
Mixed-mode interpenetrating polymer networks from polymerizable eutectics†
Alexandra L.
Mutch
 * and
Stuart C.
Thickett
*
* and
Stuart C.
Thickett
*
School of Natural Sciences – Chemistry, University of Tasmania, Hobart 7001, Tasmania, Australia. E-mail: Alexandra.mutch@utas.edu.au; stuart.thickett@utas.edu.au
First published on 12th February 2025
Abstract
The preparation of interpenetrating polymer networks (IPNs) and semi-interpenetrating polymer networks (semi-IPNs) is reported via a solvent-free approach using a binary polymerizable eutectic. N-Isopropylacrylamide (NIPAM) and ε-caprolactone (CL) were mixed in various mole ratios to prepare viscous polymerizable liquids that were stable at room temperature, based on solid–liquid equilibrium phase diagrams from differential scanning calorimetry (DSC) data. The strong degree of association between NIPAM and CL within these mixtures was confirmed via 1D and 2D NMR spectroscopy. Using an appropriate UV photoinitiator and ring opening polymerization catalyst, the orthogonal polymerization (either in a sequential or simultaneous fashion) of each component within the eutectic was achieved, with enhanced reaction kinetics for the ring opening polymerization compared to a traditional solvent (DMSO). Through the incorporation of diacrylate and bis(carbonate) crosslinkers into the resin mixture, IPNs and semi-IPNs were realised in a one-pot two-step approach from polymerizable eutectics for the first time. These networks possessed thermoresponsive swelling behaviour in water, and retained their structural integrity in good solvents for both phases. This binary eutectic was also shown to be suitable as a resin for stereolithography 3D printing on a benchtop printer through the inclusion of a RAFT agent, achieving semi-IPN printed objects in a two-step approach.
Introduction
Synthetic methods in polymer science to achieve orthogonal polymerizations within a single system have expanded significantly in recent years.1,2 Orthogonal polymerizations are based on chemical selectivity of each polymerization process, where the reactants (e.g. initiator, catalyst, monomer, amongst others) for each step can be controlled in an independent fashion – in any order, or simultaneously, without impacting on the other chemical step(s). Ideally, orthogonal polymerizations can be performed in one pot, with no intermediates or purification required. Several recent reviews exist on the topic,2–4 covering advances including wavelength-orthogonal photopolymerizations,5 “click” chemistry for post-polymerization modification6 and gel network synthesis. One of the drivers for orthogonal polymerizations is to achieve targeted material properties in a single system not achievable with one class of polymerization; for example “dual cure” or multi-material 3D printing resins that enable selective spatial polymerization of materials with significantly different mechanical behaviour (e.g. acrylates and epoxy resins).7,8One of the most popular combinations pursued in the design of orthogonal polymerization systems is the combination of radical polymerization (either free-radical or reversible-deactivation radical polymerization) and ring opening polymerization (ROP), to realise a variety of polymer architectures including block9–16 and graft17–19 copolymers. In the case of block copolymers, orthogonal polymerization of a vinyl monomer and a lactone is most typically achieved using a bi-functional initiator or catalyst. Some specific examples have been demonstrated, such as using the nitroxide-mediated polymerization of styrene and ROP of ε-caprolactone (CL),10 atom transfer radical polymerization (ATRP) of various acrylates and methacrylates combined with the ROP of CL and lactide,11,16 and the reversible addition–fragmentation chain transfer (RAFT) polymerization of N-isopropylacrylamide (NIPAM) in combination with the ROP of lactide.12 These processes were typically performed at elevated temperatures, under oxygen-free conditions, to generate the targeted block or graft copolymers.
More recently, there has been a shift towards enabling polymerization processes that yield complex morphologies to proceed under relatively benign reaction conditions, including in the presence of air. Yu et al.13 demonstrated the RAFT block copolymer synthesis of CL and N-vinylcaprolactam (as well as N-vinylcaprolactam/N-vinylpyrrolidone copolymers) at 30 °C in air, through the strategic choice of initiator and catalyst for both the radical and ring opening polymerization steps. Villarroya et al.14 performed simultaneous ATRP and enzymatically mediated ROP of methyl methacrylate (MMA)/2-hydroxyethyl methacrylate with CL to generate block-graft copolymers, this time in supercritical carbon dioxide under relatively mild reaction temperatures (35 °C). The advent of photoinduced electron/energy transfer RAFT (PET-RAFT) polymerization, as developed by Boyer's group,20–22 has further continued this trend by utilising the energy from visible or UV light to facilitate radical polymerization in a controlled fashion. Their group has demonstrated orthogonal block copolymerization of CL with methyl acrylate under ambient conditions, either in a sequential or simultaneous fashion, through the use of blue light and an iridium(III) photocatalyst (for PET-RAFT) and diphenyl phosphate (DPP) as ROP catalyst.23 In a similar vein, visible-light driven photoacid generation to catalyse ROP of CL in an orthogonal manner to PET-RAFT polymerization was reported by the same authors.24
The above-mentioned examples of preparing complex polymer architectures are all based on polymerization methods in an appropriate solvent (typically an organic solvent) for both blocks or resulting polymeric phases. However as noted by Reese et al.,2 there is a desire in many materials applications to perform polymerizations in neat or solventless conditions – for example to reduce harmful byproducts and chemical waste, or enabling the transformation of a liquid monomeric resin into a solid polymer of particular shape, such as in the case of 3D printing technologies. With respect to replacing traditional and potentially harmful solvents, deep eutectic solvents (DESs)25–27 represent an exciting class of mixtures that have shown potential to act as powerful solvents in chemical synthesis, as well as separation science,28,29 energy storage,30,31 and biotechnology,32 amongst others. DESs are most typically binary mixtures of hydrogen bond donating and accepting compounds, forming a mixture with significantly lower melting point than either starting material.33 The interpretation of what is a DES remains an important debate, with a mixture where favourable interactions between components result in enthalpic-driven negative deviations from ideal mixing a preferred general definition.34 With advantageous physical and chemical properties including low volatility, tuneable chemical composition and viscosity, low flammability and potential to be recycled and recovered, DESs have been used extensively to prepare materials such as polymeric gels (“eutectogels”),35 monoliths36,37 and molecularly imprinted polymers.38–43 When one or more component of the DES is polymerizable, which we term “polymerizable eutectics” (PEs),44,45 the solvent-free polymerization of these mixtures is readily achieved. This has been demonstrated in the context of photocured eutectogels,46–48 frontal polymerization,49–52 and various types of 3D printing technologies.53–56 Our group has also recently shown the synthesis of block copolymers prepared directly from a PE based on NIPAM and CL, in essence a solvent-free block copolymerization using a bifunctional RAFT agent.57
Recent advances in the design of PEs based on lactones for ROP,58–60 as well as PEs consisting of monomers polymerizable via different mechanisms, inspired us to design and prepare PE-based interpenetrated polymer networks through a combination of radical and ring opening polymerizations. Interpenetrating polymer networks (IPNs) are systems that consist of two crosslinked polymer networks which are physically entangled but not chemically connected to each other;61 the mechanical properties and swelling behaviour of IPNs is typically highly distinct in comparison to single networks.62 IPNs (or semi-IPNs, whereby one cross-linked network is physically entangled with a linear polymer) are most typically prepared by a (i) sequential method where a single network is swollen with a second monomer and cross-linker, then polymerized, or (ii) a simultaneous method, requiring orthogonal polymerization chemistry. Various IPNs or semi-IPNs incorporating poly(ε-caprolactone) (PCL) have been reported,63–66 however these are almost always prepared sequentially, and from either linear or multi-arm PCL architectures. Herein, we demonstrate the first synthesis of IPNs and semi-IPNs based on PEs composed of a vinyl monomer (NIPAM) and a lactone (CL) to facilitate orthogonal polymerization. PNIPAM is a well-known thermoresponsive polymer,67 enabling the design of stimuli-responsive polymer networks, whilst polyesters such as PCL are readily amenable to chemical degradation (e.g. via hydrolysis) and can be cross-linked through the inclusion of appropriate bis(carbonate) or bis(lactone) cross-linkers.68 Through cross-linking one or both phases, we demonstrate the solvent-free, one-pot synthesis of network polymers with unique polymer topologies and swelling capacity. These PEs are further demonstrated in the context of stereolithography 3D printing on a commercially available benchtop printer.
Experimental
Materials
N-Isopropylacrylamide (NIPAM, 97%), ε-caprolactone (CL, 97%), poly(ethylene glycol) diacrylate (PEGDA, average Mn 700), tin(II) 2-ethyl hexanoate (TEHA), methanesulfonic acid (MSA, ≥99.0%), diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (TPO, 97%), di(trimethylolpropane) (97%), ethyl chloroformate (97%) were purchased from Sigma-Aldrich. δ-Valerolactone (VL, 98%) was purchased from Combi-Blocks. 2,2′-[Carbonothioylbis(thio)]bis[2-methylpropanoic acid] (bisPAT) RAFT agent was purchased from Boron Molecular. Zinc acetate (Zn(OAc)2, ≥99.0%) was purchased from Ajax Chemicals. Benzyl alcohol (BnOH, 99.6%), hexanes (≥95%) and dimethyl sulfoxide (DMSO, ≥99.0%) were purchased from Chem Supply. Tetrahydrofuran (THF, ≥99.9%) and dimethylformamide (DMF), was purchased from RCI Labscan. Deuterated chloroform (CDCl3, 99.8%) was purchased from Cambridge Isotope Laboratories. A bis(carbonate) crosslinker (2,2′-bis(trimethylene carbonate-5-yl)-butylether) was prepared following the protocol reported by Fortman et al.69 A 1H NMR spectrum of the purified crosslinker is shown in Fig. S1.†Preparation of polymerizable eutectics
Polymerizable eutectics were prepared by weighing the appropriate mass of monomers (NIPAM and CL or VL) into a closed screw-cap vial to achieve a total mass of approximately 0.6 g, followed by stirring at 60 °C for at least one hour. The mixtures were then cooled to room temperature and subsequently characterized by NMR, DSC and TGA, or used for polymerization experiments. The following lactone mole fractions were used to prepare PEs; 0.25, 0.33, 0.50, 0.67, 0.75, 0.85. For polymerization experiments the mole fraction of CL was maintained at 0.67 (NIPAM-CL0.67).Polymerization of NIPAM and CL homopolymers and polymer blends
PE mixtures were polymerized by adding the relevant initiators and catalysts directly to the NIPAM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CL0.67 PE, followed by irradiation and/or thermal polymerization in ambient air in a closed vial. NIPAM polymerization was initiated by irradiation of TPO with a UV lamp (SUN 54 W UV LED lamp, λmax = 370 nm) for up to 5 minutes. A stock solution was prepared of TPO in DMF (0.01 g mL−1) and this solution was pipetted directly into PE mixtures prior to irradiation to achieve an initial ratio of [NIPAM]:[TPO] of 100:0.08. CL polymerization was catalysed by TEHA or MSA, each with BnOH as an initiator. TEHA-catalysed experiments were carried out at 90 °C, and the initial ratio of [CL]:[BnOH]:[TEHA] was 200:5:5, while MSA-catalysed experiments were performed at room temperature (20–23 °C) where the initial ratio of [CL]:[BnOH]:[MSA] was 200:5:10. For sequential polymerization experiments where NIPAM was polymerized first, all reagents were added to the PE prior to UV irradiation except TEHA or MSA. For simultaneous and sequential experiments where CL was polymerized first, all reagents were added at the beginning.
