
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermoresponsive lactate amide acrylic polymers developed from PLA bags†
Marc
Palà
 a,
Alina
Ismagilova
b,
Adrian
Moreno
a,
Jorge
Plaza
a,
Juan C.
Ronda
a,
Marina
Galià
a,
Lauri
Vares
b and
Gerard
Lligadas
*a
a,
Alina
Ismagilova
b,
Adrian
Moreno
a,
Jorge
Plaza
a,
Juan C.
Ronda
a,
Marina
Galià
a,
Lauri
Vares
b and
Gerard
Lligadas
*a
aUniversity Rovira i Virgili, Departament de Química Analítica i Química Orgànica, Laboratory of Sustainable Polymers, Tarragona 43007, Spain. E-mail: gerard.lligadas@urv.cat
bInstitute of Technology, University of Tartu, Nooruse 1, Tartu 50411, Estonia
First published on 27th February 2025
Abstract
The growing global demand for sustainable products, driven by the depletion of fossil resources and mounting environmental concerns, has amplified interest in transforming lignocellulosic biomass into bio-based solvents, fine chemicals, and polymers. Among these, lactic acid has emerged as a pivotal platform chemical for synthesizing high-value derivatives. The chemical depolymerization of polylactic acid (PLA) into lactate esters and amides represents a straightforward and efficient strategy for upcycling PLA waste into specialty polymers. In this study, we developed a mini-library of lactate amide-based acrylic monomers using commercially available PLA bags as feedstock. These monomers were polymerized into homo, statistical, and block copolymers via Cu(II)Br2/Me6TREN-mediated polymerization under UV light. The resulting polymers exhibited water solubility adjustable through amide N-substitution combined with low ecotoxicity. This innovative approach not only advances sustainable PLA waste management but also opens new possibilities for designing advanced thermoresponsive polymers with single or double phase separation behaviors—an underexplored frontier in biobased synthetic polymer research.
Introduction
The global demand for sustainable products has surged in recent years, driven by the gradual depletion of fossil resources and growing environmental concerns related to their extraction and the disposal of fossil-derived materials. In response, various synthetic strategies have been developed to convert lignocellulosic biomass into bio-based platforms for value-added chemicals, leading to a diverse range of products.1–3 Among these, lactic acid obtained through carbohydrate fermentation or chemocatalytic processes has emerged as a key platform chemical.4–6 Of particular interest are lactic acid-derived alkyl esters (lactate esters, LEs) and amides (lactate amides, LAs), which have gained attention as competitive green biosolvents to replace fossil-based solvents in chemical reactions, coatings, paints, cosmetics and hair care products due to their favorable properties, such as high boiling points, low ecotoxicity profile, and strong solvency.7–12However, the most exploited line of lactic acid processing is in the production of polylactic acid (PLA), one of the most promising biopolymers.13,14 PLA is valued for its excellent biocompatibility, ease of processing, and favorable mechanical properties, making it suitable for a wide range of applications, including disposable plastics, textiles, and food packaging.15,16 However, PLA degradation into products such as H2O and CO2 occurs only under controlled industrial conditions, requiring a lengthy process to convert degradation products back into PLA.17,18 This process involves multiple stages, including crop growth, fermentation, and polymerization. Consequently, the recycling and upcycling of end-of-life PLA have become critical challenges in both academic and industrial contexts.
Chemical depolymerization offers a promising solution by converting PLA into valuable chemicals through the use of alcohols and amines.19,20 These processes are efficient and operate under mild conditions, facilitating PLA upcycling into fine chemicals, high-value solvents, and specialty polymers.21–23 Although alcoholysis of PLA to produce LEs, such as ethyl lactate (EL), is well established, recent studies have highlighted the potential of aminolysis-mediated PLA depolymerization as a promising route for generating original polymeric products. For example, reaction of PLA with ethanolamine, followed by derivatization of the resulting LA with methacrylic anhydride, has been shown a promising approach to produce photocurable resins for 3D printing.19 Alternatively, the use of diamines has been explored to synthesize valuable diol derivatives, which, when reacted with dicarboxylic acids, yield poly(ester-amide) structures with tunable properties.24 Another notable development involved the synthesis of N-tetrahydrofurfuryl LA from EL, which was acrylated and subsequently well-controlled polymerized in water in our laboratory.25 The resulting rubbery polymer was water-soluble only at low temperatures and exhibited a clear phase separation upon heating, with a lower critical solution temperature (LCST) below room temperature. By replacing the secondary amide group with a tertiary amide bearing methyl substituents, the polymer, i.e. poly(N,N-dimethyl LA acrylate), became glassy and remained water-soluble up to high temperatures.26–28 These initial studies revealed interesting structure-dependent aqueous solution properties for N- and N,N-substituted LA polyacrylates, prompting further exploration of alternative side-chain moieties.
In this study, we build upon these findings by focusing on LA biosynthon as platform chemical for synthesizing acrylic polymers with thermoresponsive behavior. We synthesized a mini-library of ten LA-based acrylic monomers with varying N-substitution patterns using PLA bags as feedstock. Well-defined homo-, statistical-, and block-copolymers were then synthesized at ambient temperature using Cu(II)Br2/Me6TREN as a catalytic system under UV light. These polymers were systematically evaluated for their ecotoxicity, thermal properties and solubility behavior, and used as building blocks to develop thermoresponsive block copolymers (BCPs) with a single or multistep phase separation behaviors. Although similar LA acrylic homopolymers produced via free radical polymerization have been reported in earlier patent literature,29 this study represents the first detailed study on these neoteric acrylic polymers derived from lactic acid. This innovative approach not only provides a sustainable method for managing PLA waste but also paves the way for creating advanced biobased polymers with specific water-solubility characteristics, which are critical in a wide range of commercial applications.
Experimental section
Materials
The PLA bags used in this study were sourced from Sobres España and utilized without further modification. Polymerization initiator 1-(dimethylamino)-1-oxopropan-2-yl 2-bromo-2-methylpropanoate (DMLBr) was prepared from AGNIQUE® AMD 3L (2-hydroxy-N,N-dimethyl propanamide, DML) as described previously.25 DML was kindly donated by BASF and used as received. EL was prepared from PLA. The following chemicals were purchased from Merck and used as received: acryloyl chloride (97%), ammonia solution (NH3, 7 N in methanol), α-bromoisobutyryl bromide (BiBB, 98%), copper(II) bromide (CuBr2, 99%), dialysis tube benzoylated (MWCO 2000), diethylamine (≥99.5%), tris[2-(dimethylamino)ethyl]amine ligand (Me6TREN), ethylamine (2 M in THF), isopropylamine (≥99.5%), 2-methoxyethylamine (99%), morpholine (≥99%), propylamine (99%), pyrrolidine (≥99%), 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 98%) and triethylamine (TEA, ≥99%). The following chemicals were purchased from Scharlab and used as received: ethyl acetate (AcOEt, EssentQ), hexane (Hx, EssentQ), basic aluminum oxide (Al2O3), acetone (EssentQ), and deuterium oxide (D2O, 99.8%). 2-(Aminomethyl) tetrahydrofuran (THFA, 97%) were purchased from Thermo Fischer Scientific. Deuterated chloroform (CDCl3, 99.8%) and acetone (acetone-d6, 99.8%) were purchased from Eurisotop. Dichloromethane (DCM, EssentQ, Scharlab) and triethylamine (TEA, ≥99%, Merck) were distilled prior to use from CaH2. Tetrahydrofuran (THF, EssentQ, Scharlab) were distilled prior to use from sodium/benzophenone. Toxi-Chromo Test™ (Escherichia coli) and the WaterTOX™ STD (Aliivibrio fischeri) tests, respectively, were purchased from Environmental Bio-Detection Products Inc. (Ontario, Canada). The Thamnotoxkit F (Thamnocephalus platyurus) test was purchased from the Microbiotests (Gent, Belgium).General procedure for LA3, 5, 7 and 9 syntheses from PLA
PLA (4 g, 0.05 mol of ester, 1.0 eq.) and corresponding amine (0.06 mol, 1.2 eq.) were dissolved in anhydrous DCM (60 mL) under Ar atmosphere in a pressure tube reactor with a magnetic stirrer. Subsequently, TBD (7.5 mmol, 0.15 eq.) was added and the reaction mixture heated up to 75 °C and keep under continuous stirring for 2 hours. After that, the reaction mixture was cooled down to room temperature, and the crude was purified by vacuum distillation to afford the desired N- or N,N-substituted LA (results are summarized in Table 1).| Entry | Feedstock | LA structure | LA code | Amine (eq.) | TBD (eq.) | T (°C) | Time (h) | Conv.a (%) | Yield (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Determined by 1H NMR. | |||||||||
