
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLarge aromatic amide helices via living polycondensation†
Dinh Phuong Trinh
Nguyen
 ,
Saquib
Farooq
,
Nicoleta
Meyer
and
Andreas F. M.
Kilbinger
*
,
Saquib
Farooq
,
Nicoleta
Meyer
and
Andreas F. M.
Kilbinger
*
Department of Chemistry, University of Fribourg, Chemin du Musée 9, 1700 Fribourg, Switzerland. E-mail: andreas.kilbinger@unifr.ch
First published on 2nd May 2025
Abstract
We employ a living polymerization strategy and a crescent-shaped monomer to synthesize large aromatic amide helices with cavity sizes exceeding 1 nm. These polymeric foldamer helices are stabilized by a continuous strand of three-center hydrogen bonds, ensuring structural integrity. Our method efficiently yields polymeric helices of varying lengths while also producing macrocycles as side products when targeting higher molecular weights. The isolation and characterization of a 7-mer macrocycle provided key insights into the number of repeat units required to complete a full turn of the corresponding polymeric helix. Additionally, macrocycles were obtained in greater quantities by performing the polycondensation in the absence of an initiator. This straightforward and versatile approach paves the way for the development of novel materials with potential applications in host–guest chemistry, catalysis, and molecular transport.
Introduction
Evolution, nature's tool, has shaped and developed many biological systems in which biopolymers play a crucial role. Amongst them are numerous hollow macromolecules with well-defined structures and sizes, but they are also difficult to recreate synthetically. In an effort to acquire a variety of nanocavities, many folding oligomers and polymers (foldamers) with non-natural backbones were developed and synthesized.1–5 Aromatic amides, or aramids, are of particular interest thanks to their rigid backbone consisting of alternating aromatic and amide groups. Many families of aromatic oligoamide foldamers were developed by different groups over the years.6–9 They have been proven to adopt persistent helical conformations in solution and in the solid-state with a hydrophilic internal cavity thanks to the amide oxygen atoms pointing towards the inside of the helix.10–14 These tubular structures can form because of a rigid intramolecular three-center hydrogen bonding system involving the amide N–H bonds and aromatic ether oxygens.15 The meta-substitution pattern of the phenyl ring is also critical in forcing the polymer to adopt a crescent conformation. Aramid-based foldamers were reported for their application in host–guest molecular recognition, molecules and ions transport, as well as the crossing of cell membranes, among other uses.9,16–21 Most helical aramids were synthesized using variations of meta-aminobenzoic acid derivatives and combinations of meta-diamine and meta-diacid derivatives.5 In 2002, Gong et al. demonstrated that incorporating para-substituted derivatives into the backbone of the aramid oligomers could alter the curvature of the backbone, leading to the formation