
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLuminescent iridium(III) 2-cyanobenzothiazole complexes as site-specific labels to afford peptide-based phosphorogenic probes and hydrogels for enzyme activity sensing, cancer imaging and photodynamic therapy†
Jun-Wen
Xu
 a,
Lawrence Cho-Cheung
Lee
a,
Alex Man-Hei
Yip
ab,
Guang-Xi
Xu
a,
Peter Kam-Keung
Leung
ac and
Kenneth Kam-Wing
Lo
*ac
a,
Lawrence Cho-Cheung
Lee
a,
Alex Man-Hei
Yip
ab,
Guang-Xi
Xu
a,
Peter Kam-Keung
Leung
ac and
Kenneth Kam-Wing
Lo
*ac
aDepartment of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, P. R. China. E-mail: bhkenlo@cityu.edu.hk
bLaboratory for Synthetic Chemistry and Chemical Biology Limited, Units 1503–1511, 15/F, Building 17 W, Hong Kong Science Park, New Territories, Hong Kong, P. R. China
cState Key Laboratory of Terahertz and Millimetre Waves, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong, P. R. China
First published on 28th January 2025
Abstract
Site-specific modification of biomolecules is crucial to the development of functional constructs for biomedical applications, offering precise control over the number and location of the functional handles incorporated. In this work, we designed, synthesised and characterised three luminescent cyclometallated iridium(III) complexes bearing a 2-cyanobenzothiazole (CBT) moiety [Ir(N^C)2(bpy-CBT)](PF6) (HN^C = 2-(2,4-difluorophenyl)pyridine (Hdfppy) (1), 2-phenylpyridine (Hppy) (2), methyl 2-phenyl-4-quinolinecarboxylate (Hpqe) (3); bpy-CBT = 4-(2-cyanobenzo[d]thiazol-6-yl)oxymethyl-4′-methyl-2,2′-bipyridine) as site-specific labels for N-terminal cysteine (NCys). These complexes displayed high reactivity and selectivity towards NCys, enabling facile peptide conjugation via the CBT–NCys condensation reaction. Complex 2 was used to prepare a peptide-based phosphorogenic probe 2-MMP-QSY7 for matrix metalloproteinase-2/9 (MMP-2/9) activity sensing and photocytotoxic applications. The conjugate showed substantial emission enhancement (I/Io = 9.8) in the presence of MMP-2/9, which allowed for the sensitive detection of MMP-2/9 activity in live cells and the facile differentiation of cancer and normal cells. The conjugate also exhibited controllable singlet oxygen generation and thereby photocytotoxicity in these cell lines. Additionally, complex 2 was utilised to fabricate two types of hydrogels: a non-biodegradable hydrogel Gel-1 as a cell culture scaffold integrated with MMP-2/9 sensing capability for examining the enzyme activity in 3D cell culture; and a biodegradable hydrogel Gel-2 as an MMP-2/9-sensitive carrier for selective delivery of luminescent iridium(III) complexes into cancer cells for imaging and photocytotoxic applications. The results of this work will contribute to the development of site-specific bioconjugation reagents with interesting photophysical properties, facilitating the construction of photofunctional peptide conjugates and biomaterials for biosensing, bioimaging, phototherapy and drug delivery applications.
10th anniversary statementAs a scientist working at the interface of inorganic chemistry and biology, I am pleased to contribute to the 10th-anniversary collection for Inorganic Chemistry Frontiers. Since the journal was established in 2014, my group has published two Review Articles on developments in transition metal complexes used as cellular probes, intracellular sensors, bioimaging reagents and (photo)therapeutic agents. Over the past 10 years, I have observed this journal provide a platform for innovative research and reviews, fostering new ideas and collaborations within our community. Congratulations to Inorganic Chemistry Frontiers on this milestone. I look forward to seeing the journal continue to support research in inorganic chemistry. |
Introduction
Bioconjugation reactions that enable site-specific modification of peptides and proteins are important to the interrogation of the functions of these biomolecules and their associated biological processes as well as to the development of novel conjugates for biomedical applications.1,2 Among the various strategies developed, the modification of N-terminal cysteine (NCys) is a highly attractive approach due to the relatively low natural abundance of cysteine (Cys) in natural peptides and proteins, and the distinctive reactivity of its 1,2-aminothiol group that allows for site-specific labelling while retaining the modifiability of other side chains, including the internal and C-terminal Cys residues.3–5 Several reagents have been developed for NCys modification, including thioesters,6 2-cyanobenzothiazole (CBT),7 2-formylphenylboronic acid (FPBA),8–10 2-((alkylthio)(aryl)methylene)malononitrile (TAMM)11 and cyclopropenone (CPO).12 In particular, the firefly luciferin-inspired click reaction between CBT and NCys has gained significant attention due to its high efficiency, selectivity and biocompatibility, and this reaction has been widely applied in protein labelling, molecular imaging and nanomaterial fabrication.13–15There has been increasing interest in photofunctional transition metal–peptide conjugates for imaging and therapeutic applications.16–18 This interest stems from the intriguing photophysical properties of transition metal complexes, including intense and long-lived emission, efficient reactive oxygen species (ROS) photosensitisation and high photostability. Peptide conjugation can endow these photoactive complexes with interesting biological behaviour such as receptor targeting,19–22 cell penetrating,23–25 organelle targeting,26–29 enzyme-responsiveness30 and self-assembly capabilities.31 Our group has exploited various chemoselective reactions in the development of metal–peptide conjugates for organelle staining, enzyme sensing, cancer imaging and photocytotoxic applications.32–37 We anticipate that the incorporation of a CBT moiety into iridium(III) complexes will lead to new site-specific labels for the construction of peptide-based reagents and biomaterials for biomedical applications.
Herein, we report the design, synthesis and characterisation of three luminescent cyclometallated iridium(III) polypyridine complexes bearing a CBT moiety [Ir(N^C)2(bpy-CBT)](PF6) (HN^C = 2-(2,4-difluorophenyl)pyridine (Hdfppy) (1), 2-phenylpyridine (Hppy) (2), methyl 2-phenyl-4-quinolinecarboxylate (Hpqe) (3); bpy-CBT = 4-(2-cyanobenzo[d]thiazol-6-yl)oxymethyl-4′-methyl-2,2′-bipyridine) (Scheme 1). These complexes displayed high reactivity and selectivity towards NCys, enabling facile peptide conjugation via the CBT–NCys condensation reaction. Complex 2 was employed to prepare a peptide-based phosphorogenic probe for enzyme sensing and controllable photocytotoxic applications (Fig. 1a). This complex was also used to fabricate two types of enzyme-responsive hydrogels for the in situ detection of enzyme activity in 3D cell culture models (Fig. 1b), as well as for cancer-specific imaging and photocytotoxic applications (Fig. 1c).
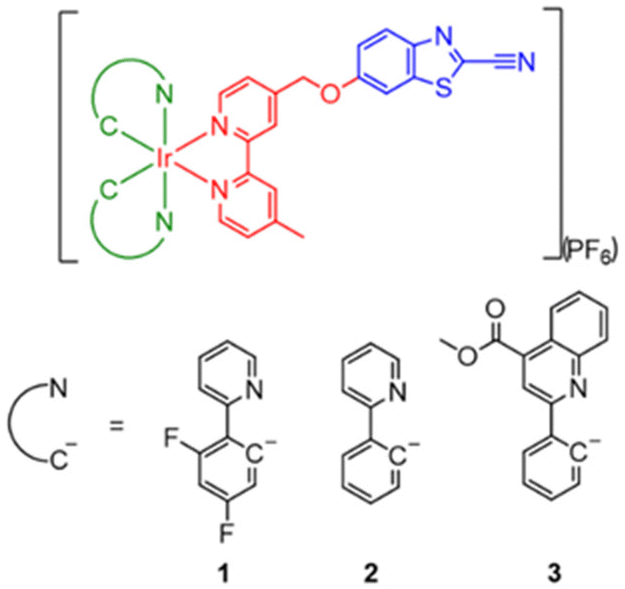 | ||
| Scheme 1 Structures of complexes 1–3. | ||
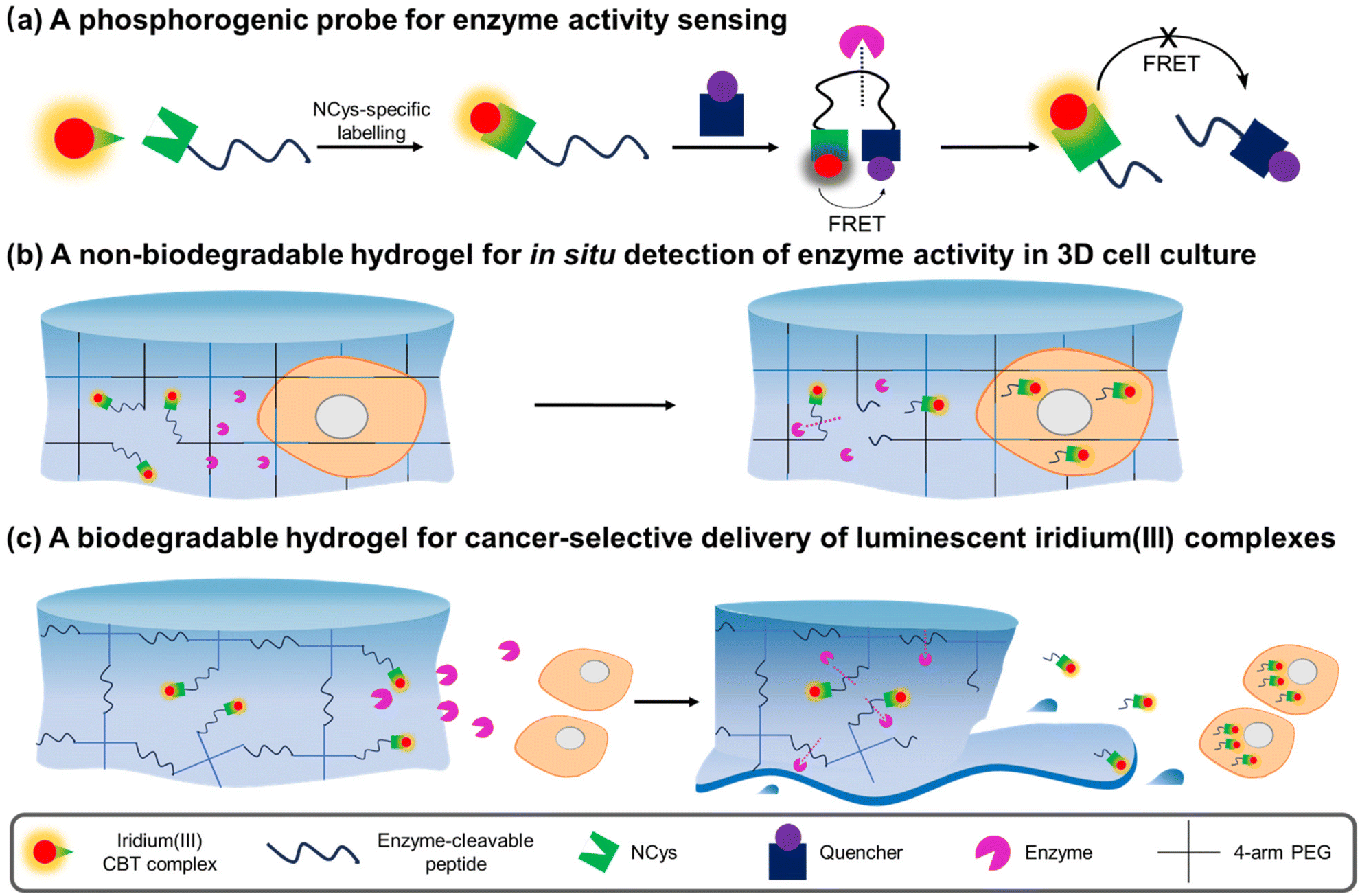 | ||
| Fig. 1 Schematic illustration of the utilisation of iridium(III) CBT complexes in the construction of (a) a FRET-based phosphorogenic probe for enzyme activity sensing; (b) a non-biodegradable hydrogel for in situ detection of enzyme activity in 3D cell culture; and (c) a biodegradable hydrogel for cancer-selective delivery of luminescent iridium(III) complexes. | ||
Results and discussion
Synthesis and characterisation
The synthesis of the diimine ligand bpy-CBT involved the reaction of 6-hydroxybenzo[d]thiazole-2-carbonitrile with 4-bromomethyl-4′-methyl-2,2′-bipyridine (bpy-Br). The iridium(III) complexes 1–3 were prepared from the reaction of respective iridium(III) dimers [Ir2(N^C)4Cl2] with bpy-CBT in CH2Cl2/CH3OH, followed by anion exchange with KPF6, purification by column chromatography and recrystallisation from CH2Cl2/Et2O. All the complexes were characterised by high-resolution ESI-MS, 1H and 13C NMR and IR spectroscopy. Detailed synthetic procedures and characterisation data are included in the ESI.†Photophysical properties
Complexes 1–3 displayed intense spin-allowed intraligand (1IL) (π → π*) (bpy-CBT and N^C) absorption features in the UV region (ca. 252–352 nm, ε on the order of 104 dm3 mol−1 cm−1) and weaker spin-allowed metal-to-ligand charge transfer (1MLCT) (dπ(Ir) → π*(bpy-CBT and N^C)) absorption shoulders or bands in a lower energy region (ca. 378–440 nm) (Table S1 and Fig. S1, ESI†).38–40 The weaker absorption tailing beyond 467 nm is assigned to spin-forbidden 3MLCT (dπ(Ir) → π*(bpy-CBT and N^C)) transitions. Upon irradiation, all the complexes exhibited intense and long-lived green to red emission in fluid solutions under ambient conditions and in low-temperature alcohol glass (Table 1 and Fig. S2, ESI†). The complexes showed broad and featureless emission bands with positive solvatochromism in fluid solutions at 298 K and their emission maxima displayed significant blue shifts upon cooling the samples to 77 K, suggestive of a predominant 3MLCT (dπ(Ir) → π*(bpy-CBT and N^C))/triplet ligand-to-ligand charge transfer (3LLCT) (π(N^C) → π*(bpy-CBT)) emissive state.38–40 Additionally, all the complexes exhibited high singlet oxygen (1O2) generation efficiencies in aerated CH3CN (ΦΔ = 0.59–0.85) (Table S2, ESI†).| Complex | Medium (T/K) | λ em /nm | τ o /μs | Φ em |
|---|---|---|---|---|
a
λ
ex = 350 nm.
