
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe X-ray structure of the adduct formed upon reaction of aurothiomalate with apo-transferrin: gold binding sites and a unique transferrin structure along the apo/holo transition pathway†
Romualdo Troisi a,
Francesco Galardoa,
Luigi Messorib,
Filomena Sicaa and
Antonello Merlino*a
a,
Francesco Galardoa,
Luigi Messorib,
Filomena Sicaa and
Antonello Merlino*a
aDepartment of Chemical Sciences, University of Naples Federico II, Complesso Universitario di Monte Sant'Angelo, Via Cintia, I-80126, Napoli, Italy. E-mail: antonello.merlino@unina.it
bDepartment of Chemistry “Ugo Schiff”, University of Florence, Via della Lastruccia 3, 50019 Sesto Fiorentino, Italy
First published on 31st January 2025
Abstract
The interaction of sodium aurothiomalate with the apo form of human serum transferrin (hTF) was studied by X-ray crystallography. The protein binds gold ions close to the side chains of His25, Asp63 and His249, His207 and Tyr238, and His273, His289, His300, His473, His585, His598, His606, and His642; the thiomalate ligand is released. Notably, the N- and C-lobes of one of the two hTF molecules in the asymmetric unit of the Au-hTF adduct crystals exhibit conformations that are slightly different from those observed in the “fully opened” apo-hTF, with the N-lobe that is intermediate between the “partially opened” form observed in the structure of hTF with Bi3+ and the “fully opened” form of apo-hTF. Thus, our data provide relevant information about the binding of gold centers to hTF and on a unique hTF structure along the apo-hTF/holo-hTF transition pathway.
Introduction
Aurothiomalate (Na2[AuL] where L is the thiomalate ligand [O2CCH2CH(S)CO2]3−) (Fig. 1) is a water-soluble prescription anti-arthritic gold drug1 used, alone or in combination with other drugs, to retard progressive cartilage destruction,2 but is currently being reconsidered as a candidate drug for cancer (ClinicalTrials.gov ID: NCT00575393, NCT01383668)3 and HIV4,5 treatments.6,7 Aurothiomalate has also been proposed as a colistin adjuvant for combating mcr-1-positive bacterial infections.8 Indeed, X-ray investigations reveal that, like in the case of other gold-based compounds, the gold ion of aurothiomalate can occupy the active site of the enzyme mcr-encoding transmembrane pEtN transferase (denoted as MCR-1), replacing the cofactor Zn2+.8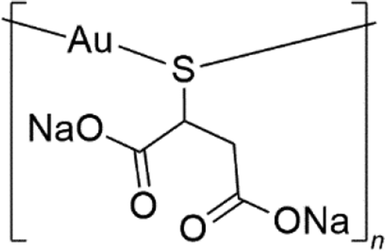 | ||
| Fig. 1 Chemical structure of sodium aurothiomalate. | ||
Commercial aurothiomalate is a variable mixture of the monosodium [NaAu(LH)] and disodium [Na2Au(L)] salts.9,10 Electrospray ionization mass spectrometry (ESI MS) studies on the commercial product showed that the molecule forms tetrameric Au4 species (Au4L4Na9+). Peaks attributed to Au4L4Na102+, AuL2Na6+ and AuL2Na5H+ are also observed in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 water–methyl cyanide solutions, plus ammonia,11 while Au4L4NaH8+, Au4L4Na2H7+, Au4L4Na3H6+, Au4L4Na4H5+, Au4L4Na5H4+, and Au4L4Na6H3+ species are also observed in ammonium acetate buffer at pH 6.8.12 Indeed, Na2[AuL] is polymeric in the solid state. Crystallographic and EXAFS studies indicated that, in the molecular structure, the gold(I) atom is bonded to two (bridging) S atoms forming two interpenetrating spirals, with an approximate 4-fold helical symmetry.13
:50 water–methyl cyanide solutions, plus ammonia,11 while Au4L4NaH8+, Au4L4Na2H7+, Au4L4Na3H6+, Au4L4Na4H5+, Au4L4Na5H4+, and Au4L4Na6H3+ species are also observed in ammonium acetate buffer at pH 6.8.12 Indeed, Na2[AuL] is polymeric in the solid state. Crystallographic and EXAFS studies indicated that, in the molecular structure, the gold(I) atom is bonded to two (bridging) S atoms forming two interpenetrating spirals, with an approximate 4-fold helical symmetry.13
As the biological effects of gold-based drugs seem to be mainly mediated by their recognition by proteins, studies on the interaction of aurothiomalate with these biological macromolecules are essential to discover its mechanism of action.14–16 According to DrugBank online (https://go.drugbank.com/drugs/DB09276), once administered, 85–90% of aurothiomalate is protein bound.
Within this framework, some of us evaluated the metalation pattern induced by aurothiomalate on some small model proteins, i.e. bovine pancreatic ribonuclease, horse heart cytochrome c and hen egg white lysozyme.12 ESI MS analysis revealed a non-covalent interaction of up to three tetrameric Au4 species ([AuL]48−) on the surface of these three proteins and the binding of monometallic gold ions (Au+ and [AuL]2−) or of gold-coordinated glutathione (GSH) molecules ([Au(GSH)2]) in the presence of GSH.12 Along the same line, ESI MS experiments carried out by Larabee, Hocker and Hanas17 showed that Au+ ions from Na2[AuL] coordinate the Sp1–3 peptide via one Cys thiolate (S−) and one Cys thiol (SH).
It has been suggested that the anticancer activity of aurothiomalate could be due to its recognition by a cysteine residue of the PB1 domain of protein kinase C.18 It has also been demonstrated that aurothiomalate binds and inhibits cathepsins K and S (that play central roles in the inflammatory and erosive components of rheumatoid arthritis).19 The X-ray structure of the adduct that is formed upon reaction of aurothiomalate with cathepsin K revealed that a gold atom binds the protease active site; the L ligand is missing.20 In contrast, Isab studied the interaction of aurothiomalate with ergothionine and showed that the compound can retain the metal ligand when it reacts with this molecule.21
Recently, 282 target proteins for aurothiomalate, including glutathione S-transferase omega 1, aminoacyl tRNA synthetase complex interacting multifunctional protein 2 (AIMP2), fatty acid synthase (FASN), protein disulfide isomerase family A member 3 (PDIA3), and thioredoxin reductase 1 (TXNRD1) have been identified by competitive activity-based protein profiling carried out using HeLa cell lysates.22
The interaction of aurothiomalate with serum proteins has also been studied. Iqbal and coworkers23 revealed that after 24 h incubation of the gold drug with blood, albumin (HSA) is the most metalated protein. This datum agrees with that obtained by Herrlinger and Weikert24 who found that 94% of gold is bound to HSA in patients treated with aurothiomalate. It has been proposed that gold atoms from Na2[AuL] could bind the side chain of Cys34 of HSA and that additional weaker binding could occur via non-covalent interactions on the protein surface.25 This view is in agreement with the idea that the major binding site for gold drugs in plasma is the Cys34 free thiol of HSA26 and with the crystallographic observation that Cys34 of bovine serum albumin binds gold ions.27 Recently, Cosottini et al.28 used ESI MS and ICP-OES to characterize the adduct formed when apoferritin reacts with aurothiomalate. This adduct contains approximately three gold atoms per ferritin subunit, arranged in a small cluster linked to Cys90 and Cys102.
Little is known about the interaction between aurothiomalate and another important serum protein: transferrin (hTF). hTF (679 residues, 78 kDa) is involved in the iron binding and delivery to all biological tissues.29,30 hTF contains two homologous domains (C-lobe and N-lobe), which are organized into two subdomains (N1: residues 1–92 and 247–330, N2: residues 93–246; C1: residues 340–425 and 573–679, C2: residues 426–572). Each lobe comprises an iron-binding site (residues Asp63, Tyr95, Tyr188, and His249 in the N-lobe and residues Asp392, Tyr426, Tyr517, and His585 in the C-lobe). In healthy humans, the serum concentration of hTF is about 25–50 μM with approximately 39% of the apo form (apo-hTF), 23% of the monoferric form with iron bound to the N-lobe (FeN-hTF), 11% monoferric form with iron bound to the C-lobe (FeC-hTF) and 27% diferric holo form (FeNFeC-hTF).31 Given its ability to be recognized by transferrin receptor 1 (TfR1), which is overexpressed in cancer cells, hTF has also been considered a potential metallodrug delivery system.32–35 It is known that hTF undergoes conformational changes upon iron binding and release: both N- and C-lobes are “fully opened” in the apo-hTF structure, while they are “fully closed” in the holo form.36–38 Three structures of intermediates along this pathway are known: the structures of two diferric-hTF (PDB codes 3QYT39 and 6CTC,40 FeN*FeC-hTF) and the structure of a mono-bismuth form of hTF (PDB code 4H0W,39 BiN*FeC-hTF). All three of these structures adopt a “partially opened” conformation of the N-lobe. Interestingly, FeN*FeC-hTF of the PDB code 6CTC and the BiN*FeC-hTF, in which only the Tyr188 binds the metal ion, have a more open conformation than the FeN*FeC-hTF of the PDB code 3QYT, where the Fe3+ coordinates not only with Tyr188 but also with Tyr95.
