
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly(β-hydroxyalkanoate)/polymethacrylate self-assembled architectures by ring-opening polymerization (ROP)/reversible addition–fragmentation chain-transfer (RAFT) polymerization and polymerization-induced self-assembly (PISA)†
Julien
Rosselgong
 *,
Ali
Dhaini
,
Manon
Rochedy
,
Lourdes Mónica
Bravo-Anaya
,
Jean-François
Carpentier
and
Sophie M.
Guillaume
*
*,
Ali
Dhaini
,
Manon
Rochedy
,
Lourdes Mónica
Bravo-Anaya
,
Jean-François
Carpentier
and
Sophie M.
Guillaume
*
Univ. Rennes, CNRS, Institut des Sciences Chimiques de Rennes, UMR 6226, 35042 Rennes, France. E-mail: julien.rosselgong@univ-rennes.fr; sophie.guillaume@univ-rennes.fr
First published on 27th January 2025
Abstract
Self-assembled poly(β-hydroxyalkanoate) (PHA)-based block copolymers are attractive materials for biomedical applications due to the biocompatibility and (bio)degradability of the PHA segment. Herein, we report the synthesis and formation of self-assemblies based on PHAs: namely, poly(3-hydroxybutyrate) (PHB) was prepared by ring-opening polymerization (ROP) of racemic β-butyrolactone (rac-β-BL) using a discrete yttrium-based catalyst in the presence of a hydroxy-terminated trithiocarbonate (TTC-OH) as initiator. The resulting TTC end-capped PHB prepolymer next promoted the controlled reversible addition–fragmentation chain-transfer (RAFT) polymerization of 2-hydroxy ethyl methacrylate (HEMA). When performed in THF, in which the initial solvophilic PHB-TTC segment and HEMA monomer are both fully soluble, this second step resulted in a polymerization-induced self-assembly (PISA) leading to the formation of nanoparticles, as the solvophobic PHEMA precipitated in the dispersed medium. The effective extension of the PHB block by a PHEMA segment, as evidenced by SEC and NMR analyses, highlighted the efficiency of the PHB-TTC macro-RAFT agent. This ROP/RAFT/PISA strategy revealed successful at various polyester (DP = 45–90) and polymethacrylate (DP = 200–500) block lengths. The size, polydispersity index (PDI) and morphology of the resulting self-assembled PHBx-b-PHEMAy particles were assessed by dynamic light scattering (DLS) measurements, transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS). Depending on both segments chain-lengths, DLS enabled identifying particles in suspension having hydrodynamic diameters (DH) varying from 56 to 194 nm, with narrow polydispersity index, i.e. PDI < 0.140. SAXS measurements and TEM observations revealed vesicles morphology for specific PHBx-b-PHEMAy samples presenting apparent diameters ranging from 134 to 316 nm. These morphologies support the successful copolymerization through a PISA process, first reported herein for the elaboration of PHA-based objects that may be valuable nano-vehicles of active ingredients for biomedical applications.
Introduction
Polyhydroxyalkanoate (PHA)-based block copolymers are valuable polyester materials for applications in nanomedicine and/or drug delivery.1–8 While PHAs are most commonly associated with other polyesters (typically another PHA or poly(ethylene glycol)), growing a second poly(meth)acrylic block from a PHA segment by reversible-deactivation radical polymerization (RDRP) is less documented.9–11 For more than two decades, RDRP techniques such as nitroxide-mediated polymerization (NMP), atom transfer radical polymerization (ATRP) or reversible addition–fragmentation chain-transfer (RAFT) polymerization have revealed key synthetic tools to access well-defined (co)polymers, including block copolymers.12–15 For instance, poly((R)-3-hydroxybutyrate) (PHB)-based triblock copolymers poly((R)-3-hydroxybutyrate)-b-poly(N-isopropylacrylamide)-b-[[poly(methyl ether methacrylate)-g-poly(ethylene glycol)]-co-[poly(methacrylate)-g-poly(propylene glycol)]], a multi-arm star copolymer providing a thermo-responsive hydrogel, was prepared by ATRP for thermogels outcomes.16 The double thermo-responsive behavior due to the (meth)acrylate blocks enabled the copolymer self-assembly at room temperature, and the subsequent delivery of a drug at body temperature. Also, RAFT enabled preparing novel PHB macro-RAFT agents subsequently used to grow an N-isopropyl acryl amide (NIPAM) segment, ultimately offering thermo-responsive block copolymers.17 In this latter example, the macro-RAFT agent was synthesized in two steps from esterification of an ω-hydroxylated PHB - first made by ring-opening polymerization (ROP) of racemic β-butyrolactone (rac-β-BL) with a RAFT agent, namely 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid used as initiator combined with a yttrium catalyst-, using 4-dimethylaminopyridine (DMAP) and N,N′-dicyclohexylcarbodiimide (DCC) as coupling agents.Growing in situ a hydrophobic chain from a soluble first-block to recover an amphiphilic block copolymer, is a process referred to as polymerization-induced self-assembly (PISA). Provided a formulation including a soluble polymer segment is chain-extended using a monomer of which the corresponding homopolymer is insoluble in the reaction solvent, PISA results in nano-objects such as micelles, worms and/or vesicles.18–23 In this process, the growing second-block becomes insoluble when reaching a critical degree of polymerization (DP), eventually leading to an in situ self-assembly phenomenon. Such systems are essentially based on non-solvents such as water,24–31 alcohols,32–36 or non-polar solvents.37–41 While a wide variety of PISA formulations have been established, to our knowledge, there is no report of PISA involving a PHA polyester block.
Recently, aqueous ROP and PISA were successfully combined to form N-carboxyanhydrides (NCAs)-based copolymers by ROPISA, as reported by Bonduelle and co-workers.42,43 An α-amino end-capped poly(ethylene oxide) macroinitiator was used in order to protect the NCA monomers from hydrolysis during their ROP performed in water. The spontaneous self-assembly of the amphiphilic poly(ethylene oxide)/polypeptide block copolymers thus led to a PISA formulation returning needle-like nanoparticles. Such a NCA ROPISA approach was further investigated by Thornton and co-workers using a polysarcosine macroinitiator to form only poly(amino acid)-based (L-phenylalanine-NCA and alanine-NCA) rod-like nanoparticles, and also by Heise and co-workers to access amino acid-based (L-proline-NCA) worm like micelles nanostructures loaded with dyes.44,45
More recently, Armes and coworkers reported the efficient synthesis of hydrolytically degradable block copolymer nanoparticles by reverse PISA in aqueous media.46 A poly(ε-caprolactone) (PCL)-based macroinitiator and a suitable water-miscible monomer (N,N′-dimethylacrylamide, DMAC) afforded PCL-b-PDMAC and PDMAC-b-PCL-b-PDMAC diblock and triblock copolymers, respectively. These copolymers were insoluble in water, thus leading to the formation of polyester/polyacrylamide micelles with sizes ranging from 20 to 120 nm. Later on, the same authors combined PISA with crystallization-driven self-assembly to form anisotropic nanoparticles from poly(L-lactide)-b-PDMAC copolymers. They obtained either rod-like nanoparticles or diamond-like platelets that have potential applications as sustainable pickering emulsifiers.47 Further on, the same group reported the RAFT dispersion polymerization of 2-hydroxyethyl methacrylate in non-polar media.48 Using a poly(lauryl methacrylate) (PLMA) precursor, a PISA process then generated PLMA-PHEMA nanoparticles forming well-defined nearly monodispersed spheres.
Herein, we report the formation of PHB-based self-assembled architectures through a combined ROP, RAFT and PISA process. As inspired by our previous work on the yttrium catalyzed ROP of β-lactones performed in the presence of an exocyclic alcoholic initiator, the ROP of racemic β-BL catalyzed by a discrete yttrium complex, in the presence of a RAFT agent acting as an alcohol moiety, afforded the PHB prepolymer that subsequently promoted the RAFT of 2-hydroxyethyl methacrylate (HEMA) by dispersion PISA performed in THF.49,50 The PHB block length and the degree of polymerization (DP) of the second PHEMA segment of the resulting PHB-b-PHEMA nano-objects, were assessed by size exclusion chromatography (SEC). Particle size and morphology were determined using dynamic light scattering (DLS) measurements, transmission electron microscopy (TEM) and small-angle X-ray scattering (SAXS) analyses. The corresponding pseudo-phase diagram established by DLS revealed an increase of the hydrodynamic diameter (DH) of particles formed with the increase of the DP of the HEMA second block and with the initial co-monomer and PHB macro-RAFT agent concentration. These results thus established a PISA process applied for the first time to elaborate PHA/polymethacrylate objects.
Experimental section
Materials and methods
All experiments involving organometallic catalysts were performed under an inert atmosphere (argon, <5 ppm O2 and H2O) using standard Schlenk, vacuum line, and glovebox techniques. Racemic β-butyrolactone (rac-β-BL) (>95%, Aldrich) was distilled twice from CaH2 prior to use. 2-Hydroxyethyl methacrylate (HEMA) (>99%, Aldrich; containing traces of ethylene glycol dimethacrylate cross-linker as established by GC-MS analyses, Fig. S1†)51 was purified prior to use, upon passing it through a basic alumina column to remove the monomethyl ether hydroquinone inhibitor. The RAFT chain transfer agent (CTA) 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanol (97%, Aldrich, trithiocarbonate (TTC), TTC-OH) was dried under vacuum at 60 °C prior to use. 2,2′-Azobis(2-methylpropionitrile) (AIBN, >98%, Aldrich) was used as received. The {ONNOMe2}H2 proligand and Y[N(SiHMe2)2]3(THF)2 metallic precursor, from which the discrete amido yttrium complex was prepared in situ, were synthesized according to the reported procedure.50 THF and toluene were freshly distilled from Na-benzophenone under argon and degassed thoroughly by freeze–thaw-vacuum cycles prior to use. All other reagents were used as received.Instrumentation and measurements
1H (500 and 400 MHz), 13C{1H} (125 and 100 MHz) and 2D (COSY, HMBC, HSQC, and DOSY) NMR spectra were recorded on Bruker Avance AM 500 or Ascend 400 spectrometers at 25 °C. 1H and 13C{1H} NMR spectra were referenced internally relative to SiMe4 (δ 0 ppm) using the residual solvent resonances. The analyses were recorded in CDCl3 for the PHB-TTC macro-RAFT agent and in DMSO-d6 for the PHB-b-PHEMA diblock copolymers.Number-average molar mass (Mn,SEC), weight-average molar mass (Mw,SEC) and dispersity (ĐM = Mw/Mn) values of the copolymers were determined by size exclusion chromatography (SEC) in THF at 20 °C for the PHB homopolymers, or in N,N-dimethylformamide (DMF + LiBr 1.0 g L−1) at 20 °C for the PHB-b-PHEMA diblock copolymers. SEC in THF was stabilized with BHT at 30 °C (flow rate = 1.0 mL min−1) on an Agilent 1260 Infinity II system equipped with a refractive index detector (dRI), a photo-diode array detector (DAD) and a set of two ResiPore (300 × 7.5 mm) columns. Polymer samples of PHB-TTC were dissolved in THF (1.0 mg mL−1). All elution curves were calibrated with polystyrene standards (Agilent Easivial kit of polystyrene standards; Mn from 162 to 364![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1). The Mn,SEC values of the PHBs were not corrected for the difference in hydrodynamic radius vs. those of polystyrene. SEC measurements in DMF were performed on an Ultimate 3000 system from Thermoscientific equipped with a DAD. The system also includes a multi-angle light scattering detector (MALS with 8 angles) and a dRI, both from Wyatt technology. Polymer samples of PHB-b-PHEMA were dissolved in DMF (1.0 mg mL−1) and were separated with a set of three columns (GF-1G 7B (7.5 × 50 mm), GF 310 (7.5 × 300 mm) GF510 (7.5 × 300 mm); exclusion limits from 500 to 300000 g mol−1) at a flow rate of 0.5 mL min−1. The columns were maintained at 50 °C. Agilent Easivial kit of polystyrene was used as standard (Mn from 162 to 364000 g mol−1). SEC DMF experiments were performed through the platform POLYCAR of the University of Bordeaux.