b The lifetimes were measured at the emission maxima (λex = 355 nm).
c The emission quantum yields were determined using [Ru(bpy)3]Cl2 (Φem = 0.04 in aerated H2O, λex = 455 nm) as a reference.
d EtOH/CH3OH (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v). :1, v/v).
|
||||
| 1 | CH2Cl2 (298) | 523 | 4.04 | 0.90 |
| CH3CN (298) | 526 | 3.40 | 0.46 | |
| Glassd (77) | 450 (max), 483, 516 sh | 4.06 | ||
| 2 | CH2Cl2 (298) | 584 | 0.18 | 0.13 |
| CH3CN (298) | 589 | 0.10 | 0.04 | |
| Glassd (77) | 510, 527 sh | 4.00 | ||
| 3 | CH2Cl2 (298) | 621 | 0.46 | 0.09 |
| CH3CN (298) | 638 | 0.20 | 0.03 | |
| Glassd (77) | 598, 645 sh | 4.87 | ||
Reactivity and selectivity towards NCys
We first studied the reaction of complexes 1–3 with NCys by using L-Cys as a model (Fig. 2a). The complexes (100 μM) were incubated with L-Cys (250 μM) in phosphate-buffered saline (PBS)/CH3CN (9:1, v/v) containing tris(2-carboxyethyl)phosphine (TCEP) (100 μM) at 37 °C. Reversed-phase high-performance liquid chromatography (RP-HPLC) results indicated that all the complexes were converted to their corresponding products 1-Cys–3-Cys within 4 h (Fig. S3, ESI†). The formation of the iridium(III)–Cys conjugates was confirmed by ESI-MS (Fig. S4, ESI†). Complex 2 was selected as an example for further studies due to its better solubility in aqueous solutions. To examine its chemoselectivity, the complex was incubated with various biologically relevant nucleophiles, including glutathione, histidine, lysine, serine and threonine. As revealed by ESI-MS analysis, no reactions were detected and the complex remained intact (Fig. S5, ESI†). Furthermore, we utilised a breast cancer cell-targeting peptide RGDPAYQGRFL41 as a model and modified it with a Cys residue at the N-terminus (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) RGDPAYQGRFL), middle (RGDPAYQGRFL) and C-terminus (RGDPAYQGRFL) to investigate the regioselectivity of the complex. A new peak (tR = 13.0 min) was observed in the HPLC chromatogram only upon incubation of complex 2 with the NCys-containing peptide RGDPAYQGRFL (Fig. 2b). The formation of the peptide conjugate was further verified by ESI-MS (Fig. 2c). These results indicate that the CBT complexes displayed high reactivity as well as excellent chemo- and regioselectivity towards NCys.
RGDPAYQGRFL), middle (RGDPAYQGRFL) and C-terminus (RGDPAYQGRFL) to investigate the regioselectivity of the complex. A new peak (tR = 13.0 min) was observed in the HPLC chromatogram only upon incubation of complex 2 with the NCys-containing peptide RGDPAYQGRFL (Fig. 2b). The formation of the peptide conjugate was further verified by ESI-MS (Fig. 2c). These results indicate that the CBT complexes displayed high reactivity as well as excellent chemo- and regioselectivity towards NCys.
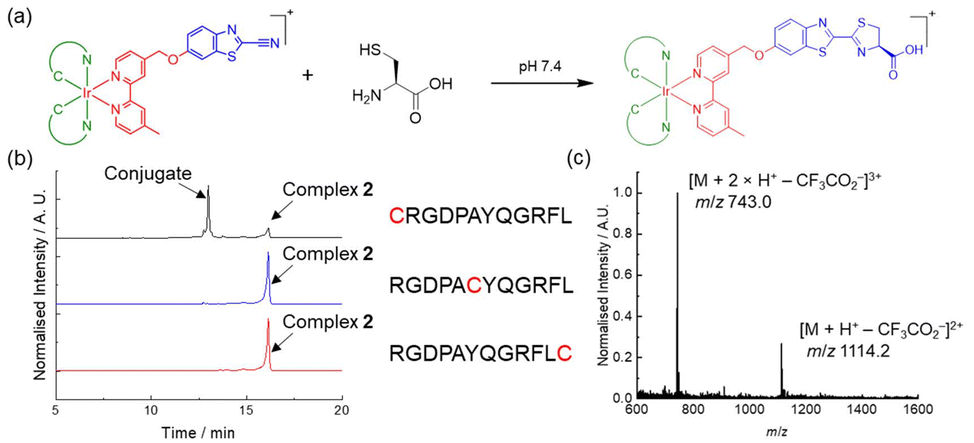 | ||
| Fig. 2 (a) Reaction of L-Cys with iridium(III) CBT complexes 1–3. (b) HPLC chromatograms (λabs = 350 nm) of the reaction mixtures of complex 2 (150 μM), peptide (150 μM), and TCEP (100 μM) in PBS/CH3CN (9:1, v/v) at 298 K for 4 h. Peptide: RGDPAYQGRFL (black), RGDPAYQGRFL (blue) and RGDPAYQGRFL (red). (c) ESI mass spectrum of the eluent collected at tR = 13.0 min of the reaction of complex 2 and RGDPAYQGRFL. | ||
Preparation of phosphorogenic probes for MMP-2/9 sensing
Given the high reactivity as well as excellent chemo- and regioselectivity of the CBT complexes towards NCys, we explored the application of these complexes in the construction of peptide-based phosphorogenic probes for sensing enzyme activity. Matrix metalloproteinases (MMPs) are a group of zinc-dependent endopeptidases present in the extracellular matrix (ECM), which are responsible for the regulation of ECM degradation and remodelling that is crucial for physiological processes such as organogenesis, wound healing and angiogenesis.42,43 Notably, MMPs are often overexpressed in tumour tissues and implicated in tumour growth, invasion and metastasis.44,45 The development of molecular probes capable of monitoring MMP activity in live cells can advance our understanding of the roles and functions of MMPs in pathophysiological processes. Thus, we modified an MMP-2/9-cleavable peptide sequence, VPMS↓MRGG (↓ indicates the cleavage site), with a Cys residue at the N-terminus and a lysine residue at the C-terminus for the conjugation with a luminescent iridium(III) complex and an emission quencher QSY-7, respectively. The peptide CVPMSMRGGK was first reacted with complex 2via the CBT–NCys condensation reaction. The resultant conjugate 2-MMP was purified (Fig. S6 and S7, ESI†) and reacted with the N-hydroxysuccinimide (NHS) ester of QSY-7 to afford conjugate 2-MMP-QSY7 as a phosphorogenic substrate for MMP-2/9 (Fig. 3a). The conjugate was purified by RP-HPLC and characterised by ESI-MS (Fig. S6 and S7, ESI†). Remarkably, conjugate 2-MMP-QSY7 displayed a substantially lower emission quantum yield in degassed buffer solutions (Φem < 0.005; Table S3, ESI†) than its QSY-7-free counterpart 2-MMP (Φem = 0.07; Table S3, ESI†). The significant emission quenching is attributed to efficient Förster resonance energy transfer (FRET) from complex 2 (λem = 580 nm) to QSY-7 (λabs = 520–600 nm) (Fig. S8, ESI†).33,36 Analysis on the FRET parameters revealed a Förster distance (R0) of 40.0 Å and high quenching efficiency (Ecalc) of 0.99, which is close to the experimentally determined value (Eexpt = 0.93) (Table S4, ESI†). The 1O2 generation efficiency of conjugate 2-MMP-QSY7 (ΦΔ = 0.06; Table S2, ESI†) was also lower than that of conjugate 2-MMP (ΦΔ = 0.52; Table S2, ESI†) due to efficient quenching by the appended QSY-7 moiety. Notably, upon incubation of conjugate 2-MMP-QSY7 (5 μM) with MMP-2 (0.002 mg mL−1) in an aerated MMP reaction buffer for 12 h, the solution exhibited substantial emission enhancement (I/Io = 9.8; Fig. 3b) due to enzymatic cleavage of the peptide sequence that separates the quenching QSY-7 moiety from the iridium(III) polypyridine unit, which was validated by RP-HPLC and ESI-MS analysis (Fig. S9, ESI†).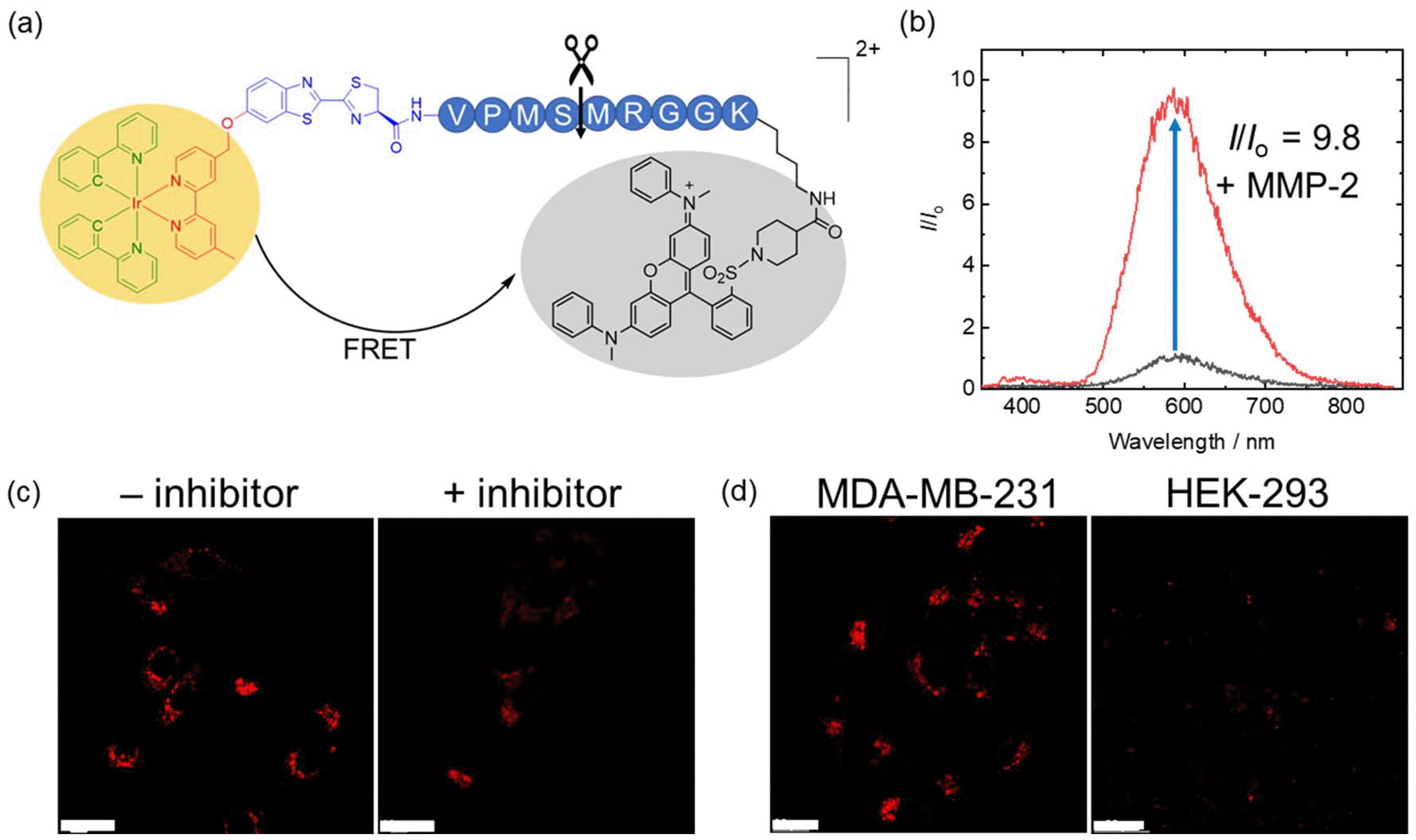 | ||
| Fig. 3 (a) Structure of conjugate 2-MMP-QSY7. (b) Emission spectra of conjugate 2-MMP-QSY7 (5 μM, 12 h) in the absence (black) and presence of MMP-2 (0.002 mg mL−1) (red) in aerated MMP reaction buffer/DMSO (99:1, v/v) at 37 °C. (c) LSCM images of live MDA-MB-231 cells incubated with conjugate 2-MMP-QSY7 (7 μM, 6 h; λex = 405 nm, λem = 550–650 nm) without (left) and with (right) pretreatment with MMP-2/9 inhibitor I (20 μM, 2 h) at 37 °C. Scale bar = 10 μm. (d) LSCM images of live MDA-MB-231 (left) and HEK-293 cells (right) incubated with conjugate 2-MMP-QSY7 (7 μM, 6 h; λex = 405 nm, λem = 550–650 nm) at 37 °C. Scale bar = 20 μm. | ||