Here, in aiming to obtain information on the interaction between aurothiomalate and hTF, we registered intrinsic fluorescence and circular dichroism spectra of apo-hTF in the presence of aurothiomalate and solved the crystal structure of the adduct obtained upon the treatment of apo-hTF crystals with this gold-based drug (Au-hTF). The elucidation of the Au/hTF interaction is also important since hTF has been used to form protein corona on the surface of gold nanoparticles41,42 to improve the tumor targeting properties.43 The Au-hTF structure allows information on the gold binding sites of hTF to be obtained and provides details on the apo-hTF/holo-hTF transition pathway. Indeed, in one of the two hTF chains in the asymmetric unit of Au-hTF crystals, both the N-lobe and C-lobe have structures slightly different from that of the apo-form, with the N-lobe adopting a conformation that is intermediate between the “partially opened” forms, observed for BiN*FeC-hTF39 and the FeN*FeC-hTF of the PDB code 6CTC,40 and the “fully opened” form of apo-hTF.44
Results and discussion
Aurothiomalate binds apo-hTF in solution
First, we verified that the gold drug is able to bind apo-hTF by recording the intrinsic fluorescence spectra of the protein at 25 °C, upon excitation at 280 and 295 nm, in the presence of increasing amounts of aurothiomalate in 10 mM HEPES at pH 7.5. The spectra, reported in Fig. S1,† show that the fluorescence intensity of apo-hTF dropped steadily with increasing concentration of aurothiomalate. This finding indicates that the gold drug binds the protein.Circular dichroism spectra of apo-hTF in the presence of aurothiomalate in 10 mM HEPES at pH 7.5 were then recorded to evaluate if the binding of the gold-based drug could have an effect on the protein secondary structure. Superimposition of the spectra of the protein recorded in the absence and in the presence of the metal compound after 16 h (Fig. S2A†) and 5 days (Fig. S2B†) of incubation at 20 °C suggests that apo-hTF retains its secondary structure upon metal compound binding.
Overall structures of the adduct formed upon reaction of aurothiomalate with hTF
The reaction of aurothiomalate with apo-hTF was then studied in the solid state. Crystals of the Au-hTF adduct belong to the space group P212121; thus, they are isomorphous to those of apo-hTF (PDB code 2HAV44) and contain two molecules in the asymmetric unit, hereafter denoted as chain A and chain B (Fig. 2). The structure refines to Rfactor and Rfree values of 0.243 and 0.296, respectively, and contains 10649 atoms, including a 2-acetamido-2-deoxy-β-D-glucopyranoses (NAG) covalently linked to Asn413 (modeled only in chain B) (Fig. S3†), 8 citrate ions and 8 water molecules. The overall folding of the two protein chains does not appear to be significantly affected by the treatment with the gold compound: the two chains of Au-hTF show the typical bilobal nature of hTF superfamily members with the N-lobe (residues 1–330) and C-lobe (residues 340–679) separated by the peptide linker (residues 331–339) (Fig. 2). Root-mean-square deviation (rmsd) between Cα atoms of the two chains in the adducts is 1.39 Å, while the rmsd calculated against Cα atoms of apo-hTF (PDB code 2HAV44) is within the range of 0.96–1.77 Å. As expected, the analysis of the iron binding residues reveals the absence of coordinated Fe3+ ions (Fig. S4†).
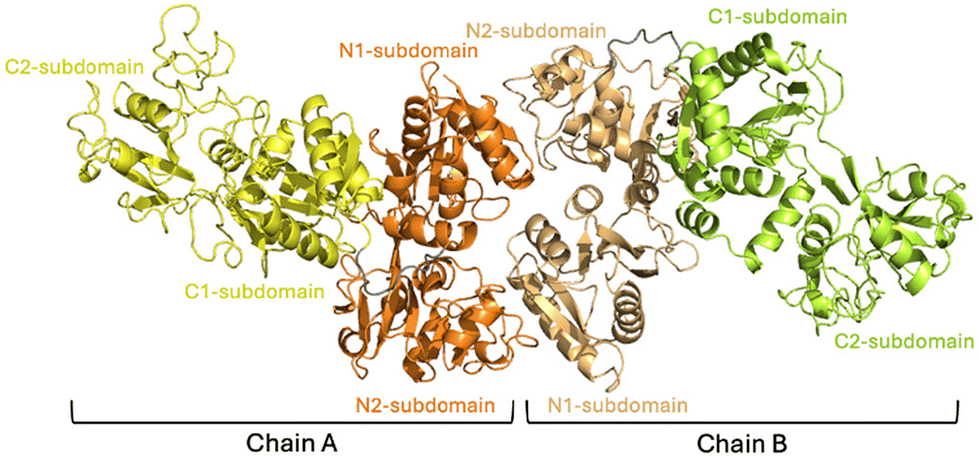 | ||
| Fig. 2 Representation of the asymmetric unit of Au-hTF. Lobes N and C of chain A are in orange and yellow, respectively. Lobes N and C of chain B are in light orange and light green, respectively. | ||
The N-lobe and C-lobe of Au-hTF chain A have non-canonical structures
It is known that binding and release of Fe3+ by hTF are accompanied by an opening/closure of the N- and C-lobes. In the apo and holo forms of hTF, the lobes adopt a “fully opened” or a “fully closed” conformation.In the N-lobe, upon Fe3+ binding, the N-terminal α-helix (α5, residues 129–135) pivots over α11 (residues 317–328), driving a rotation of the N1 subdomain over the N2 subdomain. Two structural intermediates between the “fully opened” and the “fully closed” forms of the N-lobe of hTF have been characterized while solving the structures of FeN*FeC-hTF and BiN*FeC-hTF.39
The structures of the N-lobes of the two Au-hTF chains are not identical (rmsd = 0.47 Å). A comparison with the N-lobes of apo-hTF, holo-hTF, and intermediate structures reported in the literature is shown in Fig. 3 and Fig. S5.† In Fig. 3A, the N2 subdomain of Au-hTF chain A is superimposed onto those of FeN*FeC-hTF (PDB code 3QYT39), BiN*FeC-hTF (PDB code 4H0W39), apo-hTF (PDB code 2HAV44) and holo-hTF (PDB code 3V8345). Details of the superimposition reporting the key residues in the N-lobe iron binding center are shown in Fig. 3B. Superimposition of the N2 subdomain of Au-hTF chain B onto the same structures is shown in Fig. S5.†
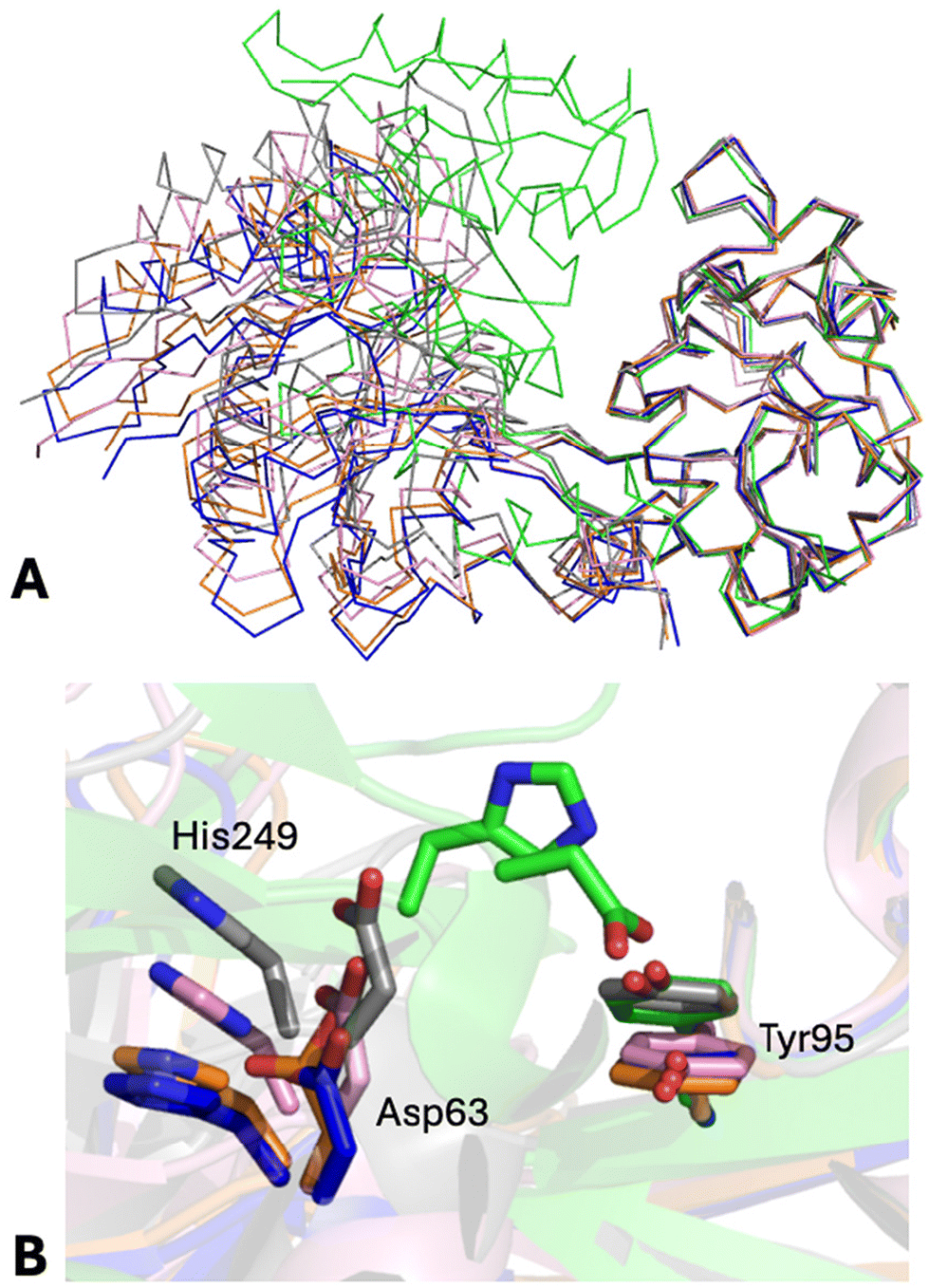 | ||
| Fig. 3 (A) Representation of the Cα trace of the N-lobes of Au-hTF chain A, apo-hTF (PDB code 2HAV, chain A), FeN*FeC-hTF (PDB code 3QYT), BiN*FeC-hTF (PDB code 4H0W), and holo-hTF (PDB code 3V83, chain A) after superimposition of the N2 subdomain. “Fully closed” (holo-hTF) and “fully opened” (apo-hTF) conformations are in green and blue, respectively. The “partially opened” conformations of hTF observed in FeN*FeC-hTF and BiN*FeC-hTF structures are in gray and pink, respectively. The structure of Au-hTF chain A is in orange. (B) Position shifts of the key residues in the N-lobe iron binding center are shown as stick models. | ||