| 1 |

|

|
LA3 | 1.2 | 0.15 | 75 | 2 | 100 | 77 |
| 2 |

|
LA5 | 1.2 | 0.15 | 75 | 2 | 100 | 86 | |
| 3 |

|
LA7 | 1.2 | 0.15 | 75 | 2 | 100 | 85 | |
| 4 |

|
LA9 | 1.2 | 0.15 | 75 | 2 | 100 | 79 | |
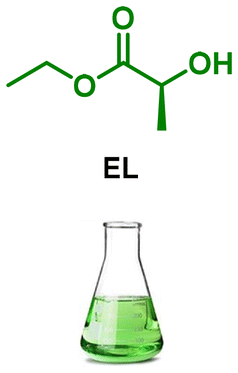
| 5 |
|

|
LA1 | 1.2 | 0 | r.t. | 24 | 100 | 98 |
| 6 |

|
LA2 | 1.2 | 0.15 | 75 | 24 | 68 | 45 | |
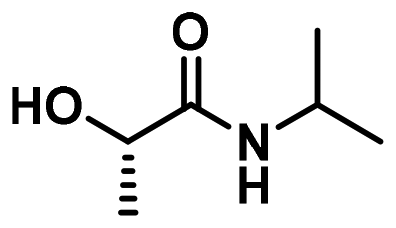
| 7 |
|
LA4 | 1.2 | 0.15 | 75 | 16 | 100 | 70 | |
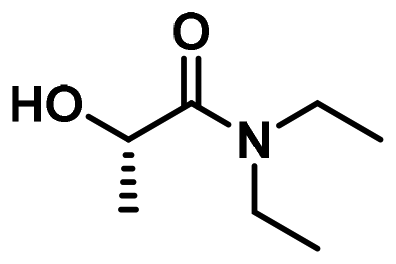
| 8 |
|
LA6 | 1.2 | 0.3 | 75 | 72 | 100 | 82 | |
| 9 |

|
LA8 | 1.2 | 0.15 | 75 | 16 | 100 | 81 | |
General procedure for LA2, 4, 6, and 8 syntheses from EL
EL (1 eq.) and a small amount of TBD (0.15 eq.) were added to a round-bottom flask under Ar atmosphere. Next, the corresponding amine (1.2 eq.) was added carefully, and the flask was sealed and heated to 75 °C. After that reaction time, the reaction mixture was cooled to room temperature, and the formed ethanol was evaporated under reduced pressure. Finally, the crude was purified by vacuum distillation to afford the desired N- or N,N-substituted LA (results are summarized in Table 1).Procedure for LA1 synthesis from EL
EL (10 mL, 87 mmol) was added into a round-bottomed flask, and ammonia solution 7 N in methanol (15 ml, 105 mmol) was added under ice-water bath and the reaction mixture was stirred for 24 hours at room temperature. The mixture was concentrated under reduced pressure to afford LA1 (7.7 g, 98%) as a colorless oil. The product was used without further purification.General procedure for LAA monomers syntheses (LAA0–9)
The desired N-substituted LA (1 eq.) and anhydrous TEA (1.5 eq.) were dissolved in anhydrous THF (LA concentration 0.5 g mL−1 aprox.) under a positive flow of Ar. The solution was stirred for 15 min in an ice bath before adding dropwise a solution of acryloyl chloride (1.2 eq.) in anhydrous THF (acryloyl chloride concentration 0.5 g mL−1 aprox.). The reaction was allowed to proceed overnight at room temperature. The mixture was then filtered, and THF was removed under reduced pressure. To remove remains of triethyl ammonium salts, the crude was re-diluted with the minimum amount of THF, filtered and the solvent was removed under reduced pressure. The final residue was purified by vacuum distillation in the presence of 5% w/w of hydroquinone, unless otherwise stated. Finally, the obtained LA acrylate was filtered throughout a small column of basic alumina to remove hydroquinone traces. Solid monomers were previously dissolved in DCM which was removed under reduced pressure after filtration.General procedure for the controlled Cu(II)Br2/Me6TREN-mediated polymerization of LAA monomers under UV light
The homopolymerization of (S)-2-hydroxy-N-isopropylpropanamide (LAA4) under the conditions [LAA4]0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[DMLBr]0:[Cu(II)Br2]0:[Me6TREN]0 = 40:1:0.02:0.12 is reported as example. This procedure is generic for all the photohomopolymerizations conducted herein. LAA4 (401.8 mg, 2.17 mmol) and DMLBr (14.5 mg, 54 μmol) were introduced into a vial containing a small Teflon-coated stirring bar, followed by the addition of 0.4 mL of stock solution containing Cu(II)Br2 (0.25 mg, 1.1 μmol) and Me6TREN (1.73 μL, 6.5 μmol) in DMSO. The flask was then sealed with a rubber septum and degassed by Ar bubbling for 15 min. Next, the vial was placed under UV light irradiation (4 × 9 W, λ = 365 nm) while stirring. The reaction was allowed to proceed 2.5 h at room temperature. Samples were taken to follow monomer conversion by 1H NMR. When high conversion was achieved, the reaction was quenched opening the vial to the air. After that, the reaction mixture was dialyzed (MWCO 2000) against acetone, refreshing the solvent 3 to 4 times for 2 days. Finally, the solvent was removed to recover the synthetized homopolymer as a white solid.
:[DMLBr]0:[Cu(II)Br2]0:[Me6TREN]0 = 40:1:0.02:0.12 is reported as example. This procedure is generic for all the photohomopolymerizations conducted herein. LAA4 (401.8 mg, 2.17 mmol) and DMLBr (14.5 mg, 54 μmol) were introduced into a vial containing a small Teflon-coated stirring bar, followed by the addition of 0.4 mL of stock solution containing Cu(II)Br2 (0.25 mg, 1.1 μmol) and Me6TREN (1.73 μL, 6.5 μmol) in DMSO. The flask was then sealed with a rubber septum and degassed by Ar bubbling for 15 min. Next, the vial was placed under UV light irradiation (4 × 9 W, λ = 365 nm) while stirring. The reaction was allowed to proceed 2.5 h at room temperature. Samples were taken to follow monomer conversion by 1H NMR. When high conversion was achieved, the reaction was quenched opening the vial to the air. After that, the reaction mixture was dialyzed (MWCO 2000) against acetone, refreshing the solvent 3 to 4 times for 2 days. Finally, the solvent was removed to recover the synthetized homopolymer as a white solid.
General procedure for BCP synthesis from LAA monomers
The polymerization of PLAA040-block-PLAA340 (BCP1) under the conditions [LAA0]0-block-[LAA3]0:[DMLBr]0:[Cu(II)Br2]0:[Me6TREN]0 = 40-block-40:1:0.02:0.12 is reported as example. This procedure is generic for all the photocopolymerizations conducted herein. LAA0 (404.3 mg, 2.36 mmol) and DMLBr (15.2 mg, 57 μmol) were introduced into a vial containing a small Teflon-coated stirring bar, followed by the addition of 0.4 mL of stock solution containing Cu(II)Br2 (0.26 mg, 1.2 μmol) and Me6TREN (1.83 μL, 7.0 μmol) in DMSO. The flask was then sealed with a rubber septum and degassed by Ar bubbling for 15 min. Next, the vial was placed under UV light irradiation (4 × 9 W, λ = 365 nm) while stirring. The reaction was allowed to proceed during 2 h at room temperature. The monomer conversion was followed by 1H NMR. When high conversion was achieved, a previously degassed solution of LAA3 (432.4 mg, 2.34 mmol) in 0.3 mL DMSO was transferred to the polymer-containing vial via cannula. The reaction was allowed to proceed for another 3 h at room temperature. Samples were taken to follow monomer conversion by 1H NMR. When high conversion was achieved, the reaction was quenched opening the vial to the air. After that, the reaction mixture was dialyzed (MWCO 2000) against acetone, refreshing the solvent 3 to 4 times for 2 days. Finally, the solvent was removed to recover the synthetized copolymer as a white solid.
General self-assembly procedure for amphiphilic BCPs
Nanoparticles from amphiphilic copolymers (BPC1, BCP1a, BCP1b, and BCP2, see Fig. S21†) were prepared using the solvent–exchange method. Briefly, each copolymer (20 mg) was dissolved in 1 mL THF. To this solution, 2 mL of DI water was progressively added at a rate of 0.3 mL h−1 using a master dual pump from Worked Precision Instruments. The final mixture was diluted with 2 mL of DI water to freeze the self-assembled structures (final ratio THF:H2O of 1:4). Excess of THF was removed through dialysis (MWCO 2000) against DI water, refreshing the DI water every 2 h for 12 h. The final polymer concentration was approximately 5 mg mL−1. The suspensions were finally characterized by DLS and TEM.
General self-assembly procedure for double hydrophilic BCP3
Nanoparticles from the double hydrophilic BCP3 were prepared by direct dissolution of the copolymer (20 mg) in 4 mL of DI water at 5 °C. The resultant solution was stirred and kept in the fridge (5 °C) for 4 hours to ensure complete dissolution. Then, the solution was heated to the desired temperature and characterized by DLS and TEM.Ecotoxicity tests
All tests were performed according to the operational procedure prescribed for each kit (see ESI†) and according to the ISO standards.30–32 The level of toxicity of a substance was determined by establishing the half-maximum effective concentration, EC50, i.e., the concentration of substances in the environment that will affect 50% of the organisms in the test population under specified conditions. The EC50 values were measured for monomers in the [0.1–8000] mg L−1 range of concentration, and for polymers, the range was [0.1–20000] mg L−1. The toxicity evaluation followed the toxicological categories adopted by the European Commission.33 According to this classification, the categories of aquatic toxicity are the following: very toxic: EC50 < 1 mg L−1, toxic: EC50 = 1–10 mg L−1, moderately toxic: EC50 = 10–100 mg L−1, practically harmless: EC50 = 100–1000 mg L−1 and harmless compounds with EC50 > 1000 mg L−1. All samples were tested in triplicate for each assay to ensure test reproducibility; the EC50 data were represented by mean values and standard deviation.
