
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparison of PGSE NMR and ESI-MS measurements on methylaluminoxane†
Luca Rocchigiani *a,
Scott Collinsbc,
J. Scott McIndoeb and
Mikko Linnolahti*c
*a,
Scott Collinsbc,
J. Scott McIndoeb and
Mikko Linnolahti*c
aDepartment of Chemistry, Biology and Biotechnology and CIRCC, University of Perugia, Via Elce di Sotto 8, 06123, Perugia, Italy. E-mail: luca.rocchigiani@unipg.it
bDepartment of Chemistry, University of Victoria, 3800 Finnerty Road, Victoria, BC V8P 5C2, Canada
cDepartment of Chemistry and Sustainable Technology, University of Eastern Finland, Joensuu Campus, Yliopistokatu 7, FI-80100, Joensuu, Finland. E-mail: mikko.linnolahti@uef.fi
First published on 22nd March 2025
Abstract
PGSE NMR and ESI-MS studies of different grades of hydrolytic methylaluminoxane (MAO) demonstrate a correlation between the average dimensions and anion distribution of MAO. This correlation is revealed through aging and gelation studies of a commercial formulation of MAO. Formulations featuring an anion distribution skewed to higher m/z ratios have been observed to contain significantly higher activator contents as measured by 1H NMR spectroscopy, despite being otherwise very similar. PGSE NMR studies in toluene vs. chlorobenzene media demonstrate that the average hydrodynamic dimensions of MAO are largely unaffected by solvent polarity, although aggregation behavior is somewhat sensitive to solvent polarity. As for catalyst activation and ion-pair speciation, the situation in polar solvents is complicated by the formation of solvated cations (and anions) in chlorobenzene which dramatically lower dimensions. DFT studies of model aluminoxane structures in the size range of MAO featuring a variety of architectures, molar masses and Me3Al contents reveal a linear correlation between Dt, as measured by PGSE NMR, and molar mass using simple relationships and estimates of molecular volume if suitable high molecular weight standards are available. There is reasonable agreement in molar mass with available ESI-MS data, recognizing that MAO is not monodisperse.
Introduction
The structure and properties of methylaluminoxane (MAO)1,2 continue to attract interest in the context of olefin polymerization using metallocene catalysts.3–5 After nearly 50 years of research, the basic mechanism of action of MAO is clear, yet the structure of the reactive components and activation mechanisms are not fully understood.Theory suggests that the bulk of MAO consists mainly of relatively unreactive structures built up of tetrahedral O3AlMe and trigonal O groups in six-membered or larger (MeAlO)n rings.6,7 Reactive structures consist of strained cages, analogous to those adopted by t-butylaluminoxanes,8 especially those with vicinal (MeAlO)2 rings,9 extended cages,10 nanotubes11,12 and two-dimensional sheet models13 with reactive sites incorporating structural Me3Al.14
Recently, a component of this complex mixture has been finally isolated from hydrolytic MAO (h-MAO) and characterized by X-ray crystallography.15 It has a sheet structure with the formula (MeAlO)26(Me3Al)9 (hereinafter 26,9) and a molecular weight of 2156 g mol−1 and was used to activate metallocenes.
The average molecular weight and presumably the molecular weight distribution of h-MAO16 are known to be important in metallocene catalyst activation. Low MW species such as (Me2AlOAlMe2)n (n = 3 to 4)17 are known to be ineffective,18 while early synthetic methods emphasize the formation of higher MW materials as confirmed through cryoscopic measurements.19 Cryoscopic measurements of commercial h-MAO (W. R. Grace)20 suggest a (number) average MW (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n) of ca. 1100 g mol−1 while small angle neutron scattering (SANS) studies of h-MAO (Chemtura/Lanxess) suggest a higher (weight) average MW (w) of 1803 ± 55 g mol−1 with a spread in values between 1400 and 2800 g mol−1 depending on the free Me3Al content (w = 1803 corresponds to ca. 75% of the total Me3Al content for the sample in question).21 The same study suggested a roughly spherical shape for bulk MAO based upon rg = 9.3 and rH = 11.7 Å as measured by SANS and PGSE NMR, respectively. Both estimates were in good agreement with earlier studies of T1 relaxation (rg = 9.7 Å)22 and [Cp2ZrMe2AlMe2][MAO(Me)] (1) ion-pair size (rH = 12–12.5 Å) by PGSE NMR.23 However, the interpretation of the PGSE NMR data in terms of molar mass (50–60 Al atoms for MAO21 vs. 150–200 Al atoms for ion-pair 1
n) of ca. 1100 g mol−1 while small angle neutron scattering (SANS) studies of h-MAO (Chemtura/Lanxess) suggest a higher (weight) average MW (w) of 1803 ± 55 g mol−1 with a spread in values between 1400 and 2800 g mol−1 depending on the free Me3Al content (w = 1803 corresponds to ca. 75% of the total Me3Al content for the sample in question).21 The same study suggested a roughly spherical shape for bulk MAO based upon rg = 9.3 and rH = 11.7 Å as measured by SANS and PGSE NMR, respectively. Both estimates were in good agreement with earlier studies of T1 relaxation (rg = 9.7 Å)22 and [Cp2ZrMe2AlMe2][MAO(Me)] (1) ion-pair size (rH = 12–12.5 Å) by PGSE NMR.23 However, the interpretation of the PGSE NMR data in terms of molar mass (50–60 Al atoms for MAO21 vs. 150–200 Al atoms for ion-pair 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23) differed significantly between the two studies.
23) differed significantly between the two studies.
The source and Me3Al content of MAO are important variables as revealed by PGSE NMR studies on h-MAO (Sigma Aldrich). In particular, dried or chemically modified MAO (DMAO and MAO-BHT) and the ion-pairs formed from Cp2ZrMe2 and DMAO had significantly larger dimensions than the h-MAO itself.24 Limiting values of rH = 8.6 and 10.3 Å for this MAO and ion-pair 1 were significantly lower than estimates from other studies where the sources of h-MAO were different.
PGSE NMR experiments yield translational diffusion coefficients Dt in the solvent used but require the use of an internal standard, either deliberately added or the solvent itself, to extract size information using the Stokes–Einstein equation or variations (e.g. eqn (1), where k = Boltzmann constant, T = temperature, c and fs are the size and shape-dependent factors, η = solution viscosity, and rH = hydrodynamic radius).25 However, the relationship between the hydrodynamic volume and molar mass is never straightforward.
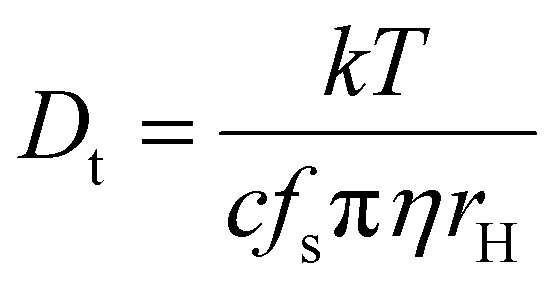 | (1) |
A more useful approach is to directly relate the measured diffusion coefficients to molar mass, by establishing empirical correlations between Dt and species of known molar mass and similar density.26 In the case of lower MW solutes, a relationship incorporating both size and molar mass has been established (eqn (2)),27 and it appears to be applicable to MAO.28
 | (2) |
Here D1 and D2 refer to the diffusion coefficients of the solute and solvent (or standard), while ri represents the van der Waals radii of these two molecules and mi represents the corresponding molar masses. It should be noted from the original study establishing the correlation in eqn (2) that the masses of the solutes and solvents were on the same order of magnitude.27 This is generally not true for MAO solutions.
The widespread availability of NMR spectrometers with pulse gradient capability suggests an accessible and powerful approach to MAO or ion-pair speciation,29 particularly if the results could be directly related to absolute molar mass.
Mass spectrometric characterization of MAO dates back to early methods of its synthesis, but only volatile, lower molecular weight components were detected using EI-MS.30 Detection and characterization of high MW ions in h-MAO using ESI-MS31 and a donor such as octamethyltrisiloxane (OMTS) in a polar solvent (fluorobenzene) were reported in 2013.32
This initial study involved the analysis of h-MAO from Sigma Aldrich (Fig. 1a). The Lewis base donor OMTS reacts with MAO according to Scheme 110,14,33 where n,m and [n,m]− are shorthand notations for (MeAlO)m(Me3Al)n and [(MeAlO)m(Me3Al)nMe]−, respectively.