As it can be clearly seen from Fig. 3, the N-lobe of Au-hTF chain A adopts a conformation that is intermediate between those of apo-hTF and BiN*FeC-hTF forms, thus representing a new intermediate in the apo-hTF/holo-hTF transition pathway, which describes the first motion step of the N-lobe subdomains, while the N-lobe of Au-hTF chain B adopts a conformation that is rather similar to those of apo-hTF N-lobes. To analyze the differences between the N-lobes of the above-mentioned structures, pairwise comparisons were performed by calculating the rmsd and the rotation angle (χ) that are required to optimally superimpose N2 subdomains after the best fitting of N1 subdomains. Initial analyses conducted by comparing rmsd values confirm that the N-lobe of Au-hTF chain B is more similar to the N-lobes of apo-hTF (rmsd = 0.44–0.52 Å) when compared to the N-lobe of chain A (rmsd = 0.64–0.74 Å). The rmsd values of the N-lobe of chain A with the apo-hTF N-lobe are intermediate (rmsd = 0.64–0.74 Å) between those found when comparing the N-lobes of the apo-hTF structure (rmsd = 0.47–0.62 Å) and those against the “partially opened” structures BiN*FeC-hTF (rmsd = 0.91 Å) and FeN*FeC-hTF (rmsd = 1.62 Å).
The analysis of the χ values agrees with these observations (Table S1†). Indeed, the χ value for the apo-hTF/holo-hTF transition for the N-lobes is about 60°; the values observed for BiN*FeC-hTF to apo-hTF and for FeN*FeC-hTF to apo-hTF are ∼7° and ∼13°, respectively, while that of Au-hTF to apo-hTF is 4° in chain A and about 2° in chain B, further supporting the idea that the N-lobe of Au-hTF chain A has structural features that are intermediate between the “fully opened” apo-hTF and the “partially opened” hTF structures, while the N-lobe of chain B is almost “fully opened”. As reference values, it should be considered that the comparison between the N-lobes of the apo-hTF structures deposited with the PDB leads to an average χ value of 1.1° ± 0.3° and the rmsd values between the N-lobes of apo-hTF structures are within the range of 0.47–0.62 Å. In this respect, it should be recalled that in our structure a “fully opened” and a “partially opened” form of the N-lobe coexist in the asymmetric unit of the same crystal, under identical experimental conditions.
A similar analysis has also been carried out for the C-lobes. Upon Fe ion binding, hTF C-lobes adopt a “fully closed” conformation with a rotation of the C1 subdomain over the C2 subdomain, which is 42.1° ± 0.5° for the monoferric FeC-hTF forms and 50.3 ± 0.4° for the “fully closed” holo-hTF conformations (Table S1†). Up to now there are no described hTF models with a C-lobe adopting a structure intermediate between the apo and the holo forms, excluding the complex between hTF and the membrane protein TbpA, where the C-lobe adopts a conformation midway between “opened” and “closed”, with a 24° rotation required to align C1 and C2 subdomains with holo-hTF.45
The C-lobes of Au-hTF present similar rmsd values to those of the structure of the apo forms (rmsd = 0.72–0.91 Å). However, a deeper inspection of the structures based on the best fitting of the C2 subdomain after superimposition of C1 suggests that the C-lobe of Au-hTF chain A is not identical to that of apo-hFT. Indeed, the χ angle needed to superimpose the C2 subdomain after the best fitting of C1 in the case of Au-hTF chain A to apo-hTF is ∼4°. The same value is 2° in chain B. In this respect, it is useful to recall that the average χ angle value obtained comparing the C-lobes of the apo-hTF structures deposited with the PDB is 1.2° ± 0.7° (Table S1†).
Interestingly, the inspection of a recently released structure of the adduct formed by apo-hTF with cisplatin (PDB code 9H49)35 shows that, differently from the Au-hTF adduct, in the presence of the Pt-based drug the protein preserves the “fully opened” conformation of both the N-lobe and the C-lobe (Table S1†).
Gold binding sites
To try to understand the structural determinants that could trigger the subtle differences in the structure of Au-hTF when compared to apo-hTF forms, we evaluated the differences in the investigated structures at the level of gold binding sites and tertiary structure features.The Fourier difference and the anomalous difference electron density maps of Au-hTF contain large peaks corresponding to gold binding sites close to the side chains of His25, Asp63 and His249, His207 and Tyr238, His273, His300, His473, His598, His606, and His642 in chain A (Fig. 4) and close to His25, Asp63 and His249, His273, His289, His473, His585, His598, His606, and His642 in chain B (Fig. 5). The binding of Au+ ions to the protein has been confirmed by comparing the electron density maps of the Au binding sites with those of more than one crystal structure of the apo-hTF obtained independently under the same conditions, i.e., from data measured on crystals that were not treated with aurothiomalate, and by comparing the electron density maps of the Au binding sites with those of the cisplatin adduct of apo-hTF35 (data not shown). His side chains have been identified as Au binding sites in other proteins, including cyclophilin 3,46 hen egg white lysozyme,47,48 bovine pancreatic ribonuclease,49,50 ferritin from different organisms,51–55 urease,56 trypanothione reductase,57 and in the nucleosome core particle,58 by crystallographic studies and complementary techniques.
