
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of phenyl-substituted open-cage silsesquioxane-pendant polysiloxanes and their thermal and optical properties†
Miku
Kosaka
a,
Kenji
Kanaori
a,
Hiroaki
Imoto
 a and
Kensuke
Naka
*ab
a and
Kensuke
Naka
*ab
aFaculty of Molecular Chemistry and Engineering, Graduate School of Science and Technology, Kyoto Institute of Technology, Kyoto 606-8585, Japan. E-mail: kenaka@kit.ac.jp
bMaterials Innovation Lab, Kyoto Institute of Technology, Goshokaido-cho, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan
First published on 11th February 2025
Abstract
We prepared phenyl-substituted corner-opened type polyhedral oligomeric silsesquioxanes (CO-POSSs) bearing tris(dimethoxysilyl)-groups with variable linker lengths at the opening vertex; that is, tris(dimethoxysilyl-ethyl-dimethylsiloxy)- and tris(dimethoxysilyl-propylthioethyl-dimethylsiloxy)-heptaphenyl-substituted CO-POSSs (Ph-H and Ph-V). Optically transparent free-standing films of phenyl-substituted open-cage silsesquioxane-pendant polysiloxanes (PolyPh-H) were prepared by optimizing the sol–gel reaction conditions for Ph-H. Polycondensation of Ph-V afforded an optically transparent and flexible phenyl-substituted CO-POSS-pendant polysiloxane film (PolyPh-V). The polycondensations of Ph-H and Ph-V were fully completed even at 50 °C for 6 h under vacuum. 29Si cross-polarization magic angle spinning (CP-MAS) NMR analysis suggests that the films included cyclotrisiloxane (D3) and linear siloxane (Dlinear) structures. The effects of the polysiloxane structures on the thermal and mechanical properties were studied. The highest temperature at which the sample lost 5 wt% of the original mass (Td5) under N2 (381 °C) was obtained for PolyPh-V, even though it contained a flexible linker unit. The predominant linear siloxane structures may provide increase higher thermal stability. The UV-vis spectra of the resulting transparent films were mostly unchanged even after six days of exposure to UV irradiation in air. The present study shows that phenyl-substituted CO-POSS-pendant polysiloxanes represent alternative UV-resistant, optically transparent materials with higher heat resistance.
Introduction
Silicone resins derived from hydrolysis and condensation reactions of alkoxysilanes and chlorosilanes are industrially important organic–inorganic hybrid materials due to their superior thermal, mechanical, optical, and electrical properties.1,2 Combinations of various siloxane monomeric units are indicated using “M” (R3SiO0.5), “D” (R2SiO), “T” (RSiO1.5), and “Q” (SiO2), with a wide variety of organic substituents offering a broad range of material properties. With the increasing performance of electronic devices and power electronics, the development of multifunctional silicone materials that are optically transparent and can be obtained with low processing temperatures is an important task.3–5 Depending on their purpose, such materials require several properties to be combined, including high thermal conductivity, insulation, heat resistance, UV resistance, flexibility, high refractive index, and lightweight character. These requirements often have a trade-off relationship, and trial-and-error studies are typically required to combine these characteristics. The usual sol–gel reaction that is used to obtain silicone resins requires a high-curing temperature to facilitate the condensation reaction. However, unreacted alkoxy and silanol groups can remain, which affect the physical properties and functions of the materials, and complete consumption is difficult to achieve under typical sol–gel reaction condition.5,6 In addition, although the molecular level structures of the siloxane linkages significantly affect the physical properties of the materials, controlling the structures of silicone resins at the molecular level is difficult with standard sol–gel processing.7–9 Correlations between three-dimensional well-defined molecular-level structures and bulk properties remain unclear.To address these issues, the concept of “polymeric materials based on element-blocks” is expected to facilitate new research and lead to new ideas for materials design with well-defined molecular-level structures in three-dimensions.10 Polyhedral oligomeric silsesquioxanes (POSSs), particularly, cage octasilsesquioxanes (T8 cages), have attracted growing interest over the past several decades as well-defined and important element-blocks.11,12 The use of POSS derivatives as monomers provides an attractive approach to the design of organic–inorganic hybrid polymers. Among the POSS family, open-cage silsesquioxanes; that is, incompletely condensed POSSs (IC-POSSs), represent promising element-blocks for building organic–inorganic hybrid materials.13 Corner-opened type POSSs (CO-POSS), termed trisilanols, are easily prepared via a one-step reaction from the corresponding silane coupling agents. One of the advantages of CO-POSSs is that variations in substituents such as isobutyl, phenyl, trifluoropropyl, and cyclohexyl groups at the seven Si corners are possible, and various functional groups can be easily introduced to the opened moiety of the CO-POSSs. Compared to completely condensed POSSs, while their thermal stabilities are comparable, the crystallinity of CO-POSSs is significantly reduced due to their reduced symmetry.13 CO-POSS is a suitable backbone for cross-linking agents in network polymers because three reactive sites can be easily introduced into their open moieties.14–16 Trisilanols are also suitable monomers for double cyclopolymerization because three functional groups are configured in the axial position and fixed in one direction.17 Previously, we designed trifluoropropyl-substituted CO-POSS bearing tris(dimethoxysilyl) groups and found that trifluoropropyl-substituted CO-POSS-pendant polysiloxanes were obtained by hydrolysis and polycondensation under mild conditions; that is, the polycondensation of the starting materials was fully achieved even at 50 °C within 6 h.18,19 These materials can be seen as DT resins with well-defined three-dimensional molecular-level structures. The resulting films exhibit excellent UV resistance, good flexibility, and optical transparency.
The properties of POSS compounds such as their crystallinity, solubility, compatibility, and thermal behavior are highly dependent on the type of organic substituents they harbor. Among them, phenyl-substituted POSS structures have higher thermal stability than saturated aliphatic POSSs, and such materials generally have a higher refractive index.20 Here, we prepared tris(dimethoxysilyl-ethyl-dimethylsiloxy)- and tris(dimethoxysilyl-propylthioethyl-dimethylsiloxy)-heptaphenyl-substituted CO-POSSs (Ph-H and Ph-V, respectively) as phenyl-substituted CO-POSS derivatives with tris(dimethoxysilyl)-groups. We optimized the sol–gel reaction to obtain free-standing films of the phenyl-substituted CO-POSS-pendant polysiloxanes. We found that the polycondensation proceeded to completion without any unreacted silanol groups in a low-temperature solution process at 50 °C for 6 h under vacuum. The effects of the polysiloxane structures on the thermal and mechanical properties were also studied.