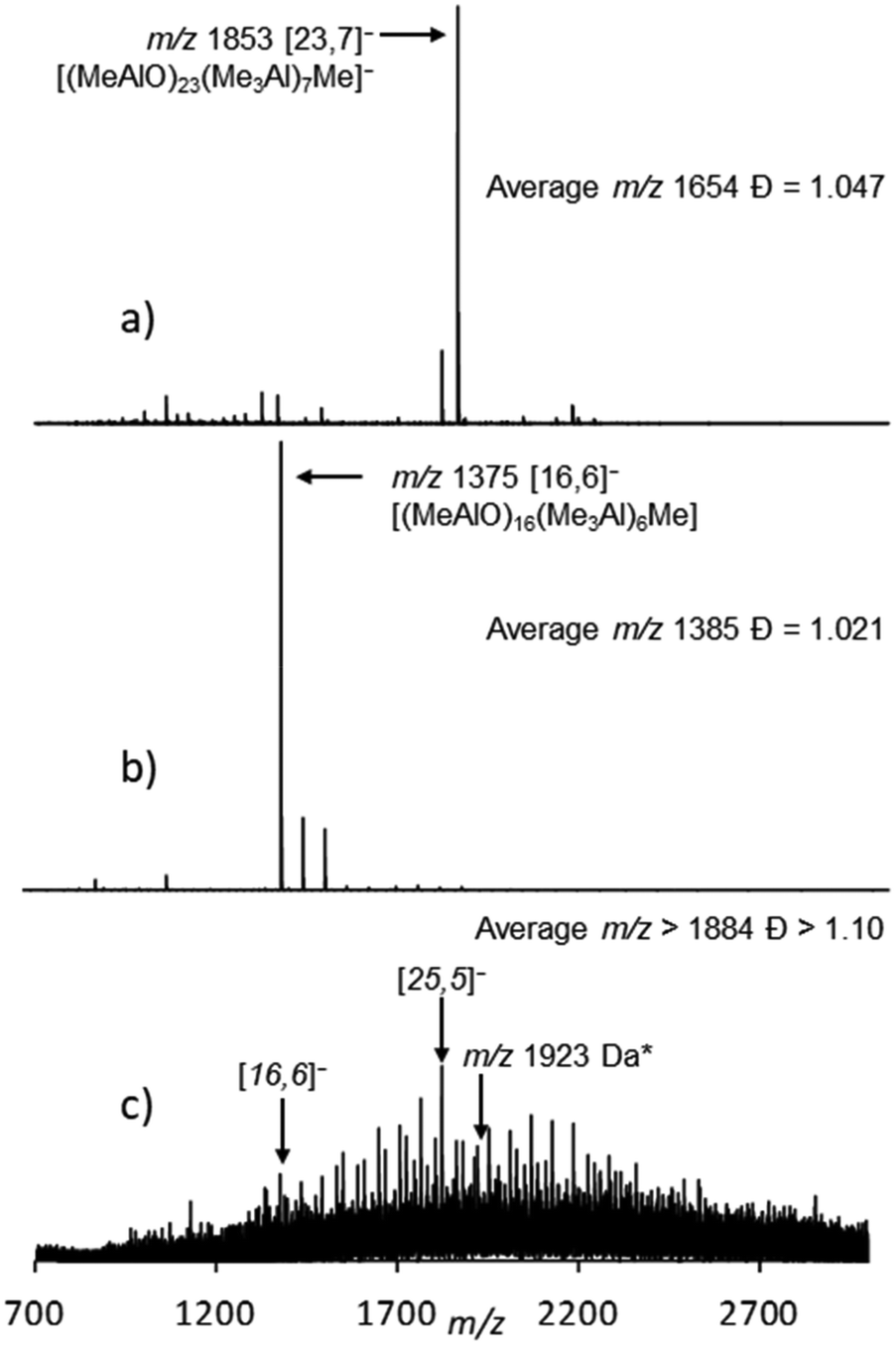 | ||
| Fig. 1 Negative ion mass spectra of hydrolytic MAO: (a) 10 wt% Sigma Aldrich, (b) 10 wt% W. R. Grace and (c) MMAO 12 (10 wt% Sigma Aldrich) in the presence of 10.0 mol% OMTS in PhF solution with [Al] ≈ 0.01 M. Assignments are based on MS/MS experiments and nominal mass (a and b) or just nominal mass (c). * The peak with m/z 1923 Da is assigned to the [(MeAlO)24(n-C8H17AlO)(Me3Al)5Me]− anion. For details, see ref. 32, 34 and 39. | ||
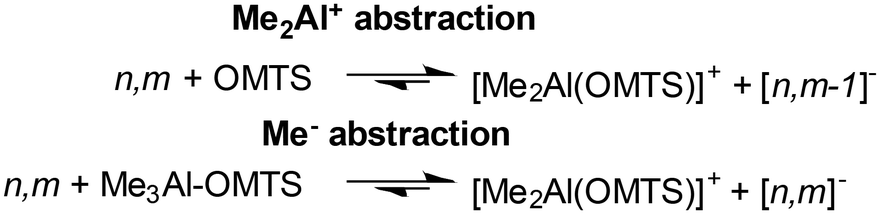 | ||
| Scheme 1 Ion-pair formation involving MAO and OMTS. | ||
Both the cations and anions formed can be separately detected by ESI-MS. The individual anions can be analyzed by MS–MS to determine their formula (i.e. [n,m]−). In the case of Sigma Aldrich MAO, the spectrum was dominated by an anion with m/z 1853 with the formula [23,7]−. We also provide here the number-average m/z ratio  and polydispersity
and polydispersity  of this mixture where (m/z)i and Ii are taken from the raw MS data files of ion intensity (I) vs. m/z ratio. The average m/z ratio and principal anion present in MAO from Sigma Aldrich were in good agreement with SANS estimates of w for h-MAO from Chemtura.21
of this mixture where (m/z)i and Ii are taken from the raw MS data files of ion intensity (I) vs. m/z ratio. The average m/z ratio and principal anion present in MAO from Sigma Aldrich were in good agreement with SANS estimates of w for h-MAO from Chemtura.21
More recently, 10 wt% h-MAO from W. R. Grace was analyzed by ESI-MS and the results were quite different.34 At the same additive level (10 mol% OMTS) the Grace material featured significantly lower m/z anions (Fig. 1b) with a significantly lower average m/z ratio, while the appearance of these spectra was very sensitive to the amount of OMTS present. This study also revealed that if h-MAO solutions from W. R. Grace were stored at room temperature, the anion distribution slowly changed from that shown in Fig. 1b to one similar to that shown in Fig. 1a.
Storage of h-MAO solutions at room temperature is accompanied by gelation and eventual loss of activity (aging).1,2,35 The stability to and extent of gelation are dependent on the total Al concentration and the amount of Me3Al present. With the benefit of hindsight, the material from Sigma Aldrich (which always required filtering to remove gel particles prior to analysis by ESI-MS) had been aged at room temperature prior to and during shipment, as confirmed by this vendor in 2014 for this material sold in North America.36
Much more recently, MMAO 12 (Sigma Aldrich), a 95:5 copolymer of methyl- and n-octylaluminoxane,37 was analyzed by low- and high-resolution MS using an inert atmosphere sampling technique.38 Families of ions with m/z ratios <1000 Da were detected and they featured hydrolysis, possible pyrolysis, and no evidence for incorporation of n-octyl groups.
The same material was also earlier analyzed by ESI-MS using 10 mol% OMTS.39 A broad continuum of anions40 with m/z ranging from ∼1000 to >3000 Da were detected with evidence of n-octyl substitution (Fig. 1c) while the corresponding positive ion spectrum showed the [Me2Al(OMTS)]+ and [(n-C8H17)MeAl(OMTS)]+ cations in relative amounts (ca. 97:3) consistent with the composition of MMAO 12.
It is evident from the foregoing discussion that a variety of different MAO samples have been analyzed by PGSE NMR, ESI-MS and other techniques (e.g. SANS) which are sensitive to or measure molar mass. Unfortunately, none of these studies involved the analysis of the same sample of MAO by all of these techniques. Also, given the difficulty in relating hydrodynamic dimensions from PGSE NMR directly to molar mass, we wished to establish a linear correlation between Dt and MW for MAO as in the work of Zaccaria et al. for other materials.
To address these issues we analyzed MAO from W. R. Grace by PGSE NMR spectroscopy as we have extensive experience with the ESI-MS analysis of that material, both 10- and 30 wt% formulations.10,34 These formulations have very similar ESI-MS spectra at similar OMTS levels, while different batches also have very similar spectra as long as they are shipped and stored refrigerated (as they were in all of our prior work – see the ESI† for examples).
