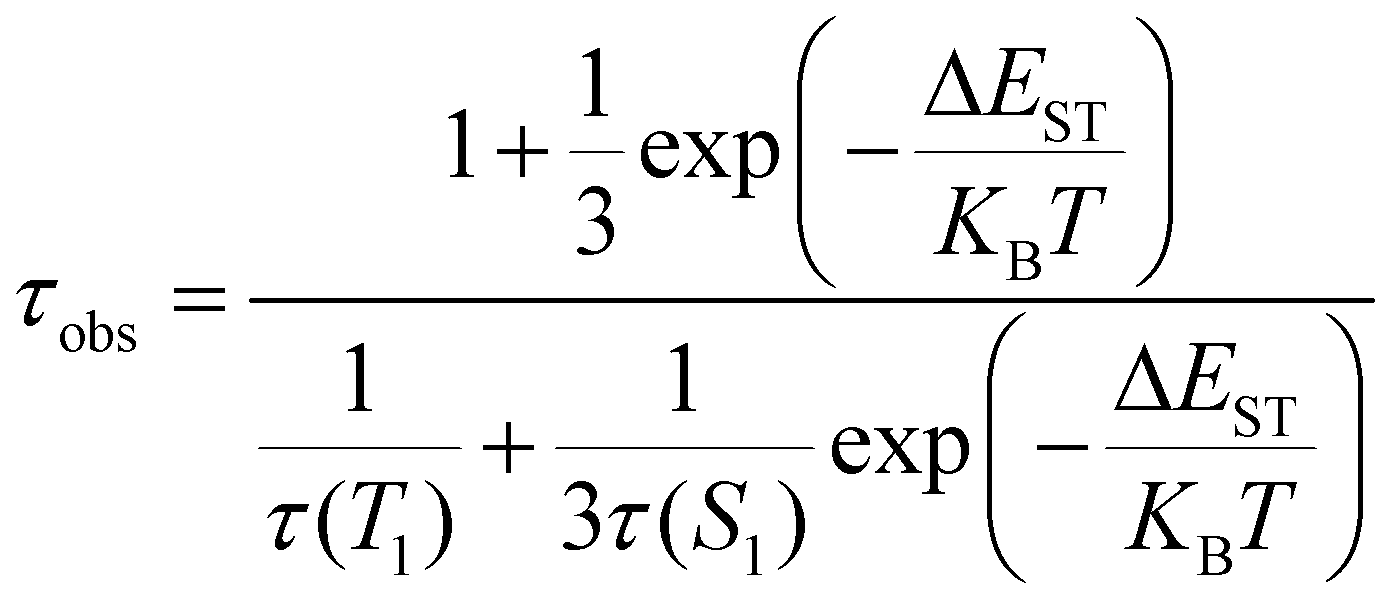DOI:
10.1039/D4QI02415J
(Research Article)
Inorg. Chem. Front., 2025,
12, 1139-1155
High-performance TADF-OLEDs utilizing copper(I) halide complexes containing unsymmetrically substituted thiophenyl triphosphine ligands†
Received
24th September 2024
, Accepted 12th December 2024
First published on 13th December 2024
Abstract
Highly efficient OLEDs fabricated with thermally activated delayed fluorescent Cu(I) complexes have attracted significant attention. However, achieving both high quantum efficiency and short decay lifetimes remains a considerable challenge. Herein, we reported the successful synthesis and characterization of two rigid triphosphine ligands, each containing two unsymmetrically substituted thiophenyl rings, and their corresponding mononuclear copper(I) halide complexes, CuX(L1) and CuX(L2) [L1 = (2-PPh2-C4H2S)2(3-PPh), X = I (1), Br (2), Cl (3); L2 = (2-PPh2-5-SiMe3-C4HS)2(3-PPh), X = I (4), Br (5), Cl (6)]. The structures and photophysical properties of these complexes were thoroughly investigated. At room temperature, the powder samples of complexes 1–6 exhibited intense delayed fluorescence, ranging from yellow-green to yellow in color (λem = 553–581 nm, τ = 3.8–9.4 μs, and Φ = 0.19–0.29 for 1–3; λem = 565–589 nm, τ = 2.2–7.6 μs, and Φ = 0.36–0.61 for 4–6). The incorporation of two trimethylsilyl groups into the unsymmetrically substituted thiophenyl rings significantly improved the photoluminescence quantum yield (PLQY) and allowed for fine-tuning of the light-emitting color of the complexes. Among them, complex 4 displayed a high PLQY of 0.61 and a short decay lifetime of 2.2 μs. The radiative decay rate (kr) was 2.8 × 105 s−1, comparable with that of Ir(III) complexes. Vacuum-deposited organic light-emitting devices incorporating complex 4 exhibited yellow emission, achieving a maximum external quantum efficiency (EQE) of 14.57% and a current efficiency of 33.44 cd A−1.
1 Introduction
Luminescent Cu(I) complexes, as an important class of thermally activated delayed fluorescence (TADF) emitters, represent a highly promising category of emitters for applications in electroluminescent devices since they facilitate the efficient conversion of all electrically generated excitons into light.1–6 TADF operates through a singlet harvesting mechanism, which significantly shortens the emission decay time at ambient temperature. This results in the suppression of efficiency roll-off as the current density increases. To achieve TADF in Cu(I) complexes, it is essential that the energy gap (ΔEST) between the first singlet excited state (S1) and the first triplet excited state (T1) be small.7–11 Furthermore, Cu(I) complexes should exhibit a high photoluminescence quantum yield (PLQY). Therefore, rigidifying the molecular structure either through the use of sterically demanding ligands or by creating a rigid environment is crucial. This strategy helps to reduce the radiative deactivation of the luminescence, which can be caused by structural distortions at the copper center in the excited state.
Tsuboyama's group synthesized a rigid diphosphine ligand, dppb (1,2-bis(diphenylphosphino)benzene), and its Cu(I) halide complexes. The dinuclear, four-coordinated, halogen-bridged structure of these complexes resulted in a PLQY of up to 80%.12 Osawa et al. reported a diphosphine ligand, dtpb (2-bis(o-ditolylphosphino)benzene), with a methyl substituent at the ortho position of the peripheral phenyl group. The o-methyl group in dtpb was essential for the formation of three-coordinate Cu(I) halide complexes.13 The trigonally planar coordination geometry of the Cu(I) halide complexes results in high PLQY of up to 71%. Furthermore, a vapor-deposited OLED incorporating these complexes exhibited a maximum EQE of 21.3%, which is comparable to that of cyclometalated iridium(II)-based devices.
Apart from rigid diphosphine ligands that can be used to construct highly emissive Cu(I) halide complexes, a rigid triphosphine ligand (TTPP = 2,2′-(phenylphosphinediyl)bis(2,1-phenylene)bis(diphenylphosphine)) was firstly reported by Xu's group.14 Complexes CuX(TTPP) show PLQY of as high as 85%, and provided 16.3% for EQE.
Thiophene is an electron-rich heteroaryl ring, which is widely used as a donor material in solar cells.15 It was also selected to construct dinuclear Cu(I) halide complexes containing diphosphine16,17 and mononuclear heteroleptic Cu(I) halide complexes containing diphosphine and monophosphine18,19 by our group and Lu's group. The replacement of benzene by more electron-rich thiophene usually results in the blue-shifting emission of the complexes,16 emission color can be largely tuned from deep blue to green.18 Moreover, Cu(I) halide complexes [Cu(I-2)X]2 containing unsymmetrically substituted thiophenyl diphosphine I-2 displayed higher quantum efficiency than [Cu(I-1)X]2-containing symmetrically substituted thiophenyl diphosphine I-1 (Fig. 1).17
 |
| | Fig. 1 Previously reported symmetrically substituted thiophenyl diphosphine (I-1) and triphosphine (I-3 and I-4), unsymmetrically substituted thiophenyl diphosphine (I-2) and their corresponding Cu(I) halide complexes and the present work. | |
Recently, two symmetrically substituted thiophenyl rings were used to replace two benzene rings to construct rigid triphosphine (Fig. 1, I-3 and I-4) and the corresponding Cu(I) halide complexes [Cu(I-3)X] and [Cu(I-4)X] by our group20 (Fig. 1), apart from tuning of the emission color can, good solubility of the complexes makes it suitable to fabricate solution-processed OLEDs. To the best of our knowledge, rigid triphosphine Cu(I) halide complexes are rarely reported, and the focus has been mainly on symmetrically substituted aromatic or heteroaromatic rings, while unsymmetrically substituted heteroaromatic triphosphine Cu(I) halide complexes have not been reported so far. With our great interests in Cu(I) halide complexes with excellent photophysical properties,21–28 here, we described the synthesis, structures and photophysical properties of two unsymmetrically substituted thiophenyl triphosphine ligands (L1 and L2), and six mononuclear Cu(I) halide complexes 1–6 (Fig. 1). Vacuum-deposited OLED devices based on complex 4 were fabricated and their electroluminescent behaviors were studied.