We then examined the phosphorogenic response of the conjugate towards MMP-2/9 in live cells using human breast cancer (MDA-MB-231) and human embryonic kidney (HEK-293) cells as model cell lines. As revealed by laser-scanning confocal microscopy (LSCM), MDA-MB-231 cells incubated with conjugate 2-MMP-QSY7 (7 μM, 6 h) showed intense intracellular emission, which was substantially reduced in intensity when the cells were pretreated with MMP-2/9 inhibitor I (N-((1,1′-biphenyl)-4-ylsulfonyl)-D-phenylalanine) (20 μM, 2 h) (Fig. 3c). Notably, when HEK-293 cells were incubated with the conjugate, only negligible intracellular emission was observed (Fig. 3d) due to the lower expression levels of MMP-2/9 in normal cells.44,45 These results indicate that the conjugate can serve as a phosphorogenic probe for MMP-2/9 and differentiate cancer and normal cells based on MMP-2/9 activity. We further studied the (photo)cytotoxicity of conjugate 2-MMP-QSY7 towards MDA-MB-231 and HEK-293 cells. The conjugate displayed low cytotoxicity towards both cell lines in the dark (IC50,dark > 40 μM; Table S5, ESI†). Remarkably, it exhibited higher photocytotoxicity towards cancerous MDA-MB-231 cells (IC50,light = 1.93 μM) than normal HEK-293 cells (IC50,light = 10.17 μM) (Table S5, ESI†). The selective photocytotoxicity of the conjugate towards MDA-MB-231 cells is attributed to its distinct cellular uptake between the two cell lines (MDA-MB-231: 0.38 fmol, HEK-293: 0.03 fmol; Table S6, ESI†). We argue that the elevated MMP-2/9 secretion in cancer cells44,45 promotes the cleavage of conjugate 2-MMP-QSY7 into its cleavage product 2-CVPMS, which has a lower formal charge (+1) and a smaller molecular size, in the ECM, resulting in more efficient cellular uptake in cancer cells over normal cells. Thus, conjugate 2-MMP-QSY7 can serve as a phosphorogenic probe and an activatable photosensitiser for cancer cells overexpressing MMP-2/9.
Preparation of enzyme-responsive hydrogels for detecting MMP-2/9 activity in 3D cell culture
Hydrogels are water-swollen networks of polymers that are highly suitable for mimicking soft tissue due to their adjustable mechanical properties, achieved through chemical and physical cross-linking.46 They have been widely applied in cell culture and tissue engineering.47,48 In particular, 3D hydrogel-based cell culture models can retain surrounding ECM, preserving cell–cell and cell–ECM interactions and reproducing key biochemical and mechanical features of mammalian tissues.49 In this regard, we designed an MMP-responsive hydrogel, termed Gel-1, with both cell encapsulation and MMP sensing capabilities for the in situ detection of MMP activity in 3D cell culture models (Fig. 4a). The gel was constructed using four components: (1) a 4-arm PEG vinylsulfone (C(PEG-VS)4); (2) a 4-arm PEG thiol (C(PEG-SH)4); (3) an MMP-sensitive iridium(III)–peptide conjugate 2-VPMS, which was prepared from the reaction of complex 2 with CRDVPMSMRGGDRC (VPMS); and (4) an integrin-targeting peptide containing a Cys residue, c(RGDfC) (Fig. 4a). Poly(ethylene glycol) (PEG) is the most common macromonomer for the design of hydrogels due to its inert, non-reactive and non-degradable properties.50 The rapid thiol–ene Michael addition reaction between the two 4-arm PEG components can enable fast gelation in aqueous solutions. Additionally, the incorporation of conjugate 2-VPMS into the hydrogel framework can impart luminescence properties and MMP sensitivity, while that of c(RGDfC) can endow the hydrogel with cell-adhesion capability to facilitate cell encapsulation. Cells expressing elevated levels of MMPs are anticipated to trigger an increased cleavage of the VPMS peptide, thereby releasing the iridium(III) complexes that will be taken up by the cells. Thus, the intracellular emission intensity can be correlated with MMP activity, enabling the detection of enzyme activity by LSCM. An MMP-insensitive hydrogel Gel-1C was also prepared for comparison studies using an iridium(III)–peptide conjugate 2-VMPS, where the iridium(III) complex was conjugated to the NCys residue of a scrambled peptide CRDVMPSRMGGDRC (VMPS).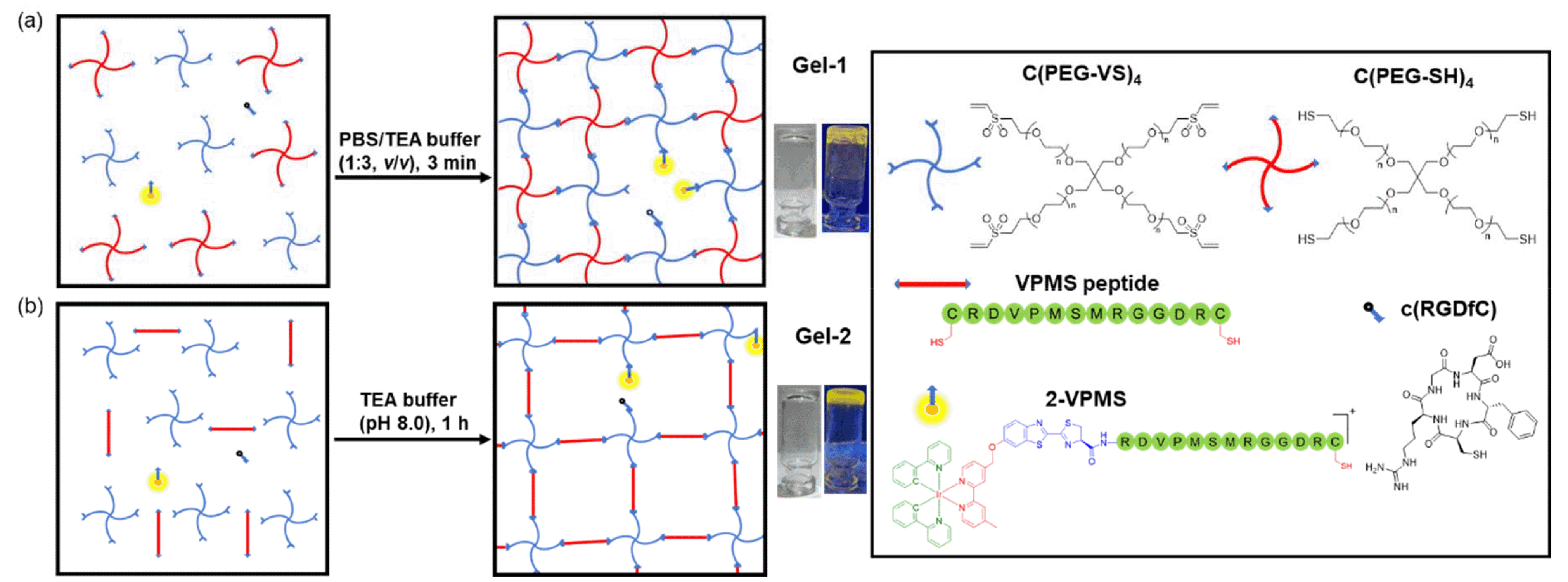 | ||
| Fig. 4 Illustration of the construction of (a) Gel-1 and (b) Gel-2. | ||
Upon the addition of C(PEG-SH)4 (30 wt%, 20 μL) in PBS (pH = 7.4, 27 μL) to a mixture of C(PEG-VS)4 (30 wt%, 20 μL), 2-VPMS (4 mM, 1 μL) and c(RGDfC) (8 mM, 5 μL) in triethanolamine (TEA) buffer (pH 8.0, 27 μL), gelation occurred within 3 min, affording Gel-1 ([Ir] = 40 μM, 12 wt% of PEG, 100 μL). The morphology and elemental composition of Gel-1 were studied by scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDS), revealing a highly porous 3D structure that is crucial for cell encapsulation (Fig. S10, ESI†). Although the iridium content in Gel-1 was too low to be accurately determined, the hydrogel displayed intense yellow emission upon photoirradiation at 365 nm (Fig. 5a), suggesting the successful incorporation of the iridium(III) complex into the hydrogel structure. Notably, when the hydrogel was treated with MMP-2 (0.002 mg mL−1 in aerated buffer), it remained in the gel state for 24 h (Fig. 5a) due to the stable cross-links formed between the two PEG monomers. This was further confirmed by transmission electron microscopy (TEM) analysis, which showed that the porous structure of the hydrogel was largely retained after the MMP treatment (Fig. 5b). The stability of Gel-1 in cell media was also studied, and no significant morphological changes were observed upon incubation in the dark for 72 h (Fig. S11a, ESI†).
 | ||
| Fig. 5 (a) Photographs of Gel-1 under UV light (λex = 365 nm) before and after the treatment with MMP-2 (0.002 mg mL−1 in PBS) for 24 h. (b) TEM images of Gel-1 before and after the treatment with MMP-2 (0.002 mg mL−1 in PBS) for 24 h. Scale bar = 0.5 μm (left) or 0.2 μm (right). | ||
Given the short gelation time of Gel-1 and its high structural integrity in the presence of MMP, we then examined its application in monitoring MMP activity in 3D cell culture. An MDA-MB-231 cell suspension (ca. 10000 cells) was first added to a PBS solution of C(PEG-SH)4, and then mixed with a TEA buffer solution containing C(PEG-VS)4, 2-VPMS and c(RGDfC) on a confocal dish. After 10 min, a hydrogel was formed, and the cells embedded in the gel were imaged after 48 h of incubation. Under the light microscope, it was observed that the cells appeared in focus at different focal lengths, indicating that the cells were successfully encapsulated into the hydrogel to form a 3D structure (Fig. S12, ESI†). After the Gel-1-embedded MDA-MB-231 cells were incubated for 72 h, the viability of the cells was studied using the live/dead cell double staining assay. As revealed by LSCM, the treated cells exhibited high viability (Fig. S13, ESI†). Notably, MDA-MB-231 cells encapsulated in Gel-1 exhibited substantially higher emission intensity than those treated with MMP-2/9 inhibitor I and those encapsulated in the MMP-insensitive hydrogel Gel-1C (Fig. 6a). The gradual increase in the emission intensity of MDA-MB-231 cells within Gel-1 at different time points indicates the gradual cleavage of the VPMS peptide by MMP-2/9 and the release of the iridium(III) complex over time (Fig. S14, ESI†). The cell encapsulation capability of Gel-1 was also tested using HEK-293 cells. While the cells were encapsulated in Gel-1 and showed high viability (Fig. S13, ESI†), only extremely weak intracellular emission was observed (Fig. 6b), indicating minimal cleavage of the peptide due to the significantly lower expression levels of MMP-2/9 in normal cells and thus reduced release and cellular uptake of the iridium(III) complex. These results collectively demonstrate that Gel-1 can serve as a versatile scaffold for 3D cell culture while simultaneously reporting on MMP-2/9 activity.
