
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStoichiometric effects on bulk stress relaxation to enhance reprocessability in covalent adaptable networks†
Jaehyun
Cho
a,
Santanu
Ghosh
c,
Mridula
Nandi
c,
Heejoon
Jeon
 b,
Liang
Yue
d,
H. Jerry
Qi
d,
M. G.
Finn
c and
Blair
Brettmann
*ab
b,
Liang
Yue
d,
H. Jerry
Qi
d,
M. G.
Finn
c and
Blair
Brettmann
*ab
aSchool of Chemical and Biomolecular Engineering, Georgia Institute of Technology, Atlanta, GA, USA. E-mail: blair.brettmann@chbe.gatech.edu
bSchool of Materials Science and Engineering, Georgia Institute of Technology, Atlanta, GA, USA
cSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA, USA
dThe George W. Woodruff School of Mechanical Engineering, Georgia Institute of Technology, Atlanta, GA, USA
First published on 22nd January 2025
Abstract
Achieving maximum crosslinking density is often thought to be crucial for high performance and mechanical strength in conventional thermoset covalent adaptable networks (CANs), although some network defects can enhance properties such as toughness and reprocessability. Controlling functional group stoichiometry and crosslinking density is therefore key to better understanding and programming the properties and reprocessability of CANs. Despite efforts to systematically control CAN properties, many previous studies show inconsistent trends in stress relaxation rates, hindering a clear understanding of the factors affecting reprocessability. We show that stoichiometry is a critical factor influencing bulk stress relaxation for a dissociative type of CAN based on the aza-Michael addition reactions utilizing triacrylate monomer and two polymeric amines (Jeffamine and polyethyleneimine, PEI). Relative functional group reactivity was characterized by Fourier transform infrared spectroscopy, and four different stoichiometries were analyzed. Thermal reprocessing demonstrated the stability of mechanical properties over multiple cycles, notably in stoichiometrically balanced systems, consistent with the general principle that mechanical properties and crosslinking density reach a maximum with stoichiometric equivalence. These findings underscore the importance of careful stoichiometric design in tailoring viscoelastic properties and mechanical behavior in dissociative type CAN systems, offering insights into polymer material design for applications requiring dynamic properties and reprocessability.
Introduction
Polymeric materials are traditionally classified into two categories: thermoplastics and thermosets, distinguished by their molecular structures and thermal behavior. Thermoplastics are characterized by weak intermolecular forces, often making them prone to low heat resistance and relatively low mechanical strength, whereas thermosets form cross-linked networks that are challenging to recycle. Recent advances in polymer science have introduced ‘reversible’ bonds to bridge the gap between these two distinct types of materials, resulting in a novel type of crosslinked polymer that can undergo reprocessing under heat. The reversible behavior is achieved by incorporating dynamic covalent bonds1–3 and/or dynamic noncovalent interactions4,5 into polymer networks. Dynamic noncovalent interactions include ionic,6 telechelic,7 metal-coordinate,8 and supramolecular interactions9,10via hydrogen bonding10 or pi–pi stacking,9 each generally less robust than covalent bonds (bond strengths <100 kJ mol−1vs. >350 kJ mol−1, respectively).11,12 Thus, covalent adaptable networks (CANs), which consist of reversible covalent bonds, possess better mechanical strength than noncovalent systems.5 Many types of covalent dynamic bonds have been introduced over the last decade, with CANs based on exchange reactions involving boronate esters,13 thioesters,14,15 Diels–Alder adducts,5 disulfide bonds,16 aza-Michael adducts,17,18 thia-Michael adducts,19 urea linkages,20 and imine bonds.21 Due to their transient bond exchange reactions, CANs are widely explored for applications such as self-healing for spontaneous damage repair,20,22,23 shape memory effects,24 and reprocessing and recycling of used materials.25,26Controlling the network design and chemistry of CANs is crucial as these factors determine the final properties of CAN-based materials. For instance, the use of external catalysts to promote network exchange poses challenges due to potential toxicity and poor catalyst thermal stability in reprocessing,27,28 prompting the development of catalyst-free CAN systems.29 And the glass transition temperature of the CAN material can be tuned through the stoichiometric ratios of the monomers, as was shown for epoxy thermoset polymers.30 Likewise, due to the thermal sensitivity of CANs, their mechanical properties can change with heat and humidity variations during product use by consumers, which are important considerations for fabrics,31 adhesives,32 sensors,33 and drug delivery systems,34 where these environmental changes are likely. It is therefore important to understand how the polymer chemistry and network structure impact the dynamic properties of CAN systems if one is to effectively design such materials.
The promotion of faster bond exchange rates is essential to increasing the efficiency of reprocessing and recycling of catalyst-free CANs,35,36 as these materials reach a new equilibrium more quickly. This leads to a viscosity drop, making the reprocessing more efficient. In other words, faster bond exchange rates facilitate the reprocessing of materials. In addition to the intrinsic stabilities of the exchangeable bonds,37 other influential factors can include the physical properties of the polymer (backbone stiffness), the nature of the polymer network (degree of crosslinking), and the concentration of the participating functional groups. The effective number of molecular moieties available for exchange nearby decreases with higher backbone rigidity, higher crosslinking density, and slow diffusion of the exchangeable groups.29,38,39 While these factors have been addressed in some efforts to tune the rate of reprocessing, the complexity of the systems has led to few structured studies of targeted molecular design parameters.
Examples of improvement in bond exchange rate by intrinsic chemical factors have included the development of new types of chemical pairs that enable thermodynamically favored bond exchanges,40 the utilization of small molecules as internal catalysts to accelerate exchange reactions,22,41 and neighboring group participation of the kind widely adopted in organic chemistry.38,39 Explorations of molecular network factors have involved variation in crosslinking density, molecular movement, and flexibility, suggesting that a slower relaxation is likely to happen when the polymer chains positioned near exchangeable covalent bonds are restricted in their movements.39,42 Often, the relative importance of each type of contribution is unclear.42–44 For example, some reports indicate that faster bond exchanges occur under conditions of relatively high chain mobility,45–47 while other polymer sets show fast bond exchanges even with highly dense and crosslinked networks that restrict chain mobility.42,48 However, there is a lack of systematic research on how these diverse chemical factors and molecular structures integrally influence the mechanical properties and reprocessability, which would enable design of more efficient reprocessing and recycling.
Prior work on CANs has highlighted the stoichiometry between the functional groups participating in the exchange reactions as a crucial factor influencing the bond exchange and material properties.30,45,49–55 In a majority of cases, the nominal stoichiometry was taken as the true stoichiometry and not experimentally measured.30,49–53,55 This can be a reasonable assumption for simple, small molecule reactants, but for oligomeric and polymeric monomers, the actual functionality can differ greatly from the nominal functionality.56,57 Because slight changes in composition can significantly impact the bond relaxation and material properties,54 use of nominal rather than measured functionality in designing stoichiometry of CANs from polymeric/oligomeric monomers can lead to performance that differs significantly from expected behavior and difficulty in identifying trends. Thus, here, we examine the effect of stoichiometry on the bond exchange rates, material properties and reprocessability using experimentally determined functionalities of the monomers to select on-stoichiometry, excess-amine and excess-acrylate compositions. To our knowledge, there is only one prior study of stoichiometry in CANs that experimentally measures the functionality, specifically that of furan-modified rubber via NMR, to select compositions of varying stoichiometric ratios, in that case with bismaleimide.45 However, in that work only the excess furan case was studied and the work was not carried through to reprocessing studies, as the focus was on the relaxation mechanism of the CAN polymers.
In this work, we focused on the stoichiometry of aza-Michael crosslinking groups using branched polyethyleneimine (PEI) as a multifunctional and branched molecular crosslinker, Jeffamine as a linear chain extender to modify flexibility, and a triacrylate for the crosslinked network (Fig. 1A). We assessed the bond exchange rate, bulk stress relaxation, and reprocessability for four sets of compositions: (1) ‘on-stoichiometry’, where the total number of amine and acrylate groups is precisely matched, assuming full consumption of bond exchangeable units, (2) ‘excess amine’ with PEI, (3) ‘excess acrylate’ formed by reducing the overall amine content relative to acrylate with both Jeffamine and PEI concurrently changing, and (4) ‘excess acrylate’ with constant Jeffamine content and varying amounts of PEI. We found consistently increased rates of stress relaxation for ‘off-stoichiometry’ compositions (both excess amine and excess acrylate), illustrating a fundamental design tradeoff between mechanical strength and reprocessability accessible through convenient adjustment of the material composition.