000 g mol−1). The Mn,SEC values of the PHBs were not corrected for the difference in hydrodynamic radius vs. those of polystyrene. SEC measurements in DMF were performed on an Ultimate 3000 system from Thermoscientific equipped with a DAD. The system also includes a multi-angle light scattering detector (MALS with 8 angles) and a dRI, both from Wyatt technology. Polymer samples of PHB-b-PHEMA were dissolved in DMF (1.0 mg mL−1) and were separated with a set of three columns (GF-1G 7B (7.5 × 50 mm), GF 310 (7.5 × 300 mm) GF510 (7.5 × 300 mm); exclusion limits from 500 to 300000 g mol−1) at a flow rate of 0.5 mL min−1. The columns were maintained at 50 °C. Agilent Easivial kit of polystyrene was used as standard (Mn from 162 to 364000 g mol−1). SEC DMF experiments were performed through the platform POLYCAR of the University of Bordeaux.
The molar mass of PHB samples was also determined by 1H NMR analysis in CDCl3 from the relative intensities of the signals of the methine hydrogen from the repeating unit (δ 5.03–5.53 ppm, –OCHCH3–CH2–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and of the methylene signal of the TTC end-group (δ 3.29–3.38 ppm, CH2–S–CS). Monomer conversion was calculated from 1H NMR spectra of the crude polymer samples in CDCl3 by using the integration (Int.) ratios [Int.PHB/PHEMA/(Int.β-BL/HEMA + Int.PHB/PHEMA)] of the methine hydrogens of PHB/PHEMA (vide supra) and of the monomers (δ 4.32 ppm, β-BL; δ 4.17 ppm, HEMA).
O) and of the methylene signal of the TTC end-group (δ 3.29–3.38 ppm, CH2–S–CS). Monomer conversion was calculated from 1H NMR spectra of the crude polymer samples in CDCl3 by using the integration (Int.) ratios [Int.PHB/PHEMA/(Int.β-BL/HEMA + Int.PHB/PHEMA)] of the methine hydrogens of PHB/PHEMA (vide supra) and of the monomers (δ 4.32 ppm, β-BL; δ 4.17 ppm, HEMA).
High resolution (error < 25 ppm) matrix-assisted laser desorption ionization – time of flight (MALDI-ToF) mass spectrometry (MS) was performed using an ULTRAFLEX III TOF/TOF spectrometer (Bruker Daltonik Gmbh, Bremen, Germany) in positive ionization mode at the Centre Régional de Mesures Physiques de l'Ouest (CRMPO, ScanMAT UAR 2025, CNRS-Université de Rennes). MS spectra were recorded using the reflectron mode and an accelerating voltage of 25 kV. A mixture of a freshly prepared solution of the polymer in CH2Cl2 (HPLC grade, 10 mg.mL−1) and trans-2-(3-(4-tert-butylphenyl)-2-methyl-2-propenylidene)-malononitrile (DCTB), and an acetonitrile solution of the cationizing agent (sodium trifluoroacetate, 10 mg mL−1) were prepared. The solutions were combined in a ratio 1:1:1 v/v/v of matrix-to-sample-to-cationizing agent. The resulting solution (ca. 0.25–0.5 μL) was deposited onto the sample target and air or vacuum dried.
Dynamic light scattering (DLS) measurements were performed in THF using a Malvern Zetasizer NanoZS instrument equipped with a standard HeNe laser emitting at 632.8 nm (Malvern, U.K.), at 25 °C, and at an angle of 173°. The correlation functions were averaged from three independent measurements of two runs of 60 s each. The equilibration time for this measurement was 60 s. Non-negative least squares and cumulants algorithms were applied to extract the intensity-weighted particle size distribution and the intensity-weighted mean hydrodynamic size, respectively. The hydrodynamic diameter (DH) was determined using the Stokes–Einstein equation proposed for spherical particles.52 THF viscosity at the selected temperature was taken into account for the calculation of DH. Measurements for each sample were at least triplicated. The size and PDI data of the objects are presented as average values along with the calculated standard deviation. Samples for DLS observations were diluted at 0.5 wt% of solids vs. THF.
Imaging was performed with a JEM-1400 transmission electron microscope (TEM, JEOL Ltd, Tokyo, Japan) operating at an accelerating voltage of 120 kV. Images were recorded with a Gatan SC200 Orius® CCD camera at 25000 magnification and set up with the imaging software Gatan Digital MicrographTM (Gatan, Pleasanton, USA); images were acquired on at least three different regions. Samples for TEM observations were diluted at 0.5 wt% of solids vs. THF; they were prepared by direct deposition of a PHB-b-PHEMA suspension droplet (7 μL) on carbon grids (300 mesh Cu-300LD from Pacific Grid Tech) and THF was evaporated at room temperature over 20 min. The particle size distribution was determined one-by-one from two different TEM-micrographs using Image-J software and displayed with a standard deviation.
Small angle X-Ray Scattering (SAXS) experiments were performed on PHB83-b-PHEMA500 suspensions at 5 wt% and 7.5 wt% on the SWING beamline at Synchrotron SOLEIL, Gif-sur-Yvette, France. The SAXS setup included a wavelength of λ = 1.033 Å−1 and an energy of 12 keV, with a 17 × 17 cm2 low-noise Aviex CCD detector positioned at distances of 0.518 and 6.136 m from the sample, yielding a q-range of 0.00104–2.06 Å−1, where q = 4πsinθ/λ. The copolymer suspensions were placed into borosilicate capillaries (1.5 mm optical path, WJM-Glas/Müller GmbH, Germany). The intensity was collected after 1000 ms exposition (waiting gap: 2000 ms) in a vertical scan of 10 steps of 1 mm each. All 10 integrated images were averaged to obtain one I(q) profile per sample. A background correction was applied on the I(q) profiles by subtracting the THF scattering, and the unit was set to cm−1 after multiplying the measured scattering by 0.02 × 1.5/capillary diameter. The resulting data was analyzed using a spherical vesicle model on SASView software, with the scattering length density of THF and PHEMA500, as previously calculated, being set.
CH–CO–), 5.81 (d, J = 14 Hz, 0.2H, CH3–CHCH–CO–), 5.24 (m, 82H, CO–O–CHCH3–CH2–CO), 4.18 (m, 0.6H, CO–O–CHCH3–OH), 4.12 (t, J = 5 Hz, 2H, –CHCH3–CH2-CO–O–CH2–CH2–), 3.32 (t, J = 8 Hz, 2H, CH3–(CH2)10–CH2–S–CS–), 2.58 (42 H, CO–O–CHCH3–CH2–CO), 2.45 (42H, CO–O–CHCH3–CH2–CO), 2.23 (m, 4.6 H, –S–CS–CCNCH3–CH2–), 1.87 (s, 3H, –S–CS–CCNCH3–CH2–), 1.84 (d, J = 7 Hz, 1.2H, CH3–CH–CH–CO–), 1.27 (m, 249H, CO–O–CHCH3–CH2–CO), 0.89 (m, 3H, CH3–(CH2)10–CH2–S–CS–) (Fig. 1). 13C J-MOD NMR (125 MHz, CDCl3, 25 °C) δ (ppm): 217.4 (CH3–(CH2)10–CH2–S–![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) S–), 171.5 (S–CS–(CN)(CH3)–CH2–), 169.2 (CO–O–CHCH3–CH2–O), 124.8 (CH3–H–CH–CO–), 122.7 (CH3–CH–H–CO–), 119.3 (S–CS–C(N)(CH3)–CH2–), 67.7 (CO–O–HCH3–CH2–CO), 64.0 (S–CS–C(CN)(H3)–CH2–), 63.2 (S–CS–C(CN)(CH3)–CH2–CH2–H2–O), 40.7 (O–O–CHCH3–H2–CO), 19.8 (CO–O–CHH3–CH2–CO) (Fig. S2†).
S–), 171.5 (S–CS–(CN)(CH3)–CH2–), 169.2 (CO–O–CHCH3–CH2–O), 124.8 (CH3–H–CH–CO–), 122.7 (CH3–CH–H–CO–), 119.3 (S–CS–C(N)(CH3)–CH2–), 67.7 (CO–O–HCH3–CH2–CO), 64.0 (S–CS–C(CN)(H3)–CH2–), 63.2 (S–CS–C(CN)(CH3)–CH2–CH2–H2–O), 40.7 (O–O–CHCH3–H2–CO), 19.8 (CO–O–CHH3–CH2–CO) (Fig. S2†).