Results and discussion
Ethylene-linked tris(dimethoxysilyl)-heptaphenyl-substituted open-cage silsesquioxane (Ph-H) was prepared via hydrosilylation of 3,7,14-tris(dimethylsilylsiloxy)heptaphenyltricycloheptasiloxane with dimethoxymethylvinylsilane (Fig. S1†). The dimethoxysilyl groups of Ph-H were hydrolyzed with 1 M HCl in THF (Scheme 1). After concentrated the reaction mixture under reduced pressure, the complete hydrolysis of the methoxy groups in Ph-H was confirmed by 1H NMR analysis of the residual products (Fig. S2a†). The residues were then re-dissolved in THF and the sol solution was cast onto a substrate, air-dried for one day, and fully condensed at 50 °C for 6 h under reduced pressure to afford phenyl-substituted CO-POSS-pendant polysiloxane films (PolyPh-H). When the concentration of the re-dissolved hydrolyzed material in THF was 0.16 g mL−1, only a fragile film of PolyPh-Ha was obtained and the resulting product was difficult to peel from the substrate (Fig. 1A). On the other hand, when the re-dissolved concentration of hydrolyzed Ph-H was increased to 0.88 g mL−1 gave a transparent, rigid, free-standing film of PolyPh-Hb (Fig. 1B). With the same concentration, when the condensation temperature was increased to 100 °C for 6 h, PolyPh-Hc was formed as a fragile film (Fig. 1C). Although PolyPh-Ha and PolyPh-Hc were partially soluble in acetone, chloroform, and THF, PolyPh-Hb was insoluble in these organic solvents. The optical transmittances of all the films were over 97% in the visible region (Fig. S3†).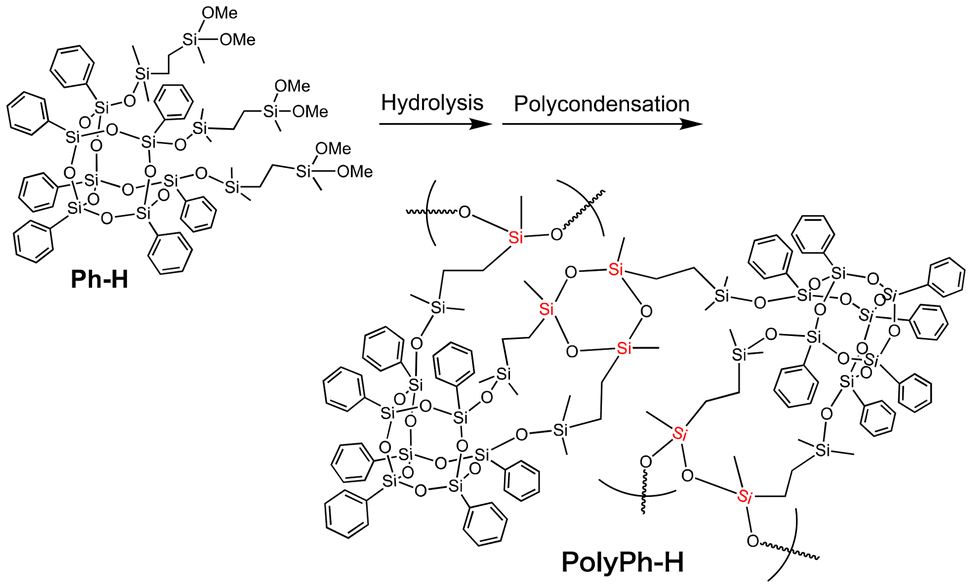 | ||
| Scheme 1 Preparation of phenyl-substituted open-cage silsesquioxane-pendant polysiloxane PolyPh-H from Ph-H. | ||
 | ||
| Fig. 1 Appearances of (A) PolyPh-Ha, (B) PolyPh-Hb, (C) PolyPh-Hc, and (D) PolyPh-V. | ||
29Si cross-polarization magic-angle spinning (CP-MAS) NMR analysis of PolyPh-Ha, PolyPh-Hb, and PolyPh-Hc, showed characteristic resonance peaks at 9 ppm and between −66 and −75 ppm corresponding to the M units, (CH3)2(![[S with combining low line]](https://www.rsc.org/images/entities/i_char_0053_0332.gif)
![[i with combining low line]](https://www.rsc.org/images/entities/i_char_0069_0332.gif) O0.5)X, and T units, CF3CH2CH2(O1.5), in the CO-POSS structure, respectively (Fig. 2). The results confirmed that the –SiOMe and SiOH groups, which should be observed at −0.61 ppm and −4.64 ppm, respectively, were absent, indicating complete polycondensation. Furthermore, no adsorption at around 2835 cm−1 or 3625 cm−1 corresponding to the –SiOMe and SiOH groups, respectively, was observed in the Fourier-transform infrared (FT-IR) spectra of the condensed products (Fig. S4†), which also indicated that condensation was complete even at 50 °C. No detectable adsorption derived from the –SiOH groups in the FT-IR analysis after storage for three months at room temperature (Fig. S5†). The 29Si CP-MAS NMR spectra of the films also included two new peaks at approximately −10 and −20 ppm ascribed to D units formed upon polycondensation. These peaks were attributed to cyclotrisiloxane structure (D3) and linear siloxane structures (Dlinear), respectively.21,22 Sample PolyPh-Ha exhibited predominant peaks at −10 ppm, corresponding to the D3 structure. The content ratio of the Dlinear to the total D units was 28% according to the integral ratio. Increasing the concentration of hydrolyzed Ph-H to 0.88 g mL−1 led to a decrease in the signal at −10 ppm and an increase in the signal at −25 ppm. The intensity ratio of the linear siloxane structures relative to the total D units in PolyPh-Hb increased to 39%. Increasing the condensation temperature to 100 °C for PolyPh-Hc, led to a decrease in the content of the linear siloxane structure to 31%. Decreasing the content of the linear siloxane structures may inhibit the formation of the free-standing-films.