:CL0.67 PE, followed by irradiation and/or thermal polymerization in ambient air in a closed vial. NIPAM polymerization was initiated by irradiation of TPO with a UV lamp (SUN 54 W UV LED lamp, λmax = 370 nm) for up to 5 minutes. A stock solution was prepared of TPO in DMF (0.01 g mL−1) and this solution was pipetted directly into PE mixtures prior to irradiation to achieve an initial ratio of [NIPAM]:[TPO] of 100:0.08. CL polymerization was catalysed by TEHA or MSA, each with BnOH as an initiator. TEHA-catalysed experiments were carried out at 90 °C, and the initial ratio of [CL]:[BnOH]:[TEHA] was 200:5:5, while MSA-catalysed experiments were performed at room temperature (20–23 °C) where the initial ratio of [CL]:[BnOH]:[MSA] was 200:5:10. For sequential polymerization experiments where NIPAM was polymerized first, all reagents were added to the PE prior to UV irradiation except TEHA or MSA. For simultaneous and sequential experiments where CL was polymerized first, all reagents were added at the beginning.
Homopolymer control experiments were carried out with the reagent ratios listed above for NIPAM (either using DMSO as a solvent or in a NIPAM-CL0.67 eutectic mixture), and for CL (neat CL, with DMSO as a solvent, or in a NIPAM-CL0.67 eutectic mixture). For experiments with monomer dissolved in DMSO the mass fraction of the monomer was equivalent to the NIPAM:CL0.67 eutectic mixtures. Linear homopolymers and polymer blends were isolated for analysis by precipitation from THF to hexanes.
Preparation of crosslinked polymer networks
Crosslinked homopolymer networks were prepared in the same manner as for linear polymers detailed above with the addition of PEGDA for NIPAM crosslinking and bis(carbonate) for CL crosslinking. The molar ratio of monomer to relevant crosslinker was maintained at 20:1. For CL crosslinking with TEHA, approximately 0.3 g of reaction mixture was placed in a glass vial (11 mm internal diameter) which was degassed with N2 for 10 minutes prior to heating at 90 °C in an oil bath overnight. For NIPAM crosslinking approximately 0.3 g of reaction mixture was placed in a rectangular mould (flat embedding PTFE mould with metal frame from Ted Pella Inc. Cavity dimensions: 14 × 6.9 × 3 mm) and the sample was irradiated with a UV lamp (370 nm) for 2 minutes on each side open to air. Interpenetrating polymer networks (IPNs) were prepared sequentially by adding all initiators, catalysts and crosslinkers to the NIPAM-CL0.67 mixture in a closed glass vial and then either exposing to UV light in air first (reaction (1), PNIPAMx-PCLx), or degassing and heating first (reaction (2), PCLx-PNIPAMx), followed by the other treatment. Semi-IPN samples were prepared by first polymerising CL with MSA in the PTFE mould open to air, followed by UV irradiation open to air (2 min on each side) to crosslink the NIPAM (PCL-PNIPAMx).
Preparation of PE resins and 3D printing
The rapid UV photocuring of NIPAM was applied to masked stereolithography (MSLA) 3D printing. NIPAM-CL eutectic mixtures were prepared and crosslinkers, initiators and catalysts were added directly to the mixture as described above. For MSLA 3D printing, the loading of TPO and PEGDA were each 2 wt% relative to the mass of NIPAM. A chain transfer agent (bisPAT) was also added to the mixture at 0.5 wt% relative to NIPAM to act as a photoblocker and to control the network morphology during printing. 3D printing was carried out using a benchtop printer (ELEGOO Mars 3 4K LCD printer, 405 nm) with the following print parameters: layer thickness = 50 μm, 5 bottom layers, bottom layer exposure time = 15 s, regular layer exposure time = 10 s. Hollow cube structures were printed (10 × 10 × 10 mm3 external dimensions) for swelling analysis.Mixed-mode PE resins were prepared following the protocol above, with the addition of Zn(OAc)2 in the resin (initial ratio of [CL]:[Zn(OAc)2] = 200:1) to achieve semi-IPNs via 3D printing. 3D printing of hollow cubes was carried out with the following parameters: bottom layer exposure time = 15 s, regular layer exposure time = 12 s, with all other parameters as listed above. After printing the material was placed in a sealed glass vial and heated to 100 °C in air overnight for polymerization of CL resulting in PNIPAMx-PCL semi-IPNs.
Swelling investigation of crosslinked PE networks
To investigate the swelling capacity of PE gel networks and IPNs at room temperature (20–23 °C), gels were dried, and the dry mass recorded (md). The dried gels were then placed in vials containing approximately 15 mL of solvent (deionized water or chloroform) and the solvated mass (ms) was recorded after swelling the gels for at least 24 hours. To investigate the thermoresponsive swelling properties of PE gels containing PNIPAM, the equilibrium swelling was also recorded in water at 45 °C. The swelling ratio of gels was calculated according to the following equation:where ms is the solvated mass of the gel and md is the dry mass.
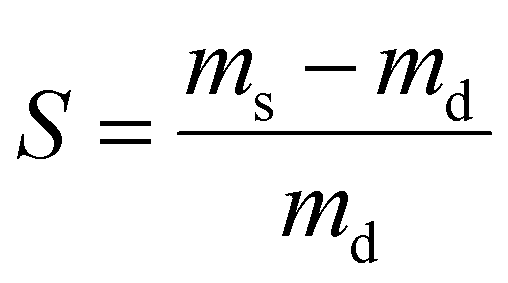
Characterisation
944000 g mol−1.
Results and discussion
Preparation and characterization of polymerizable eutectics
We commenced our investigation with a detailed analysis of preparing polymerizable eutectics from NIPAM-CL and NIPAM-VL mixtures. A range of mixtures were prepared at differing mole ratios of NIPAM:lactone, to construct a binary solid–liquid equilibrium (SLE) phase diagram of melting point(s) as a function of composition. Compositions that were stable liquids at room temperature were targeted for subsequent polymerization. Upon visual inspection of the mixtures upon heating and stirring then cooling to room temperature, stable liquids formed for all compositions xCL > 0.5 and xVL > 0.5 (Fig. 1A). Stable mixtures can also be prepared at low NIPAM/high lactone mole fractions at room temperature without the need for heating, however for consistency we retain a heating and stirring protocol for all mixtures in this work.
