
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFactors modulating the hydrolysis of Nylon-6,6 by a nylon hydrolase enzyme†
Vera
Bocharova
 *a,
Erin E.
Drufva
b,
John F.
Cahill
b,
Ivan
Popov
c,
Isaiah T.
Dishner
a,
Muchu
Zhou
a,
Gang Seob
Jung
d,
Andrew M.
Ullman
a,
Dana L.
Carper
b,
Joshua T.
Damron
a,
Jong K.
Keum
ef,
Catalin
Gainaru
a,
Serena H.
Chen
d,
Jeffrey C.
Foster
*a and
Joshua K.
Michener
*b
*a,
Erin E.
Drufva
b,
John F.
Cahill
b,
Ivan
Popov
c,
Isaiah T.
Dishner
a,
Muchu
Zhou
a,
Gang Seob
Jung
d,
Andrew M.
Ullman
a,
Dana L.
Carper
b,
Joshua T.
Damron
a,
Jong K.
Keum
ef,
Catalin
Gainaru
a,
Serena H.
Chen
d,
Jeffrey C.
Foster
*a and
Joshua K.
Michener
*b
aChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA. E-mail: bocharovav@ornl.gov; fosterjc@ornl.gov; michenerjk@ornl.gov
bBiosciences Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
cUniversity of Tennessee-Oak Ridge Innovation Institute, University of Tennessee, Knoxville, TN 37996, USA
dComputational Sciences and Engineering Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
eCenter for Nanophase Materials Science, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
fNeutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
First published on 21st March 2025
Abstract
The enzymatic hydrolysis of polyamides offers a promising approach to reduce the environmental impact of chemical recycling by enabling lower reaction temperatures, eliminating toxic organic solvents, and enhancing product selectivity. Achieving this goal will require increasing the low overall yield of enzymatic hydrolysis. In this work, we studied the mechanism of hydrolysis of commercial Nylon-6,6 polymer with a thermostable Nylon hydrolyzing enzyme and identified the substrate characteristics that influence the efficiency and deconstruction product yield. These results will guide the development of effective substrate pre-treatment methods to improve the yield of valuable oligoamide building blocks via enzymatic hydrolysis.
Introduction
Polyamides, also known as Nylons,1 have become essential industrial materials, particularly in the textile and automotive sectors,2 due to their mechanical robustness. Crystallinity enhances their mechanical performance but also increases their melting points,3 complicating their re-processing and recycling. As a result, current mechanical recycling methods for polyamides require substantial thermal energy and produce recycled material relegated to lower-grade applications.4,5 Chemical recycling can overcome these limitations,6 re-generating the original monomers from polymer waste to enable re-synthesis of the native materials. However, environmental concerns associated with chemical recycling often offset its advantages. Given the energy-intensive and environmentally taxing nature of these existing methods of deconstruction of Nylons, there is a pressing need to explore alternative approaches to polyamide recycling that are both efficient and sustainable.Successful enzymatic cleavage has been demonstrated for polyethylene terephthalate (PET),7–9 where efficient re-polymerization of the resulting monomers (e.g., terephthalic acid) has been shown. However, the enzymatic decomposition of other plastics has lagged behind. The development of a highly efficient biocatalyst capable of hydrolyzing polyamides could offer a more sustainable route to Nylon recycling, generating selective deconstruction products under mild conditions. A variety of enzymes have been identified with some capability to partially degrade Nylons, including proteases, cutinases, and amidases.8 Among these enzyme classes, the amidases have been particularly studied. The first amidase capable of hydrolysis of oligomeric Nylons, known as NylC, was discovered and characterized in a bacterium now known as Arthrobacter sp. KI72.10 It was shown to hydrolyze oligomeric Nylon-6 and Nylon-6,6 substrates.11 However, its activity has not been studied on commercial Nylons, which typically possess relatively high molecular weight (MW) that limit their water solubility. The differences in physical state and solubility between the water-soluble catalyst and the often insoluble substrate represent one of the main challenges in the enzymatic deconstruction of various polymers, thereby limiting the efficiencies of the reaction and affecting the reaction yield. Thus, to mitigate this substrate–enzyme incompatibility issue, several enzyme engineering approaches to tune the properties of enzymes have been demonstrated,12–14 with only minimal efforts focused on enzymes hydrolyzing Nylons.15,16 Another strategy involves identifying novel enzymes and microbes with enhanced polymer decomposition activity through advanced rapid screening assays.17–20 While these approaches hold significant potential, establishing a robust and comprehensive knowledge base for their implementation requires considerable time and effort.
An alternative approach to overcome the inherent incompatibility of synthetic polymers with the conditions of enzymatic reactions focuses on the pretreatment of the polymer substrate.21 Various pretreatment techniques including melt processing, mechanical grinding, cryogrinding, micro-pelletization, amorphization, micronization, and foaming have demonstrated good potential as pretreatment options.22 The choice of effective substrate pretreatment protocols often depends on understanding the extent to which various material properties—such as chain mobility, crystallinity, surface area, hydrophobicity/hydrophilicity, and the presence of additives—influence enzymatic efficiency. Some knowledge about the impact of substrate properties on enzymatic deconstruction exists in the literature. For example, minimally crystalline or/and fully amorphous plastics were shown to be more easily degraded by enzymes, leading to higher deconstruction rates and product yields of polyethylene terephthalate (PET).7,23 In contrast, high crystalline PET substrates inhibited enzymatic activity.24 In general, reliable approaches for enzymatic hydrolysis of highly crystalline polymer substrates remain limited.25 Furthermore, the enzymatic hydrolysis rates were shown to improve with increased PET substrate surface area.26 Finally, increased hydrophilicity of substrate was shown to promote biodegradability of polyesteramides.27 While various parameters have been extensively studied in the context of enzymatic cleavage of PET, their role in Nylon degradation remains largely unexplored. Recent studies on Nylon hydrolysis pretreated with formic acid28 suggest that enzymes are highly active on low MW oligomers, but the distinct properties of high MW polymers hinder complete hydrolysis. This highlights the importance of understanding substrate characteristics to develop more effective pretreatment methods beyond partial formic acid hydrolysis.
In this contribution, we examined the impact of polymer characteristics on the extent of enzymatic hydrolysis of commercial Nylon-6,6 by a thermostable, quadruple-mutant Nylon hydrolyzing enzyme, NylC-GYAQ29 (referred as NylC in the text below). We hypothesized that reducing substrate molecular weight, increasing surface area, and reducing crystallinity would enhance hydrolysis product yields. To address this hypothesis, a systematic analysis of enzymatic degradation of Nylon-6,6 substrates was conducted using various experimental techniques. Due to the strong interdependence of the molecular weight, surface area, and crystallinity, their roles in the formation of the reaction products were evaluated using a simplified model that provided initial insights into the reaction mechanism. These results lay the groundwork for developing substrate pretreatment methods to enhance enzymatic efficiency in Nylon hydrolysis.