O0.5)X, and T units, CF3CH2CH2(O1.5), in the CO-POSS structure, respectively (Fig. 2). The results confirmed that the –SiOMe and SiOH groups, which should be observed at −0.61 ppm and −4.64 ppm, respectively, were absent, indicating complete polycondensation. Furthermore, no adsorption at around 2835 cm−1 or 3625 cm−1 corresponding to the –SiOMe and SiOH groups, respectively, was observed in the Fourier-transform infrared (FT-IR) spectra of the condensed products (Fig. S4†), which also indicated that condensation was complete even at 50 °C. No detectable adsorption derived from the –SiOH groups in the FT-IR analysis after storage for three months at room temperature (Fig. S5†). The 29Si CP-MAS NMR spectra of the films also included two new peaks at approximately −10 and −20 ppm ascribed to D units formed upon polycondensation. These peaks were attributed to cyclotrisiloxane structure (D3) and linear siloxane structures (Dlinear), respectively.21,22 Sample PolyPh-Ha exhibited predominant peaks at −10 ppm, corresponding to the D3 structure. The content ratio of the Dlinear to the total D units was 28% according to the integral ratio. Increasing the concentration of hydrolyzed Ph-H to 0.88 g mL−1 led to a decrease in the signal at −10 ppm and an increase in the signal at −25 ppm. The intensity ratio of the linear siloxane structures relative to the total D units in PolyPh-Hb increased to 39%. Increasing the condensation temperature to 100 °C for PolyPh-Hc, led to a decrease in the content of the linear siloxane structure to 31%. Decreasing the content of the linear siloxane structures may inhibit the formation of the free-standing-films.
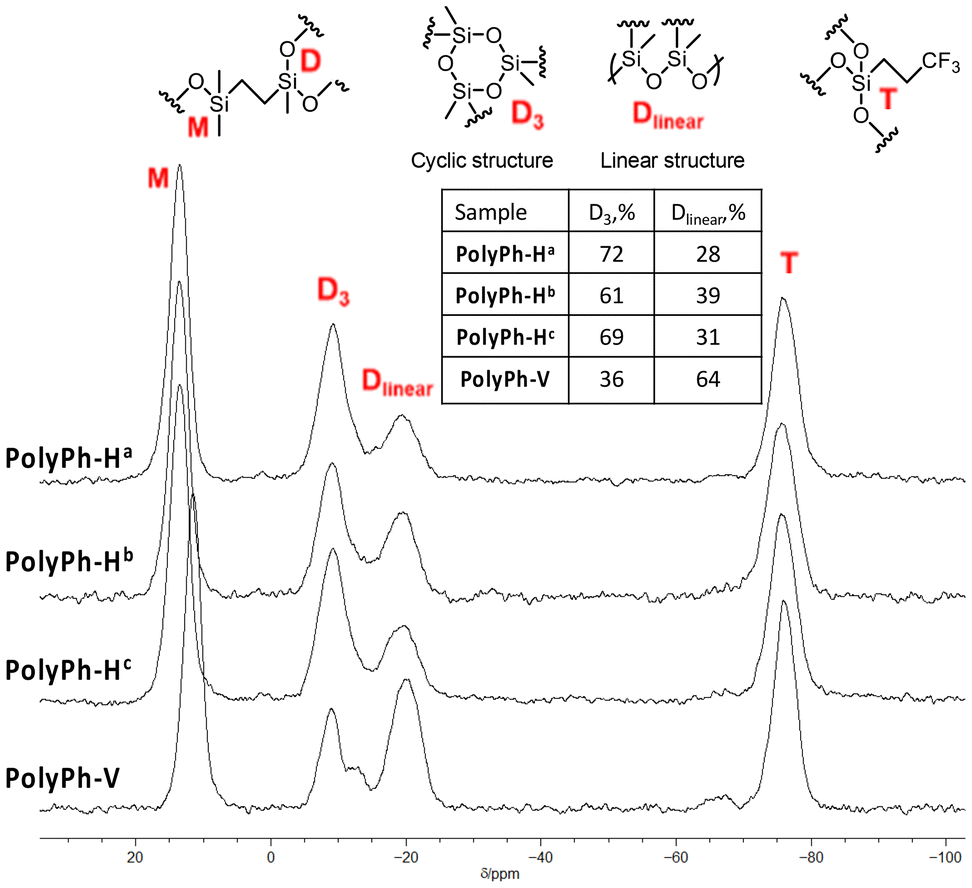 | ||
| Fig. 2 29Si CP-MAS (119 MHz) NMR spectra for PolyPh-Ha, PolyPh-Hb, PolyPh-Hc, and PolyPh-V. The insert shows the content ratio of the D3 and Dlinear units against the total D unit according to the integral ratio. | ||
The dimethoxysilyl groups of the propylenethioethyl-linked tris(dimethoxysilyl)-heptaphenyl-substituted CO-POSS (Ph-V) (Fig. S6†) were also hydrolyzed and partially condensed with 1 M HCl in THF (Scheme 2). The reaction solution was then concentrated, and the residual products were re-dissolved in THF to a concentration of 0.88 g mL−1. Subsequently, the sol solution was cast onto a substrate, air-dried for one day, and fully condensed at different temperatures under reduced pressure for polycondensation to afford an optically transparent and flexible phenyl-substituted CO-POSS-pendant polysiloxane film (PolyPh-V) (Fig. 1D). The 29Si CP-MAS NMR spectrum of the film PolyPh-V showed predominantly peaks at −25 ppm, corresponding to the linear siloxane structure. The intensity ratio of the linear siloxane structures relative to the total D units was 64%. Complete polycondensation and the absence of –SiOMe and SiOH groups were confirmed (Fig. 2 and S4†). The resulting freestanding film was sufficiently flexible to bend without cracking. The sample of PolyPh-V was insoluble in common organic solvents. SEM analysis of PolyPh-V showed smooth surface and no aggregation in the order of several micrometers (Fig. S7†).
