and K. Matyjaszewski, Prog. Polym. Sci., 2016, 62, 73–125 CrossRef CAS.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 CrossRef CAS PubMed.
- S. Dadashi-Silab, S. Droan and Y. Yagci, Chem. Rev., 2016, 116, 10212–10275 CrossRef CAS PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- D. A. Corbin and G. M. Miyake, Chem. Rev., 2022, 122, 1830–1874 CrossRef CAS PubMed.
- Q. Michaudel, V. Kottisch and B. P. Fors, Angew. Chem., Int. Ed., 2017, 56, 9670–9679 CrossRef CAS PubMed.
- Y. Zhou, J. Li, Y. Wu and Z. Luo, Chem. Rev., 2020, 120, 2950–3048 CrossRef CAS PubMed.
- J. E. Poelma, B. P. Fors, G. F. Meyers, J. W. Kramer and C. J. Hawker, Angew. Chem., Int. Ed., 2013, 52, 6844–6848 CrossRef CAS PubMed.
- C. W. Pester, B. Narupai, K. M. Mattson, D. P. Bothman, D. Klinger, K. W. Lee, E. H. Discekici and C. J. Hawker, Adv. Mater., 2016, 28, 9292–9300 CrossRef CAS PubMed.
- E. H. Discekici, C. W. Pester, N. J. Treat, J. Lawrence, K. M. Mattson, B. Narupai, E. P. Toumayan, Y. Luo, A. J. McGrath, P. G. Clark, J. Read de Alaniz and C. J. Hawker, ACS Macro Lett., 2016, 5, 258–262 CrossRef CAS PubMed.
- J. Yan, X. Pan, M. Schmitt, Z. Wang, M. R. Bockstaller and K. Matyjaszewski, ACS Macro Lett., 2016, 5, 661–665 CrossRef CAS PubMed.
- E. H. Discekici, A. Anastasaki, J. Read De Alaniz and C. J. Hawker, Macromolecules, 2018, 51, 7421–7434 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 CrossRef CAS.
- D. Konkolewicz, K. Schroder, J. Buback, S. Bernhard and K. Matyjaszewski, ACS Macro Lett., 2012, 1, 1219–1223 CrossRef CAS PubMed.
- J. Mosnáček and M. Ilčíková, Macromolecules, 2012, 45, 5859–5865 CrossRef.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2014, 136, 1141–1149 CrossRef CAS PubMed.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- X. Pan, N. Malhotra, J. Zhang and K. Matyjaszewski, Macromolecules, 2015, 48, 6948–6954 CrossRef CAS.
- S. Dadashi-Silab, X. Pan and K. Matyjaszewski, Macromolecules, 2017, 50, 7967–7977 CrossRef CAS.
- K. Parkatzidis, S. Boner, H. S. Wang and A. Anastasaki, ACS Macro Lett., 2022, 11, 841–846 CrossRef CAS PubMed.
- G. M. Miyake and J. C. Theriot, Macromolecules, 2014, 47, 8255–8261 CrossRef CAS.
- C. Aydogan, G. Yilmaz and Y. Yagci, Macromolecules, 2017, 50, 9115–9120 CrossRef CAS.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- X. Pan, M. Lamson, J. Yan and K. Matyjaszewski, ACS Macro Lett., 2015, 4, 192–196 CrossRef CAS PubMed.
- Y. C. Zhao, H. H. Gong, K. M. Jiang, S. J. Yan, J. Lin and M. Chen, Macromolecules, 2018, 51, 938–946 CrossRef CAS.
- H. Zhou, L. Zhang, P. Wen, Y. Zhou, Y. Zhao, Q. Zhao, Y. Gu, R. Bai and M. Chen, Angew. Chem., Int. Ed., 2023, 62, e202304461 CrossRef CAS PubMed.
- J. Theriot, C.-H. Lim, H. Yang, M. Ryan, C. Musgrave and G. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- C.-H. Lim, M. D. Ryan, B. G. McCarthy, J. C. Theriot, S. M. Sartor, N. H. Damrauer, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 348–355 CrossRef CAS PubMed.
- R. M. Pearson, C.-H. Lim, B. G. McCarthy, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2016, 138, 11399–11407 CrossRef CAS PubMed.
- B. G. McCarthy, R. M. Pearson, C. H. Lim, S. M. Sartor, N. H. Damrauer and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 5088–5101 CrossRef CAS PubMed.
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel and L. S. Park, Nat. Catal., 2018, 1, 794–804 CrossRef CAS.
- C. Kutahya, A. Allushi, R. Isci, J. Kreutzer, T. Ozturk, G. Yilmaz and Y. Yagci, Macromolecules, 2017, 50, 6903–6910 CrossRef CAS.
- B. L. Buss, C. H. Lim and G. M. Miyake, Angew. Chem., Int. Ed., 2020, 59, 3209–3217 CrossRef CAS PubMed.
- Q. Ma, J. Song, X. Zhang, Y. Jiang, L. Ji and S. Liao, Nat. Commun., 2021, 12, 429 CrossRef CAS PubMed.
- H. Shao, S. Li, Y. Jiang, J. Song, X. Zhang, J. Chen and S. Liao, Polym. Chem., 2024, 15, 4134–4140 RSC.