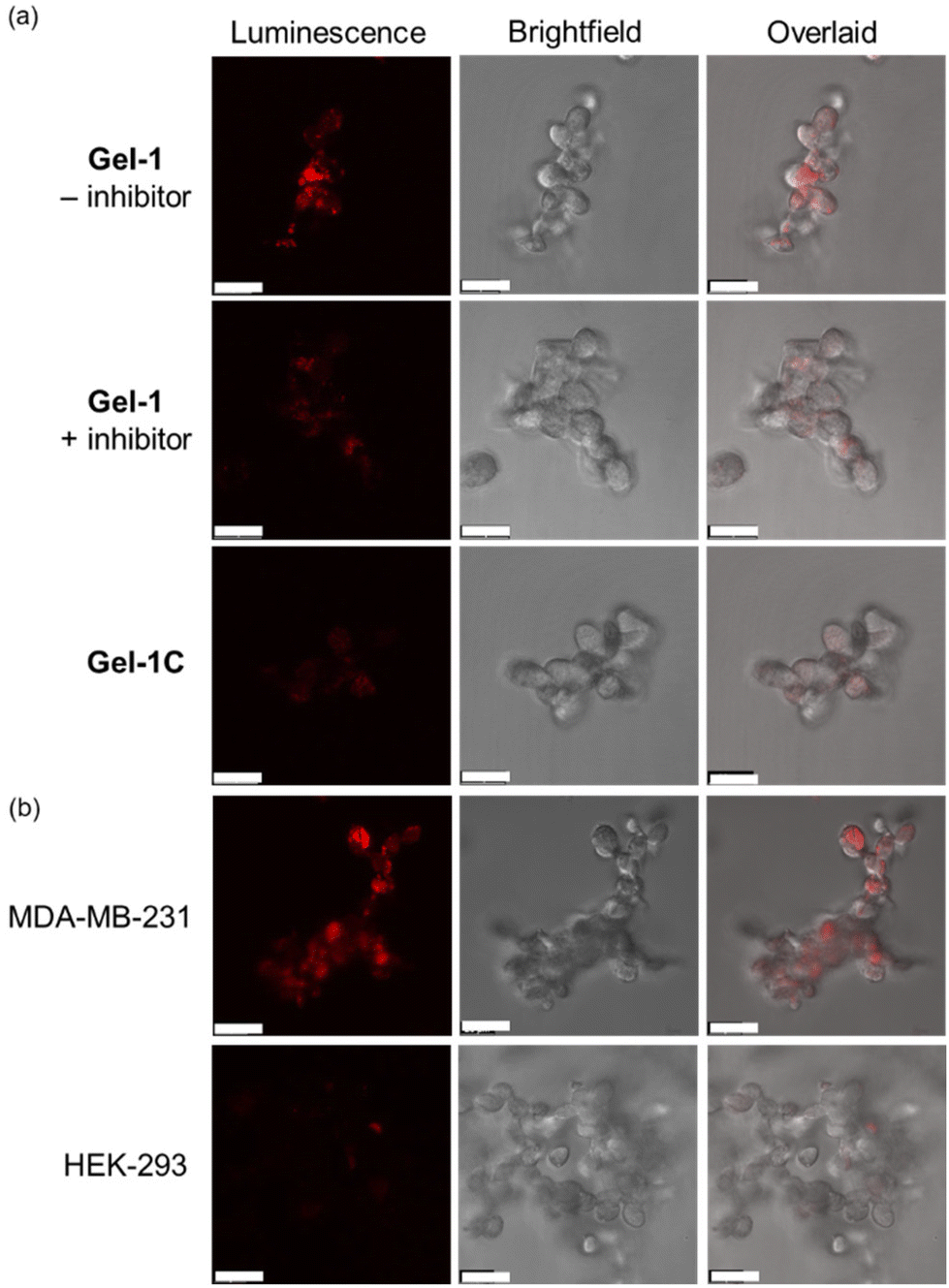 | ||
| Fig. 6 (a) LSCM images of MDA-MB-231 cells encapsulated in Gel-1 ([Ir] = 40 μM, 24 h; λex = 405 nm, λem = 570–620 nm) without (top) and with (middle) pretreatment of MMP-2/9 inhibitor I (20 μM, 4 h), and MDA-MB-231 cells encapsulated in Gel-1C ([Ir] = 40 μM, 24 h; λex = 405 nm, λem = 570–620 nm) (bottom) at 37 °C. Scale bar = 20 μm. (b) LSCM images of MDA-MB-231 (top) and HEK-293 cells (bottom) encapsulated in Gel-1 ([Ir] = 40 μM, 24 h; λex = 405 nm, λem = 570–620 nm) at 37 °C. Scale bar = 20 μm. | ||
Preparation of enzyme-responsive hydrogels for cancer-specific imaging and photocytotoxic applications
Stimuli-responsive hydrogels can alter their network structure when exposed to external stimuli, making them ideal for applications in drug delivery, bioimaging, sensing and biomaterial fabrication.51,52 This type of hydrogel allows for the controlled release of the encapsulated drugs at the targeted tumour site, enabling the selective induction of cytotoxicity towards tumour cells while minimising damage to surrounding normal tissues. Thus, we designed another MMP-responsive hydrogel, termed Gel-2, as a cancer-targeting and biodegradable carrier for the selective delivery of iridium(III) complexes to cancer cells for imaging and photocytotoxic applications. The synthetic protocol of Gel-2 was similar to that of Gel-1, except that the MMP-2/9-sensitive peptide VPMS that contains a Cys residue at its two termini was used as a crosslinker instead of C(PEG-SH)4 (Fig. 4b). This design can confer the hydrogel with highly controllable degradability, a feature that is crucial for regulating the release rate of the anchored iridium(III) complexes and also allows for complete clearance of the hydrogel from the body after the treatment. An MMP-insensitive hydrogel Gel-2C was also prepared using the scrambled peptide VMPS as the crosslinker and the corresponding conjugate 2-VMPS for comparison studies.Upon the addition of the peptide VPMS (20 mM, 20 μL) to a mixture of C(PEG-VS)4 (10 wt%, 25 μL), 2-VPMS (4 mM, 2 μL) and c(RGDfC) (8 mM, 5 μL) in TEA buffer (pH 8.0, 48 μL), gelation was observed within 1 h to afford Gel-2 ([Ir] = 80 μM, 2.5 wt% of PEG, 100 μL). As revealed by the SEM image (Fig. S15, ESI†), Gel-2 exhibited a denser structure compared to Gel-1 (Fig. S10, ESI†), attributed to the use of the short peptide VPMS as the crosslinker instead of the 4-arm macromer C(PEG-SH)4. Similar to Gel-1, Gel-2 showed high stability in cell culture media and remained in the gel state for up to 72 h (Fig. S11b, ESI†). However, upon treatment with MMP-2 (0.002 mg mL−1 in aerated buffer), it underwent complete gel–sol transition within 12 h to form a liquid solution (Fig. 7a). This is further supported by TEM analysis, showing that the hydrogel was completely degraded by MMP-2 into small scattered pieces (Fig. 7b). Similar experiments were performed using the control hydrogel Gel-2C, and no significant morphological changes were observed upon the MMP-2 treatment (Fig. 7c) due to the insensitivity of the scrambled peptide VMPS to MMPs.
 | ||
| Fig. 7 (a) Photographs of Gel-2 under UV light (λex = 365 nm) before and after the treatment with MMP-2 (0.002 mg mL−1 in PBS) for 12 h. (b) TEM images of Gel-2 before (left) and after (right) the treatment with MMP-2 (0.002 mg mL−1 in PBS) for 12 h. (c) Photographs of Gel-2C under UV light (λex = 365 nm) before and after the treatment with MMP-2 (0.002 mg mL−1 in PBS) for 12 h. | ||
We then examined the utilisation of Gel-2 for the controlled delivery of the iridium(III) complexes anchored to the hydrogel framework into cancer cells based on MMP activity. We placed Gel-2 on one side of a confocal dish seeded with MDA-MB-231 cells and allowed it to stand for 12 h (Fig. 8a). After the treatment, the hydrogel was removed and the cells were thoroughly washed with PBS prior to imaging with LSCM. Remarkably, cells located near Gel-2 exhibited significantly higher intracellular emission intensity than the cells that were relatively distant from Gel-2 (ca. 1 cm) (Fig. 8b), attributed to increased cleavage of the VPMS peptide linker in the hydrogel that led to enhanced release of the iridium(III) complex into the cells at its local surroundings. Co-staining experiments with LysoTracker Deep Red (100 nM, 30 min) confirmed the specific accumulation of the released iridium(III) complex in the lysosomes (Pearson's correlation coefficient = 0.92) after cellular uptake (Fig. 8c). However, the cells exhibited negligible intracellular emission when they were pretreated with MMP-2/9 inhibitor I (Fig. 8d), as the inhibition of MMP-2/9 activity suppressed the VPMS peptide cleavage and thereby the release of the iridium(III) complex. Similarly, cells treated with Gel-2C for 12 h displayed extremely weak emission even when they were located in a close proximity to the hydrogel (Fig. 8d), as the hydrogel was not degraded by the MMPs secreted from the cells. The MMP-mediated release of the iridium(III) complex from Gel-2 was also studied in normal HEK-293 cells. Notably, while MDA-MB-231 cells showed intense punctate staining after treatment with Gel-2, the treated HEK-293 cells exhibited negligible intracellular emission (Fig. 8e), which is in accordance with the higher expression levels of MMPs in breast cancer cells compared to normal cells. All these results indicate that the MMP-sensitive Gel-2 enabled the controlled release of the incorporated iridium(III) complexes into cancer cells.