 | ||
| Fig. 1 1H NMR spectrum (500 MHz, CDCl3, 25 °C) of PHB83-TTC prepared by ROP of racemic β-butyrolactone catalyzed by Y{ONNOMe2} in the presence of TTC-OH (Table 1, entry 1). | ||
Results and discussion
The amphiphilic PHB-b-PHEMA diblock copolymers synthesized in THF comprise a solvophilic aliphatic polyester block, PHB, and a solvophobic methacrylic polymer block, PHEMA. Two different synthetic approaches were explored to first prepare the PHB-TTC macro-RAFT agent (Scheme 1).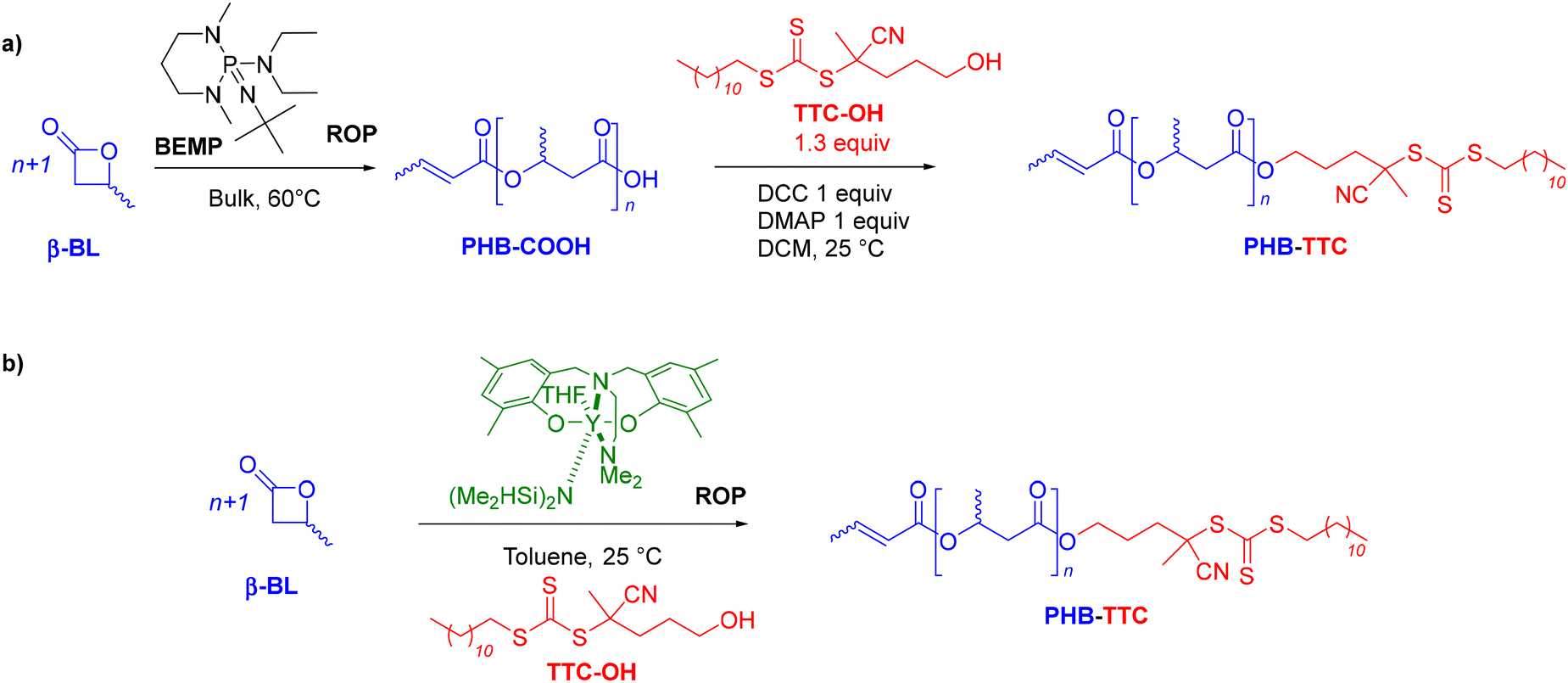 | ||
| Scheme 1 Synthesis of PHB-TTC by ROP of racemic β-butyrolactone (rac-β-BL): (a) ROP promoted by an organic phosphazene base, BEMP, followed by esterification with TTC-OH, and, (b) ROP catalyzed by a discrete yttrium catalyst in the presence of TTC-OH as initiator. | ||
Synthesis and characterization of the PHB-TTC precursor
A first strategy relying on the previously established organocatalyzed ROP of rac-β-BL mediated by the phosphazene base 2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine (BEMP), was first attempted. This was aimed at preparing an α-COOH, ω-crotonate telechelic PHB that would further be functionalized into a PHB-trithiocarbonate (TTC) by esterification with the TTC-OH chain transfer agent (Scheme 1a).53 However, although the desired PHB-COOH polymer was initially formed, the following Steglich esterification in the presence of dicyclohexylcarbodiimide (DCC) and either dimethylaminopyridine (DMAP) or 2,6-lutidine, prompted the degradation of the resulting PHB-TTC, as monitored by 1H NMR spectroscopy (Fig. S4†). We assumed that the DMAP or 2,6-lutidine base most likely altered the PHB stability. This contrasted with the previously reported successful esterification of the related poly(3-hydroxyoctanoate) using a similar Steglich esterification protocol.17Alternatively, we implemented the well-known ROP of β-lactones mediated by yttrium-based catalytic systems in the presence of a hydroxyl end-capped TTC as initiating species.1,9,16,49,50,54 The synthesis of the PHB first block was successfully achieved by ROP of rac-β-BL performed in toluene in the presence of a discrete yttrium catalyst supported by a tetradentate {ONNOMe2} dianionic ligand, and initiated by 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanol (TTC-OH) (Scheme 1b). This approach is similar to the recently reported ROP of L-lactide (LLA) catalyzed by DMAP in combination with an hydroxyl-terminated dithiocarbonate RAFT agent, affording a PLLA macroinitiator end-capped with the RAFT moiety, subsequently used for the copolymerization with various methacrylates.55 The methyl-substituted Y{ONNOMe2} catalyst system was purposely selected so as to obtain atactic PHB segments that are typically more soluble than the more crystalline iso- or syndio-tactic ones. The work reported herein thus did not aim at varying the tacticity of the PHB precursor. PHB-TTC was synthesized with three different targeted degrees of polymerization (DP = 45, 70, 90), as tuned by the [β-BL]0/[yttrium catalyst]0/[TTC-OH]0 molar ratio, with [TTC-OH]0 = [yttrium catalyst]0. The most relevant results are gathered in Table 1. High monomer conversions (85–96%) were reached, affording the expected telechelic PHB α-end capped by the TTC RAFT agent moiety and ω-functionalized by either a crotonate or hydroxy group, as evidenced by 1H and 13C NMR (Fig. 1 and Fig. S2, S5, S6,† for PHBn = PHB83, PHB48, and PHB57, respectively, with n corresponding to the number of lactone repeating units), DOSY NMR (Fig. 2 and Fig. S7, and S8† for PHB83, PHB48, and PHB57, respectively), as well as MALDI-ToF MS (Fig. 3, and Fig. S9† for PHB83 and PHB48, respectively), and SEC (Fig. 4 and Fig. S10, and S11† for PHB83, PHB48, and PHB57, respectively).
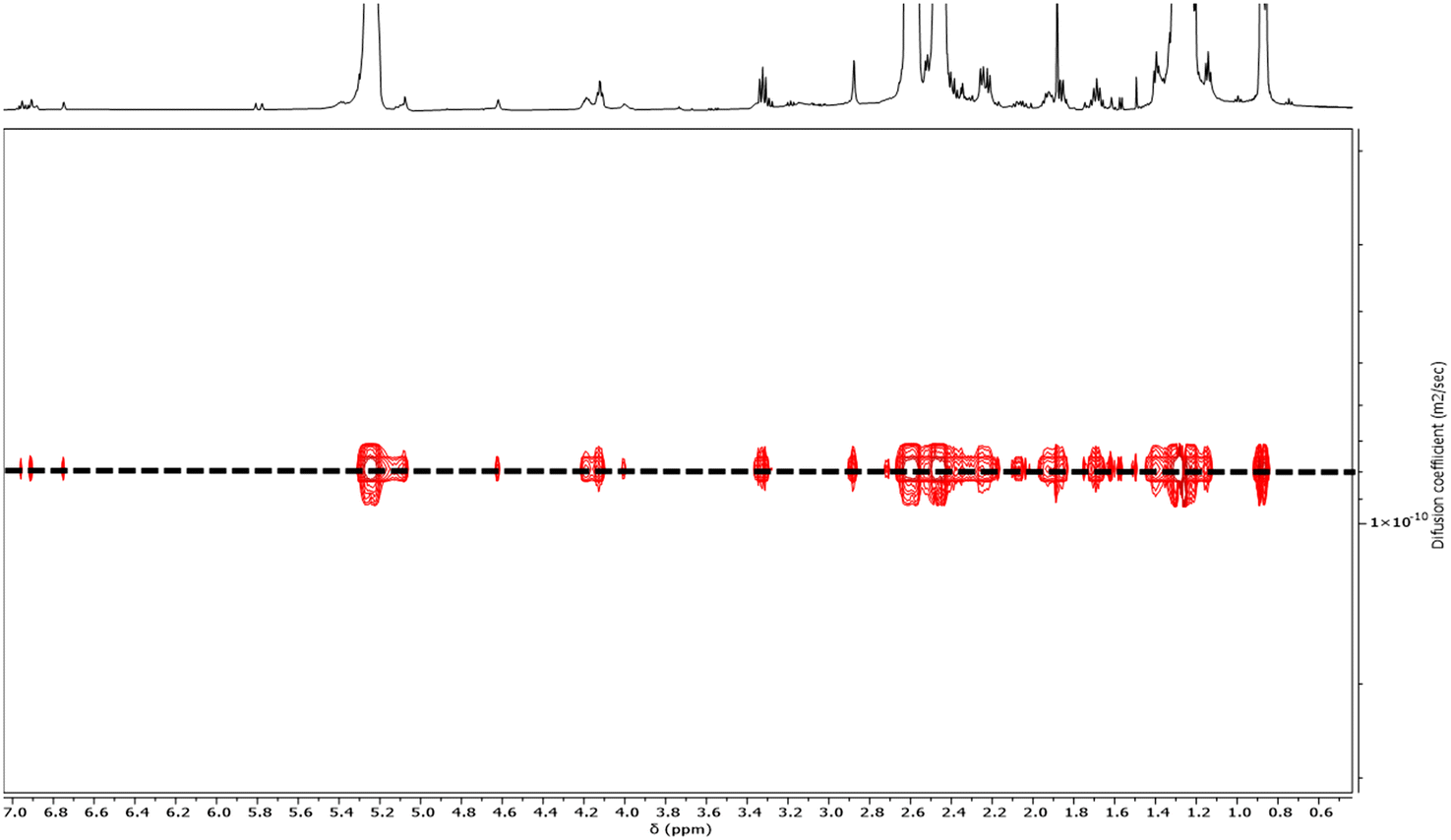 | ||
| Fig. 2 DOSY NMR spectrum (500 MHz, CDCl3, 25 °C) of PHB83-TTC prepared by ROP of racemic β-butyrolactone catalyzed by Y{ONNOMe2} and initiated by TTC-OH (Table 1, entry 1). | ||
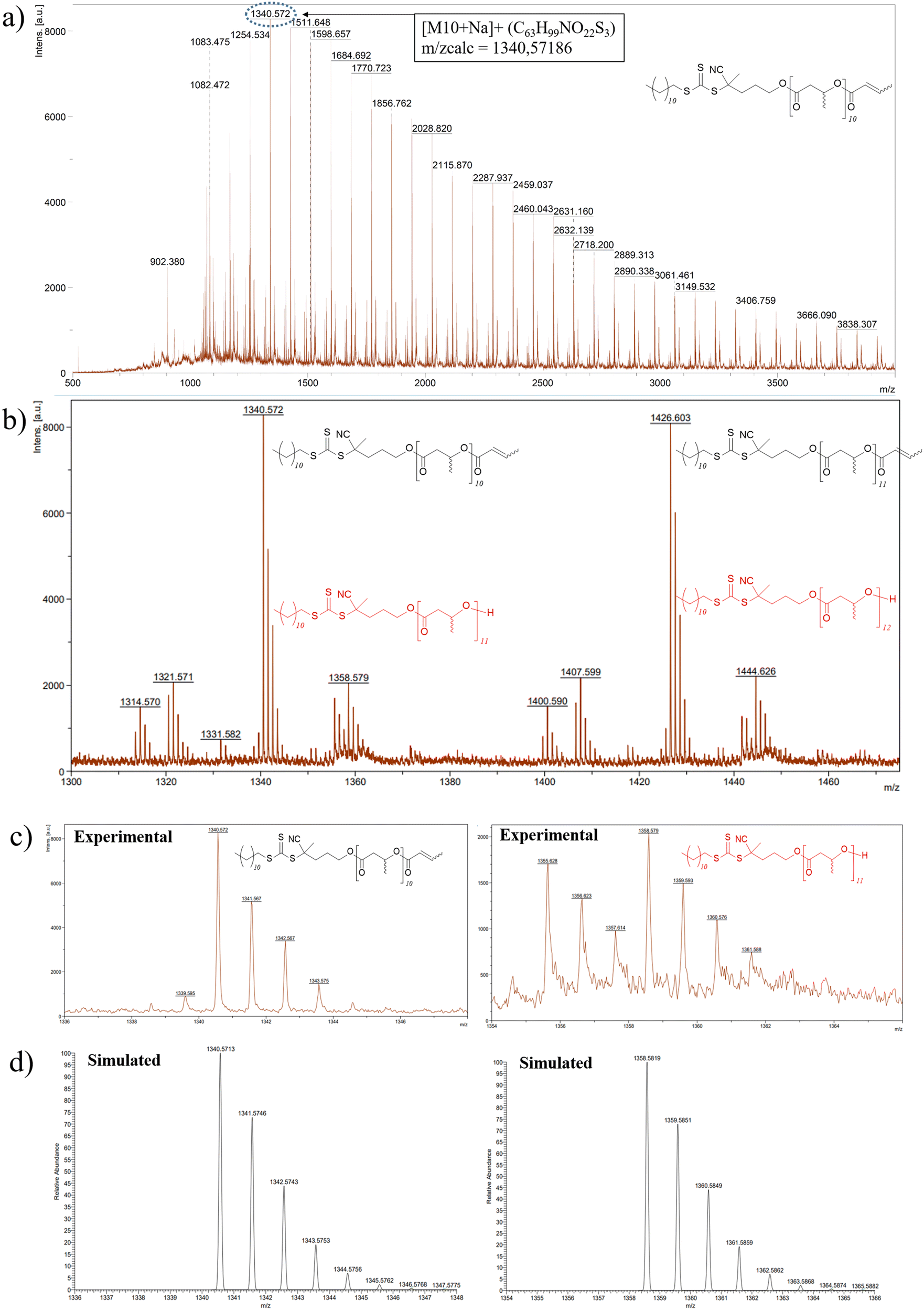 | ||
| Fig. 3 (a) High resolution MALDI-ToF mass spectrum (DCTB matrix, ionized by Na+), of PHB83-TTC prepared by ROP of racemic β-butyrolactone catalyzed by Y{ONNOMe2} and initiated by TTC-OH (Table 1, entry 1). The lower molar mass macromolecules are observed with m/z experimental (m/zexp) vs. calculated (m/zcalc) values, respectively, for the crotonate-PHB83-TTC for: DP = 9: 1254.534 vs. 1254.535; DP = 10: 1340.572 vs. 1340.572; DP = 11: 1426.603 vs. 1426.609; DP = 20: 2200.909 vs. 2200.939, (b) zoom and assignment of the two major populations crotonate-PHB83-TTC and HO-PHB83-TTC, (c) zoom of the major population crotonate-PHB83-TTC (DP = 10 and DP = 11) and the minor population HO-PHB83-TTC (DP = 11 and DP = 12), (d) simulation of the above major and minor populations, respectively. | ||
