
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclometalated half-sandwich iridium(III) and rhodium(III) complexes as efficient agents against cancer stem-cell mammospheres†
Dana
Josa
ab,
Piedad
Herrera-Ramírez
a,
Xiao
Feng
c,
Albert
Gutiérrez
 ab,
David
Aguilà
ab,
Arnald
Grabulosa
*ab,
Manuel
Martínez
a,
Kogularamanan
Suntharalingam
*c and
Patrick
Gamez
*ade
ab,
David
Aguilà
ab,
Arnald
Grabulosa
*ab,
Manuel
Martínez
a,
Kogularamanan
Suntharalingam
*c and
Patrick
Gamez
*ade
aDepartament de Química Inorgànica i Orgànica, Secció Química Inorgànica, Facultat de Química, Universitat de Barcelona, Martí i Franquès 1-11, E-08028 Barcelona, Spain. E-mail: arnald.grabulosa@ub.edu; patrick.gamez@ub.edu
bInstitute of Nanoscience and Nanotechnology (IN2UB), Universitat de Barcelona, 08028 Barcelona, Spain
cSchool of Chemistry, University of Leicester, Leicester, UK. E-mail: k.suntharalingam@leicester.ac.uk
dInstitute of Biomedicine of the University of Barcelona (IBUB), Avda. Diagonal, 643, Edifici Prevosti, planta-1, Barcelona, Spain
eCatalan Institution for Research and Advanced Studies (ICREA), Passeig Lluís Companys 23, 08010 Barcelona, Spain
First published on 28th January 2025
Abstract
Four cyclometalated complexes, namely [IrCl(η5-pentamethylcyclopendadienyl)(k2C-diphenyl(1-pyrenyl)phosphane)] (1) and [RhCl(η5-pentamethylcyclopendadienyl)(k2C-diphenyl(1-pyrenyl)phosphane)] (2), and their DMSO-coordinated counterparts [Ir(η5-pentamethylcyclopendadienyl)(kS-dmso)(k2C-diphenyl(1-pyrenyl)phosphane)]PF6 (1·DMSO) and [Rh(η5-pentamethylcyclopendadienyl)(kS-dmso)(k2C-diphenyl(1-pyrenyl)phosphane)]PF6 (2·DMSO), were synthesized and fully characterized, including their single-crystal X-ray structures. DNA-interacting 1 and 2 exhibits IC50 values in the range 0.53–0.79 μM against bulk breast cancer cells and breast cancer stem cells (CSCs), i.e., HMLER and HMLER-shEcad cells. The complexes are up to seven times more active than salinomycin and up to nine times more active than cisplatin. Moreover, 1 and 2 are very effective (in the micromolar range) against mammospheres obtained from single cell suspensions of HMLER-shEcad cells, 1 being thrice more toxic than 2 and up to 4.5-fold more potent than cisplatin and salinomycin. In depth mechanistic studies revealed that 1 induces necrosis, which is potentially dependent on necrosome formation and independent of ROS concentration. The efficacy of 1 against breast CSCs can be enhanced by co-treatment with PARP-1 inhibitors.
10th anniversary statementI had the honour of writing the first ‘review’ for the journal, the theme of which was ‘anion–π interactions’. This ‘review’ was very important to me because this type of supramolecular contact was not fully accepted by the scientific community at the time. Inorganic Chemistry Frontiers agreed to publish my discussion on this ‘new non-bonding contact’, which is now considered one of the possible π interactions. |
Introduction
The global cancer burden has been growing over the past few decades, and the World Health Organization (WHO) predicts over 35 million new cancer cases in 2050, which represents a 77% increase compared to incidences in 2022.1 Among the possible cancer treatments, chemotherapy is the second most commonly used option.2 Cisplatin is a chemotherapeutic drug that is widely used against solid tumours of various cancer types.3 Since the discovery of the anticancer properties of cisplatin,4,5 metal-containing drugs have received a great deal of attention from the medicinal inorganic chemistry community.6 Thus, numerous cytotoxic coordination compounds have been designed,7–10 as possible alternatives to clinically used metal-based drugs that may exhibit undesirable side effects or/and are prone to resistance.11 In this context, a number of ruthenium complexes with promising cytotoxic properties have been reported,12–14 including half-sandwich ruthenium(II)–arene complexes,15 such as RAPTA-C (i.e., [RuCl2(η6-p-cymene)(pta)] where pta = 1,3,5-triaza-7-phosphaadamantane),16 [RuCl(η6-fluorene)(ethylenediamine)]PF6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 17 and [RuCl(η6-5,6-dihydrophenanthrene)(ethylenediamine)]PF6.18
17 and [RuCl(η6-5,6-dihydrophenanthrene)(ethylenediamine)]PF6.18
Over the last few years, we have developed a series of piano-stool Ru(II) and Os(II) complexes of formula [MX2(η6-arene)(P(1-pyrenyl)R2R3)] (M = Ru or Os; η6-arene = p-cymene or methyl benzoate; R2 and R3 = alkyl or aryl groups), which are based on bulky monophosphane ligands (viz., containing a 1-pyrenyl moiety).19 Most of these organometallic compounds exhibited remarkable cytotoxic properties towards various cancer cell lines grown in monolayers, with IC50 values in the submicromolar range.20–22 Interestingly, the corresponding cyclometalated complexes of the type [MCl(η6-arene)(k2C-P(1-pyrenyl)R2R3)]PF6 (M = Ru or Os) were more potent than the parent non-cyclometalated compounds.20,21
In the present study, the sterically hindered ligand diphenyl(1-pyrenyl)phosphane (L), previously used to prepare Ru(II) and Os(II) complexes, was employed to generate cyclometalated Ir(III) and Rh(III) complexes of formulae [MClCp*(k2C-L)] and [MCp*(kS-dmso)(k2C-L)]PF6 (with M = Ir or Rh; Cp* = pentamethylcyclopentadienyl), to add two additional members of the “platinum metals” to our family of complexes, and evaluate the metal influence on the biological properties. It can be stressed here that some cyclometalated half-sandwich iridium(III) and rhodium(III) complexes with interesting cytotoxic behaviours have been reported in the literature.23–26
The interaction of compounds [IrClCp*(k2C-L)] (1) and [RhClCp*(k2C-L)] (2) with DNA was investigated and solution studies in water and DMSO were carried out. As 1 and 2 are very slowly converted in their DMSO solvato complexes [1·DMSO]+ and [2·DMSO]+, respectively, at room temperature in DMSO, biological studies were conducted with freshly prepared DMSO solutions of 1 and 2. The cytotoxic effects of 1 and 2 towards bulk breast cancer cells and breast cancer stem cells (i.e., CSCs, which clearly play an important role in breast tumour progression, drug resistance and metastasis) was investigated and their ability to inhibit the formation and viability of three-dimensionally cultured mammospheres (produced from breast CSCs) was subsequently examined. Finally, the mode of cell death, i.e. apoptosis or necrosis, induced by 1 and 2 was studied in detail, for instance using specific inhibitors of biomolecules involved in the two different pathways.
Results and discussion
Synthesis of the organometallic complexes
The synthesis of the ligand diphenyl(1-pyrenyl)phosphane (L) was reported earlier.22,27Cyclometalated complexes 1 and 2 were typically prepared by reaction of the corresponding pentamethylcyclopentadienyl metal dichloride dimer ([Cp*MCl2]2; M = Ir or Rh; Cp* = pentamethylcyclopentadienyl) with two equivalents of ligand L (Scheme 1, top) and four equivalents of sodium acetate, in methanol at room temperature for two hours. The cationic complexes containing an S-coordinated DMSO molecule, i.e.1·DMSO and 2·DMSO, were also prepared by reaction of respectively 1 and 2 with 10 equivalents of DMSO and 1.1 equivalents of thallium hexafluorophosphate in dichloromethane overnight at room temperature (Scheme 1, bottom).