2 Experimental section
2.1 Synthesis of ligand L1
Under nitrogen at a temperature of −90 °C, n-BuLi (2.5 M in hexane, 9.16 mL, 22.90 mmol) was added dropwise into a dry THF (30 mL) and diethyl ether (30 ml) solution containing bis(3-bromothiophen-2-yl)(phenyl)phosphine (4.50 g, 10.41 mmol). The resulting mixture was stirred at −90 °C for 0.5 h, then PPh2Cl (5.05 g, 22.90 mmol) was added and the mixture was stirred at −90 °C for 0.5 h. After that, the mixture naturally warmed up to room temperature. Water (20 mL) was added to quench the reaction. The organic phase was separated, and the aqueous phase was extracted using CH2Cl2 (3 × 20 mL). The organic phase was combined and then dried with magnesium sulfate. After removing the solvent from the filtrate, the remaining substance was purified by column chromatography over silica gel using PE (petroleum ether)/DCM(dichloromethane) (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v:v) as the eluent. The product was obtained as a white solid.
:1, v:v) as the eluent. The product was obtained as a white solid.
L1 (yield: 3.70 g, 55.3%).
1H NMR (400 MHz, d6-DMSO), δ 7.81 (d, J = 4.0 Hz, 2H), 7.33–7.26 (m, 15H), 7.23–7.19 (m, 2H), 7.16–7.08 (m, 8H), 6.75–6.73 (m, 2H). 31P NMR (162 MHz, CDCl3), δ −25.68 (1P), −26.39 (1P), −42.71 (1P, JP–P = 114.21 Hz). 13C NMR (101 MHz, CDCl3), δ 147.02, 146.73, 146.61, 146.33, 144.91, 144.75, 144.62, 144.46, 137.91, 137.80, 137.63, 137.54, 133.55, 133.38, 133.18, 133.01, 131.32, 128.95, 128.39, 128.35, 128.28, 128.21, 128.13. Anal. calcd for C38H29P3S2: C, 71.02; H, 4.55, found: C, 72.03; H, 4.54. HRMS (ESI): m/z calcd for [C38H30P3S2]+, 643.1002, found: 643.1009.
2.2 Synthesis of the ligand L2
Under nitrogen at 0–5 °C, n-BuLi (2.5 M in hexane, 3.83 mL, 9.58 mmol) was added dropwise into a dry THF (20 mL) and diethyl ether (20 ml) solution containing the ligand L1 (2.80 g, 4.36 mmol). The resulting mixture was stirred at 0–5 °C for 15 min, then trimethylchlorosilane (TMSCl, 1.04 g, 9.58 mmol) was added and the mixture was stirred at 0–5 °C for 20 min. After that, the mixture naturally warmed up to room temperature. Water (15 mL) was added to quench the reaction. The organic phase was separated, and the aqueous phase was extracted using CH2Cl2 (3 × 15 mL). The organic phase was combined and then dried with magnesium sulfate. The solvent from the filtrate was removed and the residue was purified by column chromatography over silica gel using petroleum ether/dichloromethane (7:1, v:v) as the eluent. The product was obtained as a white solid.
L2 (yield: 3.00 g, 87.5%).
1H NMR (400 MHz, d6-DMSO), δ 7.36–7.26 (m, 17H), 7.14–7.09 (m, 8H), 6.89 (d, J = 2.0 Hz, 2H), 0.12 (s, 18H, 2SiMe3). 31P NMR (162 MHz, CDCl3), δ −25.81 (1P), −26.52 (1P), −41.62 (1P, JP–P = 115.02 Hz). 13C NMR (101 MHz, CDCl3), δ 152.13, 151.85, 151.72, 151.44, 147.10, 144.91, 144.75, 144.62, 144.46, 139.53, 139.49, 138.25, 138.13, 137.74, 133.66, 133.63, 133.43, 133.37, 133.18, 128.85, 128.25, 128.18, 128.06, 0.08. Anal. calcd for C44H45P3S2Si2: C, 67.15; H, 5.76, found: C, 67.16; H, 5.75. HRMS (ESI): m/z calcd for [C44H46P3S2Si2]+, 787.1792, found: 787.1789.
2.3 Synthesis of complexes 1–6
Complexes 1–6 were prepared according to the following method. At room temperature, a solution of ligand L1 (0.96 g, 1.50 mmol) or L2 (1.18 g, 1.50 mmol) in 25 mL of dichloromethane, copper(I) halide (CuI: 0.29 g, 1.50 mmol; CuBr: 0.22 g, 1.50 mmol; CuCl: 0.15 g, 1.50 mmol) was prepared, and the mixture was stirred for 4 h. Then, the suspension was filtered off, and the filtrate was collected and concentrated in a vacuum to form a yellow solid. The yellow solid was dissolved in dichloromethane and n-hexane was added, and the precipitate formed was collected and the solvent was removed to obtain the pure product. Yellow crystals for 1–6 were obtained by dissolving solid in solvents (CH2Cl2/CH3CN for 1, 4, 5 and 6, CH2Cl2/n-hexane for 2, and CH2Cl2/ethanol for 3) and slow evaporation at room temperature.
Complex 1 (yield: 1.12 g, 89.6%).
1H NMR (400 MHz, d6-DMSO), δ 8.26 (d, J = 4 Hz, 2H), 7.85–7.75 (m, 4H), 7.59 (t, J = 8 Hz, 2H), 7.48–7.32 (m, 9H), 7.34 (t, J = 4 Hz, 2H), 7.16 (t, J = 8 Hz, 4H), 7.10–7.06 (m, 2H), 6.85–6.70 (m, 4H). 31P NMR (162 MHz, CDCl3), δ −21.27 (s, 2P), −41.56 (t, JP–P = 142.56 Hz, 1P). Anal. calcd for C38H29CuIP3S2: C, 54.78; H, 3.51, found: C, 54.79; H, 3.50. HRMS (ESI): m/z calcd for [C38H29CuP3S2]+, 705.0220, found: 705.0206.
Complex 2 (yield: 1.03 g, 87.3%).
1H NMR (400 MHz, d6-DMSO), δ 8.26 (d, J = 4 Hz, 2H), 7.96–7.87 (m, 4H), 7.66–7.59 (m, 2H), 7.51–7.42 (m, 10H), 7.32 (t, J = 8 Hz, 2H), 7.12 (t, J = 8 Hz, 6H), 6.66–6.59 (m, 3H). 31P NMR (162 MHz, CDCl3), δ −21.29 (d, JP–P = 105.3 Hz, 2P), −48.08 (1P). Anal. calcd for C38H29CuBrP3S2: C, 58.06; H, 3.72, found: C, 58.08; H, 3.70. HRMS (ESI): m/z calcd for [C38H29CuP3S2]+, 705.0220, found: 705.0219.
Complex 3 (yield: 0.94 g, 84.5%).
1H NMR (400 MHz, d6-DMSO), δ 8.25 (d, J = 4 Hz, 2H), 8.01–7.90 (m, 4H), 7.60–7.54 (m, 2H), 7.53–7.43 (m, 10 H), 7.31 (t, J = 8 Hz, 2H), 7.11 (t, J = 8 Hz, 6H), 6.62–6.56 (m, 3H). 31P NMR (162 MHz, CDCl3), δ −21.23 (d, JP–P = 102.06 Hz, 2P), −44.93 (t, JP–P = 134.46 Hz, 1P). Anal. calcd for C38H29CuClP3S2: C, 61.54; H, 3.94, found: C, 61.52; H, 3.96. HRMS (ESI): m/z calcd for [C38H29CuP3S2]+, 705.0220, found: 705.0175.
Complex 4 (yield: 1.30 g, 88.7%).
1H NMR (400 MHz, d6-DMSO), δ 7.90–7.75 (m, 5H), 7.55–7.40 (m, 10H), 7.33 (t, J = 8.0 Hz, 2H), 7.22 (d, J = 2.4 Hz, 2H), 7.11 (t, J = 8 Hz, 4H), 6.75–6.65 (m, 4H), 0.27 (s, 18H, 2SiMe3). 31P NMR (162 MHz, CDCl3), δ −22.10 (2P), −44.56 (t, JP–P = 162 Hz, 1P). Anal. calcd for C44H45CuIP3S2Si2: C, 54.06; H, 4.64, found: C, 54.07; H, 4.62. HRMS (ESI): m/z calcd for [C44H45CuP3S2Si2]+, 849.1010, found: 849.1057.
Complex 5 (yield: 1.16 g, 83.1%).
1H NMR (400 MHz, d6-DMSO), δ 7.95–7.87 (m, 4H), 7.77–7.69 (m, 2H), 7.46 (t, J = 8.0 Hz, 9H), 7.33 (t, J = 8.0 Hz, 2H), 7.22 (d, J = 2.8 Hz, 2H), 7.12 (t, J = 8.0 Hz, 4H), 6.74–6.62 (m, 4H), 0.26 (s, 18H, 2SiMe3). 31P NMR (162 MHz, CDCl3), δ −20.90 (d, JP–P = 102.06 Hz, 2P), −43.53 (t, JP–P = 152.28 Hz, 1P). Anal. calcd for C44H45CuBrP3S2Si2: C, 56.80; H, 4.87, found: C, 56.78; H, 4.86. HRMS (ESI): m/z calcd for [C44H45CuP3S2Si2]+, 849.1010, found: 849.1004.
Complex 6 (yield: 1.08 g, 81.3%).