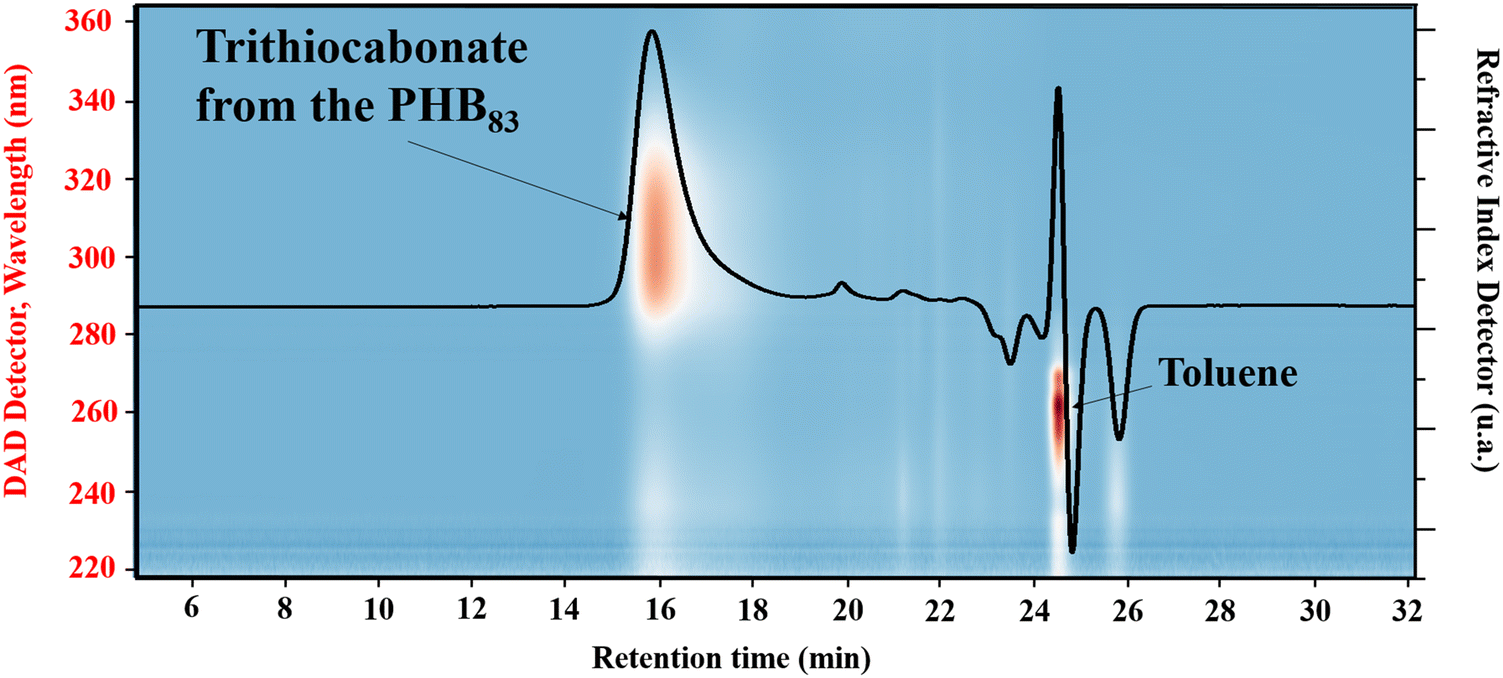 | ||
| Fig. 4 SEC chromatogram in THF superimposed with the isoabsorbance plot of PHB83-TTC prepared by ROP of racemic β-butyrolactone catalyzed by Y{ONNOMe2} in the presence of TTC-OH (Table 1 entry 1). | ||
| Entry | [β-BL]0/[Y]0/[TTC-OH]0 | Reaction time (h) | β-BL conv.a (%) |
DPtheob |
M
n,theoc (g.mol−1) |
DPNMRd |
M
n,NMRe (g.mol−1) |
M
n
,SECf (g.mol−1) |
Đg |
|---|---|---|---|---|---|---|---|---|---|
| a Monomer conversion calculated by 1H NMR analysis of the crude reaction mixture. b Theoretical degree of polymerization calculated using the β-BL conversion. c Molar mass calculated according to Mn,theo = ([β-BL]0/[TTC-OH]0 × conv.β-BL × Mβ-BL) + MTTC–OH with Mβ-BL = 86 g mol−1 and MTTC–OH = 390 g mol−1. d Experimental degree of polymerization determined by 1H NMR end-group analysis. e Molar mass determined by 1H NMR analysis of the isolated polymer, from the resonances of the terminal TTC group. f Number-average molar mass determined by SEC in THF vs. polystyrene standards. g Dispersity (Đ = Mw/Mn) determined by SEC analysis in THF. | |||||||||
| 1 | 90:1:1 |
3 | 92 | 83 | 7510 | 83 | 7500 | 9200 | 1.19 |
| 2 | 70:1:1 |
4 | 85 | 60 | 5510 | 57 | 5300 | 6600 | 1.24 |
| 3 | 45:1:1 |
3 | 96 | 43 | 4110 | 48 | 4500 | 6000 | 1.26 |
The PHB-TTC samples were characterized by detailed NMR spectroscopy. 1H and 13C NMR analyses revealed the formation of the expected α-TTC end-capped PHB, as evidenced by the TTC-RAFT agent signature (δ1H = 3.32 ppm, CH2–S–CS, δ1H = 4.12 ppm, CH2–O–CO; δ13C = 217.4 ppm, S–CS, δ13C = 119.3 ppm, CN–C) (Fig. 1 and Fig. S3, S4, S5† for PHB83, PHB48, and PHB57, respectively). Evidence of a population of PHB hydroxy-terminated was observed (δ1H = 4.14 ppm, CH(CH3)–OH; δ13C = 43.7 ppm, CH(CH3)–OH). The formation of some ω-crotonate PHB (δ1H = 6.95 ppm, CH3–CHCH, δ1H = 5.81 ppm, CH3–CHCH; δ13C = 124.8 ppm, CH3–CHCH, δ13C = 122.7 ppm, CH3–CHCH) resulted from the elimination of H2O from the above-mentioned hydroxy-end capped PHB chains, as classically observed in the ROP of β-BL.53 The DP values calculated by 1H NMR analysis, based on the CH2–S–CS methylene signal of the TTC end-group (present on all chains), were found in good agreement with the targeted values (Table 1). Moreover, the DOSY NMR spectrum displayed a single diffusion coefficient, further corroborating the purity of the PHB-TTC functional polymer thus prepared by ROP (Fig. 2).
MALDI-ToF mass spectrometry investigations enabled to gain deeper insights into the macromolecular structure and topology of the PHB-TTC. The MALDI-ToF mass spectrum, recorded using a DCTB matrix, of PHB83-TTC typically showed a main population corresponding to PHB end-capped by both the TTC and the crotonate moieties, as anticipated from the ROP of rac-β-butyrolactone catalyzed by Y{ONNOMe2} and initiated by TTC-OH (Fig. 3a and Fig. S9† for PHB83 and PHB48, respectively). Another population corresponding to the HO-PHBn-TTC with a hydroxyl end-group was also observed. These findings were supported by the good match between the corresponding experimental and simulated populations, respectively (Fig. 3c and d). The other minor populations of PHB chains could not be unambiguously assigned.
The PHB-TTC samples were fully soluble in THF at room temperature, thereby enabling their SEC analysis in this eluent (Fig. 4). The experimental molar mass values determined by NMR (Mn,NMR) matched quite well with the theoretical data (Mn,theo), while SEC values (Mn,SEC, not corrected) were found slightly higher, most likely due to the different hydrodynamic radius of the polystyrene standards used for calibration. The dispersities Đ = Mw/Mn remained lower than 1.26, indicating a good control of the ROP. Furthermore, SEC measurements of a PHB-TTC using a photo-diode array detector operating at wavelengths from 200 to 400 nm, returned an isoabsorbance plot displaying an intense elution spot at 310 nm, which corresponds to the TTC moiety of the PHB-TTC (Fig. 4 and Fig. S10, S11† for PHB83, PHB48, and PHB57, respectively).56 Rewardingly, the refractive index signal clearly matched with the UV signal, thus supporting the PHB end-capping by the TTC moiety.