Experimental
Materials
Hexamethylenediamine and ethanol (EtOH; 200 proof) was purchased from Sigma Aldrich. Adipic acid was purchased from TCI America. Propranolol and LC-MS grade Chromasolv® water, 100/0.1 (v/v) water/formic acid and acetonitrile/formic acid were purchased from Sigma Aldrich (St Louis, MO, USA). Nylon-6,6 (Akulon-S240) pellets were provided by DSM Engineering Materials B.V. (Geleen, Netherlands). Nylon-6,6 linear monomer (L1) standard was synthesized as described in the Scheme S1 and Fig. S1–S8.†Nylon-6,6 salt synthesis
In a typical reaction, a 50 wt% solution of hexamethylenediamine dissolved in water was added to a 10 wt% solution of adipic acid dissolved in ethanol at 50 °C. The reaction was stirred for an additional two hours and subsequently cooled to room temperature. The resulting white precipitate was filtered, rinsed with additional ethanol, and dried at room temperature under vacuum. 1H NMR (400 MHz, D2O) δ 3.01–2.87 (t, 4H), 2.15 (t, J = 4.0 Hz, 4H), 1.63 (p, J = 7.1 Hz, 4H), 1.50 (p, J = 3.5 Hz, 4H), 1.37 (p, J = 3.6 Hz, 4H) was used to confirm the reaction. The characteristic NMR spectra is presented in Fig. S9.†Solid-state polymerization
Synthetic Nylon-6,6 samples were prepared via a 2-step solid state polymerization procedure. Reactions were performed on a 2 g scale. Reagents were weighed into a 50 mL RBF. Then, the flask was sealed with a rubber septum and purged for 15 min with Ar gas. The mixture of hexamethylene diamine and adipic acid was initially reacted at 220 °C under a steady flow of Ar. The reaction was cooled to room temperature, and the rubber septum was replaced with a flow control adapter. The flask was evacuated under vacuum for 5 min and a chain-extension step was carried out by heating to 270 °C under dynamic vacuum. Molecular weight was controlled by the by the addition of excess HMDA/adipic acid or by varying reaction times as presented in Table 1. Size exclusion chromatography was used to characterize the molecular weight of the polymers. The data are presented in Fig. S10.†| Sample | Nylon 66 salt (equiv.) | HMDA (equiv.) | Adipic acid (equiv.) | 220° rxn time (h) | 270° rxn time (h) |
|---|---|---|---|---|---|
| I-31-Mw 9.1 kDa | 1 | — | 0.1 | 1 | 1 |
| I-49-Mw 14.3 kDa | 1 | 0.1 | — | 1 | 1 |
| I-50-Mw 11.2 kDa | 1 | — | — | 1 | 1 |
| I-51-Mw 5.1 kDa | 1 | — | — | 0.25 | — |
Synthesis and expression of enzymes
The DNA sequences of 6-aminohexanoate-oligomer endo-hydrolase (NylC) was codon-optimized for E. coli and synthesized by GenScript (Piscataway, NJ, USA) in pET-28a(+) with XbaI/EcoRI cloning sites. Plasmids were transformed into BL21(DE3) competent E. coli (New England Biolabs, Ipswich, MA, USA) according to the manufacturer's protocol. A single colony was then inoculated from the transformation plate into 10 mL of LB and grown overnight at 37 °C with shaking. The next day, 5 mL of culture was transferred into 500 mL of TB supplemented with 50 μg mL−1 kanamycin. Cells were grown for 3–4 h, until the OD600 reached 0.7. Protein expression was then induced by adding 0.25 mM IPTG and growing overnight at 20 °C with shaking. Cells were separated by centrifugation at 4000 rpm for 30 min at 4 °C and the supernatant was discarded. The cell pellet was suspended in 25 mL 20 mM KPO4 (pH 7.4) and 10% glycerol (Buffer A). Cells were then sonicated for 10 min in 10 s bursts on ice. Finally, cell debris was removed via centrifugation at 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 45 min at 4 °C. The supernatant was collected and used for enzyme purification.
000 rpm for 45 min at 4 °C. The supernatant was collected and used for enzyme purification.
Purification of Nylon-6,6 hydrolases
Enzyme purification was adapted from Negoro et al.29 Briefly, 25 mL of 4.1 M ammonium sulfate was added dropwise to the cell lysate over a period of 5 min with slow stirring on ice. After stirring an additional 30 min on ice, precipitated proteins were collected by centrifugation (10 min at 8000 rpm and 4 °C). The supernatant was discarded and the protein pellet was resuspended in Buffer A and purified on an AKTA FPLC system outfitted with a 5 mL HiTrap Q FF column (Cytiva, Marlborough, MA, USA). NylC was eluted by increasing the concentration of NaCl in Buffer A step-wise in 0.1 M increments, with enzymes eluting at 0.3 M NaCl. Enzyme elution and purity were assessed via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Enzymes were purified further by size exclusion chromatography (320 ml HiPrep Sephacryl S-200 HR preparative column, Cytiva), followed by concentration (Pierce™ Protein Concentrators PES) in Buffer A. Final purified protein concentration was determined by Bradford assay and purity was assessed by SDS-PAGE.Nylon-6,6 substrate preparation
Commercial Nylon-6,6 pellets and synthesized Nylon-6,6 were subjected to cryomilling under liquid nitrogen to produce a fine powder. In the cryomilling, the solid samples were pulverized using a SPEX Sampleprep 6775 cryo-mill. Samples were subjected to a 1 min precool, a 2 min runtime at 5 cycles per second, followed another 1 min cool time, repeated for 5 total iterations.The surface areas of milled powder were determined from gas adsorption experiments. Following milling, the commercial Nylon-6,6 powder underwent a washing process aimed at removing the low molecular weight fractions. To achieve this, CH3OH was added in excess to the polyamide powder, and the mixture was stirred at room temperature for 2 h. After the initial washing, the CH3OH was carefully decanted, and fresh CH3OH was introduced for subsequent washing. This procedure was repeated 3 times. Finally, the washed powder was dried in vacuo for a minimum of 12 h to ensure complete solvent removal.
Enzymatic assays
Reactions with purified enzymes were performed at enzyme concentrations of 0.3 mg mL−1 in buffer containing 20 mM potassium phosphate, 10 μM caffeine (reference standard) at pH 7.4. Reactions were performed using 2–10 mg of Nylon-6,6 powder and 60 μL mg−1 Nylon-6,6 reaction volume and incubated for up to 72 h at 65 °C. Completed reactions were stored at −20 °C until analysis. The liquid part of the reaction was separated for MS analysis while the solid parts were characterized with GPC.Characterization
:100 (v/v%) in HPLC grade water with 0.1% formic acid and 500 nM propranolol. 40 μL of the diluted reaction mixtures were transferred to I.DOT S.100 96-well plates. The I.DOT system was used to inject 20 nL of sample into a flow of 75/25/0.1 (v/v/v%) acetonitrile/water/formic acid, which was delivered to the electrospray ion source of a Thermo Q-Exactive HF mass spectrometer (ThermoFisher Scientific, Waltham, MA, USA). The Q-Exactive HF operated in positive ion mode with flow = 250 μL min−1, sheath gas = 80, auxiliary gas = 40, electrospray voltage = 4 kV, mass resolution = 60000, ion injection time = 50 ms, automatic gain control = 3e,6 capillary temperature = 200 °C, and mass/charge (m/z) scan range = 100–1000 m/z. In-house developed softwares were used for control of the IDOT system, extraction of data from vendor file formats, peak finding, and peak integration. Each droplet signal was background-subtracted and normalized to the propranolol signal, correcting for droplet-to-droplet variability if present. Nylon 6,6 linear and cyclic oligomer signals were confirmed by exact mass and by tandem MS (not shown). Adducts of cyclic oligomers were predominantly found as [M + K]+ adducts, while linear oligomers were [M + H]+.30 Absolute quantitation of the linear monomer (L1) incorporated a 12-point calibration curve using synthesized L1 standards. Integrated raw intensities (arbitrary units, a.u.) were reported for comparison of cyclic and linear oligomers. Each sample was measured in triplicate. In our study, we used calibration curves developed for L2, C2, and L1, which were specifically synthesized for this purpose. These calibration curves were used to obtain the exact concentration values and calculate the yield. To calculate the reaction yield of L1, we considered both the conversion of L2 to L1 and the initial concentration of L1 in each sample, with their absolute masses determined from MS calibration curves. The contribution of C2 to L1 was evaluated based on the MS intensity ratio (L1/C2 = 12.26 derived from the calibration curves), and it was found to not exceed 16% for commercial Nylon-6,6, and below 13% for synthesized Nylons. Since the contribution of C2 is relatively small, it was not accounted for in the final L1 yield.