of much larger cavities. Synthetic tubular structures with cavity diameters exceeding 1 nm are rare. In that study, a 15-unit and a 21-unit aromatic amide oligomer with alternating meta- and para-aromatic amide units were synthesized in a stepwise fashion (Scheme 1a). The authors concluded that the 15-unit oligoamide formed a flat, crescent-shaped structure, while the 21-unit oligoamide was long enough to form a 1-turn helix, based on the observed signal overlap of end-groups in NOESY NMR spectra. The modeled 21-unit oligoamide showed a cavity of over 30 Å in diameter.13 The authors also reported the synthesis of macrocycles with alternating meta- and para-residues in a one-pot reaction using meta-substituted diamine and para-substituted diacyl chloride monomers.22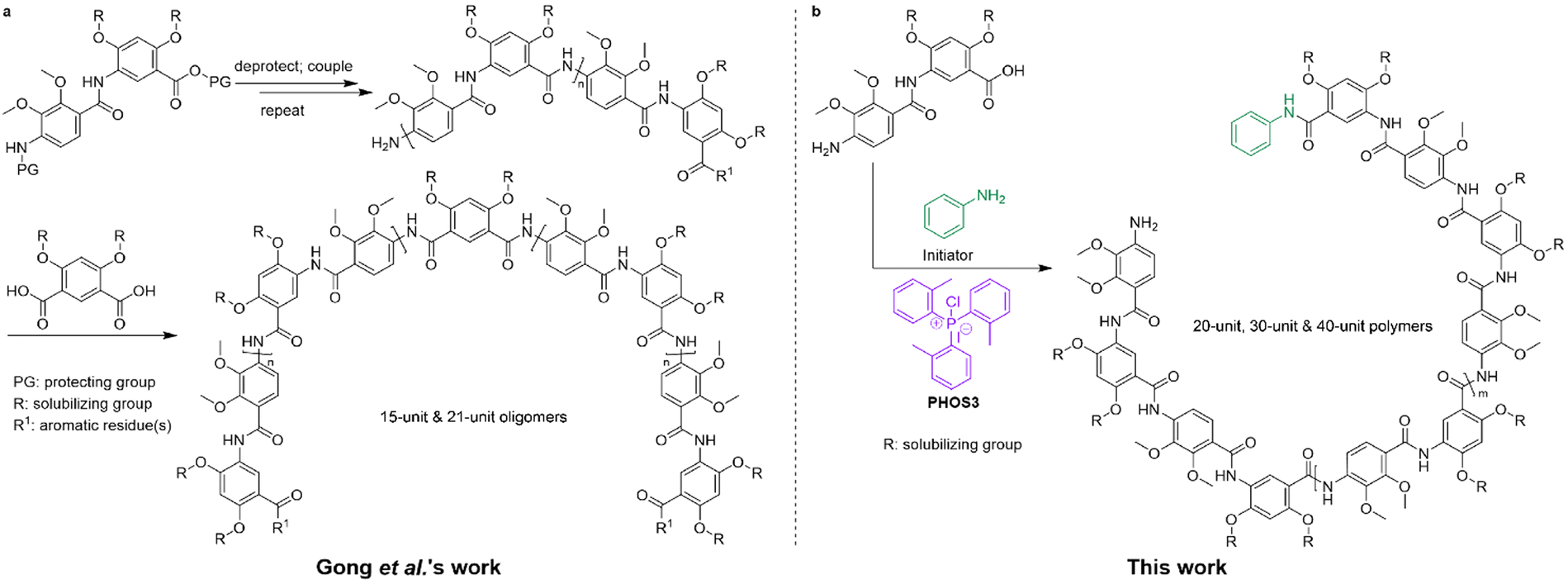 | ||
| Scheme 1 (a) General stepwise strategy developed by Gong et al. to obtain oligoamides. A 15-unit and a 21-unit aromatic amide oligomer were synthesized. (b) Polymerization strategy using an initiator and our reagent PHOS3. Several longer polymers were synthesized. | ||
Since then, to our knowledge, there were very few attempts to recreate such large cavity aramids. This could be due to the lengthy synthesis of the oligomers, because dimers, tetramers, and then heptamers must first be synthesized, protected, coupled, and deprotected to achieve an oligoamide long enough to form a full-turn helix.12 This is why, in this work, we propose another method to obtain similar giant helices in a more efficient way. A convenient living polymerization method for aromatic amides was recently developed in our group. It has been mostly used to polymerize meta-substituted aminobenzoic acids under very mild conditions using chloro tri-o-tolylphosphonium iodide, PHOS3, but it can also be utilized to polymerize para-substituted amides.21,23,24 Inspired by the monomer design of Gong and coworkers, we decided to synthesize a dimer that would consist of a para-substituted moiety, linked to a meta-substituted moiety through an amide bond. We opted for 2,4-bis((2,5,8,11-tetraoxapentadecan-15-yl)oxy)-5-(4-amino-2,3-dimethoxybenzamido)benzoic acid 1 as the dimeric unit. The synthesis starts from 2,3-dimethoxy-4-nitrobenzoic acid 10, which constitutes the para-substituted residue. This is coupled to methyl 2,4-bis((2,5,8,11-tetraoxapentadecan-15-yl)oxy)-5-aminobenzoic acid 5, the meta-substituted residue (Scheme 1). Nitro group reduction yielded the free amino-acid dimer (see ESI†). The syntheses of both individual monomers have already been reported.23,25 As the dimer would be polymerized in a living polymerization using PHOS3 (Scheme 1b), longer oligomers do not need to be synthesized, thereby greatly reducing the number of synthetic steps compared to previous efforts. This versatile method, therefore, unlocks the possibility of making helices with large cavities. This will be demonstrated in this study with the help of size-exclusion chromatography (SEC) and matrix assisted laser desorption ionization-time of flight mass spectroscopy (MALDI-ToF MS).