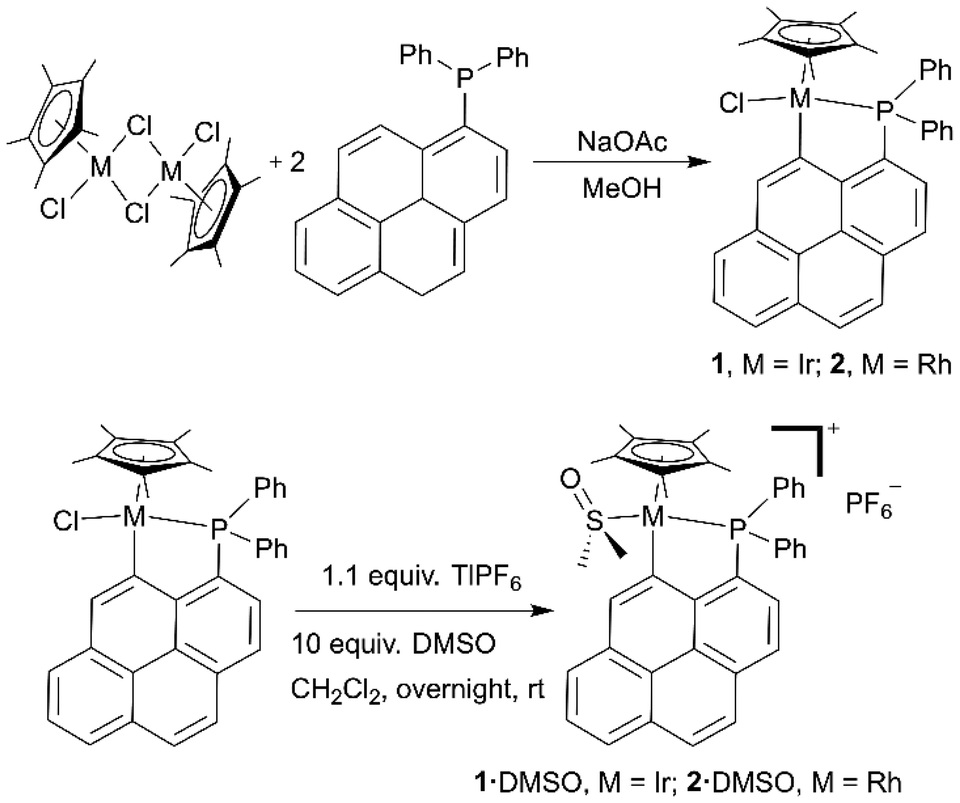 | ||
| Scheme 1 Synthetic pathways for the preparation of complexes 1 and 2 and their DMSO derivatives 1·DMSO and 2·DMSO. | ||
Crystal structures
Single crystals of 1 and 2, suitable for X-ray diffraction analysis, were obtained by layering n-hexane onto concentrated solutions of the complexes in dichloromethane. Rhodium(III) complex 2 was already described in the literature;27 hence, its crystal structure was not determined. Crystal data and structure refinement parameters for 1 are given in Table S1.† Selected bond distances and coordination angles for this iridium(III) compound are listed in Table S2.†1 crystallizes in the monoclinic space group P21/c. The solid-state structure of 1 is represented in Fig. 1.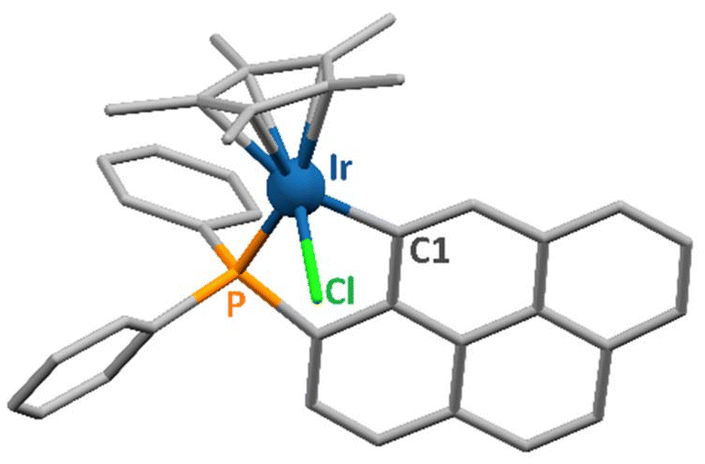 | ||
| Fig. 1 Representation of the solid-state structure of 1. The donor atoms coordinated to the metal centre are labelled. Hydrogen atoms are omitted for clarity. | ||
Complex 1 exhibits the typical coordination geometry for such organometallic compounds.28–30 The Ir–Cl, Ir–P and Ir–C bond lengths of respectively 2.407(1), 2.259(1) and 2.070(2) Å are analogous to reported values for similar complexes.31,32 The distance between the iridium atom and the centroid of the Cp* ring amounts to 1.873(1) Å, which is also in the range observed for such molecules. The coordination angles varying from 81.60(7) to 89.86(3)° for the “three legs” of the piano-stool complex and from 122.56(5) to 132.84(5)° for those involving the Cp* centroid (Table S2†) are comparable to reported values for related iridium(III) complexes.30,32
Rhodium(III) complex 2 (Fig. S1†) shows comparable coordination features, as evidenced by the published crystallographic data for this compound.27
As aforementioned, substitution of the chlorido ligand by a DMSO molecule generates complexes 1·DMSO and 2·DMSO. Single crystals of these two DMSO-containing compounds were obtained in dichloromethane/n-hexane. The crystal data and structure refinement parameters for the Ir(III) and Rh(III) complexes are listed in Tables S1 and S3,† respectively. Selected bond lengths and angles are provided in Table S2.† Representations of their X-ray structures are depicted in Fig. S2.† Replacement of the chlorides by DMSO molecules does not significantly affect the structures of the complexes. The coordination bond lengths and angles are comparable with those of the original chlorido complexes, except for the P–M–S angles which are larger than the P–M–Cl ones (viz., 97 vs. 90°; see Table S2†). These higher angle values are most likely due to steric repulsion between the methyl groups of the DMSO and the phenyls of the phosphane ligand.
Solution behaviour of the complexes in different (relevant) media
The solution behaviour of DMSO stock solutions of the complexes, as well as their stability upon aqueous dilution (final complex concentration of 5 μM), were first investigated. These studies were aimed at evaluating the stability of 1 and 2 under the cell culture conditions.Thus, DMSO solutions of 1 and 2 in the range 3–6 × 10−5 M were monitored by time-resolved UV-Vis spectroscopy at different temperatures and pressures. The observed spectral changes agreed with a first-order reaction, as expected for a solvolysis process. The Reactlab and Specfit software33,34 were used to determine the rate constants for the two complexes. As an example, Fig. 2a illustrates the spectral changes observed for iridium complex 1 at 70 °C for 21 hours. The Eyring plots obtained for the variation of the values of k with the temperature for 1 and 2 are depicted in Fig. 2b. Finally, Fig. 2c shows the lnk versus P plot for the variation of the rate constant for 1. The corresponding values for the kinetic and activation parameters for the DMSO solvolysis of complexes 1 and 2, determined from these spectroscopic data, are listed in Table 1.
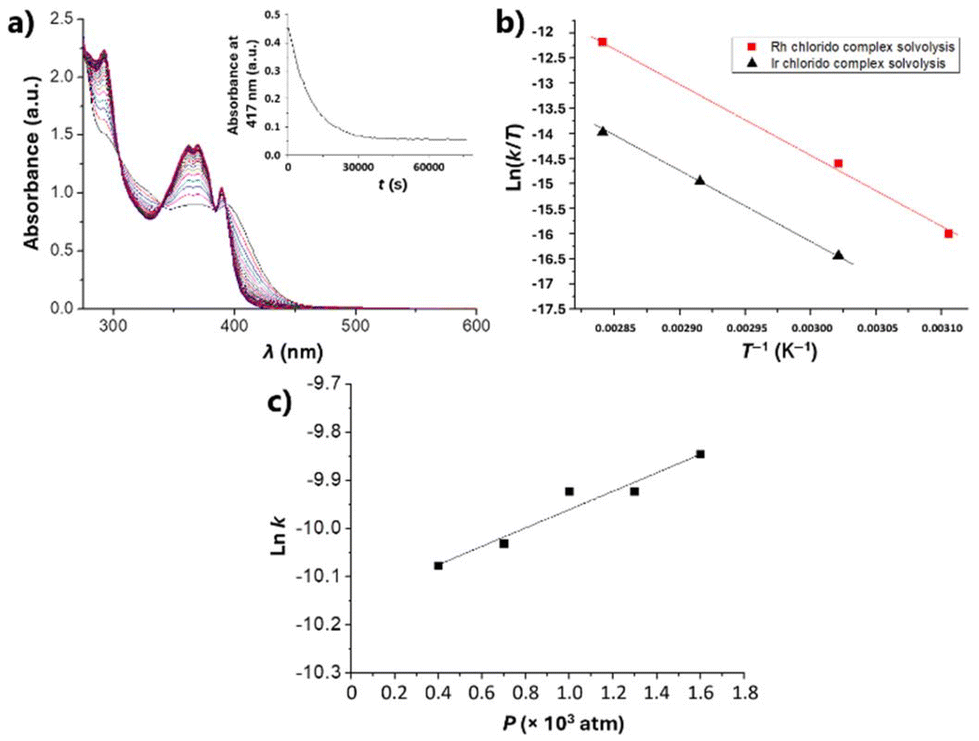 | ||
| Fig. 2 (a) UV-Vis spectra of a DMSO solution of 1 recorded at 70 °C for 21 hours, in the range 600–280 nm; insert: time evolution of the absorption at 417 nm. (b) Eyring plots of the temperature dependence of k for 1 (black triangles) and 2 (red squares) in DMSO. (c) Plots of the pressure dependence of ln k for 1 in DMSO. | ||
| Compd. | k (s−1) |
t
1/2a |
ΔH‡ (kJ mol−1) | ΔS‡ (J K−1 mol−1) | ΔV‡ (cm3 mol−1) |
|---|---|---|---|---|---|
| a Extrapolated at 310 K. b Not determined. c At 327 K. | |||||
| In DMSO | |||||
| 1 | 2.4 × 10−5 (330 K) | 160 h | 115 ± 2 | 10 ± 6 | n.d.b |
| 2 | 15 × 10−5 (330 K) | 33 h | 120 ± 6 | 39 ± 19 | −5.1 ± 0.7c |
| In H 2 O | |||||
| 1 | 0.065 (298 K) | 0.50 s | 47 ± 2 | −112 ± 12 | n.d.b |
| 2 | 0.025 (298 K) | 28 s | 50 ± 1 | −110 ± 4 | n.d.b |
| 1·DMSO | 2.7 × 10−3 (330 K) | 0.5 h | 72 ± 2 | −79 ± 9 | n.d.b |
| 2·DMSO | 1.3 × 10−3 (330 K) | 1 h | 76 ± 7 | −73 ± 22 | n.d.b |
Subsequently, stability studies of freshly prepared DMSO solutions of the chlorido complexes 1 and 2 in aqueous and PBS medium were carried out as well, as some reactivity may be expected. Clear solubility issues under these relevant conditions were evident and Fig. 3a shows the UV-Vis spectral changes observed in the minute time-scale after the addition of 2% of a 250 μM DMSO solution of 1 to neat water.
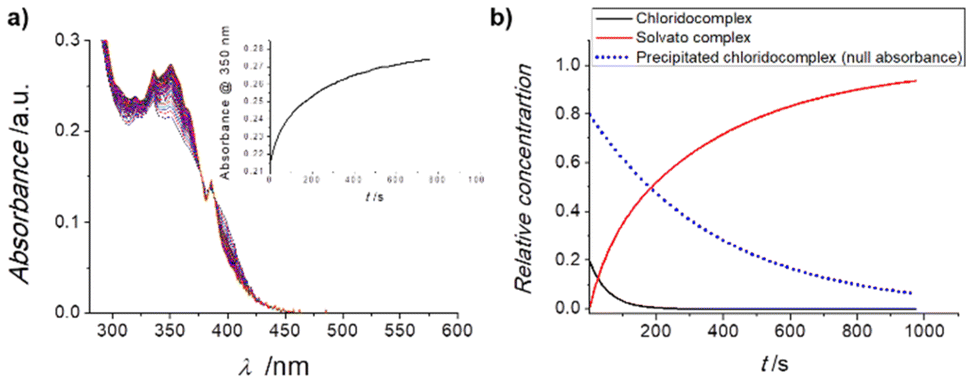 | ||
| Fig. 3 (a) UV-Vis spectra of a 2% DMSO aqueous solution of 1 recorded at 25 °C for 20 minutes, in the range 600–280 nm; insert: time evolution of the absorption at 417 nm. (b) Fitted33,34 concentration profiles for parallel A → B and precipitate → B processes of the reaction indicated. | ||
It is clear that after an initial well-behaved first-order process (as expected for a solvolysis reaction), the intensity of the spectrum keeps increasing while maintaining the same isosbestic points and overall characteristics (see inset of Fig. 3a). The same behaviour is observed for complex 2 under the same conditions. Fitting the full spectral changes to two first-order parallel processes33,34 leading to the same final solvato complex (that is an A → B and precipitate → B sequence) produces the concentration profiles indicated in Fig. 3b, which are in excellent agreement with the monitored time-resolved spectral changes.