Gel permeation chromatography-size exclusion chromatography (GPC-SEC)
Size exclusion chromatography (SEC) analysis was performed on an Agilent 1260 Infinity II LC system equipped with an Agilent PL HFIPgel guard column (9 μm, 50 × 4.6 mm) and two Agilent PL HFIPgel columns (9 μm, 250 × 4.6 mm). A 0.02 M CF3COONa/HFIP solution was used as the mobile phase at 40 °C and at a flow rate of 0.300 mL min−1. Elution time was monitored using a differential refractive index (dRI) detector, a light scattering detector operating with two angles at 90° and 15°, and a differential viscometer. Number-average molecular weights (Mn), weight-average molecular weights (Mw) and polydispersity (Đ = Mw/Mn) were calculated based on calibrations with PMMA standards using the Agilent GPC/SEC software. Dilution factors were determined for individual samples to reduce the sample-to-sample variability due to viscosity change.Nuclear magnetic resonance (NMR) spectroscopy
1H NMR spectroscopy was performed on a Bruker Avance III 400 NMR spectrometer operating at 400 MHz (1H). Chemical shifts are reported in parts per million (ppm) relative to residual protonated solvent.Differential scanning calorimetry (DSC)
DSC (Q20, TA Instruments, New Castle, DE) was used to investigate the thermal properties of the Nylon-6,6 samples, such as their glass-transition temperatures (Tg), melting temperatures (Tm), and enthalpies of melting. Approximately 2–5 mg of Nylon-6,6 powder was placed in a Tzero aluminum pan and sealed with Tzero lid. All measurements were carried out under the N2 atmosphere. The analysis of the thermograms was carried out using the TA Instruments Universal Analysis 2000 software. The Tm value was reported from the endothermic peak temperature value. The enthalpy of melting was analyzed via integration of the endothermic peak. The Tg was reported from the heating cycle as the inflection point temperature.Wide angle X-ray scattering (WAXS)
WAXS was used to confirm the crystallinity of the Nylon-6,6 samples. WAXS measurements were carried out using a Xenocs Xeuss 3.0 instrument equipped with a D2+ MetalJet X-ray source (Ga Kα, λ = 1.3414 Å). The scattered beam was recorded on a Dectris Eiger 2R 4M hybrid photon counting detector with a pixel dimension of 75 × 75 μm2. The collected 2-dimensional (2D) WAXS images were circularly averaged and expressed as intensity versus Q, where Q = (4π·sinθ)/λ, after subtraction of background scattering. The Nylon-6,6 powder samples were prepared using a washer, which held the samples between two Kapton™ films. The empty washer with Kapton™ tape on both sides was used as a control.
Density measurements
The densities of the Nylon-6,6 powders were determined by pycnometry, performed on an AccuPyc II 1340 (Micromeritics, Norcross, Georgia) at room temperature. He gas was used for analysis and the equilibration rate was set to 0.050 psig per min. Each density value was averaged over three independent measurements.Brunaner–Emmett–Teller (BET) surface area measurement
N2 adsorption isotherms were collected on a Micromeritics 3Flex Adsorption Analyzer, and the data were analyzed using the provided software (3Flex Version 5.03). All samples for N2 isotherm measurements were added to a tared sample tube set (tube and check valve) and degassed at 80 °C on a Micromeritics VacPrep for 21.5 hours. The BET surface area values were extracted from a multipoint linear regression in the p/p° range of 0.05–0.40. The correlation coefficients of the linear regressions were >0.999.Particle size estimations
The particle sizes of Nylon-6,6 samples were estimated using the equation below according to the results from density measurements and BET surface area measurements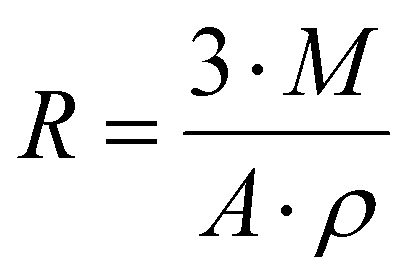 | (1) |
Molecular dynamics simulations
We conducted molecular dynamics (MD) simulations to investigate the interactions between enzyme NylC-GYAQ (the complete amino acid sequence is presented in Scheme S2†) and four Nylon-6,6 substrates: linear monomer (L1), linear dimer (L2), cyclic monomer (C1), and cyclic dimer (C2). We built the enzyme–substrate complex by positioning each Nylon substrate near the catalytic residue T267 of enzyme chain A at the interface of the enzyme chain A and chain D. Subsequently, we performed steered molecular dynamics (SMD) simulations to pull the carbonyl carbon at the center of the Nylon substrates toward the catalytic residue. We tracked the conformational changes of the Nylon substrates via the distance between the carbonyl carbon atom and the catalytic residue through conventional NPT (isothermal-isobaric ensemble) simulations.Enzyme and Nylon models
We constructed the NylC-GYAQ enzyme model based on an AlphaFold2 wild-type NylC tetramer model, with four residues in each chain (D122G, H130Y, D36A, and E263Q) mutated using the psfgen tool in VMD.34 Nylon-6,6 substrates, L1, L2, C1, and C2, were prepared using SMILES strings, from which the initial molecular geometries were generated using Open Babel software.35 All substrates were charge-neutral, with L1 and L2 edges terminated by NH3+ and CO2− groups. The point charge estimation was critical for the interaction between the substrate and enzyme. Point charges for the molecules were estimated using the AM1-BCC model36via the Antechamber tool.We used the Antechamber tool37 for the Nylon-6,6 substrates to assign General Amber ForceField (GAFF) parameters,38 which effectively capture the behavior of non-protein organic molecules.39,40 To ensure compatibility with GAFF, we employed the Amber Force Field FF19SB for the enzyme and the TIP3P water model.41 A 12 Å buffer was maintained between the enzyme and the edges of the solvent box. All systems were neutralized with NaCl at a concentration of 0.1 mol L−1 and fully solvated. Long-range electrostatic interactions were computed using the particle–particle particle–mesh (PPPM) method.42 The Nylon models and the solvated enzyme model were integrated using Moltemplate.43
The enzyme model was initially relaxed using a Langevin thermostat at 10 K for 50 ps, followed by another 50 ps relaxation with a Berendsen thermostat at 10 K. This was succeeded by a 50 ps relaxation using a Berendsen barostat at 1 atm, during which a 0.2 Å limit on position updates was applied to ensure rapid relaxation without disrupting the structure. The position update limit was then removed, and the structure was relaxed for an additional 50 ps. The temperature was gradually increased to 300 K over 50 ps, followed by relaxation at a constant 300 K for another 50 ps. Finally, the enzyme model underwent a 2 ns relaxation using the Nose–Hoover chain NPT ensemble (constant number of atoms, pressure, and temperature) with a 1 fs timestep. This configuration was applied to the enzyme/solvent model and saved in a Moltemplate-compatible format for later integration with the Nylon models. The root-mean-square deviation (RMSD) of the enzyme structure stabilized at approximately 2 Å and showed no significant changes with extended relaxation, confirming the structural stability of the enzyme model.