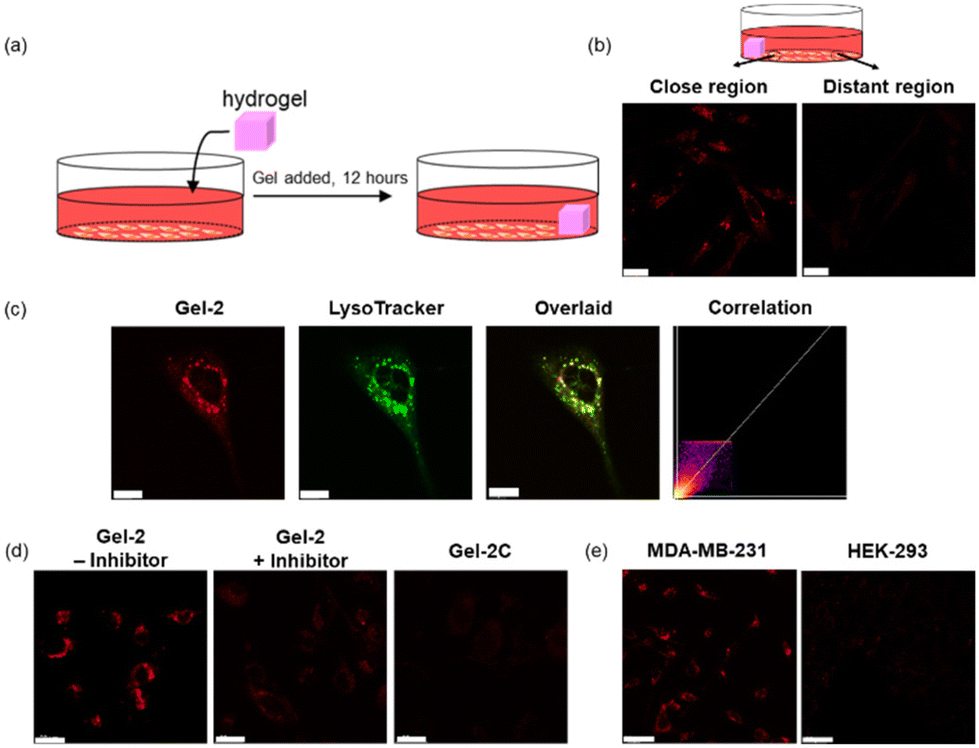 | ||
| Fig. 8 (a) A schematic diagram illustrating the treatment of cells with Gel-2. (b) LSCM images of MDA-MB-231 cells at regions that were close to (left) and distant from (right) Gel-2 ([Ir] = 80 μM, 12 h; λex = 405 nm, λem = 570–620 nm). Scale bar = 20 μm. (c) LSCM images of MDA-MB-231 cells incubated with Gel-2 ([Ir] = 80 μM, 24 h; λex = 405 nm, λem = 570–600 nm) and LysoTracker Deep Red (100 nM, 30 min; λex = 635 nm, λem = 650–700 nm) at 37 °C. Scale bar = 10 μm. (d) LSCM images of MDA-MB-231 cells incubated with Gel-2 ([Ir] = 80 μM, 12 h; λex = 405 nm, λem = 570–620 nm) without (left) and with (middle) pretreatment of MMP-2/9 inhibitor I (20 μM, 4 h), and MDA-MB-231 cells incubated with Gel-2C ([Ir] = 80 μM, 12 h; λex = 405 nm, λem = 570–620 nm) (right) at 37 °C. Scale bar = 20 μm. (e) LSCM images of MDA-MB-231 (left) and HEK-293 cells (right) incubated with Gel-2 ([Ir] = 80 μM, 12 h; λex = 405 nm, λem = 570–620 nm). Scale bar = 20 μm. All the images in (c), (d) and (e) were taken at regions that were close to Gel-2. | ||
Lysosome is an organelle that contains numerous hydrolases including cathepsins, performing cellular catabolic processes that are crucial for maintaining cell homeostasis and governing cell death.53 Since tumour cells contain abundant lysosomes, photosensitisers that can selectively target and damage lysosomes would trigger the release of cathepsins into the cytoplasm and thereby induce cell death.54 Given the high cancer selectivity of Gel-2 and the strong lysosome-targeting capability of the iridium(III) complex released from the hydrogel, we examined the application of Gel-2 in dual cancer- and lysosome-targeted photodynamic therapy (PDT). MDA-MB-231 cells were first treated with Gel-2 ([Ir] = 80 μM) for 24 h, thoroughly washed with PBS, and then irradiated at 450 nm (15 mW cm−2) for 30 min. After incubation for 24 h, the cell viability was examined using the live/dead cell double staining assay. As revealed by LSCM, the treated cells remained highly viable in the dark (Fig. 9, upper). However, upon irradiation, most of the cell populations were eradicated (Fig. 9, lower), which is likely due to the photogenerated ROS by the released iridium(III) complex causing damage to the lysosomes and ultimately triggered cell death. The elevated production of intracellular ROS upon irradiation of the cells was also confirmed by the chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) assay (Fig. S16, ESI†). All these results collectively indicate that the biodegradable hydrogel Gel-2 can function as an MMP-2/9-sensitive carrier for selective delivery of luminescent iridium(III) complexes into cancer cells, and the released complexes specifically accumulated in the lysosomes and efficiently triggered cell death upon irradiation through ROS generation.
 | ||
| Fig. 9 Analysis of live/dead MDA-MB-231 cells using Calcein-AM (1 μM, 30 min; λex = 488 nm, λem = 510–540 nm) and propidium iodide (PI) (3 μM, 30 min; λex = 532 nm, λem = 610–640 nm). Top: the cells were incubated with Gel-2 ([Ir] = 80 μM) for 24 h, then washed thoroughly with PBS, and incubated in fresh medium in the dark for 24 h. Bottom: the procedure was similar except that the cells were irradiated at 450 nm (15 mW cm−2) for 30 min before the final incubation in fresh medium in the dark for 24 h. Scale bar = 100 μm. The images were taken at regions close to Gel-2. | ||
Conclusion
In summary, we developed three luminescent cyclometallated iridium(III) CBT complexes as site-specific labels for NCys. These complexes displayed high reactivity and excellent chemo- and regioselectivity towards NCys, enabling the facile preparation of photofunctional metal–peptide conjugates via the CBT–NCys condensation reaction. One of the CBT complexes was used to prepare an MMP-2/9-responsive metal–peptide conjugate 2-MMP-QSY7 as both a phosphorogenic probe for enzyme activity sensing in live cells and an activatable photosensitiser for cancer-targeted photocytotoxic applications. The same CBT complex was also utilised to fabricate two types of hydrogels: a non-biodegradable hydrogel Gel-1 as a dual functional platform with both cell encapsulation and MMP-2/9 sensing capabilities for examining the enzyme activity in 3D cell culture; and a biodegradable hydrogel Gel-2 as an MMP-2/9-sensitive carrier for selective delivery of luminescent iridium(III) complexes into cancer cells for imaging and photocytotoxic applications. We believe that the CBT complexes will serve as a new class of site-specific bioconjugation reagents with interesting photophysical properties, facilitating the construction of photofunctional peptide conjugates and biomaterials for biosensing, bioimaging, PDT and drug delivery applications.Experimental
Materials and instruments
All solvents were of analytical reagent grade and purified according to standard procedures.55 Triethylamine (Et3N), Na2CO3, MgSO4, NaBH4, K2CO3, AgNO3, KPF6, TCEP, trifluoroacetic acid (TFA), Hdfppy, Hppy and 2-phenylquinoline-4-carboxylic acid were purchased from Acros. 6-Hydroxybenzo[d]thiazole-2-carbonitrile was purchased from Energy Chemical. 4,4′-Dimethyl-2,2′-bipyridine, SeO2, Na2S2O5 and IrCl3·3H2O were obtained from Aldrich. The peptides RGDPACYQGRFL, RGDPAYQGRFLC, CRGDPAYQGRFL, CVPMSMRGGK, CRDVPMSMRGGDRC (VPMS), c(RGDfC) and CRDVMPSRMGGDRC (VMPS) were purchased from GL Biochem. Polymers C(PEG-VS)4 and C(PEG-SH)4 were purchased from JenKem Technology USA. All these chemicals were used without further purification. 4-Carboxylaldehyde-4′-methyl-2,2′-bipyridine (bpy-CHO),56 bpy-Br,57 Hpqe,58 and the iridium(III) dimers [Ir2(N^C)4Cl2] (HN^C = Hdfppy, Hppy and Hpqe)59 were prepared according to literature procedures. All buffer components were of biological grade and used as received. Autoclaved Milli-Q H2O was used for the preparation of the aqueous solutions. MDA-MB-231 and HEK-293 cells were obtained from the American Type Culture Collection. QSY-7 carboxylic acid succinimidyl ester (QSY-7 NHS), human MMP-2 recombinant protein, Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), penicillin/streptomycin, PBS, trypsin-EDTA, CM-H2DCFDA and LysoTracker Deep Red were purchased from Invitrogen. The growth medium for cell culture contained DMEM supplemented with 10% FBS and 1% penicillin/streptomycin.1H and 13C NMR spectra were recorded on a Bruker 300 and 600 MHz AVANCE III spectrometer at 298 K using deuterated solvents. Chemical shifts (δ, ppm) were reported relative to tetramethylsilane (TMS). Positive-ion ESI mass spectra were recorded on a SCIEX API-3200 mass spectrometer at 298 K. Electronic absorption spectra were recorded on an Agilent 8453 diode array spectrophotometer. Steady-state emission spectra were recorded on a HORIBA Jobin Yvon FluoroMax-4 spectrofluorometer. Unless specified otherwise, all solutions for photophysical studies were degassed with no fewer than four successive freeze–pump–thaw cycles and stored in a 10 cm3 round-bottomed flask equipped with a side-arm 1 cm fluorescence cuvette and sealed from the atmosphere by a Rotaflo HP6/6 quick-release Teflon stopper. Luminescence quantum yields (Φem) were measured by the optically dilute method using an aerated aqueous solution of [Ru(bpy)3]Cl2 (Φem = 0.04, λex = 455 nm)60 as the standard solution. The concentrations of the standard and sample solutions were adjusted until the absorbance at 455 nm was 0.1. Emission lifetimes were measured on an Edinburgh Instruments LP920 Laser Flash Photolysis spectrometer using the third harmonic output (355 nm; 6–8 ns fwhm pulse width) of a Spectra-Physics Quanta-Ray Q-switched LAB-150 pulsed Nd:YAG laser (10 Hz) as the excitation source. HPLC was performed on an Agilent 1260 Infinity II system coupled with a diode array detector WR and 1260 Infinity II fluorescence detector using H2O containing 0.1% (v/v) TFA (solvent A), CH3CN containing 0.1% (v/v) TFA (solvent B) and CH3OH containing 0.1% (v/v) TFA (solvent C) as the solvents. The diode array detector was set at 220, 350 and 560 nm.
:1, v/v) as the eluent. The solvent was removed under reduced pressure to afford the product as a white solid. Yield: 204 mg (85%). 1H NMR (300 MHz, (CD3)2SO, 298 K, TMS): δ 8.70 (d, J = 4.9 Hz, 1H, H6 of bpy), 8.55 (d, J = 5.0 Hz, 1H, H6′ of bpy), 8.49 (s, 1H, H3 of bpy), 8.26 (s, 1H, H3′ of bpy), 8.21 (d, J = 9.1 Hz, 1H, H5 of benzothiazole), 8.00 (s, 1H, H7 of benzothiazole), 7.54 (d, J = 4.6 Hz, 1H, H5 of bpy), 7.49 (d, J = 9.1 Hz, 1H, H4 of benzothiazole), 7.31 (d, J = 4.4 Hz, 1H, H5′ of bpy), 5.44 (s, 2H, CH2O), 2.43 (s, 3H, CH3 of bpy). IR (KBr) ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) /cm−1: 2227 (C
/cm−1: 2227 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N). ESI-MS: m/z 359 [M + H+]+.
:1, v/v) as the eluent. The solvent was removed under reduced pressure to afford a yellow solid. Subsequent recrystallisation of the solid from CH2Cl2/Et2O afforded complex 1 as yellow crystals. Yield: 35 mg (78%). 1H NMR (300 MHz, (CD3)2CO, 298 K, TMS): δ 9.06 (s, 1H, H3 of bpy), 8.83 (s, 1H, H3′ of bpy), 8.37 (d, J = 8.4 Hz, 2H, H6 pyridyl ring of dfppy), 8.25–8.19 (m, 2H, H4 and H7 of benzothiazole), 8.11–8.06 (m, 3H, H6 of bpy and H5 of pyridyl ring of dfppy), 7.97–7.95 (m, 3H, H6′ of bpy and H3 of pyridyl ring of dfppy), 7.89 (d, J = 4.9 Hz, 1H, H5 of bpy), 7.62 (d, J = 5.6 Hz, 1H, H5′ of bpy), 7.52 (dd, J = 9.1 and 3.0 Hz, 1H, H5 of benzothiazole), 7.30–7.24 (m, 2H, H4 of pyridyl ring of dfppy), 6.83–6.74 (m, 2H, H5 of phenyl ring of dfppy), 5.81 (m, 2H, H3 of phenyl ring of dfppy), 5.63 (s, 2H, CH2O), 2.63 (s, 3H, CH3 of bpy). 13C NMR (150 MHz, (CD3)2SO, 298 K): δ 163.3, 158.7, 155.9, 155.0, 152.8, 150.9, 150.4, 150.0, 147.2, 140.5, 137.9, 134.9, 130.3, 128.0, 127.4, 126.5, 126.0, 124.9, 123.9, 123.4, 119.4, 113.9, 113.7, 106.4, 99.5, 68.5, 21.4. IR (KBr) /cm−1: 2227 (CN), 843 (PF6−). HR-ESI-MS (positive-ion mode, m/z): 931.1423 [M − PF6−]+.