Characterization
Proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectra were recorded on a 400 MHz (for 1H) and 100.6 MHz (for 13C) Varian VNMR-S400 NMR instrument at 25 °C in indicated deuterated solvent. All chemical shifts are quoted on the δ scale in ppm using the residual solvent as internal standard (1H NMR: CDCl3 = 7.26, D2O = 4.79, (CD3)2CO = 2.05, and 13C NMR: CDCl3 = 77.16, (CD3)2CO = 29.84, (CD3)2CO = 206.26). Molecular weight analysis was performed via size exclusion chromatography (SEC) using an Agilent 1200 series system equipped with a precolumn (PLgel 5 μm Guard column) and a two-serial column system (2× PLgel 5 μm MIXED-D) and with an Agilent 1100 series refractive index detector. Chromatograms were carried out in N,N-dimethyl formamide (DMF), HPLC grade, containing 0.05% (w/w) of LiBr with a flow rate of 1 mL min−1 at 50 °C. Samples (ca. 5 mg mL−1) were filtered through 0.22 μm Teflon syringe filter and 20 μL of the polymer solution was injected using a manual sample injector Rheodyne Model 7125. The calibration curves for SEC analysis were obtained with poly(methyl methacrylate) (PMMA) standards purchased from PSS Polymer Standards Service GmbH. The molecular weights were calculated using the universal calibration principle and Mark–Houwink parameters. Toluene was used as flow rate marker. Differential scanning calorimetry (DSC) measurements were carried out on a Mettler DSC3+ instrument using N2 as a purge gas (50 mL min−1) at a scanning rate of 20 °C min−1 in the −80 to 150 °C temperature range for tree cycles, and the second heating curve was used to determine the glass transition temperature (Tg) in the middle of the step transition. Polymer (5 to 10 mg) were encapsulated in aluminium pans before measurements. Calibration was made using an indium standard (heat flow calibration) and an indium-lead-zinc standard (temperature calibration). Thermal stability studies were carried out with a Mettler TGA2 with N2 as the purge gas with a scanning rate of 10 °C min−1 in the 30 to 600 °C temperature range. Dynamic light scattering (DLS) measurements were carried out at targeted temperatures using Zetasizer Ultra from Malvern Instruments equipped with a He–Ne laser. Optical rotation values were measured using a PerkinElmer 241 MC polarimeter with a path length of 10 cm. The phase separation temperature or Tcp of the synthetized polymers was analyzed on a UV-2401PC UV-VIS Recording Spectrophotometer using a wavelength of 500 nm in transmittance mode at temperature range of 5–90 °C, in which the Tcp was determined at 50% change of the transmittance. Mass Spectroscopy (MS) were run on an Exactive Orbitrap™ mass spectrometer from Thermo Scientific (Bremen, Germany), with a heated electrospray ionization (HESI) source and a higher-energy collisional dissociation (HCD) cell to fragment the analytes. Samples were ionized in positive mode using the following parameters: spray voltage, 2 kV; skimmer voltage, 20 V; capillary voltage, 25 V; and tube lens voltage, 20 V. Gas flow rates were: sheath gas, 20 AU (adimensional units) and auxiliary gas, 10 AU; and the heater and capillary temperatures were 350 °C. The probe position settings were side to side 0, vertical C, and micrometer 0.5. Transmission electron microscopy (TEM) images were recorded using a JEOL JEM-1011 TEM microscope after sample staining with phosphotungstic acid (2 wt%, pH 7.5). Diluted nanoparticles suspensions were added onto a carbon-coated copper grid. After 10 min to allow the polymer to settle down, the excess of polymer solution was taken away using a piece of filtration paper. Then, the grid was air dried for 30 min. To stain the sample, a drop of staining solution was applied to the grid surface and, after 30 seconds, blotted off with filtration paper. This procedure was repeated twice. Finally, the grid was allowed to dry under ambient conditions before analysis. Static contact angles (CA) of water drops on polymer films were measured using Dataphysics OCA 15EC. Films were prepared by deposition a small drop of methanol polymer solution (10 mg mL−1) over silicon surface and posterior spin-coating for 60 seconds at 3000 rpm. The contact angle was measured immediately after placing the water drop (3 μL) on coated surface and the contact angle was calculated from a digital image by SCA software included in the apparatus; for each test reported, at least three drops of water were used.Results and discussion
Monomer synthesis via PLA depolymerization and acrylation
In this report, a mini-library of ten LA-based acrylic monomers with various N-substitution patterns was prepared from a biobased consumer product (Fig. 1a). With the exception of LAA0, which was prepared from AGNIQUE® AMD 3L (2-hydroxy-N,N-dimethylpropanamide, DML)12 solvent following a previously reported procedure,26 the rest of monomers were synthesized in two or three steps, using PLA plastic bags as starting material (Fig. S1†) as depicted in Fig. 1b. The 1H NMR spectrum of this commercial product did not show the presence of impurities or additives, at least at the detection limits of this technique. On the other hand, SEC and DSC analysis was used to characterize molecular weight distribution and thermal properties, respectively (Fig. S2–S4†). | ||
| Fig. 1 (a) Mini-library of LAA monomers synthesized in this work. (b) Synthesis of monomers via depolymerization of PLA by TBD-catalyzed aminolysis or alcoholysis, followed by acrylation and subsequent controlled polymerization mediated by Cu(II)Br2/Me6TREN under UV-light. (c) Amines investigated for the amidation process. | ||
In a first set of experiments, aminolysis reaction was applied to commercial PLA plastic bags dissolved in DCM, using propylamine, 2-methoxyethylamine, tetrahydrofurfuryl amine, and morpholine (A3, A5, A7, and A9 in Fig. 1c). To expedite the reaction, 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) was introduced as organocatalyst (15 mol%). As summarized in Table 1 entries 1–4, regardless the chemical structure of the amine used, PLA depolymerization was quantitative in all cases after 2 h at 75 °C as revealed SEC and NMR analysis (Fig. S5 and S6†). However, when using more sterically hindered amines, such as isopropylamine (A4), diethylamine (A6), and pyrrolidine (A8), or amines not commercially available in pure liquid format, such as ammonia (A1) and ethylamine (A2), the aminolysis procedure proved unsuccessful due to limited depolymerization conversion, even after extended reaction times (data not shown).
Alternatively, we opted for EL as the starting material, which was obtained from PLA using a general organocatalyzed ethanolysis procedure described in the literature.34 Next, aminolysis of EL was conducted in bulk at 75 °C with A4 and A8 in the presence of 15 mol% TBD to quantitatively yield the corresponding LAs within 16 hours (entries 7 and 9, Table 1). However, when using diethylamine (A6), the reaction was notably slower, requiring 72 hours to reach complete conversion, even with the addition of 30 mol% TBD (entry 8). On contrary, limited amidation degree (68%) was achieved in the case of ethylamine (A2) (entry 6). This could be attributable to lower amine concentration since A2 is commercialized in THF solution and to its gaseous nature at the reaction temperature. The synthesis of LA1 also proceeded particularly slowly, as it was carried out under diluted conditions at room temperature, utilizing an ammonia solution in methanol without the aid of a catalyst (entry 5). After purification through vacuum distillation, the LA1–9 precursors were obtained as viscous, clear liquids. These products were thoroughly characterized using NMR spectroscopy before proceeding to the next step. Acrylation of the LA products was conducted by reaction with acryloyl chloride in the presence of triethylamine in THF. The optically active acrylic monomers were isolated after high vacuum distillation in yields from 50 to 70%, except for LAA1, which was isolated by column chromatography (63% yield) due to its high boiling point. The structure of the ten acrylic monomers was confirmed by 1H and 13C NMR (Fig. S7–S16†), and high-resolution mass spectrometry (HRMS) analyses.