We were also interested in whether the metallocene ion-pairs formed from this material had similar dimensions as seen in earlier work, and whether their dimensions were sensitive to solvent polarity as the ESI-MS work, unlike previous PGSE NMR studies, was conducted in a polar solvent.
Finally, we have also studied the aging/gelation of MAO by both ESI-MS and PGSE NMR and show that this process can be studied by either technique. We also establish that h-MAO with different anion distributions can have significantly different activator content.
Results and discussion
Relationship between molar mass and Dt for MAO
It is evident, based upon both this work and previous literature studies of MAO and the ion-pairs derived from it, that a reliable estimate of molar mass from Dt values is needed to relate PGSE NMR experiments to the limited ESI-MS (or MS) data that are available. The linear correlation reported in ref. 26 is not appropriate in the sense that the density of the compounds used to construct it were all ≤1 g mL−1 while MAO and the ion-pairs derived from it are expected to have higher densities. For example, the crystalline density of 26,9 is 1.26 g mL−1 although toluene is incorporated into the lattice.15For the calibration, we selected representative structures for MAO comprising reasonably stable sheets, cages and nanotubes varying in molar mass between 750 and 2000 g mol−1, including two unreported sheet and cage structures corresponding to the neutral compound 23,7 and some 16,4 tube structures located at the M06-2X/TZVP level of theory. We also included the structure of the 26,9 sheet15 after establishing the correlation reported here. The structures of any unreported aluminoxane models are depicted in Fig. 2. We use the polyhedral octasilsesquioxane (POSS) compounds studied previously by PGSE NMR21,26 as standards based on their similar shapes, high molar mass and calculated densities.
 | ||
| Fig. 2 Structures of some new models of MAO (Al pink, O red, C gray) with H-atoms omitted for clarity. Tetrahedral AlO4 groups are highlighted in light blue for the cage 23,7 structure. For coordinate files and energies, see ESI.† | ||
The properties of the aluminoxane models, along with those of the POSS standards are summarized in Table 1. Their van der Waals volumes (VvdW) and those of the reference compounds were estimated using the Connolly method41 with a probe radius of 1.4 Å as implemented in Chem3D.42 We chose this convenient approach as the Connolly solvent excluded volumes of the compounds, cholesterol, camphor, neopentane and methane were in very good agreement with the van der Waals volumes previously estimated by the method of Bondi.43 The calculated densities for the aluminoxane models including that for sheet 26,9 vary between 1.55 and 1.72 g mol−1 (Table 1, entries 1–14) while the POSS standards have somewhat lower calculated densities (entries 15–17).
| Entry | Structure | ΔEa (kJ mol−1) | MW (g mol−1) | VvdWb (Å3) |
ρcalc.c (g mL−1) |
Dtd |
|---|---|---|---|---|---|---|
| a Electronic energy differences at the M06-2X/TZVP level of theory. For complete energies, see Table S1 in the ESI† and for coordinates, see ESI, Table S1.xyz.†b van der Waals volume calculated using a probe radius of 1.4 Å and Chem3D.c Density calculated from molar mass and VvdW.d Experimental or calculated diffusion coefficient (10−10 m2 s−1) estimated from eqn (2) using Ti[O2(i-Bu8Si8O11)]2 as a reference.e Molar mass and volume data from ref. 21. | ||||||
| 1 | 8,4 5C-sheet | 30.2 | 753.48 | 792 | 1.58 | 10.5 |
| 2 | 8,4 4C-sheet | 48.1 | 753.48 | 809 | 1.55 | 10.4 |
| 3 | 8,4-cage | 24.3 | 753.48 | 760 | 1.65 | 10.8 |
| 4 | 8,4-tube | 0.0 | 753.48 | 786 | 1.59 | 10.6 |
| 5 | 12,5 sheet | 7.3 | 1057.63 | 1112 | 1.58 | 7.5 |
| 6 | 12,5 cage | 0.0 | 1057.63 | 1061 | 1.65 | 7.8 |
| 7 | 16,4 3,1 tube | 18.6 | 1216.59 | 1201 | 1.68 | 6.8 |
| 8 | 16,4 2,2 tube | 55.3 | 1216.59 | 1171 | 1.72 | 6.9 |
| 9 | 16,4 4,0 tube | 93.7 | 1216.59 | 1202 | 1.68 | 6.8 |
| 10 | 16,6 sheet | 9.4 | 1360.77 | 1367 | 1.65 | 5.9 |
| 11 | 16,6 cage | 0.0 | 1360.77 | 1439 | 1.57 | 5.7 |
| 12 | 23,7 sheet | 77.2 | 1838.96 | 1895 | 1.61 | 4.1 |
| 13 | 23,7 cage | 0.0 | 1838.96 | 1835 | 1.66 | 4.2 |
| 14 | 26,9 sheet15 | 2156 | 2253 | 1.59 | 3.7 | |
| 15 | (i-BuSi)8O11(OH)2 | 891.6 | 1095 | 1.35 | 5.3 | |
| 16 | (i-C8H17Si)8O1245 |
1493.6 | 1771 | 1.25 | 4.0 | |
| 17e | (i-C8H17Si)8O12 | 1332.5 | 1662 | 1.33 | 4.0 | |
| 18 | Ti[O2(i-Bu8Si8O11)]2 | 1828.1 | 1934 | 1.57 | 4.0 | |
It should be noted that the experimental liquid densities reported for (i-BuSi)8O12 and (i-OctSi)8O12 are 1.13 and 1.01 g mol−1.44 Our calculated densities are significantly higher as the VvdW value is smaller than molar volume and does not include free volume.43
The best correlation was obtained using the Ti[O2(i-Bu8Si8O11)]2 standard (R2 = 0.9985, Fig. 3) although good correlations with R2 = 0.9979 or 0.9876 were also obtained using (i-C8H17Si)8O1245 or (i-BuSi)8O11(OH)2, respectively. Only the Ti POSS complex actually lies close to the correlation line for the MAO models due to its similar calculated density.
 | ||
| Fig. 3 Log(Dt) vs. log(MW) for some models of MAO using Ti[O2(i-Bu8Si8O11)]2 as a reference to construct the red calibration line. Data for POSS compounds reported in ref. 21 and 26 are shown in blue with the correlation established in ref. 26 shown as a dashed blue line. | ||
Going forward we will make use of this correlation both with respect to PGSE NMR studies on MAO from W. R. Grace and discussion of prior literature work where appropriate. Typical experimental errors in the determination of Dt (and thus rH) are about ±3.0% so one can expect the same error in calculated MW.
As an example, original PGSE NMR data for 30 wt% Chemtura MAO had Dt = 3.1 ± 0.1 × 10−10 m2 s−1 at a limiting concentration of 380 mM (see the next section for studies of the effect of MAO concentration). Our correlation gives MW = 2865 ± 92 g mol−1 which is statistically lower but in reasonable agreement with the earlier estimate based on VH (ca. 3360 g mol−1).21 Both estimates are much higher than either the number- or weight-average estimates given in the literature, though the latter depends on free vs. associated Me3Al content,21 while the former is dependent on the amount of low MW materials present including Me3Al.20
It is important to mention that the average MW determined by PGSE NMR spectroscopy (or other techniques used to study diffusion, such as light scattering in the case of large macromolecules) is different for polydisperse materials such as synthetic polymers, including MAO. Studies of synthetic polymers have shown that the diffusion average MW (D) lies between the weight- and Z-average values of the molecular weight distribution but lies closer to w.46 Thus, it is not unexpected that an D value for commercial MAO is higher than either the number- or weight-average values measured by other techniques.
Characterization of 10 wt% MAO by PGSE NMR spectroscopy
A 10 wt% W. R. Grace sample was analyzed in toluene-d8 solution by preparing samples upon serial dilution of the commercial material. As in earlier work, MAO aggregates at higher concentrations lead to an increase in the calculatedD. The data obtained are summarized in Table 1, which includes the results obtained earlier for the material from Sigma Aldrich using the same techniques and instrumentation. Fig. 3 shows the changes in D as a function of [AlMe] concentration as measured by NMR. Within the error of these experiments, the material from W. R. Grace is marginally of higher MW than the Sigma Aldrich h-MAO sample studied previously, although not at the 95% confidence level. The tendency towards aggregation of the two commercial samples is also quite similar. In contrast, DMAO, which features a lower Me3Al content than either of the commercial samples (ESI, Fig. S1†), shows a much higher tendency to aggregate at the same concentrations, and this feature is responsible for its higher average MW.