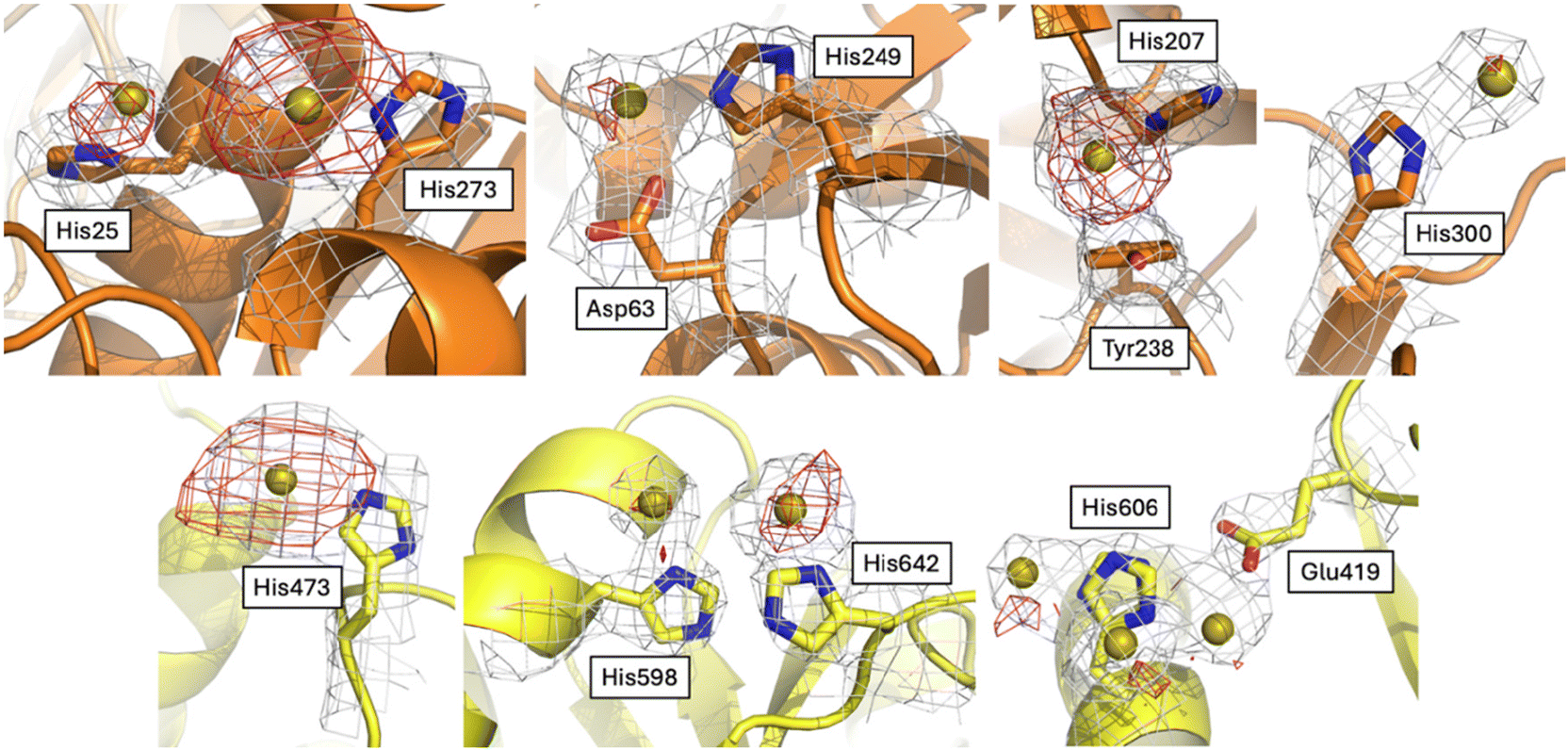 | ||
| Fig. 4 Gold binding sites close to His25, Asp63 and His249, His207 and Tyr238, His273, His300, His473, His598, His606, and His642 in chain A. 2Fo − Fc (1.0σ) and anomalous difference (2.5σ) electron density maps are in gray and red, respectively. Lobes N and C are in orange and yellow, respectively. | ||
 | ||
| Fig. 5 Gold binding sites close to His25, Asp63 and His249, His273, His289, His473, His585, His598, His606, and His642 in chain B. 2Fo − Fc (1.0σ) and anomalous difference (2.5σ) electron density maps are in gray and red, respectively. Lobes N and C are in light orange and light green, respectively. | ||
His25, Asp63 and His249, His273, His289 and His300 are in the N1-sudomain; His207 and Tyr238 are in the N2-subdomain; His606, His585, His598 and His642 are in the C1-subdomain; and His473 is in the C2-subdomain. Asp63 has been considered as a conserved putative metal binding residue in all TF superfamily members39 and together with His249 is involved in Fe3+ recognition.59 His273 and His289 have been previously identified as Ru3+ and Ru3+/Os3+ binding sites (Ru3+/Os3+ binding sites: His14/His289, His273, His349/His350, Glu507/Lys489–490, and His578/Arg581), respectively.60 The other gold binding sites have not been identified in previous structures of hTF with metal ions or metallodrugs.61
At all of these binding sites, metal ligands are missing. This is not surprising since it is known that the metal ligand is detached from the gold ion of aurothiomalate within a few hours in aqueous solutions,62 and that gold ions without the original gold ligand have been found to be bound to cathepsin K20 and MCR-1-S8 in the X-ray structures of the adducts that these proteins form upon reaction with the gold drug.
The comparison between the Au-hTF chains A and B shows that the number and location of the gold binding sites are not identical. In particular, gold ions were observed close to the side chains of His207 and Tyr238, and close to the His300 side chain only in chain A (Fig. 4), while a gold ion is found close to His289 and His585 side chains only in chain B (Fig. 5). The gold binding close to His289 of chain A is hampered by crystal packing (Fig. S6†). His300 is solvent accessible and very mobile. A small peak of the electron density map is visible close to this residue in chain B, thus we cannot exclude a low occupancy binding of a gold ion to this site. The reason for the absence of a gold ion close to the side chains of His207 and Tyr238 in chain B and close to the side chain of His585 in chain A is less clear. At the former site, the metal ion binding is associated with a small conformational variation of the side chains of residues His207 and Tyr238 (Fig. S7†). The side chains of His207 and Tyr238 are almost parallel in chain B, while they face each other in chain A, where an Au⋯π interaction between the metal and the aromatic electrons is formed. This interaction has already been reported in previous studies.53,63,64
Conformational variations that are associated with the gold ion binding can be evidenced by comparing the structure of Au-hTF with that of apo-hTF (PDB code 2HAV44). Small conformational variations, which are strictly related to the presence of Au centers, were observed at almost all gold binding sites. Significant conformational variations (in both chains A and B) were found at the level of His473. The side chain of this residue rotates by ca. 140° to bind the gold ion in chain A and by ca. 170° in chain B. In chain A, this rotation is accompanied by a significant conformational variation of both main chain and side chain atoms of Asn472 and Arg475 (Fig. 6A and Fig. S8A†). The side chains of Ser21, His25, Val29, His273 and Glu281 also change their conformation in the structure of Au-hTF when compared to that adopted in apo-hTF. In chain A, these structural variations are associated with a change in the main chain atom conformation of Val29 (Fig. 6B and Fig. S8B†). A structural modification in the C-lobe occurs close to Glu603, His606, Leu607 and Asn611 (Fig. 6C and Fig. S8C†).
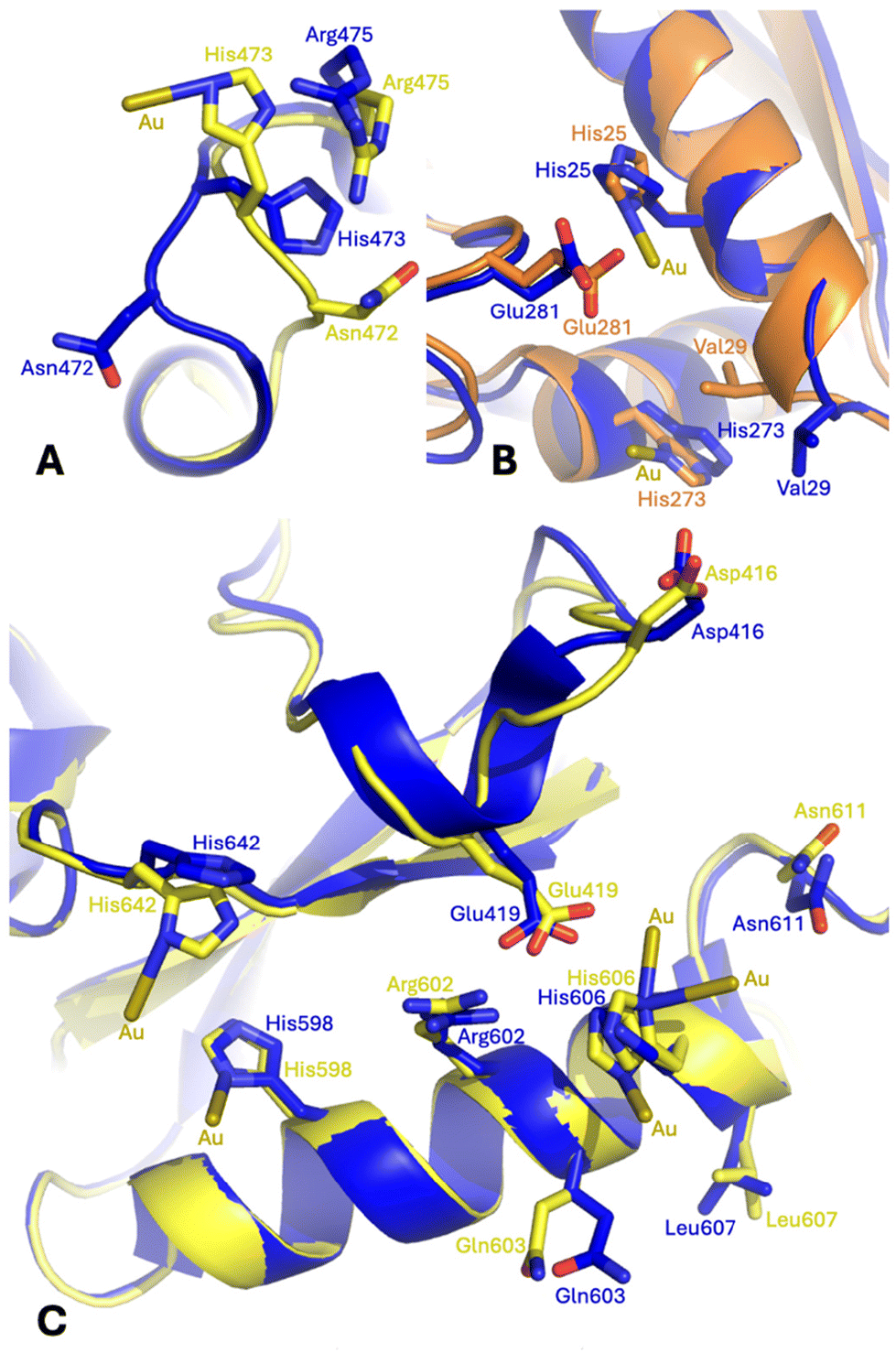 | ||
| Fig. 6 Gold ion binding close to the side chains of (A) His473, (B) His25 and His273, and (C) His598, His606 and His642 in the Au-hTF chain A (orange for N-lobe and yellow for C-lobe) superimposed onto the corresponding residues of apo-hTF chain A (blue, PDB code 2HAV). Residues 461–470 are superimposed in panel A; residues 13–25 are superimposed in panel B; and residues 594–608 are superimposed in panel C. | ||
Tertiary structure features and interlobe communication
Here, we analyzed other tertiary structural features that could play a role in the dynamics of hTF.According to Woodworth and coworkers,65 the closed conformation of the N-lobe is stabilized by a di-lysine hydrogen bridge (Lys206–Lys296) close to the N-lobe iron binding site. Mutation of these residues allows hTF to retain iron at a pH value 1 unit lower than that of the wild-type protein, and release iron much more slowly.66 The di-lysine interaction is found in the “fully closed” holo-hFT structure (NZ–NZ distances between 3.1 and 3.4 Å), while it is absent in apo-hTF (NZ–NZ distances = 8.7 and 9.6 Å in the two chains of 2HAV), FeN*FeC-hTF (NZ–NZ distance = 10.9 Å), BiN*FeC-hTF (NZ–NZ distance = 6.0), and Au-hTF structures (NZ–NZ distances = 8.2 and 9.0 Å in the two chains) (Fig. S9†). In the two chains of Au-hTF, the side chain of Lys206 is close to a citrate ion, similar to that found in the apo-hTF. In the B chain, the side chain of Lys296 forms a strong salt bridge with the side chain of Asp63, keeping the side chain of this residue in the right position to bind a gold ion, together with the His249 side chain (Fig. 7). In the A chain, this interaction is less strong and more similar to that found in the apo-hTF (Fig. 7 and Fig. S10†). This difference might contribute to explaining the observed difference in the N-lobe conformation of chains A and B.