Controlled synthesis of PLAA polymers
Poly(lactate amide acrylates) (PLAAs) were prepared at ambient temperature using Cu(II)Br2/Me6TREN as catalytic system in the presence of UV light (λmax ≈ 365 nm) (Table 2).35 In our previous work, we demonstrated the suitability of this methodology for the preparation of well-defined homo- and block copolymers (BCPs) from EL acrylate and LAA0.26 Polymerization reactions were initiated from a DML-derived monofunctional initiator (DMLBr, Fig. 1b) using DMSO as reaction media. Initial feed ratio was [DMLBr]0:[CuBr2]0:[Me6TREN]0 = 1:0.02:0.12, targeting a degree of polymerization (DP) of 40. Photopolymerization of LAA4 is presented as example of the performed homopolymerizations. 1H NMR analysis after 2.5 h revealed almost complete conversion according to disappearing of the vinylic proton Ha (Fig. S17†). The structure of PLAAs was confirmed by 1H NMR after dialysis (Fig. S18–S28†).
| Entry | Polymer | Conv.b (%) |
M
thc |
M
NMRd |
M
SECne |
Đ |
T
gf (°C) |
CA (°) |
T
cpg (°C) |
|---|---|---|---|---|---|---|---|---|---|
|
a Cu(II)Br2/Me6TREN-mediated polymerization under UV light, reaction conditions [LAA]0:[DMLBr]0:[Cu(II)Br2]0:[Me6TREN]0 = 40:1:0.02:0.12, reaction time 2.5 h.
b Determined by 1H NMR using eqn (S1).†
c
M
th = MLAA × ([LAA]0/[DMLBr]0) × conv. + 266.14.
d Determined by 1H NMR using eqn (S2).†
e Determined by SEC using PMMA standards.
f Determined by DSC at 20 °C min−1.
g Determined by UV/Vis spectroscopy (5 mg mL−1).
h UCST-type transition.
i Not determined due to overlapping signals.
|
|||||||||
| 1 | PLAA0 | 100 | 7100 | —i | 7900 | 1.08 | 71 | 36 ± 2 | Soluble |
| 2 | PLAA1 | 95 | 6200 | 6100 | 8600 | 1.17 | 86 | 54 ± 8 | 55h |
| 3 | PLAA2 | 100 | 7200 | 7100 | 7900 | 1.20 | 83 | 75 ± 3 | 11 |
| 4 | PLAA3 | 100 | 7600 | 7500 | 8700 | 1.16 | 73 | 76 ± 1 | Insoluble |
| 5 | PLAA4 | 98 | 7400 | 6600 | 8900 | 1.13 | 92 | 70 ± 1 | Insoluble |
| 6 | PLAA5 | 100 | 8300 | 8000 | 13700 |
1.10 | 44 | 33 ± 2 | 62 |
| 7 | PLAA6 | 100 | 7800 | 8100 | 7900 | 1.11 | 50 | 39 ± 1 | 24 |
| 8 | PLAA7 | 100 | 9600 | 9100 | 10900 |
1.12 | 64 | 41 ± 1 | 18 |
| 9 | PLAA8 | 100 | 7900 | 8900 | 7200 | 1.13 | 73 | 25 ± 2 | 48 |
| 10 | PLAA9 | 100 | 7900 | 11100 |
8700 | 1.10 | 78 | 32 ± 9 | 81 |
| 11 | PELA | 100 | 7000 | 6500 | 6900 | 1.16 | −4 | 88 ± 3 | Insoluble |
As depicted in Fig. S29,† SEC analysis of PLAA4 after dialysis revealed monomodal distribution (i.e., narrow dispersity of 1.13) for a number average molecular weight (MSECn) of 8900 g mol−1, which was in concordance with the theoretical value (Mth) and the weight calculated by NMR (MNMR). As summarized in Table 2, similar results were observed for the remaining monomers (Fig. S29†). All synthesized polymers were soluble in a large variety of organic solvents of strongly differing polarities, such as methanol, acetonitrile, N-methylpyrrolidone, DMSO, ethyl acetate, dioxane, THF, chloroform and dichloromethane. Only diethylether and linear hydrocarbons such as hexane were found to be non-solvents. Solubility in water will be discussed specifically in another subchapter.
Thermal properties
After establishing an efficient synthetic route to prepare LA-based acrylic polymers from a PLA, we investigated their bulk thermal properties using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) (Fig. S30 and S31†). After an initial weight loss, associated with some humidity absorbed from the environment, all synthesized LA-based acrylic polymers exhibited stepwise decomposition, with two distinct degradation stages centered at around 300 °C and 400 °C, as observed in the DTGA curves. The onset of decomposition for polymers with secondary and tertiary amide groups occurred at approximately 300 °C, while PLAA1, containing pendant primary amide groups, was somewhat less thermally stable. However, it remained more stable than PELA (MSECn = 6900, Đ = 1.16), which was synthesized and analyzed for comparison (see entry 11, Table 2).DSC measurements revealed an amorphous nature for all the synthesized polymers and a relatively high glass transition temperatures (Tg) compared to poly(alkyl lactate)s with comparable side chain lengths (see Table 2).36 For example, PLAA2 exhibited a Tg at approximately 80 °C, which is considerably higher than that of PELA which is well below room temperature. The higher Tg can be explained by the presence of secondary amide moieties in the side chain and the associated hydrogen-bonding capability, as shown for other monomers with hydrogen-bonding capabilities.37 As Tg depends primarily on the flexibility of the polymer backbone, increasing the length of the side-group favors the movement of the individual repeating units. This led to an inefficient packing relaxation of the chain backbones; thus, Tg is observed at lower temperatures. Accordingly, Tg was registered for PLAA2, PLAA3 and PLAA5 in the following order: 83, 73 and 44 °C. The same generalization can be made for tertiary amide PLAAs with linear substituents; PLAA0 (Tg = 71 °C) vs. PLAA6 (Tg = 50 °C). Another generalization can be made with regards to Tg and the presence of bulky and stiff group. As the isopropyl side group in PLAA4 presents a bulky and stiff group, the barrier for segmental motion is higher than that for the linear analogue. Thus, PLAA4 exhibited a Tg at approximately 90 °C, which is considerably higher than that of PLAA3 (73 °C). Similarly, for tertiary amide PLAAs, the presence of rings in PLAA8 and PLAA9 greatly increased the Tg as compared to homopolymers bearing linear substituents.
Hydrophilicity and thermoresponsive behavior
To gain an initial understanding of the hydrophilicity of the synthesized polymers, surface property analysis was conducted using water contact angle (CA) measurements. Uniform thin polymer films of each homopolymer were prepared on silicon slides using a spin-coating technique. At room temperature, PLAA1, PLAA2, PLAA3, and PLAA4 stood out for their high contact angle values (54°, 75°, 76°, and 70°, respectively) comparable even to that of PELA (88°). In line with these observations, these samples were insoluble in water, although PLAA2 proved soluble at low temperatures, and PLAA1 dissolved upon increasing the solution temperature (see details below). The remaining polymers exhibited contact angles of 40 degrees or less, indicative of their strong affinity for water.To further investigate the aqueous solubility of the prepared polymers, optical transmission measurements were conducted on their aqueous solutions (5 mg mL−1) over a temperature range of 5–90 °C (Fig. 2). The thermoresponsive behavior of the polymers is predominantly influenced by the hydrophilic/hydrophobic balance, as well as polymer–solvent and polymer–polymer hydrogen bonding interactions.38 Homopolymers containing secondary amide pendant groups, such as PLAA3 and PLAA4, were turbid throughout the entire temperature range, confirming their high hydrophobicity, water insolubility, and lack of thermosensitive behavior. This can be attributed to strong polymer–polymer hydrogen bonding interactions involving N–H donors and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O acceptors. In contrast, PLAA2, PLAA5, and PLAA7 exhibited clear aqueous solutions at low temperatures that became turbid upon heating, demonstrating a sharp LCST-type transition. These polymers displayed Tcp values of 11 °C, 62 °C, and 18 °C, respectively (Fig. 2a). This behavior is associated with either a reduction in the hydrophobicity of the N-substituent by decreasing the number of carbons (PLAA2) or enhanced polymer–water hydrogen bonding interactions due to the incorporation of an oxygen atom (PLAA5 and PLAA7).
O acceptors. In contrast, PLAA2, PLAA5, and PLAA7 exhibited clear aqueous solutions at low temperatures that became turbid upon heating, demonstrating a sharp LCST-type transition. These polymers displayed Tcp values of 11 °C, 62 °C, and 18 °C, respectively (Fig. 2a). This behavior is associated with either a reduction in the hydrophobicity of the N-substituent by decreasing the number of carbons (PLAA2) or enhanced polymer–water hydrogen bonding interactions due to the incorporation of an oxygen atom (PLAA5 and PLAA7).