Since we were interested in the influence of a polar solvent on these results, we also diluted the W. R. Grace h-MAO sample in chlorobenzene-d5. This solvent has a similar dielectric constant to PhF used in the ESI-MS work. In fact, chlorobenzene has been used as a solvent for ESI-MS experiments of this type.47 The Dt of this solvent could not be measured due to the overlap of residual 1H signals with toluene. Instead, the decay of the toluene Me resonances was used to estimate the Dt of toluene in this solvent assuming the viscosity of the mixture was the same as that of pure solvent (0.75 cP). Furthermore, the known cfsrH properties of toluene and the Chen equation48 were used to estimate cfs (fs assumed the same for both the solvent and solute) and thus rH for MAO in this solvent using the decay of the AlMe resonances.
The correlation we developed for D vs. Dt is not applicable in this more polar solvent. The results suggest that the hydrodynamic dimensions of MAO do not differ significantly in the more polar solvent (Table 2, entries 8–10 vs. 11–13), yet the higher D values derived from Dt (which are significantly smaller in this solvent compared with toluene) would suggest otherwise. We do note that the slope of VH vs. [AlMe] is significantly different in the two solvents (ESI†) suggesting that the aggregation tendency of MAO is increased in the more polar solvent.
| Sample NMR solvent | Entry | [AlMe]a (mM) | Dtb (10−10 m2 s−1) | rHb (Å) | Db (g mol−1) |
|---|---|---|---|---|---|
| a AlMe concentration calculated from the 1H NMR spectra.b Errors in Dt, rH and D are ±3.0%.c Data from ref. 24. DMAO was prepared by removal of excess Me3Al in vacuo.d Dt and rH were determined as described in the text. |
|||||
| Sigma Aldrich 10 wt% | 1 | 17.4 | 4.02 | 8.57 | 1860 |
| Benzene-d6c |
2 | 60.5 | 3.81 | 8.99 | 2040 |
| 3 | 184.5 | 3.89 | 8.82 | 1970 | |
| 4 | 299.3 | 3.63 | 9.41 | 2200 | |
| Sigma Aldrich DMAO | 5 | 10.0 | 3.58 | 9.52 | 2160 |
| Benzene-d6c |
6 | 55.0 | 3.47 | 9.79 | 2380 |
| 7 | 127 | 3.26 | 10.4 | 2640 | |
| W. R. Grace 10 wt% | 8 | 11.0 | 3.78 | 9.79 | 2060 |
| Toluene-d8 | 9 | 85.5 | 3.66 | 10.1 | 2180 |
| 10 | 377.0 | 3.54 | 10.4 | 2300 | |
| W. R. Grace 10 wt% | 11 | 31.2 | 3.30 | 9.23 | — |
| Chlorobenzene-d5d |
12 | 241.8 | 3.06 | 9.90 | — |
| 13 | 521.3 | 2.85 | 10.6 | — | |
Characterization of MAO-based ion-pairs by PGSE NMR spectroscopy
Table 3 summarizes the PGSE NMR results for the ion-pairs formed from the 10 wt% W. R. Grace sample and Cp2ZrMe2 including those reported earlier using DMAO derived from the Sigma Aldrich material.24 As shown in Scheme 2, both contact (2) and outer-sphere (1) ion-pairs are formed and are involved in slow exchange on the 1H NMR chemical shift time scale. | ||
| Scheme 2 Equilibria between Cp2ZrMe2, MAO and inner and outer-sphere ion-pairs 2 and 1. | ||
| Sample | |||||
|---|---|---|---|---|---|
| Entry | Species | Conc. (mM) | Dtb (10−10 m2 s−1) | rHb (Å) | Db (g mol−1) |
| a Excess Me3Al removed in vacuo, samples prepared in benzene-d6 ref. 24.b Errors in Dt, rH and D are ±3.0%.c Sample diluted with toluene-d8.d Sample diluted with chlorobenzene-d5; Dt and rH were determined as described in the text. |
|||||
| Sigma Aldrich DMAOa | |||||
| 1 | OSIP 1 | 0.63 | 2.89 | 12.3 | 3220 |
| 2 | ISIP 2 | 5.07 | 3.04 | 11.1 | 2960 |
| 3 | DMAO | 333 | 2.89 | 11.6 | 3220 |
| 4 | OSIP 1 | 0.10 | 3.22 | 10.3 | 2690 |
| 5 | ISIP 2 | 1.25 | 3.51 | 9.7 | 2330 |
| 6 | DMAO | 84 | 3.25 | 10.3 | 2650 |
| W. R. Grace 10 wt%c |
|||||
| 7 | OSIP 1 | 0.12 | 3.28 | 11.2 | 2610 |
| 8 | ISIP 2 | 1.23 | 3.69 | 10.0 | 2150 |
| 9 | MAO | 167 | 3.47 | 10.6 | 2380 |
| W. R. Grace 10 wt%d |
|||||
| 10 | OSIP 1 | 0.22 | 4.06 | 7.69 | — |
| 11 | ISIP 2 | 0.88 | 3.51 | 8.73 | — |
| 12 | MAO | 122 | 2.86 | 10.5 | — |
Chem3D Connolly volume calculations on both a cage and sheet model for OSIP 149 indicate that they have VvdW of 1660 and 1740 Å3 and have comparable densities of ρcalc. = 1.70 and 1.62 g mL−1 with models for MAO itself (Table 1) and so we have calculated D for these species from their measured Dt.
The results for the inner sphere ion-pair (ISIP) Cp2ZrMe-μ-Me-MAO 2 appear largely independent of the source of the MAO in use. The rh of ISIP 2 correlates with that of the MAO in use and the extent to which MAO aggregates at higher concentrations leads to parallel increases in the apparent size of 2 at higher concentrations as seen in earlier work.24 However, by comparing entry 5 with entry 8 the D of ISIP 2 is only slightly different at similar concentrations.
Outer-sphere ion-pair (OSIP) [Cp2ZrMe2AlMe2][Me(MAO)] 1 is much more prone to aggregation at higher concentrations to form ion quadruples and larger aggregates, as are all related ion-pairs of this type.24 The D of OSIP 1 is also not influenced by the source of the MAO in use (entry 4 vs. 7) though it would appear that the solvated dimensions are slightly different in toluene-d8 vs. benzene-d6 solution.
Although the dimensions of MAO appear largely unaffected by solvent polarity (Table 2), the same is not true for ion-pairs formed from MAO and Cp2ZrMe2. In chlorobenzene-d5, the dimensions of both ion-pairs are dramatically reduced, even though the size of h-MAO is largely unaffected (Table 3, entries 10–12). The size reduction is much more pronounced for OSIP 1 than for ISIP 2.
These results can be partially explained with reference to the equilibria shown in Scheme 2. While ion-pairs 1 and 2 are involved in slow exchange on the NMR time scale, diffusion is an even slower process, and the diffusion of each species influences the diffusion of all others related by the equilibria shown. In polar media, one can thus expect that OSIP 1 will be in exchange with solvated cations (and anions).50
Since the [Cp2ZrMe2AlMe2]+ cation has a much smaller size, the averaged dimensions of ion-pair 1 will be significantly reduced compared to the situation in toluene. The effect will even be seen for ISIP 2 but to a lesser extent as it is indirectly coupled to the third equilibrium. Alternately, this ion-pair is in direct equilibrium with Cp2ZrMe2 and MAO and the larger size of the latter species will counter the effect of the third equilibrium. In other studies,51 discrete contact ion-pairs like 2 were shown to be in equilibrium with σ-chlorobenzene complexes52 so it is possible that this equilibrium may also reduce the average dimensions of ion-pair 2 as well.
Comparison of ESI-MS results with PGSE NMR spectroscopy results
The negative ion ESI-MS spectra of 10 or 30 wt% MAO from W. R. Grace and metallocene complexes are also dominated by the same anion with m/z 1375 and formula [16,6]−.53–55 In positive ion mode using Cp2ZrMe2 one sees mainly the [Cp2ZrMe2AlMe2]+ cation with m/z 307 at Al:Zr ratios of 100:1 or greater.53,54 We also note that the average m/z ratio of the anion distribution is sensitive to the Al:Zr ratio.54,55 For ratios of 100:1 vs. 1000:1 the values are 1395 and 1484 Da, respectively (ESI, Fig. S2†). This means the nominal mass of the ion-pair 1 should vary between 1702 and 1791 Da, while that of ISIP 2, which is not directly detected by ESI-MS, would be between 1630 and 1719 g mol−1.