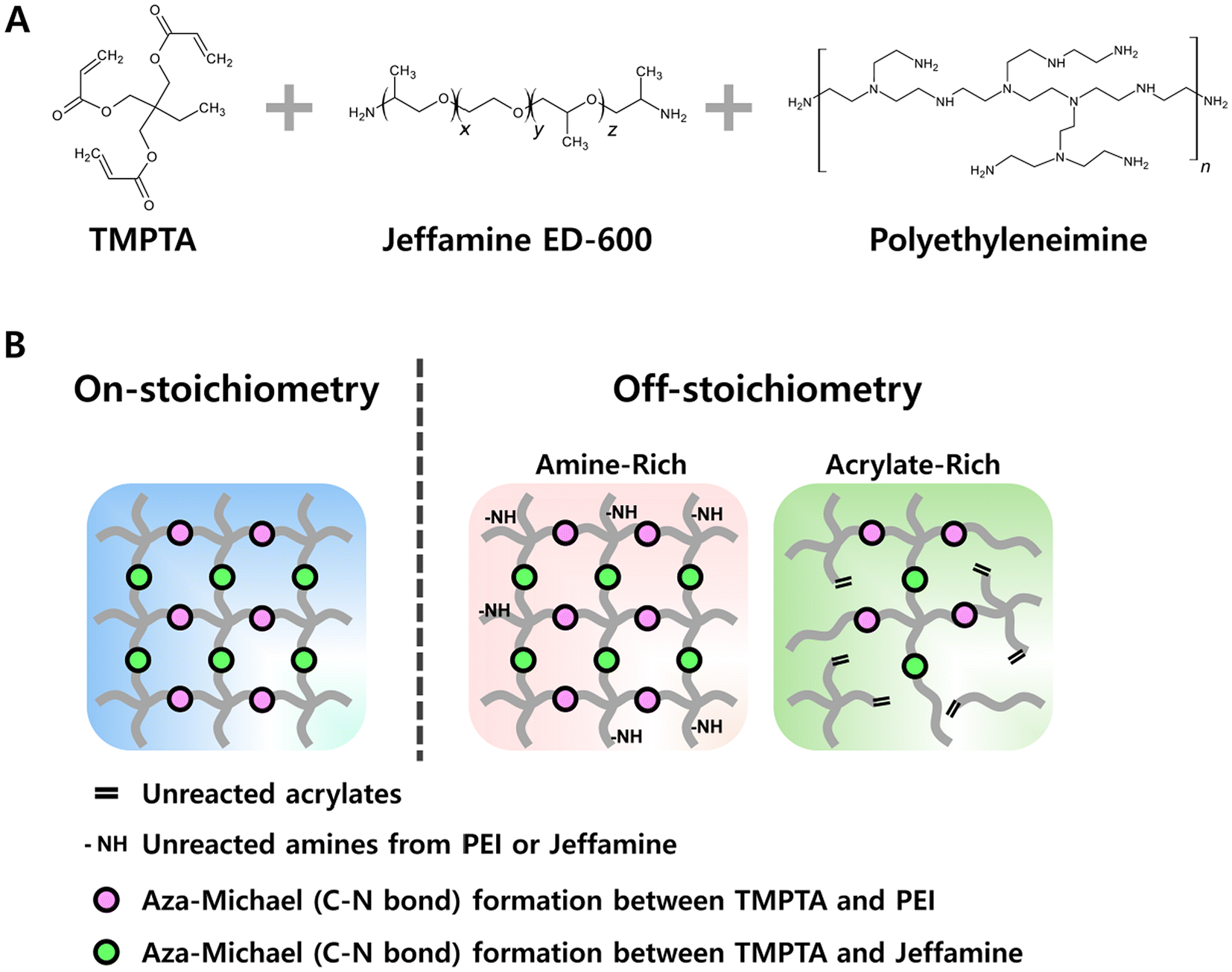 | ||
| Fig. 1 (A) Chemical schematic of the three-components in the aza-Michael reaction-based design and (B) illustrations of the CANs showing the differences between ‘on-stoichiometry’ and ‘off-stoichiometry’, representing excess amine and excess acrylate scenarios. | ||
Results and discussion
A dissociative type of CAN based on the aza-Michael addition reaction (attack of an amine nucleophile on an acrylate electrophile) was designed with a triacrylate, trimethylolpropane triacrylate (TMPTA), and polymeric amines, Jeffamine and PEI (Fig. 1A). Jeffamine provides a flexible backbone, which produces CAN films that are not brittle and can be conveniently handled. We chose PEI as our primary bond exchanging moiety because PEI contributes a higher number of exchangeable units (primary and secondary amines) along with tertiary amine sites that can function as catalysts of aza-Michael addition and reversion, facilitating faster bond exchange. The secondary amine groups of PEI are expected to participate in exchange reactions at different rates than primary amines.58In branched materials, the effective functionality of the reactive centers often deviates from the theoretical values due to steric hindrance, chain mobility, and differences in reactivity of different amine species (i.e., primary, secondary, and tertiary).59 We therefore focused on functional stoichiometry by empirically measuring the reactivity of the component materials (in terms of functionality parameter, f) using attenuated total reflectance Fourier transform infrared spectroscopy (ATR-FTIR) to measure the disappearance of acrylate groups during or after the aza-Michael addition reaction of different compositions (ESI Fig. S1–S5†). Starting with a fixed amount of TMPTA and using the nominal stoichiometric reactivity of Jeffamine based on product data sheets, we prepared a series of samples differing in both directions from theoretical stoichiometric equivalence (i.e., TMPTA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Jeffamine molar ratio = 1:0.75 to 1:1.5). The extent of these reactions was determined by measurement of the peak areas of characteristic acrylate peaks normalized to the C
:Jeffamine molar ratio = 1:0.75 to 1:1.5). The extent of these reactions was determined by measurement of the peak areas of characteristic acrylate peaks normalized to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band (Fig. S3B–3G and S5B–5G†) and the expected shift in the frequency of that band from 1729 to 1735 cm−1 (ESI Fig. S4†). The stoichiometric point is taken as the TMPTA:Jeffamine ratio at which the peak area has decreased by at least 80% of the initial peak area and this was used to estimate f. Using this FTIR method, the empirical f values for the reactive groups per molecule of Jeffamine and PEI were estimated to be in the range of f = 2.1–2.3 and f = 10.7–12, respectively. Interestingly, these values can differ significantly from those reported in the literature and commercially available data.17 For example, commercially available data sheets (from Huntsman Jeffamine Polyetheramine ED-600) list the amine hydrogen equivalent weight of Jeffamine as 132 g per eq (f = 4.55), while our FTIR analysis suggests it to be 273 g per eq (f = 2.2; within 2.1–2.3), and 1H NMR estimates it to be as high as 310–330 g per eq (f = 1.85–1.9, data shown in ESI Fig. S6†), close to the f values estimated by FTIR analysis.
O stretching band (Fig. S3B–3G and S5B–5G†) and the expected shift in the frequency of that band from 1729 to 1735 cm−1 (ESI Fig. S4†). The stoichiometric point is taken as the TMPTA:Jeffamine ratio at which the peak area has decreased by at least 80% of the initial peak area and this was used to estimate f. Using this FTIR method, the empirical f values for the reactive groups per molecule of Jeffamine and PEI were estimated to be in the range of f = 2.1–2.3 and f = 10.7–12, respectively. Interestingly, these values can differ significantly from those reported in the literature and commercially available data.17 For example, commercially available data sheets (from Huntsman Jeffamine Polyetheramine ED-600) list the amine hydrogen equivalent weight of Jeffamine as 132 g per eq (f = 4.55), while our FTIR analysis suggests it to be 273 g per eq (f = 2.2; within 2.1–2.3), and 1H NMR estimates it to be as high as 310–330 g per eq (f = 1.85–1.9, data shown in ESI Fig. S6†), close to the f values estimated by FTIR analysis.
Using this determination of quantitative reactivity of the component monomers, we designed four different scenarios to explore the effect of stoichiometry on material properties such as stress relaxation, crosslinking density, and mechanical strength. Table 1 summarizes these scenarios and the range of compositions tested. The first category, labeled as ‘on-stoichiometry’, set an exact balance of the total number of acrylate and reactive amines. The second category, labeled as ‘excess PEI’, represents the amine-rich case achieved by adding more PEI to a stoichiometrically matched mixture of TMPTA and Jeffamine. The last two composition categories were designed to have excess acrylate by reducing the amount of Jeffamine; in one case changing the Jeffamine:PEI ratio to ensure that the total number of amines remains the same, and the other in which the Jeffamine concentration was fixed while increasing the PEI concentration. Fig. 1B shows idealized representations of these scenarios with dots representing the dynamic C–N linkages.
| Composition category | Sample code | Molar ratio | Functional group | |||||
|---|---|---|---|---|---|---|---|---|
| TMPTA (mol) | JEFFAMINE (mol) | PEI (mol) | CCTMPTA (eq) |
NHJeffamine (eq) | NHPEI (eq) | |||
| On-stoichiometry | 3J + 0P | 1 | 1.36 | 0 | 3 | 3 | 0 | |
| 2J + 1P | 0.91 | 0.08 | 2 | 1 | ||||
| 1J + 2P | 0.45 | 0.15 | 1 | 2 | ||||
| Off-stoichiometry | Excess amine | 3J + 0.5P | 1.36 | 0.04 | 3 | 0.5 | ||
| 3J + 0.9P | 1.36 | 0.07 | 3 | 0.9 | ||||
| Excess acrylate (decreasing Jeffamine and increasing PEI) | 2J + 0P | 0.75 | 0 | 2 | 0 | |||
| 1J + 1P | 0.5 | 0.07 | 1 | 1 | ||||
| 0.5J + 1.5P | 0.25 | 0.13 | 0.5 | 1.5 | ||||
| Excess acrylate (fixed Jeffamine and increasing PEI) | 2J + 0.4P | 0.75 | 0.03 | 2 | 0.4 | |||
| 2J + 0.7P | 0.75 | 0.05 | 2 | 0.7 | ||||
Each of the compositions shown in Table 1 were prepared and characterized as described in detail in the methods section. We first examined the stress relaxation of the CANs by dynamic mechanical analysis (DMA). In this test, applied stress diminishes over time through molecular rearrangements, and thus the rate of relaxation is directly relevant to the rates of dynamic bond exchange and network movement.43,60 These factors are expected to be different when more free reactive groups are present. Fig. 2 shows the characteristic time for stress relaxation of each composition, defined as the time when the relaxation modulus reaches 1/e of its initial value; based on the single mode Maxwell relationship, this is considered full relaxation (The stress relaxation raw data is provided in ESI Fig. S7.†).