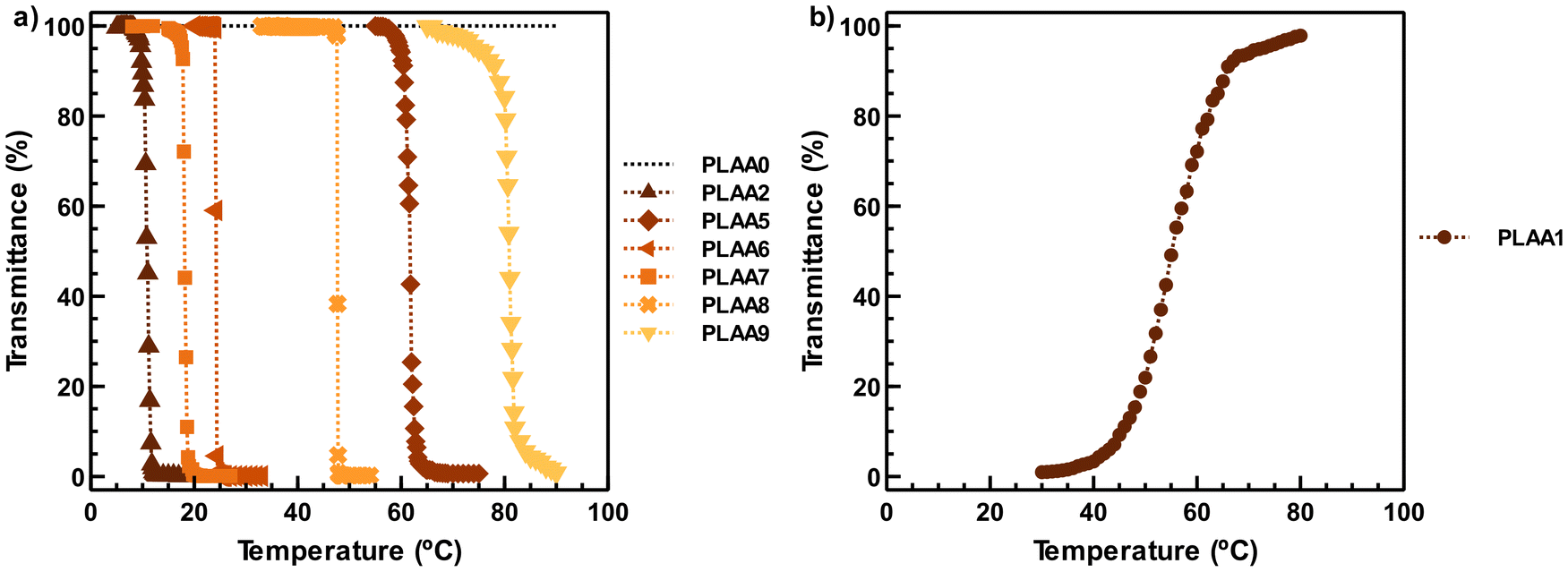 | ||
| Fig. 2 Temperature-dependent transmittance of (a) LCST-type and (b) UCST-type PLAAs (c = 5 mg mL−1). See Table 2 for polymerization results. | ||
Homopolymers with tertiary amide groups, such as PLAA6, PLAA8, and PLAA9, also showed LCST-type transitions, with Tcp of 24, 48, and approximately 81 °C, respectively. In contrast, PLAA0 solutions remained clear across the tested temperature range (5–90 °C). However, it is important to note that in previous studies,27 cloud points between 86 and 98 °C were observed for PLAA0x, where x > 50. Interestingly, PLAA1 displayed a unique UCST-type (upper critical solution temperature) behavior, transitioning from turbid at low temperatures to clear upon heating, with a TUCST of 55 °C (Fig. 2b). This phenomenon is likely due to strong polymer–polymer hydrogen bonding from its two N–H donors and CO acceptors, which are disrupted upon heating. Similar behavior has been reported for other polymers containing primary amide pendant units.39,40 In addition, precisely tunning up and down the LCST of PLAAs over a wide range of temperatures should also be possible through controlled statistical copolymerization of hydrophilic and hydrophobic LAAs.
As a proof of concept, a series of copolymerizations between LAA5 (Tcp = 62 °C for the corresponding homopolymer) and LAA7 (Tcp = 18 °C for the corresponding homopolymer) were conducted to deliver a set of statistical PLAA5-stat-PLAA7 copolymers (see Table S1†). Solutions with different comonomer composition were placed under UV light in the presence of Cu(II)Br2/Me6TREN catalytic system and DMLBr initiator. Statistical copolymers ranging from 32 mol% (SCP1) to 70 mol% (SCP5) of LAA5 were obtained with molecular weight around 10000 g mol−1 and narrow polydispersity (Đ ≈ 1.15). Due to its similar chemical structure, it is expected similar reactivity ratios and thus good agreement between the monomer feed ratio and the polymer comonomer composition, which was confirmed by 1H NMR after dialysis (Fig. S32†). DSC analysis revealed the amorphous nature of the synthesized copolymers, with their Tg decreasing from 53 to 49 °C with the increasing of LAA5 comonomer content. All copolymers aqueous solutions (5 mg mL−1) were clear at low temperatures and become turbid while heating. Measured optical transmittance decreased sharply at specific temperatures, thus indicating LCST-type behavior (Fig. 3). Measured Tcp was dependent on comonomer composition and increased linearly upon increasing LAA5 content. Hence, allowing accurate control on Tcp values over the 18–62 °C temperature range (which correspond to Tcp of each homopolymer) for specific applications (e.g., body temperature). In conclusion, turbidity measurements demonstrated the exceptional potential of LAA monomers in the design of thermoresponsive polymers with precisely tunable LCSTs.
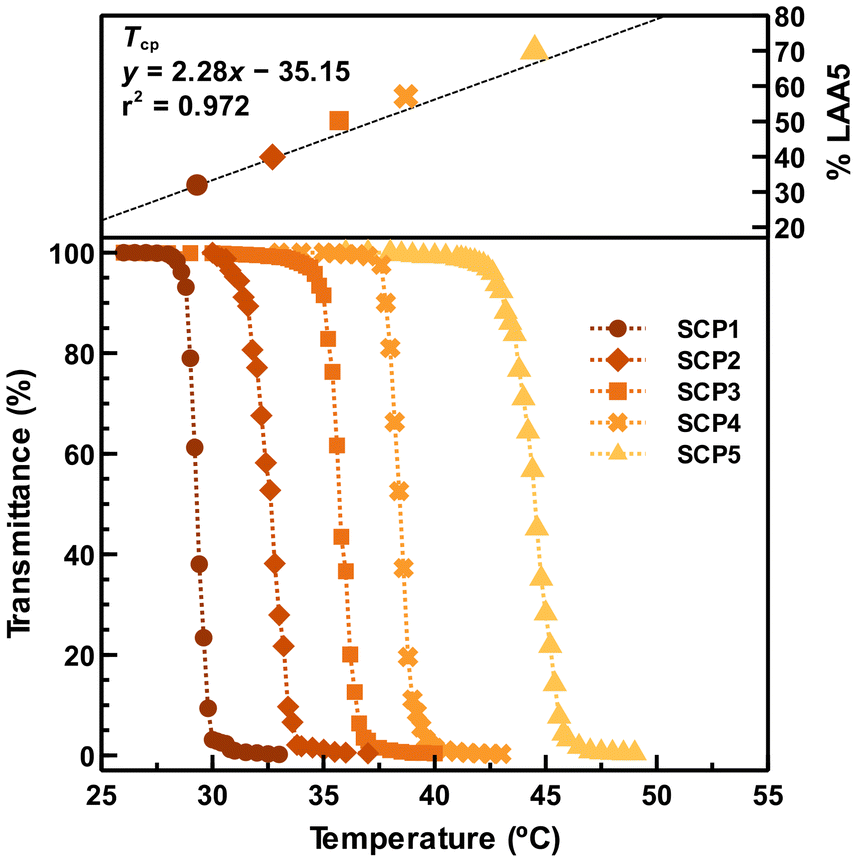 | ||
| Fig. 3 Temperature-dependent transmittance of SCPs (c = 5 mg mL−1) and relationship between Tcp values and LAA5 comonomer content. See Table S1† for statistical copolymerization conditions and results. | ||
This versatility can be achieved either through strategic monomer design or by utilizing statistical copolymerization with a diverse range of monomers. Such adaptability underscores LAAs as powerful building blocks for creating more complex polymer architectures, paving the way for the development of advanced smart materials that respond to specific thermal and environmental stimuli.