In summary, the DMSO solutions of the chlorido complexes 1 and 2 are stable for days at room temperature with no significant changes. However, when they are added to aqueous solutions, they evolve within minutes to new solvato species, provided solubility is achieved. To examine this fast process, the monitoring of the UV-Vis spectral changes of 10 μM aqueous (and PBS) solutions of the complexes in 20% DMSO was conducted. In these cases, a perfect first-order fitting of the time-resolved changes is obtained (Fig. S3a†) and the corresponding Eyring activation parameters for the reactions can be derived (Fig. S3b†). These data are also collected in Table 1. The values of the thermal activation parameters are rather different from those determined for the DMSO solvolysis, while ΔH‡ decreases to half the value, the entropy of activation becomes very negative. The data for the chlorido-to-water substitution seem thus to fit to a clear associative interchange mechanism, opposite to that observed for the chlorido-to-DMSO substitution. Even so, the disagreement may suggest that both processes are, in fact dissociatively activated, but with a large electrostriction component that produces a higher ordering in the more polar solvent (i.e., water) upon going through a charge separated Mδ+⋯Clδ− transition state, thus leading to a more enthalpicaly favoured reaction.
Given the data obtained for 1 and 2, the possible aqueous reactivity of complexes 1-DMSO or 2-DMSO was also examined. Aqueous dilution of a DMSO solution of the 1-DMSO or 2-DMSO complexes at the 5–10 μM concentration level (final solution containing 20% DMSO) was conducted and monitored via time-resolved UV-Vis spectroscopy. Provided solubility issues are dealt with (filtering was needed for the highest concentrations), the time-resolved spectral changes indicated very small, but consistent and reproducible, changes in the hours’ time-scale at room temperature (Fig. S4a†); these were associated to a rather small-shifted solvolysis equilibrium. The temperature dependence of these equilibrium aquation reactions produced the kinetic and thermal activation parameters indicated in Table 1, as derived from the Eyring plots shown in Fig. S4b.† Clearly, the DMSO derivatives of the parent compounds undergo a partial aquation process that is limited by solubility issues. The values determined for ΔH‡ are larger than those obtained for the aquation of the chlorido ligands, agreeing with a dissociatively activated process from a stronger M-SO(CH3)2 bond (as expected for the soft nature of the metals involved). As for the ΔS‡ values, these are much less negative than for the solvolysis of the parent chlorido complexes 1 and 2 in DMSO solution, as the above-mentioned electrostriction factors are at a minimum and only hydrogen-bonding interactions may be relevant.
Time course ESI mass spectrometry studies were performed with 1 and 2 to provide insight into the solvato species formed in aqueous solution. The ESI mass spectrum of 1 and 2 (500 μM) in H2O:DMSO (5:1) was recorded over the course of 72 h at 37 °C (Fig. S5 and S7†). The positive mode of the ESI mass spectra of 1 and 2 displayed distinctive signals for the aquated species [1-Cl + H2O + Na]+ (m/z = 754.3) and [2-Cl + H2O + Na]+ (m/z = 664.3), respectively (Fig. S5 and S7†) throughout the incubation period. This suggests that 1 and 2 undergo aquation in the presence of water and the resulting aquated species remains stable for at least 72 h. The ESI mass spectrum of 1 and 2 (500 μM) in H2O:DMSO (5:1) in the presence of glutathione (500 μM) was also recorded over the course of 72 h at 37 °C (Fig. S6 and S8†). The resultant ESI mass spectra of 1 and 2 were dominated by signals associated to the aquated species for 1 and 2, [1-Cl + H2O + Na]+ (m/z = 754.3) and [2-Cl + H2O + Na]+ (m/z = 664.3), respectively (Fig. S6 and S8†). This implies that the aquated species of 1 and 2 (formed in the presence of water) are resistant to reaction with glutathione.
Interaction with DNA
The interaction of 1 and 2 with pBR322 plasmid DNA was examined by agarose gel electrophoresis. Aliquots of different concentrations of the complexes, viz. from 0.1 to 20 μM, were used with 15 μM DNA (in base pairs). The corresponding gel electrophoresis images are shown in Fig. 4. For free DNA, only form I (supercoiled DNA) is observed (lane 1), whereas great amounts of form II are detected for DNA samples treated with the highest concentrations of the complexes (lanes 2–9). For instance, complex 1 induces the formation of 70–99% form II (see lanes 3–9 in Fig. 4 top), using concentrations ranging from 0.5 to 20 μM. For complex concentrations <0.1 μM, only form I is seen in the agarose gel (lane 2 in Fig. 4 top). Comparable features are observed for complex 2 (Fig. 4 bottom). However, lower amounts of form II are comparatively found for concentrations of 2 ranging from 1 to 15 μM (see Fig. S9†). Hence, complex 2 appears to be a slightly less DNA-damaging agent than complex 1.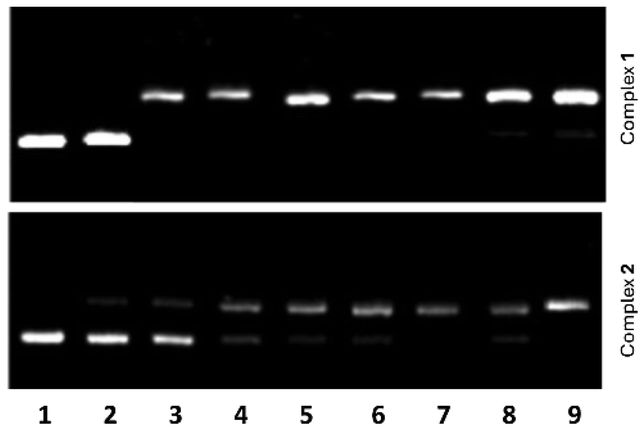 | ||
| Fig. 4 Agarose gel electrophoresis images of pBR322 plasmid DNA ([DNA]bp = 15 μM) incubated with decreasing concentrations (20–0.1 μM) of 1 (top) and 2 (bottom) for 1 h minutes at 37 °C. Lane 1: DNA control; lanes 3–9: DNA and complexes at concentrations 0.1, 0.5, 1, 2.5, 5, 10, 15, 20 μM. | ||
Cytotoxicity towards breast cancer cells
The cytotoxicity of the cyclometalated complexes 1 and 2 toward bulk breast cancer cells (HMLER) and breast CSCs (HMLER-shEcad) cultured in monolayers was assessed using the colorimetric MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. The IC50 values (concentrations required to reduce cell viability by 50%) were derived from dose–response curves (Fig. S10 and S11†) and are summarised in Table 2.The cyclometalated complexes 1 and 2 displayed similar IC50 values towards bulk breast cancer cells and breast CSCs, within the sub-micromolar range. This suggests that the metal in the cyclometalated complexes does not affect their potency towards bulk breast cancer cells and breast CSCs. Notably the cyclometalated complexes 1 and 2 exhibited up to 7-fold and 9-fold higher potency towards breast CSCs than salinomycin (a gold standard anti-breast CSC agent) and cisplatin (a metallodrug used in certain breast cancer treatment regimens), respectively.35,36 To shed light on the therapeutic potential of 1 and 2, their cytotoxicity towards non-cancerous bronchial epithelial BEAS-2B cells was determined (Fig. S12†). The iridium(III) complex 1 displayed similar toxicity towards BEAS-2B cells as HMLER and HMLER-shEcad cells whereas the rhodium(III) complex 2 displayed significantly (p < 0.05) lower toxicity towards BEAS-2B cells compared to HMLER and HMLER-shEcad cells (up to 3.5-fold higher IC50 value).
Mammosphere studies
Given the promising potencies of 1 and 2 toward two-dimensionally cultured breast CSCs, their ability to inhibit the formation and to lessen the viability of three-dimensionally cultured breast CSCs was probed using established mammosphere-based experiments.37 The incubation of 1 and 2 within single cell suspensions of HMLER-shEcad cells (IC20 value for 5 days) significantly (p < 0.05) reduced the number and size of mammospheres formed (Fig. 5).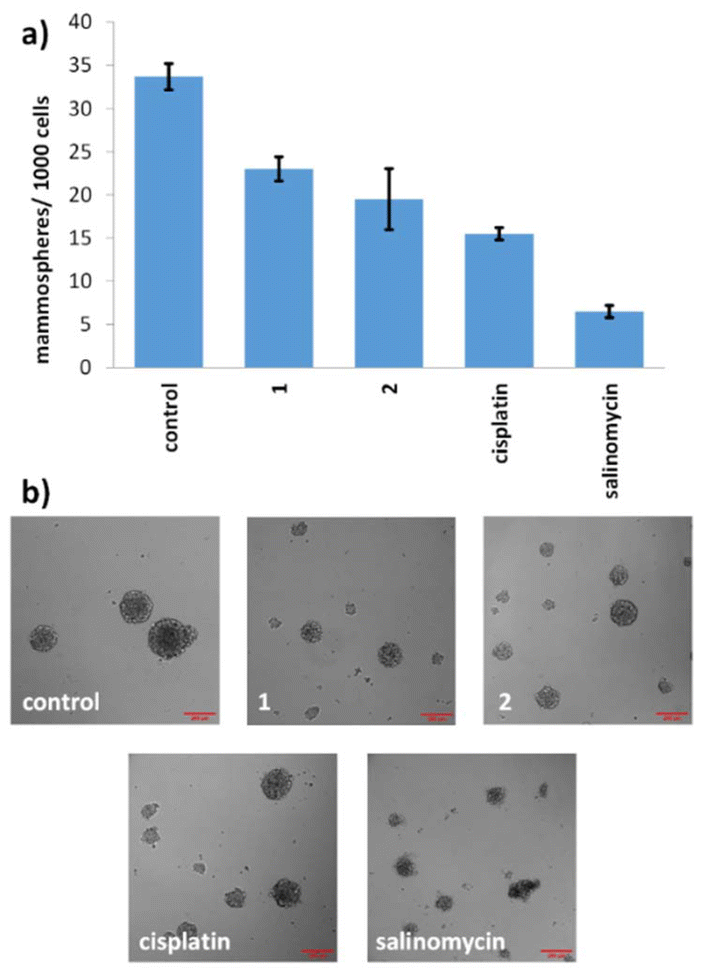 | ||
| Fig. 5 (a) Quantification of mammosphere formation with HMLER-shEcad cells untreated and treated with 1, 2, cisplatin, or salinomycin (at their IC20 values, 5 days). Error bars represent standard deviations. (b) Representative bright-field images (×10) of HMLER-shEcad mammospheres in the absence and presence of 1, 2, cisplatin, or salinomycin (at their IC20 values, 5 days). | ||
Under identical conditions cisplatin and salinomycin inhibited the formation of mammospheres from single cell suspensions of HMLER-shEcad cells to a slightly greater extent than 1 and 2 (with respect to the number and size of mammospheres formed) (Fig. 5). To gauge the ability of 1 and 2 to reduce the viability of mammospheres, the colorimetric resazurin-based reagent, TOX8 was used (Fig. S13 and S14†). The IC50 value of 1 and 2 was in the micromolar range (IC50 value for 1 = 4.11 ± 0.13 μM and IC50 value for 2 = 12.60 ± 0.62 μM). Interestingly, the potency of iridium(III) complex 1 was 3-fold higher than rhodium(III) complex 2, implying that the metal influences mammosphere potency, possibly as the result of the observed difference in aquation rates (see Table 1). The potency of 2 towards mammospheres was comparable to the potencies reported for cisplatin (IC50 value = 13.50 ± 2.34 μM) and salinomycin (IC50 value = 18.50 ± 1.50 μM) under identical conditions, however the potency of 1 towards mammospheres was up to 4.5-fold greater than cisplatin and salinomycin.38 Overall, the mammosphere studies indicate that both 1 and 2 are able to effectively reduce mammosphere formation and that 1 is able to reduce mammosphere viability significantly (p < 0.05) greater than cisplatin and salinomycin.