The generated Nylon models were initially self-entangled, requiring adjustments to their configurations. For the cyclic substrates (C1 and C2), we enforced a circular shape by fixing four points in the models to minimize inter-chain atomic interactions. The approximate radius was estimated, and multiple energy minimizations were performed to relieve undesired strains. For the linear substrates (L1 and L2), we ensured they were straightened and applied energy minimizations to eliminate residual strain. The relaxed coordinates were saved in a Moltemplate-compatible format. MD simulations were conducted using the LAMMPS package44 to investigate the conformations and relative position between substrates and the enzyme.
Conformational changes of Nylon substrates in the presence of enzyme
We combined the Nylon and solvated enzyme models using Moltemplate to generate LAMMPS input data. This approach provided flexibility in adjusting the initial positions of the substrates while maintaining consistent, relaxed enzyme structures across different Nylon models. Since the Nylon models were inserted into the solution, the initial configurations could produce unphysically high forces. To relieve this steric effect, we performed an additional short relaxation of the enzyme/solution system for 50 ps, applying a positional update limit of 0.2 Å. The relaxation was conducted using a Langevin thermostat, with the temperature gradually increased from 10 K to 300 K.We then conducted SMD simulations to guide the catalytic residue T267 of the enzyme toward the carbonyl carbon of the Nylon amide bond. During the pulling process, L1 and L2 were constrained using an imaginary spring with a constant of 10.0 kcal mol−1 to minimize self-entanglement. A second spring with a constant of 20.0 kcal mol−1 Å−1 was applied between the carbonyl carbon and T267, with a pulling rate of 1 × 10−5 Å fs−1. This relatively strong spring constant was selected to precisely control the SMD steps required to bring the catalytic group within ∼4 Å of the target residue, a distance empirically determined.
Through modeling, we aimed to understand and compare interactions between different Nylon substrates and the enzyme, recognizing that the precise binding structures have not yet been identified. To ensure consistency across models, we followed a uniform process to evaluate how variations in the Nylon substrate influenced its interaction with the enzyme. The structures were further relaxed for 50 ps using a spring with a reduced constant of 10.0 kcal mol−1 Å−1 between the catalytic residue and the Nylon's carbonyl carbon. Following this relaxation, we monitored the distance between the Nylon's carbonyl carbon atom and the enzyme's catalytic residue over a 5 ns simulation.
Results and discussion
To better understand the substrate limitations of enzymatic hydrolysis of Nylon-6,6, we first performed a detailed characterization of the starting material. The Nylon-6,6 was cryomilled to increase surface area, and the resulting powder was washed with methanol. This washing procedure was necessary to remove low MW Nylon-6,6 oligomers, which are byproducts of synthesis in the commercial samples45 and could be degraded by NylC. To evaluate the efficacy of washing, soluble compounds extracted from the powdered Nylon-6,6 were analyzed by mass spectrometry (MS) before and after washing (Fig. S11a†). Substantial amounts of cyclic monomer (C1), cyclic dimer (C2), linear monomer (L1), and linear dimer (L2) were found in the sample before washing. After washing, concentrations of all low MW species were significantly reduced. The MW of the washed Nylon-6,6 powder was measured by SEC prior to deconstruction, yielding a weight average MW, Mw, of 76.3 kDa with Đ = 2.2.We then incubated the washed Nylon-6,6 powder (16.7 mg ml−1) with 0.3 mg mL−1 NylC for 72 h at 65 °C and measured changes in the residual polymer and soluble products by SEC and MS, respectively, compared to samples processed similarly but without the addition of enzyme (Fig. 1a and b). As measured by SEC, the control sample contained peaks at both low and high MWs or high and low retention times (RTs), which changed after incubation with NylC. The low MW peak increased in amplitude, while the high MW peak showed changes in amplitude and width. Overlapping error bars suggest these changes may not be significant. Analysis of peak areas (Fig. S11b†) confirms the low MW peak change is significant at the 80% confidence level, while the high MW peak variation is not. Importantly, no intermediate MW species were detected in the SEC data after 72 h incubation, as evidenced by the absence of broadening in the high MW peak and the absence of any new peaks in the retention time interval from 18–22 min. This observation could suggest that either products with intermediate MW (L3–L4) were not generated or that such intermediate products, once formed, were rapidly hydrolyzed to the observed products with low MW (L1–L2).
 | ||
| Fig. 1 Hydrolysis of washed commercial Nylon-6,6 by NylC. (a) Normalized SEC traces averaged over three biological replicates, with standard deviation (SD) included and (b) MS intensities for hydrolysis of commercial Nylon-6,6 incubated with (red) or without NylC (control, black) for 72 h. (c) L1 yield obtained from MS plotted vs. time. Inset: L1 to enzyme mole ratio for each kinetic point. The fitted line defines the linear regime of the reaction with the slope representing the reaction rate. | ||
To distinguish between these two scenarios, the kinetics of product formation during enzymatic hydrolysis of washed Nylon-6,6 were studied with MS. As above, a kinetic control experiment was also conducted in the absence of enzyme for comparison. The MS intensity profile for L1–L4 species for each kinetic point are shown in Fig. S11(c).† Only the L1 product increased in abundance over time, while the L2–L4 products were either not detected or remained below the limit of quantitation (<1 × 104 a.u., at this intensity the concentration is considered equal to zero). Furthermore, after 72 h of incubation with NylC (Fig. 1b), the level of C1 remained nearly constant, the C2 signal was eliminated, and the signal for L1 increased substantially (by over 1000-fold). The minimal change in C1 intensity suggested that it was not a preferred substrate for the enzyme. In contrast, the disappearance of C2 pointed to an enzymatic ring-opening of this substrate to L2 and its subsequent scission to form L1. It is important to note that we looked for all possible hydrolysis products from L2 (e.g., L1.5), but no significant signals were observed for other products than L1 which is suggestive that not all amides are equally available for cleavage by this enzyme. The contribution of C2 to final L1 yield, calculated from MS calibration, did not exceed 16%, suggesting that most of the generated L1 originated from polymer chain cleavage.
Since intermediate MW products were not detected by either SEC or MS, we hypothesized that hydrolysis of commercial Nylon-6,6 by NylC proceeded via a predominately exo-cleavage mechanism. This mechanism involves enzymatic cleavage exclusively at the chain ends, with L1 (monomer) being the sole product of the hydrolysis. In contrast, endo-cleavage enzymes operate by random chain scission and would be characterized by a broad MW distribution that rapidly evolves towards low MW. To understand the relative hydrolysis rate of NylC reaction, the yield of L1, as determined by MS calibration with a chemically synthesized L1 standard, was plotted at each time point (Fig. 1c). The hydrolysis reaction was largely complete after 72 h, with no significant change in yield between 72 h and 240 h, affording a total turnover number of ca. 20 (inset, Fig. 1c). We note that the formation of L1 with respect to time would be expected to increase exponentially for an endo-cleavage mechanism,46 with minimal L1 formation at the early stages of the hydrolysis reaction. As such, these kinetic data further implicate an exo-cleavage mechanism.