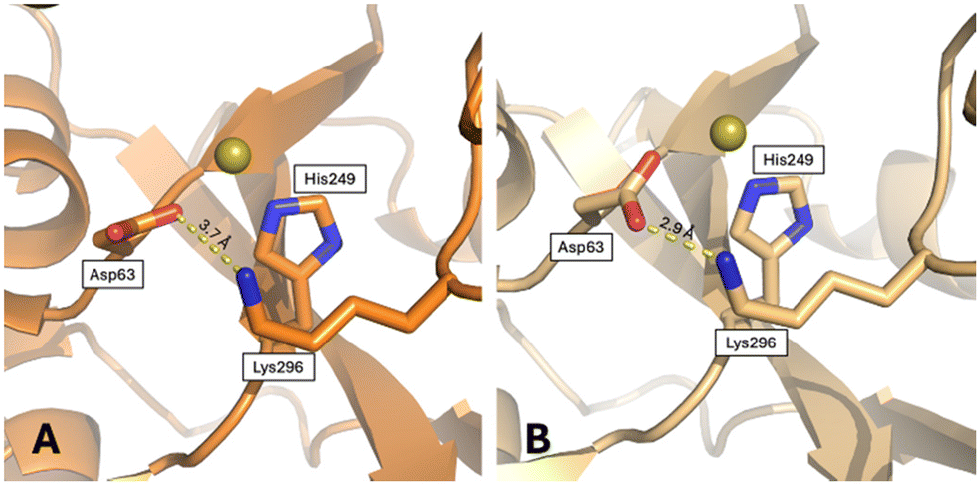 | ||
| Fig. 7 Salt bridge formed by Lys296 and Asp63, close to the gold ion binding site, in (A) chain A and (B) chain B. | ||
The hTF N- and C-lobes are connected by two salt bridges, which are formed by residues Arg308 and Asp376 and by residues Asp240 and Arg678.
The salt bridge formed by Arg308 and Asp376 is conserved in all the investigated structures (distance between the two side chains = 2.7 and 2.7 Å in the two chains of apo-hTF, between 3.2 and 3.7 Å in the structure of holo-hTF, 2.6 Å in BiN*FeC-hTF, 2.4 Å in FeN*FeC-hTF, and 3.2 and 2.8 Å in the two chains of Au-hTF).
The salt bridge Asp240–Arg678 is observed in apo-hTF (distance between the two side chains d240–678 = 2.5 and 3.1 Å), BiN*FeC-hTF (d240–678 = 2.9 Å), and Au-hTF (d240–678 = 2.6 Å in the two chains), while it is not found in FeN*FeC-hTF and holo-hTF. In FeN*FeC-hTF, the Arg678 side chain undergoes a rotation of about 110° to form hydrogen bonds with main chain atoms of Pro91 and side chain atoms of Gln92 and is far from the side chain of Asp240 (d240–678 = 12.7 Å). In holo-hTF, residue 678 has not been modeled. Rangel and coworkers59 claim that in this structure, an additional salt bridge is formed by side chains of Lys312 and Glu385 (the distance between the two side chains d312–385 is between 3.0 and 4.6 Å).
This interaction is also found in Au-hTF (d312–385 = 2.8 and 3.2 Å in the two chains), FeN*FeC-hTF (d312–385 = 2.7 Å), and BiN*FeC-hTF (d312–385 = 3.7 Å), while it is observed only in one of the two chains of apo-hTF (d312–385 = 5.1 and 3.4 Å in the two chains).
Experimental
Materials
Human serum transferrin was purchased from Sigma-Aldrich (Merck) in its iron-free (apo) form (apo-hTF, T4382–100MG, in the lyophilized form). Sodium aurothiomalate was purchased from Sigma-Aldrich (Merck, code number: 157201) with the following characteristics: purity ≥ 98% (HPLC), 1H NMR (D2O; ppm): 4.08 (m; 1H); 2.86 (dd; J2 = 15.84 Hz; J3 = 8.08 Hz; 1H); 2.69 (dd; J2 = 15.92 Hz; J3 = 6.80 Hz; 1H); 13C NMR (D2O; ppm): 182.19; 179.71; 48.60; 48.22; logP: −1.8; UV/vis (H2O): broadband with absorption between 240 and 280 nm.
In solution studies
The fluorescence spectra of apo-hTF were recorded using a Jasco FP-8700 spectrofluorometer with a 1.0 cm path length quartz cell and a thermostat bath at 25 °C using excitation wavelengths of 280 and 295 nm. Emission spectra were recorded using the protein at a concentration of 0.5 μM in 10 mM HEPES buffer at pH 7.5 in the absence and in the presence of aurothiomalate at a concentration ranging from 0 to 10 μM.Far-UV circular dichroism spectra were recorded at 25 °C using a 3 μM apo-hTF sample in 10 mM HEPES buffer at pH 7.5 in the absence and in the presence of aurothiomalate in 1:1, 1:3, 1:5 and 1:20 protein-to-metal molar ratios and after 16 h or 5 days of incubation at 20 °C. Spectra were recorded using a Jasco J-1500 spectropolarimeter equipped with a Peltier thermostat and using a 0.1 cm quartz cell (bandwidth of 1 nm).