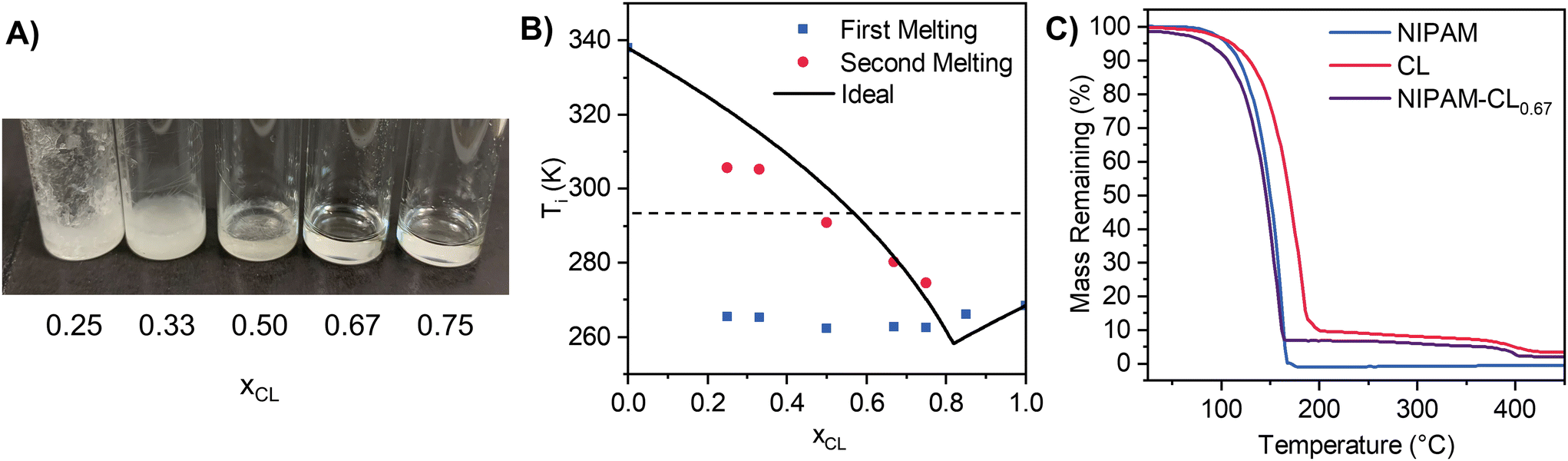 | ||
| Fig. 1 (A) Photo of NIPAM-CL eutectic mixtures with the CL mole fraction listed below each vial. (B) NIPAM-CL phase diagram showing experimental onset melting temperatures as well as ideal melting points calculated using eqn (1). All error bars are smaller than the symbol size (n = 2). The dotted line represents approximate room temperature. (C) TGA thermograms of monomers and NIPAM-CL0.67 eutectic mixture. | ||
When constructing a binary SLE phase diagram, experimental melting points can be compared to the melting point depression predicted by the isobaric mixing of two pure compounds, namely:34,70
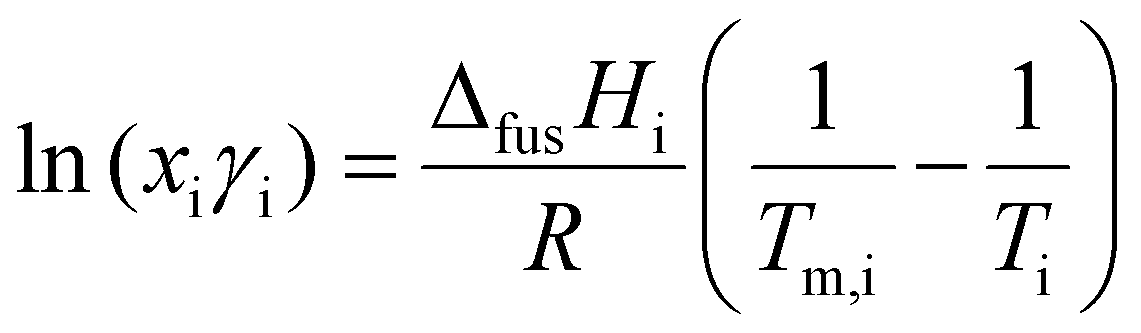 | (1) |
:2 choline chloride:ethylene glycol mixture) exhibiting close to ideal phase behaviour.73 The terms “eutectic solvents”35 or “low transition temperature mixtures”26 are recommended by some researchers, with the most important aspect being that these are functional supramolecular liquids with unique physical and chemical properties.
Differential scanning calorimetry (DSC) was used to quantify enthalpies of fusion of our starting materials, as well as the melting point onset of our mixtures. We use the melt onset from heating loops to construct binary SLE diagrams (Fig. 1B), given that a reasonably significant freezing point hysteresis was observed in the cooling loops regardless of scan rate (see Fig. S2, ESI†), which may be in part due to a supercooling effect under DSC conditions.74,75 For our pure compounds, excellent agreement with literature melting points was found when using the melt onset, justifying our choice. The SLE phase diagram is shown in Fig. 1B along with the predicted liquidus line assuming ideality, which was determined from eqn (1) using experimental data for pure compounds (NIPAM: ΔfusH = 15 526 J mol−1, m.p. = 337.95 K; CL: ΔfusH = 11272 J mol−1, m.p. = 261.15 K). We observe two melting transitions for all compositions where xCL < 0.75, where the first melt (blue squares) is attributed to the solidus line of the phase diagram. The average temperature of the first melt (264 ± 2 K) is close to the predicted ideal eutectic temperature (258.3 K), supporting the notion that these mixtures exhibit near-to-ideal mixing. Similar behaviour is observed for NIPAM-VL mixtures (see Fig. S3 & S4, ESI†). Targeting a polymerizable eutectic that is both a stable room temperature liquid and a relatively high proportion of each monomer for subsequent polymerization, the composition xCL = 0.67 was chosen for further analysis. Thermal stability of this mixture (purple line, Fig. 1C) as determined by TGA was shown to exhibit characteristics of both components; the maximum decomposition temperature was similar to that of NIPAM, with a residual mass at higher temperatures similar to that of CL.
The NIPAM-CL0.67 PE was characterized by 1D and 2D NMR spectroscopy following the same protocol as previous reports.48,57,76 A high degree of association was present in these mixtures as evidenced through 1D and 2D 1H NMR spectroscopic measurements; the 1D spectrum exhibits broader resonances that are shifted downfield relative to the pure starting materials (Fig. 2D). 1H-1H ROESY data demonstrate a number of off-diagonal cross-peaks that are consistent with close association of the two PE components through space (Fig. 2E). There are distinct correlations between the vinyl protons of NIPAM and the β and γ protons in CL and these signals have been identified with black boxes in Fig. 2E. 2D DOSY NMR measurements (Fig. S5, ESI†) indicate a separate diffusion coefficient for each monomer in the PE (approximately 10−10.0 m2 s−1 for NIPAM and 10−9.8 m2 s−1 for CL). We have previously shown that the self-diffusion coefficient for NIPAM in water is ∼10−9.1 m2 s−1,76 demonstrating that the monomers in our PE have significantly reduced mobilities compared to those in a conventional solvent. The viscosity of the NIPAM-CL0.67 PE was measured to be 8.5 cP at 25 °C, similar to previously reported acrylate and acrylamide-based PEs.48,57 PCL as an impurity is sometimes observed in the PE mixtures at low concentrations (5 mol% relative to total CL) prior to the addition of ROP catalysts, which we also see by DOSY NMR (see Fig. S5, ESI†).
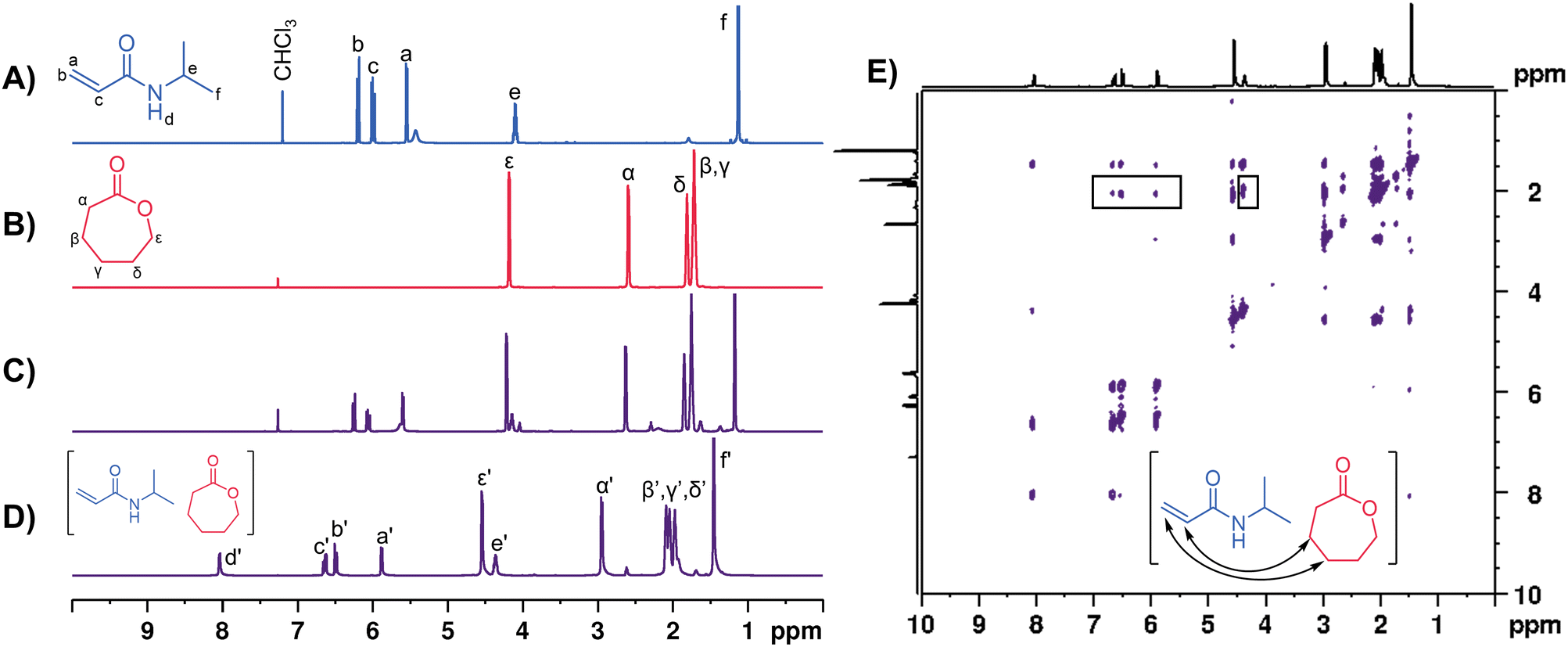 | ||
| Fig. 2 (A–D) 1H NMR spectra of (A) NIPAM, (B) CL, (C), NIPAM-CL0.67 dissolved in CDCl3, (D) neat NIPAM-CL0.67 PE containing a capillary filled with CDCl3. (E) 1H-1H ROESY NMR spectrum of NIPAM-CL0.67 with discernible correlations identified. | ||
Orthogonal polymerization of PEs
Preliminary studies were performed to evaluate the orthogonality of the polymerization of each component within our NIPAM:CL PEs where xCL = 0.67. These reactions were carried out in the absence of cross-linkers, to fully characterise the linear polymers formed. NIPAM was polymerized using TPO as a UV-active Type I photoinitiator upon exposure to 370 nm UV light; CL was polymerized via one of two common ROP catalysts, namely methanesulfonic acid (MSA) at room temperature, or tin(II) 2-ethylhexanoate (TEHA) at 90 °C, each using benzyl alcohol (BnOH) as an initiator. These processes are shown in Scheme 1A.