1H NMR (400 MHz, d6-DMSO), δ 8.05–7.97 (m, J = 8.0 Hz, 4H), 7.75–7.68 (m, 2H), 7.52–7.44 (m, 9H), 7.31 (t, J = 8.0 Hz, 2H), 7.24 (d, J = 2.4 Hz, 2H), 7.09 (t, J = 8.0 Hz, 4H), 6.62–6.54 (m, 4H), 0.26 (s, 18H, 2SiMe3). 31P NMR (162 MHz, d6-DMSO), δ −19.79 (d, JP–P = 157.14 Hz, 2P), −42.12 (t, JP–P = 162 Hz, 1P). Anal. calcd for C44H45CuClP3S2Si2: C, 59.64; H, 5.12, found: C, 59.65; H, 5.11. HRMS (ESI): m/z calcd for [C44H45CuP3S2Si2]+, 849.1010, found: 849.1052.
3 Results and discussion
3.1 Syntheses and characterization
Scheme 1 presents the synthetic pathways to ligands L1, L2 and complexes 1–6. The ligand L1 was synthesized in a mixture of bis(3-bromothiophen-2-yl)(phenyl)phosphine and n-BuLi to which PPh2Cl was added to form a mole ratio of 1:2.2:2.2 in THF/Et2O at −90 °C under N2. L2 was synthesized from a mixture of L1, n-BuLi and TMSCl at a mole ratio of 1:2.2:2.2 in THF/Et2O at 0–5 °C under N2. L1 and L2 were obtained in 55.3% and 87.5% yields, respectively. Complexes 1–6 were synthesized in 81.3%−89.6% yields from the mixture of CuX and 1 equivalent of the ligand L1 or L2 in CH2Cl2. The structures of complexes 1–6 were identified (Fig. 2 and Fig. S1–S26†). They are stable in the air (Fig. S27†) and dissolved in dichloromethane, chloroform, ethyl acetate and THF.
 |
| | Scheme 1 Synthetic routes of the ligands L1 and L2 and complexes 1–6. (1) n-BuLi, PPh2Cl, THF/Et2O, −90 °C; (2) n-BuLi, TMSCl, THF/Et2O, 0–5 °C; and (3) CuX, DCM, r.t. | |
 |
| | Fig. 2 ORTEP diagrams of complexes 1–6. H atoms were omitted for clarity. | |
The Oak Ridge thermal ellipsoid plots (ORTEP) diagrams of complexes 1–6 show that one copper(I) center is tetrahedrally coordinated with one halogen atom and three P atoms of ligand L1 or L2 (Fig. 2). There is one solvent CH2Cl2 molecule in the complex 2 (Tables 1 and S1†). The bond length of Cu–X increases as the X radius increases (1 > 2 > 3 and 4 > 5 > 6). Under the same halogen, the bond lengths of Cu–X in 4–6 are shorter than those in 1–3 due to the introduction of two trimethylsilyl groups. The bond lengths of Cu–X in 1–3 are very close to those in Cu(I) halide complexes [Cu(I-4)X] with the symmetrically substituted thiophenyl triphosphine I-420 (Fig. 1, Cu–I: 2.5631(4), Cu–Br: 2.3956(5) and Cu–Cl: 2.2670(6) Å), while the bond lengths of Cu–X in 4–6 are shorter than those in Cu(I) halide complexes [Cu(I-3)X] with symmetrically substituted dimethylthiophenyl triphosphine I-320 (Fig. 1, Cu–I: 2.5261(4), Cu–Br: 2.3599(5) and Cu–Cl: 2.2389(6) Å). The average bond lengths of Cu–P in 1–3 (2.2797–2.2883 Å) are shorter than those in 4–6 (2.3036–2.3091 Å), suggesting that L1 has stronger coordinating ability with Cu(I) than L2. The bond angles of P1–Cu–P2 and P1–Cu–P3 in 1–3 (89.50–90.56°) are very close to those in Cu(I) halide complexes [Cu(I-4)X] with symmetrically substituted thiophenyl triphosphine I-4 (89.00–91.56°)20 larger than that in 4–6 (86.11–88.33°), showing more compact configuration around Cu(I) center in 4–6 than that in 1–3. Complexes 4–6 exhibit slightly stronger π–π interactions between the two thiophenyl rings than in complexes 1–3, the centroid–centroid distances between the two thiophenyl rings in complexes 4–6 are 4.599–4.774 Å in comparison to 4.858–4.929 Å in 1–3 (Fig. S28–S33†).
Table 1 Selected bond lengths (Å) and angles (°) for complexes 1–6
| Complex |
1
|
2·CH2Cl2 |
3
|
4
|
5
|
6
|
| Cu–X |
2.5698(18) |
2.4034(3) |
2.2828(6) |
2.5480(4) |
2.3782(3) |
2.2510(4) |
| Cu–P1 |
2.298(3) |
2.2798(5) |
2.2875(7) |
2.3051(6) |
2.3073(5) |
2.3144(4) |
| Cu–P2 |
2.265(3) |
2.2807(5) |
2.2786(6) |
2.2949(7) |
2.3182(5) |
2.2947(5) |
| Cu–P3 |
2.276(3) |
2.2794(6) |
2.2988(6) |
2.3108(7) |
2.2944(5) |
2.3181(5) |
| P1–Cu–P2 |
89.50(12) |
89.575(19) |
90.38(2) |
88.33(2) |
86.217(17) |
87.978(16) |
| P1–Cu–P3 |
89.66(11) |
90.379(19) |
90.56(2) |
86.43(2) |
88.166(17) |
86.112(16) |
| P2–Cu–X |
115.34(10) |
108.606(17) |
116.49(2) |
119.75(2) |
118.336(16) |
120.076(18) |
| P3–Cu–X |
109.97(9) |
117.578(17) |
114.12(2) |
117.41(2) |
119.792(16) |
118.261(18) |
3.2 Photophysical properties, electrochemical properties and computational studies
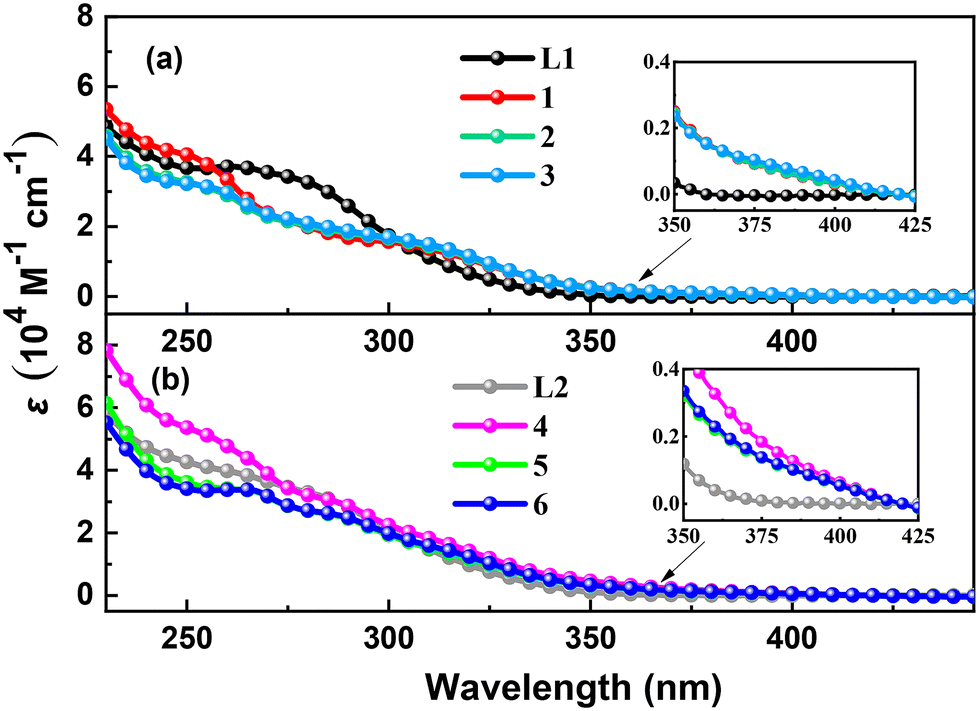
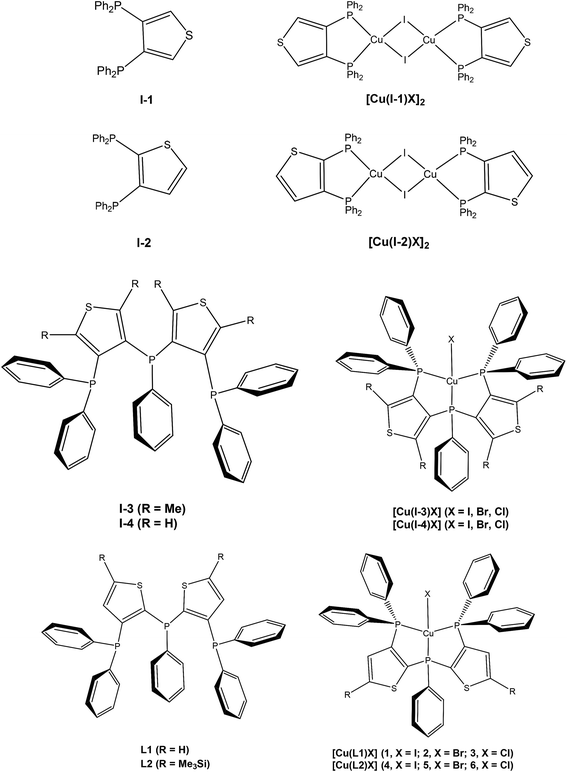
UV-vis absorption spectra of ligands L1, L2 and complexes 1–6 in CH2Cl2 at room temperature are shown in Fig. 3. Ligand L1 shows strong absorption peaks at 263 and 278 nm, and ligand L2 exhibits more intense and red-shifted absorption peaks at 264 and 285 nm due to the introduction of the trimethylsilyl groups, which correspond to π → π* and n → π* transitions of phenylphosphine and thiophenylphosphine, respectively.18 The red shift of the absorption peaks is due to the introduction of the trimethylsilyl groups, which shows σ–π hyperconjugated effect.29 Complexes 1–3 display intense absorption bands (251–257 nm), broad bands (276–281 and 305–307 nm) and tails (365–420 nm), and complexes 4–6 exhibit more intense and red-shifted absorption bands (256–266 nm), two broad bands (286–287 and 314–317 nm) and tails (370–420 nm). The weak low-energy absorption tails, which are not observed in the corresponding free ligands spectra, might be ascribed to the charge transfer transitions including metal-to-ligand charge transition (MLCT) and halogen-to-ligand charge transition (XLCT).30
 |
| | Fig. 3 Absorption spectra of (a) ligand L1 and complexes 1–3 (b) ligand L2 and complexes 4–6 in CH2Cl2 at room temperature. | |