Crystallization and cryocooling
The protein solution was prepared by dissolving the lyophilized sample in water and concentrating it to 80 mg ml−1 in 10 mM NaHCO3 and 10 mM HEPES pH 7.5 buffer. Apo-hTF crystals were grown in 20% w/v PEG 3350 and 0.2 M trisodium citrate (condition 94 of the Hampton Research's Index kit), using the hanging drop vapor diffusion method. 1 μL of protein solution was mixed with 1 μL of reservoir solution at 20 °C. The drop containing the apo-hTF crystals was saturated with aurothiomalate 5 days before they were flash-cooled in liquid N2. During this procedure aurothiomalate was directly added to the drop. The crystals were cryoprotected by adding 20% v/v glycerol to the crystallization solution. Attempts to obtain crystals of the adduct by co-crystallization failed.Data collection, structure resolution, refinement, and structural analysis
Diffraction data were collected at the ID23-2 beamline of the European Synchrotron Radiation Facility (ESRF) at 3.02 Å resolution, using λ = 0.8731 Å (360° rotation). The dataset was processed using the autoPROC program.67–71 Initial phases were determined by molecular replacement using Phaser MR70,72 and the PDB entry 7Q1L37 as a template, although the structure could also be solved by the Fourier difference method. REFMAC570,73 and Coot74 programs were used for refinement and model building, respectively. The presence of Au ions in the structure was verified by visual inspection of Fourier difference (2Fo − Fc, Fo − Fc) and anomalous difference electron density maps in Coot74 and comparing the electron density with those of apo-hTF obtained using more than one crystal of apo-hTF not treated with aurothiomalate and solved at a resolution comparable or even better than that of Au-hTF. Although the wavelength used for the data collection is not the best one for Au identification, Au binding sites can be clearly identified. The occupancies of Au+ were evaluated by trying to minimize the Fourier difference Fo − Fc electron density maps at metal centers and to obtain the best Rfactor and Rfree values. However, the occupancy value should be considered with caution because of the low resolution of the data used for the refinement. The Au-hTF adduct structure was validated using the Coot routines74 and the PDB validation server (https://validate-rcsb-1.wwpdb.org/). Data collection and refinement statistics are reported in Table S2.† The final coordinates and the structural factors were deposited with the Protein Data Bank (PDB code: 9H4V). Root-mean-square deviations (rmsd) and the rotation angles needed to superimpose N2 subdomains after best fitting N1 subdomains, and C2 subdomains after best fitting C1 subdomains (χ angles), were calculated using the Superpose program from the CCP4 package.70 Molecular graphics figures were rendered using PyMOL (DeLano Scientific, Palo Alto, CA, USA).Conclusions
In conclusion, herein we have verified that aurothiomalate binds apo-hTF in solution and have studied the interaction of the gold drug with the protein using X-ray crystallography. Given the essential role of hTF in metallodrug trafficking, understanding the interaction of the gold-based drug with this protein at the atomic level provides a solid basis for the design of more efficient aurotherapeutics. Our data indicate that gold atoms bind the protein mainly close to residues that are not involved in iron transportation. Along with the discovery of gold binding sites on the transferrin structure, our findings offer a structural basis for the potential role played by hTF as a gold-based drug delivery system. Given that hTF is recognized by the dimeric TfR1 overexpressed by cancer cells,75 our data suggest that gold-hTF adducts could be used to transport Au+ ions within the cancer cell cytosol. Of course, the possible transport of Au+ in real cell environments must be further investigated. However, given that gold binding sites are distinct from those observed for Pt33,35 and other metals,39,76–78 our data suggest the possibility of using hFT to transport more than one metallodrug at the same time.Equally important, the crystal structure reported here occupies a unique position along the apo-hTF/holo-hTF transition pathway, adopting conformations of the N- and C-lobes that are slightly distinct when compared to those adopted by apo-hTF (Fig. S11†). Our structure provides the first example of a small rigid-body motion of C-lobe subdomains starting from the apo-hTF structure and shows an N-lobe conformation that is intermediate between the “partially opened” form of the protein described when the structure of BiN*FeC-hTF was solved39 and the “fully opened” apo-structure,44 thus providing a fine sampling of the first steps of the apo/holo-hTF transition. Overall, these structural results indicate that apo-hTF crystals have high plasticity, allowing subtle, but significant, conformational variations of both N- and C-lobes, thus pushing us to solve further structures of hTF with ligands in this crystal form.
Author contributions
Romualdo Troisi: investigation, data curation, writing – review & editing, resources, and formal analysis. Francesco Galardo: investigation and data curation. Luigi Messori: writing – review & editing, resources, and formal analysis. Filomena Sica: writing – review & editing, formal analysis, and conceptualization. Antonello Merlino: writing – original draft, writing – review & editing, formal analysis, resources, supervision, and conceptualization.Data availability
Atomic coordinates and structure factors have been deposited with the Protein Data Bank under accession code 9H4V.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the European Synchrotron Radiation Facility (ESRF) for beamtime (proposal MX-2464), and the staff of the ID23-2 beamline for technical assistance during data collection. R. T. was supported with a research fellowship by #NEXTGENERATIONEU (NGEU) and the Ministry of University and Research (MUR), the National Recovery and Resilience Plan (NRRP), project MNESYS (PE0000006)—A Multiscale integrated approach to the study of the nervous system in health and disease (DN. 1553 11.10.2022). A. M. and L. M. thank MIUR PRIN 2022-Cod. 2022JMFC3X, “Protein Metalation by Anticancer Metal-based Drugs” for financial support.References
- R. T. Mertens, S. Gukathasan, A. S. Arojojoye, C. Olelewe and S. G. Awuah, Next Generation Gold Drugs and Probes: Chemistry and Biomedical Applications, Chem. Rev., 2023, 123, 6612–6667, DOI:10.1021/acs.chemrev.2c00649.
- R. C. Elder, K. Ludwig, J. N. Cooper and M. K. Eidsness, EXAFS and WAXS structure determination for an antiarthritic drug, sodium gold(I) thiomalate, J. Am. Chem. Soc., 1985, 107, 5024–5025, DOI:10.1021/ja00303a049.
- M. Stallings-Mann, L. Jamieson, R. P. Regala, C. Weems, N. R. Murray and A. P. Fields, A novel small-molecule inhibitor of protein kinase Ciota blocks transformed growth of non-small-cell lung cancer cells, Cancer Res., 2006, 66, 1767–1774, DOI:10.1158/0008-5472.CAN-05-3405.
- K. Yamaguchi, H. Ushijima, M. Hisano, Y. Inoue, T. Shimamura, T. Hirano and W. E. Müller, Immunomodulatory effect of gold sodium thiomalate on murine acquired immunodeficiency syndrome, Microbiol. Immunol., 2001, 45, 549–555, DOI:10.1111/j.1348-0421.2001.tb02657.x.
- P. N. Fonteh, F. K. Keter and D. Meyer, HIV therapeutic possibilities of gold compounds, BioMetals, 2010, 23, 185–196, DOI:10.1007/s10534-010-9293-5.
- S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Gold compounds as anticancer agents: chemistry, cellular pharmacology, and preclinical studies, Med. Res. Rev., 2010, 30, 550–580, DOI:10.1002/med.20168.
- A. Casini, R. W.-Y. Sun and I. Ott, Medicinal Chemistry of Gold Anticancer Metallodrugs, Met. Ions Life Sci., 2018, 18 DOI:10.1515/9783110470734-013 , https://books/9783110470734/9783110470734-007/9783110470734-013.xml.
- Q. Zhang, M. Wang, X. Hu, A. Yan, P.-L. Ho, H. Li and H. Sun, Gold drugs as colistin adjuvants in the fight against MCR-1 producing bacteria, JBIC, J. Biol. Inorg. Chem., 2023, 28, 225–234, DOI:10.1007/s00775-022-01983-y.
- S. Budavari, The Merck index: an encyclopedia of chemicals, drugs, and biologicals, 12th edn, 1996 Search PubMed.
- Medical Economics Company, Physicians’ Desk Reference 1998, 52nd edn, 1998 Search PubMed.
- H. E. Howard-Lock, D. J. LeBlanc, C. J. L. Lock, R. W. Smith and Z. Wang, Concerning the nature of the gold-containing anti-arthritic drug, myochrysine, Chem. Commun., 1996, 1391–1392, 10.1039/CC9960001391.
- F. Darabi, T. Marzo, L. Massai, F. Scaletti, E. Michelucci and L. Messori, Reactions of model proteins with aurothiomalate, a clinically established gold(I) drug: The comparison with auranofin, J. Inorg. Biochem., 2015, 149, 102–107, DOI:10.1016/j.jinorgbio.2015.03.013.
- R. Bau, Crystal Structure of the Antiarthritic Drug Gold Thiomalate (Myochrysine): A Double-Helical Geometry in the Solid State, J. Am. Chem. Soc., 1998, 120, 9380–9381, DOI:10.1021/ja9819763.
- B. Bertrand and A. Casini, A golden future in medicinal inorganic chemistry: the promise of anticancer gold organometallic compounds, Dalton Trans., 2003, 43, 4209–4219, 10.1039/c3dt52524d.
- V. Fernández-Moreira, R. P. Herrera and M. C. Gimeno, Anticancer properties of gold complexes with biologically relevant ligands, Pure Appl. Chem., 2019, 91, 247–269, DOI:10.1515/pac-2018-0901.
- B. Possato, L. F. Dalmolin, L. M. Pereira, J. Q. Alves, R. T. C. Silva, R. V. Gelamo, A. P. Yatsuda, R. F. V. Lopez, S. de Albuquerque and N. B. Leite, et al., Gold(III) complexes with thiosemicarbazonate ligands as potential anticancer agents: Cytotoxicity and interactions with biomolecular targets, Eur. J. Pharm. Sci., 2021, 162, 105834, DOI:10.1016/j.ejps.2021.105834.
- J. L. Larabee, J. R. Hocker and J. S. Hanas, Mechanisms of aurothiomalate-Cys2His2 zinc finger interactions, Chem. Res. Toxicol., 2005, 18, 1943–1954, DOI:10.1021/tx0501435.
- E. Erdogan, T. Lamark, M. Stallings-Mann, J. Lee, M. Pellecchia, E. A. Thompson, T. Johansen and A. P. Fields, Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota, J. Biol. Chem., 2006, 281, 28450–28459, DOI:10.1074/jbc.M606054200.