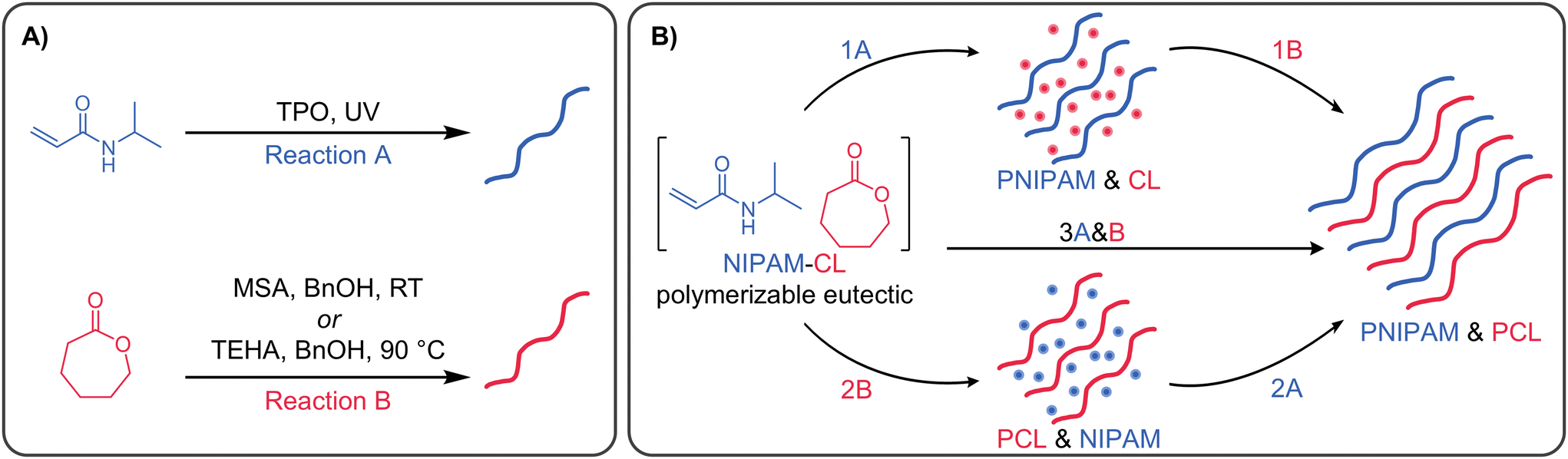 | ||
| Scheme 1 (A) Schemes indicating reaction conditions used to prepare PNIPAM and PCL linear polymers. (B) Scheme showing sequential (reaction (1) & (2)) and simultaneous (reaction (3)) preparation of PNIPAM and PCL polymer blends from a PE mixture. Reactions labelled ‘A’ use photoinitiator and UV irradiation. Reactions labelled ‘B’ use an ROP catalyst & initiator. | ||
We commenced by performing control experiments where either NIPAM or CL was polymerized, either in the as-prepared PE or, as comparison, in the bulk or a traditional solvent at the same mass fraction of monomer. The results are shown in Fig. 3. The photopolymerization of NIPAM in the PE is rapid, achieving a conversion of ∼65% in 2 minutes (red triangles, Fig. 3A). The reaction proceeds at a comparable rate and final conversion to that performed in DMSO (green squares), and the polymerization is not inhibited by the presence of TEHA catalyst for the ROP process. The ROP of CL was examined in neat CL, solution (DMSO) and eutectic polymerization via two catalysts (Fig. 3B and C). As anticipated due to the higher monomer concentration, the neat polymerization of CL was the fastest process. Encouragingly, the polymerization was much faster in the PE compared to in DMSO, for both sets of catalysts studied, with quantitative conversion of monomer to polymer. Our group has observed similar increases in reaction rates previously, which we attribute to both the higher viscosity of PEs compared to traditional solvents, which decreases the rate of chain stoppage events in the reaction medium.57,76 A small increase in polymerization rate at low conversion/early time is observed for the TEHA catalyzed polymerization of CL in the presence of TPO (red triangles and purple diamonds, Fig. 3B), the mechanistic origins of which are unclear at this point.
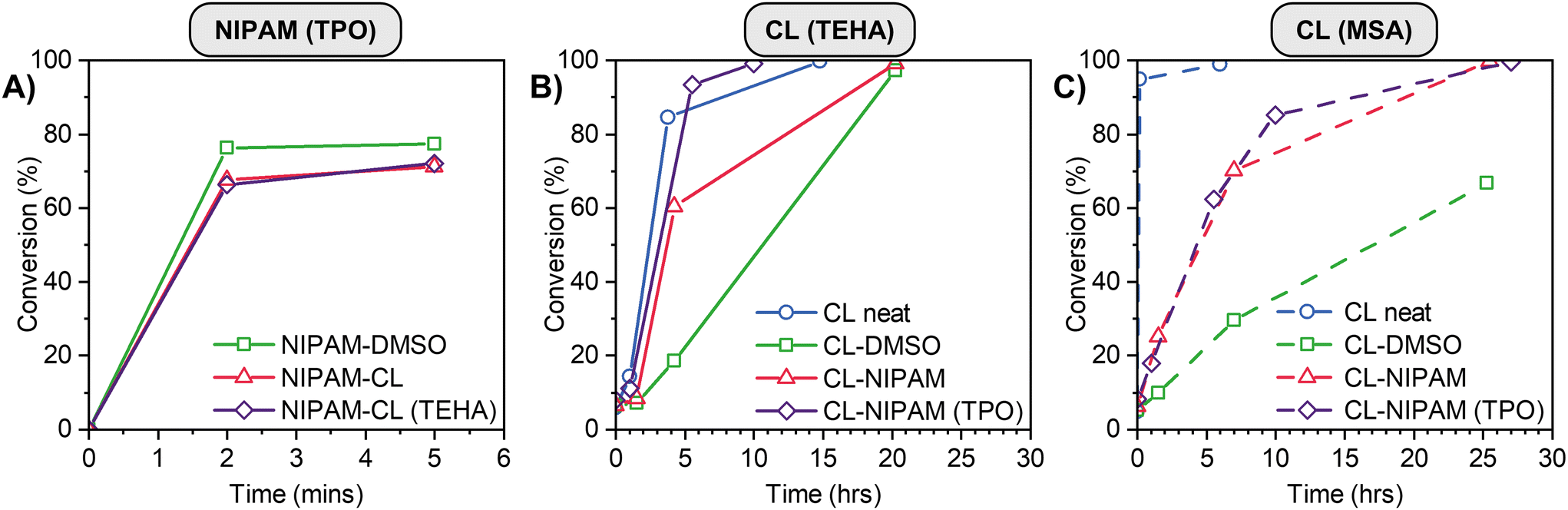 | ||
| Fig. 3 Homopolymer control experiment conversion data for (A) NIPAM initiated by TPO, (B) CL catalysed by TEHA and (C) CL catalysed by MSA. | ||
Various orthogonal polymerizations of our PEs were then performed according to the reaction pathways described in Scheme 1B. For Reaction 1 the ROP catalyst was added to the PE mixture after polymerization of NIPAM, while for reaction (2) and (3) all initiators and catalysts were present in the PE mixture prior to any polymerization. Fig. S6C (ESI†) shows representative 1H NMR spectra with integration regions used to calculate monomer conversion. In order to calculate conversion when both polymers are present, the integration of the PCL peak at 2.30 ppm is subtracted from the integration of the overlapping PNIPAM and PCL signals at 4.00 ppm to determine the PNIPAM signal area. When NIPAM is polymerized first, followed by CL using TEHA as catalyst at 90 °C (Fig. 4A), PNIPAM is formed very quickly in the timescale of the reaction, and to a comparable conversion to the data in Fig. 3A. The presence of linear PNIPAM and the corresponding increase in sample viscosity does not impede the full conversion of CL over 24 h. In the reverse process (CL first followed by NIPAM, Fig. 4B), the polymerization of CL proceeds smoothly to full conversion in 6 h, with zero conversion of NIPAM (even at the elevated reaction temperature of 90 °C). After polymerization of CL, the reaction mixture becomes slightly opaque, however this does not affect the next step; upon irradiation with UV light, the conversion of NIPAM is essentially quantitative over a few minutes. The simultaneous polymerization (Fig. 4C) is ‘the best of both worlds’, with rapid conversion of NIPAM and rapid polymerization of CL, achieving >80% conversion in 5 hours. The use of MSA as catalyst at room temperature (Fig. 4D–F) gives inferior results in some instances – when CL is polymerized last, or sequentially (Fig. 4D and F), the conversion is much lower compared to the use of TEHA. This is attributed to the lower reaction temperature (90 °C vs. room temperature), which greatly influences the reaction viscosity (in particular, when a high mass fraction of PNIPAM is present). We subsequently focus on the TEHA catalysed systems due to the high conversion of both monomers, however room temperature reactions can be advantageous in numerous instances, which we discuss later in the manuscript.