 | ||
| Fig. 2 Characteristic time extracted from the stress relaxation measurements by DMA for the four compositional categories shown in Table 1: (A) ‘on-stoichiometry’, (B) excess amine, (C) excess acrylate relative to constant total amine concentration, changing Jeffamine:PEI ratio, (D) excess acrylate relative to constant Jeffamine concentration and increasing PEI concentration. Lighter lines and points indicate higher temperatures. The standard deviation of 3 samples is indicated by the shaded region. | ||
For the ‘on-stoichiometry’ sets, no significant change was observed in the characteristic relaxation time with changing Jeffamine:PEI ratio at any temperature (Fig. 2A). This suggests that the tertiary amines (present in PEI but not Jeffamine) are not acting as self-catalysts or their effect is countered by the more restricted movement of the branched structure of PEI. When there is an excess of amines overall, a small but significant decrease in the characteristic network relaxation time was found with increasing amounts of amines provided by PEI (Fig. 2B). Interestingly, the impact of the availability of more amine functional groups on the characteristic time was smaller at higher temperatures, likely due to greater chain mobility, which can overshadow contributions from covalent bond exchange to network relaxation.
A similar effect, but of much slower relaxation rate, was observed in the presence of excess acrylate groups. Interestingly, this was seen for both cases tested: constant concentration of total reactive amines with increasing fraction provided by PEI (Fig. 2C) and increasing concentration of total reactive amines provided by PEI, with constant Jeffamine composition (Fig. 2D). In both systems, a relatively large increase in network relaxation rate (decrease in characteristic time) was observed as PEI was added, at least at the lowest temperature (150 °C). At higher temperatures, the decrease was not always statistically significant, possibly due to the masking of PEI-related effects by higher temperature-dependent chain mobility. Since relaxation time decreases with increasing PEI concentration in both cases (constant or increasing overall number of amine groups), features of the PEI structure compared to Jeffamine are important in the stress relaxation phenomenon. The relevant factors could include higher reactivity provided by tertiary amine catalysis, inhibition of network relaxation by limitation of chain mobility due to the branched nature of the PEI structure, or changes in how the overall network is formed with PEI vs. Jeffamine. The relative contributions of these factors cannot be separated, but point to an interesting direction for further study. From a practical design perspective, however, it is clear that off-stoichiometry formulations with either excess amine (Fig. 2B) or excess acrylate (Fig. 2D) consistently provide for faster stress relaxation of CANs.
Fig. 3 shows the temperature dependence of stress relaxation, which allowed us to extract a simple estimate of activation energy (Ea) for this process in each case. Acrylate-amine based materials comprise so-called dissociative CANs because the molecular event required for bond exchange first requires a bond disconnection (retro-Michael elimination). For such systems, if two materials exhibit similar activation energies but show different stress relaxation behavior, attention should turn to their viscosity profiles (indicative of chain entanglement) and potential differences in the reactivity of the debonded state, which is ordinarily not rate-limiting. However, especially at temperatures high enough for bond dissociation to become fast and the equilibrium to shift toward the debonded state (endothermic side), re-bonding can become important to the overall stress relaxation rate and therefore to the overall bulk relaxation behavior.61,62 For the materials studied here, no significant differences were found in activation energy values within experimental error; extracted enthalpies and entropies of activation (ESI Fig. S8†) were similarly consistent.
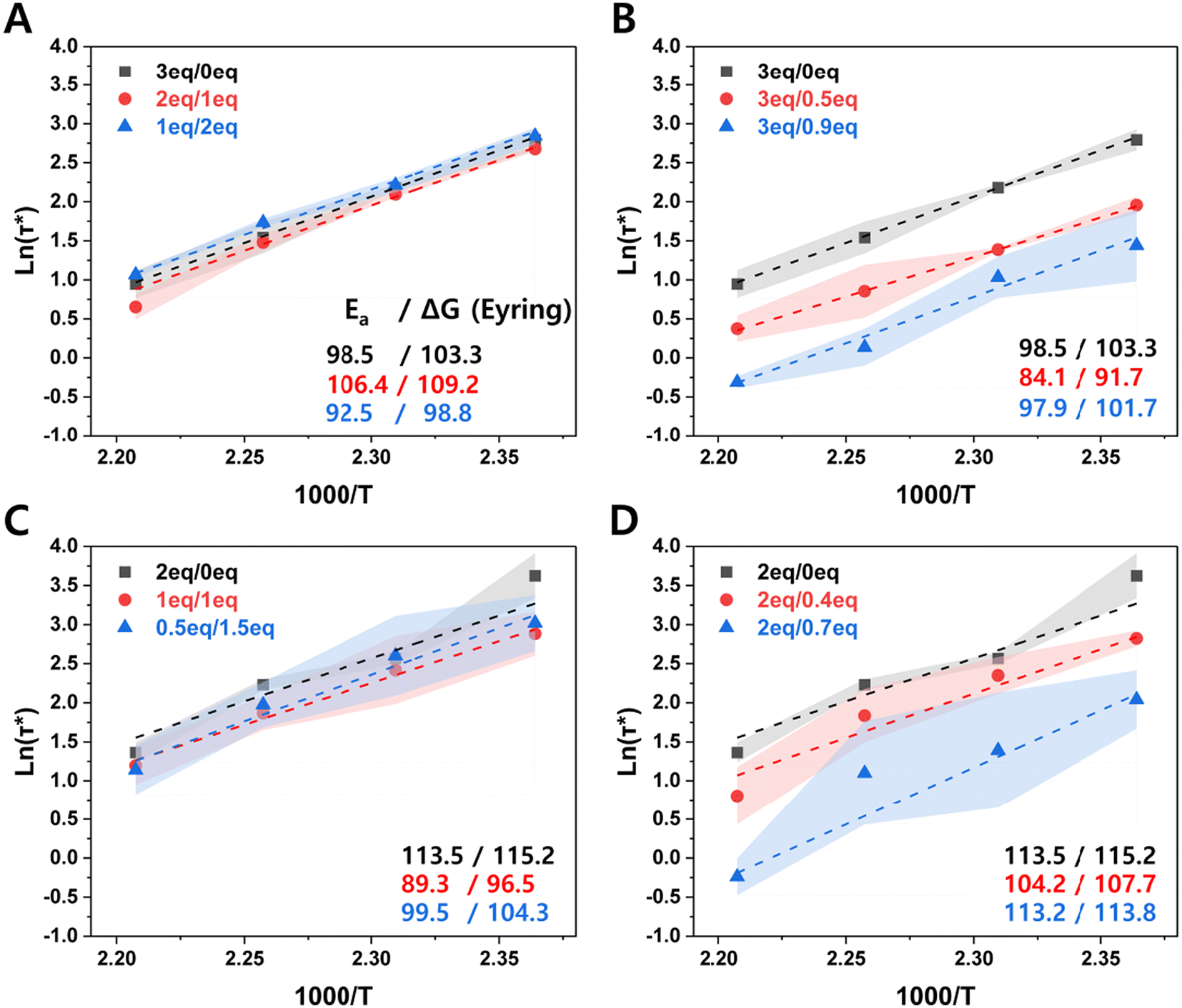 | ||
| Fig. 3 Arrhenius plots relating characteristic stress relaxation time to temperature. (A) ‘on-stoichiometry’, (B) excess amine, (C) excess acrylate relative to constant total amine concentration, changing Jeffamine:PEI ratio, (D) excess acrylate relative to constant Jeffamine concentration and increasing PEI concentration. Dashed lines indicate linear fits to the experimental data with the activation energy calculated from the slope; the standard deviation of 3 samples is indicated by the shaded region. | ||
The similar activation energies suggest one of three possibilities: (i) one particular process (among covalent bond exchange reactivity, chain mobility, and chain entanglement) is rate-determining in all cases, (ii) more than one process contributes with similar temperature-dependent kinetic parameters, or (iii) the contributions counterbalance each other to yield a similar average rate in each case. The first is unlikely, since all of the reactivity and structural parameters should change significantly among the sample set. For example, chain mobility will be dominated by the Jeffamine chains as the component containing by far the greatest number of rotatable bonds, and the concentration of Jeffamine in the various samples can vary greatly. The second possibility also seems unlikely, as the activation energy for the rate-determining step of bond exchange (amine elimination) will be very different from the activation energy of any process involving only chain motion. This leaves the third possibility as the most likely, albeit one for which we are as yet unable to offer detailed mechanistic insights. The measured activation energies in this work of 84–114 kJ mol−1 are on the low end of bulk activation energy values for aza-Michael CANs reported in the literature, many of which are based solely on monomers with different crosslinking densities or the addition of an internal catalyst.17,18,49 (Note that these values are for overall network relaxation, sometimes called the “apparent activation energy” or “flow activation energy”). For example, Taplan et al. designed aza-Michael CANs with β-amino esters and achieved activation energies ranging from 110–242 kJ mol−1,17 while a fluro-containing CAN from bio-based bis-hydroxylated amine and bis-trifluoromethylacrylate, described by Berne et al., showed activation energies of 99–191 kJ mol−1.63 A study specifically designed to improve reprocessability through a lower activation energy without a significant increase in creep used liquid crystals in the CAN matrix to produce activation energies on the order of 100 kJ mol−1.64 In our case, PEI clearly plays an important role in enhancing bond exchange rates within these materials.