- A. V. Raevsky, M. Sharifi, V. Pinchuk and A. Klegeris, In silico design of novel gold-phosphate containing compounds as selective inhibitors of cathepsin B in neuroinflammation, Neurosciences, 2018, 5, 33, DOI:10.20517/2347-8659.2018.34.
- E. Weidauer, Y. Yasuda, B. K. Biswal, M. Cherny, M. N. G. James and D. Brömme, Effects of disease-modifying anti-rheumatic drugs (DMARDs) on the activities of rheumatoid arthritis-associated cathepsins K and S, Biol. Chem., 2007, 388, 331–336, DOI:10.1515/BC.2007.037.
- A. A. Isab, The 13C NMR study of the binding of gold(I) thiomalate with ergothionine in aqueous solution, J. Inorg. Biochem., 1992, 45, 261–267, DOI:10.1016/0162-0134(92)84014-e.
- X.-F. Cheng, N. Wang, Z. Jiang, Z. Chen, Y. Niu, L. Tong, T. Yu and B. Tang, Quantitative Chemoproteomic Profiling of Targets of Au(I) Complexes by Competitive Activity-Based Protein Profiling, Bioconjugate Chem., 2022, 33, 1131–1137, DOI:10.1021/acs.bioconjchem.2c00080.
- M. S. Iqbal, S. G. Taqi, M. Arif, M. Wasim and M. Sher, In vitro distribution of gold in serum proteins after incubation of sodium aurothiomalate and auranofin with human blood and its pharmacological significance, Biol. Trace Elem. Res., 2009, 130, 204–209, DOI:10.1007/s12011-009-8330-0.
- J. D. Herrlinger and W. Weikert, Protein binding of gold in serum of patients treated with different gold preparations, Z. Rheumatol., 1982, 41, 230–234 CAS.
- C. Frank Shaw III, N. A. Schaeffer, R. C. Elder, M. K. Eidsness, J. M. Trooster and G. H. M. Calis, Bovine serum albumin-gold thiomalate complex: gold-197 Moessbauer, EXAFS and XANES, electrophoresis, sulfur-35 radiotracer, and fluorescent probe competition studies, J. Am. Chem. Soc., 1984, 106, 3511–3521, DOI:10.1021/ja00324a019.
- J. Christodoulou, P. J. Sadler and A. Tucker, A new structural transition of serum albumin dependent on the state of Cys34. Detection by 1H-NMR spectroscopy, Eur. J. Biochem., 1994, 225, 363–368, DOI:10.1111/j.1432-1033.1994.00363.x.
- A. Pratesi, D. Cirri, D. Fregona, G. Ferraro, A. Giorgio, A. Merlino and L. Messori, Structural Characterization of a Gold/Serum Albumin Complex, Inorg. Chem., 2019, 58, 10616–10619, DOI:10.1021/acs.inorgchem.9b01900.
- L. Cosottini, A. Geri, V. Ghini, M. Mannelli, S. Zineddu, G. Di Paco, A. Giachetti, L. Massai, M. Severi and T. Gamberi, et al., Unlocking the Power of Human Ferritin: Enhanced Drug Delivery of Aurothiomalate in A2780 Ovarian Cancer Cells, Angew. Chem., Int. Ed., 2024, e202410791, DOI:10.1002/anie.202410791.
- P. Aisen and I. Listowsky, Iron transport and storage proteins, Annu. Rev. Physiol., 1980, 49, 357–393, DOI:10.1146/annurev.bi.49.070180.002041.
- N. C. Andrews and P. J. Schmidt, Iron homeostasis, Annu. Rev. Biochem., 2007, 69, 69–85, DOI:10.1146/annurev.physiol.69.031905.164337.
- J. Williams and K. Moreton, The distribution of iron between the metal-binding sites of transferrin human serum, Biochem. J., 1980, 185, 483–488, DOI:10.1042/bj1850483.
- H. Li and Z. M. Qian, Transferrin/transferrin receptor-mediated drug delivery, Med. Res. Rev., 2002, 22, 225–250, DOI:10.1002/med.10008.
- R. Troisi, F. Galardo, G. Ferraro, F. Sica and A. Merlino, Cisplatin Binding to Human Serum Transferrin: A Crystallographic Study, Inorg. Chem., 2023, 62, 675–678, DOI:10.1021/acs.inorgchem.2c04206.
- C. Li, L. Zhou and X. Yin, Pathophysiological aspects of transferrin-A potential nano-based drug delivery signaling molecule in therapeutic target for varied diseases, Front. Pharmacol., 2024, 15, 1342181, DOI:10.3389/fphar.2024.1342181.
- R. Troisi, F. Galardo, G. Ferraro, R. Lucignano, D. Picone, A. Marano, M. Trifuoggi, F. Sica and A. Merlino, Cisplatin/Apo-Transferrin Adduct: X-ray Structure and Binding to the Transferrin Receptor 1, Inorg. Chem., 2025, 64, 761–765, DOI:10.1021/acs.inorgchem.4c04435.
- D. R. Gumerov, A. B. Mason and I. A. Kaltashov, Interlobe communication in human serum transferrin: metal binding and conformational dynamics investigated by electrospray ionization mass spectrometry, Biochemistry, 2003, 42, 5421–5428, DOI:10.1021/bi020660b.
- C. Campos-Escamilla, D. Siliqi, L. A. Gonzalez-Ramirez, C. Lopez-Sanchez, J. A. Gavira and A. Moreno, X-ray Characterization of Conformational Changes of Human Apo- and Holo-Transferrin, Int. J. Mol. Sci., 2021, 22, 13392, DOI:10.3390/ijms222413392.
- H. Ochner, S. Szilagyi, M. Edte, T. K. Esser, S. Rauschenbach, L. Malavolti and K. Kern, Imaging conformations of holo- and apo-transferrin on the single-molecule level by low-energy electron holography, Sci. Rep., 2023, 13, 10241, DOI:10.1038/s41598-023-37116-x.
- N. Yang, H. Zhang, M. Wang, Q. Hao and H. Sun, Iron and bismuth bound human serum transferrin reveals a partially-opened conformation in the N-lobe, Sci. Rep., 2012, 2, 999, DOI:10.1038/srep00999.
- R. Pratt, G. J. Handelman, T. E. Edwards and A. Gupta, Ferric pyrophosphate citrate: interactions with transferrin, BioMetals, 2018, 31, 1081–1089, DOI:10.1007/s10534-018-0142-2.
- R. Barbir, R. R. Jiménez, R. Martín-Rapún, V. Strasser, D. Domazet Jurašin, S. Dabelić, J. M. de la Fuente and I. Vinković Vrček, Interaction of Differently Sized, Shaped, and Functionalized Silver and Gold Nanoparticles with Glycosylated versus Nonglycosylated Transferrin, ACS Appl. Mater. Interfaces, 2021, 13, 27533–27547, DOI:10.1021/acsami.1c04063.
- S. Khan, W. C. Cho, A. Hussain, S. Azimi, M. M. N. Babadaei, S. H. Bloukh, Z. Edis, M. Saeed, T. L. M. ten Hagen and H. Ahmadi, et al., The interaction mechanism of plasma iron transport protein transferrin with nanoparticles, Int. J. Biol. Macromol., 2023, 240, 124441, DOI:10.1016/j.ijbiomac.2023.124441.
- J.-L. Li, L. Wang, X.-Y. Liu, Z.-P. Zhang, H.-C. Guo, W.-M. Liu and S.-H. Tang, In vitro cancer cell imaging and therapy using transferrin-conjugated gold nanoparticles, Cancer Lett., 2009, 274, 319–326, DOI:10.1016/j.canlet.2008.09.024.
- J. Wally, P. J. Halbrooks, C. Vonrhein, M. A. Rould, S. J. Everse, A. B. Mason and S. K. Buchanan, The Crystal Structure of Iron-free Human Serum Transferrin Provides Insight into Inter-lobe Communication and Receptor Binding, J. Biol. Chem., 2006, 281, 24934–24944, DOI:10.1074/jbc.M604592200.
- N. Noinaj, N. C. Easley, M. Oke, N. Mizuno, J. Gumbart, E. Boura, A. N. Steere, O. Zak, P. Aisen and E. Tajkhorshid, et al., Structural basis for iron piracy by pathogenic Neisseria, Nature, 2012, 483, 53–58, DOI:10.1038/nature10823.
- J. Zou, P. Taylor, J. Dornan, S. P. Robinson, M. D. Walkinshaw and P. J. Sadler, First Crystal Structure of a Medicinally Relevant Gold Protein Complex: Unexpected Binding of [Au(PEt3)]+ to Histidine, Angew. Chem., Int. Ed., 2000, 39, 2931–2934, DOI:10.1002/1521-3773(20000818)39:16<2931::AID-ANIE2931>3.0.CO;2-W.
- L. Messori, F. Scaletti, L. Massai, M. A. Cinellu, C. Gabbiani, A. Vergara and A. Merlino, The mode of action of anticancer gold-based drugs: a structural perspective, Chem. Commun., 2013, 49, 10100–10102, 10.1039/C3CC46400H.