/cm−1: 2227 (CN), 843 (PF6−). HR-ESI-MS (positive-ion mode, m/z): 859.1797 [M − PF6−]+.
/cm−1: 2227 (CN), 841 (PF6−). HR-ESI-MS (positive-ion mode, m/z): 1075.2212 [M − PF6−]+.
N). ESI-MS: m/z 359 [M + H+]+.
:1, v/v) as the eluent. The solvent was removed under reduced pressure to afford a yellow solid. Subsequent recrystallisation of the solid from CH2Cl2/Et2O afforded complex 1 as yellow crystals. Yield: 35 mg (78%). 1H NMR (300 MHz, (CD3)2CO, 298 K, TMS): δ 9.06 (s, 1H, H3 of bpy), 8.83 (s, 1H, H3′ of bpy), 8.37 (d, J = 8.4 Hz, 2H, H6 pyridyl ring of dfppy), 8.25–8.19 (m, 2H, H4 and H7 of benzothiazole), 8.11–8.06 (m, 3H, H6 of bpy and H5 of pyridyl ring of dfppy), 7.97–7.95 (m, 3H, H6′ of bpy and H3 of pyridyl ring of dfppy), 7.89 (d, J = 4.9 Hz, 1H, H5 of bpy), 7.62 (d, J = 5.6 Hz, 1H, H5′ of bpy), 7.52 (dd, J = 9.1 and 3.0 Hz, 1H, H5 of benzothiazole), 7.30–7.24 (m, 2H, H4 of pyridyl ring of dfppy), 6.83–6.74 (m, 2H, H5 of phenyl ring of dfppy), 5.81 (m, 2H, H3 of phenyl ring of dfppy), 5.63 (s, 2H, CH2O), 2.63 (s, 3H, CH3 of bpy). 13C NMR (150 MHz, (CD3)2SO, 298 K): δ 163.3, 158.7, 155.9, 155.0, 152.8, 150.9, 150.4, 150.0, 147.2, 140.5, 137.9, 134.9, 130.3, 128.0, 127.4, 126.5, 126.0, 124.9, 123.9, 123.4, 119.4, 113.9, 113.7, 106.4, 99.5, 68.5, 21.4. IR (KBr) /cm−1: 2227 (CN), 843 (PF6−). HR-ESI-MS (positive-ion mode, m/z): 931.1423 [M − PF6−]+.
/cm−1: 2227 (CN), 843 (PF6−). HR-ESI-MS (positive-ion mode, m/z): 859.1797 [M − PF6−]+.
/cm−1: 2227 (CN), 841 (PF6−). HR-ESI-MS (positive-ion mode, m/z): 1075.2212 [M − PF6−]+.
where Φ is 1O2 generation quantum yield, I is excitation intensity, B is 1–10−AL, A is the absorbance at the excitation wavelength, L is path length in cm, n is the refractive index of the solvent and D is integrated emission intensity.
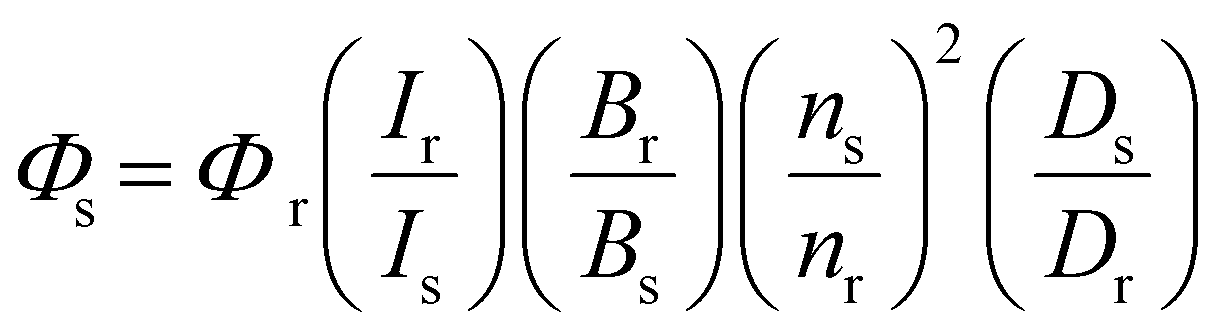 :1, v/v, 1 mL) containing TCEP (100 μM) was stirred at 37 °C in the dark for 4 h. The reaction mixture was analysed by analytical RP-HPLC. The HPLC analyses were carried out using an Agilent analytical column (ZORBAX Eclipse Plus C18: 4.6 × 150 mm, 5 μm) with a linear gradient of 10–100% B over 25 min and a flow rate of 1 mL min−1.
:1, v/v, 1 mL) containing TCEP (3 μmol) was stirred at 37 °C in the dark for 4 h. The resulting mixture was purified by RP-HPLC. The HPLC purification was performed on an Agilent semi-preparative column (ZORBAX Eclipse XDB-C18 column: 9.4 × 250 mm, 5 μm) using solvent A and solvent B with a linear gradient of 30–100% B over 30 min and a flow rate of 2 mL min−1. The fractions containing the product were combined and lyophilised. The purified product was characterised by analytical RP-HPLC and ESI-MS. The HPLC analyses were carried out using an Agilent analytical column (ZORBAX Eclipse Plus C18: 4.6 × 150 mm, 5 μm) with a linear gradient of 10–100% B over 25 min and a flow rate of 1 mL min−1. 2-MMP. Yield: 5.4 mg (90%). tR = 25.1 min. ESI-MS: m/z 637 [M + 2 × H+ − CF3CO2−]3+, 955 [M + H+ − CF3CO2−]2+. 2-VPMS. Yield: 6.7 mg (88%). tR = 13.2 min. ESI-MS: m/z 808 [M + 2 × H+ − CF3CO2−]3+, 1212 [M + H+ − CF3CO2−]2+. 2-VMPS. Yield: 6.2 mg (81%). tR = 13.2 min. ESI-MS: m/z 808 [M + 2 × H+ − CF3CO2−]3+, 1212 [M + H+ − CF3CO2−]2+.
:1, v/v, 1 mL) containing TCEP (100 μM) was stirred at 37 °C in the dark for 4 h. The reaction mixture was analysed by analytical RP-HPLC. The HPLC analyses were carried out using an Agilent analytical column (ZORBAX Eclipse Plus C18: 4.6 × 150 mm, 5 μm) with a linear gradient of 10–100% B over 25 min and a flow rate of 1 mL min−1.
:1, v/v, 1 mL) containing TCEP (3 μmol) was stirred at 37 °C in the dark for 4 h. The resulting mixture was purified by RP-HPLC. The HPLC purification was performed on an Agilent semi-preparative column (ZORBAX Eclipse XDB-C18 column: 9.4 × 250 mm, 5 μm) using solvent A and solvent B with a linear gradient of 30–100% B over 30 min and a flow rate of 2 mL min−1. The fractions containing the product were combined and lyophilised. The purified product was characterised by analytical RP-HPLC and ESI-MS. The HPLC analyses were carried out using an Agilent analytical column (ZORBAX Eclipse Plus C18: 4.6 × 150 mm, 5 μm) with a linear gradient of 10–100% B over 25 min and a flow rate of 1 mL min−1. 2-MMP. Yield: 5.4 mg (90%). tR = 25.1 min. ESI-MS: m/z 637 [M + 2 × H+ − CF3CO2−]3+, 955 [M + H+ − CF3CO2−]2+. 2-VPMS. Yield: 6.7 mg (88%). tR = 13.2 min. ESI-MS: m/z 808 [M + 2 × H+ − CF3CO2−]3+, 1212 [M + H+ − CF3CO2−]2+. 2-VMPS. Yield: 6.2 mg (81%). tR = 13.2 min. ESI-MS: m/z 808 [M + 2 × H+ − CF3CO2−]3+, 1212 [M + H+ − CF3CO2−]2+.
For conjugate 2-MMP-QSY7, a mixture of the purified conjugate 2-MMP (1 μmol), QSY7-NHS (1 μmol) and Et3N (1.1 μmol) in DMF (1 mL) was stirred at 37 °C under an inert atmosphere of nitrogen in the dark for 18 h. The solvent was removed under reduced pressure, and the residual solid was purified by RP-HPLC. 2-MMP-QSY7. Yield: 1.3 mg (50%). tR = 18.6 min. ESI-MS: m/z 849 [M + H+ − CF3CO2−]3+.
where κ2 is a factor describing the relative orientation in space of the transition dipoles of the D and the A and is assumed to be 2/3; n is the refractive index of the solvent; ΦD is the emission quantum yield of conjugate 2-MMP; J(λ) is the overlap integral of the donor 2-MMP emission and the acceptor QSY-7 absorption spectra.
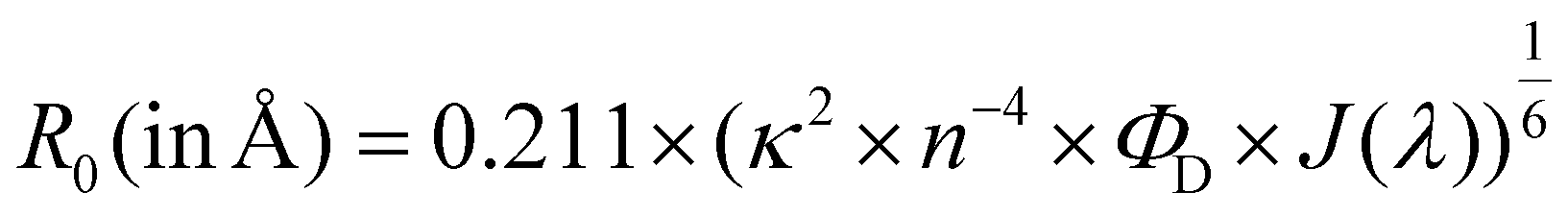
Calculation of J(λ) was based on the equation below:
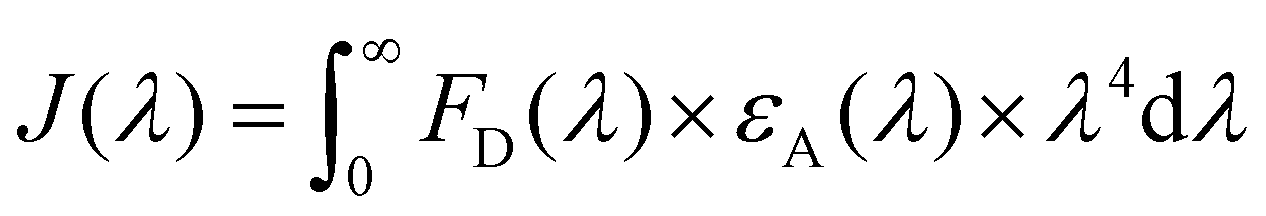
Calculated energy transfer efficiency (Ecalc) based on Förster's theory was determined according to the following equation:
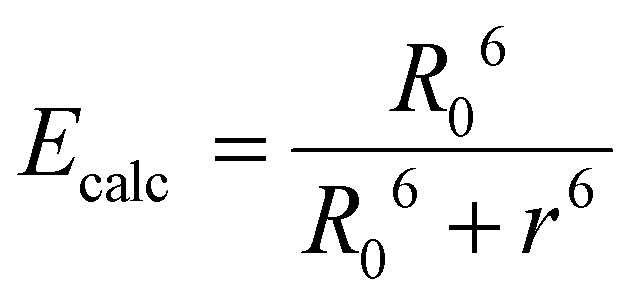
Experimentally determined energy transfer efficiency (Eexpt) was determined on the basis of the emission quantum yields of conjugates 2-MMP and 2-MMP-QSY7 according to the following equation:
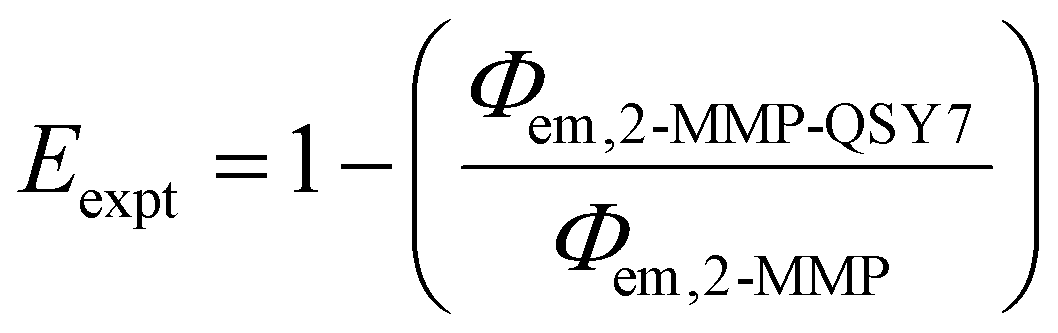 :1, v/v) containing conjugate 2-MMP-QSY7 (7 μM). After incubation for 6 h, the cells were washed with PBS (1 mL × 3) and imaging was performed using a Leica TCS SPE (inverted configuration) confocal microscope and a 63× oil-immersion objective lens. In the negative control experiments, MDA-MB-231 cells were pretreated with MMP-2/9 inhibitor I (20 μM) for 2 h prior to incubation with conjugate 2-MMP-QSY7 or HEK-293 cells was used as the control cell line. The excitation wavelength of conjugate 2-MMP-QSY7 was 405 nm.