Mode of cell death
To provide insight into the mode of breast CSC death induced the cyclometalated complexes 1 and 2, the Annexin V-propidium iodide assay was conducted. This assay probes the presences of phosphatidylserine residues on the exterior of cells (as sign of early- and late-stage apoptosis) and the uptake of propidium iodide (as sign of late-stage apoptosis and necrosis).39 Upon treatment of HMLER-shEcad cells with 2 (4 × IC50 value for 72 h) there was no statistically significant change in the population of apoptotic or necrotic cells (Fig. 6).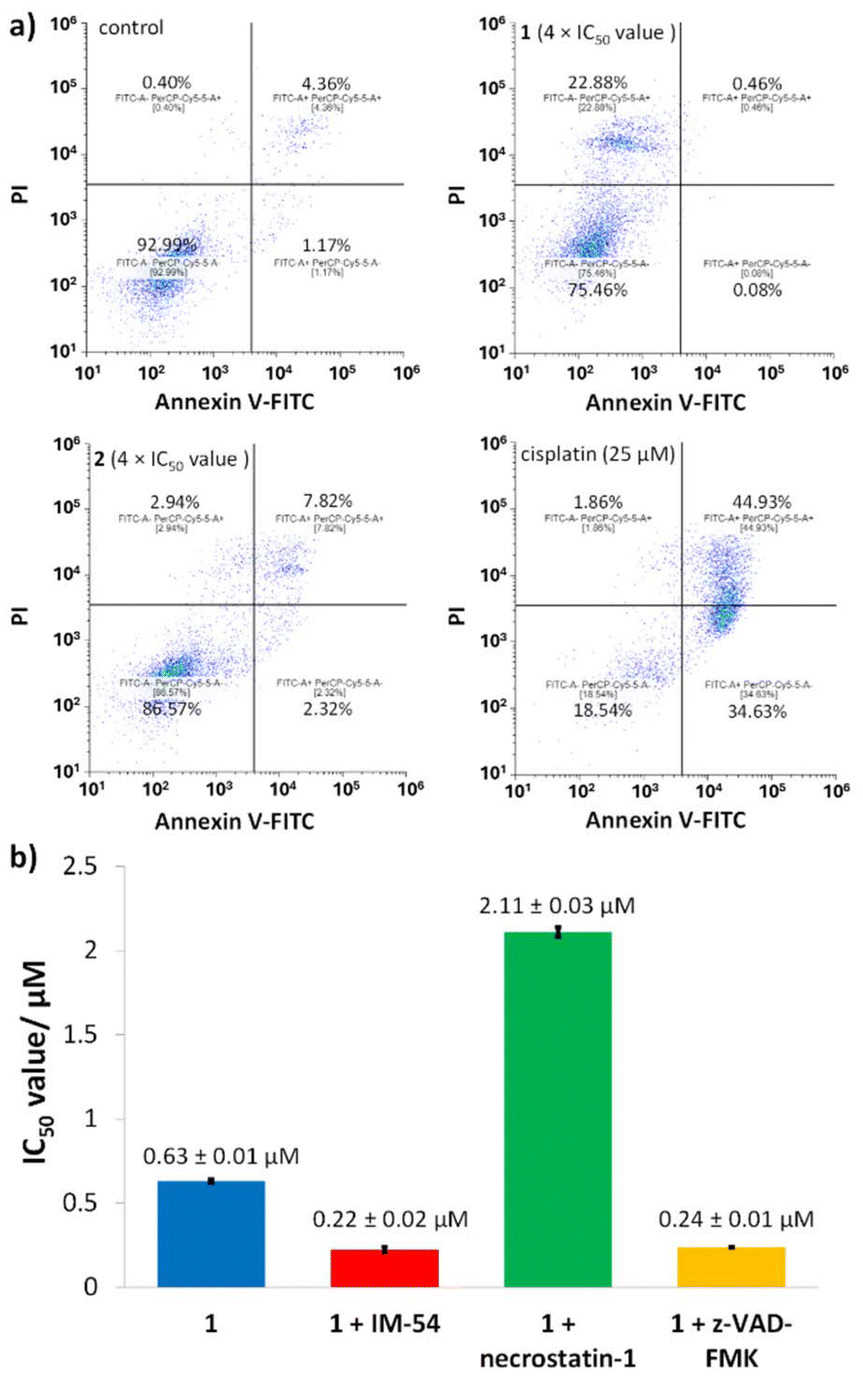 | ||
| Fig. 6 (a) FITC Annexin V-propidium iodide binding assay plots of untreated HMLER-shEcad cells and HMLER-shEcad cells treated with 1 (4 × IC50 value for 72 h) or 2 (4 × IC50 value for 72 h) or cisplatin (25 μM for 72 h). (b) Graphical representation of the IC50 values of 1 towards HMLER-shEcad cells in the absence and presence of IM-54 (10 μM), necrostatin-1 (20 μM) or z-VAD-FMK (5 μM). Error bars represent standard deviations. | ||
In contrast, incubation of HMLER-shEcad cells with 1 (4 × IC50 value for 72 h) induced a significant population of cells to take up propidium iodide (but not expose phosphatidylserine on the cell membrane exterior), indicative of necrosis (Fig. 6). As expected, treatment of HMLER-shEcad cells with cisplatin (25 μM for 72 h) induced large populations to express early- and late-stage apoptosis features (Fig. 6).
To further validate the mode of cell death induced by 1, cytotoxicity studies were carried out in the presence of necrosis inhibitors. Necrosis can occur in an unregulated, random manner as well as a regulated ordered form called necroptosis which relies on the formation of necrosomes (the amalgamation of three protein kinases; RIP1, RIP3, and MLKL).40,41 Co-treatment of HMLER-shEcad cells with 1 and IM-54 (10 μM), an inhibitor of unregulated necrosis,42 led to an increase in potency (rather than a decrease) implying that 1 does not induce unregulated, random necrosis (Fig. 6b and S15†).
Similar studies in the presence of necrostatin-1 (20 μM), a RIP1 inhibitor that attenuates necroptosis,43 led to a significant decrease (3-fold) in the potency of 1 (Fig. 6b and S15†). This suggests that 1 may induce necrosome-mediated necroptotic cell death of breast CSCs. In the presence of z-VAD-FMK (10 μM), a caspase-dependent apoptosis inhibitor,44 the potency of 1 increased (Fig. 6 and S15†). This implies that that inhibiting caspase-dependent apoptosis in breast CSCs promotes 1-mediated cell death. Comparable increases in potency in the presence of z-VAD-FMK have been reported for bona fide necroptosis inducers.45,46 Complementary immunoblotting studies showed that the expression of the necrosome components RIP1, RIP3, and MLKL remained largely unaltered in HMLER-shEcad cells treated with increasing concentrations of 1 (0.32–1.26 μM for 72 h) (Fig. S16†). This suggests that 1-induced necroptosis is dependent on the formation of the necrosome complex (association of RIP1, RIP3, and MLKL) and not on the perturbation of the expression of its individual protein kinase components. Necroptosis is associated with a number of morphological changes such as the increase in volume of certain organelles, overall cell volume expansion, and the preservation of the nuclear membrane but disintegration of the plasma membrane.47 Bright-field microscopy studies of HMLER-shEcad cells treated with 1 (4 × IC50 value for 24 h) showed no clear cell membrane but a preserved nuclear membrane (Fig. 7), consistent with necroptotic cell death.