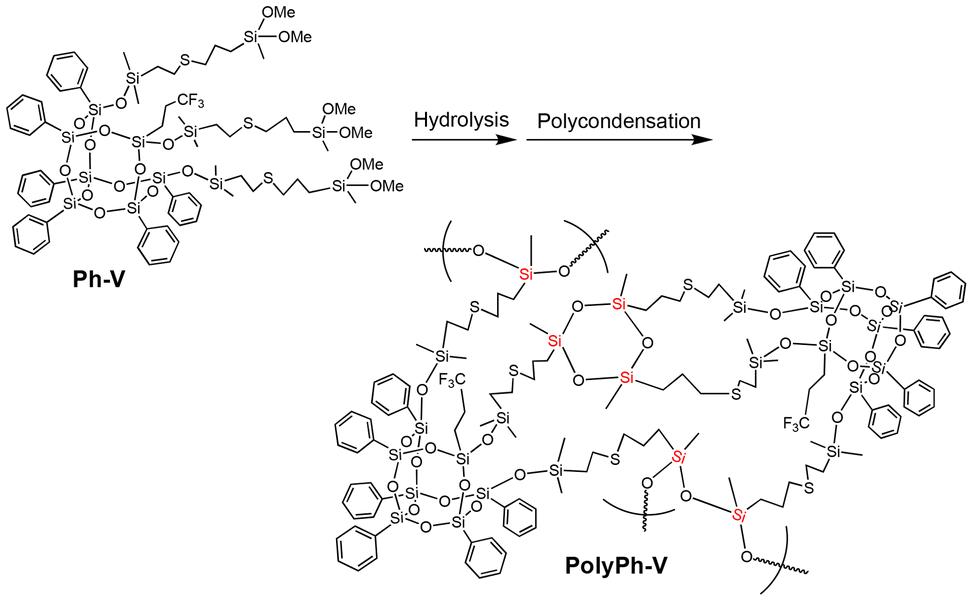 | ||
| Scheme 2 Preparation of phenyl-substituted open-cage silsesquioxane-pendant polysiloxane (PolyPh-V) from Ph-V. | ||
The densities of PolyPh-Ha, PolyPh-Hb, PolyPh-Hc were 1.2132 ± 0.0046 g cm−3, 1.2220 ± 0.0031 g cm−3, and 1.233 ± 0.005 g cm−3, respectively, based on gas displacement pycnometry analysis (Table 1). The higher density of PolyPh-Hb compared to that of PolyPh-Ha suggests that a greater proportion of linear units increased the density of the material. Furthermore, the higher density of PolyPh-Hc than those of the other materials, suggests that higher curing temperatures also increased the density. The density of PolyPh-V (1.2019 ± 0.005 g cm−3) was lower than those of PolyPh-H due to the increase in the organic content.
| Sample | ρ (kg cm−3) | C p (J (kg K)−1) | α (m2 s−1) | λ (W (m K)−1) |
|---|---|---|---|---|
| a Average values of ten independent measurements. b Average values of three independent measurements. c Average of two independent film productions and five measurements for each film. | ||||
| PolyPh-Ha | (1.2132 ± 0.0046) × 103 | (1.37 ± 0.05) × 103 | (0.89 ± 0.06) × 10−7 | 0.15 ± 0.01 |
| PolyPh-Hb | (1.2220 ± 0.0031) × 103 | (1.37 ± 0.01) × 103 | (1.30 ± 0.20) × 10−7 | 0.22 ± 0.03 |
| PolyPh-Hc | (1.2330 ± 0.0050) × 103 | (1.18 ± 0.03) × 103 | (0.80 ± 0.30) × 10−7 | 0.12 ± 0.04 |
| PolyPh-V | (1.2019 ± 0.0013) × 103 | (1.19 ± 0.02) × 103 | (0.99 ± 0.08) × 10−7 | 0.14 ± 0.01 |
| Ran-B | (1.2075 ± 0.0009) × 103 | (1.30 ± 0.06) × 103 | (1.07 ± 0.03) × 10−7 | 0.17 ± 0.01 |
| PolyF-H | (1.3765 ± 0.0036) × 103 | (1.12 ± 0.02) × 103 | (0.80 ± 0.02) × 10−7 | 0.12 ± 0.004 |
| PolyB-H | (1.0843 ± 0.0036) × 103 | (1.45 ± 0.02) × 103 | (0.77 ± 0.06) × 10−7 | 0.12 ± 0.01 |
The thermal stabilities of all the products were estimated from thermogravimetric analysis (TGA) data obtained under N2 (Fig. 3). The temperatures at which samples of PolyPh-Ha, PolyPh-Hb, PolyPh-Hc, and PolyPh-V lost 5 wt% of their original mass (Td5) under N2 were 360, 366, 376 °C, and 381 °C, respectively. The Td5 values for PolyPh-H were correlated with the density. Even considering the presence of the flexible linker unit, the highest Td5 value was obtained for PolyPh-V. In this case, predominant linear siloxane structures may provide increase higher thermal stability.
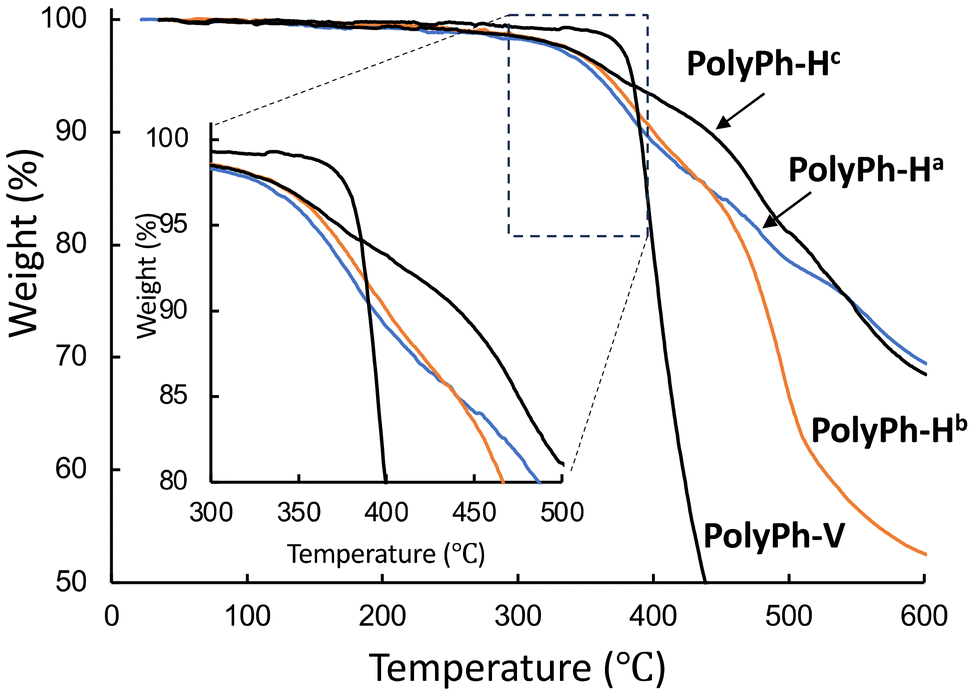 | ||
| Fig. 3 TGA thermograms of PolyPh-Ha, PolyPh-Hb, PolyPh-Hc, and PolyPh-V at a heating rate of 10 °C min−1 in N2 flow. | ||
Differential scanning calorimetry (DSC) analysis of PolyPh-Ha, PolyPh-Hb, and PolyPh-Hc revealed glass transition temperatures (Tg) under N2 of 67, 73, and 82 °C, respectively (Fig. S8†). The Tg values for PolyPh-H also correlated with the density. DSC analysis of PolyPh-V revealed a Tg value under N2 of 54 °C. No softening was observed above the Tg for PolyPh-Ha, PolyPh-Hb, and PolyPh-Hc, whereas softening of PolyPh-V was observed above its Tg. Dynamic mechanical analysis (DMA) measurements were performed on the freestanding films of PolyPh-V (Fig. S9†) showed that the Tg was manifested as a gentle step in E′, a broad peak in E′′, and a clear peak in tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ. The measured Tg was consistent with that of the Tg observed in the DSC analysis.