Results and discussion
Synthesis of the dimer and polymerization
Methyl 2,4-bis((2,5,8,11-tetraoxapentadecan-15-yl)oxy)-5-aminobenzoic acid 5, the meta-residue was first synthesized following an established protocol (see ESI†).23 This monomer and its homopolymers are not only soluble in an array of common solvents such as tetrahydrofuran, methanol or chloroform, but they are also soluble in water due to their triethylene glycol derivative side chains. Similarly to Gong et al.'s 4,6-bis(2-[2-(2-methoxyethoxy)ethoxy]ethoxy)-1,3-benzenedicarboxylic acid,13 this monomer should help to solubilize the synthesized giant helices aimed at here. It will also provide the necessary crescent shape to form the curvature of the helical backbone. 2,3-Dimethoxy-4-nitrobenzoic acid 10, the para-substituted residue, was synthesized from 2-hydroxy-6-methoxybenzaldehyde following a modified procedure of Kishimoto et al.25 Before undergoing Pinnick oxidation, 2-hydroxy-3-methoxy-4-nitrobenzaldehyde was subjected to methylation using methyl iodide (see ESI†). The resulting para-nitrobenzoic acid derivative 10 was then reacted with oxalyl chloride in dichloromethane in the presence of a catalytic amount of dimethylformamide to form the corresponding acyl chloride. After the removal of excess oxalyl chloride and the solvent, the product was reacted further with meta-aminobenzoic acid derivative 5 to obtain dimer 11. Finally, the reduction of the nitro group lead to the formation of the desired dimer 1, as illustrated in Scheme 2. Dimer 1 was fully characterized by 1H NMR, 13C NMR, and HR-ESI (Fig. S11, S13 and S26†). The 1H NMR spectrum showed very small traces of non-fully converted compound 1, which became important during polymerization to some extent (see below in the MALDI-ToF MS analysis section). Dimer 1 was insoluble in water, and, surprisingly, only slightly soluble in methanol. | ||
Scheme 2 Top: Synthesis route for dimer 1. (a) 1. Oxalyl chloride, DCM, 1 drop DMF. 2. Methyl 2,4-bis((2,5,8,11-tetraoxapentadecan-15-yl)oxy)-5-aminobenzoic acid 5, DCM. (b) H2 (40 bar), 40 °C, Pd/C, MeOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc. Bottom: Summarized polymerization conditions. Different size polymers and macrocycles as side-products were obtained. :EtOAc. Bottom: Summarized polymerization conditions. Different size polymers and macrocycles as side-products were obtained. | ||
The polymerization was performed using a method previously developed in our group, while using a mixture of DCM:DMAc (3:2) to ensure the solubility of dimer 1 and of the growing polymers (see ESI† and Scheme 2).23,24 Based on Gong et al.'s work, a full turn required around 20 aromatic amide residues,13i.e. ten dimeric units. This is why, for our first attempt, we aimed at a 1-turn giant helix, or 10-mer helix, when polymerizing P1. 10 eq. of dimer 1 were dissolved in a DCM:DMAc mixture (3:2) and added slowly (0.08 mL h−1) at room temperature to the reaction mixture, which contained 1 eq. of aniline I as the initiator, an excess of reagent PHOS3, and an excess of pyridine (see ESI†). The rate of addition was even slower than in the original report23 as we could show that the dispersity of the polymers was directly related to the rate of addition (see ESI, Table 4† of ref. 23). The SEC elugram in DMF of the crude polymer solution already showed an excellent control over the size and size distribution of P1 (Mn, theo = 7.7 kDa, Mn, SEC (DMF) = 7.5 kDa, Đ = 1.14, Fig. S1†). The polymer was then purified by recycling gel permeation chromatography in chloroform. The DMF SEC elugram of purified P1 confirmed the size and size distribution (Mn, theo = 7.7 kDa, Mn, SEC (DMF) = 8.7 kDa, Đ = 1.16, Fig. S2†) (Fig. 1a). The 1H NMR spectrum of P1 also supported the success of the polymerization; the ratio between the aromatic protons of the aniline initiator and the protons of the polymer's repeating units matched a 10-mer polymer (Fig. S15†). This encouraging result prompted us to target higher molecular weight polymers. Using the same polymerization protocol, we aimed to synthesize a 2-turn 20-mer helix, with a ratio [1:I, 20:1]. SEC analysis in DMF of the crude polymer P2 solution