Obviously, there is a significant discrepancy between the MS results in fluorobenzene and the PGSE NMR results in toluene, well outside the experimental errors of either technique. While it is tempting to convert the Dt values in chlorobenzene-d5 to D values, OSIP 1 would end up lower in molar mass than ISIP 2 (1830 vs. 2330 g mol−1), while both would be much lower than that of the MAO in use (3270 g mol−1). This result is certainly not supported by any earlier work and as we have shown the correlation involving MAO does not appear to be applicable in this polar solvent.
The effect of polydispersity should also apply to the PGSE NMR results and thus one would expect D would be larger than that determined by ESI-MS. However, the ESI-MS results suggest that the polydispersity of the MAO-based anions, even in aged materials (e.g. Fig. 1a vs. Fig. 1b), is very narrow with Đ = w/n = 1.047 to 1.021 (see also ESI MAO.xlsx†). This suggests that the D values for both ion-pairs 1 and 2 should be larger but not much larger than those estimated by ESI-MS. Clearly this is not the case, so we discuss possible reasons for this discrepancy.
The number average m/z ratio (and any higher averages as well as Đ) calculated from the raw MS data is dependent on all anions being detected with the same sensitivity.56 For higher m/z anions, this cannot be true in a general sense given the intrinsic bias of MS towards lower m/z ions.40 This leads to a pronounced narrowing of the MWD and a bias towards lower m/z averages as measured by low resolution MS vs. other methods such as GPC.56 Even in the case of high-resolution techniques such as FT-ICR MS, the polydispersity of narrow MWD synthetic polymers is still underestimated by mass spectrometry.57
If there are also differences in surface activity between homologous ions even more pronounced intensity differences can be observed.58 For an example obtained under the same conditions as we use for the analysis of MAO, see ESI Fig. S3† where the sensitivity difference for [n-Bu4N]+ (m/z 242) vs. [(n-C18H37)4N]+ (m/z 1026) is ca. 150-fold at equivalent concentrations. If differences of this magnitude also pertain to the aluminoxane anions, particularly those with higher m/z ratios, it is easy to understand why n, w, etc. are underestimated using the ESI-MS data.
It is also possible that the higher D values measured using PGSE NMR reflect weak but reversible aggregation of the ion-pairs with other neutral components of MAO, even at the lowest concentrations examined. These interactions might be simply too weak to be detected in the ESI-MS experiment under the conditions used for the analysis of these mixtures (see the Experimental section).
In any event, the PGSE NMR results for the Grace 10 wt% MAO are in good agreement with the MW of an isolated, neutral component of that mixture,15 while the NMR estimates of MW reported here for the ion-pairs 1 and 2 seem reasonable given that for MAO itself. In particular, our NMR measurements and the correlation between Dt and D do not support the conclusion that OSIP 1 is very high in MW or very different from the MAO in use.
Aging of MAO determined by ESI-MS and PGSE NMR
Earlier we obtained ESI-MS and 1H NMR spectra of a commercial and a proprietary sample of h-MAO with nearly identical Al (13.7 and 13.6 wt%) and similar Me3Al contents (5.0 and 4.1 wt%), in the presence of OMTS and THF-d8 donors, respectively. Representative negative ion mass spectra are shown in Fig. S4† while Me3Al and [Me2Al(THF)2]+ contents as measured by NMR (Fig. S5†)21,59 are summarized in Table S2† as a function of storage time at room temperature.A 30 wt% commercial formulation ages much faster than a 10 wt% formulation (vide infra) and is accompanied by noticeable gel formation after only 2–3 weeks at room temperature. The proprietary sample with higher m/z anions gels faster under these conditions even though the anion distribution is not changing rapidly, a seemingly counter-intuitive result. That the two different anion distributions change at different rates is shown by comparing the average m/z ratio of each mixture vs. time (Fig. 5a, open vs. closed symbols).
If one recognizes that the proprietary sample has an initial composition (average m/z = 1709 Da) that is similar to the commercial sample at three weeks (average m/z = 1710 Da), the slower rate of aging of the former can be rationalized as being further located along the same growth curve (dashed line in Fig. 5a). Presumably the onset of gelation is also dependent on w60 of the material present and thus more rapid in the material with higher m/z anions.
Although we do not have spectra of these two samples over longer time periods, due to their pronounced tendency to gel, we have included some ESI-MS spectra of the 10 wt% W. R. Grace sample at six weeks and six months (Fig. S6†)34 which shows that this material has an anion distribution that resembles the aged proprietary formulation at longer times.
NMR experiments in THF-d8 demonstrate that the proprietary formulation with higher m/z anions has ca. 50% higher activator content based on the intensity of signals due to the [Me2Al(THF)2]+ cation. This difference is maintained for the aged samples, as both the sol and gel phases are soluble in excess THF or sufficient amounts of OMTS (ca. 2 mol% in PhF).
The only time dependent change seen is the total Me3Al and toluene content, which decreases in both samples (Fig. S5†), because these two volatile materials evaporate from screw-top vials inside the glovebox over the three-week period of these experiments.
To compare with these results, we monitored the aging of 10 wt% Grace formulation, which is much less prone to gelation, by PGSE NMR. In Table 4 we show Dt and size data at 0 to 140 days at room temperature with D values based on the correlation developed. In these experiments, undiluted MAO was employed and so even in unaged materials the dimensions and MW of the MAO are much larger due to reversible aggregation.24 Based on the data in Table 4 (entries 1–4) the increase in D scales in a linear fashion with [AlMe] (R2 = 0.997).
| Entry | Time (days) | [AlMe] (mM) | Dtb (10−10 m2 s−1) |
rHb (Å) | VHb (Å3) | Db (g mol−1) |
|---|---|---|---|---|---|---|
| a Samples diluted with toluene-d8 except where noted.b Errors in Dt, rH and D are ±3.0%, VH ±10.0%.c Undiluted sample.d Using a viscosity of 0.7 cP corresponding to a 10% wt MAO solution in toluene. |
||||||
| 1 | 0 | 11.0 | 3.78 | 9.79 | 3930 | 2070 |
| 2 | 0 | 85.0 | 3.66 | 10.09 | 4300 | 2180 |
| 3 | 0 | 377 | 3.54 | 10.40 | 4710 | 2300 |
| 4 | 0 | 2020c | 2.78d | 11.5 | 6370 | 3410 |
| 5 | 30 | 1980 | 2.65d | 12.0 | 7340 | 3690 |
| 6 | 60 | 1920 | 2.60d | 12.3 | 7750 | 3810 |
| 7 | 97 | 1800 | 2.60d | 12.3 | 7800 | 3810 |
| 8 | 140 | 1630 | 2.55d | 12.4 | 8100 | 3940 |
Based on the integral ratio of aluminoxane and Me3Al to toluene in this sample, the amount of soluble aluminoxane decreased by about 20% over 140 days due to gelation. Interestingly, methane formation61,62 was not detectable over this period in this (sealed) sample (Fig. S7†) suggesting that gelation occurs without chemical change at room temperature. Indeed, since the gel can be redissolved in a donor solvent, initial gelation must be a process involving non-covalent interactions which makes sense given the sheet structure for an isolated component of MAO. However, it seems plausible that physical gelation may be complicated by chemical cross-linking in samples where water is not completely excluded, for example, if exposed to the atmosphere in a glovebox with ppm background levels of water for long periods of time.
Over the 140-day period, the molar mass of the sol phase increases by about 15% which is lower than the 22% increase in the average m/z ratio for a 30 wt% formulation of the same MAO over 21 days. This is to be expected given the difference in concentrations. A comparison of the ESI-MS and PGSE NMR data is shown in Fig. 5b, where the dashed lines correspond to growth curves fit to the experimental data.
It is clear that the same process is being monitored by the two techniques although there is a systematic discrepancy, by more than a factor of two for the 10 wt% solution, between D and the average m/z ratio of these mixtures. Given that the m/z ratio is expected to track the n, while the critical MW for gelation (<3400 g mol−1 for the sol phase) will be correlated with w,60 and thus D, the magnitude of the discrepancy, is what one might expect for a polydisperse sample, bearing in mind the concentration-dependent aggregation of MAO in solution.
Conclusions
As outlined here, PGSE NMR and ESI-MS provide reasonably consistent data as to the average molar mass of MAO viz a viz the ion-pairs derived from this material and reactive donors. The most pronounced discrepancy is related to outer-sphere ion-pairs, which are known to be prone to self-aggregation24 and may also be susceptible to aggregation with the additional, neutral components of MAO. Future work should examine this hypothesis since it is one that can be experimentally tested, especially given the recent isolation of a reactive component of MAO.The differences between formulations can be detected using either technique, while we expect that 2D techniques like DOSY28 will provide information on molecular weight distribution, especially when complemented by data from ESI-MS (i.e. MMAO 12 vs. hydrolytic MAO).