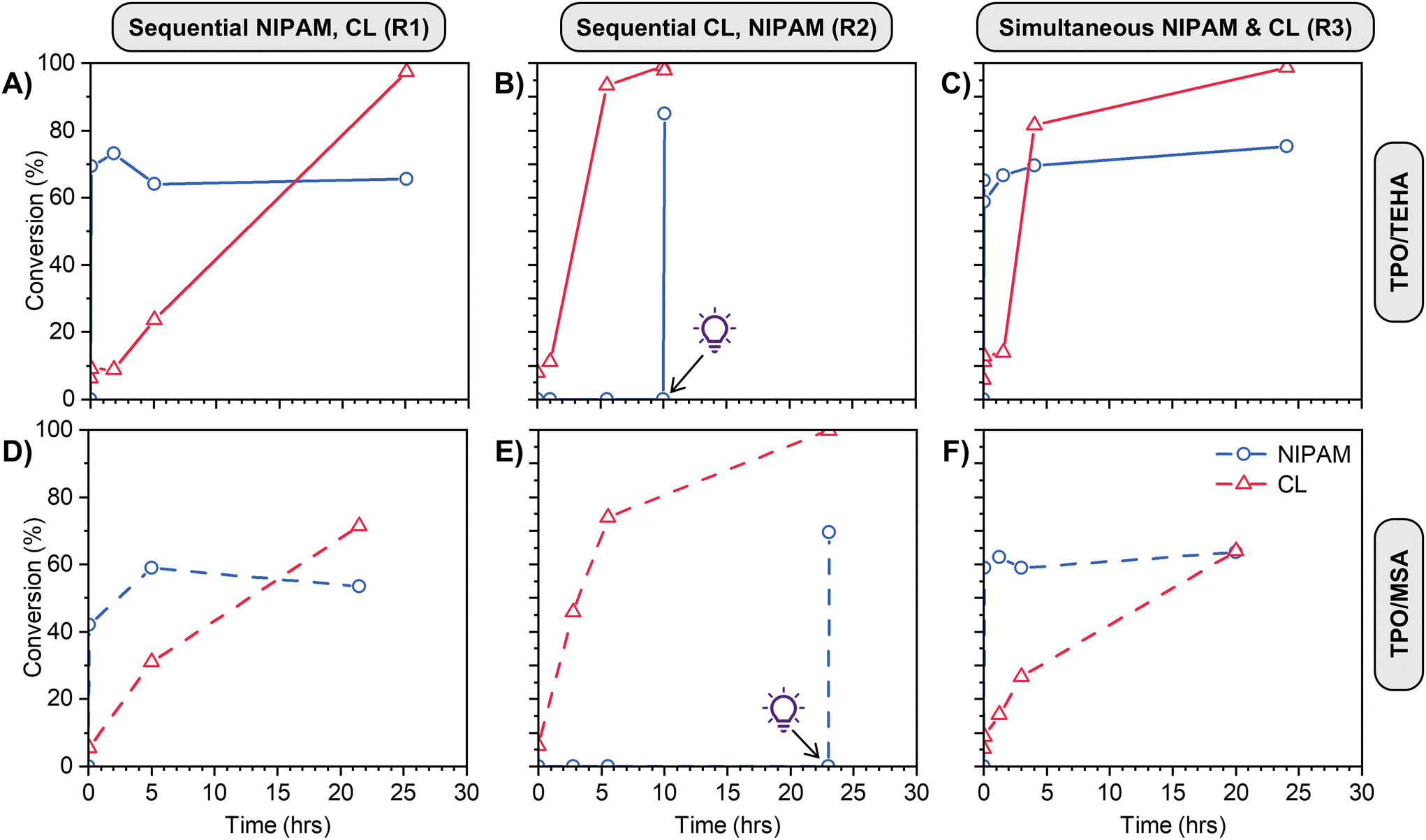 | ||
| Fig. 4 Mixed-mode conversion of NIPAM (blue) and CL (red) for (A), (B) and (C) TPO/TEHA systems, (D), (E) and (F) TPO/MSA systems. (A) & (D) Sequential polymerization NIPAM then CL (reaction (1)). (B) & (E) Sequential polymerization CL then NIPAM (reaction (2)). (C) & (F) Simultaneous polymerization NIPAM & CL (reaction (3)). For panels (A), (C), (D) and (E) UV irradiation occurs for the first 5 min, for (B) and (E) light bulb symbol indicates when 5 min UV exposure started. | ||
Linear homopolymers and polymer blends were isolated by precipitation and were analysed via TGA and GPC. Select TGA thermograms and GPC chromatograms are displayed in Fig. 5 comparing PNIPAM and PCL homopolymers each prepared from a eutectic mixture, with PNIPAM-PCL polymer blends prepared via reaction (3) (the simultaneous pathway). TGA thermograms and GPC chromatograms for all samples in this work are shown in Fig. S7 and S8,† respectively, and summarised in Table 1. Homopolymer control experiments for both PNIPAM and PCL show that both the molar mass distribution and the mass loss profile by TGA are comparable between reactions performed in the eutectic and those performed either in the bulk or in DMSO; the polymers have comparable molar mass distributions with relative Mn values between 3–7 kDa; given the high initiator loading these molar mass distributions are not unexpected. The thermal stability and number average molar mass for PCL is higher when MSA is used as a catalyst compared to TEHA (see entries 3–5 & 6–8, Table 1); the value of Tmax increases by close to 100 °C when MSA is used, which is attributed to the differing end-groups as a result of the choice of catalyst influencing the thermal stability of the polymer.77 The TGA profile of our simultaneous polymerization (using TPO/TEHA, entry 11, Table 1; Fig. 5A, central panel) has distinct features with a two-step mass loss profile that can be directly attributed to PCL and PNIPAM respectively. Assuming the mass change in each section of the TGA profile corresponds to homopolymer decomposition of either PNIPAM or PCL, the PCL mass fraction in each system was 0.72, 0.72 and 0.76 (for reaction (1), reaction (2) and reaction (3) respectively), which is in good agreement with the mass fraction of the original composition of the eutectic.
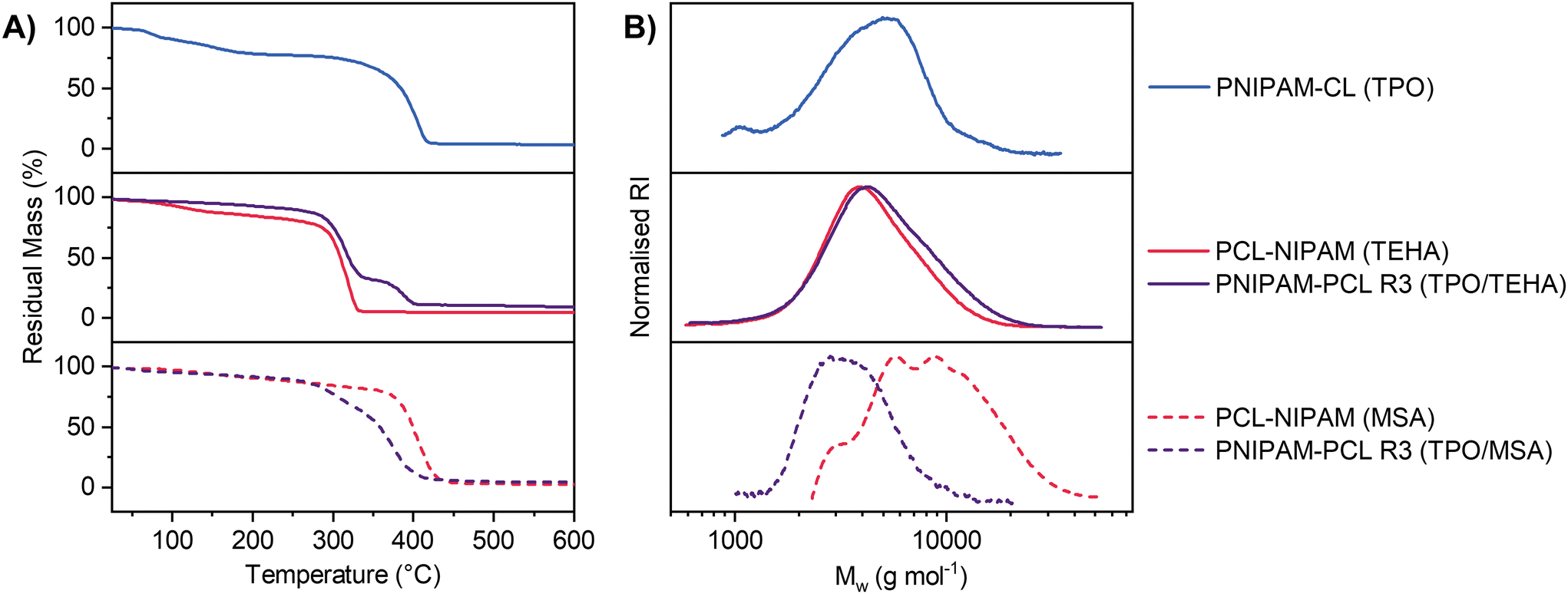 | ||
| Fig. 5 (A) TGA thermograms and (B) GPC chromatograms for PNIPAM (blue) and PCL (red) homopolymer controls as well as PNIPAM-PCL copolymers (purple). In each graph the homopolymers were prepared from the eutectic mixture, and copolymers were polymerized simultaneously (reaction (3)). | ||
| Entry # | Sample | Initiator/catalyst | T max (°C) |
M
na (g mol−1) |
Đ |
|---|---|---|---|---|---|
| a Relative to PMMA standards by THF GPC. | |||||
| 1 | PNIPAM-DMSO | TPO | 414.1 | 3930 | 1.5 |
| 2 | PNIPAM-CL | TPO | 405.4 | 3280 | 1.6 |
| 3 | PCL neat | TEHA | 309.5 | 4390 | 1.4 |
| 4 | PCL-DMSO | TEHA | 308.4 | 3870 | 1.4 |
| 5 | PCL-NIPAM | TEHA | 318.1 | 3520 | 1.4 |
| 6 | PCL neat | MSA | 407.3 | 7260 | 1.9 |
| 7 | PCL-DMSO | MSA | 405.0 | 4140 | 1.6 |
| 8 | PCL-NIPAM | MSA | 405.6 | 6950 | 1.4 |
| 9 | PNIPAM-PCL R1 | TPO/TEHA | 314.5 | 3620 | 1.2 |
| 10 | PNIPAM-PCL R2 | TPO/TEHA | 321.8 | 3300 | 1.5 |
| 11 | PNIPAM-PCL R3 | TPO/TEHA | 316.7 | 3720 | 1.5 |
| 12 | PNIPAM-PCL R1 | TPO/MSA | 380.8 | 2880 | 1.1 |
| 13 | PNIPAM-PCL R2 | TPO/MSA | 397.7 | 3410 | 1.2 |
| 14 | PNIPAM-PCL R3 | TPO/MSA | 371.7 | 3150 | 1.2 |
Interpenetrating and semi-interpenetrating networks from PEs
Our NIPAM-CL PEs with xCL = 0.67 were subsequently used for the solvent-free preparation of IPNs and semi-IPNs through the cross-linking of one or both components (see Fig. 6A). IPNs were achieved in a sequential fashion (reaction (1) or (2)), whereby CL was cross-linked through the use of a bis(carbonate) compound prepared as per the literature,69 and NIPAM was cross-linked with commercially available PEGDA. Each cross-linker was present at a mole fraction of 5% relative to its respective monomer. PEs present a technical advantage for the preparation of IPNs as both monomers are present at the start of the reaction, avoiding the need for sequential synthetic steps such as purification and isolation of the first network for swelling with the second monomer. For cross-linking of PCL only the TEHA ROP catalyst was used as the MSA catalyst was not compatible with the bis(carbonate) crosslinker, whereby no cross-linked networks were formed. In this section the subscript ‘x’ refers to a cross-linked phase. As both monomers are present through the formation of the initial PE, the first network does not need to be swollen with additional monomer to form an IPN; furthermore the orthogonality of each polymerization enables the network structure in each step to be studied independently.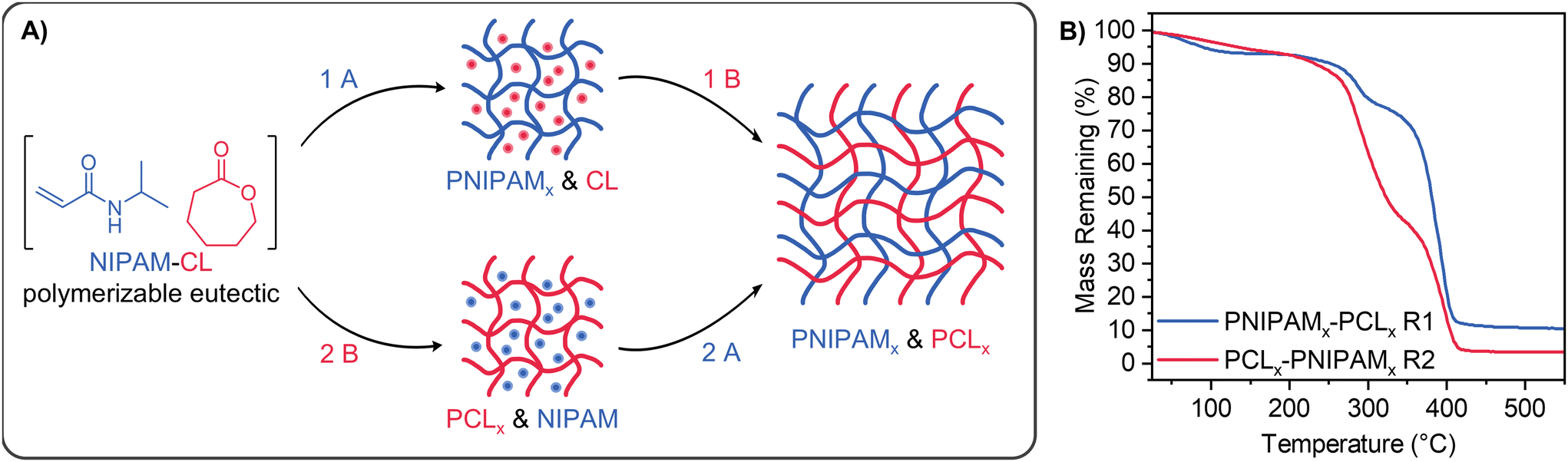 | ||