Considering the relatively slow reaction rate of hydrolysis and the proposed exo-cleavage mechanism, we hypothesized that increasing the number of chain ends (i.e., reducing substrate MW) should increase the yield of L1. To test this hypothesis, Nylon-6,6 with different MWs were synthesized (see Table S1† for MW characterization). These polymers were incubated with NylC for 72 h and hydrolysis was monitored by SEC and MS (Fig. 2). The differences between the control and hydrolyzed samples in SEC traces for synthesized polymers with higher MWs (e.g. Mw = 11.2 kDa and Mw = 14.3 kDa) qualitatively aligned with those observed for commercial Nylon-6,6 (Fig. 1a and Fig. 2a). Specifically, the low MW peak showed an increase in amplitude, while the broadness and width of the high MW peak remained barely changed. Samples with lower initial substrate MW showed larger changes in their MW distributions, as these variations extended beyond the error range. In each case, the low MW peak at 21 min, which was consistent with the L1 standard, increased in amplitude in the presence of the enzyme (Fig. 2b). Simultaneously, the width of the high MW peak at 18.5 min decreased compared to the control, due to the consumption of chains at the low MW end of the distribution. As in the case of commercial Nylon-6,6, no intermediate MW products were observed during hydrolysis of the synthesized polymers.
 | ||
| Fig. 2 Enzymatic hydrolysis of synthesized Nylon-6,6 with lower MW. (a) and (b) Normalized SEC traces averaged over two biological replicates with included SD of washed Nylon-6,6 after incubation with NylC for 72 h, a control sample treated under the same conditions but with no added enzyme, and pure L1. Panels a and b show the differences in product evolution when starting from different MW Nylon-6,6. (c) MS data for enzymatic hydrolysis of synthesized Nylon-6,6 after 72 h with the indicated MWs. | ||
The concentrations of small, soluble compounds were also monitored by MS, focusing on the polymers with Mw = 5.1 kDa and 11.2 kDa (Fig. 2c). The post-reaction mixtures contained L1 and C1, suggesting that C2 and L2 originally present in the control sample were converted to the L1 product. The contribution of C2 to final L1 yield was found not to exceed 13%. C1 was not consumed during the reaction, in line with our observations for commercial Nylon-6,6 substrate. There was no L3 or L4 found before or after the reaction which further corroborated an exo-cleavage mechanism for enzymatic hydrolysis of these samples.
To understand the observed differences in NylC activity on the various oligomeric constituents of Nylon-6,6 (C1, C2, L1, and L2), we conducted molecular dynamics (MD) simulations, where enzymatic activity was evaluated based on substrate dynamics in the presence of the enzyme. The simulation results are presented in Fig. 3. The conformation of C1 was highly strained within the enzyme binding region. The smaller radius of C1 constrained its flexibility to adopt an elliptical shape, leading to its escape from the enzyme binding region with the largest distance between the T267 catalytic site and the amide bond (Fig. 3a, Fig. S12 and S13†). In contrast, C2 adopted a stable elliptical shape within the enzyme binding region with the amide bond reaching close to the T267 catalytic site (Fig. 3b). This allowed favorable binding of this substrate to the enzyme. Similarly to C1, L1 remained stretched, with its amide group positioned farther from the catalytic residue, thus making L1 binding unfavorable (Fig. 3c). In contrast, L2 was shown to be stable near the catalytic residue by adopting a bent conformation (Fig. 3d, Fig. S14 and S15), easing enzyme-catalyzed hydrolysis of this substrate. Our experimental and MD findings demonstrate that NylC exhibits both exo- and endo-cleavage activity, as shown by its ability to aid hydrolysis of the L2 and C2 substrates, respectively. While an endo-cleavage mechanism has been proposed in the literature for this enzyme,47 our results further revealed that the enzyme's activity is highly substrate-specific, determined by the compatibility between the substrate's shape and the enzyme's binding region. This specificity rendered substrates L1 and C1 unsuitable for enzymatic hydrolysis. Additionally, substrate properties of the high MW Nylon-6,6, such as crystallinity and low water solubility likely hindered the proper positioning of the polymer within the binding site, thus, favoring hydrolysis at the more accessible for the enzyme terminal regions of the chain.
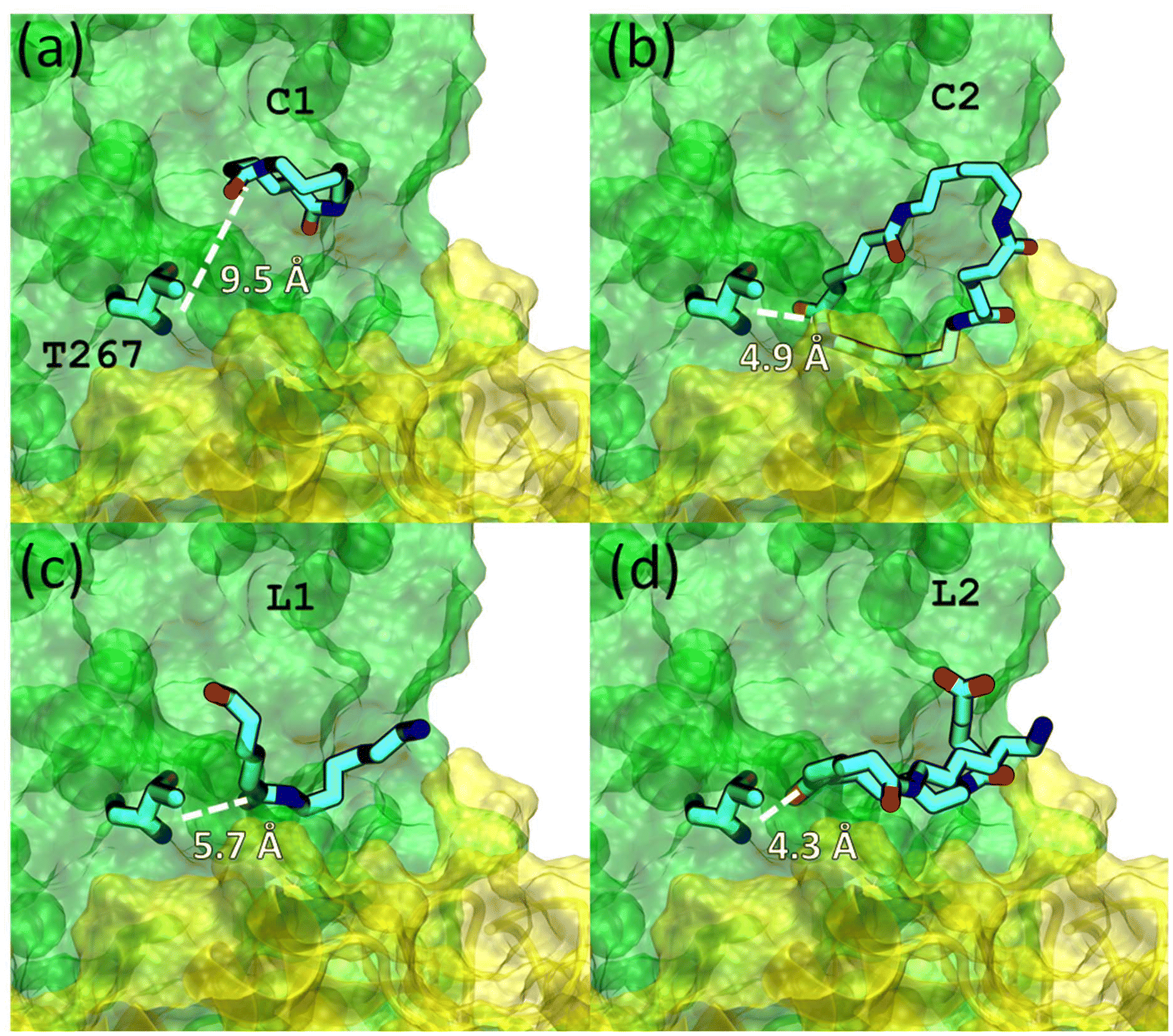 | ||
| Fig. 3 MD simulated binding of NylC-GYAQ with C1 (a), C2 (b), L1 (c), and L2 (d). NylC-GYAQ is shown in surface, with chain A in green and chain D in yellow. Chains B and C are omitted for clarity. The catalytic residue T267 and Nylon-6,6 substrates are in stick. Distances between T267 and carbonyl carbon of Nylon-6,6 substrates are labeled. Note the close proximity of C2 and L2 to T267. The snapshots are taken from the last conformations of 5 ns trajectories. | ||
L1 yields obtained in the enzymatic reaction with different MWs from MS were used to probe the mechanism of enzymatic hydrolysis. We first assessed the impact of initial polymer MW on hydrolytic yield (Fig. 4a). On a double logarithmic scale, this behavior was described by a linear relationship with a slope close to 1/Mw, suggesting that L1 yield strongly depended on the number of chains and, hence, the number of chain ends, supporting our initial hypothesis. However, deviations from this relationship (e.g. averaged slope is <−1) indicated the involvement of additional substrate limitations. We therefore investigated the impact of other substrate properties, specifically crystallinity, and surface area (Fig. 4b and c), on L1 yields.