Of the two methods, PGSE NMR is the more practical technique based on the availability of suitable instrumentation and expertise. Also, as it is a non-destructive solution technique, it can be used to study weak aggregation phenomena that are less easily studied in most mass spectrometric experiments.
Finally, we have established a useful, internally consistent, and reasonably precise relationship between the molar mass of MAO and Dt, now fully consistent with the experimental data which we hope will allow routine characterization of this material by NMR spectroscopy in non-polar solvents going forward.
Experimental section
Methylaluminoxane solution (10 or 30 wt% in toluene [Al] ≈ 1.8 or 5.4 M) was obtained from W. R. Grace and stored at −20 °C in a glove-box freezer. A sample of anhydrous and degassed THF-d8 was also provided by W. R. Grace for NMR studies of MAO aging. Octamethyltrisiloxane (OMTS) and Cp2ZrMe2 were obtained from Sigma Aldrich and used as received. Fluorobenzene (PhF) was obtained from Oakwood Chemicals Ltd, refluxed and distilled from CaH2 and stored over activated molecular sieves (4 Å) prior to use.Aging and activator content of 30 wt% MAO
A solution of 30 wt% MAO in toluene was added to 10 screw top vials containing 0.1 mL (ESI-MS) or 0.25 mL (1H NMR) of solution. The vials were sealed with Parafilm™ and analyzed after 0, 1-, 7-, 14- and 21-days of storage at room temperature in the glovebox.For the ESI-MS experiments, the contents of one vial were diluted with 1.0 mL of a solution of OMTS (21.2 mg in 10 mL of PhF). A 0.1 mL aliquot of the homogeneous solution was then diluted by a factor of 10 with dry PhF prior to analysis by ESI-MS: this solution was analyzed using a QTOF Micro spectrometer via pumping it at ca. 40 μL min−1 through PTFE tubing (1/16′′ o.d., 0.005′′ i.d.) to the ESI-MS probe and source using a syringe pump. The capillary voltage was set at 2900 V with source and desolvation gas temperatures set at 85 °C and 150 °C, respectively with the desolvation gas flow at 400 L h−1.
Raw MS data files of m/z ratio vs. ion intensity (counts) were imported into Excel and analyzed to calculate number- and weight-average m/z ratios and dispersity.56,57 Some results are deposited as ESI† for a sample of MMAO 12 and 30 wt% h-MAO at the different times analyzed (MAO.xlsx†).
For the NMR experiments, the contents of another vial were diluted with 1.0 mL of THF-d8 with stirring to mix the contents. After one hour, 0.6 mL of the diluted sample was then transferred to a 5 mm NMR tube equipped with an air-tight Teflon™ valve and analyzed by 1H NMR spectroscopy at 500 MHz on a Bruker Avance 500 instrument, using a 30° pulse-width and 5 s relaxation delay averaging 64 transients. The spectra were integrated before and after baseline subtraction of the broad MAO resonance as described in the literature21,59 to determine the total Al, Me3Al and [Me2Al(THF)2]+ contents.
PGSE NMR experiments
1H PGSE NMR measurements were performed using a double stimulated echo pulse sequence (dstegp3s1d) on a Bruker AVANCE III HD 400 spectrometer equipped with a SmartProbe™ with a Z-gradient coil, at 298 K without spinning. For polydisperse species, the dependence of the resonance intensity (I) on a constant waiting time and on a varied gradient strength (G) is described using the equationwhere I = intensity of the observed spin echo, (I0)i = intensity of the spin echo in the absence of a gradient corresponding to the i-th species, Dt = self-diffusion coefficient of the i-th species, Δ = delay between the midpoints of the gradients, δ = length of the gradient pulse, and γ = magnetogyric ratio. The shape of the gradients was rectangular, their lengths were 4–5 ms, and their strengths G were varied during the experiments. In the present case, we tested both monoexponential (i = 1) and biexponential (i = 2) treatments by fitting the decays of I versus G with a non-linear regression algorithm and the best model for each sample was chosen by comparing the fitting errors and R values (see the ESI† for the data). All spectra were recorded using 64 K points and a spectral window of 6400 Hz and they were processed with a line broadening of 1.0. Different values of Δ, G, and numbers of transients were used for different samples.
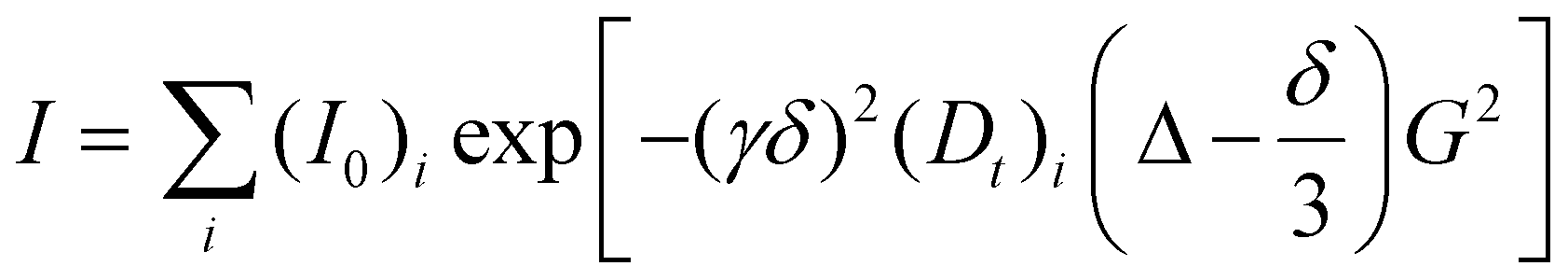
The self-diffusion coefficient Dt, which is directly proportional to the slope (m) of the exponential fitting obtained by plotting I versus G was estimated by evaluating the proportionality constant using a sample of HDO (5%) in D2O (known diffusion coefficients in the range 274–318 K)63 under identical conditions as for the sample of interest. The solvent was taken as an internal standard in the case of toluene-d8 measurements. In the case of chlorobenzene-d5, the overlap between aromatic signals hampered the precise estimation of Dt and hydrodynamic dimensions were obtained using dissolved toluene as an internal standard. Dt data were treated as described in the literature in order to derive the hydrodynamic dimensions.25 Experimental errors in rH are ±3.0% and ±10% for VH.
NMR samples for PGSE NMR measurements were prepared inside a glovebox by successive dilution. After having inserted the sample into the NMR probe, the sample was equilibrated for at least 30 minutes before starting the experiment to achieve optimal thermal stability of the solution. The actual concentration was estimated by integration relative to an external standard. The nominal concentration refers to Al–Me groups and considers both MAO and AlMe3.
Computational studies
DFT studies of aluminoxane structures employed the M06-2X density functional64 and TZVP basis set65 for geometry optimization. Gaussian 1666 was used for all calculations and stationary points were confirmed as minima using frequency calculations. Electronic energies and thermodynamic quantities for all structures are summarized in Table S1† while coordinate files of structures in Fig. 4 are deposited as the ESI (Table S1.xyz).†
 | ||
| Fig. 4 Diffusion average molecular weight (error bars are ±3.0%) vs. AlMe concentration for different MAO samples as measured by PGSE NMR. Data for the Sigma Aldrich samples are from ref. 24. | ||
 | ||
| Fig. 5 (a) Change in average m/z ratio of hydrolytic MAO vs. time (days). Commercial 30 wt% MAO (open symbols) and a proprietary formulation (black symbols) at 0, 1, 7, 14 and 21 days. The average m/z ratio was calculated from the raw ESI-MS data files of m/z (50–3000 Da) vs. intensity (counts). (b) Average m/z ratio (left hand y axis) and diffusion average MW (right hand y axis) vs. time for 30- and 10 wt% MAO from Grace. A function of the form y = a + b(1 − exp(−ct)) was fit to each set of data using non-linear regression. | ||
Author contributions
L. R. carried out PGSE NMR experiments. S. C. carried out ESI-MS and 1H NMR analyses of commercial and proprietary MAO formulations. S. C. and M. L. conducted quantum chemical calculations. S. C. devised the linear correlation between Dt and molar mass. The manuscript was written through contributions of all authors.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank W. R. Grace for providing all hydrolytic MAO samples studied here and for permission to publish the aging and composition study of 30 wt% MAO. We also thank Dr Jamie Strickler of W. R. Grace for providing information on the physical properties of MAO solutions and their gelation. L. R. acknowledges the financial support from the University of Perugia and the European Union–NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041−VITALITY. S. C. acknowledges the University of Victoria for a Visiting Scientist position and Prof. J. S. McIndoe for the use of facilities in connection with the aging study conducted as a consultant to Albemarle. M. L. acknowledges the support of the Research Council of Finland, decision 357509. DFT computations were made possible by the use of the Finnish Grid and Cloud Infrastructure resources (urn:nbn:fi:research-infras-2016072533). J.S.M. thanks NSERC (Strategic Project Grant #478998-15) and NOVA Chemicals Centre for Applied Research for operational funding and CFI, BCKDF, and the University of Victoria for infrastructural support.References
- H. S. Zijlstra and S. Harder, Methylalumoxane – History, Production, Properties, and Applications, Eur. J. Inorg. Chem., 2015, 19–43 CrossRef CAS.