In order to clarify the transition character of these weak low-energy absorption tails, we carried out the time-dependent density functional theory (TD-DFT) calculations for complexes 1–6 on the basis of their optimized S0 geometries. All the simulated UV-vis absorption spectra showed a similar profile to the corresponding experimental ones, indicating the validity of our calculation method (Fig. 4 and Fig. S34†). According to the calculation, the absorption tails can be ascribed to the electronic transitions of the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) due to its overwhelming contribution (≥96%) (Tables S2–S7†). According to the orbital composition analysis (Table 2, Fig. 5 and Fig. S37–S42†), it can be seen that the Cu, halogen, P of triphosphine ligand L1 or L2 contribute significantly to HOMO. The LUMO of the complexes is mainly localized on the thiophenyl rings of the ligand L1 or L2 (ca. 99%). Therefore, the absorption tails of complexes 1–6 in the experiment can reasonably be assigned to (metal and halogen)-to-ligand charge transitions (M + X)LCT and intraligand charge transitions (ILCT). The ratio of charge transfer contribution at optimized S0 geometry of complexes 1–6 shows that the contribution from ILCT is much higher than MLCT and XLCT (Table 2). As the halogen atomic number decreases, the ratio of ILCT and MLCT further increases. According to the MO distribution patterns of complexes 1–6 in Fig. 5 and the orbital compositions analysis in Fig. S37–S42,† the trimethylsilyl group linked to the LUMO-dominating thiophenyl rings of complexes 4–6 can slightly extend the LUMO distribution and show σ–π hyperconjugated effect, leading to a stabilized LUMO energy level. Hence, complexes 4–6 show lower LUMO energy levels than those of 1–3. Therefore, complexes 4–6 show slightly red-shifted absorption tails relative to those of 1–3.
Table 2 TD-DFT results of complexes 1–6 on the basis of their optimized S0 geometries
| Complex |
MO |
Contribution ratios of metal, halogen, and triphosphine ligand to MOs (%) |
Main configuration of S0 → S1 excitation/Ecal/λcal/fa |
| Cu |
X |
L1 or L2 |
|
H → L denotes the transition from HOMO to LUMO. Ecal, λcal and f denote calculated excitation energy, absorption wavelength and oscillator strength, respectively.
|
|
1
|
H |
26.12 |
29.28 |
44.60 |
H → L (97%) |
| L |
0.91 |
0.12 |
98.97 |
3.21 eV/385.9 nm/0.0185 |
|
2
|
H |
31.59 |
14.19 |
54.22 |
H → L (97%) |
| L |
0.90 |
0.10 |
99.00 |
3.24 eV/383.1 nm/0.0201 |
|
3
|
H |
33.91 |
8.61 |
57.48 |
H → L (98%) |
| L |
0.90 |
0.08 |
99.02 |
3.24 eV/383.1 nm/0.0204 |
|
4
|
H |
25.96 |
29.08 |
44.96 |
H → L (96%) |
| L |
0.88 |
0.12 |
99.00 |
3.17 eV/391.2 nm/0.0250 |
|
5
|
H |
31.16 |
14.16 |
54.68 |
H → L (98%) |
| L |
0.88 |
0.10 |
99.02 |
3.19 eV/388.1 nm/0.0273 |
|
6
|
H |
33.44 |
8.61 |
57.95 |
H → L (98%) |
| L |
0.89 |
0.09 |
99.02 |
3.19 eV/388.1 nm/0.0280 |
 |
| | Fig. 4 Experimental UV-vis absorption spectra and simulated spectra with oscillator strength for complexes 1 and 4. | |
 |
| | Fig. 5 Molecular orbital (MO) distribution patterns of complexes 1–6 based on their optimized S0 geometries. | |
The electrochemical properties of complexes 1–6 were studied by cyclic voltammetry (CV). Cyclic voltammograms of complexes 1–6 in CH2Cl2 (2 × 10−3 M) and TBAPF6 (0.1 M) are provided in Fig. 6, and the electrochemical data are shown in Table S8.† The first oxidation peak potentials (Eox1) of complexes 1–3 were found to be 0.65–0.78 V, higher than those of 4–6 (0.62–0.72 V), indicating easier oxidation by the electron-rich trimethylsilyl moieties in 4–6. The first oxidation stages of all the complexes correspond to the oxidation of Cu+ ions. The onset oxidation potentials (Eon-s) of the complexes were found to be 0.10–0.21 V vs. Fc/Fc+ (Table S8†), and the energy levels for their HOMOs were calculated to be −4.84–−4.95 eV (EHOMO = −4.74 − Eon-s). In association with the values of their energy gaps (ΔEg), which were estimated from the onset wavelengths of their absorption spectra (Fig. 3), the energy levels of their LUMOs were calculated to be −1.92–−2.00 eV (ELUMO = EHOMO + ΔEg).
 |
| | Fig. 6 Cyclic voltammograms of 2 × 10−3 M solutions of complexes 1–6 in DCM containing 0.1 M TBAPF6 using the saturated calomel electrode as a reference electrode. | |
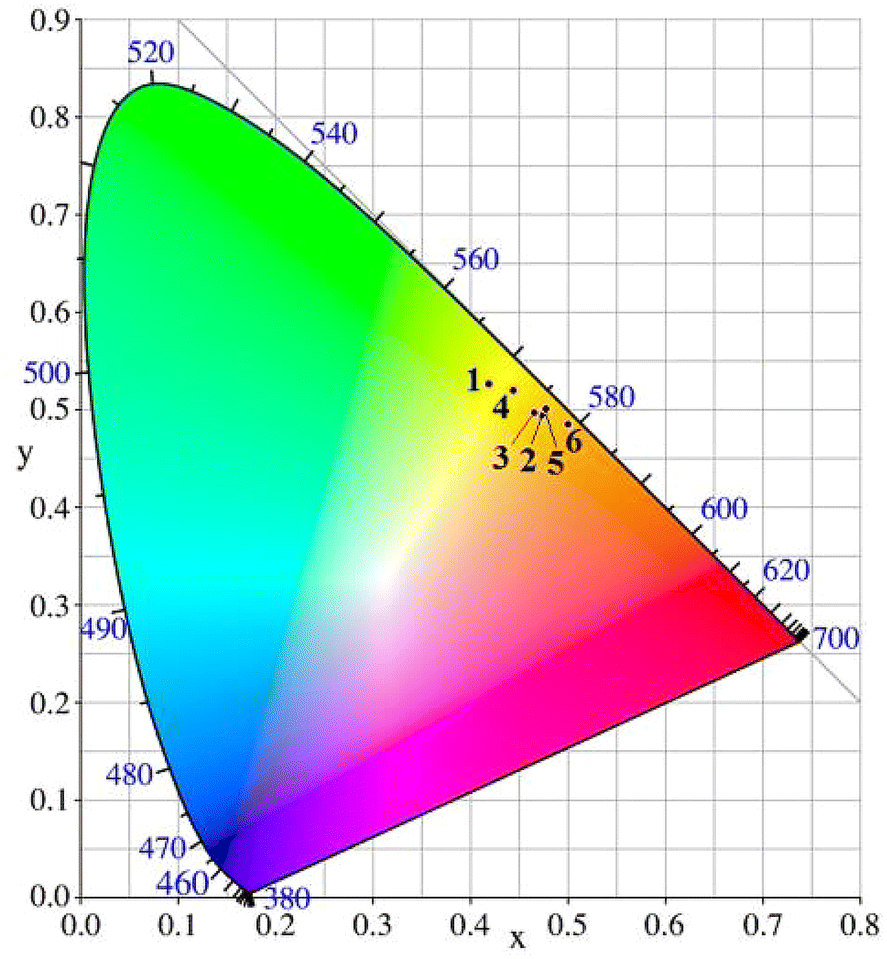
At 297 K, complexes 1–6 in powder exhibit intense yellow-green to yellow emissions (1 → 6: λem: 553, 581, 577, 565, 578 and 589 nm; τ: 8.0, 3.8, 9.4, 2.2, 6.4 and 7.6 μs; Φ: 0.29, 0.19, 0.27, 0.61, 0.38 and 0.36, Fig. 7, Fig. S36† and Table 3). The maximum emission wavelengths of complexes decrease (except complex 2) with the decrease of the field strength of the halogen (I < Br < Cl).12 Compared with complexes 1 and 3, the maximum emission wavelengths of complexes 4 and 6 are red-shifted by 12 nm due to the introduction of two trimethylsilyl groups into two thiophenyl rings. The σ–π hyperconjugation effect between trimethylsilyl and thiophenyl rings, as well as slightly stronger π–π interactions between the two thiophenyl rings (see crystal discussion part), lead to a lower LUMO energy level compared to that in 1–3. Compared with the previously reported Cu(I) halide complexes [Cu(I-4)X] containing two symmetrically substituted thiophenyl triphosphine I-4 (I → Cl: λem: 550, 559 and 507 nm; PLQY: 0.09, 0.04, 0.27),20 the emission wavelengths of 1–3 are red-shifted by 3–70 nm, and the PLQYs of 1 and 2 increase by 3.2–4.8 times. The Commission Internationale de L'Eclairage (CIE) color coordinates of complexes 1–6 at 297 K are (0.42, 0.53), (0.47, 0.49), (0.47, 0.50), (0.44, 0.52), (0.48, 0.50), and (0.50, 0.49), respectively (Fig. 8).