Synthesis and characterization of the PHB-b-PHEMA block copolymers
Chain extension of the PHB83-TTC acting as the macro-RAFT agent with HEMA was performed in THF, a good solvent of the RAFT-macroinitiator, at 60 °C in the presence of AIBN (typical PISA operating conditions: [AIBN]0/[PHB-TTC]0 = 1:5; Scheme 2 and Table 2). HEMA was selected as the methacrylate monomer as it satisfies the PISA requirements, namely the solubility as a monomer in the copolymerization solvent, and the insolubility of the corresponding (co)polymer segment in that same solvent.57 Thus, at the early stage of the copolymerization, all components were soluble and, as the PISA proceeded, the reaction mixture became cloudier-to-turbid, clearly visually showing a dispersion of the copolymer in THF (Fig. S12†).
 | ||
| Scheme 2 Synthesis of PHB-b-PHEMA diblock copolymers by RAFT dispersion polymerization of HEMA from PHB-TTC macro-RAFT agents. | ||
| Entry | [HEMA + PHB-TTC]a (wt%) |
DPHEMA targetedb |
ConvHEMAc (%) |
DPHEMAd Calc |
M
n,SECe (g.mol−1) |
Đf |
DH,DLSg (nm) |
PDIDLSh |
Dapp,TEMi (nm) |
SDTEMj (nm) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Initial HEMA monomer weight added to PHB-TTC macro-RAFT agent weight vs. THF weight in wt%. b Targeted degree of polymerization of HEMA. c HEMA monomer conversion as calculated by 1H NMR analysis. d Calculated degree of polymerization of PHEMA as established from DPHEMA,targeted × XHEMA. e Number-average molar mass as determined by SEC in DMF vs. polystyrene standards. f Dispersity as determined by SEC in DMF. g Hydrodynamic diameter of the particles formed from PHB-b-PHEMA, as measured by DLS. h Polydispersity index of the particles formed from PHB-b-PHEMA as measured by DLS. i Apparent diameter of the particles formed from PHB-b-PHEMA as measured from TEM. j Standard deviation of the apparent diameter of the particles formed from PHB-b-PHEMA as measured from two TEM micrographs by Image J (Fig. S13–S22†). k Micrograph not taken by TEM. | ||||||||||
| 0 | PHB83 | — | — | 8300 | 1.20 | — | — | — | — | |
| 1 | 5 | 200 | 78 | 157 | 38480 |
1.75 | 56 | 0.09 | 208 | 106 |
| 2 | 5 | 350 | 80 | 281 | 72200 |
1.71 | 83 | 0.07 | 179 | 90 |
| 3 | 5 | 500 | 77 | 387 | 104300 |
1.81 | 164 | 0.03 | 316 | 128 |
| 4 | 7.5 | 200 | 94 | 189 | 39530 |
1.77 | 62 | 0.04 | 171 | 59 |
| 5 | 7.5 | 350 | 95 | 331 | 64580 |
1.79 | 85 | 0.10 | 252 | 90 |
| 6 | 7.5 | 500 | 86 | 431 | 84240 |
1.88 | 176 | 0.11 | 163 | 44 |
| 7 | 10 | 200 | 84 | 168 | 66000 |
1.66 | 72 | 0.06 | N/Ak | N/Ak |
| 8 | 10 | 350 | 91 | 319 | 86160 |
1.83 | 83 | 0.07 | N/Ak | N/Ak |
| 9 | 10 | 500 | 90 | 450 | 108400 |
1.72 | 178 | 0.03 | 134 | 30 |
| 10 | 15 | 200 | 84 | 168 | 53040 |
1.60 | 85 | 0.14 | 151 | 84 |
| 11 | 15 | 350 | 91 | 319 | 84150 |
1.82 | 123 | 0.12 | 197 | 57 |
| 12 | 15 | 500 | 91 | 455 | 109500 |
2.55 | 194 | 0.02 | 255 | 78 |
Four series of PHB83-b-PHEMAy were synthesized in THF at 60 °C, by varying the length of the polymethacrylate block with 200, 350, or 500 y repeating units, at different initial PHB-TTC macro-RAFT agent and HEMA concentrations, the latter ranging from 5 to 15wt% (Table 2). The HEMA comonomer conversion, as determined by 1H NMR in DMSO-d6 of the final mixture recovered after 20 h, revealed greater than 77% in all cases. At the lowest comonomer and PHB-TTC macro-RAFT agent composition of 5wt%, the HEMA conversion ranged from 77 to 80% which is the lowest conversion detected for the whole range of composition investigated, as anticipated. On the other hand, at the higher compositions of 7.5, 10 or 15wt%, the HEMA conversion significantly increased (86–95%), returning copolymers with a longer polymethacrylate segment.
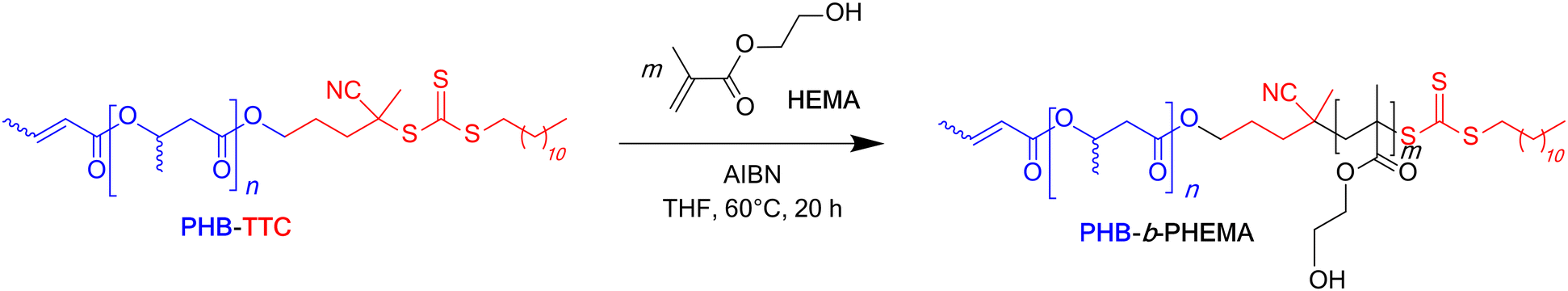
The corresponding SEC chromatograms of the PHB-b-PHEMA samples analyzed in DMF clearly showed the shift of the trace of the PHB83-TTC to lower elution volumes, proportionally to the increase of the DPHEMA ranging from 200 to 500 units, respectively. This demonstrated the successful chain extension from the macro-RAFT agent upon copolymerizing with HEMA (Table 2 and Fig. 5). All the chromatograms were not ideally monomodal, showing small low and high molar mass shoulders, especially the one (green trace) corresponding to DPHEMA of 500 at 15wt% (Table 2, entry 12; Fig. 5). Accordingly, the dispersity values were slightly enlarged. This suggested a non-fully optimized control of the RAFT polymerization, possibly due to a slower initiation as compared to the propagation and/or to some undesirable side reactions (typically termination reactions). This may arise from the fact that the block copolymerization were performed in THF, a solvent from which residual traces of peroxides may to some extent be deleterious to the TTC group.58 The latter above-mentioned shoulder at high molar mass suggested a branched architecture, which most likely arose from residual ethylene glycol dimethacrylate impurity within HEMA.59,60 This monomer contaminant most probably acted as a branching agent and accounted for the high dispersity values observed for the corresponding diblock copolymers (1.60 < Đ < 1.88), except for the composition targeting 500 HEMA units at 15 wt% (Đ = 2.55) (Table 2, entry 12). Besides, the presence of a low molar mass shoulder in the SEC traces could arise from the presence of a few PHB segments void of the RAFT terminal/initiating group in the prepolymer; however, these could not be unambiguously identified in the MALDI-ToF mass spectrum. In any case, the shift of the retention time between the black trace of the TTC end-capped PHB prepolymer and the three other colored traces of the PHBx-b-PHEMAy copolymers clearly suggested that a block copolymerization occurred as mediated by RAFT, thereby supporting that the polymerization was controlled to some extent.
 | ||
| Fig. 5 SEC chromatograms (DMF, 25 °C) of PHB83-TTC macro-RAFT agent (black trace) and of the dispersions made at four different initial concentrations of HEMA i.e. 5 wt%, 7.5 wt%, 10 wt% and 15 wt% for different targeted degrees of polymerization, DP = 200 (red trace), 350 (blue trace) and 500 (green trace). | ||
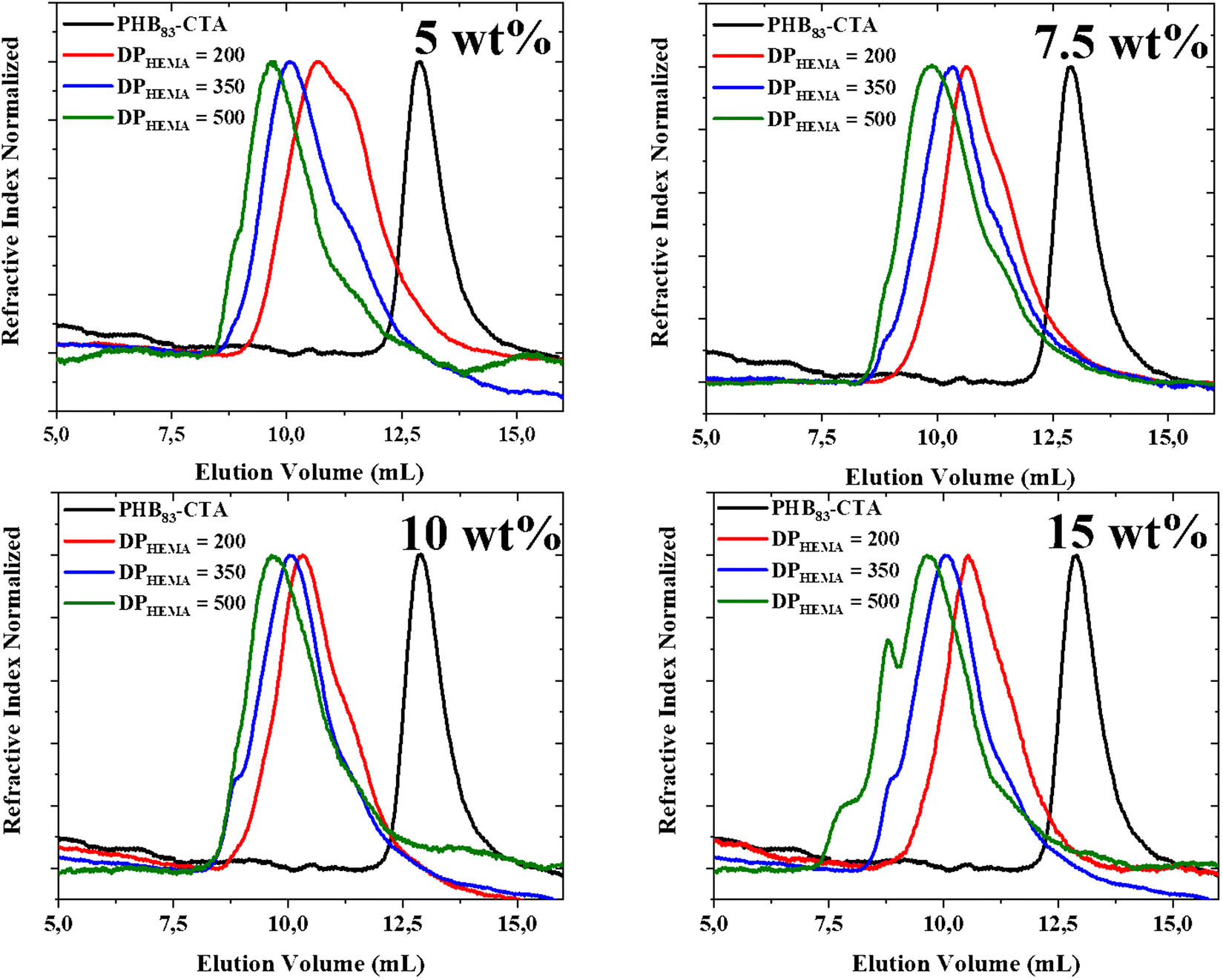
The dispersions of PHB83-b-PHEMAy diblock copolymers in THF were further analyzed using dynamic light scattering (DLS) (Table 2). Narrow monomodal distributions of the scattered intensity as a function of hydrodynamic diameters with PDI < 0.14 were recorded for all dispersions. These are gathered in a pseudo-phase diagram showing the evolution of the hydrodynamic diameter with the initial weight percentage of comonomer and PHB-TTC vs. THF as the x-axis and to the targeted DP of HEMA as the y-axis (Fig. 6). The hydrodynamic diameter DH of the particles was found to increase monotonously, according to the targeted DPHEMA and also to the initial weight percentage of comonomer and PHB-TTC along the pseudo-phase diagram, as expected. The DH values varied from 56 to 194 nm along with a relatively low polydispersity index (0.02 < PDI < 0.14) (Table 2, entries 1–12).61 This indicated that the size of PHB83-b-PHEMAy particles were narrowly dispersed in THF.