δ. The measured Tg was consistent with that of the Tg observed in the DSC analysis.
Thermal conductivities (λ) of the films were calculated by multiplying the corresponding thermal diffusivity (α), density (ρ), and specific heat (Cp); the results for all the tested films are summarized in Table 1. The highest thermal conductivity of 0.22 W m−1 K−1 was observed for PolyPh-Hb, with PolyPh-Ha and PolyPh-Hc showing lower thermal conductivities. Increasing the number of linear siloxane structures thus increased the thermal conductivity, especially the thermal diffusivity. A higher thermal diffusivity was observed for PolyPh-V, which may be due to the predominant content of linear siloxane structures. Decreasing the density and volumetric heat capacity reduced the thermal conductivity of PolyPh-V.
To study the effect of the CO-POSS structure, random polysilsesquioxanes were prepared from 1-(methyldimethoxysilyl)-2-(dimethylmethoxysilyl)ethane (MDME) and phenyltrimethoxysilane (PTMS) using the same components and conditions used for 1 with a MDME/PTMS molar ratio of 3:7, via hydrolysis and condensation performed under the same conditions as those used to generate PolyPh-Hb and PolyPh-Hc (Scheme 3). The reaction solution was concentrated, and the residual product was re-dissolved in THF to 0.88 g mL−1 and then condensed at either 50 °C for 6 h or 100 °C for 6 h to afford Ran-A and Ran-B, respectively. TGA analysis of Ran-A and Ran-B revealed Td5 values of 249 and 279 °C, respectively (Fig. S10†). Increasing the condensation temperature increased the Td5 value. This low condensation temperature resulted in unreacted alkoxy groups and silanol groups remaining, thereby reducing the thermal stability. The density of Ran-B (1.2075 ± 0.0009 g cm−3) was lower than that of PolyPh-Hb. The CO-POSS based-structure promoted complete polycondensation without –SiOMe and SiOH groups. A higher thermal diffusivity was observed for Ran-B than for PolyPh-Ha and PolyPh-Hc, but lower than that for PolyPh-Hb.
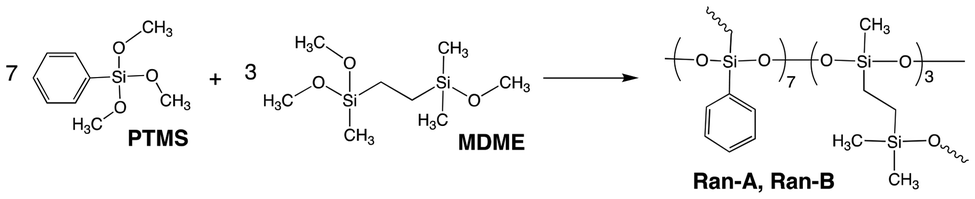 | ||
| Scheme 3 Synthesis of random polysilsesquioxanes (Ran-A and Ran-B) from 1-(methyldimethoxysilyl)-2-(dimethylmethoxysilyl)ethane (MDME) and phenyltrimethoxysilane (PTMS). | ||
Tris(dimethoxysilyl-ethyl-dimethylsiloxy)-heptatrifluoropropyl-substituted open-cage silsesquioxane (F–H) (Fig. 4) and tris(dimethoxysilyl-ethyl-dimethylsiloxy)-heptaisobutyl-substituted open-cage silsesquioxane (B–H) were hydrolyzed and condensed to afford isobutyl- and trifluoropropyl-substituted open-cage silsesquioxane-pendant polysiloxanes PolyF-H and PolyB-H under the same conditions as PolyPh-H, except using toluene in the case of PolyB-H. The relative intensity ratios of the linear siloxane structures to the total D units are 52% for PolyF-H and 62% for PolyB-H, respectively (Fig. S11†). The thermal conductivities and thermal diffusivities were lower than those of PolyPh-Hb.