 |
| | Fig. 7 Normalized emission spectra of complexes 1–6 in powder with λex = 375 nm at (a) 297 K and (b) 77 K. | |
 |
| | Fig. 8 CIE graph of complexes 1–6 at 297 K. | |
Table 3 Photophysical data of complexes 1–6 in the powder form
| |
λ
max
(nm) |
τ (μs)b |
Φ
297 K |
k
r
(104 s−1) |
k
nr
(104 s−1) |
E(S1)e (eV) |
E(T1)e (eV) |
ΔE(S1 − T1)e (eV) |
λ
(nm) |
| 297 K |
77 K |
297 K |
77 K |
297 K |
|
Emission peak wavelength.
Decay lifetime.
Absolute emission photoluminescence quantum yield.
Radiative decay rate constant. kr = Φ/τ.
E(S1) and E(T1) are the adiabatic excitation energies, ΔE(S1 − T1) = E(S1) − E(T1).
Calculated adiabatic emission wavelengths according to the optimized S1 and S0 geometries.
|
|
1
|
553 |
544 |
8.0 |
155 |
0.29 |
3.6 |
8.9 |
2.152 |
2.088 |
0.064 |
576 |
|
2
|
581 |
580 |
3.8 |
933 |
0.19 |
5.0 |
21.3 |
2.196 |
2.118 |
0.078 |
565 |
|
3
|
577 |
555 |
9.4 |
1130 |
0.27 |
2.9 |
7.8 |
2.197 |
2.123 |
0.074 |
564 |
|
4
|
565 |
554 |
2.2 |
67.6 |
0.61 |
27.7 |
17.7 |
2.136 |
2.057 |
0.079 |
580 |
|
5
|
578 |
573 |
6.4 |
720 |
0.38 |
5.9 |
9.7 |
2.183 |
2.084 |
0.099 |
568 |
|
6
|
589 |
584 |
7.6 |
1550 |
0.36 |
4.7 |
8.4 |
2.185 |
2.089 |
0.096 |
568 |
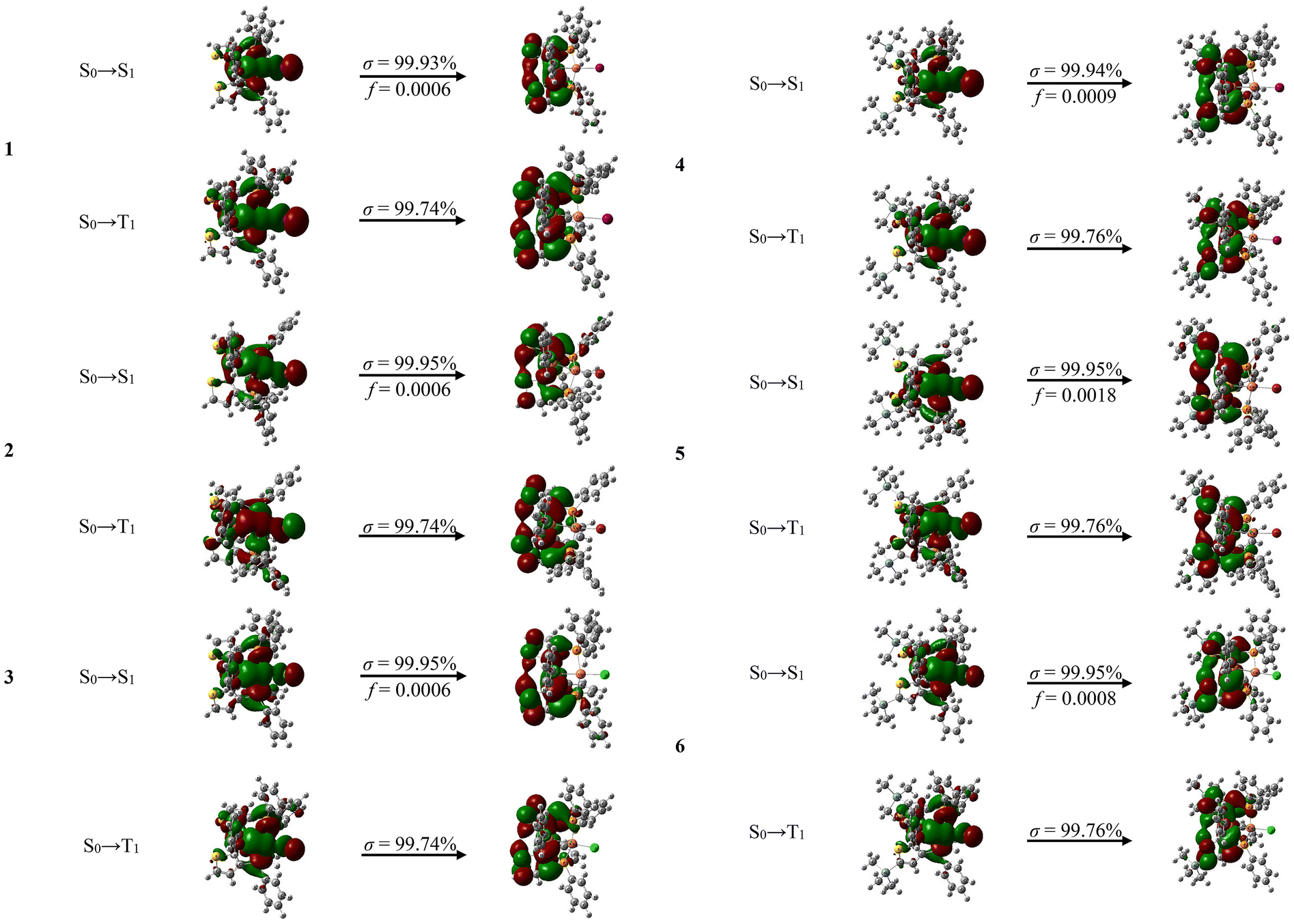
Based on the TD-DFT calculations, the simulated emission wavelengths of complexes 1–6 are close to the experimental values (Table 3), showing the validity of our calculation method. Natural transition orbital (NTO) analysis was performed to investigate the singlet transition characteristics of complexes 1–6 (Fig. 9 and Table 4). For the S0 → S1 excitation, the “hole” is mainly localized on Cu, X and P of triphosphine ligand L1 or L2, and the “electron” is mostly confined on two thiophenyl rings of L1 or L2. Therefore, the emissions of complexes 1–6 mainly originate from MLCT, XLCT and ILCT. Compositions of the frontier natural transition orbitals at the optimized S1 geometry of complexes 1–6 are shown in Fig. S43–S48,† among them, the highest contribution ratio of MLCT and XLCT results in the shortest decay lifetime (2.2 μs) of complex 4, which is the record for the shortest decay lifetime among the reported copper halide complexes containing the triphosphine ligand.14,20
 |
| | Fig. 9 NTO patterns of both S0 → S1 and S0 → T1 excitations based on the optimized S1 and T1 geometries for complexes 1–6. σ and f refer to the associated weight and oscillator strength, respectively. | |
Table 4 NTO results for complexes 1–6 based on the optimized S1 and T1 geometries
| Complex |
NTOa |
Contribution ratios of metal, halogen, and the triphosphine ligand to NTOs (%) |
| S1 |
T1 |
| Cu |
X |
L1 or L2 |
Cu |
X |
L1 or L2 |
|
H and E imply NTO hole and electron orbital, respectively.