 | ||
| Fig. 6 Intensity-averaged size distribution obtained by DLS for each PHB83-b-PHEMAy dispersion, showing the evolution of the hydrodynamic diameters (DH in nm) and PDI, with the increase of the DPHEMA and with the initial weight concentration of the PHB83-TTC macro-RAFT agent added to the monomer weight vs. THF (wt% copolymer concentration) for the PHB83-TTC macro-RAFT agent. DLS measurements were performed in triplicate for each sample. | ||
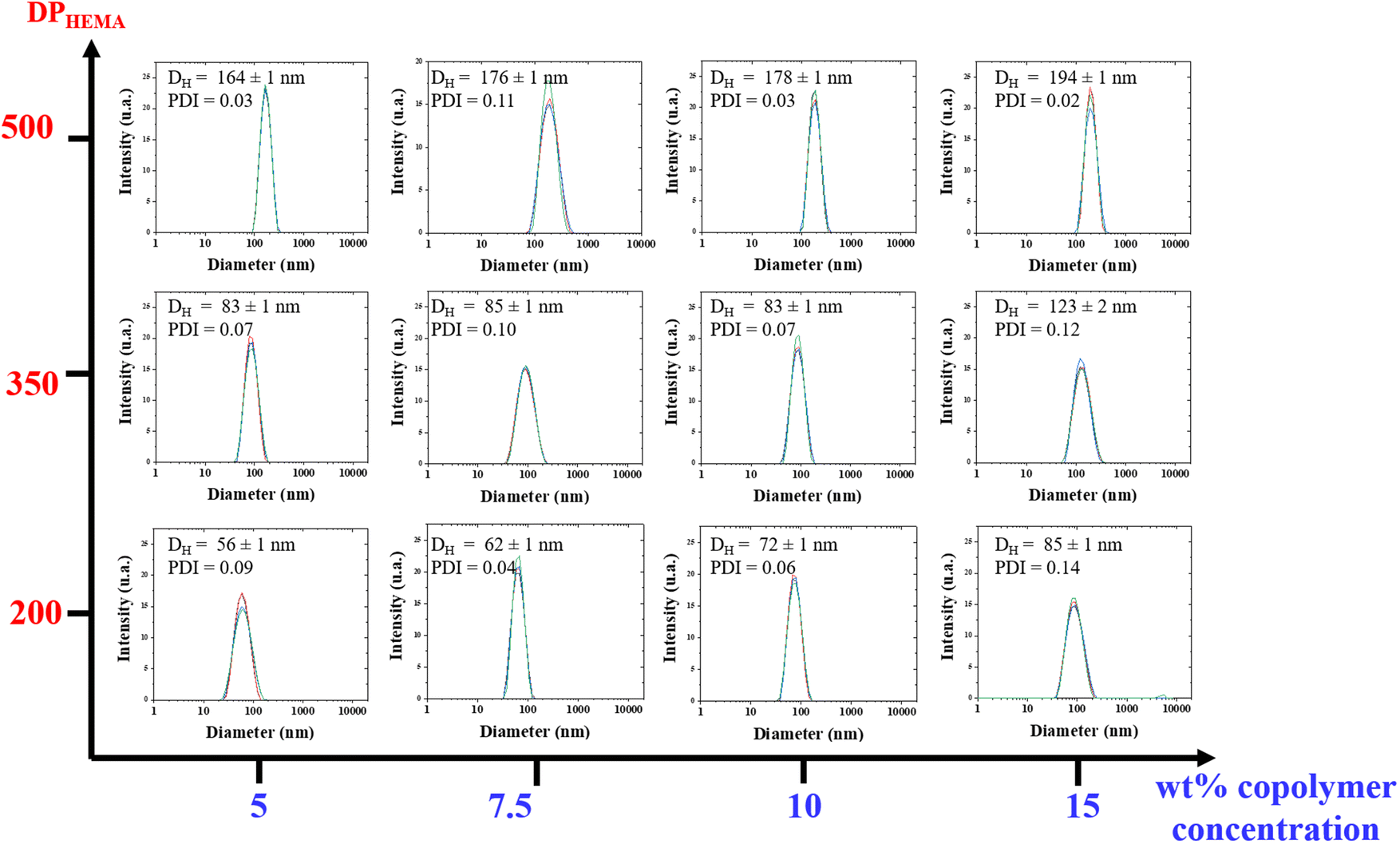
The morphology of the particles generated within the dispersions were investigated by transmission electron microscopy (TEM) analyses. TEM analysis of such type of PHA-based copolymer dispersions revealed difficult due to the unusual solvent used, i.e. THF. Typical micrographs are illustrated in the ESI (Fig. S13–S22†). As a general trend, the apparent hydrodynamic diameter Dapp (apparent diameter ranging from 134 to 316 nm) determined by Image J on two different micrographs, taking at least 50 particles, returned values higher than DDLS ones (56 to 194 nm) (Table 2). Larger and more polydisperse particles were observed by TEM as compared to the corresponding DLS measurements, most likely arising from the sample preparation and from the different physical states between the solution analyzed by DLS and the dried sample captured by TEM. Hence, this difference may result from the adsorption of these particles onto the surface, which may cause a slight spreading and, consequently, an increase in their lateral dimension, as previously observed in other polymer systems.62 Furthermore, some spheres exhibited a vesicular shape, with thin membranes being clearly distinguished (Fig. 7a and Fig. S13, S14, S15, S20†), suggesting the coexistence of vesicles and micelles in the dried samples prepared for TEM. These results further demonstrated that a PISA process was indeed proceeding smoothly during the copolymerization of HEMA from the PHB83-TTC macro-RAFT agent in THF. As stated in the literature, PISA offers a highly effective approach for designing and producing a diverse array of block copolymer nano-objects, including spheres, worms, and vesicles, in different media.18–41
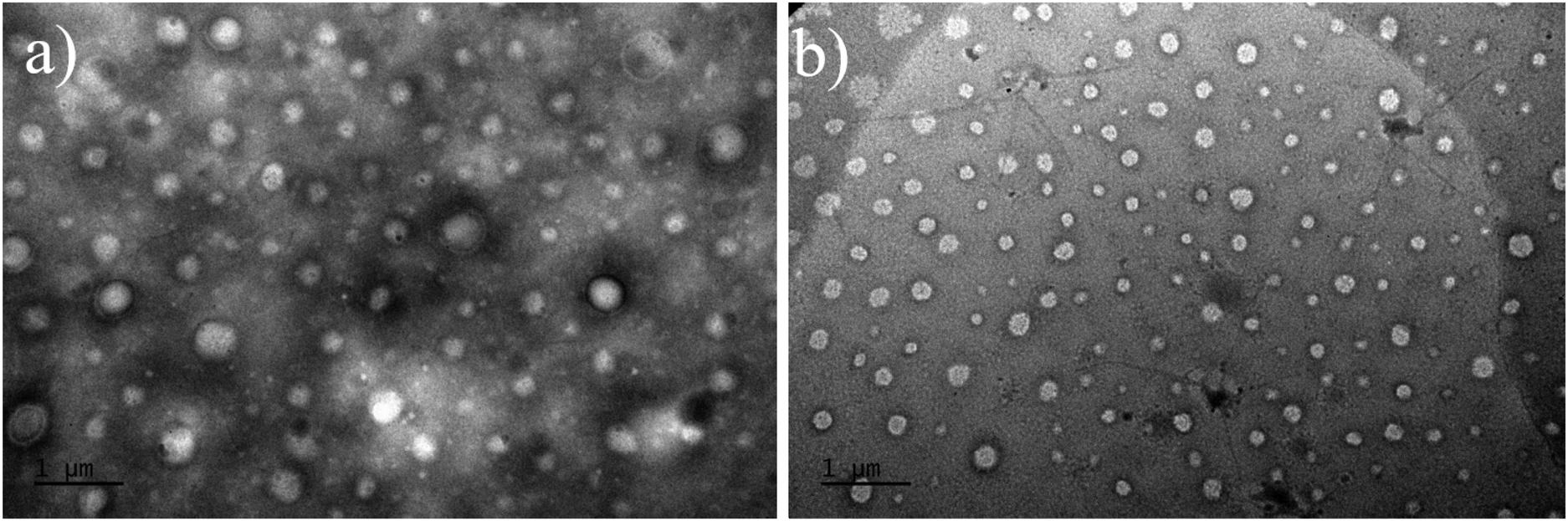 | ||
| Fig. 7 TEM micrographs of PHB83-b-PHEMA500 made at (a) 5 wt% and (b) 7.5 wt% of HEMA comonomer and PHB83-TTC macro-RAFT agent concentration. | ||
SAXS experiments were performed on two samples in order to better understand the morphology of the particles. Fig. 8 shows the SAXS patterns of PHB83-b-PHEMA500 at 5 and 7.5 wt% of HEMA comonomer and macro-RAFT agent concentration. The gradient in the low q region of both patterns is about −2.5, exhibiting the presence of vesicles in the suspension, as already stated in the literature.63 This result is in agreement with the TEM images showing spherical particles with morphologies most-likely corresponding to those of vesicles (Fig. 7). After fitting the data by using a spherical vesicle model, the two local minima of both patterns were investigated.64 The minima observed at 1.3–1.4 × 10−2 Å−1 gives information about the vesicle membrane thickness, and the one around 3 × 10−3 Å−1 highlights the overall vesicle size. It was found that for both compositions, the membrane thickness is about 50 nm, and the overall vesicle diameter around 195–200 nm. These particle sizes are in the same range as the hydrodynamic diameters obtained by DLS (Fig. 6).