showed a molecular weight distribution closer to a 15-mer polymer (Mn, theo = 15 kDa, Mn, SEC (DMF) = 12 kDa, Đ = 1.19, Fig. S3†). SEC traces of purified polymer P2 gave the same molecular weight range with a slightly broader dispersity (Mn, theo = 15 kDa, Mn, SEC (DMF) = 11 kDa, Đ = 1.25, Fig. S4†) (Fig. 1a). The 1H NMR spectrum supported this, with an initiator-to-repeating units signal integration ratio showing the formation of a 15-mer polymer (Fig. S18†). The SEC trace also revealed a smaller distribution on the lower molecular weight side of the main distribution, which could explain the lower Mn and broader dispersity that were obtained. We tried another polymerization and, this time, aimed directly at a four-turn, i.e. 40-mer helix, for which we expected an Mn, theo of 31 kDa. The same method as above was performed with a dimer 1 to initiator I ratio [40:1]. The crude DMF SEC trace, in this instance, revealed a bi-modal distribution. The analysis showed a higher molecular weight species (approximately 53% of the integrated area) had a molecular weight Mn of 18 kDa with a dispersity Đ = 1.06, while a lower molecular weight compound (approximately 47% of the integrated area) showed an Mn of 5.7 kDa with a dispersity Đ = 1.03 (Fig. S5†). These molecular weights and dispersities were inaccurate, however, since the peaks were slightly merged. We decided to use recycling gel permeation chromatography to separate both distributions and were able to look at both groups of compounds individually. The higher molecular weight compound in the SEC trace was attributed to a polymer, P3, which had a molecular weight of 14 kDa with a dispersity Đ = 1.07 (Fig. S6†). This correlates to a 20-mer size polymer. The lower molecular weight compound in the SEC trace had a molecular weight of 5.4 kDa with a dispersity Đ = 1.04 (Fig. 1c). The observed molecular weight was consistent with either a linear 7-mer or a 7-mer macrocycle, with MALDI-ToF MS analysis suggesting the latter.
 | ||
| Fig. 1 (a) SEC (GPC) traces of purified P1, P2, and P3 polymers in DMF. (b) Isotopically resolved MALDI-ToF mass spectrum (DCTB, NaTFA) of polymer P1. (c) DMF SEC (GPC) trace of the isolated mixture of M1, M2 (most abundant), and M3 macrocycles in DMF. (d) Isotopically resolved MALDI-ToF mass spectrum (DCTB, NaTFA) of purified 7-mer macrocycle M2. | ||
MALDI-ToF MS analysis
First, the isotopically resolved MALDI-ToF mass spectrum of polymer P1 was examined. All analyses were done with DCTB as the matrix and NaTFA as the ionizing salt. A main distribution can be observed with matching repeating units and the monoisotopic mass confirmed the polymer structure with the expected aniline initiator and free amine end-groups (Fig. 1b). However, there were smaller distributions that showed some polymer chains were end-capped with the small traces of non-fully converted dimer 1, which, by analyzing the MALDI-ToF mass spectrum, corresponded to the intermediate nitrosobenzoic acid species (Fig. S27†). SEC traces and MALDI-ToF analysis showed that the presence of this undesired compound did not impact the polymerization significantly. The isotopically resolved MALDI-ToF MS of polymer P2 also revealed the same patterns of mass distribution, but the peaks were much less intense (Fig. S28†). What was interesting to note was the prominent peak at 5390.38 m/z. Upon further inspection, the monoisotopic mass (5387.33) corresponded to the theoretical mass (5387.71) of a seven-membered macrocycle (M2) consisting of self-initiated dimer 1. Some smaller distributions were also assigned to a six-membered macrocycle (M1) and an eight-membered macrocycle (M3) (Fig. S28†). Considering the presence of these macrocycles, it was safe to assume the smaller distribution in the SEC trace of purified polymer P2 corresponded to these self-initiated macrocyclic species (approximately 16% of the integrated area), while the bigger distribution could be attributed to the targeted aniline-initiated polymer (approximately 84% of the integrated area). These polymers could probably not be ionized very well at higher molecular weights, explaining why the main mass distribution intensity in the MALDI-ToF mass