|
|
1
|
H |
27.58 |
27.51 |
44.91 |
26.68 |
22.43 |
50.89 |
| E |
0.66 |
0.11 |
99.23 |
1.00 |
0.23 |
98.77 |
|
2
|
H |
31.07 |
18.47 |
50.46 |
29.52 |
16.46 |
54.02 |
| E |
0.53 |
0.03 |
99.44 |
1.02 |
0.20 |
98.78 |
|
3
|
H |
33.36 |
14.47 |
52.17 |
31.43 |
12.96 |
55.61 |
| E |
0.51 |
0.03 |
99.46 |
1.04 |
0.17 |
98.79 |
|
4
|
H |
27.36 |
29.25 |
43.39 |
26.36 |
23.47 |
50.17 |
| E |
0.76 |
0.16 |
99.08 |
1.06 |
0.26 |
98.68 |
|
5
|
H |
30.61 |
19.36 |
50.03 |
29.43 |
17.08 |
53.49 |
| E |
0.71 |
0.10 |
99.19 |
1.08 |
0.23 |
98.69 |
|
6
|
H |
34.22 |
15.66 |
50.12 |
31.37 |
13.30 |
55.33 |
| E |
0.69 |
0.09 |
99.22 |
1.10 |
0.19 |
98.71 |
At 77 K, the emission maxima of complexes 1–6 are 544, 580, 555, 554, 573 and 584 nm, respectively, which are blue-shifted than those at 297 K due to the suppression of the energy relaxation of the excited state by vibrations and rotations.31 The decay lifetimes of 1–6 at 77 K (155, 933, 1130, 67.6, 720 and 1550 μs, Table 3 and Fig. S36†) are 19–246 times longer than those at 297 K, respectively, indicating that complexes 1–6 could emit thermally activated delayed fluorescence (TADF). Furthermore, the ΔE(S1 − T1) values calculated by TD-DFT for complexes 1–6 are very low (0.064–0.099 eV, Table 3), which are smaller than those previously reported for Cu(I) halide complexes containing two symmetric thiophenyl triphosphine,20 exhibiting efficient TADF. The radiative rate constants kr of complexes 1–6 were calculated to be 2.9 × 104 to 2.8 × 105 s−1, much larger than those previously reported for Cu(I) halide triphosphine complexes containing two symmetrically substituted thiophenyl rings (5.2 × 103 to 2.3 × 104 s−1).20 Especially, the kr value of complex 4 is as high as 2.8 × 105 s−1, which is 3.7 times higher than that previously reported for complex CuI(TTPP) (kr = 7.5 × 104 s−1),14 and comparable to that of phosphorescent Ir(III) metal complexes. NTO analysis displays that for the S0 → T1 excitation, the “hole” is mainly focused on Cu, X and P of the triphosphine ligand L1 or L2, and the “electron” is primarily concentrated on the two thiophenyl rings of L1 or L2. Compositions of the frontier natural transition orbitals at optimized T1 geometry of complexes 1–6 are shown in Fig. S49–S54,† the lower contribution ratio of MLCT and XLCT than that at optimized S1 geometry, results in the much longer decay lifetimes of complexes 1–6 at 77 K relative to those at 297 K.
To prove the existence of TADF in the complexes, the temperature-dependent emission spectra of complexes 1–6 were measured at 77–297 K in the powder state (Fig. 10). As the temperature decreases, the luminescence intensity of the complexes increases, indicating that the decrease in temperature reduces the non-radiative decay process. The relationship between different temperatures (77 K–297 K) and decay times (τobs) of complexes 1–6 were also studied. The decay time (τobs) is described by a Boltzmann-type relation1 as follows:
| |  | (1) |
 |
| | Fig. 10 Temperature-dependent emission spectra of complexes 1–6 at 77–297 K in the powder state. | |
Based on eqn (1), a fitted curve is presented in Fig. 11 (KB: Boltzmann constant; T: absolute temperature; τ(S1): lifetime of S1; τ(T1): lifetime of T1; ΔEST: the energy separation between S1 and T1). The fitted results show that the τ(S1) and τ(T1) values of complexes 1–6 are in the ranges of 0.1117–0.7827 and 68.3–1590 μs, respectively, and the ΔEST values are 0.048–0.076 eV. The fitted τ(T1) values are close to the experimental decay times at 77 K. The fitted ΔEST values (0.048–0.076 eV) are very small, close to the calculated values (0.064–0.099 eV). The experimental decay times of complexes 1–6 at 297 K are 2.2–9.4 μs, indicating that they represent a delayed fluorescence. At 77 K, complexes 1–6 mainly emit phosphorescence. As the temperature increases, the phosphorescence decreases and TADF increases.
 |
| | Fig. 11 Experimental and fitted decay times of complexes 1–6 at different temperatures. The values of the fitted results τ(S1), τ(T1) and ΔEST are indicated. | |
Fig. 12 shows the geometries of complexes 1–6 with Cu(I) as the core under optimized S0, S1, and T1. The small change in the bond angles of P1–Cu–P2 and P1–Cu–P3 at S0, S1 and T1 geometries indicates the rigidity of the ligands L1 and L2, while significant changes in bond angles of P2–Cu–X and P3–Cu–X were found. The changes in the sum of the angles around the Cu(I) center in S0 and S1 geometries in 1–6 are 37.84° for 1, 41.70° for 2, 40.09° for 3, 36.85° for 4, 35.35° for 5 and 36.88° for 6. The change in the bond angles of complexes 1–3 is greater than those in complexes 4–6, showing a more compact configuration around the Cu(I) center in 4–6 due to the steric hindrance caused by two trimethylsilyl groups, which is also consistent with the result in the section on crystal discussion. A more compact configuration of 4–6 results in smaller Jahn–Teller distortion of the excited states and higher PLQYs relative to 1–3. In addition, the PLQY of complex 4 is the highest, which is also related to the lowest contribution ratio of MLCT (Table 4).
 |
| | Fig. 12 The core structures in the optimized S0, S1, and T1 geometries for complexes 1–6. | |
3.3. Thermal properties
Thermal properties of complexes 1–6 in powder were investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) under a stream of nitrogen (Fig. 13). From the onset of the TGA curves, complexes 1, 2 and 4 show good thermal stability with their Tdec values of 443, 435 and 364 °C, respectively, the small weight loss of ca. −4% below 177 °C is ascribed to the removal of solvent molecules. At 153 °C, complex 3 shows weight loss of ca. 9%, which is ascribed to the removal of solvent molecules and halogen. From 200 to 368 °C, complex 5 gradually removes all halogen with weight loss of ca. 9%. For complex 6, the small weight loss of ca. −4% below 160 °C is ascribed to the removal of solvent molecules. Starting from 253 °C, complex 6 gradually loses halogen until the weight loss reaches ca. 9% at 327 °C, corresponding to the loss of all the halogen. A large weight loss of ca. 61%−73% for complexes 1–6 was observed, which is due to the removal of the organic and/or halide ligands. The DSC curves of complexes 1–6 show that the Tm (melting point) values are 315 °C for complex 1, 291 °C for complex 2 and 295 °C for complex 4. For complexes 3, 5 and 6, the Tm values are 292 °C, 265 °C and 283 °C, respectively, accompanied by partial decomposition, especially for complex 6, where evident weight loss can be observed.
 |
| | Fig. 13 TGA and DSC (inset) curves of complexes 1–6. | |
3.4 Electroluminescent properties
Due to the highest quantum efficiency among complexes 1–6 and good thermal stability with the onset decomposition temperature (Tdec) 364 °C, complex 4 was selected to construct OLED devices by vacuum deposition with the structure of indium tin oxide (ITO)/HAT-CN (5 nm)/TAPC (50 nm)/TCTA (10 nm)/TCTA:DPEPO:x nm complex 4 (10%)/DPEPO (10 nm)/TPBi (40 nm)/LiF (1 nm)/Al (100 nm). In these devices, HAT-CN, TAPC, TPBi and LiF were used for hole injection, hole transport, electron transport and electron injection, respectively. TCTA was used as the hole buffer and host material, and DPEPO was used as exciton blocking and the host material. TCTA:DPEPO = w/w 1:1, and complex 4 was doped with 10 wt% in the emissive layer (EML) with thicknesses of 20, 25, 30 and 35 nm, respectively (Fig. 14).
 |
| | Fig. 14 Device configuration and molecular structures of the materials for the OLEDs. | |
The EL characteristics and the key data of OLEDs are shown in Fig. 15 and Table 5, respectively. The excitons are recombined and confined in the emitting layer to exhibit yellow light with a peak wavelength of 584 nm for complex 4, which is red-shifted compared to the photoluminescence maximum emission wavelength of 565 nm for complex 4. A similar EL bathochromic shift was also observed for other copper(I) halide complexes and attributed to the microcavity effect.12 No emission from TCTA and DPEPO was observed, implying that the emission peak is indeed derived from complex 4 and good energy transfer occurred between the host material and complex 4. When the thickness of EML increases from 20 nm to 25 nm, the maximum EQE value increases from 10.93% to 14.57%, indicating that the thicker EML can expand the exciton recombination region, reduce exciton density, and effectively alleviate exciton annihilation. However, when the thickness of EML increases to 30 and 35 nm, the maximum EQE value decreases to 11.76% and 11.78%, respectively, indicating that as the thickness of EML increases, the migration distance of charge carriers relatively increases, which increases the transport resistance of charge carriers and leads to a decrease in device efficiency.
 |
| | Fig. 15 EL characteristics of OLEDs based on complex 4: (a) EL spectra, (b) J–V–L properties, (c) curves of EQE vs. luminance, and (d) curves of CE and PE vs. luminance. | |
Table 5 EL data for the vacuum deposition devices
| Device |
Emitter (thickness of EML) |
λ
em (nm) |
CIE 1931 |
V
(V) |
L
max (cd m−2) |
EQEb (%) |
CEb (cd A−1) |
PEb (lm W−1) |
|
Driving voltages (V) in the order of at luminance of 1, and 100 cd m−2.