Love et al. measured activation energies from 22.2–69.9 kJ mol−1 for the aza-Michael exchange of a number of reacting pairs in solution using NMR.65 We attempted similar experiments using monovalent reactants resembling the molecular motifs in Jeffamine, PEI, and TMPTA, as shown in Fig. 4. Under a variety of conditions, including in the presence and absence of solvent and catalyst at temperatures up to 150 °C, the desired exchange reactions were not observed (ESI Fig. S9A–9D†). Only the reaction of 1 with acrylate 5, a simple mimic of the ‘excess acrylate’ network condition, gave a new product: adduct 6 by virtue of conjugate addition with the secondary amine group in 1 (Fig. 4). This suggests that direct β-aminoacrylate exchange may not play a large role in material relaxation, although we cannot be sure that the conditions of the model reactions adequately represent those of the bulk material, the key difference being the presence of nearby basic nitrogen atoms in the materials, which can serve as catalysts for amine elimination in ways that cannot be matched by monomeric reactants even at high concentration.
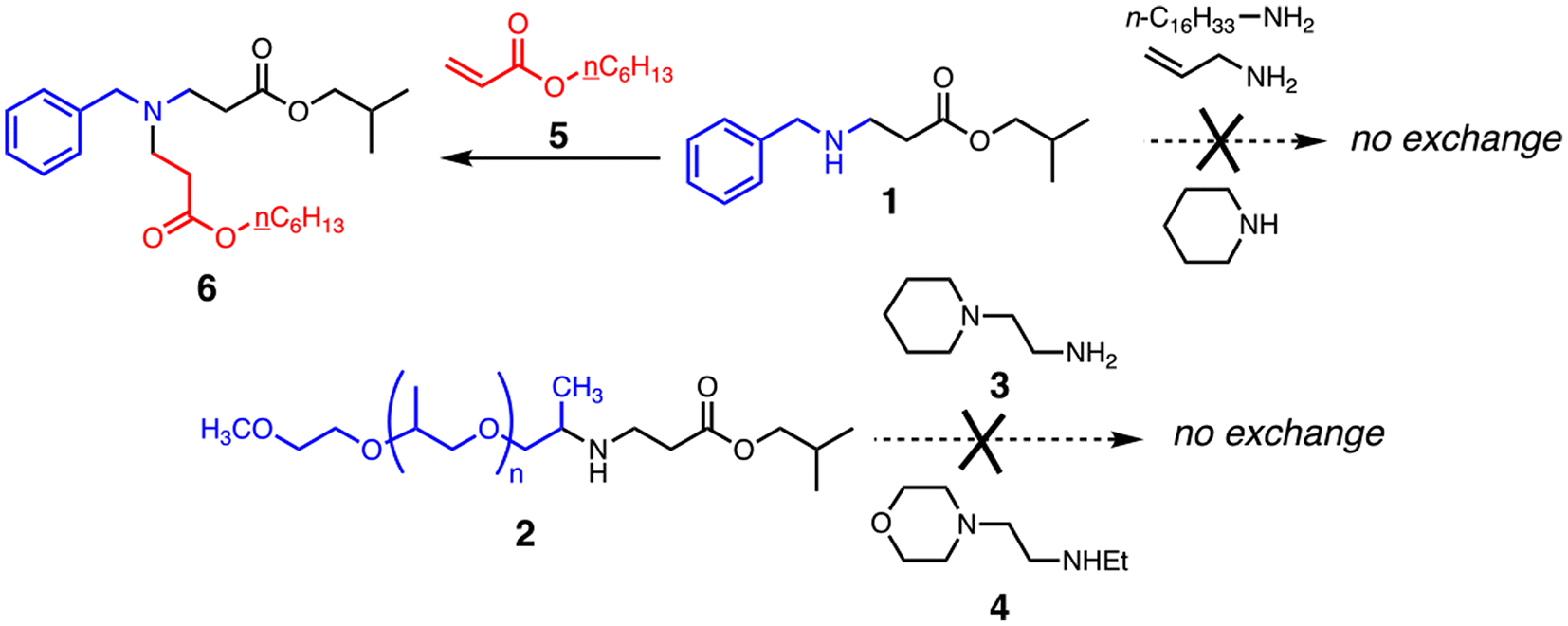 | ||
| Fig. 4 Attempted model exchange reactions. | ||
Fig. 5 shows swelling ratio and glass transition temperature (Tg) values of the bulk CAN materials for each composition. Even though dissociative networks can dissolve in a good solvent under favorable conditions61 (e.g., high temperature) where crosslinked chains are highly mobile and solvent diffusion is sufficient to penetrate all crosslinked points, the samples studied here did not dissolve after 48 hours of immersion at room temperature. Consistently decreased swelling and increased Tg were observed with increasing PEI content, whether stoichiometrically balanced or not. We attribute this trend to the creation of additional crosslinking nodes from the branched structure of the polyvalent amine. Furthermore, the magnitude of swelling was considerably greater for materials having excess acrylate groups (Fig. 5C and D) than the on-stoichiometry and excess amine cases (Fig. 5A and B), and the excess amine materials also exhibited much less variation in Tg than the other groups (Fig. 5B). The greater degree of crosslinking produced by stoichiometric matching should naturally lead to decreased swelling, and the presence of excess amine also ensures maximum use of the tripodal acrylate connectors, which can also be expected to be very important in setting the crosslinking density. These are also consistent with the generally lower characteristic relaxation times shown in Fig. 2A and B for the ‘on-stoichiometry’ and excess amine cases. We note that all of the Tg's are well below 0 °C, which means that these materials are softened at room temperature and may not have sufficient mechanical stability for some applications. We did not aim to correct this deficiency or optimize for other material properties, focusing instead on the fundamental relationships between stress relaxation and related properties vs. composition.
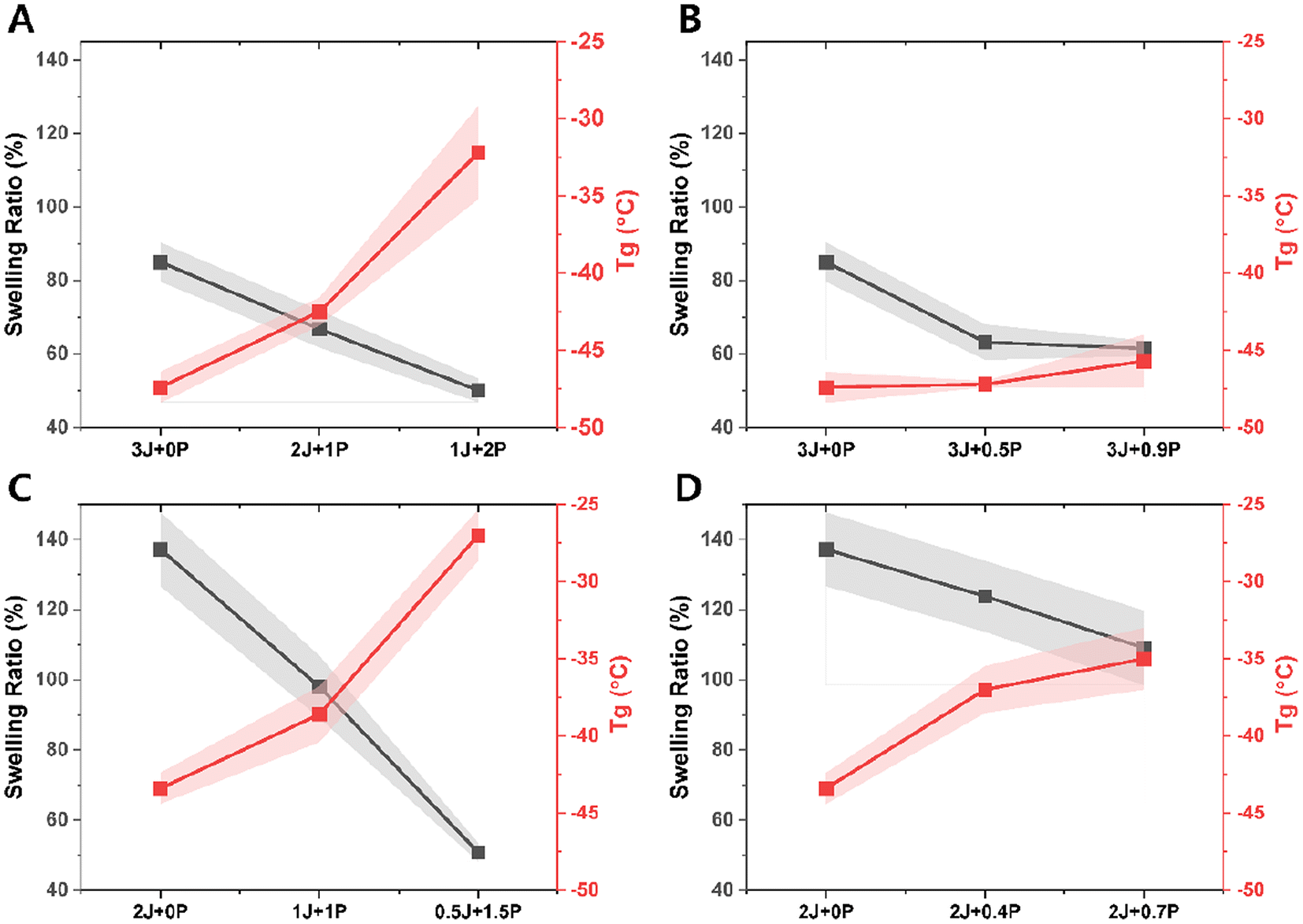 | ||
| Fig. 5 Swelling ratios after 24 h in dimethyl formamide (black color) and glass transition temperature measurement by differential scanning calorimetry (red color). (A) ‘on-stoichiometry’, (B) excess amine, (C) excess acrylate relative to constant total amine concentration, changing Jeffamine:PEI ratio, (D) excess acrylate relative to constant Jeffamine concentration and increasing PEI concentration. The standard deviation of 3 samples is indicated by the shaded region. | ||
To assess reprocessability of the materials used here, small pieces of the polymer films were placed in a mold and subjected to thermal compression at 180 °C and 1 MPa pressure for 40 minutes. Starting films were made directly by solution casting of the mixture of monomers in dimethyl formamide (DMF), followed by vacuum drying and heating to 70 °C to complete crosslinking by aza-Michael reaction. The as-synthesized and reprocessed films for ‘on-stoichiometry’ with only the Jeffamine (3J, 0P) samples are shown in Fig. 6. We observed that there are slight color changes (i.e. yellowing) as these materials are repeatedly reprocessed. Analysis by FTIR showed no significant changes in peak intensities from the first to third reprocessing step (ESI Fig. S10†), demonstrating that the samples were chemically stable within this reprocessing window. We did note some changes in peak intensity in N–H (3000–3500 cm−1) and CO (1735 cm−1) stretching vibrations between freshly-prepared and reprocessed materials, attributed to the different thermal history of sample fabrication (the pristine sample was cured at 70 °C in a vacuum oven while the reprocessed sample was fabricated at 180 °C under compression).