| Fig. 6 (A) Scheme showing the sequential preparation of PNIPAMx & PCLx IPNs. Reactions A and B have the same conditions as in Scheme 1A with the addition of PEGDA for reaction (A), and bis(carbonate) for reaction (B). PCL-PNIPAMx semi-IPNs were prepared following reaction (2) without bis(carbonate) crosslinker present. (B) TGA thermograms of IPNs. | ||
Initially, cross-linked PNIPAM and PCL homopolymer gels were prepared from both PEs and a conventional solvent (DMSO), as well as in the bulk for CL for comparison. PNIPAMx-DMSO gels were optically transparent, while PNIPAMx-CL gels prepared under the same conditions were opaque (Fig. S9A, ESI†). All PCL gels (PCLx neat, PCLx-DMSO, PCLx-NIPAM) were opaque with a slight yellow colour due to the bis(carbonate) crosslinker (Fig. S9B, ESI†). PCLx-NIPAM gels were better at retaining their original shape after washing and drying compared with PCLx neat or PCLx-DMSO. To evaluate their level of cross-linking, the swelling of dried homopolymer gels was studied in water (for PNIPAMx) and chloroform (for PCLx). This data is shown in Table 2. The swelling ratio of PCL networks in chloroform had a large standard deviation compared with swelling in water due to the rapid evaporation of chloroform from these samples. Additionally, PCLx neat samples lacked structural integrity after swelling and some samples crumbled into several pieces. PNIPAMx gels showed reversible thermoresponsive swelling in water (Table 2, entries 1 and 2), as anticipated due to the lower critical solution temperature (LCST) of PNIPAM at ∼32 °C, resulting in a volume phase transition temperature (VPTT) of these networks. The swelling ratio of PNIPAMx is similar when prepared either in DMSO or in CL, indicating a comparable level of cross-linking. The effective cross-link density qeff of these networks was estimated via Flory–Rehner theory78 (see ESI† and Table 2). PCLx essentially undergoes no swelling in water (entries 3–5), but swells substantially in chloroform; the gel prepared from the PE (entry 5) has the lowest cross-link density and highest swelling capacity. Notably, the cross-link density of the PCL networks is significantly lower than the predicted value (qpredeff = 0.1, based on 5 mol% of cross-linker in the mixture and 2 cross-link sites formed per molecule), suggesting a relatively low fraction of the bis(carbonate) crosslinker incorporated into the network structure.
| Entry # | Sample | S water (RT) | S water (45 °C) | S CHCl3 | q eff |
|---|---|---|---|---|---|
| a Denotes sample crumbled when immersed in solvent. RT = 20–23 °C. n = 2 for all samples except those denoted *n = 3, ^n = 1. | |||||
| 1 | PNIPAMx-DMSO | 2.29 ± 0.04* | 0.40 ± 0.03* | x | 0.17 |
| 2 | PNIPAMx-CL | 2.14 ± 0.07* | 0.37 ± 0.01* | x | 0.19 |
| 3 | PCLx bulk | 0.04 ± 0.01 | — | 8.31 ± 0.02 | 0.01 |
| 4 | PCLx-DMSO | 0.041 ± 0.008 | — | 12.7 ± 0.9 | 0.005 |
| 5 | PCLx-NIPAM | 0.044 ± 0.006 | — | 15.2 ± 0.2 | 0.004 |
| 6 | PNIPAMx-PCLx R1 IPN | 1.44^ | 0.25^ | 3.74^ | |
| 7 | PCLx-PNIPAMx R2 IPN | x | — | — | |
| 8 | PCL-PNIPAMx semi-IPN | 1.95 ± 0.01 | 0.3 ± 0.1 | — | |
IPNs were subsequently prepared from our PEs using two sequential one-pot approaches, where either NIPAM (reaction(1)) or CL (reaction (2)) was the first phase to be cross-linked (entries 6, 7, Table 2 and Fig. 6B). While the conversion of CL with TEHA was similar regardless of reaction order for linear polymerization (Fig. 4A and C), the relative mass fraction PNIPAMx and PCLx in the resulting IPNs based on TGA data was dependent on the order of cross-linking (Table S1, ESI†). The behaviour of these networks in water and chloroform was also studied. When PCL was cross-linked first followed by PNIPAM (rection (2), entry 7), the network lacked the structural integrity to remain intact when immersed in water. In comparison, PNIPAM crosslinking followed by PCL worked well (reaction (1), entry 6), forming a robust IPN. The network showed thermoresponsive swelling in water as well as swelling in chloroform, however the equilibrium swelling capacity in each solvent was reduced compared to homopolymer gels. This is attributed to both the inter-entanglement of cross-linked PCL and PNIPAM domains within the IPN and unfavourable polymer-solvent interactions (e.g. PCLx and water) that favour a contraction of the network. Semi-IPNs were also successfully prepared (reaction (2), entry 8) where linear PCL was first formed, followed by polymerization and cross-linking of NIPAM resulting in a semi-2 type IPN (linear polymer prepared first, followed by a crosslinked network).79 For this process CL was polymerized in situ using MSA as catalyst in air at room temperature, enabling the entire process to be performed in an open PTFE mould with no complicated equipment (see Fig. S11, ESI†). PCL-PNIPAMx semi-IPNs were washed with water and then chloroform to remove any unreacted monomer and linear PCL before drying; the thermoresponsive swelling capacity of this PNIPAMx network in water was reduced in comparison to homopolymer PNIPAM networks prepared in the absence of PCL.