EQE, CE, and PE in the order of the maximum value and at 100 cd m−2.
|
| A1 |
4 (20 nm) |
584 |
(0.46, 0.49) |
4.0/5.0 |
3913 |
10.93/10.82 |
24.55/24.32 |
17.14/15.25 |
| A2 |
4 (25 nm) |
584 |
(0.46, 0.50) |
4.0/5.7 |
844.9 |
14.57/14.11 |
33.44/32.41 |
21.63/17.85 |
| A3 |
4 (30 nm) |
584 |
(0.46, 0.50) |
4.0/6.1 |
3036 |
11.76/10.87 |
27.12/25.23 |
20.62/13.43 |
| A4 |
4 (35 nm) |
584 |
(0.45, 0.50) |
4.0/5.6 |
3475 |
11.78/11.57 |
27.50/27.05 |
19.20/15.20 |
Device A2 (the concentration of 4 in the EML is 10% and the thickness of EML is 25 nm) afforded the maximum EQE of 14.57% (operation voltage = 6 V). The maximum luminance obtained was 844.9 cd m−2 at 8.38 mA cm−2, and the maximum current efficiency and power efficiency were 33.44 cd A−1 and 21.63 lm W−1, respectively. The EQE, CE and PE were maintained as high as 14.11%, 32.41 cd A−1, and 17.85 lm W−1 with very low−efficiency roll-off at a luminance of 100 cd m−2, which suggests that the holes and electrons can be completely confined in the EML and current-induced exciton quenching is also effectively suppressed. As far as we know, this is one of the best performances for the OLEDs fabricated using mononuclear copper halide complexes as emitters (Table 6).13,14,31
Table 6 Reported performances of vacuum-deposited devices based on mononuclear Cu(I) halide complexes
| The structure of Cu(I) complexes |
k
r (105 s−1) |
λ
EL (nm) |
EQE (%) |
Ref. |

|
R = Me, X = Cl |
0.82 |
527 |
21.1 |
13 and 31 |
| R = Me, X = Br |
0.69 |
517 |
21.3 |
| R = Me, X = I |
0.83 |
513 |
21.2 |
| R = Et, X = Br |
0.12 |
529 |
22.5 |
| R = iPr, X = Br |
1.07 |
515 |
18.6 |

|
X = I |
0.75 |
584 |
16.3 |
14
|
| X = Br |
0.49 |
584 |
12.4 |
| X = Cl |
0.40 |
584 |
9.6 |

|
X = I |
1.20 |
521 |
16.4 |
32
|

|
X = I |
0.46 |
613 |
3.4 |
33
|
|
X = I |
0.84 |
645 |
2.68 |
34
|

|
X = I |
2.77 |
584 |
14.57 |
This work |
4 Conclusions
In this work, two rigid triphosphine ligands with unsymmetrically substituted thiophenyl were synthesized, and single crystals of six corresponding mononuclear four-coordinate copper(I) halide complexes were successfully obtained. Two trimethylsilyl groups displayed σ–π hyperconjugation effect with thiophenyl rings, which lowers the LUMO level and results in the red-shifted absorption and emission of complexes 4–6 in comparison with complexes 1–3. On the other hand, the large steric hindrance of the trimethylsilyl group makes the structure of the complexes more compact, thereby reducing the geometric distortion of the copper center and suppressing the Jahn–Teller effect, which improves PLQYs of complexes 4–6. These complexes exhibit thermally activated delayed fluorescence at room temperature due to the very small ΔE(S1 − T1) values <0.1 eV. The emissions mainly originate from MLCT, XLCT and ILCT. Among them, complex 4 in the powder form displayed the highest PLQY (0.61) due to the compact geometry and the lowest ratio of MLCT contribution. NTO analysis shows that the highest contribution ratio of MLCT and XLCT results in the shortest decay lifetime (2.2 μs) of complex 4, which is the record for the shortest decay lifetime among the reported copper halide complexes containing the triphosphine ligand. High PLQY and short decay lifetime of complex 4 results in the high kr of 2.8 × 105 s−1, which is comparable to that of phosphorescent Ir(III) metal complexes. Complex 4-based vacuum-deposited devices exhibited yellow light with an EQE of 14.57%, which is one of the best performances of the fabricated OLEDs using mononuclear copper halide complexes as emitters. Our work indicates that mononuclear Cu(I) halide complexes containing unsymmetrically substituted heteroaryl triphosphine have the advantages of red-shifted emission and elevated PLQY compared to symmetrically substituted heteroaryl triphosphine. By introducing functional groups such as trimethylsilyl groups into unsymmetrically substituted thiophenyl rings, the PLQYs of the complexes are effectively improved and the decay lifetimes are shortened. High PLQYs and short decay lifetimes provide the Cu(I) halide complexes with rigid triphosphine great potential as abundant and inexpensive luminescent materials for application in TADF-OLEDs.
Data availability
All the data have been included in the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Li Liu thanks the financial support from the National Natural Science Foundation of China (21671061) and the application foundation frontier special project from Wuhan Science and Technology Bureau (2019010701011414). Thanks to senior engineer Mingxing Chen (Peking University) for the photophysical measurements.
References
- M. J. Leitl, D. M. Zink, A. Schinabeck, T. Baumann, D. Volz and H. Yersin, Copper(I) Complexes for Thermally Activated Delayed Fluorescence: From Photophysical to Device Properties, Top. Curr. Chem., 2016, 374, 25 CrossRef PubMed
 .
.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Thermally Activated Delayed Fluorescence Materials Towards the Breakthrough of Organoelectronics, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS .
- M. Wallesch, D. Volz, D. M. Zink, U. Schepers, M. Nieger, T. Baumann and S. Bräse, Bright Coppertunities: Multinuclear CuI Complexes with N–P Ligands and Their Applications, Chem. – Eur. J., 2014, 20, 6578–6590 CrossRef CAS PubMed .
- D. Volz, M. Wallesch, C. Fléchon, M. Danz, A. Verma, J. M. Navarro, D. M. Zink, S. Bräse and T. Baumann, From Iridium and Platinum to Copper and Carbon: New Avenues for More Sustainability in Organic Light-emitting Diodes, Green Chem., 2015, 17, 1988–2011 RSC .
- F. Dumur, Recent Advances in Organic Light-Emitting Devices Comprising Copper Complexes: A Realistic Approach for Low-Cost and Highly Emissive Devices?, Org. Electron., 2015, 21, 27–39 CrossRef CAS .
- X. Li, Y. Xie and Z. Li, Diversity of Luminescent Metal Complexes in OLEDs: Beyond Traditional Precious Metals, Chem. – Asian J., 2021, 16, 2817–2829 CrossRef CAS .
- Q. Zhang, T. Komino, S. Huang, S. Matsunami, K. Goushi and C. Adachi, Triplet Exciton Confinement in Green Organic Light Emitting Diodes Containing Luminescent Charge-Transfer Cu(I) Complexes, Adv. Funct. Mater., 2012, 22, 2327–2336 CrossRef CAS .
- B. Huitorel, H. E. Moll, R. Utrera-Melero, M. Cordier, A. Fargues, A. Garcia, F. Massuyeau, C. Martineau-Corcos, F. Fayon, A. Rakhmatullin, S. Kahlal, J. Y. Saillard, T. Gacoin and S. Perruchas, Evaluation of Ligands Effect on the Photophysical Properties of Copper Iodide Clusters, Inorg. Chem., 2018, 57, 4328–4339 CrossRef CAS PubMed .
- S. Evariste, A. M. Khalil, M. E. Moussa, A. K. W. Chan, E. Y. H. Hong, H. L. Wong, B. L. Guennic, G. Calvez, K. Costuas, V. W. W. Yam and C. Lescop, Adaptive Coordination-Driven Supramolecular Syntheses Toward New Polymetallic Cu(I) Luminescent Assemblies, J. Am. Chem. Soc., 2018, 140, 12521–12526 CrossRef CAS PubMed .
- M. E. S. Moussa, A. M. Khalil, S. Evariste, H. L. Wong, V. Delmas, B. L. Guennic, G. Calvez, K. Costuas, V. W. W. Yam and C. Lescop, Intramolecular Rearrangements Guided by Adaptive Coordination-Driven Reactions Toward Highly Luminescent Polynuclear Cu(I) Assemblies, Inorg. Chem. Front., 2020, 7, 1334–1344 RSC .
-
Highly Efficient OLEDs-Materials Based on Thermally Activated Delayed Fluorescence, ed. H. Yersin, Wiley-VCH, Weinheim, Germany, 2019 Search PubMed .