 | ||
| Fig. 6 Images of as-synthesized films and films after three reprocessing steps of ‘on-stoichiometry’ with only the Jeffamine (3J, 0P) samples. | ||
Furthermore, we assessed the stress relaxation and mechanical properties of the reprocessed films to determine whether the reprocessing changed their material properties. Fig. 7A and B show the characteristic time measured at a fixed temperature of 160 °C for on-stoichiometry and excess amine cases, respectively (stress relaxation and storage modulus raw data in ESI Fig. S11†), for the neat films and after the 1st and 3rd reprocesing step. We selected 160 °C because it was one of the temperatures that showed a significant decrease in characteristic time for the excess amine case (Fig. 2B) and we plotted the results on the same y-axis scale as Fig. 2 to enable comparison. We see that the measured characteristic time does not change significantly from the as-synthesized to the first reprocessing step or from the first to the third reprocessing step. This indicates that the reprocessed materials maintain their ability to relax applied stress through at least 3 cycles, likely through both bond relaxation and chain mobility mechanisms.
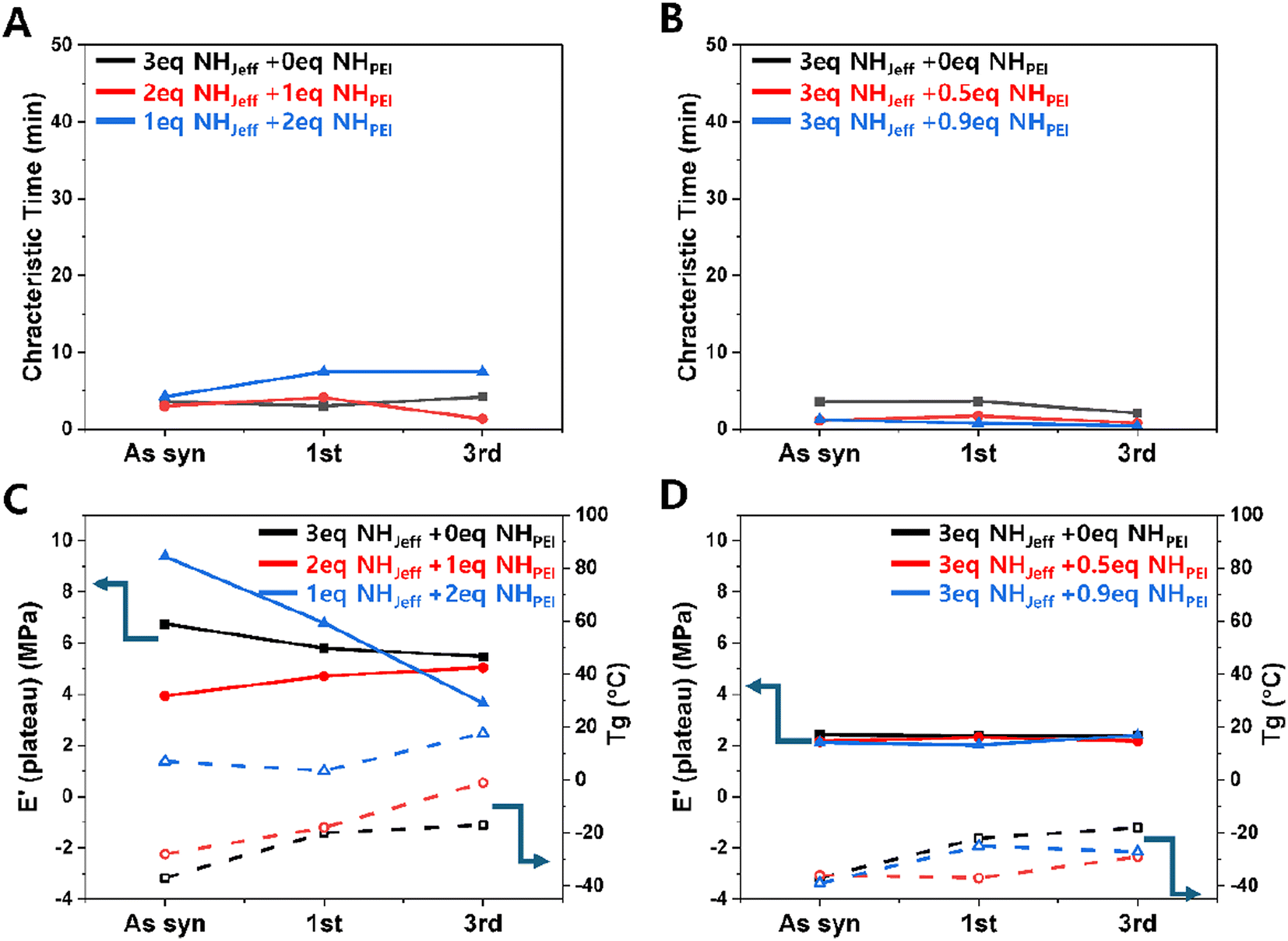 | ||
| Fig. 7 Characteristic times for stress relaxation obtained by DMA at 160 °C for (A) the ‘on-stoichiometry’ and (B) the ‘off-stoichiometry’ excess amine cases. (C) Plateau modulus at 100 °C and the glass transition temperature determined by the peak tan delta value for the ‘on-stoichiometry’ cases. (D) Plateau modulus at 100 °C and Tg for ‘off-stoichiometry’ samples with excess amine. “As syn” refers to freshly-prepared films; “1st” and “3rd” refer to materials reprocessed by heat one or three times, respectively. | ||
Fig. 7C and D present DMA results aimed at exploring the impact of reprocessing on crosslinked network formation, focusing on the rubbery plateau values after each thermal reprocessing cycle and the Tg. The results also indicate that the material system can withstand three cycles of reprocessing without detrimental effects on these material properties, as they remain practically unchanged compared to the original values. The only sample showing a significant change in rubber modulus was the 1 Jeffamine + 2 PEI in ‘on-stoichiometry’, which showed a decreasing value of rubber modulus with reprocessing up to 3 cycles. The reason for this is unclear; however, this change in modulus and slight increase in glass transition temperature did not affect exchange kinetics for the current course of reprocessing.
Tensile properties were assessed to investigate changes in mechanical behavior over multiple reprocessing cycles. Fig. 8 plots specific mechanical property parameters extracted from stress–strain curves including the Young's modulus, tensile strength at break, and elongational strain at break for both the ‘on-stoichiometry’ sets (A–C) and excess amine sets (D–F) over three cycles of heat-press reprocessing. While a statistically significant decrease in Young's modulus and tensile strength were observed between the as-synthesized and 1st reprocessing samples, which is attributed to the different thermal history of sample fabrication as noted above, we were most interested in the impact of multiple thermal reprocessing steps on the material properties. In most on-stoichiometry cases, no significant change was observed in the mechanical properties from the first to the third reprocessing cycles (only the Young's modulus for the 2 Jeffamine + 1 PEI material showed a statistically significant difference), indicating that for the on-stoichiometry samples the mechanical properties were maintained well during reprocessing. In contrast, there was a statistically significant decrease in mechanical properties from the first to the third reprocessing step at the highest amount of PEI in excess amine cases (Fig. 8D–F, blue). Taken together with stress relaxation measurements (excess amines give rise to relatively fast relaxation) and network characterization (low degree of swelling but also low glass transition temperature), we suggest that the presence of relatively large amounts of the mobile Jeffamine component leads to significant rearrangement during reprocessing that induces irreversible changes in the network, but this would need to be confirmed through additional study.