- W. Kaminsky, Discovery of methylaluminoxane as cocatalyst for olefin polymerization, Macromolecules, 2012, 45, 3289–3297 Search PubMed.
- F. Zaccaria, L. Sian, C. Zuccaccia and A. Macchioni, Chapter One - Ion pairing in transition metal catalyzed olefin polymerization, in Advances in Organometallic Chemistry, ed. P. J. Perez, Academic Press, Cambridge, MA, 2020, vol. 73, pp. 1–78 Search PubMed.
- M. Bochmann, The chemistry of catalyst activation: The case of group 4 polymerization catalysts, Organometallics, 2010, 29, 4711–4740 CrossRef CAS.
- E. Y.-X. Chen and T. J. Marks, Cocatalysts for metal-catalyzed olefin polymerization: activators, activation processes, and structure− activity relationships, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed.
- E. Zurek and T. Ziegler, Theoretical studies of the structure and function of MAO (methylaluminoxane), Prog. Polym. Sci., 2004, 29, 107–148 CrossRef CAS.
- M. Linnolahti, T. N. P. Luhtanen and T. A. Pakkanen, Theoretical studies of aluminoxane chains, rings, cages, and nanostructures, Chem. – Eur. J., 2004, 10, 5977–5987 CAS.
- M. R. Mason, J. M. Smith, S. G. Bott and A. R. Barron, Hydrolysis of tri-tert-butylaluminum: the first structural characterization of alkylalumoxanes [(R2Al)2O]n and (RAlO)n, J. Am. Chem. Soc., 1993, 115, 4971–4984 CrossRef CAS.
- Z. Falls, N. Tyminska and E. Zurek, The Dynamic Equilibrium Between (AlOMe)n Cages and (AlOMe)n(AlMe3)m Nanotubes in Methylaluminoxane (MAO): A First-Principles Investigation, Macromolecules, 2014, 47, 8556–8569 CrossRef CAS.
- H. S. Zijlstra, A. Joshi, M. Linnolahti, S. Collins and J. S. McIndoe, Interaction of neutral donors with methylaluminoxane, Eur. J. Inorg. Chem., 2019, 2346–2355 CrossRef CAS.
- M. Linnolahti, J. R. Severn and T. A. Pakkanen, Are aluminoxanes nanotubular? Structural evidence from a quantum chemical study, Angew. Chem., Int. Ed., 2006, 45, 3331–3334 CrossRef CAS PubMed.
- M. Linnolahti, J. R. Severn and T. A. Pakkanen, Formation of nanotubular methylaluminoxanes and the nature of the active species in single-site α-olefin polymerization catalysis, Angew. Chem., Int. Ed., 2008, 47, 9279–9283 CrossRef CAS PubMed.
- M. Linnolahti and S. Collins, Formation, structure, and composition of methylaluminoxane, ChemPhysChem, 2017, 18, 3369–3374 CrossRef CAS PubMed.
- F. Zaccaria, C. Zuccaccia, R. Cipullo, P. H. M. Budzelaar, A. Macchioni, V. Busico and C. Ehm, On the Nature of the Lewis Acidic Sites in “TMA-Free” Phenol-Modified Methylaluminoxane, Eur. J. Inorg. Chem., 2020, 1088–1095 CrossRef CAS . and references therein.
- L. Luo, J. M. Younker and A. V. Zabula, Structure of methylaluminoxane (MAO): Extractable [Al(CH3)2]+ for precatalyst activation, Science, 2024, 384, 1424–1428 CrossRef CAS PubMed.
- To our knowledge, characterization of MAO by GPC has been attempted just once. D. Cam, E. Albizzati and P. Cinquina, Characterization of methylalumoxane by means of gel permeation chromatography, Makromol. Chem., 1990, 191, 1641–1647 CrossRef CAS.
- K. Kacprzak and J. Serwatowski, Organoboron water, part I: Synthesis and multinuclear magnetic resonance studies on the structure of tetramethyldialuminoxane, Appl. Organomet. Chem., 2004, 18, 394–397 CrossRef CAS.
- H. Sinn, I. Schimmel, M. Ott, N. von Thienen, A. Harder, W. Hagendorf, B. Heitmann and E. Haupt, Formation, structure and mechanism of oligomeric methylaluminoxanes (MAO), in Metalorganic Catalysts for Synthesis and Polymerization, ed. W. Kaminsky, Springer, Berlin, 1999, pp. 105–122 Search PubMed.
- W. Kaminsky and H. Hähnsen, Process for the preparation of oligomeric aluminoxanes, Eur. Patent Appl EP0108339A, 1984, 11 pp Search PubMed.
- D. W. Imhoff, L. S. Simeral, D. R. Blevins and W. R. Beard, Determination of Trimethylaluminum and Characterization of Methylaluminoxanes Using Proton NMR in Olefin Polymerization: Emerging Frontiers, in ACS Symposium Series, 1999, vol. 749, ch. 12, pp. 177–191 Search PubMed.
- F. Ghiotto, C. Pateraki, J. Tanskanen, J. R. Severn, N. Luehmann, A. Kusmin, J. Stellbrink, M. Linnolahti and M. Bochmann, Probing the structure of methylalumoxane (MAO) by a combined chemical, spectroscopic, neutron scattering, and computational approach, Organometallics, 2013, 32, 3354–3362 CrossRef CAS.
- E. W. Hansen, R. Blom and P. O. Kvernberg, Diffusion of Methylaluminoxane (MAO) in Toluene Probed by 1H NMR Spin–Lattice Relaxation Time, Macromol. Chem. Phys., 2001, 202, 2880–2889 CrossRef CAS.
- D. E. Babushkin and H.-H. Brintzinger, Activation of Dimethyl Zirconocene by Methylaluminoxane (MAO) - Size Estimate for Me-MAO− Anions by Pulsed Field-Gradient NMR, J. Am. Chem. Soc., 2002, 124, 12869–12873 CrossRef CAS PubMed.
- L. Rocchigiani, V. Busico, A. Pastore and A. Macchioni, Probing the interactions between all components of the catalytic pool for homogeneous olefin polymerisation by diffusion NMR spectroscopy, Dalton Trans., 2013, 42, 9104–9111 RSC.
- A. Macchioni, G. Ciancaleoni, C. Zuccaccia and D. Zuccaccia, Determining accurate molecular sizes in solution through NMR diffusion spectroscopy, Chem. Soc. Rev., 2008, 37, 479–489 RSC.
- F. Zaccaria, C. Zuccaccia, R. Cipullo and A. Macchioni, Extraction of reliable molecular information from diffusion NMR spectroscopy: hydrodynamic volume or molecular mass?, Chem. – Eur. J., 2019, 25, 9930–9937 CrossRef CAS PubMed.
- K. Rah, S. Kwak, B. C. Eu and M. Lafleur, Relation of tracer diffusion coefficient and solvent self-diffusion coefficient, J. Phys. Chem. A, 2002, 106, 11841–11845 CrossRef CAS.
- J. L. Eilertsen, R. W. Hall, L. S. Simeral and L. G. Butler, Tools and strategies for processing diffusion-ordered 2D NMR spectroscopy (DOSY) of a broad, featureless resonance: an application to methylaluminoxane (MAO), Anal. Bioanal. Chem., 2004, 378, 1574–1578 CrossRef CAS PubMed.
- L. Rocchigiani and A. Macchioni, Disclosing the multi-faceted world of weakly interacting inorganic systems by means of NMR spectroscopy, Dalton Trans., 2016, 45, 2785–2790 RSC.
- H. W. Sinn, W. R. Kaminsky, H.-J. C. Vollmer and R. O. H. H. Woldt, Preparing ethylene polymers using Ziegler catalyst comprising cyclodienyl compound of zirconium, U.S. Patent 4404344, 1983, 6 pp Search PubMed.