Ecotoxicity of monomers and polymers
The toxicity of selected water-soluble monomers (i.e., LAA0, LAA5, LAA7, and LAA9) and its polymers was evaluated on representative bacteria (A. fischeri), vacular plant (S. polyrhiza), and invertebrate (T. platyaurus). The obtained results (Table S2†) are summarized in Fig. 4. All tested monomers exhibited harmless or practically harmless behavior toward bacteria, and moderately toxic and toxic behavior toward plants and invertebrates, respectively. It has been demonstrated that LEs and LAs and, such as EL and DML are non-toxic,12,41 hence observed toxicity could be attributable to the acrylate segment, as reported in many studies.42,43 Active double bounds of the acrylate group can interact with biological systems and metabolic pathways, which can cause cellular toxicity.44,45 Moreover, no significant difference in toxicity was observed regarding the N-substituted moiety on the evaluated monomers. Acrylic polymers are usually categorized as harmless, although some toxicity has been attributed to the presence of unreacted monomers in the polymeric material or to the release of degradation products.46 Despite almost complete conversion was achieved during polymerization (see Table 2 for polymerization results), all PLAAs tested were dialyzed against acetone to ensure no unreacted monomers and reagents remains in the evaluated polymers. All tested polymers resulted to be harmless or practically harmless to the tested organisms. Again, the pendant units’ structure did not have a significant impact on the measured EC50 values. | ||
| Fig. 4 Mean effective concentration (EC50, mg L−1) of the tested monomers and polymers on bacteria (A. Fischeri) vascular plant (S. Polyrhiza) and invertebrate (T. Platyurus). Toxicity levels were stated as follows: harmless (EC50 > 1000 mg L−1), practically harmless (100 < EC50 < 1000 mg L−1), moderately toxic (10 < EC50 < 100 mg L−1) and toxic (1 < EC50 < 10 mg L−1). See Table S2† for ecotoxicity results. | ||
Synthesis of single and double thermoresponsive block copolymers
As mentioned above, combinations of LAAs hold significant promise for the synthesis of complex macromolecular structures, such as amphiphilic BCP nanoassemblies, which can display controlled morphologies and finely tuned LCST behavior. Furthermore, the non-toxic nature of these polymers significantly enhances their appeal for a wide range of potential applications, particularly in fields like drug delivery or biocompatible coatings. Accordingly, a series of BCPs were synthesized using Cu(II)Br2/Me6TREN-mediated controlled photopolymerization, with the ultimate goal of generating thermosensitive micellar nanoaggregates. These micelles are designed to be highly effective for targeted and controlled drug delivery applications, offering the potential for precise and responsive therapeutic interventions. The chemical structure and characterization data for the targeted copolymers is depicted in Fig. S33 and Table S3,† respectively. Initially, LAA0 and LAA3 were copolymerized at equimolar ratio [LAA3]:[LAA0] = 40:40 to produce an amphiphilic BCP1 having LA0 bearing chains as hydrophilic block and LA3 as hydrophobic one. Briefly, LAA0 was first polymerized to high conversion in DMSO, after which chain extension with degassed LAA3 in DMSO was performed to form the second block. Monomer conversion was monitored via1H NMR spectroscopy, which confirmed the disappearance of vinyl group signals between 5.8–6.4 ppm. 1H NMR analysis of the isolated BCP1 confirmed the targeted structure (top of Fig. S34†). SEC analysis was also in agreement with a block copolymerization between LAA0 and LAA3 with good control, as evidenced by a clear shift to higher molar masses in the SEC trace after chain extension and low Đ values (top of Fig. 5a). Similarly, another amphiphilic BCP with similar ratio of hydrophobic to hydrophilic blocks was produced using an alternative combination of monomers (bottom of Fig. 5a). In BCP2, i.e., PLAA9-block-PLAA4 hydrophobic (PLAA4) and hydrophilic (PLAA9) segments were combined with the purpose to deliver nanoparticles with LCST transition closer to the physiological temperature. 1H NMR analysis of the isolated BCP2 is depicted in Fig. S35.†
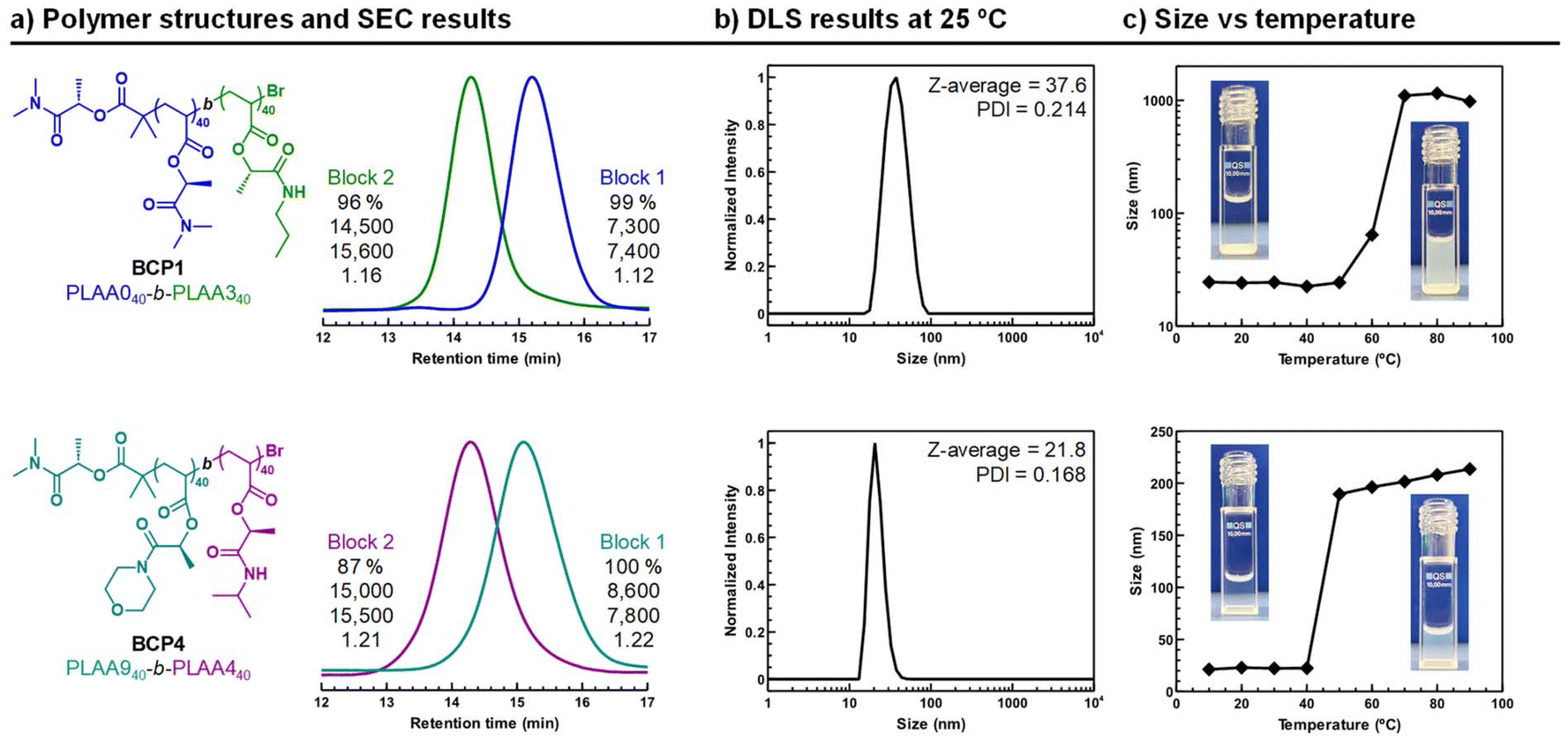 | ||
| Fig. 5 Characterization of BCP1 and BCP2 and the corresponding self-assembled nanoparticles. (a) Chemical structures of the copolymers and SEC analysis of the in situ block copolymerization (numbers shown together with SEC traces correspond to monomer conversion, Mth, MSECn and Đ, respectively, from the top to the bottom), (b) size of the BCP nanoparticles by DLS at 25 °C, and (c) temperature-dependent hydrodynamic nanoparticle size changes combined with pictures of the BCP solution taken below and above LCST. | ||
Nanoparticles of the two BCPs were prepared via solvent–exchange method. Slow-injection of water droplets into copolymer dissolved in THF was used to ensure the obtention of thermodynamically stable self-assembled structures. In both cases, particle size analysis by dynamic light scattering (DLS) at 25 °C, complemented by transmission electron microscopy (TEM) imaging, revealed the formation of micellar aggregates with an average size of 25 nm and reasonable dispersity (Fig. 5b and S37a and b†). However, it is worth noting that BCP2 particles exhibited a significantly lower tendency to cluster, which is promising, as micelles typically tend to aggregate. Other self-assembled aggregates, such as worms or vesicles, were also successfully generated by using alternative BCPs with more hydrophobic compositions (see Fig. S38†). Next, we studied the thermoresponsive behavior BCP1 and BCP2 micelles by DLS measurements in the 10–90 °C temperature range (Fig. 5c). The BCP1 micelles size persisted until T ≈ 50 °C when ∼1000 nm aggregates suddenly formed because of the dehydratation of the block-forming shell of the micelles leading to the micelle aggregation. Meanwhile, it was visualized that the opalescent solution of BCP1 in water at room temperature became opaque when the sample was heated above 50 °C, indicating the presence of a LCST-type transition temperature. Although no Tcp was observed for PLAA0 homopolymer, it is evident that its hydrophilicity is influenced and reduced by the presence of the hydrophobic block. As expected, the micelles of the BCP2 also showed heat-induced precipitation in water, but in this case the LCST transition appeared closer to the human body temperature. In this case, the polymer chains collapsed at around 40 °C. Therefore, the aggregation of such micelles, whose solvation state suddenly changes at a critical temperature, has interest for the design of smart materials including drug release systems and biosensors.
Finally, we explored the preparation of double-hydrophilic BCPs exhibiting multistep assembly behavior. We targeted a novel PLAA0-block-PLAA7 copolymer (BCP3) looking for a system displaying two discrete LCSTs (Fig. 6a). BCP3 was successfully synthesized via sequential Cu(II)Br/Me6TREN-mediated photoinduced controlled polymerization. The structure of BCP3 was confirmed by 1H NMR (Fig. S36†). The overlay of the SEC curves of the polymers before and after chain extension confirmed its successful formation with a narrow molar mass distribution. Upon heating BCP3 in water, two distinct thermal transitions were anticipated: the LCST of the PLAA0 block was expected to be higher than that of the PLAA7 block. Theoretically, the copolymer should undergo a stepwise transition from hydrophilic to amphiphilic, and ultimately to fully hydrophobic, as the temperature increases (see Fig. 6b).