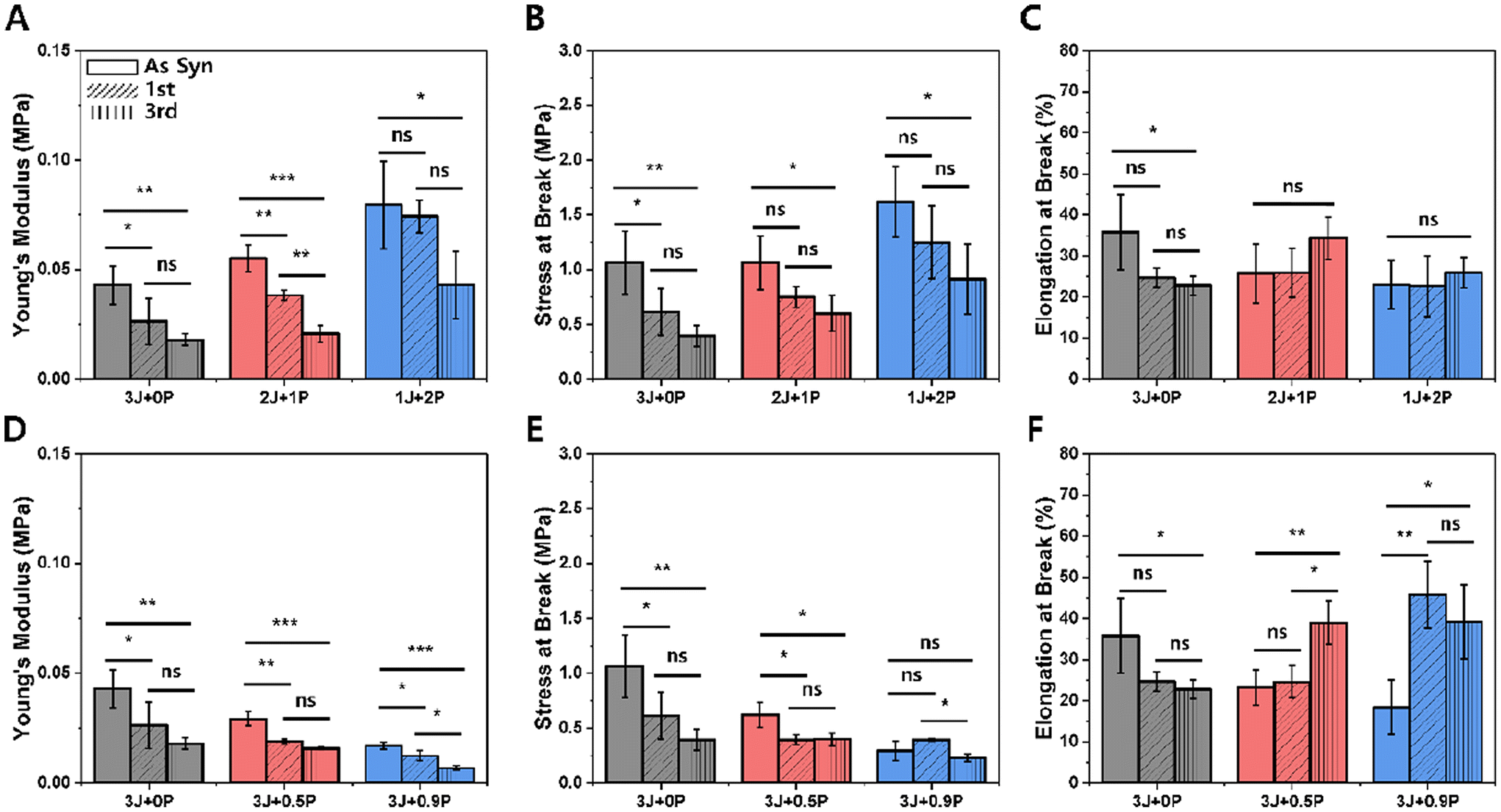 | ||
| Fig. 8 Tensile measurements of on-stoichiometry cases (A–C) and off-stoichiometry excess amine cases (D–F) for three reprocessing cycles. Data are presented as the mean ± standard deviation (n = 3–5). The statistical analysis was conducted using a multiple group comparison through one-way analysis of variance along with Tukey's post hoc test. “ns” = not-significant, * = p-value <0.05, ** = p-value <0.01, *** = p-value <0.001. | ||
Because acrylates are sensitive to further curing in thermal post-treatments and re-processing is a thermal treatment, we also examined the change in mechanical properties of the excess-acrylate case. ESI Fig. S13† presents the mechanical properties (modulus, stress at break, and elongation at break) for the three excess-acrylate materials as-synthesized and after the 1st and 3rd re-processing steps. We observe that all three properties increased from the as-synthesized state to after the first thermal reprocessing cycle and were maintained over subsequent reprocessing cycles, up to three cycles. This indicates that further reactions (post-curing or post-reaction) occurred due to the excess acrylate remaining in the material system, but only during the first cycle.
Conclusions
In this study, we targeted a specific CAN design parameter, the stoichiometry of the reactive components (amines and acrylates in aza-Michael reactions), determining the functional values by quantitative assessment of reactivity rather than assuming ideal structures and behavior. We also varied the linear-vs.-branched nature of the amine nucleophile to gain some insight into the role of network structure in controlling properties. Our assessments focused on the important material properties of stress relaxation, solvent swelling, glass transition temperature and tensile strength. We showed that stress relaxation is more sensitive to the number of amines provided by PEI (as compared to Jeffamine) when the amine-acrylate stoichiometry is not matched, but that it is insensitive to this parameter when the stoichiometry is matched. This has important implications for considering how to tune stress relaxation, as the characteristic time for relaxation could be lowered by increasing the proportion of PEI only in the off-stoichiometry cases. From a mechanistic perspective, inaccuracies in stoichiometry matching are likely to produce different contributions from associative, dissociative, and concerted bond exchange mechanisms,61,62,66,67 and thus difficulties in accurate modeling of the properties of such materials.With the long-term goal of preparing reprocessable plastic materials, we also examined the ability to maintain properties after multiple cycles of reprocessing, again finding significant differences in on- vs. off-stoichiometry materials. Thus, bulk properties changed significantly from as-synthesized (prepared at 70 °C in a vacuum oven) to first reprocessing (180 °C under compression) for off-stoichiometry cases, but mechanical properties and stress relaxation did not change significantly for on-stoichiometry CANs when reprocessed at constant conditions. Furthermore, the incorporation of high PEI content gave rise to more dramatic degradation in the mechanical properties in off-stoichiometry materials, suggesting a practical tradeoff between the achievement of high rates of reprocessing with off-stoichiometry formulations and maintaining good properties through multiple reprocessing cycles. These insights may be useful for catalyst-free CAN design, particularly systems with slow stress relaxation, where formulation-based approaches to increasing relaxation rate are of particular value.