- B. M. Moscato, B. Zhu and C. R. Landis, GPC and ESI-MS Analysis of Labeled Poly(1-Hexene): Rapid Determination of Initiated Site Counts during Catalytic Alkene Polymerization Reactions, J. Am. Chem. Soc., 2010, 132, 14352–14354 CrossRef CAS PubMed , and references therein.
- T. K. Trefz, M. A. Henderson, M. Wang, S. Collins and J. S. McIndoe, Mass Spectrometric Characterization of Methylaluminoxane, Organometallics, 2013, 32, 3149–3152 CrossRef CAS.
- L. Luo, S. A. Sangokoya, X. Wu, S. P. Diefenbach and B. Kneale, Aluminoxane catalyst activators derived from dialkylaluminum cation precursor agents, processes for making same, and use thereof in catalysts and polymerization of olefins, US Patent 8575284B2, 2013, 18 pp Search PubMed.
- H. S. Zijlstra, M. Linnolahti, S. Collins and J. S. McIndoe, Additive and aging effects on methylalumoxane oligomers, Organometallics, 2017, 36, 1803–1809 CrossRef CAS.
- H. S. Zijlstra, M. C. A. Stuart and S. Harder, Structural investigation of methylalumoxane using transmission electron microscopy, Macromolecules, 2015, 48, 5116–5119 CrossRef CAS.
- S. Collins, personal communication with Sigma Aldrich USA, 2014.
- D. B. Malpass, Industrial Metal Alkyls and Their Use in Polyolefin Catalysts, in Handbook of Transition Metal Polymerization Catalysts, ed. R. Hoff, John Wiley & Sons, Inc., 2018, pp. 1–30 Search PubMed.
- A. Naim, M. Hubert-Roux, V. Cirriez, A. Welle, A. Vantomme, E. Kirillov, J.-F. Carpentier, P. Giusti and C. Afonso, Characterization of modified methylaluminoxane by ion mobility spectrometry mass spectrometry and ultra-high-resolution Fourier-transform ion cyclotron resonance mass spectrometry, New J. Chem., 2023, 47, 21244–21252 RSC.
- H. S. Zijlstra, A. Joshi, M. Linnolahti, S. Collins and J. S. McIndoe, Modifying methylalumoxane via alkyl exchange, Dalton Trans., 2018, 47, 17291–17298 RSC.
- I. C. Chagunda, G. T. Russell and J. S. McIndoe, The signal-to-noise issue in mass spectrometric analysis of polymers, Polym. Chem., 2021, 12, 4451–4461 RSC.
- M. L. Connolly, Computation of Molecular Volume, J. Am. Chem. Soc., 1985, 107, 1118–1124 CrossRef CAS.
- Chem3D Pro Version 21.0.0.28 ©1998-2022, Perkin Elmer Informatics Inc. Search PubMed.
- J. T. Edward, Molecular volumes and the Stokes-Einstein equation, J. Chem. Educ., 1970, 47, 261–270 CrossRef CAS.
- C. Dearmitt, Polyhedral Oligomeric Silsesquioxanes, in Functional Fillers for Plastics, ed. M. Xanthos, Wiley-VCH Verlag GmbH & Co., 2nd edn, 2010, DOI:10.1002/9783527629848.ch23, Ch. 23.
- It should be noted that in the ESI in ref. 21 the (i-C8H17Si)8O12 standard in use was actually a mixture of (i-C8H17Si)8O12 (58 mol%), (i-C8H17Si)10O15 (34 mol%) and (i-C8H17Si)12O16 (8 mol%) as determined by GPC. Thus, the average mass and van der Waals volume were calculated as 1493.6 g mol−1 and 1980 Å3, respectively in this work.
- B. R. White and G. J. Vancso, Diffusion average molar mass of polydisperse polymers: A photon correlation spectroscopy study of polystyrene in a good and a theta solvent, Eur. Polym. J., 1992, 28, 699–702 CrossRef CAS.
- M. Vatamanu, Observation of zirconium allyl species formed during zirconocene-catalyzed propene polymerization and mechanistic insights, J. Catal., 2015, 323, 112–120 CrossRef CAS.
- H.-C. Chen and S.-H. Chen, Diffusion of crown ethers in alcohols, J. Phys. Chem., 1984, 88, 5118–5121 CrossRef CAS.
- S. Collins and M. Linnolahti, Cages versus Sheets: A Critical Comparison in the Size Range Expected for Methylaluminoxane (MAO), ChemPhysChem, 2023, 24, e202300342 CrossRef CAS PubMed.
- Indeed, ESI-MS would not even be possible without an excess of these solvated ions at the droplet interface. See, for example, K. L. Vikse and J. S. McIndoe, Ionization methods for the mass spectrometry of organometallic compounds, J. Mass Spectrom., 2018, 53, 1026–1034 CrossRef CAS PubMed.
- L. Rocchigiani, G. Ciancaleoni, C. Zuccaccia and A. Macchioni, Low-Temperature Kinetic NMR Studies on the Insertion of a Single Olefin Molecule into a Zr-C Bond: Assessing the Counterion–Solvent Interplay, Angew. Chem., Int. Ed., 2011, 50, 11752–11755 CrossRef CAS PubMed.
- F. Wu, A. K. Dash and R. F. Jordan, Structures and Reactivity of Zr(IV) Chlorobenzene Complexes, J. Am. Chem. Soc., 2004, 126, 15360–15361 CrossRef CAS PubMed.
- T. K. Trefz, M. A. Henderson, M. Linnolahti, S. Collins and J. S. McIndoe, Mass spectrometric characterization of methylaluminoxane–activated metallocene complexes, Chem. – Eur. J., 2015, 21, 2980–2991 CrossRef CAS PubMed.
- S. Collins, M. Linnolahti, M. G. Zamora, H. S. Zijlstra, M. T. R. Hernández and O. Perez-Camacho, Macromolecules, 2017, 50, 8871–8884 CrossRef CAS.
- S. Collins and M. Linnolahti, Activation of Substituted Metallocene catalysts using Methylaluminoxane, ChemCatChem, 2022, 14, e202101918 CrossRef CAS.
- B. Thomson, K. Suddaby, A. Rudin and G. Lajoie, Characterization of low molecular weight polymers using matrix assisted laser desorption time-of-flight mass spectrometry, Eur. Polym. J., 1996, 32, 239–256 CrossRef CAS.
- E. P. Maziarz III, G. A. Baker and T. D. Wood, Capitalizing on the High Mass Accuracy of Electrospray Ionization Fourier Transform Mass Spectrometry for Synthetic Polymer Characterization: A Detailed Investigation of Poly(dimethylsiloxane), Macromolecules, 1999, 32, 4411–4418 CrossRef.
- L. Omari, P. Randhawa, J. Randhawa, J. Yu and J. S. McIndoe, Structure, anion, and solvent effects on cation response in ESI-MS, J. Am. Soc. Mass Spectrom., 2019, 30, 1750–1757 CrossRef PubMed.
- D. W. Imhoff, L. S. Simeral, S. A. Sangokoya and J. H. Peel, Characterization of methylaluminoxanes and determination of trimethylaluminum using proton NMR, Organometallics, 1998, 17, 1941–1945 CrossRef CAS.
- A. Rudin and P. Choi, Gel Formation During Copolymerization and Crosslinking, in The Elements of Polymer Science and Engineering, Academic Press, 2012, ch. 9, pp. 410–412 Search PubMed.
- N. E. Khrushch and N. M. Bravaya, Interaction of zirconocenes with polymethylalumoxane. Kinetics of methane liberation, J. Mol. Catal. A: Chem., 2000, 156, 69–78 CrossRef CAS.
- W. Kaminsky and C. Strübel, Hydrogen transfer reactions of supported metallocene catalysts, J. Mol. Catal. A: Chem., 1998, 128, 191–200 CrossRef CAS.
- H. J. W. Tyrrell and K. R. Harris, Diffusion in Liquids: A Theoretical and Experimental Study, Butterworth & Co. Ltd, London, 1984 Search PubMed.
- Y. Zhao and D. G. Truhla, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. Schäfer, C. Huber and R. Ahlrichs, Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SI.pdf – [Me2Al(THF)2]+ and Me3Al content of MAO samples and representative 1H NMR spectra. Additional ESI-MS spectra. Electronic energies and thermodynamic quantities for the MAO structures. PGSE NMR signal intensity decay data and non-linear regression analyses. Table S-2.xyz – Coordinates for all new theoretical MAO structures reported. MAO.xlsx – ESI-MS m/z ratio vs. intensity (counts) and m/z averages of h-MAO and MMAO 12. See DOI: https://doi.org/10.1039/d4qi02982h |
| This journal is © the Partner Organisations 2025 |