3D printing from PE resins
Our NIPAM-CL0.67 PE was then used as a stereolithography (SLA) resin on a benchtop 3D printer. The use of a UV photoinitiator such as TPO enables these resins to be cured at the nominal wavelength of the printer (405 nm), in the absence of any traditional solvents. This is particularly advantageous for monomers such as NIPAM, that possess melting points well above room temperature and most typically used in an aqueous hydrogel format80,81 for SLA 3D printing. Using 2 wt% PEGDA as cross-linker, printing performance was evaluated using a 10 × 10 × 10 mm3 hollow cube with vertical and horizontal struts as a test design (see Fig. 7, top left panel). Without any further additives, significant overcuring was observed and a solid cube was formed. To address this problem, a symmetric RAFT agent was dissolved in the resin at a concentration of 0.5 wt% relative to NIPAM, similar to previously published work on RAFT-mediated 3D printing.48,53 The RAFT agent plays multiple roles; it acts as an in situ photoblocker to reduce unwanted light penetration during the printing process in addition to reducing residual stress in the polymer network through the degenerative transfer process whilst not compromising the thermal properties of the polymer (see Fig. S13†). A highly transparent PNIPAMx-CL hollow cube was printed as a result, with excellent shape fidelity in comparison to the target design (see Fig. 7, top right; Table S2† gives measured dimensions of the printed objects). | ||
| Fig. 7 (A) Images of PNIPAMx-CL 3D printed cube structure showing reversible thermoresponsive swelling in water. Scale bar in all figures represents 1 cm. (B) Images of PNIPAMx-CL (with zinc acetate present) and PNIPAMx-PCL semi-IPN 3D printed materials. (C) TGA thermograms of PNIPAMx-CL with zinc acetate present (blue) and PNIPAMx-PCL semi-IPN (red). | ||
Similar to our bulk cured networks, these printed objects possess thermoresponsive behaviour when immersed in water. At room temperature, the object symmetrically swells to approximately 1.5 times its original external size, with a significant increase in mass due to absorbed water, and loss of optical transparency, whilst retaining its structural features (see Table S2,† and Fig. 7, bottom right panel). When the swollen object is heated through the LCST of PNIPAM to 45 °C, we observe mass loss due to desorbed water, coupled with only a very small decreased in external size as well as loss of the smooth structural features of the original cube. A rough, “ragged/buckled” cube is formed as a result, which is fully reversible back to the “ordered” structure when allowed to cool back to room temperature in water. This observation could be attributed to the buckling of the PNIPAM network due to the osmotic pressure created as water desorbs from the outermost surface of the printed object at higher temperatures.81
As a final investigation of orthogonal preparation of IPNs from PEs, we prepared 3D printing PE resins also containing the reagents required for subsequent CL polymerization. This enables the preparation of semi-1 IPNs where linear PCL is formed within a PNIPAMx 3D-printed network. For these printing experiments the same base resin formulation was used as for the NIPAM-only polymerization discussed above. Initial attempts where TEHA was incorporated into the resin resulted in unsuccessful printing, attributed to unwanted formation of linear PCL during the print step. This altered the viscosity of the resin and required UV exposure to achieve sufficient curing throughout each layer of the print. While the linear polymerization of NIPAM and CL with TPO and TEHA was orthogonal under 370 nm irradiation (Fig. 4), this orthogonality was lost when using the different wavelength of the 3D printer (λmax = 405 nm). An alternative ROP catalyst, zinc acetate,82 was subsequently used which resulted in successful printing of PNIPAMx structures from the PE resin containing CL and zinc acetate throughout the material (Fig. 7B). This structure was placed in a sealed glass vial and heated at 100 °C to polymerize the CL; as a visual indication of PCL formation, the opacity of the material increased after heat treatment. These semi-IPN materials were studied via TGA after each step to determine the relative mass fractions of each component. Fig. 7C shows the TGA thermograms after 3D-printing, prior to ROP of CL (PNIPAMx-CL (Zn(OAc)2)) as well as after heating the material to polymerize CL (PNIPAMx-PCL semi-IPN). The as-printed material shows a significant mass loss (64.9%) around 144 °C. This is in excellent agreement with the initial mass fraction of CL in the resin (65.5%), supporting minimal PCL formation during the printing process. After heating, this first mass loss (135 °C) was greatly reduced (17% of total mass). This result indicates that a considerable amount of CL is polymerized within the PNIPAMx network structure, yielding a PNIPAMx-PCL 3D-printed semi-IPN.
Conclusions
In this work, the direct preparation of interpenetrating polymer networks (IPNs) and semi-IPNs from a binary polymerizable eutectic is reported for the first time. Using differential scanning calorimetry, a solid–liquid equilibrium phase diagram was created for mixtures of NIPAM and CL, which exhibit near-to-ideal behaviour. A high degree of association between the two monomers was demonstrated by 1D/2D NMR spectroscopy, and significantly reduced self-diffusion coefficients of both species compared to those measured in conventional solvents. The formation of a stable room temperature liquid enables orthogonal polymerization of both components in the absence of added solvents, either in a sequential or simultaneous fashion, with an increase in the polymerization rate of CL in the polymerizable eutectic compared to the same process in DMSO. Using a commercially available diacrylate cross-linker and an as-synthesised bis(carbonate) compound to participate in ring opening polymerization, both IPNs and semi-IPNs can be prepared in a one-pot process. The presence of a crosslinked PNIPAM network provides reversible thermoresponsive swelling in water for these IPNs, the preparation of which is highly dependent on the order in which the two phases are polymerized/crosslinked. The rapid polymerization of NIPAM using a UV-active photoinitiator also enables these mixtures to be used as resins for SLA 3D printing on a benchtop printer, with CL present within the cross-linked PNIPAM matrix. The printed networks give intriguing reversible morphology changes when allowed to swell and deswell in water as the temperature is varied, and upon heating the printed object to polymerize CL, semi-IPN materials can be achieved in a two-step fashion. Future work in this area will investigate the design of printed IPN and semi-IPN networks with good print fidelity. We anticipate that due to the flexibility surrounding the design of differing PEs based on monomer type and class, this approach will enable a wide range of polymer networks to be created with tailored physical and chemical properties in the future.Author contributions
A.L.M – data curation, formal analysis, investigation, methodology, project administration, visualization, writing – original draft, writing – review and editing. S.C.T – conceptualization, funding acquisition, methodology, supervision, writing – original draft, writing – review and editing.Data availability
Electronic supporting information (ESI): NMR characterisation of bis(carbonate) crosslinker, DSC traces as a function of heating/cooling rate, photographs of NIPAM-VL eutectics and phase diagram, DOSY NMR spectra, TGA and GPC data for all polymers, digital photographs of gels.Raw data for this manuscript (graphical outputs of processed DSC traces, raw mass vs. temperature data for TGA thermograms, RI signal vs. time, Mw calculated from PMMA standards for GPC elution data, 1H NMR spectra, tabulated conversion-time data from polymerizations, and .ctb slice files for 3D printing) are publicly available at the University of Tasmania Research Data Portal (https://www.rdp.utas.edu.au) and accessible at the following address: https://dx.doi.org/10.25959/18hy-nb80.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. L. M. and S. C. T.'s contributions were supported by an Australian Research Council Future Fellowship (FT220100096). The authors thank the UTAS Central Science Laboratory for providing access to NMR spectroscopy services and Dr James Horne for assistance.References
- Y. Ma, R. J. Dreiling, E. A. Recker, J.-W. Kim, S. L. Shankel, J. Hu, A. D. Easley, Z. A. Page, T. H. Lambert and B. P. Fors, ACS Cent. Sci., 2024, 10(11), 2125–2131 CrossRef CAS PubMed.
- C. J. Reese, G. M. Musgrave and C. Wang, Polym. Chem., 2024, 15, 3954–3966 RSC.
- S. Liu, K. T. Dicker and X. Jia, Chem. Commun., 2015, 51, 5218–5237 RSC.
- N. Corrigan, M. Ciftci, K. Jung and C. Boyer, Angew. Chem., Int. Ed., 2021, 60, 1748–1781 CrossRef CAS PubMed.
- V. X. Truong, J. Bachmann, A. Unterreiner, J. P. Blinco and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2022, 61, e202113076 CrossRef CAS PubMed.
- M. A. Gauthier, M. I. Gibson and H. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- G. I. Peterson, J. J. Schwartz, D. Zhang, B. M. Weiss, M. A. Ganter, D. W. Storti and A. J. Boydston, ACS Appl. Mater. Interfaces, 2016, 8, 29037–29043 CrossRef CAS PubMed.
- N. D. Dolinski, Z. A. Page, E. B. Callaway, F. Eisenreich, R. V. Garcia, R. Chavez, D. P. Bothman, S. Hecht, F. W. Zok and C. J. Hawker, Adv. Mater., 2018, 30, 1800364 CrossRef PubMed.
- Y. C. Yu, G. Li, H. U. Kang and J. H. Youk, Colloid Polym. Sci., 2012, 290, 1707–1712 CrossRef CAS.
- D. Mecerreyes, G. Moineau, P. Dubois, R. Jérôme, J. L. Hedrick, C. J. Hawker, E. E. Malmström and M. Trollsas, Angew. Chem., Int. Ed., 1998, 37, 1274–1276 CrossRef CAS PubMed.
- W. Jakubowski and K. Matyjaszewski, Macromol. Symp., 2006, 240, 213–223 CrossRef CAS.
- Y. You, C. Hong, W. Wang, W. Lu and C. Pan, Macromolecules, 2004, 37, 9761–9767 CrossRef CAS.
- Y. C. Yu, G. Li, J. Kim and J. H. Youk, Polymer, 2013, 54, 6119–6124 CrossRef CAS.
- S. Villarroya, J. Zhou, K. J. Thurecht and S. M. Howdle, Macromolecules, 2006, 39, 9080–9086 CrossRef CAS.
- N. Chagneux, T. Trimaille, M. Rollet, E. Beaudoin, P. Gérard, D. Bertin and D. Gigmes, Macromolecules, 2009, 42, 9435–9442 CrossRef CAS.
- F. F. Wolf, N. Friedemann and H. Frey, Macromolecules, 2009, 42, 5622–5628 CrossRef CAS.
- S. Zhang, N. Ren, X. Li, Y. Xiao, M. Lang and X. Zhu, Polym. Chem., 2024, 15, 1992–2001 RSC.
- M. Le Hellaye, C. Lefay, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3058–3067 CrossRef CAS.
- Y.-Y. Yuan, Q. Du, Y.-C. Wang and J. Wang, Macromolecules, 2010, 43, 1739–1746 CrossRef CAS.
- J. Xu, K. Jung and C. Boyer, Macromolecules, 2014, 47, 4217–4229 CrossRef CAS.
- J. Xu, S. Shanmugam, H. T. Duong and C. Boyer, Polym. Chem., 2015, 6, 5615–5624 RSC.
- J. Xu, K. Jung, A. Atme, S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2014, 136, 5508–5519 CrossRef CAS PubMed.