Materials and methods
Materials
Trimethylolpropane triacrylate (TMPTA), branched polyethylenimine (PEI, average Mw = 800 g mol−1 by LS, average Mn = 600 g mol−1 by GPC), and Jeffamine (ED-600) were purchased from Millipore Sigma. Dimethylformamide solvent (DMF, anhydrous, 99.9%) was purchased from Beantown Chemical. All reagents were used as received. According to the manufacturer (Millipore Sigma), the Jeffamine has an approximate value of x + z ∼ 6 and y ∼ 39. According to Grenda et al., who studied the branched PEI used here, the value of n cannot be defined, but rather, the branched PEI can have a general formulas C26H68N14 that contains approximately 20 aziridine-derived fragments in different branching patterns to give a mixture of primary, secondary, and tertiary amine sites.Synthesis
The initial state of all monomers (i.e., TMPTA, Jeffamine, and PEI) was liquid. TMPTA was stored in a closed cabinet to prevent light-induced termination of the active acrylate, while the other two monomers, Jeffamine ED-600 and PEI, were stored in a desiccator. Jeffamine was used as a chain extender (i.e., to modulate Tg) for mechanically viable samples, as the resulting crosslinked material between TMPTA and PEI is brittle. Each monomer was mixed in DMF solvent, based on the molar ratios shown in Table 1. The target concentration of the solution was adjusted by varying the amount of solvent for solution casting to produce a film thickness of 0.2 mm. After mixing on an orbital shaker (Benchtop Orbital Shaker Model 416 HP, Thermo Scientific) at 200 rpm for 30 minutes, the monomer mixture was cast into a PTFE Petri dish. To ensure complete crosslinking, the solutions were placed in a heated vacuum oven at 70 °C for 24 hours, resulting in a robust, free-standing, crosslinked film based on dynamic aza-Michael bonds.Characterization
The functionality parameter, f, was determined by using measured FTIR spectra of different ratios of TMPTA and Jeffamine or TMPTA and PEI. For Jeffamine, molar ratios from 1:0.75 to 1:1.5 were mixed at room temperature and cured overnight at 80 °C in an oven. For PEI, molar ratios from 1:0.2 to 1:0.3 were mixed at room temperature and cured overnight at 80 °C in an oven. The Jeffamine peaks in the regions 820 cm−1 to 795 cm−1, from 1440 cm−1 to 1380 cm−1, and from 1660 cm−1 to 1600 cm−1 were extracted and the baselines were added by using the tangent method in the PerkinElmer software. The PEI peaks in the regions 825 cm−1 to 790 cm−1, from 1440 cm−1 to 1380 cm−1, and from 1660 cm−1 to 1600 cm−1 were extracted and the baselines were added using the tangent method. Peak areas were calculated using the baseline-corrected peaks by the PerkinElmer software. The area was plotted vs. the TMPTA:amine oligomer ratio and the first TMPTA:amine oligomer ratio where the peak area was ≥80% less than the maximum area was taken as the stoichiometric point. 80% decrease was chosen because in most cases, the peak area did not continue decreasing from there. To calculate f from the ratio corresponding to the stoichiometric point, the following equation was used:
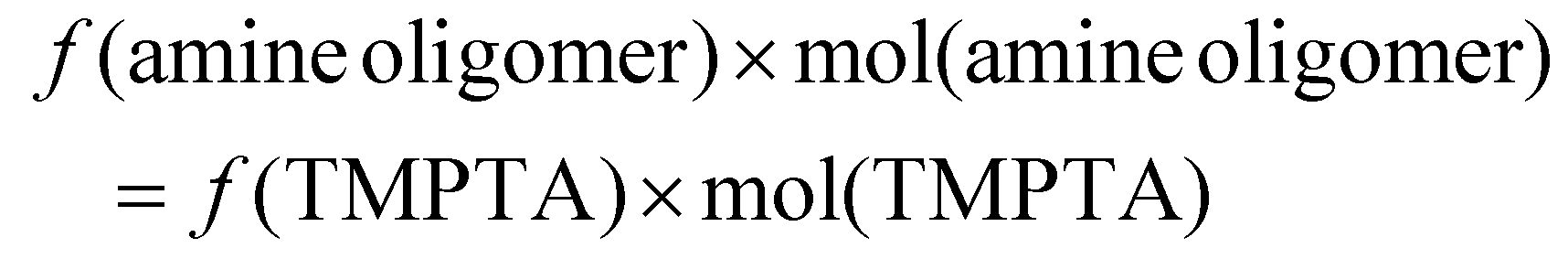
The molar extinction coefficient (ε) for composition quantification was determined using ATR-FTIR in the following manner. Different concentrations of TMPTA in DMF under dilute conditions were prepared and changes in absorbance intensities of the relevant peaks (CO and CC stretching and bending) were plotted against the solute molar concentration. The slope obtained from linear regression represents the molar extinction (or absorption) coefficient (ESI Fig. S2†).
where,
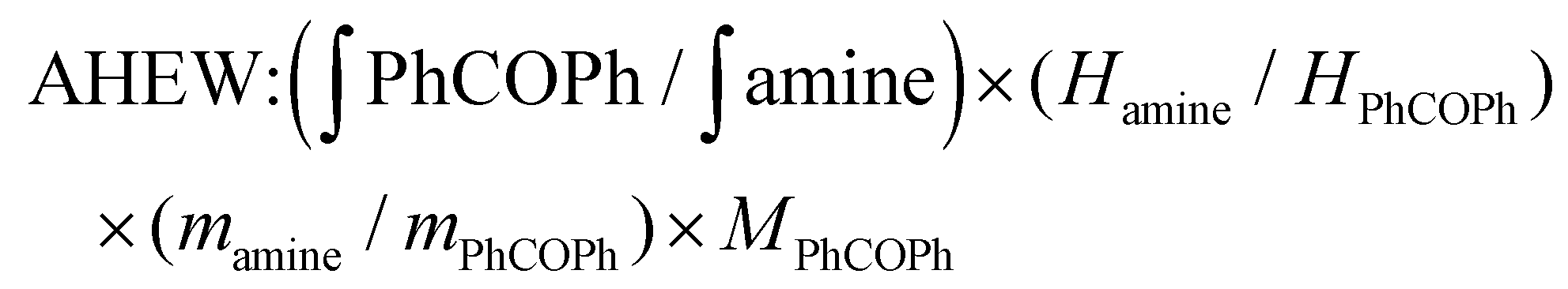
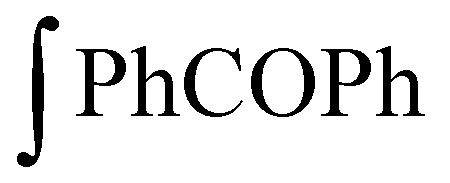 : integral of the signal from benzophenone protons;
: integral of the signal from benzophenone protons;  : integral of the signals from protons in α-position to the primary amine functionality; Hamine: number of protons in α-position to the primary amine functionality; HPhCOPh: number of benzophenone protons; mamine: mass of amine; mPhCOPh: mass of benzophenone; MPhCOPh: benzophenone molar mass.
: integral of the signals from protons in α-position to the primary amine functionality; Hamine: number of protons in α-position to the primary amine functionality; HPhCOPh: number of benzophenone protons; mamine: mass of amine; mPhCOPh: mass of benzophenone; MPhCOPh: benzophenone molar mass.
The AHEW was determined by comparing the value of the integral of the signals arising from the benzophenone protons (7.6–7.64 ppm) with the value of the integral of the signals arising from the –CH protons (2.86–3.04 ppm) in α-position to the primary amine groups in Jeffamine (ESI Fig. S6†). The measurement of AHEW was done in duplicate. The f was calculated as: (molecular weight of amine/AHEW).
To determine the free energy (ΔG) the enthalpy (ΔH‡), and entropy (ΔS‡) of activation, the Eyring equation was used. The temperature (T) and characteristic relaxation time (τ*) were obtained from the stress relaxation measurement from the DMA experiment and the activation enthalpy (ΔH‡) and activation entropy (ΔS‡) were determined using the Eyring equation:
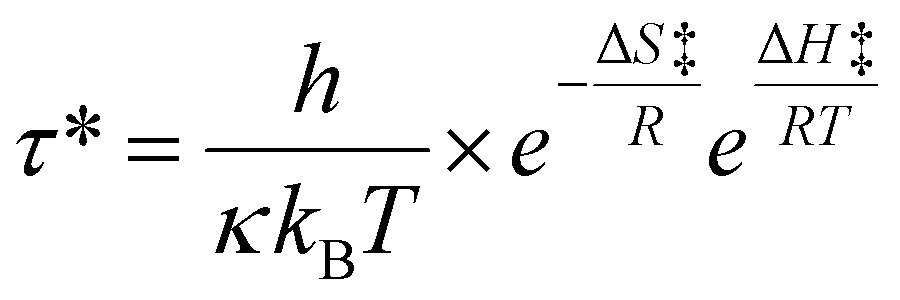
Author contributions
Jaehyun Cho: conceptualization, data curation, formal analysis, investigation, methodology, validation, visualization, writing – original draft, writing – review and editing. Santanu Ghosh: formal analysis, investigation, methodology, validation, writing – review and editing, Mridula Nandi: formal analysis, investigation, methodology, validation, writing – review and editing, Heejoon Jeon: formal analysis, investigation, methodology, validation, writing – review and editing, Liang Yue: formal analysis, investigation, methodology, validation, writing – review and editing, H. Jerry Qi: conceptualization, funding acquisition, resources, supervision, writing – review and editing, M. G. Finn: conceptualization, data curation, formal analysis, funding acquisition, investigation, supervision, writing – review and editing, Blair Brettmann: conceptualization, data curation, formal analysis, funding acquisition, investigation, supervision, writing – original draft, writing – review and editing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the Office of Naval Research through a Multidisciplinary University Research Initiative (MURI) Grant N00014-20-1-2586.References
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- C. J. Kloxin, T. F. Scott, B. J. Adzima and C. N. Bowman, Macromolecules, 2010, 43, 2643–2653 CrossRef CAS PubMed.
- Y. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- K. Liu, Y. Kang, Z. Wang and X. Zhang, Adv. Mater., 2013, 25, 5530–5548 CrossRef CAS PubMed.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698–1702 CrossRef CAS PubMed.
- Q. Chen, G. J. Tudryn and R. H. Colby, J. Rheol., 2013, 57, 1441–1462 CrossRef CAS.
- K. Xing, M. Tress, P.-F. Cao, F. Fan, S. Cheng, T. Saito and A. P. Sokolov, Macromolecules, 2018, 51, 8561–8573 CrossRef CAS.
- G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed.
- S. Burattini, B. W. Greenland, D. H. Merino, W. Weng, J. Seppala, H. M. Colquhoun, W. Hayes, M. E. Mackay, I. W. Hamley and S. J. Rowan, J. Am. Chem. Soc., 2010, 132, 12051–12058 CrossRef CAS PubMed.
- G. V. Oshovsky, D. N. Reinhoudt and W. Verboom, Angew. Chem., Int. Ed., 2007, 46, 2366–2393 CrossRef CAS PubMed.
- Y. Xu, M. Patino Gaillez, R. Rothe, S. Hauser, D. Voigt, J. Pietzsch and Y. Zhang, Adv. Healthcare Mater., 2021, 10, 2100012 CrossRef CAS PubMed.