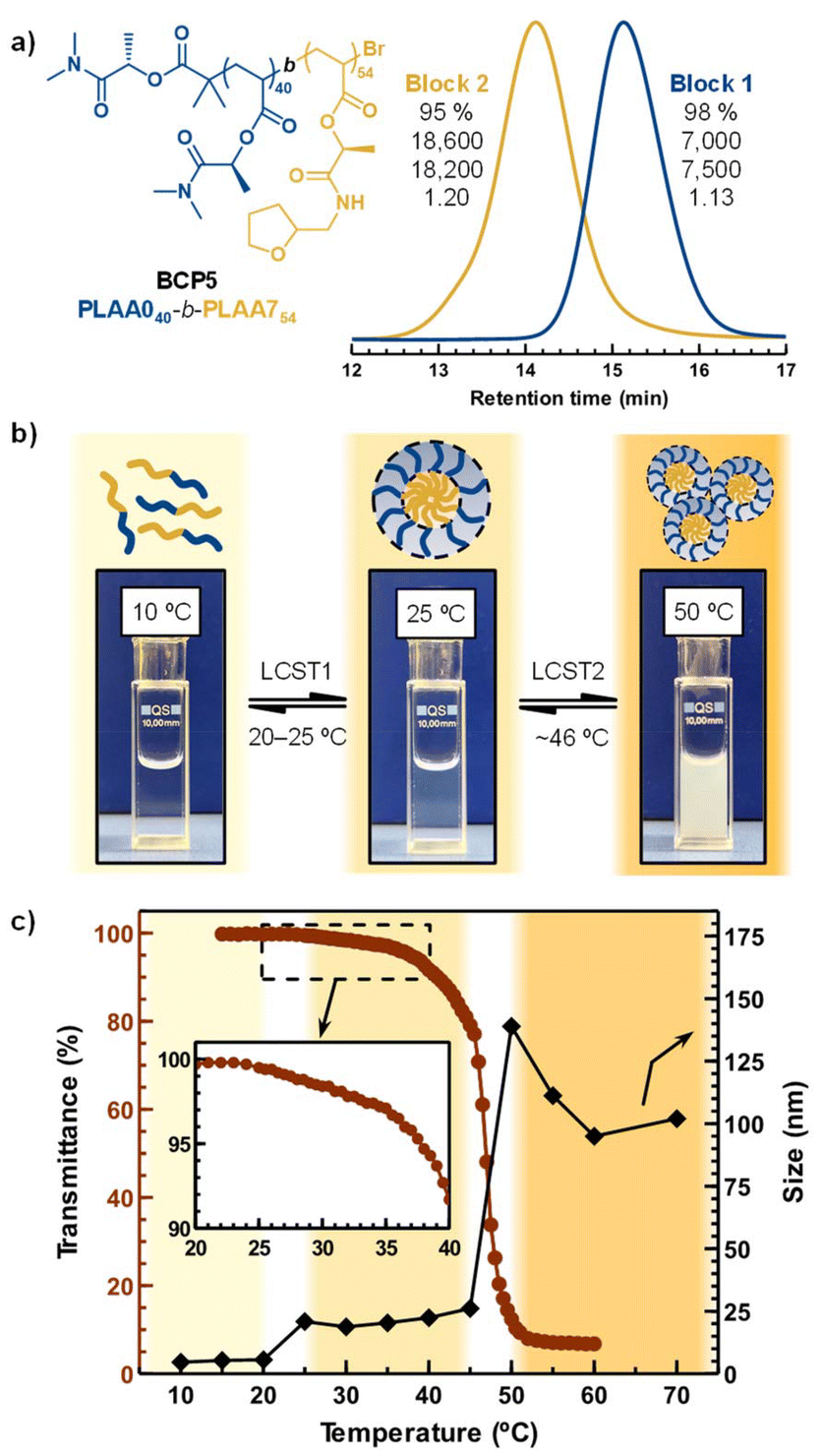 | ||
| Fig. 6 Characterization of the double hydrophilic BCP3. (a) Chemical structure of the copolymers and SEC analysis of the in situ block copolymerization (numbers shown together with SEC traces correspond to monomer conversion, Mth, MSECn and Đ, respectively, from the top to the bottom), (b) general scheme for the thermally induced self-assembly–aggregation process of the BCP3 aqueous solution combined with pictures of the BCP solution (5 mg mL−1) taken below at temperatures representative of each regime (free chains-micelles-aggregates), and (c) temperature-dependent hydrodynamic size (black diamonds) and transmittance (red circles) changes. | ||
At low temperatures, both blocks remain soluble in water. At intermediate temperatures, the PLAA7 block should dehydrate, leading to micelle formation, and at higher temperatures, the PLAA0 block should also collapse, resulting in fully hydrophobic aggregates. A visual inspection of the BCP solution at different temperatures provided an initial confirmation (see digital images in Fig. 6b). The solution remained clear below 20 °C, as expected for fully hydrated chains. Between 25 °C and 45 °C, the solution became opalescent, indicating micelle assembly. A drop of the BCP solution at room temperature was deposited on a TEM copper grid in order to visualize the micelles (Fig. S37c†). The nanoparticles observed by TEM were roughly spherical with a diameter size of ∼33 nm. Above 45 °C, the solution turned opaque due to the collapse of the polymer chains. This complex thermal behavior in water was also assessed with DLS in the 10–90 °C temperature range. In the 10–20 °C temperature region, the hydrodynamic size was ∼8 nm, indicating that both copolymer blocks were molecularly dissolved. The increased hydrodynamic diameter to 23 nm at T > 25 °C for BCP3 indicated the micelle assembly because of dehydration of the PLAA0 segments. The micelle size persisted until T ∼45 °C when larger ∼150 nm aggregates formed because of the PLAA0 micelle corona dehydration leading to the micelle aggregation. The staggered transmittance profile obtained from turbidimetry analysis also coincides with the formation and aggregation of micelles consisting of a dehydrated hydrophobic core (PLAA7) and a well-solvated hydrophilic corona (PLAA0), verifying the DLS and TEM data above presented.
Conclusions
As environmental protective polices expanded, the use of polymeric materials from sustainable resources have gained exponential interest. Among all available biopolymers from lignocellulosic biomass chemical platforms, PLA is receiving substantial attention. However, as PLA production increase, it also does the need to recover and recycle the potential long-lasting waste generated. Here, we report the chemical depolymerization of PLA via organocatalyzed aminolysis and subsequent transformation of the obtained lactate amides (LAs) into a mini-library of N-substituted lactate amide-based acrylic (LAAs) monomers. Homopolymers from N-substituted lactate amide acrylates (PLAAs) were prepared using CuBr2/Me6TREN as catalytic system under UV-light with fine control over molecular weight (MSECn ≈ 9000 g mol−1) and dispersity (Đ ≤ 1.2). Thermal analysis revealed their amorphous nature (Tg = 44–92 °C) and stability up to 300 °C where they decompose through two-stage decomposition. PLAAs exhibited variable hydrophilicity adjustable by modification of the N-moiety with different primary, secondary and tertiary amides, obtaining water insoluble polymers and a range LCST-type thermoresponsive polymers with variable cloud point temperature (Tcp = 11–81 °C). Interestingly, UCST-behavior was observed for PLAA pendant units containing primary amide (TUCST = 55 °C). Moreover, precise tunning of Tcp was also possible through statistical copolymerization with two LAA monomers. Finally, block copolymerization of different LAAs allowed us to prepare amphiphilic polymers with one- or two-phase transition events that could self-assemble in water. Therefore, due to the possibility to adjust the desired polymer hydrophilicity and their non-toxicity demonstrated with assays on bacteria, vascular plant and invertebrate, evidence the potential of these PLA-recycled smart materials for applications in the biomedical field such as drug carriers or biosensors.Author contributions
Marc Palà: methodology, validation, investigation, formal analysis, and writing – review & editing. Alina Ismagilova: investigation and formal analysis of toxicity studies and review & editing. Adrian Moreno: investigation and formal analysis of PLA aminolysis, review & editing. Jorge Plaza: investigation. Juan Carlos Ronda: review & editing. Marina Galià: funding acquisition and review & editing. Lauri Vares: methodology of the toxicity studies and review & editing. Gerard Lligadas: funding acquisition, conceptualization, methodology, supervision (lead), and writing – review & editing. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.† Raw data from monomer and polymer characterization (1H and 13C NMR spectra, SEC traces, DLS, DSC, TGA, UV/Vis, and digital images) are available at CORA. Repositori de Dades de Recerca at https://doi.org/10.34810/data1989.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by MICIU/AEI/10.13039/501100011033 and FEDER UE through projects PID2020-114098RB-I00, PID2023-149489OB-I00 (to G. L. and M. G.), “Ramón y Cajal” contract (RYC2022- 035322-I) (to A. M.). and FPI grant PRE-2021-100387 (to M. P.). G. L. acknowledges “Pla Serra Húnter” of the Government of Catalonia. L. V. acknowledges the Estonian Research Council (PRG2665) and the Estonian Ministry of Education and Research through the Centre of Excellence in Circular Economy for Strategic Mineral and Carbon Resources (TK228). The authors also thank BASF SE, Ludwigshafen, Germany (Dr O. Gronwald) for kindly supplying Agnique® AMD 3L solvent (2-hydroxy-N,N-dimethyl propanamide).References
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- I. Delidovich, P. J. C. Hausoul, L. Deng, R. Pfützenreuter, M. Rose and R. Palkovits, Chem. Rev., 2016, 116, 1540–1599 CrossRef CAS PubMed.