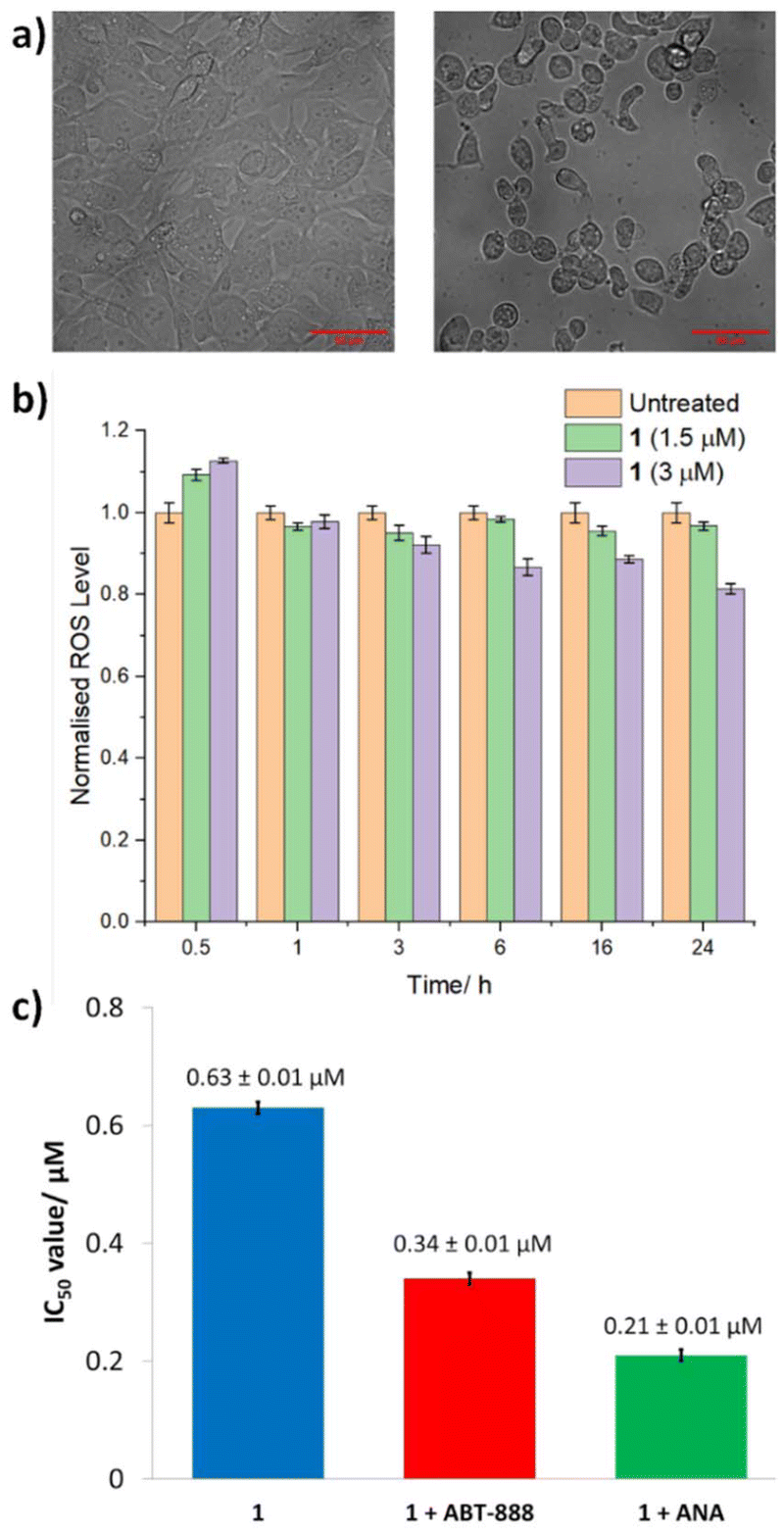 | ||
| Fig. 7 (a) Representative bright-field images (×20) of untreated HMLER-shEcad cells (left) and HMLER-shEcad cells treated with 1 (4 × IC50 value for 24 h, right). (b) Normalised ROS activity in untreated HMLER-shEcad cells and HMLER-shEcad cells treated with 1 (4 × IC50 value for 0.5, 1, 3, 6, 16, and 24 h). Error bars represent standard deviations. (c) Graphical representation of the IC50 values of 1 towards HMLER-shEcad cells in the absence and presence of ABT-888 (10 μM) or ANA (10 μM). Error bars represent standard deviations. | ||
In certain cases, the formation of necrosomes leads to downstream reactive oxygen species (ROS) generation.48–50 To determine if 1-induced necroptosis leads to ROS elevation, the established ROS indicator, 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), was used. The intracellular ROS levels in HMLER-shEcad cells dosed with 1 (1.5 μM or 3 μM) did not change significantly (p > 0.05) over the course of 24 h (Fig. 7). This implies that 1-induced necroptosis is independent of ROS elevation. Another common feature of necroptosis is the hyperactivation of PARP-1, which leads to DNA damage and the depletion of ATP and NAD levels.51,52 Cytotoxicity studies were performed to determine whether 1-induced necroptosis was related to PARP-1 hyperactivation. More specifically, the potency of 1 towards HMLER-shEcad cells was determined in the presence of well-known PARP-1 inhibitors, veliparib (ABT-888, 10 μM) and 4-amino-1,8-naphthalimide (ANA, 10 μM) (Fig. 7 and S17†).53,54 The IC50 value of 1 decreased in the presence of ABT-888 (IC50 value = 0.34 ± 0.01 μM) and ANA (IC50 value = 0.21 ± 0.01 μM), suggesting that hyperactivation of PARP-1 was not part of necroptosis mechanism evoked by 1. The cytotoxicity data also suggests that the activity of 1 toward breast CSCs can be enhanced by co-treating with PARP-1 inhibitors.
Experimental
Materials and methods
All compounds were prepared from commercially available reagents, under a dinitrogen atmosphere using standard vacuum-line techniques. The solvents were obtained from a purification system (Innovative Technology, Pure Solv) or dried applying standard procedures,55 and stored under dinitrogen. 1H, 13C{1H},5631P{1H}, 1H–13C HSQC NMR spectra were recorded at room temperature with 400 or 500 MHz spectrometers. Chemical shifts are reported downfield from standards and the coupling constants are given in Hz. All NMR data were analysed using MestRe Nova (Mnova) version 14.2.1.57 The infrared spectra were recorded using attenuated total reflection (ATR) in the range 4000–400 cm−1, and the main absorption bands are expressed in cm−1. High-resolution mass analyses (HRMS) were carried out at the Centres Cientifics i Tecnòlogics de la Universitat de Barcelona, with a time-of-flight instrument using electrospray ionization. UV-Vis spectroscopy was conducted on Cary 60, Agilent 8453 or Cary 100 instruments in thermally controlled 1 cm path-length quartz cuvettes. Kinetic runs were conducted as described previously.21Synthesis
The ligand diphenyl(1-pyrenyl)phosphane (L) was prepared as described earlier.22 The metallic dimers of formula [MCl(μ-Cl)(η5-pentamethylcyclopendadienyl)]2 (M = Ir,58 Rh59) were obtained applying reported procedures.Preparation of complexes 1 and 2, and their DMSO-coordinated derivatives 1·DMSO and 2·DMSO
 = 3045, 2816, 1572, 1434, 1374, 1095, 1028, 866, 832, 732, 619 cm−1. HRMS: calcd for [M]+ 748.1632, found 748.1598; for [M − Cl]+ 713.1943, found 713.1934.
:1). Pure 2 was obtained as an orange solid with a yield of 52% (69.0 mg). 31P{1H} NMR (162 MHz, CDCl3): δ = +66.4 (d, JPRh = 161.0) ppm (Fig. S22†). 1H NMR (400 MHz, CDCl3): δ = 8.40 (s, 1H), 8.32–8.06 (m, 5HAr), 7.99 (t, J = 7.6, 1HAr), 7.52–7.49 (m, 3H), 7.34–7.289 (m, 1H), 7.26–7.21 (m, 4H), 6.98–6.94 (m, 2H), 1.62 (d, J = 3.2, 15H) ppm (Fig. S23 and S24†). 13C{1H} NMR (101 MHz, CDCl3): δ = 164.5 (dd, J = 32.1, 10.2; C), 151.0 (d, J = 37.4, C), 136.4–122.5 (m, CAr, CHAr), 100.6 (dd, J = 4.8, 3.2, 5C, Cp*), 9.3 (s, 5CH3, Cp*) ppm (Fig. S24 and S25†).
= 3045, 2816, 1572, 1434, 1374, 1095, 1028, 866, 832, 732, 619 cm−1. HRMS: calcd for [M]+ 748.1632, found 748.1598; for [M − Cl]+ 713.1943, found 713.1934.
:1). Pure 2 was obtained as an orange solid with a yield of 52% (69.0 mg). 31P{1H} NMR (162 MHz, CDCl3): δ = +66.4 (d, JPRh = 161.0) ppm (Fig. S22†). 1H NMR (400 MHz, CDCl3): δ = 8.40 (s, 1H), 8.32–8.06 (m, 5HAr), 7.99 (t, J = 7.6, 1HAr), 7.52–7.49 (m, 3H), 7.34–7.289 (m, 1H), 7.26–7.21 (m, 4H), 6.98–6.94 (m, 2H), 1.62 (d, J = 3.2, 15H) ppm (Fig. S23 and S24†). 13C{1H} NMR (101 MHz, CDCl3): δ = 164.5 (dd, J = 32.1, 10.2; C), 151.0 (d, J = 37.4, C), 136.4–122.5 (m, CAr, CHAr), 100.6 (dd, J = 4.8, 3.2, 5C, Cp*), 9.3 (s, 5CH3, Cp*) ppm (Fig. S24 and S25†).
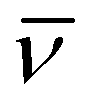 = 2964, 1568, 1472, 1436, 1294, 1261, 1181, 1046, 1013, 830 (ν(PF6)), 701, 556 cm−1. HRMS: calcd for [M − PF6]+ 791.2083, found 791.2081.
= 2964, 1568, 1472, 1436, 1294, 1261, 1181, 1046, 1013, 830 (ν(PF6)), 701, 556 cm−1. HRMS: calcd for [M − PF6]+ 791.2083, found 791.2081.
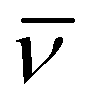 = 2990, 1568, 1471, 1436, 1385, 1309, 1292, 1184, 1046, 1099, 1013, 832 (ν(PF6)), 751, 693, 567 cm−1. HRMS: calcd for [M-PF6-dmso]+ 623.1385, found 623.1369.
= 2990, 1568, 1471, 1436, 1385, 1309, 1292, 1184, 1046, 1099, 1013, 832 (ν(PF6)), 751, 693, 567 cm−1. HRMS: calcd for [M-PF6-dmso]+ 623.1385, found 623.1369.