spectrum was very weak. Isotopically resolved MALDI-ToF analysis of isolated P3 (Fig. S29†) didn't show any matching distribution. The molecular weight of P3 could not be confirmed by 1H NMR spectroscopy, as, contrary to the 1H NMR spectra of P1 and P2, the aniline initiator signals could not be seen. The peaks were also broader compared to those observed in P1 and P2 (Fig. S21†). The MALDI-ToF analysis of the Mn, SEC (DMF) = 5.4 kDa fraction clearly revealed the presence of 6-mer (M1), 7-mer (M2), and 8-mer (M3) macrocycles, with M2 being the most abundant (Fig. S30†). M2 was separated from the mixture using preparative high-pressure liquid chromatography (HPLC), with 3–10% methanol in dichloromethane (DCM) as the mobile phase and a normal phase silica gel column as the stationary phase. The separation was successful, as shown by the isotopically resolved MALDI-ToF mass spectrum (Fig. 1d) as well as 1H NMR and 13C NMR (Fig. S22 and S23†). Ongoing attempts to prepare crystals of M2 were unsuccessful until now.Aggregation and conformation analysis
One noteworthy feature of previous prepared meta-oriented aramid helices obtained by our group was their broad 1H NMR signals in CDCl323,24 and the fact that they could not be detected by analytical chloroform SEC/GPC with UV/vis and/or refractive index detection. We assume that aggregation phenomena of individual helices were responsible for this behavior.
The 1H NMR spectrum of P3 also showed broadened signals and the signals of the aniline initiator I could not be observed at all. The 1H NMR spectra of P1, P2, and M2 on the other hand showed well-resolved repeat unit peaks and initiator I signals. The formation of very large macrocyclic side products is evidence, in our opinion, that the oligomers and polymers fold in the proposed manner and, therefore, have to form helical shapes when exceeding the molecular weight of the macrocycle.
Interestingly, SEC trace analysis afforded the following observation: it was possible to polymerize dimer 1 up to a 10-mer without any macrocycle formation. However, targeting higher molecular weights seemed to prompt a self-initiation side reaction of dimer 1 and macrocycles formation, which was confirmed by MALDI-ToF. The macrocycle formation started to occur at the point where the initiated polymer reached one full turn. We believe that the rate of propagation was reduced once the helix started to overlap with the first full turn, leading to an accumulation of the slowly added monomer 1 and, eventually, at a sufficient concentration of 1, self-initiation.
Had the growing polymers not adopted a robust helical conformation but a random coil, macrocycles would likely not have formed at all. We showed that 7-mer macrocycle M2, which had 14 aromatic amide residues, was the most stable species to form compared to smaller or larger macrocycles under these polymerization conditions. From this we concluded that these giant helices needed ca. 14 aromatic amide units to form one full turn. This was a smaller oligomer size than what Gong and coworkers proposed using models and NOESY analysis. Their 15-mer oligomer only formed a crescent shape and not a full turn, and only their 21-mer oligomer, which was slightly different from the one reported here, showed chain-ends overlapping.13,22 The polymers and macrocycles obtained here, although very similar, had a key difference in their structure, which explains this variation. Macrocycle M2 was prepared from an AB monomer while, as already described, the macrocycles Gong et al. obtained were through coupling of diamines and diacyl chlorides in an A2B2 manner. The amide bond angles play an important role in the curvature of the backbone; since they deviate from a perfect 120° and bend slightly towards the NH side of the three-centered hydrogen bond system, this creates a wider curvature in an oligomer that has an isophtalamide center, which Gong et al. synthesized.13 In other words, for the same number of alternating meta- and para-residues in the backbone, their helix would have a marginally wider curvature, thus creating a larger cavity and a need for more aromatic amide residues to form a full-turn compared to our monomer.