- G. Qiang, M. F. Ansari, Z. Sun and S. Elangovan, Adv. Synth. Catal., 2024, 366, 4805–4834 CrossRef.
- M. Dusselier, P. Van Wouwe, A. Dewaele, E. Makshina and B. F. Sels, Energy Environ. Sci., 2013, 6, 1415–1442 RSC.
- R. Datta and M. Henry, J. Chem. Technol. Biotechnol., 2006, 81, 1119–1129 CrossRef CAS.
- J. Becker, A. Lange, J. Fabarius and C. Wittmann, Curr. Opin. Biotechnol., 2015, 36, 168–175 CrossRef CAS PubMed.
- M. Palà, S. E. Woods, F. L. Hatton and G. Lligadas, Macromol. Chem. Phys., 2022, 223, 2200005 CrossRef.
- H. Pereira, Wood Fiber Sci., 1988, 20, 82–90 CAS.
- S. M. Nikles, M. Piao, A. M. Lane and D. E. Nikles, Green Chem., 2001, 3, 109–113 RSC.
- S. Uebele, K. S. Johann, T. Goetz, O. Gronwald, M. Ulbricht and T. Schiestel, J. Appl. Polym. Sci., 2021, 138, 50935 CrossRef CAS.
- A. Moreno, D. Garcia, M. Galià, J. C. Ronda, V. Cádiz, G. Lligadas and V. Percec, Biomacromolecules, 2017, 18, 3447–3456 CrossRef CAS PubMed.
- I. Fleute-Schlachter, A. Blanazs and N. Shabelina, AgroPages-BASF: Next Generation Polar Solvent for Agrochemical Formulations-Agricultural news, https://news.agropages.com/News/NewsDetail---39087.htm, (accessed December 21, 2024).
- A. Z. Naser, I. Deiab and B. M. Darras, RSC Adv., 2021, 11, 17151–17196 RSC.
- N. Simon, K. Raubenheimer, N. Urho, S. Unger, D. Azoulay, T. Farrelly, J. Sousa, H. van Asselt, G. Carlini, C. Sekomo, M. L. Schulte, P.-O. Busch, N. Wienrich and L. Weiand, Science, 2021, 373, 43–47 CrossRef CAS PubMed.
- T. A. Swetha, A. Bora, K. Mohanrasu, P. Balaji, R. Raja, K. Ponnuchamy, G. Muthusamy and A. Arun, Int. J. Biol. Macromol., 2023, 234, 123715 CrossRef CAS PubMed.
- N. Tripathi, M. Misra and A. K. Mohanty, ACS Eng. Au, 2021, 1, 7–38 CrossRef CAS.
- S. M. Emadian, T. T. Onay and B. Demirel, Waste Manage., 2017, 59, 526–536 CrossRef CAS PubMed.
- G. Kale, T. Kijchavengkul, R. Auras, M. Rubino, S. E. Selke and S. P. Singh, Macromol. Biosci., 2007, 7, 255–277 CrossRef CAS.
- L. Shao, Y.-C. Chang, C. Hao, M. Fei, B. Zhao, B. J. Bliss and J. Zhang, Green Chem., 2022, 24, 8716–8724 RSC.
- A. J. Spicer, A. Brandolese and A. P. Dove, ACS Macro Lett., 2024, 13, 189–194 CrossRef CAS PubMed.
- S. Figalla, V. Jašek, J. Fučík, P. Menčík and R. Přikryl, Biomacromolecules, 2024, 25, 6645–6655 CrossRef CAS PubMed.
- Y. Li, S. Wang, S. Qian, Z. Liu, Y. Weng and Y. Zhang, ACS Omega, 2024, 9, 13509–13521 CrossRef CAS PubMed.
- M. Palà, G. Lligadas and A. Moreno, Biomacromolecules, 2024, 25, 6338–6356 CrossRef PubMed.
- S. Liu, L. Hu, J. Liu, Z. Zhang, H. Suo and Y. Qin, Macromolecules, 2024, 57, 4662–4669 CrossRef CAS.
- M. Palà, H. El Khannaji, M. Garay-Sarmiento, J. C. Ronda, V. Cádiz, M. Galià, V. Percec, C. Rodriguez-Emmenegger and G. Lligadas, Green Chem., 2022, 24, 8314–8323 RSC.
- N. Bensabeh, A. Moreno, A. Roig, M. Rahimzadeh, K. Rahimi, J. C. Ronda, V. Cádiz, M. Galià, V. Percec, C. Rodriguez-Emmenegger and G. Lligadas, ACS Sustainable Chem. Eng., 2020, 8, 1276–1284 CrossRef CAS.
- S. E. Woods, J. D. Tinkler, N. Bensabeh, M. Palà, S. J. Martin, I. Martin-Fabiani, G. Lligadas and F. L. Hatton, ACS Sustainable Chem. Eng., 2023, 11, 9979–9988 CrossRef CAS PubMed.
- J. Englert, M. Palà, L. Witzdam, F. Rayatdoost, O. Grottke, G. Lligadas and C. Rodriguez-Emmenegger, Langmuir, 2023, 39, 18476–18485 CrossRef CAS PubMed.
- D. D. Reynolds and W. O. Kenyon, Acrylic ester-amides and polymers thereof. US2458420A, November 22, 1947, https://patents.google.com/patent/US2458420A/en, (accessed October 1, 2024).
- ISO 14380:2011 – Water quality – Determination of the acute toxicity to Thamnocephalus platyurus, European Committee for Standardization, 2011.
- ISO 20079:2005 – Water quality – Determination of toxic effect of water constituents and waste water to duckweed (Lemna minor) – Duckweed growth inhibition test, European Committee for Standardization, 2005.
- ISO 11348-3:2007 – Water quality – Determination of the inhibitory effect of water samples on the light emission of Vibrio fischeri (Luminescent bacteria test), European Committee for Standardization, 2007.
- EU classification categories for hazardous to the aquatic environment, https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2011:083:0001:0053:en:PDF, (accessed December 29, 2024).
- P. Majgaonkar, R. Hanich, F. Malz and R. Brüll, Chem. Eng. J., 2021, 423, 129952 CrossRef CAS.
- A. Anastasaki, V. Nikolaou, Q. Zhang, J. Burns, S. R. Samanta, C. Waldron, A. J. Haddleton, R. McHale, D. Fox, V. Percec, P. Wilson and D. M. Haddleton, J. Am. Chem. Soc., 2014, 136, 1141–1149 CrossRef CAS PubMed.
- M. Purushothaman, P. S. G. Krishnan and S. K. Nayak, J. Appl. Polym. Sci., 2014, 131, 40962 CrossRef.
- F. Fleischhaker, A. P. Haehnel, A. M. Misske, M. Blanchot, S. Haremza and C. Barner-Kowollik, Macromol. Chem. Phys., 2014, 215, 1192–1200 CrossRef CAS.
- C. Zhao, Z. Ma and X. X. Zhu, Prog. Polym. Sci., 2019, 90, 269–291 CrossRef CAS.
- J. Seuring and S. Agarwal, Macromolecules, 2012, 45, 3910–3918 CrossRef CAS.
- J. Seuring, F. M. Bayer, K. Huber and S. Agarwal, Macromolecules, 2012, 45, 374–384 CrossRef CAS.
- J. J. Clary, V. J. Feron and J. A. Van Velthuijsen, Regul. Toxicol. Pharmacol., 1998, 27, 88–97 CrossRef CAS PubMed.
- H. Greim, J. Ahlers, R. Bias, B. Broecker, H. Hollander, H.-P. Gelbke, S. Jacobi, H.-J. Klimisch, I. Mangelsdorf, W. Mayr, N. Schön, G. Stropp, P. Stahnecker, R. Vogel, C. Weber, K. Ziegler-Skylakakis and E. Bayer, Chemosphere, 1995, 31, 2637–2659 CrossRef CAS.
- C. A. Staples, S. R. Murphy, J. E. McLaughlin, H.-W. Leung, T. C. Cascieri and C. H. Farr, Chemosphere, 2000, 40, 29–38 CrossRef CAS.
- M. Suh, D. Proctor, G. Chappell, J. Rager, C. Thompson, S. Borghoff, L. Finch, R. Ellis-Hutchings and K. Wiench, Toxicology, 2018, 402–403, 50–67 CrossRef CAS PubMed.
- T. J. McCarthy and G. Witz, Toxicology, 1997, 116, 153–158 CrossRef CAS.
- A. Ismagilova, L. Matt, P. Jannasch, V. Kisand and L. Vares, Green Chem., 2023, 25, 1626–1634 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00070j |
| This journal is © The Royal Society of Chemistry 2025 |