- A. Tsuboyama, K. Kuge, M. Furugori, S. Okada, M. Hoshino and K. Ueno, Photophysical Properties of Highly Luminescent Copper(I) Halide Complexes Chelated with 1,2-Bis(diphenylphosphino)benzene, Inorg. Chem., 2007, 46, 1992–2001 CrossRef CAS PubMed .
- M. Hashimoto, S. Igawa, M. Yashima, I. Kawata, M. Hoshino and M. Osawa, Highly Efficient Green Organic Light-Emitting Diodes Containing Luminescent Three-Coordinate Copper(I) Complexes, J. Am. Chem. Soc., 2011, 133, 10348–10351 CrossRef CAS PubMed .
- J. Zhang, C. Duan, C. Han, H. Yang, Y. Wei and H. Xu, Balanced Dual Emissions from Tridentate Phosphine-Coordinate Copper(I) Complexes
Toward Highly Efficient Yellow OLEDs, Adv. Mater., 2016, 28, 5975–5979 CrossRef CAS .
- F. Liu, Z. C. Zhou, C. Zhang, T. Vergote, H. J. Fan, F. Liu and X. Z. Zhu, A Thieno[3,4-b]thiophene-Based Non-fullerene Electron Acceptor for High-Performance Bulk-Heterojunction Organic Solar Cells, J. Am. Chem. Soc., 2016, 138, 15523–15526 CrossRef CAS PubMed .
- Q. Wei, H. T. Chen, L. Liu, X. X. Zhong, L. Wang, F. B. Li, H. J. Cong, W. Y. Wong, K. A. Alamry and H. M. Qin, Syntheses and Photoluminescence of Copper(I) Halide Complexes Containing Dimethylthiophene Bidentate Phosphine Ligands, New J. Chem., 2019, 43, 13408–13417 RSC .
- X. Li, J. Zhang, Z. Zhao, X. Yu, P. Li, Y. Yao, Z. Liu, Q. Jin, Z. Bian, Z. Lu and C. Huang, Bluish-Green Cu(I) Dimers Chelated with Thiophene Ring-Introduced Diphosphine Ligands for Both Singlet and Triplet Harvesting in OLEDs, ACS Appl. Mater. Interfaces, 2019, 11, 3262–3270 CrossRef CAS .
- Q. Wei, R. Zhang, L. Liu, X. X. Zhong, L. Wang, G. H. Li, F. B. Li, K. A. Alamry and Y. Zhao, From Deep Blue to Green Emitting and Ultralong Fluorescent Copper(I) Halide Complexes Containing Dimethylthiophene Diphosphine and PPh3 Ligands, Dalton Trans., 2019, 48, 11448–11459 RSC .
- B. K. Guo, F. Yang, Y. Q. Wang, Q. Wei, L. Liu, X. X. Zhong, L. Wang, J. K. Gong, F. B. Li, W. Y. Wong, K. A. Alamry and Y. Zhao, Efficient TADF-OLEDs with Ultra-Soluble Copper(I) Halide Complexes Containing Non-Symmetrically Substituted Bidentate Phosphine and PPh3 Ligands, J. Lumin., 2020, 220, 116963 CrossRef CAS .
- Q. Wei, F. F. Gong, R. Zhang, L. Liu, X. X. Zhong, L. Wang, F. B. Li, W. Y. Wong and H. M. Qin, Mononuclear Cu(I) Halide Complexes with Two Thiophenyl Rings Triphosphine: Structure and Photophysical Properties, J. Lumin., 2022, 250, 119098 CrossRef CAS .
- W. J. Zhang, Z. X. Zhou, L. Liu, X. X. Zhong, A. M. Asiri, K. A. Alamry, F. B. Li, N. Y. Zhu, W. Y. Wong and H. M. Qin, Highly Efficient Blue Neutral Mononuclear Copper(I) Halide Complexes Containing Bi- and Mono-dentate Phosphine Ligands, J. Lumin., 2018, 196, 425–430 CrossRef CAS .
- X. Hong, B. Wang, L. Liu, X. X. Zhong, F. B. Li, L. Wang, W. Y. Wong, H. M. Qin and Y. H. Lo, Highly Efficient Blue-Green Neutral Dinuclear Copper(I) Halide Complexes Containing Bidentate Phosphine Ligands, J. Lumin., 2016, 180, 64–72 CrossRef CAS .
- L. P. Liu, R. Zhang, L. Liu, X. X. Zhong, F. B. Li, L. Wang, W. Y. Wong, G. H. Li, H. J. Cong, N. S. Alharbi and Y. Zhao, A New Strategy to Synthesize Three-Coordinate Mononuclear Copper(I) Halide Complexes Containing a Bulky Terphenyl Bidentate Phosphine Ligand and Their Luminescent Properties, New J. Chem., 2019, 43, 3390–3399 RSC .
- K. Xu, B. L. Chen, F. Yang, L. Liu, X. X. Zhong, L. Wang, X. J. Zhu, F. B. Li, W. Y. Wong and H. M. Qin, Largely Color-Tuning Prompt and Delayed Fluorescence: Dinuclear Cu(I) Halide Complexes with tert-Amines and Phosphines, Inorg. Chem., 2021, 60, 4841–4851 CrossRef CAS .
- L. P. Liu, Q. Li, S. P. Xiang, L. Liu, X. X. Zhong, C. Liang, G. H. Li, T. Hayat, N. S. Alharbi, F. B. Li, N. Y. Zhu, W. Y. Wong, H. M. Qin and L. Wang, Near-Saturated Red Emitters: Four-Coordinate Copper(I) Halide Complexes Containing 8-(Diphenylphosphino)quinoline and 1-(Diphenylphosphino)naphthalene Ligand, Dalton Trans., 2018, 47, 9294–9302 RSC .
- K. Xu, B. L. Chen, R. Zhang, L. Liu, X. X. Zhong, L. Wang, F. Y. Li, G. H. Li, K. A. Alamry, F. B. Li, W. Y. Wong and H. M. Qin, From a Blue to White to Yellow Emitter: A Hexanuclear Copper Iodide Nanocluster, Dalton Trans., 2020, 49, 5859–5868 RSC .
- W. Xu, B. Chen, L. Liu, X. X. Zhong, G. J. Zhou, F. B. Li and H. M. Qin, Syntheses, Structures, Photophysical Properties and Electroluminescent Applications of Dinuclear Copper(I) Halide Complexes Containing 9-Carbazolyl-Substituted 1,2-Bis(diphenylphosphino)benzene, J. Lumin., 2024, 266, 120257 CrossRef CAS .
- C. Chen, L. Liu, X. X. Zhong, F. B. Li and H. M. Qin, Mononuclear Cu(I) Halide Complexes Containing Asymmetric Diphosphine and Diphenyl Pyridyl Monophosphine: Structures and Photophysical Properties, Inorg. Chim. Acta, 2024, 560, 121839 CrossRef CAS .
- S. Kyushin and Y. Suzuki, Cooperation of σ–π and σ*–π* Conjugation in the UV/Vis and Fluorescence Spectra of 9,10-Disilylanthracene, Molecules, 2022, 27, 2241 CrossRef CAS PubMed .
- D. M. Zink, D. Volz, T. Baumann, M. Mydak, H. Flügge, J. Friedrichs, M. Nieger and S. Bräse, Heteroleptic, Dinuclear Copper(I) Complexes for Application in Organic Light-Emitting Diodes, Chem. Mater., 2013, 25, 4471–4486 CrossRef CAS .
- M. Osawa, M. Hoshino, M. Hashimoto, I. Kawata, S. Igawa and M. Yashima, Application of Three-Coordinate Copper(I) Complexes with Halide Ligands in Organic Light-Emitting Diodes That Exhibit Delayed Fluorescence, Dalton Trans., 2015, 44, 8369–8378 RSC .
- M. Klein, N. Rau, M. Wende, J. Sundermeyer, G. Cheng, C. M. Che, A. Schinabeck and H. Yersin, Cu(I) and Ag(I) Complexes with a New Type of Rigid Tridentate N,P,P-Ligand for Thermally Activated Delayed Fluorescence and OLEDs with High External Quantum Efficiency, Chem. Mater., 2020, 32, 10365–10382 CrossRef CAS .
- B. Jiao, J. Wang, J. Huang, M. Cao, C. Liu, G. Yin, Y. Zhu, B. Zhang and C. Du, Design and Synthesis of Stable Cuprous Complexes Bearing P^N-type Ligands for Vapor-Deposited Organic Light-Emitting Device, Org. Electron., 2019, 64, 158–165 CrossRef CAS .
- Y. L. Song, B. J. Jiao, C. M. Liu, X. L. Peng, M. M. Wang, Y. Yang, B. Zhang and C. X. Du, Inorg. Chem. Commun., 2020, 112, 107689 CrossRef CAS .
Footnotes |
| † Electronic supplementary information (ESI) availableNMR and mass data, molecular structures, photophysical data, and computational details (PDF). CCDC 2282047, 2285430, 2285431–2285433, 2297624. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi02415j |
| ‡ These authors contributed equally to this work. |
|
| This journal is © the Partner Organisations 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a,
Xin-Xin
Zhong
*a,
Fa-Bao
Li
*a,
Xin-Xin
Zhong
*a,
Fa-Bao
Li