Synthesis of large macrocycles
We demonstrated the synthesis of large helical polymers using PHOS3 and aniline I as the initiator. At higher target molecular weights, macrocycles also formed as side products. Notably, this was the first reported synthesis of a large aramid macrocycle with a fully unsymmetrical backbone.To investigate whether these macrocycles could be obtained in greater quantities, we conducted a modified experiment. An excess of PHOS3 and pyridine was dissolved in a DCM:DMAc (3:2) mixture without an initiator, while dimer 1 was slowly introduced via syringe pump (Fig. 2a and ESI†). In the absence of an initiator, dimer 1 was the sole reactive species, undergoing conversion to acyl chlorides before reacting with other dimer 1 molecules.
 | ||
| Fig. 2 (a) Summary of the macrocyclization conditions. Different macrocycles were obtained. (b) DMF SEC trace of crude macrocyclization solution. (c) MALDI-ToF mass spectrum (DCTB, NaTFA) of the crude macrocyclization solution. | ||
The crude macrocyclization solution, analyzed at the end of dimer 1 addition, showed a dominant species with an Mn, SEC (DMF) of 6.8 kDa and dispersity Đ = 1.13 (Fig. 2b), slightly exceeding the theoretical Mn of 5.4 kDa for a 7-mer macrocycle. A minor high-molecular-weight distribution suggested the presence of larger species. MALDI-ToF mass spectrometry confirmed that the primary product was the 7-mer macrocycle M2, with additional peaks corresponding to the 6-mer (M1), 8-mer (M3), and, for the first time, the 9-mer macrocycle M4 (Fig. 2c and S32†). No linear polymers were detected.
Conclusions
This work presents an effective strategy for synthesizing large (>1 nm diameter) helical structures with unsymmetrical backbones formed from AB-type amino-acid monomers. Using a recent polymerization method, we successfully synthesized extended helices incorporating alternating meta-/para-aromatic amide blocks from dimer 1. Specifically, we obtained 10-mer, 15-mer, and 20-mer helices (containing 20, 30, and 40 aromatic amide residues, respectively), characterized by SEC/GPC, 1H NMR spectroscopy, and MALDI-ToF mass spectrometry. Additionally, the isolation of a 7-mer macrocycle provided key insights into the number of meta-/para-residues required to complete a full helical turn in the helical polymers.Furthermore, macrocycles were synthesized in larger quantities by employing dimer 1 in a self-condensation setup in the absence of an initiator. This approach expands the foldamer library and holds promise for applications in host–guest chemistry, catalysis, and molecular transport.
Author contributions
D. P. T. N. and A. F. M. K. designed the experiments. D. P. T. N. synthesized the phosphorous reagent and most compounds and conducted all polymerizations and the macrocyclization as well as most of the polymer and molecular analysis. S. F. and N. M. synthesized some of the compounds and conducted their analyses. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. F. M. K., D. P. T. N., S. F., and N. M. thank the National Center of Competence in Research (NCCR Bioinspired Materials) for their support.References
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef PubMed.
- E. Yashima and K. Maeda, Macromolecules, 2008, 41, 3–12 CrossRef.
- C. E. Schafmeister, Z. Z. Brown and S. Gupta, Acc. Chem. Res., 2008, 41, 1387–1398 CrossRef PubMed.
- T. A. Sobiech, Y. Zhong and B. Gong, Org. Biomol. Chem., 2022, 20, 6962–6978 RSC.
- B. Gong, Acc. Chem. Res., 2008, 41, 1376–1386 CrossRef PubMed.
- Y. Ferrand and I. Huc, Acc. Chem. Res., 2018, 51, 970–977 CrossRef PubMed.