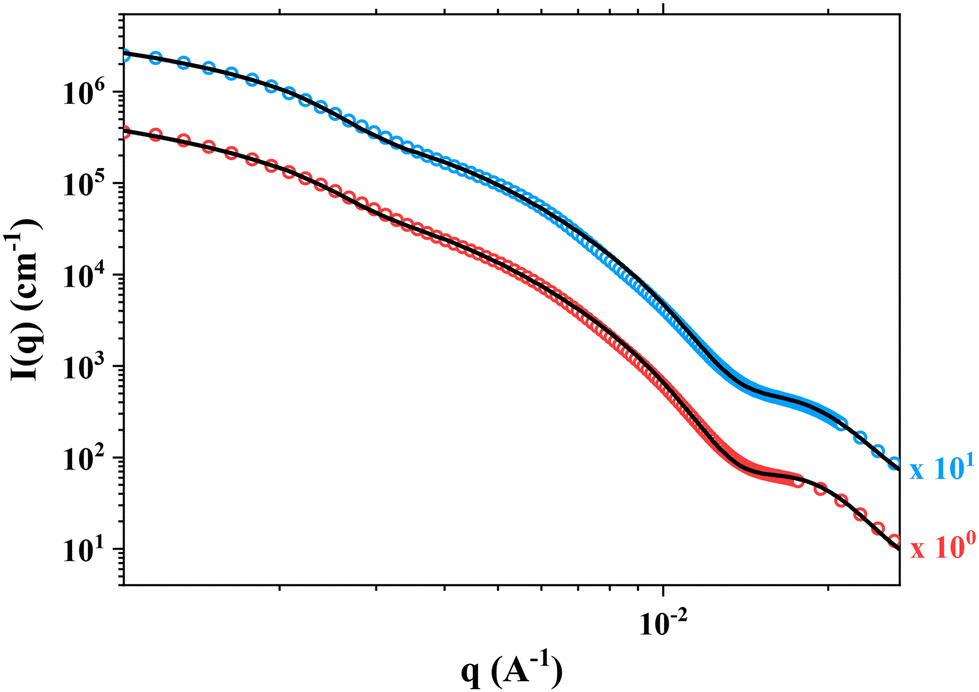 | ||
| Fig. 8 SAXS patterns of PHB83-b-PHEMA500 prepared at 5 wt% (in red) and 7.5 wt% (in blue) of HEMA comonomer and PHB83-TTC macro-RAFT agent concentration. The data fits obtained by using a spherical vesicle model is shown in black solid lines. The I(q) profiles were multiplied by a constant indicated on the Figure, so as to improve its readability. | ||
Conclusion
PHB-TTC macro-RAFT agent samples were successfully synthesized by ROP of rac-β-BL catalyzed by an in situ generated yttrium alkoxide complex in the presence of a TTC-OH initiator. Well-defined α-crotonate (and also α-OH),ω-TTC PHBs (Mn,NMR up to 7500 g mol−1, Đ ca. 1.19) recovered polyesters, as characterized by NMR spectroscopy, MALDI-ToF mass spectrometry and SEC analyses, supported a controlled polymerization. Their subsequent use as macro-RAFT agent in the synthesis of dispersion copolymers via RAFT PISA copolymerization of HEMA in THF, represents the first use of a PHA in a PISA process, to elaborate, in the present study, PHA/polymethacrylate nanoparticles. The resulting PHB-b-PHEMA copolymers were obtained with a noticeable increase of the molar mass from the PHB-TTC precursor. Particle sizes were found to increase with larger initial concentration of comonomer added to the macro-RAFT agent. The same effect was similarly observed with the increase of the length of the second PHEMA block. TEM observations confirmed the existence of particles formed through the PISA process, presenting a spherical shape corresponding to micelles, presumably, and in some cases, a vesicular shape. SAXS measurements performed on PHB83-b-PHEMA500 at 5 wt% and 7.5 wt% of HEMA comonomer and macro-RAFT agent revealed the presence of vesicles in the suspension, in good agreement with TEM observations. This ROP/RAFT/PISA process thus paves the way to our on-going investigations on dispersions formed in water upon copolymerizing from a hydrophilic PHA precursor, ultimately leading to pharmaceutical applications.Author contributions
Julien Rosselgong: investigation, formal analysis, conceptualization, writing – original draft, review and editing. Ali Dhaini: investigation, formal analysis. Manon Rochedy: SAXS analysis, data curation, writing – original draft. Lourdes Mónica Bravo-Anaya: SAXS, TEM and DLS analyses, writing – original draft, review and editing. Jean-François Carpentier: writing – original draft, review and editing, funding acquisition, formal analysis. Sophie M Guillaume: writing – original draft, review and editing, supervision, formal analysis, funding acquisition and conceptualization.Data availability
The raw data required to reproduce these findings are available from the authors. The processed data required to reproduce these findings are available in the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Jérôme Ollivier (ISCR) and Philippe Jehan (ScanMAT, UAR2025, University of Rennes) are thanked for MALDI-ToF MS analyses and fruitful discussions. Agnès Burel (UMS Biosit University of Rennes) is acknowledged for useful discussions regarding TEM images, and Ludivine Rault for TEM analyses (ScanMAT, UAR2025, University of Rennes). Ma. Lizbeth Zepeda García is acknowledged for her help in TEM images analysis. Javier Perez (Synchrotron SOLEIL, F-91192, Gif-sur-Yvette) and the BAG 20231446 (SLAMM) are warmly thanked for the opportunity to perform SAXS experiments on the SWING beamline of SOLEIL facilities. The authors acknowledge financial support from GDR2019 CNRS/INRAE “Solliciter LA Matière Molle” (SLAMM) and from the Synchrotron Soleil (beamline SWING). Prof. Steve Armes (University of Sheffield, UK) and Dr Matthew Derry (Aston University, UK) are gratefully acknowledged for fruitful discussions.References
- G. Barouti, C. G. Jaffredo and S. M. Guillaume, Advances in drug delivery systems based on synthetic poly(hydroxybutyrate) (co)polymers, Prog. Polym. Sci., 2017, 73, 1–31 CrossRef.
- J. Mai, K. Kockler, E. Parisi, C. M. Chan, S. Pratt and B. Laycock, Synthesis and physical properties of polyhydroxyalkanoate (PHA)-based block copolymers: a review, Int. J. Biol. Macromol., 2024, 263, 130204 CrossRef CAS PubMed.
- Z. Luo, Y.-L. Wu, Z. Li and X. J. Loh, Recent progress in polyhydroxyalkanoates-based copolymers for biomedical applications, Biotechnol. J., 2019, 14, 1900283 CrossRef CAS PubMed.
- M. Koller, The handbook of polyhydroxyalkanoates, CRC Press, 1st edn, 2020, vol. 1–3 Search PubMed.
- S. Taguchi, T. Iwata, H. Abe and Y. Doi, in Polymer Science: A Comprehensive Reference, ed. K. Matyjaszewski, and M. Mçller, Elsevier, London, 2012, Vol. 9, pp. 157–182 Search PubMed.
- A. H. Westlie, E. C. Quinn, C. R. Parker and E. Y.-X. Chen, Synthetic biodegradable polyhydroxyalkanoates (PHAs): Recent advances and future challenges, Prog. Polym. Sci., 2022, 134, 101608 CrossRef CAS.
- J. K. Muiruri, J. C. C. Yeo, Q. Zhu, E. Ye, X. J. Loh and Z. Li, Poly(hydroxyalkanoates): production, applications and end-of-life strategies–life cycle assessment nexus, ACS Sustainable Chem. Eng., 2022, 10, 3387–3406 CrossRef CAS.
- A. Dhaini, V. Hardouin-Duparc, A. Alaaeddine, J.-F. Carpentier and S. M. Guillaume, Recent advances in polyhydroxyalkanoates degradation and chemical recycling, Prog. Polym. Sci., 2024, 149, 101781 CrossRef CAS.
- G. Barouti, S. S. Liow, Q. Dou, H. Ye, C. Orione, S. M. Guillaume and X. J. Loh, New linear and star–shaped thermogelling poly([R]–3−hydroxybutyrate) copolymers, Chem. – Eur. J., 2016, 22, 10501–10512 CrossRef CAS PubMed.
- X. J. Loh, Z.-X. Zhang, Y.-L. Wu, T. S. Lee and J. Li, Synthesis of novel biodegradable Thermoresponsive triblock copolymers based on poly[(R)-3-hydroxybutyrate] and poly(N-isopropylacrylamide) and their formation of thermoresponsive micelles, Macromolecules, 2009, 42, 194–202 CrossRef CAS.
- X. J. Loh, W. C. D. Cheong, J. Li and Y. Ito, Novel poly(N-isopropylacrylamide)-poly[(R)-3-hydroxybutyrate]-poly(N-isopropylacrylamide) triblock copolymer surface as a culture substrate for human mesenchymal stem cells, Soft Matter, 2009, 5, 2937–2946 RSC.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Reversible-deactivation radical polymerization (Controlled/living radical polymerization): From discovery to materials design and applications, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- M. K. Georges, R. P. N. Veregin, P. M. Kazmaier and G. K. Hamer, Narrow molecular weight resins by a free-radical polymerization process, Macromolecules, 1993, 26, 2987–2988 CrossRef CAS.
- J.-S. Wang and K. Matyjaszewski, Controlled/” living” radical polymerization. Halogen atom transfer radical polymerization promoted by a Cu(I)/Cu(II) redox process, Macromolecules, 1995, 28, 7901–7910 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Living free-radical polymerization by reversible addition-fragmentation chain transfer: the RAFT process, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- G. Barouti, S. S. Liow, Q. Dou, H. Ye, C. Orione, S. M. Guillaume and X. J. Loh, New linear and star–shaped thermogelling poly([R]–3−hydroxybutyrate) copolymers, Chem. – Eur. J., 2016, 22, 10501–10512 CrossRef CAS PubMed.
- B. Hazer, M. Eren, Y. Senemoğlu, E. Renard and V. Langlois, Novel poly(3-hydroxy butyrate) macro RAFT agent. Synthesis and characterization of thermoresponsive block copolymers, J. Polym. Res., 2020, 27, 1–12 CrossRef.
- N. J. Penfold, J. Yeow, C. Boyer and S. P. Armes, Emerging trends in polymerization-induced self-assembly, ACS Macro Lett., 2019, 8, 1029–1054 CrossRef CAS PubMed.
- N. J. Warren and S. P. Armes, Polymerization-induced self-assembly of block copolymer nano-objects via RAFT aqueous dispersion polymerization, J. Am. Chem. Soc., 2014, 136, 10174–10185 CrossRef CAS PubMed.
- S. L. Canning, G. N. Smith and S. P. Armes, A critical appraisal of RAFT-mediated polymerization-induced self-assembly, Macromolecules, 2016, 49, 1985–2001 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Polymerization-induced self-assembly of block copolymer nanoparticles via RAFT non-aqueous dispersion polymerization, Prog. Polym. Sci., 2016, 52, 1–18 CrossRef CAS.
- J. Rieger, Guidelines for the synthesis of block copolymer particles of various morphologies by RAFT dispersion polymerization, Macromol. Rapid Commun., 2015, 36, 1458–1471 CrossRef CAS PubMed.
- B. Charleux, G. Delaittre, J. Rieger and F. D'Agosto, Polymerization-induced self-assembly: from soluble macromolecules to block copolymer nano-objects in one step, Macromolecules, 2012, 45, 6753–6765 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, Mechanistic insights for block copolymer morphologies: how do worms form vesicles?, J. Am. Chem. Soc., 2011, 133, 16581–16587 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. Douglas and S. P. Armes, Sterilizable gels from thermoresponsive block copolymer worms, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS PubMed.
- S. J. Byard, M. Williams, B. E. McKenzie, A. Blanazs and S. P. Armes, Preparation and cross-linking of all-acrylamide diblock copolymer nano-objects via polymerization-induced self-assembly in aqueous solution, Macromolecules, 2017, 50, 1482–1493 CrossRef CAS PubMed.
- Y. Jiang, N. Xu, J. Han, Q. Yu, L. Guo, P. Gao, X. Lu and Y. Cai, The direct synthesis of interface-decorated reactive block copolymer nanoparticles via polymerisation-induced self-assembly, Polym. Chem., 2015, 6, 4955–4965 RSC.