UV-Vis binding studies
The potential interaction of Calf Thymus DNA (ct-DNA) with complexes 1 and 2 was investigated by recording UV-Visible spectra in cacodylate/NaCl buffer solutions (1 mM sodium cacodylate, 20 mM NaCl, pH 7.3). Before the interaction with DNA, the stability of 25 μM solutions of 1 and 2 in 1 mM cacodylate–20 mM NaCl buffer solution (containing 1% DMSO) was first monitored over a period of 72 h. Subsequently, DNA-binding experiments were performed using solutions of increasing concentrations of ct-DNA (0–25 μM), which were added to a 25 μM solution of the complexes in cacodylate–NaCl buffer. The concentration of calf thymus DNA (ct-DNA) was determined spectrophotometrically at 260 nm using the nucleobase molar absorptivity of 6600 M−1 cm−1.60 Moreover, the absorbance ratio at 260 and 280 nm (A260/A280) of 1.90 indicated that the DNA was sufficiently free of protein.61Agarose gel electrophoresis
Electrophoretic studies were performed with pBR322 plasmid DNA in cacodylate buffer (10 mM sodium cacodylate and 50 mM NaCl in milliQ water). 3 mM stock solutions of 1 and 2 were prepared in DMSO. The samples at the different concentrations required for the electrophoresis (viz., 25, 15, 12.5, 10, 5, 2.5, 1 and 0.5 μM) were obtained by dilution in cacodylate buffer. pBR322 DNA was diluted as well in cacodylate buffer to obtain a DNA concentration of 15 μM (in base pairs) and incubated with distinct concentrations of the two complexes. Two types of DNA-complex samples were prepared, namely (i) the dark controls for which the complex (at the required concentration) and DNA were incubated for 30 min at 37 °C and kept in the dark at room temperature; (ii) the irradiated samples for which a solution of the complex (at a specific concentration) and DNA was first incubated for 30 min at 37 °C and subsequently irradiated for 30 min with blue light (λ = 450 nm; 1.2 J cm−2). Next, all the samples were mixed with loading buffer (viz. xylene cyanol 0.25% aqueous solution containing 30% glycerol) and loaded onto 1% agarose gels (1% in TBE buffer). The gels were run at 100 V for 1 h using a Bio-Rad horizontal tank connected to a Consort EV231 variable potential power supply. Subsequently, the DNA was stained with SYBR™ Safe overnight, and the gels were imaged with a BioRad GelDoc EZ Imager.Crystallography
Data for compound 1 were collected on a Bruker APEX II QUAZAR diffractometer equipped with a microfocus multilayer monochromator with MoKα radiation (λ = 0.71073 Å). Data for compounds 1·DMSO and 2·DMSO were collected at BL13-XALOC beamline62 of the ALBA synchrotron (λ = 0.72931 Å). Data reduction and absorption corrections were performed by using SAINT and SADABS, respectively.63 The structures were solved using SHELXT64 and refined by full-matrix least-squares on F2 with SHELXL.65 For compounds 1·DMSO and 2·DMSO, a void containing only diffuse electron density was analysed and taken into account with Olex2/Solvent Mask.66 An estimated content of one diffuse lattice CH2Cl2 molecule per formula unit was deduced for both compounds, and included in the formula. All details can be found in CCDC 2390209–2390211,† which contain the supplementary crystallographic data for this paper.Cell culture
The human mammary epithelial cell lines, HMLER and HMLER-shEcad were kindly donated by Prof. R. A. Weinberg (Whitehead Institute, MIT). HMLER and HMLER-shEcad cells were maintained in mammary epithelial cell growth medium (MEGM) with supplements and growth factors (BPE, hydrocortisone, hEGF, insulin, and gentamicin/amphotericin-B). The BEAS-2B bronchial epithelium cell line was acquired from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in RPMI 1640 medium with 2 mM L-glutamine supplemented with 1% penicillin and 10% fetal bovine serum. The cells were grown at 310 K in a humidified atmosphere containing 5% CO2.Monolayer cytotoxicity studies
Exponentially growing cells were seeded at a density of approximately 5 × 103 cells per well in 96-well flat-bottomed microplates and allowed to attach for 24 h prior to addition of compounds. Various concentrations of the test compounds (0.0004–100 μM) were added and incubated for 72 h at 37 °C (total volume 200 μL). Stock solutions of the compounds were prepared as 10 mM DMSO solutions and diluted using cell media. The final concentration of DMSO in each well was ≤1%. After 72 h, 20 μL of MTT (4 mg mL−1 in PBS) was added to each well and the plates incubated for an additional 4 h at 37 °C. The media/MTT mixture was eliminated and DMSO (100 μL per well) was added to dissolve the formazan precipitates. The optical density was measured at 550 nm using a 96-well multiscanner autoreader. Absorbance values were normalised to (DMSO-containing) control wells and plotted as concentration of compound versus % cell viability. IC50 values were interpolated from the resulting dose dependent curves. The reported IC50 values are the average of three independent experiments (n = 18).Mammosphere formation and viability studies
HMLER-shEcad cells (5 × 103) were plated in ultralow-attachment 96-well plates (Corning) and incubated in MEGM supplemented with B27 (Invitrogen), 20 ng mL−1 EGF and 4 μg mL−1 heparin (Sigma) for 5 days. Studies were also conducted in the presence of 1, 2, cisplatin, and salinomycin (0–133 μM). Mammospheres treated with 1, 2, cisplatin, and salinomycin (at their respective IC20 values, 5 days) were counted and imaged using an inverted microscope. The viability of the mammospheres was determined by addition of a resazurin-based reagent, TOX8 (Sigma). After incubation for 16 h, the fluorescence of the solutions was read at 590 nm (λex = 560 nm). Viable mammospheres reduce the amount of the oxidised TOX8 form (blue) and concurrently increase the amount of the fluorescent TOX8 intermediate (red), indicating the degree of mammosphere cytotoxicity caused by the test compound. Fluorescence values were normalised to DMSO-containing controls and plotted as concentration of test compound versus % mammosphere viability. IC50 values were interpolated from the resulting dose dependent curves. The reported IC50 values are the average of two independent experiments, each consisting of three replicates per concentration level (n = 6).Annexin V-propidium iodide assay
HMLER-shEcad cells were incubated with and without 1 and 2 (4 × IC50 value for 72 h) and cisplatin (25 μM for 72 h) at 37 °C. Cells were harvested from adherent cultures by trypsinisation. The FITC Annexin V/dead cell apoptosis kit was used. The manufacture's (Thermo Fisher) protocol was followed to carry out this experiment. Briefly, untreated and treated cells (1 × 106) were suspended in 1× Annexin binding buffer (100 μL) (10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4), then 5 μL of FITC Annexin V and 1 μL of PI (100 μg mL−1) were added to each sample and incubated at room temperature for 15 min. After which more 1× Annexin binding buffer (400 μL) was added while gently mixing. The cells were analysed using a FACSCanto II flow cytometer (BD Biosciences) (10000 events per sample were acquired) at the University of Leicester FACS Facility. The FL1 channel was used to assess Annexin V binding and the FL2 channel was used to assess PI uptake. Cell populations were analysed using Floreada.io.
Immunoblotting analysis
HMLER-shEcad cells (1 × 106) were incubated with 1 (0.32–1.26 μM for 72 h) at 37 °C. HMLER-shEcad cells were harvested and isolated as pellets. SDS-PAGE loading buffer (64 mM Tris-HCl (pH 6.8), 9.6% glycerol, 2% SDS, 5% β-mercaptoethanol, 0.01% bromophenol blue) was added to the pellets, and this was incubated at 95 °C for 10 min. Cell lysates were resolved by 4–20% sodium dodecylsulphate polyacylamide gel electrophoresis (SDS-PAGE; 200 V for 25 min) followed by electro transfer to polyvinylidene difluoride membrane, PVDF (350 mA for 1 h). Membranes were blocked in 5% (w/v) non-fat milk in PBST (PBS/0.1% Tween 20) and incubated with the appropriate primary antibodies (Cell Signalling Technology). After incubation with horseradish peroxidase-conjugated secondary antibodies (Cell Signalling Technology), immune complexes were detected with the ECL detection reagent (BioRad) and analysed using a chemiluminescence imager (Bio-Rad ChemiDoc Imaging System).Intracellular ROS assay
HMLER-shEcad cells (5 × 103) were seeded in each well of a 96-well plate. After incubating the cells overnight, they were treated with 1 (1.5 μM or 3 μM for 0.5–24 h), and incubated with 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (20 μM) for 30 min. The intracellular ROS level was determined by measuring the fluorescence of the solutions in each well at 529 nm (λex = 504 nm).Conclusions
Following earlier studies with ruthenium and osmium, the phosphane ligand diphenyl(1-pyrenyl)phosphane was used to prepare cycloiridated and cyclorhodiated half-sandwich complexes bearing a pentamethylcyclopentadienyl (Cp*) ring. The crystal structures of the organometallic compounds show features that are found in other piano-stool complexes. It can be pointed out here that 1 and 2 represent uncommon examples of Cp*-containing cyclometalated Ir(III) and Rh(III) complexes which have been structurally characterised by X-ray diffraction.27,31,32,67–70 The compounds are slowly converted into the DMSO solvato complexes, namely 1·DMSO and 2·DMSO, if dissolved in DMSO, as shown by solution studies. Moreover, 1 and 2, which are poorly soluble in water, are gradually converted into the aquated [MCp*L(H2O)]+ species, as suggested by UV-Vis studies and confirmed by mass spectrometry; as expected these new cationic species become more water soluble. Gel electrophoresis revealed that 1 and 2 interact with DNA and in vitro cytotoxicity assays showed that they are significantly cytotoxic against tumorigenic mammary epithelial HMLER (which is a genetically modified cell line representative of bulk breast cancer cells) and HMLER-shEcad (where E-cadherin was silenced, to generate breast CSC-enriched populations) cells. Remarkably, rhodium(III) complex 2 is more active against bulk breast cancer cells and breast CSCs (viz., HMLER and HMLER-shEcad cells) compared to non-tumorigenic cells (namely BEAS-2B cells), while iridium(III) complex 1 is toxic for both cancer and healthy cells. Subsequent investigation of the ability of 1 and 2 to inhibit the formation and reduce the viability of three-dimensionally cultured breast CSCs showed that 1 was thrice more efficient than 2, in contrast to the monolayer cytotoxicity data (which indicated that 1 and 2 had similar potency). Moreover, while 2 was as cytotoxic as cisplatin and salinomycin against the mammospheres, 1 was 4.5-fold more effective than cisplatin and salinomycin. In depth mechanistic studies indicated that whereas 2 was not producing any significant changes with respect to the population of apoptotic or necrotic cells, 1 was causing necrosis. This difference may be explained but the faster aquation of complex 1. It was shown that the necroptotic cell death provoked by 1 was potentially dependent on the formation of the necrosome complex and independent of intracellular ROS levels. Furthermore, hyperactivation of PARP-1 does not seem to be involved in the necroptotic pathway induced by 1; actually, our experimental data suggest that the efficiency of 1 against breast CSCs can be enhanced through co-treatment with PARP-1 inhibitors.Data availability
Data for this article are available at the Universitat de Barcelona Digital Repository at https://diposit.ub.edu/dspace/?locale=en.The data supporting this article have been included as part of the ESI.†
Crystallographic data for compounds 1, 2, 1·DMSO and 2·DMSO (CCDC 2390209–2390211†).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Agència de Gestió dels Ajuts Univeristaris i de Recerca (project 2021-SGR-01107), Spanish Ministerio de Ciencia, Innovación y Universidades (projects PID2020-115537RB-I00, PID2020-115658GB-I00 and PCI2021-122027-2B, MCIU/AEI/10.13039/501100011033 and European Union NextGenerationEU/PRTR) and the RSC (RSC Research Fund grant RF19-7147) is kindly acknowledged. P. G. thanks the Catalan Institution for Research and Advanced Studies (ICREA). K. S. is supported by an EPSRC New Investigator Award (EP/S005544/1) and the University of Leicester. We also thank the Advanced Imaging Facility (RRID:SCR_020967) at the University of Leicester for support. Crystallographic measurements were performed at BL13-XALOC beamline at ALBA Synchrotron with the collaboration of ALBA staff.References
- WHO, Global cancer burden growing, amidst mounting need for services, https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing-amidst-mounting-need-for-services, (accessed October 18th, 2024).