- C. Fu, J. Xu, M. Kokotovic and C. Boyer, ACS Macro Lett., 2016, 5, 444–449 CrossRef CAS PubMed.
- C. Fu, J. Xu and C. Boyer, Chem. Commun., 2016, 52, 7126–7129 RSC.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- M. Francisco, A. vandenBruinhorst and M. C. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- Y. Liu, J. B. Friesen, J. B. McAlpine, D. C. Lankin, S.-N. Chen and G. F. Pauli, J. Nat. Prod., 2018, 81, 679–690 CrossRef CAS PubMed.
- M. Q. Farooq, N. M. Abbasi and J. L. Anderson, J. Chromatogr. A, 2020, 1633, 461613 CrossRef CAS PubMed.
- X. Li and K. H. Row, J. Sep. Sci., 2016, 39, 3505–3520 CrossRef CAS PubMed.
- D. Julião, M. Xavier and X. Mascarenhas, Mater. Today Energy, 2024, 42, 101432 Search PubMed.
- A. Sharma, R. Sharma, R. C. Thakur and L. Singh, J. Energy Chem., 2023, 82, 592–626 Search PubMed.
- C. Florindo, F. Lima, B. D. Ribeiro and I. M. Marrucho, Curr. Opin. Green Sustainable Chem., 2019, 18, 31–36 Search PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71 Search PubMed.
- D. O. Abranches and J. A. P. Coutinho, Annu. Rev. Chem. Biomol. Eng., 2023, 14, 141–163 CrossRef CAS PubMed.
- P. A. Mercadal, A. González, A. Beloqui, L. C. Tomé, D. Mecerreyes, M. Calderón and M. L. Picchio, JACS Au, 2024, 4, 3744–3758 CrossRef CAS PubMed.
- N. Ndizeye, S. Suriyanarayanan and I. A. Nicholls, Polym. Chem., 2019, 10, 5289–5295 RSC.
- Y. Nahar, P. Wei, C. Cipriani, A. Khodabandeh, A. C. Bissember, E. B. Pentzer and S. C. Thickett, ACS Appl. Polym. Mater., 2022, 4, 8429–8440 CrossRef CAS.
- N. Fu, X. Liu, L. Li, B. Tang and K. H. Row, J. Sep. Sci., 2017, 40, 2286–2291 CrossRef CAS PubMed.
- G. Li and K. H. Row, J. Sep. Sci., 2017, 40, 4765–4772 CrossRef CAS PubMed.
- X. Li and K. H. Row, J. Chromatogr. B, 2017, 1068–1069, 56–63 CAS.
- G. Li, W. Wang, Q. Wang and T. Zhu, J. Chromatogr. Sci., 2015, bmv138 Search PubMed.
- Y. Liu, Y. Wang, Q. Dai and Y. Zhou, Anal. Chim. Acta, 2016, 936, 168–178 CrossRef CAS PubMed.
- S. A. H. Shah, M. R. Ramachandran, S. A. Mansur, N. M. Saleh and S. Asman, Polymer, 2023, 283, 126279 CrossRef CAS.
- Y. Nahar and S. C. Thickett, Polymers, 2021, 13, 447 CrossRef CAS PubMed.
- J. D. Mota-Morales, R. J. Sánchez-Leija, A. Carranza, J. A. Pojman, F. Del Monte and G. Luna-Bárcenas, Prog. Polym. Sci., 2018, 78, 139–153 CrossRef CAS.
- S. Xing, G. Zhang, A. Zhang, Z. Chang, Z. Guo, A. Dong and J. Zhang, J. Polym. Sci., 2024, 62, 925–936 CrossRef CAS.
- R. Li, G. Chen, M. He, J. Tian and B. Su, J. Mater. Chem. C, 2017, 5, 8475–8481 RSC.
- A. L. Mutch, Y. Nahar, A. C. Bissember, N. Corrigan, C. Boyer, X. Y. Oh, V. X. Truong and S. C. Thickett, Macromol. Rapid Commun., 2024, 2400268 CrossRef CAS PubMed.
- B. Li, X. Xu, Z. Hu, Y. Li, M. Zhou, J. Liu, Y. Jiang and P. Wang, RSC Adv., 2022, 12, 19022–19028 RSC.
- J. D. Mota-Morales, M. C. Gutiérrez, M. L. Ferrer, I. C. Sanchez, E. A. Elizalde-Peña, J. A. Pojman, F. D. Monte and G. Luna-Bárcenas, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1767–1773 CrossRef CAS.
- J. D. Mota-Morales, M. C. Gutiérrez, I. C. Sanchez, G. Luna-Bárcenas and F. del Monte, Chem. Commun., 2011, 47, 5328–5330 RSC.
- R. J. Sánchez-Leija, J. A. Pojman, G. Luna-Bárcenas and J. D. Mota-Morales, J. Mater. Chem. B, 2014, 2, 7495–7501 RSC.
- N. Corrigan, A. L. Mutch, C. Boyer and S. C. Thickett, RSC Appl. Polym., 2024, 2, 914–925 RSC.
- Y. Li, R. K. Kankala, L. Wu, A.-Z. Chen and S.-B. Wang, ACS Appl. Polym. Mater., 2023, 5, 991–1001 CrossRef CAS.
- G. Zhu, M. Liu, S. Weng, G. Zhang, Y. Hu, Z. Kou, C. Bo, L. Hu, S. Wu and Y. Zhou, Chem. Eng. J., 2023, 472, 144987 CrossRef CAS.
- M. Liu, G. Zhang, Y. Hu, C. Bo, Y. Dai, L. Hu, G. Zhu and Y. Zhou, Green Chem., 2024, 26, 10441–10455 RSC.
- Y. Nahar, M. K. Stanfield, A. C. Bissember and S. C. Thickett, Polym. Chem., 2023, 14, 2724–2733 RSC.
- M. Castillo-Santillan, P. Quiñonez-Angulo, D. Maniar, J. R. Torres-Lubian, M. C. Gutiérrez, T. Pelras, A. J. J. Woortman, Q. Chen, M. G. Pérez-García, K. Loos and J. D. Mota-Morales, RSC Appl. Polym., 2024, 2, 403–414 RSC.
- M. Castillo-Santillan, J. R. Torres-Lubian, A. Martínez-Richa, S. T. Huerta-Marcial, M. C. Gutierrez, K. Loos, M. G. Peréz-García and J. D. Mota-Morales, Polymer, 2022, 262, 125432 CrossRef CAS.
- M. G. Pérez-García, M. C. Gutiérrez, J. D. Mota-Morales, G. Luna-Bárcenas and F. del Monte, ACS Appl. Mater. Interfaces, 2016, 8, 16939–16949 CrossRef PubMed.
- M. S. Silverstein, Polymer, 2020, 207, 122929 CrossRef CAS.
- E. S. Dragan, Chem. Eng. J., 2014, 243, 572–590 CrossRef CAS.
- N. Firoozi, A. H. Rezayan, S. J. Tabatabaei Rezaei, M. Mir-Derikvand, M. R. Nabid, J. Nourmohammadi and J. Mohammadnejad Arough, Int. J. Polym. Mater. Polym. Biomater., 2017, 66, 805–811 CrossRef CAS.
- J. Hao, Y. Liu, S. Zhou, Z. Li and X. Deng, Biomaterials, 2003, 24, 1531–1539 CrossRef CAS PubMed.
- D. S. Jones, D. W. J. McLaughlin, C. P. McCoy and S. P. Gorman, Biomaterials, 2005, 26, 1761–1770 CrossRef CAS PubMed.
- L. N. Woodard, K. T. Kmetz, A. A. Roth, V. M. Page and M. A. Grunlan, Biomacromolecules, 2017, 18, 4075–4083 CrossRef CAS PubMed.
- B. R. Saunders, Langmuir, 2004, 20, 3925–3932 CrossRef CAS PubMed.
- T. Şucu and M. P. Shaver, Polym. Chem., 2020, 11, 6397–6412 RSC.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2019, 48, 962–982 CrossRef CAS.
- A. T. N. Fajar, T. Hanada, A. D. Hartono and M. Goto, Commun. Chem., 2024, 7, 27 CrossRef CAS PubMed.
- A. Shishov, P. Makoś-Chełstowska, A. Bulatov and V. Andruch, J. Phys. Chem. B, 2022, 126, 3889–3896 CrossRef CAS PubMed.
- V. Agieienko and R. Buchner, Phys. Chem. Chem. Phys., 2022, 24, 5265–5268 RSC.
- L. Rycerz, J. Therm. Anal. Calorim., 2013, 113, 231–238 CrossRef CAS.
- Y. Chen, F. Wang, Y. Zhang, J. Wang, C. Liu, J. Han, Z. Zhang, Q. Zhang and M. Yang, Thermochim. Acta, 2023, 729, 179607 CrossRef CAS.
- Y. Nahar, J. Horne, V. Truong, A. C. Bissember and S. C. Thickett, Polym. Chem., 2021, 12, 254–264 RSC.
- G. Gorrasi, L. Vertuccio, L. Annunziata, C. Pellecchia and D. Pappalardo, React. Funct. Polym., 2010, 70, 151–158 CrossRef CAS.
- P. J. Flory and J. Rehner, J. Chem. Phys., 1943, 11, 512–520 CrossRef CAS.
- D. A. Thomas and L. H. Sperling, in Polymer Blends, ed. D. R. Paul and S. Newman, Academic Press, 1978, pp. 1–33 Search PubMed.
- X. Sun, P. Tyagi, S. Agate, M. G. McCord, L. A. Lucia and L. Pal, Carbohydr. Polym., 2020, 234, 115898 CrossRef CAS PubMed.
- D. M. Solis and A. Czekanski, Soft Matter, 2022, 18, 3422–3429 RSC.
- R. R. Gowda and D. Chakraborty, J. Mol. Catal. A: Chem., 2010, 333, 167–172 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01456a |
| This journal is © The Royal Society of Chemistry 2025 |