000 cells per well) in growth medium (100 μL) and incubated at 37 °C under a 5% CO2 atmosphere for 48 h. The growth medium was replaced with conjugate 2-MMP-QSY7 with concentrations ranging from 1 to 300 μM in medium/DMSO (99:1, v/v) at 37 °C under a 5% CO2 atmosphere for 4 h. Wells containing untreated cells were used as blank control. After the treatment, the medium was removed, the cells were washed with PBS (100 μL), and phenol red-free growth medium was added to each wall (100 μL). One of the microplates was irradiated at 450 nm (15 mW cm−2) for 10 min in a LED cellular photocytotoxicity irradiator (PURI Materials, Shenzhen, China), and the other microplate was kept in the dark. The culture medium was then replaced with fresh medium, and the cells were incubated at 37 °C under a 5% CO2 atmosphere. After incubation for 24 h, the medium in each well was replaced with fresh medium (90 μL), and 10 μL of MTT (5 mg mL−1) in PBS was added. The medium was removed after incubation at 37 °C for 4 h, and DMSO (100 μL) was added to each well. The microplates were further incubated at 37 °C for 20 min. The absorbance of the solutions at 570 nm was measured with BioTek Powerwave XS MQX200R microplate spectrophotometer.
:1, v/v) containing conjugate 2-MMP-QSY7 (10 μM) at 37 °C under a 5% CO2 atmosphere. After incubation for 3 h, the medium was removed, and the cells were washed thoroughly with PBS (1 mL × 3). The cells were trypsinised by trypsin-EDTA (500 μL) and harvested with PBS (1 mL × 3). The harvested cells, together with the collected PBS, were digested with 65% HNO3 (1 mL) at 60 °C for 24 h. The iridium concentration in the solution was measured using a NexION 2000 ICP-MS (PerkinElmer SCIEX Instruments).
000 cells) was gently added into a PBS solution (27 μL) containing C(PEG-SH)4 (30 wt%, 20 μL). Then the two mixtures were added to a confocal dish, and the mixture was gently mixed and left for 10 min to form a hydrogel ([Ir] = 40 μM). Fresh medium (2 mL) was then added, and the cells were incubated at 37 °C under a 5% CO2 atmosphere for 48 h. The cells were then imaged using a Leica TCS SPE (inverted configuration) confocal microscope and a 63× oil-immersion objective lens. In the negative control experiments, MDA-MB-231 cells were pretreated with MMP-2/9 inhibitor I (20 μM) for 4 h prior to treatment with Gel-1 or the control hydrogel Gel-1C was used instead of Gel-1. The excitation wavelength of Gel-1 and Gel-1C was 405 nm.
:1, v/v) containing conjugate 2-MMP-QSY7 (7 μM). After incubation for 6 h, the cells were washed with PBS (1 mL × 3) and imaging was performed using a Leica TCS SPE (inverted configuration) confocal microscope and a 63× oil-immersion objective lens. In the negative control experiments, MDA-MB-231 cells were pretreated with MMP-2/9 inhibitor I (20 μM) for 2 h prior to incubation with conjugate 2-MMP-QSY7 or HEK-293 cells was used as the control cell line. The excitation wavelength of conjugate 2-MMP-QSY7 was 405 nm.
000 cells per well) in growth medium (100 μL) and incubated at 37 °C under a 5% CO2 atmosphere for 48 h. The growth medium was replaced with conjugate 2-MMP-QSY7 with concentrations ranging from 1 to 300 μM in medium/DMSO (99:1, v/v) at 37 °C under a 5% CO2 atmosphere for 4 h. Wells containing untreated cells were used as blank control. After the treatment, the medium was removed, the cells were washed with PBS (100 μL), and phenol red-free growth medium was added to each wall (100 μL). One of the microplates was irradiated at 450 nm (15 mW cm−2) for 10 min in a LED cellular photocytotoxicity irradiator (PURI Materials, Shenzhen, China), and the other microplate was kept in the dark. The culture medium was then replaced with fresh medium, and the cells were incubated at 37 °C under a 5% CO2 atmosphere. After incubation for 24 h, the medium in each well was replaced with fresh medium (90 μL), and 10 μL of MTT (5 mg mL−1) in PBS was added. The medium was removed after incubation at 37 °C for 4 h, and DMSO (100 μL) was added to each well. The microplates were further incubated at 37 °C for 20 min. The absorbance of the solutions at 570 nm was measured with BioTek Powerwave XS MQX200R microplate spectrophotometer.
:1, v/v) containing conjugate 2-MMP-QSY7 (10 μM) at 37 °C under a 5% CO2 atmosphere. After incubation for 3 h, the medium was removed, and the cells were washed thoroughly with PBS (1 mL × 3). The cells were trypsinised by trypsin-EDTA (500 μL) and harvested with PBS (1 mL × 3). The harvested cells, together with the collected PBS, were digested with 65% HNO3 (1 mL) at 60 °C for 24 h. The iridium concentration in the solution was measured using a NexION 2000 ICP-MS (PerkinElmer SCIEX Instruments).
000 cells) was gently added into a PBS solution (27 μL) containing C(PEG-SH)4 (30 wt%, 20 μL). Then the two mixtures were added to a confocal dish, and the mixture was gently mixed and left for 10 min to form a hydrogel ([Ir] = 40 μM). Fresh medium (2 mL) was then added, and the cells were incubated at 37 °C under a 5% CO2 atmosphere for 48 h. The cells were then imaged using a Leica TCS SPE (inverted configuration) confocal microscope and a 63× oil-immersion objective lens. In the negative control experiments, MDA-MB-231 cells were pretreated with MMP-2/9 inhibitor I (20 μM) for 4 h prior to treatment with Gel-1 or the control hydrogel Gel-1C was used instead of Gel-1. The excitation wavelength of Gel-1 and Gel-1C was 405 nm.
Data availability
We confirm that all the relevant research data is contained with the manuscript and ESI.† No databases have been used and no references to such databases are contained in the manuscript or ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Hong Kong Research Grants Council (Project No. CityU 11301121, CityU 11317022, CityU 11309423 and C7075-21GF) and the Hong Kong Research Grants Council and National Natural Science Foundation of China (Project No. N_CityU104/21). We also thank the funding support from “Laboratory for Synthetic Chemistry and Chemical Biology” under the Health@InnoHK Programme launched by Innovation and Technology Commission, The Government of Hong Kong SAR, P. R. China. We thank Mr Michael Wai-Lun Chiang of Department of Chemistry, City University of Hong Kong, for his support with the TEM and SEM measurements. J.-W. X. acknowledges the receipt of a Postgraduate Studentship administered by City University of Hong Kong.References
- O. Boutureira and G. J. L. Bernardes, Advances in chemical protein modification, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Site-selective modification strategies in antibody-drug conjugates, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- N. Asiimwe, M. F. Al Mazid, D. P. Murale, Y. K. Kim and J.-S. Lee, Recent advances in protein modifications techniques for the targeting N-terminal cysteine, Pept. Sci., 2022, 114, e24235 CrossRef CAS.
- H. Jiang, W. Chen, J. Wang and R. Zhang, Selective N-terminal modification of peptides and proteins: Recent progresses and applications, Chin. Chem. Lett., 2022, 33, 80–88 CrossRef CAS.
- C. B. Rosen and M. B. Francis, Targeting the N terminus for site-selective protein modification, Nat. Chem. Biol., 2017, 13, 697–705 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Synthesis of proteins by native chemical ligation, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- H. Ren, F. Xiao, K. Zhan, Y.-P. Kim, H. Xie, Z. Xia and J. Rao, A biocompatible condensation reaction for the labelling of terminal cysteine residues on proteins, Angew. Chem., Int. Ed., 2009, 48, 9658–9662 CrossRef CAS PubMed.
- K. Li, W. Wang and J. Gao, Fast and stable N-terminal cysteine modification through thiazolidino boronate mediated acyl transfer, Angew. Chem., Int. Ed., 2020, 59, 14246–14250 CrossRef CAS PubMed.
- A. Bandyopadhyay, S. Cambray and J. Gao, Fast and selective labelling of N-terminal cysteines at neutral pH via thiazolidino boronate formation, Chem. Sci., 2016, 7, 4589–4593 RSC.
- H. Faustino, M. J. S. A. Silva, L. F. Veiros, G. J. L. Bernardes and P. M. P. Gois, Iminoboronates are efficient intermediates for selective, rapid and reversible N-terminal cysteine functionalisation, Chem. Sci., 2016, 7, 5052–5058 RSC.
- X. Zheng, Z. Li, W. Gao, X. Meng, X. Li, L. Y. P. Luk, Y. Zhao, Y.-H. Tsai and C. Wu, Condensation of 2-((alkylthio)(aryl)methylene)malononitrile with 1,2-aminothiol as a novel bioorthogonal reaction for site-specific protein modification and peptide cyclization, J. Am. Chem. Soc., 2020, 142, 5097–5103 CrossRef CAS PubMed.
- A. Istrate, M. B. Geeson, C. D. Navo, B. B. Sousa, M. C. Marques, R. J. Taylor, T. Journeaux, S. R. Oehler, M. R. Mortensen, M. J. Deery, A. D. Bond, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, Platform for orthogonal N-cysteine-specific protein modification enabled by cyclopropenone reagents, J. Am. Chem. Soc., 2022, 144, 10396–10406 CrossRef CAS PubMed.
- Y. Yuan and G. Liang, A biocompatible, highly efficient click reaction and its applications, Org. Biomol. Chem., 2014, 12, 865–871 RSC.
- M. Zhang and G. Liang, Applications of CBT-Cys click reaction: Past, present, and future, Sci. China Chem., 2018, 61, 1088–1098 CrossRef CAS.
- X. Hu, R. Tang, L. Bai, S. Liu, G. Liang and X. Sun, CBT-Cys click reaction for optical bioimaging in vivo, VIEW, 2023, 4, 20220065 CrossRef CAS.
- L. C.-C. Lee, L. Huang, P. K.-K. Leung and K. K.-W. Lo, Recent development of photofunctional transition metal–peptide conjugates for bioimaging and therapeutic applications, Eur. J. Inorg. Chem., 2022, 2022, e202200455 CrossRef CAS.
- K. S. Gkika, D. Cullinane and T. E. Keyes, Metal peptide conjugates in cell and tissue imaging and biosensing, Top. Curr. Chem., 2022, 380, 30 CrossRef CAS PubMed.
- L. C.-C. Lee and K. K.-W. Lo, Shining new light on biological systems: Luminescent transition metal complexes for bioimaging and biosensing applications, Chem. Rev., 2024, 124, 8825–9014 CrossRef CAS PubMed.