- K. D. Miller, L. Nogueira, T. Devasia, A. B. Mariotto, K. R. Yabroff, A. Jemal, J. Kramer and R. L. Siegel, Cancer treatment and survivorship statistics, 2022, CA-Cancer J. Clin., 2022, 72, 409–436 CrossRef PubMed.
- S. Dasari and P. B. Tchounwou, Cisplatin in cancer therapy: Molecular mechanisms of action, Eur. J. Pharmacol., 2014, 740, 364–378 CrossRef CAS PubMed.
- A. Casini, A. Vessières and S. M. Meier-Menches, Metal-based Anticancer Agents Preface, The Royal Society of Chemistry, 2019 Search PubMed.
- R. A. Alderden, M. D. Hall and T. W. Hambley, The discovery and development of cisplatin, J. Chem. Educ., 2006, 83, 728–734 CrossRef CAS.
- S. Adhikari, P. Nath, A. Das, A. Datta, N. Baildya, A. K. Duttaroy and S. Pathak, A review on metal complexes and its anti-cancer activities: Recent updates from in vivo studies, Biomed. Pharmacother., 2024, 171, 48 CrossRef PubMed.
- A. Casini and A. Pothig, Metals in Cancer Research: Beyond Platinum Metallodrugs, ACS Cent. Sci., 2024, 10, 242–250 CrossRef CAS PubMed.
- R. Paprocka, M. Wiese-Szadkowska, S. Janciauskiene, T. Kosmalski, M. Kulik and A. Helmin-Basa, Latest developments in metal complexes as anticancer agents, Coord. Chem. Rev., 2022, 452, 21 CrossRef.
- A. K. Singh, A. Kumar, H. Singh, P. Sonawane, P. Pathak, M. Grishina, J. P. Yadav, A. Verma and P. Kumar, Metal Complexes in Cancer Treatment: Journey So Far, Chem. Biodivers., 2023, 20, 50 Search PubMed.
- P. Moharana, D. Ghosh and P. Paira, Drive to organoruthenium and organoiridium complexes from organoplatinum: Next-generation anticancer metallotherapeutics, Inorg. Chem. Commun., 2021, 124, 18 CrossRef.
- E. J. Anthony, E. M. Bolitho, H. E. Bridgewater, O. W. L. Carter, J. M. Donnelly, C. Imberti, E. C. Lant, F. Lermyte, R. J. Needham, M. Palau, P. J. Sadler, H. Y. Shi, F. X. Wang, W. Y. Zhang and Z. J. Zhang, Metallodrugs are unique: opportunities and challenges of discovery and development, Chem. Sci., 2020, 11, 31 RSC.
- S. Thota, D. A. Rodrigues, D. C. Crans and E. J. Barreiro, Ru(II) Compounds: Next-Generation Anticancer Metallotherapeutics?, J. Med. Chem., 2018, 61, 5805–5821 CrossRef CAS PubMed.
- C. Sonkar, S. Sarkar and S. Mukhopadhyay, Ruthenium(II)-arene complexes as anti-metastatic agents, and related techniques, RSC Med. Chem., 2022, 13, 22–38 RSC.
- Y. Lu, D. Zhu, Q. Y. Le, Y. J. Wang and W. Wang, Ruthenium-based antitumor drugs and delivery systems from monotherapy to combination therapy, Nanoscale, 2022, 14, 16339–16375 RSC.
- M. Bashir, I. A. Mantoo, F. Arjmand, S. Tabassum and I. Yousuf, An overview of advancement of organoruthenium(II) complexes as prospective anticancer agents, Coord. Chem. Rev., 2023, 487, 27 CrossRef.
- M. Rausch, P. J. Dyson and P. Nowak-Sliwinska, Recent Considerations in the Application of RAPTA-C for Cancer Treatment and Perspectives for Its Combination with Immunotherapies, Adv. Ther., 2019, 2, 12 Search PubMed.
- R. Carter, A. Westhorpe, M. J. Romero, A. Habtemariam, C. R. Gallevo, Y. Bark, N. Menezes, P. J. Sadler and R. A. Sharma, Radiosensitisation of human colorectal cancer cells by ruthenium(II) arene anticancer complexes, Sci. Rep., 2016, 6, 12 CrossRef PubMed.
- Y. Lin, Y. D. Huang, W. Zheng, F. Y. Wang, A. Habtemariam, Q. Luo, X. C. Li, K. Wu, P. J. Sadler and S. X. Xiong, Organometallic ruthenium anticancer complexes inhibit human glutathione-S-transferase π, J. Inorg. Biochem., 2013, 128, 77–84 CrossRef CAS PubMed.
- L. Rafols, S. Torrente, D. Aguilà, V. Soto-Cerrato, R. Pérez-Tomás, P. Gamez and A. Grabulosa, Expanding the Range of Pyrenylphosphines and Their Derived Ru(II)-Arene Complexes, Organometallics, 2020, 39, 2959–2971 CrossRef CAS.
- D. Josa, D. Aguila, P. Fontova, V. Soto-Cerrato, P. Herrera-Ramirez, L. Rafols, A. Grabulosa and P. Gamez, Cytotoxicity of osmium(II) and cycloosmated half-sandwich complexes from 1-pyrenyl-containing phosphane ligands, Dalton Trans., 2023, 52, 8391–8401 RSC.
- L. Rafols, D. Josa, D. Aguilà, L. A. Barrios, O. Roubeau, J. Cirera, V. Soto-Cerrato, R. Pérez-Tomás, M. Martínez, A. Grabulosa and P. Gamez, Piano-Stool Ruthenium(II) Complexes with Delayed Cytotoxic Activity: Origin of the Lag Time, Inorg. Chem., 2021, 60, 7974–7990 CrossRef CAS PubMed.
- R. F. Brissos, P. Clavero, A. Gallen, A. Grabulosa, L. A. Barrios, A. B. Caballero, L. Korrodi-Gregório, R. Pérez-Tomás, G. Muller, V. Soto-Cerrato and P. Gamez, Highly Cytotoxic Ruthenium(II)-Arene Complexes from Bulky 1-Pyrenylphosphane Ligands, Inorg. Chem., 2018, 57, 14786–14797 CrossRef CAS PubMed.
- W. Y. Zhang, H. E. Bridgewater, S. Banerjee, J. J. Soldevila-Barreda, G. J. Clarkson, H. Y. Shi, C. Imberti and P. J. Sadler, Ligand-Controlled Reactivity and Cytotoxicity of Cyclometalated Rhodium(III) Complexes, Eur. J. Inorg. Chem., 2020, 2020, 1052–1060 CrossRef CAS PubMed.
- Y. L. Yang, L. H. Guo, X. X. Ge, S. P. Shi, Y. T. Gong, Z. S. Xu, X. F. Zheng and Z. Liu, Structure-activity relationships for highly potent half-sandwich organoiridium(III) anticancer complexes with CN-chelated ligands, J. Inorg. Biochem., 2019, 191, 1–7 CrossRef CAS PubMed.
- Z. Liu, I. Romero-Canelon, A. Habtemariam, G. J. Clarkson and P. J. Sadler, Potent Half-Sandwich Iridium(III) Anticancer Complexes Containing C∧I-Chelated and Pyridine Ligands, Organometallics, 2014, 33, 5324–5333 CrossRef CAS PubMed.
- A. Nahaei, Z. Mandegani, S. Chamyani, M. Fereidoonnezhad, H. R. Shahsavari, N. Y. Kuznetsov and S. M. Nabavizadeh, Half-Sandwich Cyclometalated RhIII Complexes Bearing Thiolate Ligands: Biomolecular Interactions and In Vitro and In Vivo Evaluations, Inorg. Chem., 2022, 61, 2039–2056 CrossRef CAS PubMed.
- C. Sire, H. Cattey, A. Tsivery, J. C. Hierso and J. Roger, Phosphorus-Directed Rhodium-Catalyzed C-H Arylation of 1-Pyrenylphosphines Selective at the K-Region, Adv. Synth. Catal., 2022, 364, 440–452 CrossRef CAS.
- D. Brackemeyer, C. S. T. Brinke, F. Roelfes and F. E. Hahn, Regioselective C8-metalation of N-phosphine tethered adenine derivatives via, C8-H activation, Dalton Trans., 2017, 46, 4510–4513 RSC.
- W. H. Wang, Y. Wu, H. T. Wang, P. J. Qi, W. N. Lan and Q. W. Zhang, Enantioselective synthesis of P-stereogenic allenylphosphines through Ni-catalysed propargylic substitution, Nat. Synth., 2022, 1, 738–747 CrossRef CAS.
- Y. Yamamoto, K. Sugawara and X. H. Han, Novel single or double insertion of alkynes into rhodium- and iridium-oxygen or -phosphorus atom bonds and transannular addition of 1-alkynes between the rhodium atom and the ipso-carbon atom of the phosphorus ligand, J. Chem. Soc., Dalton Trans., 2002, 195–211, 10.1039/b104811m.
- J. Campos and E. Carmona, Rhodium and Iridium Complexes of Bulky Tertiary Phosphine Ligands. Searching for Isolable Cationic MIII Alkylidenes, Organometallics, 2015, 34, 2212–2221 CrossRef CAS.
- R. C. Sun, S. W. Zhang, X. D. Chu and B. L. Zhu, Synthesis, Structures, and Reactivity of Cyclometalated Complexes Formed by Insertion of Alkynes into M-C (M = Ir and Rh) Bonds, Organometallics, 2017, 36, 1133–1141 CrossRef CAS.