- B. McCarthy, S. Sartor, J. Cole, N. Damrauer and G. M. Miyake, Macromolecules, 2020, 53, 9208–9219 CrossRef CAS PubMed.
- C. Aydogan, C. Kutahya, A. Allushi, G. Yilmaz and Y. Yagci, Polym. Chem., 2017, 8, 2899–2903 RSC.
- Y. Xu, G. Li, Y. Hu and Y. Wang, Macromol. Chem. Phys., 2018, 219, 1800192 CrossRef.
- D. Wei, Y. Xu, C. Liu, Y. Zhai, H. Chen, L. Bai, H. Yang, L. Yang, W. Wang and Y. Niu, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1265–1269 CrossRef CAS.
- J. S. Yuan, W. Q. Wang, Z. F. Zhou and J. Niu, Macromolecules, 2020, 53, 5655–5673 CrossRef CAS.
- G. I. Peterson and T. L. Choi, Chem. Sci., 2020, 11, 4843–4854 RSC.
- G. Chen, L. Xia, F. Wang, Z. Zhang and Y.-Z. You, Polym. Chem., 2021, 12, 3740–3752 RSC.
- D.-Y. Zhang and D.-F. Chen, J. Funct. Polym., 2023, 36, 261–274 CAS.
- D. Pasini and D. Takeuchi, Chem. Rev., 2018, 118, 8983–9057 CrossRef CAS PubMed.
- G. B. Butler and R. J. Angelo, J. Am. Chem. Soc., 1957, 79, 3128–3131 CrossRef CAS.
- Y. Kametani, M. Nakano, T. Yamamoto, M. Ouchi and M. Sawamoto, ACS Macro Lett., 2017, 6, 754–757 CrossRef CAS PubMed.
- P. Gerdt and A. Studer, Angew. Chem., Int. Ed., 2022, 61, e202206964 CrossRef CAS PubMed.
- Y. Assem, H. Chaffey-Millar, C. Barner-Kowollik, G. Wegner and S. Agarwal, Macromolecules, 2007, 40, 3907–3913 CrossRef CAS.
- Y. Assem, A. Greiner and S. Agarwal, Macromol. Rapid Commun., 2007, 28, 1923–1928 CrossRef CAS.
- S. Erkoc and A. E. Ouchi, Macromolecules, 2008, 41, 9019–9024 CrossRef CAS.
- J. Li, M. Du, Z. Zhao and H. Liu, Macromolecules, 2016, 49, 445–454 CrossRef CAS.
- Y. Kametani, M. Sawamoto and M. Ouchi, Angew. Chem., Int. Ed., 2018, 57, 10905–10909 CrossRef CAS PubMed.
- Y. Kametani, F. Tournilhac, M. Sawamoto and M. Ouchi, Angew. Chem., Int. Ed., 2020, 59, 5193–5201 CrossRef CAS PubMed.
- H. Huang, W. Wang, Z. Zhou, B. Sun, M. An, F. Haeffner and J. Niu, J. Am. Chem. Soc., 2019, 141, 12493–12497 CrossRef CAS PubMed.
- Y. Wang, J. Du and H. Huang, Angew. Chem., Int. Ed., 2024, 63, e202318898 CrossRef CAS PubMed.
- M. Tsuji, R. Sakai, T. Satoh, H. Kaga and T. Kakuchi, Macromolecules, 2002, 35, 8255–8257 CrossRef CAS.
- S. Erkoc, L. J. Mathias and A. E. Acar, Macromolecules, 2006, 39, 8936–8942 CrossRef CAS.
- Y. Hibi, M. Ouchi and M. Sawamoto, Angew. Chem., Int. Ed., 2011, 50, 7434–7437 CrossRef CAS PubMed.
- M. Ouchi, M. Nakano, T. Nakanishi and M. Sawamoto, Angew. Chem., Int. Ed., 2016, 55, 14584–14589 CrossRef CAS PubMed.
- Y. Hibi, S. Tokuoka, T. Terashima, M. Ouchi and M. Sawamoto, Polym. Chem., 2011, 2, 341–347 RSC.
- Z. Hu, Y.-P. Xu, D. Wu, B. Li, Y.-C. Huang, J.-X. Jiang, G.-Q. Lai and T.-Y. Luh, Macromol. Chem. Phys., 2012, 213, 566–571 CrossRef CAS.
- S. Moins, J. C. Martins, A. Krumpmann, V. Lemaur, J. Cornil, N. Delbosc, A. Decroly, P. Dubois, R. Lazzaroni, J.-F. Gohy and O. Coulembier, Chem. Commun., 2017, 53, 6899–6902 RSC.
- D.-F. Chen, B. M. Boyle, B. G. McCarthy, C.-H. Lim and G. M. Miyake, J. Am. Chem. Soc., 2019, 141, 13268–13277 CrossRef CAS PubMed.
- S. Zhang, C. Cao, S. Jiang and H. Huang, Macromolecules, 2022, 55, 9411–9419 CrossRef CAS.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- W. Tang and K. Matyjaszewski, Macromolecules, 2007, 40, 1858–1863 CrossRef CAS.
- M. D. Ryan, R. M. Pearson, T. A. French and G. M. Miyake, Macromolecules, 2017, 50, 4616–4622 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c
*c