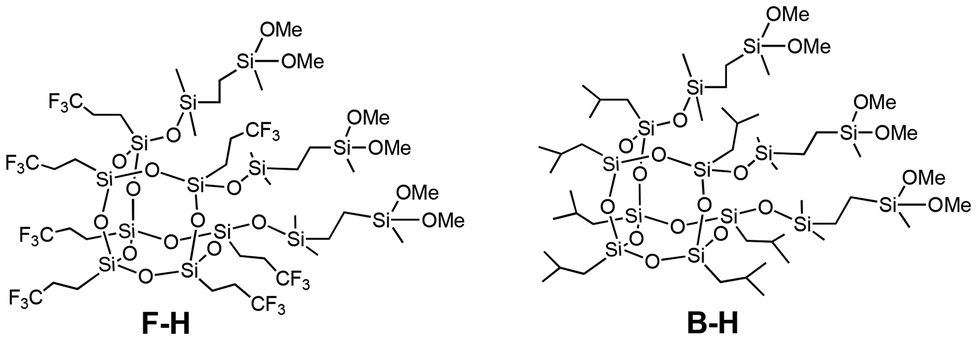 | ||
| Fig. 4 Chemical structures of F–H and B–H. | ||
The surface hydrophobicity of the films was investigated by static contact angle measurements with probe liquids such as water (θwater) and n-tetradecane (θn-tetradecane) (Fig. 5). The contact angle for PolyPh-Hc was difficult to assess because of its many cracks. The water contact angles of PolyPh-Ha, PolyPh-Hb, and PolyPh-V were 97.1°, 99.4°, and 100.7°, respectively. n-Tetradecane contact angles of PolyPh-Ha, PolyPh-Hb, and PolyPh-V were 9.4°, 5.1°, and 14.3°, respectively. The surface free energies of the cast films were estimated using the geometric mean model described by eqn (1) and (2):
| 1 + cosθ = (2/γL)[(γdLγds)0.5 + (γpLγps)0.5] | (1) |
| γds + γps = γs | (2) |
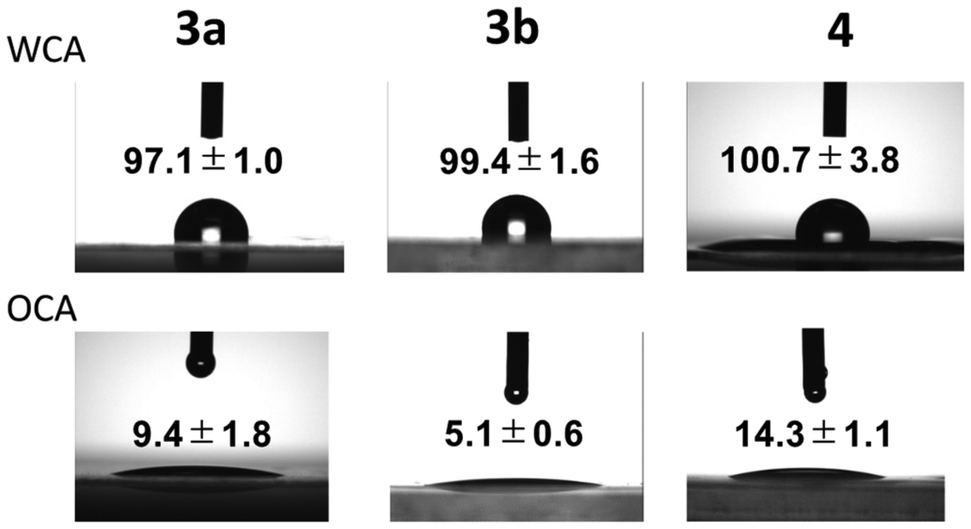 | ||
| Fig. 5 Static water and n-tetradecane contact angles of the films of PolyPh-Ha, PolyPh-Hb, and PolyPh-V. | ||
A solubility study of isobutyl- and phenyl-substituted T8 cages suggested that the bulky isobutyl groups were completely covered by the polar siloxane cage frame work.23 In contrast, part of the polar siloxane cage framework was exposed to the environment for derivatives with phenyl substituents. In this study, the surface free energies of PolyPh-Ha, PolyPh-Hb, and PolyPh-V were higher than those of PolyF-H and PolyB-H (Table 2). The surface free energy of Ran-A was lower than those of PolyPh-Ha and PolyPh-Hb, suggesting that the siloxane backbone in the cage structure with high curvature was more exposed than in the random type.
| Sample | Static contact angles (°) | Surface free energya (mN m−1) | |||
|---|---|---|---|---|---|
| θ water | θ n-tetrodecane | γ s d | γ s p | γ s | |
| a Water: γdwater = 26.4 mN m−1, γpwater = 46.4 mN m−1; n-tetradecane: γdn-tetradecaner = 26.4 mN m−1, γpn-tetradecaner = 0.0 mN m−1. | |||||
| PolyPh-Ha | 97.1 ± 1.0 | 9.4 ± 1.8 | 26.0 | 0.7 | 26.7 |
| PolyPh-Hb | 99.4 ± 1.6 | 5.1 ± 0.6 | 26.3 | 0.4 | 26.7 |
| PolyPh-V | 100.7 ± 3.8 | 14.3 ± 1.1 | 25.6 | 0.3 | 25.9 |
| Ran-A | 98.0 ± 0.5 | 21.7 ± 0.7 | 24.6 | 0.7 | 25.3 |
| PolyF-H | 96.2 ± 2.3 | 31.3 ± 4.1 | 22.7 | 1.4 | 24.1 |
| PolyB-H | 98.7 ± 1.1 | 23.2 ± 1.2 | 24.3 | 0.7 | 25.0 |
Transmittance spectra corresponding to varying irradiation times for PolyPh-Hb were used to study the UV resistance in air (Fig. 6A). The initial transmittance spectrum of PolyPh-Hb showed a high transmittance from 300 nm to 600 nm. No significant change in the transmittance was observed after PolyPh-Hb was irradiated with 365 nm UV light for 6 days. It is known that organic substituents, such as methyl groups and unreacted residual alkoxy groups in siloxane absorb UV light and generate radicals;3,5 this can result further oxidation and a reduction in transparency. Complete consumption of the alkoxy groups thus suppresses decomposition upon exposure to UV light. The transmittance of PolyPh-V at approximately 300 nm decreased upon exposure to UV irradiation for several days (Fig. 6B). The decrease in the transmittance at 300 nm was due to the absorption by sulfoxide formed by the oxidation of sulfur atoms. No decrease in the transmittance was observed at wavelength longer than 350 nm; thus 4 appeared colorless and transparent even after 6 days of UV irradiation.