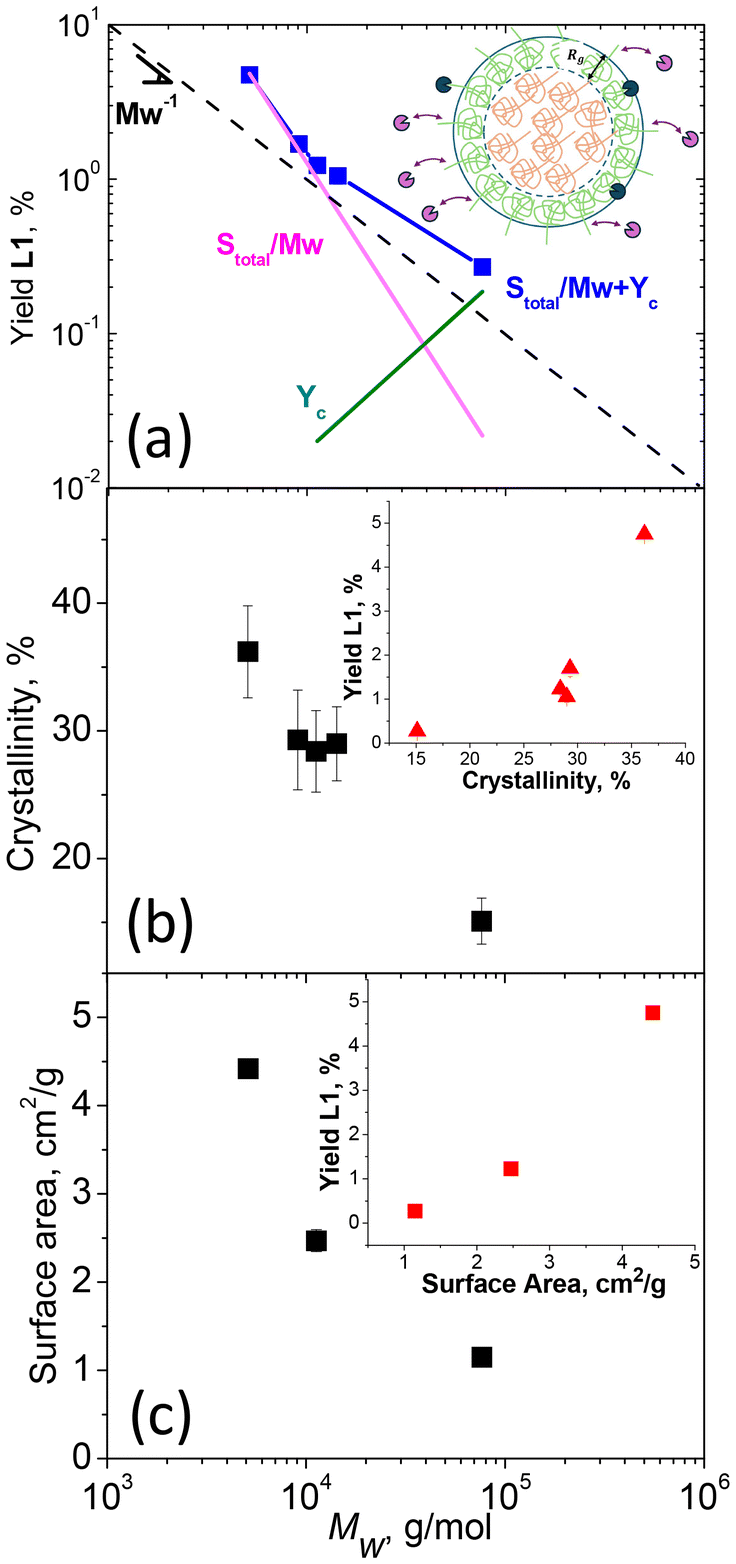 | ||
| Fig. 4 (a) Dependence of L1 yield on Mw of the polymers (squares). Two regimes are observed: (i) at a small molecular weight the yield is proportional to Stotal/Mw (pink line) suggesting enzyme hydrolyzing surface confined chains by association/dissociation mechanism, (ii) at high molecular weight, the yield dominates by Yc (green line), provided by enzymes hydrolyzing the same polymer chain; (b) crystallinity and (c) surface area plotted vs. Mw the insets are (b) crystallinity and (c) surface area plotted vs.L1 yield. | ||
The degree of crystallinity of the polymers was determined by fitting the high-q region of the WAXS data obtained for each sample (Fig. S16a†). Examples of fitting are shown in Fig. S16b and c,† with the fitting results for all samples summarized in Table S1.† Independently, changes in crystallinity were evaluated using DSC by extracting the enthalpy of melting from the heating cycles (Fig. S17†). These data, also summarized in Table S1,† were found to align with the results from WAXS. The relationship between crystallinity and Mw is presented in Fig. 4b. Specifically, crystallinity was observed to increase as Mw decreased, aligning with the general behavior found in polymers.48–50
Nitrogen adsorption isotherm analysis (Fig. S18†) was used to estimate the surface area and characteristic size of the polymer particles (eqn (1)), and these values are summarized in Table S1.† The surface area as a function of Mw is shown in Fig. 4c, revealing an increase in surface area with decreasing Mw of the polymer. Since we applied the same cryomilling procedure to all samples, the trend suggested that at higher crystallinities, the sample became more brittle, which could lead to particles breaking down into finer sizes, resulting in a net increase in surface area. As evident from insets of Fig. 4, crystallinity and surface area also contributed to the yield, as some dependence is observed in the yield with changes in both of these parameters. As a result of these contributions, the scaling with only 1/Mw failed for the points in Fig. 4a. Since MW, crystallinity, and surface area are interdependent, disentangling their individual contributions in the experiment is challenging. On the other hand, individual contribution could be revealed more easily through a model.