- B. B. Gerbelli, S. V. Vassiliades, J. E. U. Rojas, J. N. B. D. Pelin, R. S. N. Mancini, W. S. G. Pereira, A. M. Aguilar, M. Venanzi, F. Cavalieri, F. Giuntini and W. A. Alves, Macromol. Chem. Phys., 2019, 220, 1970027 CrossRef.
- A. P. Bapat, D. Roy, J. G. Ray, D. A. Savin and B. S. Sumerlin, J. Am. Chem. Soc., 2011, 133, 19832–19838 CrossRef CAS PubMed.
- A. G. Orrillo and R. L. E. Furlan, Angew. Chem., 2022, 134, e202201168 CrossRef.
- C. Wang, S. Mavila, B. T. Worrell, W. Xi, T. M. Goldman and C. N. Bowman, ACS Macro Lett., 2018, 7, 1312–1316 CrossRef CAS PubMed.
- S. P. Black, J. K. M. Sanders and A. R. Stefankiewicz, Chem. Soc. Rev., 2014, 43, 1861–1872 RSC.
- C. Taplan, M. Guerre and F. E. Du Prez, J. Am. Chem. Soc., 2021, 143, 9140–9150 CrossRef CAS PubMed.
- G. Lee, H. Y. Song, S. Choi, C. B. Kim, K. Hyun and S. Ahn, Macromolecules, 2022, 55, 10366–10376 CrossRef CAS.
- B. Zhang, Z. A. Digby, J. A. Flum, P. Chakma, J. M. Saul, J. L. Sparks and D. Konkolewicz, Macromolecules, 2016, 49, 6871–6878 CrossRef CAS.
- H. Ying, Y. Zhang and J. Cheng, Nat. Commun., 2014, 5, 3218 CrossRef PubMed.
- M. E. Belowich and J. F. Stoddart, Chem. Soc. Rev., 2012, 41, 2003–2024 RSC.
- O. R. Cromwell, J. Chung and Z. Guan, J. Am. Chem. Soc., 2015, 137, 6492–6495 CrossRef CAS PubMed.
- J. A. Neal, D. Mozhdehi and Z. Guan, J. Am. Chem. Soc., 2015, 137, 4846–4850 CrossRef CAS PubMed.
- M. M. Perera and N. Ayres, Polym. Chem., 2020, 11, 1410–1423 RSC.
- P. Chakma and D. Konkolewicz, Angew. Chem., Int. Ed., 2019, 58, 9682–9695 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- X.-L. Zhao, P.-X. Tian, Y.-D. Li and J.-B. Zeng, Green Chem., 2022, 24, 4363–4387 RSC.
- C. Hao, T. Liu, S. Zhang, W. Liu, Y. Shan and J. Zhang, Macromolecules, 2020, 53, 3110–3118 CrossRef CAS.
- L. L. Robinson, E. S. Taddese, J. L. Self, C. M. Bates, J. Read de Alaniz, Z. Geng and C. J. Hawker, Macromolecules, 2022, 55, 9780–9789 CrossRef CAS.
- K. Yu, P. Taynton, W. Zhang, M. L. Dunn and H. Jerry Qi, RSC Adv., 2014, 4, 48682–48690 RSC.
- M. K. McBride, B. T. Worrell, T. Brown, L. M. Cox, N. Sowan, C. Wang, M. Podgorski, A. M. Martinez and C. N. Bowman, Annu. Rev. Chem. Biomol. Eng., 2019, 10, 175–198 CrossRef CAS PubMed.
- C. Cui, X. Chen, L. Ma, Q. Zhong, Z. Li, A. Mariappan, Q. Zhang, Y. Cheng, G. He, X. Chen, Z. Dong, L. An and Y. Zhang, ACS Appl. Mater. Interfaces, 2020, 12, 47975–47983 CrossRef CAS PubMed.
- W. Luo, H. Yu, Z. Liu, R. Ou, C. Guo, T. Liu and J. Zhang, J. Mater. Res. Technol., 2022, 19, 1699–1710 CrossRef CAS.
- G. Zhang, X. Li, Q. Liao, Y. Liu, K. Xi, W. Huang and X. Jia, Nat. Commun., 2018, 9, 2785 CrossRef PubMed.
- N. Van Herck, D. Maes, K. Unal, M. Guerre, J. M. Winne and F. E. Du Prez, Angew. Chem., Int. Ed., 2020, 59, 3609–3617 CrossRef CAS PubMed.
- V. Zhang, B. Kang, J. V. Accardo and J. A. Kalow, J. Am. Chem. Soc., 2022, 144, 22358–22377 CrossRef CAS PubMed.
- C. H. Fox, G. M. ter Hurrne, R. J. Wojtecki, G. O. Jones, H. W. Horn, E. W. Meijer, C. W. Frank, J. L. Hedrick and J. M. García, Nat. Commun., 2015, 6, 7417 CrossRef CAS PubMed.
- F. Cuminet, S. Caillol, É. Dantras, É. Leclerc and V. Ladmiral, Macromolecules, 2021, 54, 3927–3961 CrossRef CAS.
- M. Hayashi, ACS Appl. Polym. Mater., 2020, 2, 5365–5370 CrossRef CAS.
- A. Chao and D. Zhang, Macromolecules, 2019, 52, 495–503 CrossRef CAS.
- W. C. Yount, D. M. Loveless and S. L. Craig, J. Am. Chem. Soc., 2005, 127, 14488–14496 CrossRef CAS PubMed.
- M. Hayashi and R. Yano, Macromolecules, 2020, 53, 182–189 CrossRef CAS.
- N. B. Tito, C. Creton, C. Storm and W. G. Ellenbroek, Soft Matter, 2019, 15, 2190–2203 RSC.
- M. Hayashi, Polym. J., 2021, 53, 779–788 CrossRef CAS.
- X. Kuang, G. Liu, X. Dong and D. Wang, Mater. Chem. Front., 2016, 1, 111–118 RSC.
- T. Isogai and M. Hayashi, Macromolecules, 2022, 55, 6661–6670 CrossRef CAS.
- M. Hayashi and A. Katayama, ACS Appl. Polym. Mater., 2020, 2, 2452–2457 CrossRef CAS.
- M. Hayashi, R. Yano and A. Takasu, Polym. Chem., 2019, 10, 2047–2056 RSC.
- L. Stricker, C. Taplan and F. E. Du Prez, ACS Sustainable Chem. Eng., 2022, 10, 14045–14052 CrossRef CAS.
- A. Safaei, S. Terryn, B. Vanderborght, G. Van Assche and J. Brancart, Polymers, 2021, 13, 2522 CrossRef CAS PubMed.
- N. Jarach, D. Golani, O. Asaf, H. Dodiuk, Y. Shamir, A. Goldbourt, S. Kenig and N. Naveh, Polym. Chem., 2021, 12, 3307–3320 RSC.
- Q.-A. Poutrel, J. J. Blaker, C. Soutis, F. Tournilhac and M. Gresil, Polym. Chem., 2020, 11, 5327–5338 RSC.
- F. I. Altuna, C. E. Hoppe and R. J. J. Williams, Polymers, 2018, 10, 43 CrossRef PubMed.
- J. M. Winne, L. Leibler and F. E. D. Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- M. Podgórski, S. Mavila, S. Huang, N. Spurgin, J. Sinha and C. N. Bowman, Angew. Chem., Int. Ed., 2020, 59, 9345–9349 CrossRef PubMed.
- J. L. Stanford, R. H. Still and A. N. Wilkinson, Polymer, 2003, 44, 3985–3994 CrossRef CAS.
- D. M. Kroll and S. G. Croll, Polymer, 2015, 79, 82–90 CrossRef CAS.
- F. Chen, F. Gao, X. Guo, L. Shen and Y. Lin, Macromolecules, 2022, 55, 10124–10133 CrossRef CAS.
- A. Demirci, S. Yamamoto, J. Matsui, T. Miyashita and M. Mitsuishi, Polym. Chem., 2015, 6, 2695–2706 RSC.
- R. G. Ricarte and S. Shanbhag, Polym. Chem., 2024, 15, 815–846 RSC.
- J. M. Winne, L. Leibler and F. E. D. Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- M. L. Martins, X. Zhao, Z. Demchuk, J. Luo, G. P. Carden, G. Toleutay and A. P. Sokolov, Macromolecules, 2023, 56, 8688–8696 CrossRef CAS.
- D. Berne, B. Quienne, S. Caillol, E. Leclerc and V. Ladmiral, J. Mater. Chem. A, 2022, 10, 25085–25097 RSC.
- E. Ghimire, C. A. Lindberg, T. D. Jorgenson, C. Chen, J. J. de Pablo, N. D. Dolinski and S. J. Rowan, Macromolecules, 2024, 57, 682–690 CrossRef CAS.
- D. Love, K. Kim, D. W. Domaille, O. Williams, J. Stansbury, C. Musgrave and C. Bowman, Polym. Chem., 2019, 10, 5790–5804 RSC.
- B. R. Elling and W. R. Dichtel, ACS Cent. Sci., 2020, 6, 1488–1496 CrossRef CAS PubMed.
- A. Jourdain, R. Asbai, O. Anaya, M. M. Chehimi, E. Drockenmuller and D. Montarnal, Macromolecules, 2020, 53, 1884–1900 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00013k |
| This journal is © The Royal Society of Chemistry 2025 |