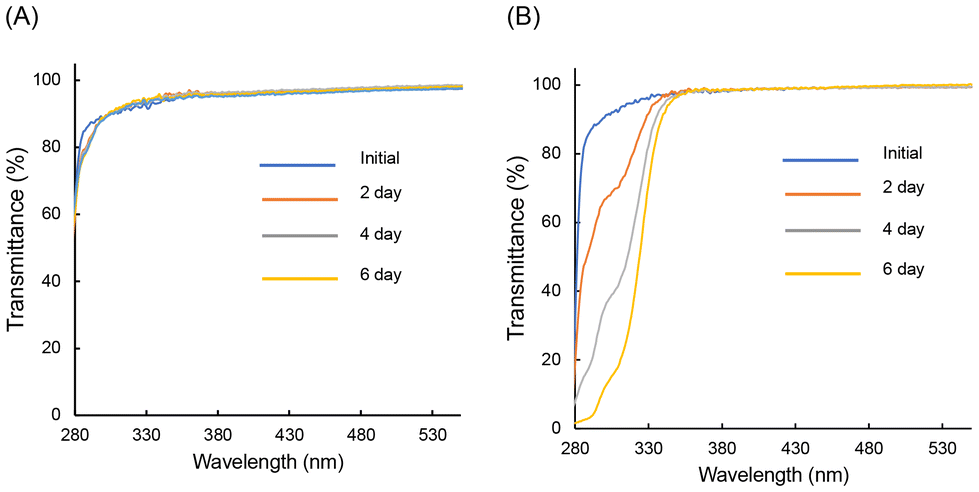 | ||
| Fig. 6 Transmittance spectra of (A) PolyPh-Hb, and (B) PolyPh-V, along with UV irradiation time in air. The thicknesses were within 39–75 μm. | ||
Conclusions
We prepared phenyl-substituted IC-POSSs bearing tris(dimethoxysilyl) groups at the open positions and optimized their sol–gel reactions to obtained optically transparent free-standing films. Polycondensation was fully completed via a low-temperature solution process, even at 50 °C for 6 h under vacuum. The Td5 values under N2 for PolyPh-Ha, PolyPh-Hb, and PolyPh-Hc (360, 366, and 376 °C, respectively) correlated with their densities. The highest Td5 value was obtained for PolyPh-V, even in the presence of the flexible linker unit. The predominant linear siloxane structures may provide increase higher thermal stability. The film of PolyPh-Hb exhibited high optical transparency, heat resistance, and UV-resistance. Optically transparent materials that are resistant to UV light have attracted considerable attention because of their important practical applications. Previously, we reported that trifluoropropyl-substituted CO-POSS-pendant polysiloxanes exhibited UV-resistant optically transparent materials. The present study shows that phenyl-substituted CO-POSS-pendant polysiloxanes are alternative UV-resistant, optically transparent materials with higher heat resistance. Whereas the flexibility of PolyPh-Hb was relatively low, the film of PolyPh-V could be bent without cracking, it was lightweight, and it exhibited high heat resistance. However, a lower Tg and reduced UV resistance were observed. This study is expected to contribute to a better understanding of silicone materials, where the relationship between molecular-level structure and bulk properties is unknown. The present study will aid in the fabrication of optically transparent materials that combine several trade-off properties, such as thermal insulation, heat resistance, UV resistance, flexibility, high refractive index, and light weight characteristics.Experimental
Materials
All solvents and chemicals were of reagent-grade quality and used without further purification. Tetrahydrofuran (super dehydrated, stabilizer-free) was purchased from Fujifilm Wako Chemicals U.S.A. Corporation. 3,7,14-Tris(dimethylsilylsiloxy)heptaphenyltricycloheptasiloxane (Ph-H) and 3,7,14-tris(dimethylvinylsilylsiloxy)heptaphenyltricycloheptasiloxane (Ph-V) were prepared according to a previous report24 from heptaphenyl-trisilanol-POSS, which were purchased from Hybrid Plastics Inc., (Hattiesburg, Mississippi, US). Tris(dimethoxysilyl-ethyl-dimethylsiloxy)-heptatrifluoropropyl-substituted open-cage silsesquioxane (F–H) and tris(dimethoxysilyl-ethyl-dimethylsiloxy)-heptaisobutyl-substituted open-cage silsesquioxane (B–H) was prepared according a previous report.18 Dimethylmethoxyvinylsilane was prepared from chlorodimethylvinylsilane and methanol in 4-methyltetrahydropyran with imidazole and purified by distillation (Fig. S12†). Its boiling temperature was similar to that reported data, 80–85 °C.25 1-(Methyldimethoxysilyl)-2-(dimethylmethoxysilyl)ethane (MDME) was prepared by hydrosilylation from dimethoxymethylsilane and dimethylmethoxyvinylsilane (Fig. S13†).Instruments
A Bruker AVANCE II 600 spectrometer was used to obtain solid-state 13C (150 MHz) and 29Si (119 MHz) cross-polarization magic angle spinning (CP-MAS) NMR spectra. Fourier transform infrared (FT-IR) spectra were recored with a JASCO FT/IR-4600 spectrometer (JASCO, Tokyo, Japan). A Jasco spectrophotometer V-670 KKN (Jasco, Tokyo, Japan) was used to record UV-vis spectra. TGA was performed using a Shimadzu DTG-60 Thermogravimetric Analyzer (Shimadzu, Kyoto, Japan) under N2 or dry air at a heating rate of 10 °C min−1. DSC was conducted using a Shimadzu DSC-60 Plus. DMA of rectangular samples (20 mm × 9.5 mm × 0.15 mm), which were cut from the cast films, was performed using a DMA7100 (Hitachi High-Tech) instrument at a heating rate of 2 °C min−1 at 1 Hz under N2 flow. An AccuPyc II 1340 gas displacement pycnometry system (Micromeritics Instrument Corporation, Norcross, CA, USA) was used to obtain density data; the sample was placed in a 0.1 cm3 chamber and the measured pressure of He was 135 kPa at 25 °C. The thermal diffusivity of the films was measured as a value in the thickness direction using an ai-phase mobile 1u (ai-phase Co. Ltd, Tokyo, Japan) based on the temperature wave analysis method. The specific heat capacity was estimated using a DSC-60 Plus (Shimadzu, Kyoto, Japan) with alumina as the reference substance. The UV irradiation tests were performed under air at room temperature using a UV lamp (365 nm) placed 1 cm from the cast films. Static contact angles of the films were measured at 25 °C using a DMs-301/401 (Kyowa Interface Science, Saitama, Japan) with water (1.0 μL) or n-tetradecane (1.0 μL) as probe fluids. The average of four independent measurements was used for each contact angle. The ultraviolet irradiation test was performed using a UV lamp (365 nm) with a distance of 1 cm to cast films under air at room temperature.Synthesis
Film preparation
To a solution of Ph-H (0.25 g, 0.17 mmol) in THF (6.47 mL) was added distilled water (0.40 mL) and 1 M HCl, (0.023 mmol) and the resulting solution was stirred at 0 °C for 0.5 h and at room temperature for 3 h to allow hydrolysis and partial condensation. The reaction mixture was then concentrated under vacuum and re-dissolved in THF (2 mL), followed by filtering through a 0.45 μm PTFE syringe filter. The solution was cast onto a clean quartz substrate or a PTFE sheet, air-dried at room temperature for one or two days to remove the most of the solvents, and heated at either 50 °C for 6 h for PolyPh-Hb or 100 °C for 6 h for PolyPh-Hc. To prepare PolyPh-Ha, the residues obtained after the hydrolysis of Ph-H under the same conditions used to prepare PolyPh-Hb and PolyPh-Hc described above, was re-dissolved in THF (1.56 mL) and heated at 50 °C for 6 h.Random polysiloxanes were prepared from MDMS and phenyltrimethoxysilane (PTMS) at a molar ratio of 3:7 under the same conditions used for 1. The reaction solutions were concentrated under vacuum and the resulting residue was redissolved in THF (1.56 mL). The solution was drop-cast onto a clean quartz substrate or a PTFE sheet. The samples were air-dried at room temperature for 1 d to remove the most of the solvents and heated at either 50 °C for 6 h (Ran-A) or 110 °C for 6 h (Ran-B).