- W. Shen, Y. Chang, G. Liu, H. Wang, A. Cao and Z. An, Biocompatible, antifouling, and thermosensitive core-shell nanogels synthesized by RAFT aqueous dispersion polymerization, Macromolecules, 2011, 44, 2524–2530 CrossRef CAS.
- C. Grazon, J. N. Rieger, N. Sanson and B. Charleux, Study of poly(N,N-diethylacrylamide) nanogel formation by aqueous dispersion polymerization of N,N-diethylacrylamide in the presence of poly(ethylene oxide)-b-poly(N,N-dimethylacrylamide) amphiphilic macromolecular RAFT agents, Soft Matter, 2011, 7, 3482–3490 RSC.
- D. Ikkene, J.-L. Six and K. Ferji, Progress in aqueous dispersion RAFT PISA, Eur. Polym. J., 2023, 188, 111848–111867 CrossRef CAS.
- S. W. Thompson, T. R. Guimarães and P. B. Zetterlund, Multiblock copolymer synthesis via aqueous RAFT polymerization-induced self-assembly (PISA), Polym. Chem., 2022, 13, 5048–5057 RSC.
- M. Semsarilar, N. J. W. Penfold, E. R. Jones and S. P. Armes, Semi-crystalline diblock copolymer nano-objects prepared via RAFT alcoholic dispersion polymerization of stearyl methacrylate, Polym. Chem., 2015, 6, 1751–1757 RSC.
- Y. Pei, N. C. Dharsana and A. B. Lowe, Ethanolic RAFT dispersion polymerization of 2-(Naphthalen-2-yloxy)ethyl methacrylate and 2-phenoxyethyl methacrylate with poly[2-(dimethylamino)ethyl methacrylate] macro-chain transfer agents, Aust. J. Chem., 2015, 68, 939–945 CrossRef CAS.
- Y. Pei, N. C. Dharsana, J. A. van Hensbergen, R. P. Burford, P. J. Roth and A. B. Lowe, RAFT dispersion polymerization of 3-phenylpropyl methacrylate with poly[2-(dimethylamino)ethyl methacrylate] macro-CTAs in ethanol and associated thermoreversible polymorphism, Soft Matter, 2014, 10, 5787–5796 RSC.
- E. R. Jones, M. Semsarilar, P. Wyman, M. Boerakker and S. P. Armes, Addition of water to an alcoholic RAFT PISA formulation leads to faster kinetics but limits the evolution of copolymer morphology, Polym. Chem., 2016, 7, 851–859 RSC.
- G. Desnos, A. Rubio, C. Gomri, M. Gravelle, V. Ladmiral and M. Semsarilar, Semi-fluorinated di and triblock copolymer nano-objects prepared via RAFT alcoholic dispersion polymerization (PISA), Polymers, 2021, 13, 2502–2515 CrossRef CAS PubMed.
- M. J. Derry, L. A. Fielding and S. P. Armes, Industrially-relevant polymerization-induced self-assembly formulations in non-polar solvents: RAFT dispersion polymerization of benzyl methacrylate, Polym. Chem., 2015, 6, 3054–3062 RSC.
- L. A. Fielding, M. J. Derry, V. Ladmiral, J. Rosselgong, A. M. Rodrigues, L. P. D. Ratcliffe, S. Sugihara and S. P. Armes, RAFT dispersion polymerization in non-polar solvents: facile production of block copolymer spheres, worms and vesicles in n-alkanes, Chem. Sci., 2013, 4, 2081–2087 RSC.
- L. Houillot, C. Bui, M. Save, B. Charleux, C. Farcet, C. Moire, J. A. Raust and I. Rodriguez, Synthesis of well-defined polyacrylate particle dispersions in organic medium using simultaneous RAFT polymerization and self-assembly of block copolymers. A strong influence of the selected thiocarbonylthio chain transfer agent, Macromolecules, 2007, 40, 6500–6509 CrossRef CAS.
- Y. Pei, L. Thurairajah, O. R. Sugita and A. B. Lowe, RAFT dispersion polymerization in nonpolar media: polymerization of 3-phenylpropyl methacrylate in n-tetradecane with poly(stearyl methacrylate) homopolymers as macro chain transfer agents, Macromolecules, 2015, 48, 236–244 CrossRef CAS.
- C. György and S. P. Armes, Recent advances in polymerization-induced self-assembly (PISA) syntheses in non-polar media, Angew. Chem., Int. Ed., 2023, 62, e202308372 CrossRef PubMed.
- C. Grazon, P. Salas-Ambrosio, E. Ibarboure, A. Buol, E. Garanger, M. W. Grinstaff, S. Lecommandoux and C. Bonduelle, Aqueous ring–opening polymerization–induced self–assembly (ROPISA) of N–carboxyanhydrides, Angew. Chem., Int. Ed., 2020, 59, 622–626 CrossRef CAS PubMed.
- C. Grazon, P. Slalas-Ambrosio, S. Antoine, E. Ibarboure, O. Sandre, A. J. Clulow, B. J. Boyd, M. W. Grinstaff, S. Lecommandoux and C. Bonduelle, Aqueous ROPISA of α-amino acid N-carboxyanhydrides: polypeptide block secondary structure controls nanoparticle shape anisotropy, Polym. Chem., 2021, 12, 6242–6251 RSC.
- A. H. Morrell, N. J. Warren and P. D. Thornton, The production of polysarcosine-containing nanoparticles by ring-opening polymerization-induced self-assembly, Macromol. Rapid Commun., 2024, 45, 2400103 CrossRef CAS PubMed.
- E. Tinajero-Díaz, N. Judge, B. Li, T. Leigh, R. D. Murphy, P. D. Topham, M. J. Derry and A. Heise, Poly(l-proline)-stabilized polypeptide nanostructures via ring-opening polymerization-induced self-assembly (ROPISA), ACS Macro Lett., 2024, 13, 1031–1036 CrossRef PubMed.
- M. A. H. Farmer, O. M. Musa and S. P. Armes, Efficient synthesis of hydrolytically degradable block copolymer nanoparticles via reverse sequence polymerization–induced self–assembly in aqueous media, Angew. Chem., Int. Ed., 2023, 62, e20230952 CrossRef PubMed.
- M. A. H. Farmer, O. M. Musa and S. P. Armes, Combining crystallization-driven self-assembly with reverse sequence polymerization-induced self-assembly enables the efficient synthesis of hydrolytically degradable anisotropic block copolymer nano-objects directly in concentrated aqueous media, J. Am. Chem. Soc., 2024, 146, 16926–16934 CrossRef CAS PubMed.
- P. Chohan, C. György, O. O. Mykhaylyk, G. M. Prentice, S. V. Filip, M. J. Payne, G. Manna and S. P. Armes, RAFT Dispersion Polymerization of 2-Hydroxyethyl Methacrylate in Non-polar Media, Macromolecules, 2024, 57, 11738–11752 CrossRef CAS PubMed.
- H. Li, R. M. Shakaroun, S. M. Guillaume and J.-F. Carpentier, Recent advances in metal-mediated stereoselective ring-opening polymerization of functional cyclic esters towards well-defined poly(hydroxy acid)s: from stereoselectivity to sequence-control, Chem. – Eur. J., 2020, 26, 128–138 CrossRef CAS PubMed.
- A. Dhaini, R. M. Shakaroun, A. Alaaeddine, J.-F. Carpentier and S. M. Guillaume, Metal-Catalyzed Stereoselective Ring-Opening Polymerization of Functional β-Lactones: Methylene-Alkoxy Fluorinated Polyhydroxyalkanoates Unveil the Role of Non-Covalent Interactions, Polym. Chem., 2024, 15, 999–1014 RSC.
- H. Kakwere and S. Perrier, Design of complex polymeric architectures and nanostructured materials/hybrids by living radical polymerization of hydroxylated monomers, Polym. Chem., 2011, 2, 270–288 RSC.
- L. Xu, F. Mallamace, Z. Yan, F. W. Starr, S. V. Buldyrev and H. Eugene Stanley, Appearance of a fractional Stokes–Einstein relation in water and a structural interpretation of its onset, Nat. Phys., 2009, 5, 565–569 Search PubMed.
- C. G. Jaffredo, J.-F. Carpentier and S. M. Guillaume, Controlled ROP of β-butyrolactone simply mediated by amidine, guanidine, and phosphazene organocatalysts, Macromol. Rapid Commun., 2012, 33, 1938–1944 CrossRef CAS PubMed.
- J.-F. Carpentier, Rare-earth complexes supported by tripodal tetradentate bis (phenolate) ligands: a privileged class of catalysts for ring-opening polymerization of cyclic esters, Organometallics, 2015, 34, 4175–4189 CrossRef CAS.
- E. Themistou, G. Battaglia and S. P. Armes, Facile synthesis of thiol-functionalized amphiphilic polylactide–methacrylic diblock copolymers, Polym. Chem., 2014, 5, 1405–1417 RSC.
- K. Skrabania, A. Miasnikova, A. M. Bivigou-Koumba, D. Zehm and A. Laschewsky, Examining the UV-vis absorption of RAFT chain transfer agents and their use for polymer analysis, Polym. Chem., 2011, 2, 2074–2083 RSC.
- J. V. M. Weaver, I. Bannister, K. L. Robinson, X. Bories-Azeau, S. P. Armes, M. Smallridge and P. McKenna, Stimulus-responsive water-soluble polymers based on 2-hydroxyethyl methacrylate, Macromolecules, 2004, 37, 2395–2403 CrossRef CAS.
- T. Gruendling, R. Pickford, M. Guilhaus and C. Barner-Kowollik, Degradation of RAFT polymers in a cyclic ether studied via high resolution ESI-MS: Implications for synthesis, storage, and end-group modification, J. Polym. Sci., Part A:Polym. Chem., 2008, 46, 7447–7461 CrossRef CAS.
- K. Liang and R. A. Hutchinson, Solvent effects in semibatch free radical copolymerization of 2-hydroxyethyl methacrylate and styrene at high temperatures, Macromol. Symp., 2013, 325–326, 203–212 CrossRef CAS.
- K. Liang, T. R. Rooney and R. A. Hutchinson, Solvent effects on kinetics of 2-hydroxyethyl methacrylate semibatch radical copolymerization, Ind. Eng. Chem. Res., 2014, 53, 7296–7304 CrossRef CAS.
- S. Bhattacharjee, DLS and zeta potential – What they are and what they are not?, J. Controlled Release, 2016, 235, 337–351 CrossRef CAS PubMed.
- P. A. Montaño-González, L. M. Bravo-Lozano, S. Chevance, F. Dole, J. Rosselgong, P. Loyer, S. Tranchimand, J.-P. Chapel, F. Gauffre, C. Schatz and L. M. Bravo-Anaya, Interactions between PEI and biological polyanions and the ability of glycosaminoglycans in destabilizing PEI/peGFP-C3 polyplexes for genetic material release, Int. J. Biol. Macromol., 2025, 301, 14035 CrossRef PubMed.
- E. E. Brotherton, F. L. Hatton, A. A. Cockram, M. J. Derry, A. Czajka, E. J. Cornel, P. D. Topham, O. O. Mykhaylyk and S. P. Armes, In situ SAXS studies during RAFT aqueous emulsion polymerization, J. Am. Chem. Soc., 2019, 141, 13664–13675 CrossRef CAS PubMed.
- A. Guinier, G. Fournet and K. L. Yudowitch, Small-Angle Scattering of X-Rays, 1955 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01447b |
| This journal is © The Royal Society of Chemistry 2025 |