To describe the relationship depicted in Fig. 4a, our model incorporated spherical particles and enzymes, as illustrated in the inset of Fig. 4a. We assumed that the accessibility of the polymer substrate for enzymatic attack would initially be limited primarily to the chain ends located on the surface of the polymer particles. In this scenario, the enzyme would periodically associate with a chain end, initiate polymer cleavage, and subsequently dissociate back into the solution. The yield associated with these association–dissociation processes would then depend on the probability of encountering chain ends on the surface of the polymer particle, which can be expressed as
 | (2) |
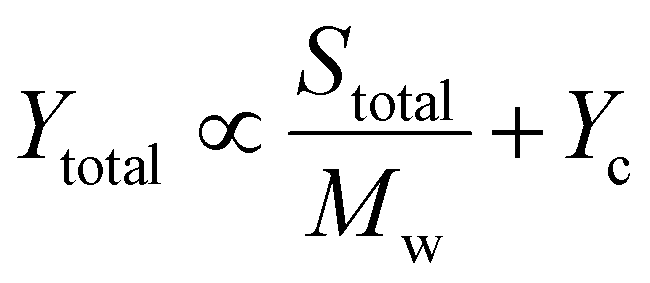 | (3) |
In this case, the more crystalline, low and medium MWs samples, characterized by more chains and higher overall surface area compared to the high MW sample, contributed to an increase in the S/Mw component with the first mechanism predominating. At high MW, the number of chains was lower, reducing the S/Mw component, but the amorphous fraction was higher, allowing the bound enzyme to turn over for a longer time before encountering a crystalline phase and dissociating. As a result, the contribution to Yc is enhanced relative to S/Mw. Furthermore, at high MW, the relative fraction of repeat units located in amorphous domains was necessarily higher compared with the low MW samples. This would further increase the relative contribution to c, shifting the behavior toward the dominance of the second mechanism. This simplified model does not account for the specific mechanisms of substrate binding to the enzyme, nor does it consider substrate heterogeneity or inhibition,51 both of which could influence final yields.
Conclusions
In this contribution, we demonstrated that the MW, surface area, and crystallinity can influence the enzymatic hydrolysis of Nylon-66, with the yield of monomer L1 changing from 0.25% to 4%. We demonstrated that although NylC is capable of endo hydrolysis of cyclic oligomers, it largely performs exo-cleavage of polymers. Consequently, MW is the primary factor influencing enzymatic reaction yield. Other polymer characteristics have indirect effects; in high MW polymers, lower crystallinity may enhance enzyme penetration into the bulk to access chain ends, while in low MW polymers, hydrolysis is likely limited to surface chain ends. Our studies provide a solid foundation for discovering the most efficient substrate pre-treatment protocols to advance the enzymatic hydrolysis of Nylon-6,6.Author contributions
VB: conceptualization, visualization, writing – original draft preparation; ED: investigation, writing – reviewing & editing; JFC: investigation, formal analysis, writing – reviewing & editing; IP, CG: methodology; ITD: investigation; MZ: investigation; GSJ, SHC: methodology, visualization, writing – reviewing & editing; AMU: investigation; DLC: investigation, formal analysis; JTD: investigation; JKK: investigation, formal analysis; JCF: conceptualization, visualization, writing – original draft preparation; JKM: conceptualization, writing – original draft preparation, Funding acquisition.Notes
This manuscript has been authored by UT-Battelle, LLC, under contract DE-AC05-00OR22725 with the US Department of Energy (DOE). The US government retains and the publisher, by accepting the article for publication, acknowledges that the US government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for US government purposes. DOE will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (https://energy.gov/downloads/doe-public-access-plan).Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI files.† Should any raw data files be needed in another format they are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Research was sponsored by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory, managed by UT-Battelle, LLC under Contract No. DE-AC05-00OR22725, for the U.S. Department of Energy. This research used resources from the Compute and Data Environment for Science (CADES) at the Oak Ridge National Laboratory, which is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC05-00OR22725 and National Energy Research Scientific Computing Center (NERSC), a DOE Office of Science User Facility for access to additional supercomputing resources. The enzyme/Nylon modeling work is part of a user project at the Center for Nanophase Materials Sciences (CNMS), a US Department of Energy, Office of Science User Facility at Oak Ridge National Laboratory. We thank Luigi Balzano from DMS Engineering Plastics and Wim Bras from Oak Ridge National Laboratory for providing the Nylon-6,6 pellets.References
- E. K. Bolton, Ind. Eng. Chem., 1942, 34, 53–58 CAS
.
-
B. Herzog, M. I. Kohan, S. A. Mestemacher, R. U. Pagilagan, K. Redmond and R. Sarbandi, in Ullmann's Encyclopedia of Industrial Chemistry, 2020, pp. 1–47, DOI:10.1002/14356007.a21_179.pub4
- H. W. Starkweather Jr, G. E. Moore, J. E. Hansen, T. M. Roder and R. E. Brooks, J. Polym. Sci., 1956, 21, 189–204 Search PubMed
- V. Hirschberg and D. Rodrigue, J. Polym. Sci., 2023, 61, 1937–1958 CAS
- G. Mondragon, G. Kortaberria, E. Mendiburu, N. González, A. Arbelaiz and C. Peña-Rodriguez, J. Appl. Polym. Sci., 2020, 137, 48442 CAS
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CAS
- V. Tournier, C. M. Topham, A. Gilles, B. David, C. Folgoas, E. Moya-Leclair, E. Kamionka, M. L. Desrousseaux, H. Texier, S. Gavalda, M. Cot, E. Guémard, M. Dalibey, J. Nomme, G. Cioci, S. Barbe, M. Chateau, I. André, S. Duquesne and A. Marty, Nature, 2020, 580, 216–219 CAS
- V. Tournier, S. Duquesne, F. Guillamot, H. Cramail, D. Taton, A. Marty and I. André, Chem. Rev., 2023, 123, 5612–5701 CrossRef CAS PubMed
- A. R. Bergeson, A. J. Silvera and H. S. Alper, Nat. Commun., 2024, 15, 4715 CrossRef CAS PubMed
- S. Negoro, N. Shibata, Y.-H. Lee, I. Takehara, R. Kinugasa, K. Nagai, Y. Tanaka, D.-i. Kato, M. Takeo, Y. Goto and Y. Higuchi, Sci. Rep., 2018, 8, 9725 CrossRef PubMed
-
S. Negoro, D.-i. Kato, T. Ohki, K. Yasuhira, Y. Kawashima, K. Nagai, M. Takeo, N. Shibata, K. Kamiya and Y. Shigeta, in Methods in Enzymology, ed. G. Weber, U. T. Bornscheuer and R. Wei, Academic Press, 2021, vol. 648, pp. 357–389 Search PubMed
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Nature, 2022, 604, 662–667 CrossRef CAS PubMed
- L. Pfaff, J. Gao, Z. Li, A. Jäckering, G. Weber, J. Mican, Y. Chen, W. Dong, X. Han, C. G. Feiler, Y.-F. Ao, C. P. S. Badenhorst, D. Bednar, G. J. Palm, M. Lammers, J. Damborsky, B. Strodel, W. Liu, U. T. Bornscheuer and R. Wei, ACS Catal., 2022, 12, 9790–9800 CrossRef CAS PubMed
- Y. Cui, Y. Chen, X. Liu, S. Dong, Y. e. Tian, Y. Qiao, R. Mitra, J. Han, C. Li, X. Han, W. Liu, Q. Chen, W. Wei, X. Wang, W. Du, S. Tang, H. Xiang, H. Liu, Y. Liang, K. N. Houk and B. Wu, ACS Catal., 2021, 11, 1340–1350 Search PubMed
- H. Puetz, C. Janknecht, F. Contreras, M. Vorobii, T. Kurkina and U. Schwaneberg, ACS Sustainable Chem. Eng., 2023, 11, 15513–15522 CrossRef CAS
- E. L. Bell, G. Rosetto, M. A. Ingraham, K. J. Ramirez, C. Lincoln, R. W. Clarke, J. E. Gado, J. L. Lilly, K. H. Kucharzyk, E. Erickson and G. T. Beckham, Nat. Commun., 2024, 15, 1217 Search PubMed
- J. de Witt, M.-E. Ostheller, K. Jensen, C. A. M. R. van Slagmaat, T. Polen, G. Seide, S. Thies, B. Wynands and N. Wierckx, Green Chem., 2024, 26, 9911–9922 RSC
- E. E. Drufva, J. F. Cahill, P. M. B. Saint-Vincent, A. N. Williams, V. Bocharova, N. Capra, F. Meilleur, D. L. Carper, C. Bourgery, K. Miyazaki, M. Yonemura, Y. Shiraishi, J. M. Parks, M. Zhou, I. T. Dishner, J. C. Foster, S. J. Koehler, H. R. Valentino, A. Sedova, V. Kertesz, D. P. Vasileva, L. H. Hochanadel, C. A. Figg, S. Negoro, D.-i. Kato, S. H. Chen and J. K. Michener, bioRxiv, 2024. DOI:10.1101/2024.11.14.623603.