- X. Ma, J. Jia, R. Cao, X. Wang and H. Fei, Histidine–iridium(III) coordination-based peptide luminogenic cyclisation and cyclo-RGD peptides for cancer-cell targeting, J. Am. Chem. Soc., 2014, 136, 17734–17737 CrossRef CAS PubMed.
- K. Vellaisamy, G. Li, W. Wang, C.-H. Leung and D.-L. Ma, A long-lived peptide-conjugated iridium(III) complex as a luminescent probe and inhibitor of the cell migration mediator, formyl peptide receptor 2, Chem. Sci., 2018, 9, 8171–8177 RSC.
- Z. Zhao, X. Zhang, C.-E. Li and T. Chen, Designing luminescent ruthenium prodrug for precise cancer therapy and rapid clinical diagnosis, Biomaterials, 2019, 192, 579–589 CrossRef CAS PubMed.
- W. Wang, K.-J. Wu, K. Vellaisamy, C.-H. Leung and D.-L. Ma, Peptide-conjugated long-lived theranostic imaging for targeting GRPr in cancer and immune cells, Angew. Chem., Int. Ed., 2020, 59, 17897–17902 CrossRef CAS PubMed.
- C. A. Puckett and J. K. Barton, Fluorescein redirects a ruthenium−octaarginine conjugate to the nucleus, J. Am. Chem. Soc., 2009, 131, 8738–8739 CrossRef CAS PubMed.
- A. Byrne, C. S. Burke and T. E. Keyes, Precision targeted ruthenium(II) luminophores; highly effective probes for cell imaging by stimulated emission depletion (STED) microscopy, Chem. Sci., 2016, 7, 6551–6562 RSC.
- S. Ji, X. Yang, X. Chen, A. Li, D. Yan, H. Xu and H. Fei, Structure-tuned membrane active Ir-complexed oligoarginine overcomes cancer cell drug resistance and triggers immune responses in mice, Chem. Sci., 2020, 11, 9126–9133 RSC.
- A. Martin, A. Byrne, C. S. Burke, R. J. Forster and T. E. Keyes, Peptide-bridged dinuclear Ru(II) complex for mitochondrial targeted monitoring of dynamic changes to oxygen concentration and ROS generation in live mammalian cells, J. Am. Chem. Soc., 2014, 136, 15300–15309 CrossRef CAS PubMed.
- C. S. Burke, A. Byrne and T. E. Keyes, Highly selective mitochondrial targeting by a ruthenium(II) peptide conjugate: Imaging and photoinduced damage of mitochondrial DNA, Angew. Chem., Int. Ed., 2018, 57, 12420–12424 CrossRef CAS PubMed.
- C. S. Burke, A. Byrne and T. E. Keyes, Targeting photoinduced DNA destruction by Ru(II) tetraazaphenanthrene in live cells by signal peptide, J. Am. Chem. Soc., 2018, 140, 6945–6955 CrossRef CAS PubMed.
- A. H. Day, M. H. Übler, H. L. Best, E. Lloyd-Evans, R. J. Mart, I. A. Fallis, R. K. Allemann, E. A. H. Al-Wattar, N. I. Keymer, N. J. Buurma and S. J. A. Pope, Targeted cell imaging properties of a deep red luminescent iridium(III) complex conjugated with a c-Myc signal peptide, Chem. Sci., 2020, 11, 1599–1606 RSC.
- Q. Wu, K. Y. Zhang, P. Dai, H. Zhu, Y. Wang, L. Song, L. Wang, S. Liu, Q. Zhao and W. Huang, Bioorthogonal “labelling after recognition” affording an FRET-based luminescent probe for detecting and imaging caspase-3 via photoluminescence lifetime imaging, J. Am. Chem. Soc., 2020, 142, 1057–1064 CrossRef CAS PubMed.
- C. Jin, G. Li, X. Wu, J. Liu, W. Wu, Y. Chen, T. Sasaki, H. Chao and Y. Zhang, Robust packing of a self-assembling iridium complex via endocytic trafficking for long-term lysosome tracking, Angew. Chem., Int. Ed., 2021, 60, 7597–7601 CrossRef CAS PubMed.
- L. C.-C. Lee, A. W.-Y. Tsang, H.-W. Liu and K. K.-W. Lo, Photofunctional cyclometalated iridium(III) polypyridine complexes bearing a perfluorobiphenyl moiety for bioconjugation, bioimaging, and phototherapeutic Applications, Inorg. Chem., 2020, 59, 14796–14806 CrossRef CAS PubMed.
- P. K.-K. Leung, L. C.-C. Lee, T. K.-Y. Ip, H.-W. Liu, S.-M. Yiu, N. P. Lee and K. K.-W. Lo, Luminescent rhenium(I) perfluorobiphenyl complexes as site-specific labels for peptides to afford photofunctional bioconjugates, Chem. Commun., 2021, 57, 11256–11259 RSC.
- L. Huang, P. K.-K. Leung, L. C.-C. Lee, G.-X. Xu, Y.-W. Lam and K. K.-W. Lo, Photofunctional cyclometallated iridium(III) polypyridine methylsulfone complexes as sulfhydryl-specific reagents for bioconjugation, bioimaging and photocytotoxic applications, Chem. Commun., 2022, 58, 10162–10165 RSC.
- J. Shum, L. C.-C. Lee, M. W.-L. Chiang, Y.-W. Lam and K. K.-W. Lo, A concerted enzymatic and bioorthogonal approach for extra- and intracellular activation of environment-sensitive ruthenium(II)-based imaging probes and photosensitizers, Angew. Chem., Int. Ed., 2023, 62, e202303931 CrossRef CAS PubMed.
- L. Huang, L. C.-C. Lee, J. Shum, G.-X. Xu and K. K.-W. Lo, Construction of photofunctional peptide conjugates through selective modification of N-terminal cysteine with cyclometallated iridium(III) 2-formylphenylboronic acid complexes for organelle-specific imaging, enzyme activity sensing and photodynamic therapy, Chem. Commun., 2024, 60, 6186–6189 RSC.
- E. C.-L. Mak, Z. Chen, L. C.-C. Lee, P. K.-K. Leung, A. M.-H. Yip, J. Shum, S.-M. Yiu, V. W.-W. Yam and K. K.-W. Lo, Exploiting the potential of iridium(III) bis-nitrone complexes as phosphorogenic bifunctional reagents for phototheranostics, J. Am. Chem. Soc., 2024, 146, 25589–25599 CrossRef CAS PubMed.
- M. S. Lowry, W. R. Hudson, R. A. Pascal and S. Bernhard, Accelerated luminophore discovery through combinatorial synthesis, J. Am. Chem. Soc., 2004, 126, 14129–14135 CrossRef CAS PubMed.
- K. Y. Zhang, H.-W. Liu, M.-C. Tang, A. W.-T. Choi, N. Zhu, X.-G. Wei, K.-C. Lau and K. K.-W. Lo, Dual-emissive cyclometalated iridium(III) polypyridine complexes as ratiometric biological probes and organelle-selective bioimaging reagents, Inorg. Chem., 2015, 54, 6582–6593 CrossRef CAS PubMed.
- K. P. S. Zanoni, A. Ito, M. Grüner, N. Y. Murakami Iha and A. S. S. de Camargo, Photophysical dynamics of the efficient emission and photosensitization of [Ir(pqi)2(NN)]+ complexes, Dalton Trans., 2018, 47, 1179–1188 RSC.
- S. Ahmed, A. S. Mathews, N. Byeon, A. Lavasanifar and K. Kaur, Peptide arrays for screening cancer specific peptides, Anal. Chem., 2010, 82, 7533–7541 CrossRef CAS PubMed.
- A. Jabłońska-Trypuć, M. Matejczyk and S. Rosochacki, Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs, J. Enzyme Inhib. Med. Chem., 2016, 31, 177–183 CrossRef PubMed.
- A. Page-McCaw, A. J. Ewald and Z. Werb, Matrix metalloproteinases and the regulation of tissue remodelling, Nat. Rev. Mol. Cell Biol., 2007, 8, 221–233 Search PubMed.
- R. Roy, J. Yang and M. A. Moses, Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer, J. Clin. Oncol., 2009, 27, 5287–5297 CrossRef CAS PubMed.
- M. Egeblad and Z. Werb, New functions for the matrix metalloproteinases in cancer progression, Nat. Rev. Cancer, 2002, 2, 161–174 CrossRef CAS PubMed.
- S. A. Fisher, A. E. G. Baker and M. S. Shoichet, Designing peptide and protein modified hydrogels: Selecting the optimal conjugation strategy, J. Am. Chem. Soc., 2017, 139, 7416–7427 Search PubMed.
- S. R. Caliari and J. A. Burdick, A practical guide to hydrogels for cell culture, Nat. Methods, 2016, 13, 405–414 Search PubMed.
- V. Nele, J. P. Wojciechowski, J. P. K. Armstrong and M. M. Stevens, Tailoring gelation mechanisms for advanced hydrogel applications, Adv. Funct. Mater., 2020, 30, 2002759 Search PubMed.
- C. Jensen and Y. Teng, Is it time to start transitioning from 2D to 3D cell culture?, Front. Mol. Biosci., 2020, 7, 513823 Search PubMed.
- T. R. Hoare and D. S. Kohane, Hydrogels in drug delivery: Progress and challenges, Polymer, 2008, 49, 1993–2007 Search PubMed.
- X. Ma, K. P. C. Sekhar, P. Zhang and J. Cui, Advances in stimuli-responsive injectable hydrogels for biomedical applications, Biomater. Sci., 2024, 12, 5468–5480 RSC.
- H. Zhou, Y. Zhu, B. Yang, Y. Huo, Y. Yin, X. Jiang and W. Ji, Stimuli-responsive peptide hydrogels for biomedical applications, J. Mater. Chem. B, 2024, 12, 1748–1774 RSC.
- L. Schneider and J. Zhang, Lysosomal function in macromolecular homeostasis and bioenergetics in Parkinson's disease, Mol. Neurodegener., 2010, 5, 14 CrossRef PubMed.
- M. Ghosh, F. Carlsson, A. Laskar, X.-M. Yuan and W. Li, Lysosomal membrane permeabilization causes oxidative stress and ferritin induction in macrophages, FEBS Lett., 2011, 585, 623–629 CrossRef CAS PubMed.
- W. L. F. Armarego and C. Chai, Purification of laboratory chemicals, 6th edn, Butterworth-Heinemann, Oxford, 2009 Search PubMed.
- B. M. Peek, G. T. Ross, S. W. Edwards, G. J. Meyer, T. J. Meyer and B. W. Erickson, Synthesis of redox derivatives of lysine and related peptides containing phenothiazine or tris(2,2′-bipyridine)ruthenium(II), Int. J. Pept. Protein Res., 1991, 38, 114–123 Search PubMed.
- B. D. Sherman, Y. Xie, M. V. Sheridan, D. Wang, D. W. Shaffer, T. J. Meyer and J. J. Concepcion, Light-driven water splitting by a covalently linked ruthenium-based chromophore–catalyst assembly, ACS Energy Lett., 2017, 2, 124–128 CrossRef CAS.
- T. S.-M. Tang, K.-K. Leung, M.-W. Louie, H.-W. Liu, S. H. Cheng and K. K.-W. Lo, Phosphorescent biscyclometallated iridium(III) ethylenediamine complexes functionalised with polar ester or carboxylate groups as bioimaging and visualisation reagents, Dalton Trans., 2015, 44, 4945–4956 RSC.
- M. Nonoyama, Benzo[h]quinolin-10-yl-N iridium(III) complexes, Bull. Chem. Soc. Jpn., 1974, 47, 767–768 CrossRef CAS.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Reevaluation of absolute luminescence quantum yields of standard solutions using a spectrometer with an integrating sphere and a back-thinned CCD detector, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- J. N. Demas and G. A. Crosby, Measurement of photoluminescence quantum yields. Review, J. Phys. Chem., 1971, 75, 991–1024 CrossRef.
- A. A. Abdel-Shafi, P. D. Beer, R. J. Mortimer and F. Wilkinson, Photosensitised generation of singlet oxygen from ruthenium(II)-substituted benzoaza-crown-bipyridine complexes, Phys. Chem. Chem. Phys., 2000, 2, 3137–3144 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qi03276d |
| This journal is © the Partner Organisations 2025 |