Author contributions
M. Kosaka: synthesis, measurement, data curation; K. Kanaori: measurement, data curation, review and editing; H. Imoto: conceptualization, investigation, writing – review and editing, supervision; K. Naka: conceptualization, investigation, writing – original draft, writing – review and editing, funding acquisition project administration, supervision.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Raw data were generated at Kyoto Institute of Technology. Derived data supporting the findings of this study are available from the corresponding author on request.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grant-in-Aid for Scientific Research (No. 19H02764 and 23K17944) from the Ministry of Education, Culture, Sports, Science, and Technology, Government of Japan. We thank Prof. Tsuyoshi Kawai, Ms Yoshiko Nishikawa, and Ms Mieko Yamagaki of Nara Institute of Science and Technology for performing MALDI-TOF-MS supported by ARIM.References
- A. M. Sawvel, J. C. Crowhurst, H. E. Mason, J. S. Oakdale, S. Ruelas, H. V. Eshelman and R. S. Maxwell, Macromolecules, 2021, 54, 4300–4312 CrossRef CAS.
- C. Robeyns, L. Picard and F. Ganachaud, Prog. Org. Coat., 2018, 125, 287–315 CrossRef CAS.
- J.-Y. Bae, H.-Y. Kim, Y.-W. Lim, Y.-H. Kim and B.-S. Bae, RSC Adv., 2016, 6, 26826–26834 RSC.
- J.-S. Kim, S.-C. Yang and B.-S. Bae, Chem. Mater., 2010, 22, 3549–3555 CrossRef CAS.
- J.-Y. Bae, H.-Y. Kim, Y.-B. Kim, J. Jin and B.-S. Bae, ACS Appl. Mater. Interfaces, 2015, 7, 1035–1039 CrossRef CAS PubMed.
- H. R. Fischer, C. Semprimoschnig, C. Mooney, T. Rohr, E. R. H. van Eck and M. H. W. Verkuijlen, Polym. Degrad. Stab., 2013, 98, 720–726 CrossRef CAS.
- H. W. Ro, E. S. Park, C. L. Soles and D. Y. Yoon, Chem. Mater., 2010, 22, 1330–1339 CrossRef CAS.
- L.-H. Lee, W.-C. Chen and W.-C. Liu, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1560–1571 CrossRef CAS.
- J. H. Lee, W. C. Kim, S. K. Min, H. W. Ree and D. Y. Yoon, Macromol. Mater. Eng., 2003, 288, 455–461 CrossRef.
- Y. Chujo and K. Tanaka, Bull. Chem. Soc. Jpn., 2015, 86, 633–643 CrossRef.
- R. M. Laine, J. Mater. Chem., 2005, 15, 3725–3744 RSC.
- D. B. Cordes, P. D. Lickiss and F. Rataboul, Chem. Rev., 2010, 110, 2081–2173 CrossRef CAS PubMed.
- H. Imoto, Y. Nakao, N. Nishizawa, S. Fujii, Y. Nakamura and K. Naka, Polym. J., 2015, 47, 609–615 CrossRef CAS.
- M. Miyasaka, Y. Fujiwara, H. Kudo and T. Nishikubo, Polym. J., 2010, 42, 799–803 CrossRef CAS.
- H. Imoto, A. Ishida, M. Hashimoto, Y. Mizoue, S. Yusa and K. Naka, Chem. Lett., 2019, 48, 1266–1269 CrossRef CAS.
- L. Li, H. Imoto and K. Naka, J. Appl. Polym. Sci., 2021, 138, 50167 CrossRef CAS.
- T. Nakano, K. Okamoto, H. Imoto and K. Naka, Polym. J., 2023, 55, 193–201 CrossRef CAS.
- L. Li, H. Imoto, A. Okada, K. Kanaori and K. Naka, ACS Appl. Polym. Mater., 2021, 3, 1368–1375 CrossRef CAS.
- M. Kosaka, T. Nakano, K. Kanaori, H. Imoto and K. Naka, Polym. J., 2024, 56, 481–489 CrossRef CAS.
- A. Fina, D. Tabuani, F. Carniato, A. Frache, E. Boccaleri and G. Caminoa, Thermochim. Acta, 2006, 440, 36–42 CrossRef CAS.
- Z. Zhang, B. P. Gorman, H. Dong, R. A. Orozco-Teran, D. W. Mueller and R. F. Reidy, J. Sol-Gel Sci. Technol., 2003, 28, 159–165 CrossRef CAS.
- T. B. Casserly and K. K. Gleason, J. Phys. Chem. B, 2005, 109, 13605–13610 CrossRef CAS PubMed.
- A. J. Guenthner, K. R. Lamison, L. M. Lubin, T. S. Haddad and J. M. Mabry, Ind. Eng. Chem. Res., 2012, 51, 12282–12293 CrossRef CAS.
- S. Yuasa, Y. Sato, H. Imoto and K. Naka, Bull. Chem. Soc. Jpn., 2019, 92, 127–132 CrossRef CAS.
- P. R. Jones, T. F. O. Thomas, M. L. McBee and R. A. Pierce, J. Organomet. Chem., 1987, 159, 99–110 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra, MALDI-TOF-MS spectra, FT-IR spectra, DSC and DMA analyses. See DOI: https://doi.org/10.1039/d4py01460j |
| This journal is © The Royal Society of Chemistry 2025 |