- E. Erickson, J. E. Gado, L. Avilán, F. Bratti, R. K. Brizendine, P. A. Cox, R. Gill, R. Graham, D.-J. Kim, G. König, W. E. Michener, S. Poudel, K. J. Ramirez, T. J. Shakespeare, M. Zahn, E. S. Boyd, C. M. Payne, J. L. DuBois, A. R. Pickford, G. T. Beckham and J. E. McGeehan, Nat. Commun., 2022, 13, 7850 CAS
- E. R. Hoffman, A. M. M. Rangaswamy, M. E. Cleveland, J. W. Keillor and G. W. Howe, Angew. Chem., Int. Ed., 2025, 64, e202414842 CrossRef CAS PubMed
- R. Wei, G. Weber, L. M. Blank and U. T. Bornscheuer, Nat. Chem. Eng., 2025, 2, 110–117 Search PubMed
- B. Ciuffi, E. Fratini and L. Rosi, Polym. Degrad. Stab., 2024, 222, 110698 CrossRef CAS
- Å. M. Ronkvist, W. Xie, W. Lu and R. A. Gross, Macromolecules, 2009, 42, 5128–5138 CrossRef
- T. B. Thomsen, C. J. Hunt and A. S. Meyer, New Biotechnol., 2022, 69, 28–35 CAS
- S. Kaabel, J. P. D. Therien, C. E. Deschênes, D. Duncan, T. Friščić and K. Auclair, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2026452118 CAS
- R. K. Brizendine, E. Erickson, S. J. Haugen, K. J. Ramirez, J. Miscall, D. Salvachúa, A. R. Pickford, M. J. Sobkowicz, J. E. McGeehan and G. T. Beckham, ACS Sustainable Chem. Eng., 2022, 10, 9131–9140 CrossRef CAS
- C. Park, E. Y. Kim, Y. T. Yoo and S. S. Im, J. Appl. Polym. Sci., 2003, 90, 2708–2714 CrossRef CAS
- Y. Shiraishi, D.-i. Kato, K. Miyazaki, M. Yonemura, Y. Furuno, R. Yokoyama, Y. Yokogawa, S. Nonaka, Y. Kaneko, K. Ebata, Y. Himeda and S. Negoro, PLoS One, 2025, 20, e0318641 CAS
- S. Negoro, N. Shibata, Y. Tanaka, K. Yasuhira, H. Shibata, H. Hashimoto, Y.-H. Lee, S. Oshima, R. Santa, S. Oshima, K. Mochiji, Y. Goto, T. Ikegami, K. Nagai, D.-i. Kato, M. Takeo and Y. Higuchi, J. Biol. Chem., 2012, 287, 5079–5090 CAS
- J. F. Cahill, V. Kertesz, P. Saint-Vincent, H. Valentino, E. Drufva, N. Thiele and J. K. Michener, J. Am. Soc. Mass Spectrom., 2023, 34, 1383–1391 CAS
- G. J. Van Berkel, V. Kertesz and H. Boeltz, Bioanalysis, 2017, 9, 1667–1679 Search PubMed
- G. J. Van Berkel and V. Kertesz, Rapid Commun. Mass Spectrom., 2015, 29, 1749–1756 CAS
- V. Kertesz, D. L. Carper and J. F. Cahill, Rapid Commun. Mass Spectrom., 2024, 38, e9687 CAS
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CAS
- N. M. O'Boyle, M. Banck, C. A. James, C. Morley, T. Vandermeersch and G. R. Hutchison, J. Cheminf., 2011, 3, 33 Search PubMed
- A. Jakalian, D. B. Jack and C. I. Bayly, J. Comput. Chem., 2002, 23, 1623–1641 CAS
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Mol. Graphics Modell., 2006, 25, 247–260 CAS
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CAS
- G. G. Jang, G. S. Jung, J. Seo, J. K. Keum, M. Yoon, J. T. Damron, A. K. Naskar, R. Custelcean, A. Kasturi, S. Yiacoumi and C. Tsouris, ChemSusChem, 2024, 17, e202300735 CAS
- H. Song, L. Vogt-Maranto, R. Wiscons, A. J. Matzger and M. E. Tuckerman, J. Phys. Chem. Lett., 2020, 11, 9751–9758 CAS
- D. J. Price and C. L. Brooks, III, J. Chem. Phys., 2004, 121, 10096–10103 CAS
-
R. W. Hockney and J. W. Eastwood, Computer Simulation Using Particles (1st ed.), CRC Press, 2021 Search PubMed
- A. I. Jewett, D. Stelter, J. Lambert, S. M. Saladi, O. M. Roscioni, M. Ricci, L. Autin, M. Maritan, S. M. Bashusqeh, T. Keyes, R. T. Dame, J.-E. Shea, G. J. Jensen and D. S. Goodsell, J. Mol. Biol., 2021, 433, 166841 CAS
- S. Plimpton, J. Comput. Phys., 1995, 117, 1–19 CAS
- A. Schweighuber, J. Fischer and W. Buchberger, Polymers, 2021, 13, 2032 CAS
- H. S. Wang, N. P. Truong, Z. Pei, M. L. Coote and A. Anastasaki, J. Am. Chem. Soc., 2022, 144, 4678–4684 CAS
- S. Kakudo, S. Negoro, I. Urabe and H. Okada, Appl. Environ. Microbiol., 1993, 59, 3978–3980 CAS
- A. Fernández-Tena, R. A. Pérez-Camargo, O. Coulembier, L. Sangroniz, N. Aranburu, G. Guerrica-Echevarria, G. Liu, D. Wang, D. Cavallo and A. J. Müller, Macromolecules, 2023, 56, 4602–4620 Search PubMed
- R. A. Pérez, M. E. Córdova, J. V. López, J. N. Hoskins, B. Zhang, S. M. Grayson and A. J. Müller, React. Funct. Polym., 2014, 80, 71–82 Search PubMed
- L. L. Schega, D. Hesse-Hornich, R. Helwing, R. Scholz, F. Walther and U. A. Handge, Polymer, 2024, 300, 127012 CAS
- J. A. Thoma, Biochemistry, 1966, 5, 1365–1374 CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00023h |
| This journal is © The Royal Society of Chemistry 2025 |