- J. Shen, R. Ye, Z. Liu and H. Zeng, Angew. Chem., Int. Ed., 2022, 61, e202200259 CrossRef PubMed.
- H. Zhao, S. Sheng, Y. Hong and H. Zeng, J. Am. Chem. Soc., 2014, 136, 14270–14276 CrossRef PubMed.
- J.-L. Hou, C. Li, H.-P. Yi and Z.-T. Li, Chem. – Asian J., 2006, 1, 766–778 CrossRef PubMed.
- L. Yuan, H. Zeng, K. Yamato, A. R. Sanford, W. Feng, H. S. Atreya, D. K. Sukumaran, T. Szyperski and B. Gong, J. Am. Chem. Soc., 2004, 126, 16528–16537 CrossRef PubMed.
- Y. Zhong, B. Kauffmann, W. Xu, Z.-L. Lu, Y. Ferrand, I. Huc, X. C. Zeng, R. Liu and B. Gong, Org. Lett., 2020, 22, 6938–6942 CrossRef CAS PubMed.
- B. Gong, H. Zeng, J. Zhu, L. Yua, Y. Han, S. Cheng, M. Furukawa, R. D. Parra, A. Y. Kovalevsky, J. L. Mills, E. Skrzypczak-Jankun, S. Martinovic, R. D. Smith, C. Zheng, T. Szyperski and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11583–11588 CrossRef CAS.
- Y.-X. Lu, Z.-M. Shi, Z.-T. Li and Z. Guan, Chem. Commun., 2010, 46, 9019–9021 RSC.
- R. D. Parra, H. Zeng, J. Zhu, C. Zheng, X. C. Zeng and B. Gong, Chem. – Eur. J., 2001, 7, 4352–4357 CrossRef CAS.
- K. Ziach, C. Chollet, V. Parissi, P. Prabhakaran, M. Marchivie, V. Corvaglia, P. P. Bose, K. Laxmi-Reddy, F. Godde, J.-M. Schmitter, S. Chaignepain, P. Pourquier and I. Huc, Nat. Chem., 2018, 10, 511–518 CrossRef CAS.
- K. Yamato, L. Yuan, W. Feng, A. J. Helsel, A. R. Sanford, J. Zhu, J. Deng, X. C. Zeng and B. Gong, Org. Biomol. Chem., 2009, 7, 3643–3647 RSC.
- Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang and I. Huc, Science, 2011, 331, 1172–1175 CrossRef CAS PubMed.
- E. R. Gillies, F. Deiss, C. Staedel, J.-M. Schmitter and I. Huc, Angew. Chem., Int. Ed., 2007, 46, 4081–4084 CrossRef CAS.
- Y. Zhong, T. A. Sobiech, B. Kauffmann, B. Song, X. Li, Y. Ferrand, Y. Huc and B. Gong, Chem. Sci., 2023, 14, 4759–4768 RSC.
- S. Farooq, J. A. Malla, M. Nedyalkova, R. V. M. Freire, I. Mandal, A. Crochet, S. Salentinig, M. Lattuada, C. T. McTernan and A. F. M. Kilbinger, Angew. Chem., Int. Ed., 2025 DOI:10.1002/anie.202504170.
- W. Feng, K. Yamato, L. Yang, J. S. Ferguson, L. Zhong, S. Zou, L. Yuan, X. C. Zeng and B. Gong, J. Am. Chem. Soc., 2009, 131, 2629–2637 CrossRef CAS PubMed.
- S. Pal, D. P. T. Nguyen, A. Molliet, M. Alizadeh, A. Crochet, R. D. Ortuso, A. Petri-Fink and A. F. M. Kilbinger, Nat. Chem., 2021, 13, 705–713 CrossRef.
- S. Pal, L. Hong, R. V. M. Freire, S. Farooq, S. Salentinig and A. F. M. Kilbinger, Macromolecules, 2023, 56, 7984–7992 CrossRef.
- S. Kishimoto, S. Nishimura, M. Hatano, M. Igarashi and H. Kakeya, J. Org. Chem., 2015, 80, 6076–6082 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5py00275c |
| This journal is © The Royal Society of Chemistry 2025 |