- M. Maeder and P. King, ReactLab, 2009 Search PubMed.
- R. A. Binstead, A. D. Zuberbuhler and B. Jung, SPECFIT32, 2005 Search PubMed.
- J. N. Boodram, I. J. McGregor, P. M. Bruno, P. B. Cressey, M. T. Hemann and K. Suntharalingam, Breast Cancer Stem Cell Potent Copper(II)-Non-Steroidal Anti-Inflammatory Drug Complexes, Angew. Chem., Int. Ed., 2016, 55, 2845–2850 CrossRef CAS PubMed.
- A. Eskandari and K. Suntharalingam, A reactive oxygen species-generating, cancer stem cell-potent manganese(II) complex and its encapsulation into polymeric nanoparticles, Chem. Sci., 2019, 10, 7792–7800 RSC.
- G. Dontu, W. M. Abdallah, J. M. Foley, K. W. Jackson, M. F. Clarke, M. J. Kawamura and M. S. Wicha, In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells, Genes Dev., 2003, 17, 1253–1270 CrossRef CAS PubMed.
- A. Eskandari, A. Kundu, S. Ghosh and K. Suntharalingam, A Triangular Platinum(II) Multinuclear Complex with Cytotoxicity Towards Breast Cancer Stem Cells, Angew. Chem., Int. Ed., 2019, 58, 12059–12064 CrossRef CAS PubMed.
- P. J. Quinn, Plasma Membrane Phospholipid Asymmetry, Subcell. Biochem., 2002, 36, 39–60 CAS.
- T. Vanden Berghe, A. Linkermann, S. Jouan-Lanhouet, H. Walczak and P. Vandenabeele, Regulated necrosis: the expanding network of non-apoptotic cell death pathways, Nat. Rev. Mol. Cell Biol., 2014, 15, 134–146 CrossRef PubMed.
- P. Vandenabeele, L. Galluzzi, T. Vanden Berghe and G. Kroemer, Molecular mechanisms of necroptosis: an ordered cellular explosion, Nat. Rev. Mol. Cell Biol., 2010, 11, 700–714 CrossRef CAS PubMed.
- K. Dodo, M. Katoh, T. Shimizu, M. Takahashi and M. Sodeoka, Inhibition of hydrogen peroxide-induced necrotic cell death with 3-amino-2-indolylmaleimide derivatives, Bioorg. Med. Chem. Lett., 2005, 15, 3114–3118 CrossRef CAS PubMed.
- A. Degterev, J. Hitomi, M. Germscheid, I. L. Ch'en, O. Korkina, X. Teng, D. Abbott, G. D. Cuny, C. Yuan, G. Wagner, S. M. Hedrick, S. A. Gerber, A. Lugovskoy and J. Yuan, Identification of RIP1 kinase as a specific cellular target of necrostatins, Nat. Chem. Biol., 2008, 4, 313–321 CrossRef CAS PubMed.
- E. A. Slee, H. J. Zhu, S. C. Chow, M. MacFarlane, D. W. Nicholson and G. M. Cohen, Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32, Biochem. J., 1996, 315, 21–24 CrossRef CAS PubMed.
- K. Moriwaki, J. Bertin, P. J. Gough, G. M. Orlowski and F. K. M. Chan, Differential roles of RIPK1 and RIPK3 in TNF-induced necroptosis and chemotherapeutic agent-induced cell death, Cell Death Dis., 2015, 6, 11 Search PubMed.
- M. Flamme, P. B. Cressey, C. X. Lu, P. M. Bruno, A. Eskandari, M. T. Hemann, G. Hogarth and K. Suntharalingam, Induction of Necroptosis in Cancer Stem Cells using a Nickel(II)Dithiocarbamate Phenanthroline Complex, Chem. – Eur. J., 2017, 23, 9674–9682 CrossRef CAS PubMed.
- J. Seo, Y. W. Nam, S. Kim, D. B. Oh and J. Song, Necroptosis molecular mechanisms: Recent findings regarding novel necroptosis regulators, Exp. Mol. Med., 2021, 53, 1007–1017 CrossRef CAS PubMed.
- Y. Cho, S. Challa, D. Moquin, R. Genga, T. D. Ray, M. Guildford and F. K. M. Chan, Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation, Cell, 2009, 137, 1112–1123 CrossRef CAS PubMed.
- C. W. Davis, B. J. Hawkins, S. Ramasamy, K. M. Irrinki, B. A. Cameron, K. Islam, V. P. Daswani, P. J. Doonan, Y. Manevich and M. Madesh, Nitration of the mitochondrial complex I subunit NDUFB8 elicits RIP1-and RIP3-mediated necrosis, Free Radical Biol. Med., 2010, 48, 306–317 CrossRef CAS PubMed.
- D. W. Zhang, J. Shao, J. Lin, N. Zhang, B. J. Lu, S. C. Lin, M. Q. Dong and J. H. Han, RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis, Science, 2009, 325, 332–336 CrossRef CAS PubMed.
- J. Sosna, S. Voigt, S. Mathieu, A. Lange, L. Thon, P. Davarnia, T. Herdegen, A. Linkermann, A. Rittger, F. K. M. Chan, D. Kabelitz, S. Schutze and D. Adam, TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death, Cell. Mol. Life Sci., 2014, 71, 331–348 CrossRef CAS PubMed.
- X. S. Xu, C. C. Chua, M. Zhang, D. Q. Geng, C. F. Liu, R. C. Hamdy and B. H. L. Chua, The role of PARP activation in glutamate-induced necroptosis in HT-22 cells, Brain Res., 2010, 1343, 206–212 CrossRef CAS PubMed.
- C. K. Donawho, Y. Luo, Y. P. Luo, T. D. Penning, J. L. Bauch, J. J. Bouska, V. D. Bontcheva-Diaz, B. F. Cox, T. L. DeWeese, L. E. Dillehay, D. C. Ferguson, N. S. Ghoreishi-Haack, D. R. Grimm, R. Guan, E. K. Han, R. R. Holley-Shanks, B. Hristov, K. B. Idler, K. Jarvis, E. F. Johnson, L. R. Kleinberg, V. Klinghofer, L. M. Lasko, X. S. Liu, K. C. Marsh, T. P. McGonigal, J. A. Meulbroek, A. M. Olson, J. P. Palma, L. E. Rodriguez, Y. Shi, J. A. Stavropoulos, A. C. Tsurutani, G. D. Zhu, S. H. Rosenberg, V. L. Giranda and D. J. Frost, ABT-888, an orallyactive poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models, Clin. Cancer Res., 2007, 13, 2728–2737 CrossRef CAS PubMed.
- M. Banasik, H. Komura, M. Shimoyama and K. Ueda, Specific Inhibitors of Poly(ADP-ribose) Synthetase and Mono(ADP-ribosyl) Transferase, J. Biol. Chem., 1992, 267, 1569–1575 CrossRef CAS.
- W. L. F. Armarego, Purification of Laboratory Chemicals, Elsevier, 8th edn, 2017 Search PubMed.
- M. R. Plutino, L. M. Scolaro, A. Albinati and R. Romeo, Atropisomerization, C-H activation, and dissociative substitution at some biphenyl platinum(II) complexes, J. Am. Chem. Soc., 2004, 126, 6470–6484 CrossRef CAS PubMed.
- NMR software, MestreNova, 2021 Search PubMed.
- E. T. Chang, D. B. Green and K. R. Brereton, Microwave-assisted synthesis of pentamethylcyclopentadienyl iridium dihalide dimers, Polyhedron, 2022, 226, 6 CrossRef.
- J. Tönnemann, J. Risse, Z. Grote, R. Scopelliti and K. Severin, Efficient and Rapid Synthesis of Chlorido-Bridged Half-Sandwich Complexes of Ruthenium, Rhodium, and Iridium by Microwave Heating, Eur. J. Inorg. Chem., 2013, 2013, 4558–4562 CrossRef.
- J. K. Barton, J. M. Goldberg, C. V. Kumar and N. J. Turro, Binding Modes and Base Specificity of Tris(phenanthroline)ruthenium(II) Enantiomers with Nucleic Acids. Tuning the Stereoselectivity, J. Am. Chem. Soc., 1986, 108, 2081–2088 CrossRef CAS.
- S. Satyanarayana, J. C. Dabrowiak and J. B. Chaires, Tris(phenanthroline)ruthenium(II) Eantiomer Interactions with DNA. Mode and Specificity of Binding, Biochemistry, 1993, 32, 2573–2584 CrossRef CAS PubMed.
- J. Juanhuix, J. Nicolás, G. Cuni, C. Colldelram, A. Rubio, J. Avila, N. González, A. Enrique, I. Ramos, G. Jover, D. Fullà, F. Gil and J. Benach, New developments and operation of the MX beamline XALOC at ALBA synchrotron, Acta Crystallogr., Sect. A, 2014, 70, C1744–C1744 Search PubMed.
- G. M. Sheldrick, SAINT and SADABS, 2012 Search PubMed.
- G. M. Sheldrick, SHELXT - Integrated space-group and crystal-structure determination, Acta Crystallogr., Sect. A, 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. F. Espada, A. C. Esqueda, J. Campos, M. Rubio, J. López-Serrano, E. Alvarez, C. Maya and E. Carmona, Cationic (η5-C5Me4R)RhIII Complexes with Metalated Aryl Phosphines Featuring η4-Phosphorus plus Pseudo-Allylic Coordination, Organometallics, 2018, 37, 11–21 CrossRef CAS.
- J. Campos, E. Alvarez and E. Carmona, Synthesis and reactivity of half-sandwich (η5-C5Me5)Ir(III) complexes of a cyclometallated aryl phosphine ligand, New J. Chem., 2011, 35, 2122–2129 RSC.
- L. E. E. Broeckx, S. Güven, F. J. L. Heutz, M. Lutz, D. Vogt and C. Müller, Cyclometalation of Aryl-Substituted Phosphinines through C-H-Bond Activation: A Mechanistic Investigation, Chem. – Eur. J., 2013, 19, 13087–13098 CrossRef CAS PubMed.
- J. Campos, J. López-Serrano, E. Alvarez and E. Carmona, Cationic Ir(III) Alkylidenes Are Key Intermediates in C-H Bond Activation and C-C Bond-Forming Reactions, J. Am. Chem. Soc., 2012, 134, 7165–7175 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2390209–2390211. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi02763a |
| This journal is © the Partner Organisations 2025 |