- I. Russo Krauss, L. Messori, M. A. Cinellu, D. Marasco, R. Sirignano and A. Merlino, (2014). Interactions of gold-based drugs with proteins: the structure and stability of the adduct formed in the reaction between lysozyme and the cytotoxic gold(III) compound Auoxo3, Dalton Trans., 2003, 43, 17483–17488, 10.1039/c4dt02332c.
- L. Messori, F. Scaletti, L. Massai, M. A. Cinellu, I. R. Krauss, G. Martino, A. di Vergara, L. Paduano and A. Merlino, Interactions of gold-based drugs with proteins: crystal structure of the adduct formed between ribonuclease A and a cytotoxic gold(III) compound, Metallomics, 2014, 6, 233–236, 10.1039/C3MT00265A.
- M. Serratrice, L. Maiore, A. Zucca, S. Stoccoro, I. Landini, E. Mini, L. Massai, G. Ferraro, A. Merlino and L. Messori, et al., Cytotoxic properties of a new organometallic platinum(II) complex and its gold(I) heterobimetallic derivatives, Dalton Trans., 2015, 45, 579–590, 10.1039/C5DT02714D.
- C. A. Butts, J. Swift, S.-G. Kang, L. Di Costanzo, D. W. Christianson, J. G. Saven and I. J. Dmochowski, Directing noble metal ion chemistry within a designed ferritin protein, Biochemistry, 2008, 47, 12729–12739, DOI:10.1021/bi8016735.
- M. Suzuki, M. Abe, T. Ueno, S. Abe, T. Goto, Y. Toda, T. Akita, Y. Yamada and Y. Watanabe, Preparation and catalytic reaction of Au/Pd bimetallic nanoparticles in Apo-ferritin, Chem. Commun., 2009, 4871–4873, 10.1039/B908742G.
- G. Ferraro, D. M. Monti, A. Amoresano, N. Pontillo, G. Petruk, F. Pane, M. A. Cinellu and A. Merlino, Gold-based drug encapsulation within a ferritin nanocage: X-ray structure and biological evaluation as a potential anticancer agent of the Auoxo3-loaded protein, Chem. Commun., 2016, 52, 9518–9521, 10.1039/c6cc02516a.
- B. Maity, S. Abe and T. Ueno, Observation of gold sub-nanocluster nucleation within a crystalline protein cage, Nat. Commun., 2017, 8, 14820, DOI:10.1038/ncomms14820.
- D. M. Monti, G. Ferraro, G. Petruk, L. Maiore, F. Pane, A. Amoresano, M. A. Cinellu and A. Merlino, Ferritin nanocages loaded with gold ions induce oxidative stress and apoptosis in MCF-7 human breast cancer cells, Dalton Trans., 2017, 46, 15354–15362, 10.1039/C7DT02370G.
- L. Mazzei, M. N. Wenzel, M. Cianci, M. Palombo, A. Casini and S. Ciurli, Inhibition Mechanism of Urease by Au(III) Compounds Unveiled by X-ray Diffraction Analysis, ACS Med. Chem. Lett., 2019, 10, 564–570, DOI:10.1021/acsmedchemlett.8b00585.
- A. Ilari, P. Baiocco, L. Messori, A. Fiorillo, A. Boffi, M. Gramiccia, T. Di Muccio and G. Colotti, A gold-containing drug against parasitic polyamine metabolism: the X-ray structure of trypanothione reductase from Leishmania infantum in complex with auranofin reveals a dual mechanism of enzyme inhibition, Amino Acids, 2012, 42, 803–811, DOI:10.1007/s00726-011-0997-9.
- Z. Adhireksan, G. Palermo, T. Riedel, Z. Ma, R. Muhammad, U. Rothlisberger, P. J. Dyson and C. A. Davey, Allosteric cross-talk in chromatin can mediate drug-drug synergy, Nat. Commun., 2017, 8, 14860, DOI:10.1038/ncomms14860.
- A. M. N. Silva, T. Moniz, B. de Castro and M. Rangel, Human transferrin: An inorganic biochemistry perspective, Coord. Chem. Rev., 2021, 449, 214186, DOI:10.1016/j.ccr.2021.214186.
- M. Wang, H. Wang, X. Xu, T.-P. Lai, Y. Zhou, Q. Hao, H. Li and H. Sun, Binding of ruthenium and osmium at non-iron sites of transferrin accounts for their iron-independent cellular uptake, J. Inorg. Biochem., 2022, 234, 111885, DOI:10.1016/j.jinorgbio.2022.111885.
- J. B. Vincent and S. Love, The binding and transport of alternative metals by transferrin, Biochim. Biophys. Acta, Gen. Subj., 2012, 1820, 362–378, DOI:10.1016/j.bbagen.2011.07.003.
- E. Jellum, E. Munthe, G. Guldal and J. Aaseth, Fate of the gold and the thiomalate part after intramuscular administration of aurothiomalate to mice, Ann. Rheum. Dis., 1980, 39, 155–158, DOI:10.1136/ard.39.2.155.
- M. Piña, L. N. de, A. Frontera and A. Bauzá, Regium-π Bonds Are Involved in Protein-Gold Binding, J. Phys. Chem. Lett., 2020, 11, 8259–8263, DOI:10.1021/acs.jpclett.0c02295.
- A. Giorgio and A. Merlino, Gold metalation of proteins: Structural studies, Coord. Chem. Rev., 2020, 407, 213175, DOI:10.1016/j.ccr.2019.213175.
- Q. Y. He, A. B. Mason, B. M. Tam, R. T. MacGillivray and R. C. Woodworth, Dual role of Lys206-Lys296 interaction in human transferrin N-lobe: iron-release trigger and anion-binding site, Biochemistry, 1999, 38, 9704–9711, DOI:10.1021/bi990134t.
- D. Nurizzo, H. M. Baker, Q. Y. He, R. T. MacGillivray, A. B. Mason, R. C. Woodworth and E. N. Baker, Crystal structures and iron release properties of mutants (K206A and K296A) that abolish the dilysine interaction in the N-lobe of human transferrin, Biochemistry, 2001, 40, 1616–1623, DOI:10.1021/bi002050m.
- P. Evans, Scaling and assessment of data quality, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2006, 62, 72–82, DOI:10.1107/S0907444905036693.
- W. Kabsch, XDS, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2010, 66, 125–132, DOI:10.1107/S0907444909047337.
- C. Vonrhein, C. Flensburg, P. Keller, A. Sharff, O. Smart, W. Paciorek, T. Womack and G. Bricogne, Data processing and analysis with the autoPROC toolbox, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2011, 67, 293–302, DOI:10.1107/S0907444911007773.
- M. D. Winn, C. C. Ballard, K. D. Cowtan, E. J. Dodson, P. Emsley, P. R. Evans, R. M. Keegan, E. B. Krissinel, A. G. W. Leslie and A. McCoy, et al., Overview of the CCP4 suite and current developments, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2011, 67, 235–242, DOI:10.1107/S0907444910045749.
- P. R. Evans and G. N. Murshudov, How good are my data and what is the resolution?, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2013, 69, 1204–1214, DOI:10.1107/S0907444913000061.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, Phaser crystallographic software, J. Appl. Crystallogr., 2007, 40, 658–674, DOI:10.1107/S0021889807021206.
- G. N. Murshudov, P. Skubák, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, REFMAC5 for the refinement of macromolecular crystal structures, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2011, 67, 355–367, DOI:10.1107/S0907444911001314.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Features and development of Coot, Acta Crystallogr., Sect. D:Biol. Crystallogr., 2010, 66, 486–501, DOI:10.1107/S0907444910007493.
- Y. Niitsu, Y. Kohgo, T. Nishisato, H. Kondo, J. Kato, Y. Urushizaki and I. Urushizaki, Transferrin receptors in human cancerous tissues, Tohoku J. Exp. Med., 1987, 153, 239–243, DOI:10.1620/tjem.153.239.
- M. Wang, T. P. Lai, L. Wang, H. Zhang, N. Yang, P. J. Sadler and H. Sun, “Anion clamp” allows flexible protein to impose coordination geometry on metal ions, Chem. Commun., 2015, 51, 7867–7870, 10.1039/c4cc09642h.
- A. D. Tinoco, M. Saxena, S. Sharma, N. Noinaj, Y. Delgado, E. P. Quiñones González, S. E. Conklin, N. Zambrana, S. A. Loza-Rosas and T. B. Parks, Unusual Synergism of Transferrin and Citrate in the Regulation of Ti(IV) Speciation, Transport, and Toxicity, J. Am. Chem. Soc., 2016, 138, 5659–5665, DOI:10.1021/jacs.6b01966.
- C. M. Petersen, K. C. Edwards, N. C. Gilbert, J. B. Vincent and M. K. Thompson, X-ray structure of chromium(III)-containing transferrin: First structure of a physiological Cr(III)-binding protein, J. Inorg. Biochem., 2020, 210, 111101, DOI:10.1016/j.jinorgbio.2020.111101.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi03184a |
| This journal is © the Partner Organisations 2025 |