
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring protonation sites with cation-responsive polyethylene glycol (PEG) tethers in [FeFe]-hydrogenase mimics†‡
Alejandro Torres ab,
Sergio Aguadoab,
Alba Colladobcd,
Elena Sáeze,
Mar Gómez-Gallegoab and
Miguel A. Sierra*ab
ab,
Sergio Aguadoab,
Alba Colladobcd,
Elena Sáeze,
Mar Gómez-Gallegoab and
Miguel A. Sierra*ab
aDepartamento de Química Orgánica, Facultad de Química, Universidad Complutense, 28040-Madrid, Spain. E-mail: sierraor@ucm.es
bCentro de Innovación en Química Avanzada (ORFEO-CINQA), Departamento de Química Orgánica, Facultad de Química, Universidad Complutense, 28040-Madrid, Spain
cDepartamento de Química Inorgánica, Universidad Autónoma de Madrid, 28049 Madrid, Spain
dInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
eUnidad de Resonancia Magnética, Facultad Ciencias Químicas, Universidad Complutense, 28040-Madrid, Spain
First published on 26th March 2025
Abstract
Mimics of [FeFe]-hydrogenases having two [(μ-adt)Fe2(CO)6] moieties linked through 1,2,3-triazole rings with polyethylene glycol (PEG) chains [(–OCH2CH2O–)4 (7) and (–OCH2CH2O–)5 (8)] are able to coordinate to alkali ions (Na+, K+) via the O-PEG atoms and the triazole-N3 positions. Electrocatalytic studies in trifluoroacetic acid (TFA) demonstrate that their catalytic performance is affected by the presence of Na+ and K+ salts. The addition of NaPF6 decreases the electrocatalytic activity of 7 and 8 (about 50% reduction of the TOF values). As, in TFA, 7 and 8 could be protonated in both the triazole and adt-amino groups, the reduction in TOF values suggests that NaPF6 inhibits the contribution of the triazolium species to the electrocatalytic process, likely due to the involvement of the triazole-N3 positions in Na+ binding. However, the addition of KPF6 either does not change (7) or increases the TOF values (8). 1H NMR titration experiments demonstrate that, despite the presence of K+ ions in the media, triazolium salts are formed. Therefore, the TOF values should reflect the contribution of species protonated in both the triazole and adt-amino groups to the HER process.
Introduction
[FeFe]-hydrogenases ([FeFe]-H2ases) are metalloenzymes featuring a bimetallic complex composed of two iron atoms linked by an azadithiolate (adt) bridged ligand, to which a [Fe4S4] cluster is attached through cysteine sulfur atoms (1 in Fig. 1).1 The metal core completes the coordination sphere with CO and CN− ligands.2 While the [Fe4S4] cluster plays a fundamental role in electron transport, the adt bridge is responsible for providing/removing protons due to the basic properties of the secondary amine.3 Diiron hexacarbonyl complexes of general formula [(μ SR)2Fe2(CO)6] (2) are considered [FeFe]-H2ase mimics, as they exhibit significant structural similarities with the active center of enzymes 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 and also show an analogous acid–base and redox behavior.5 In mimics 2, the iron centers of the [FeFe] subsite have a formal oxidation state of +1, form a single Fe–Fe bond (∼2.5 Å)6 and complete their coordination sphere with CO ligands. Depending on the substituents on the dithiolate bridgehead, these complexes belong to two different structural classes: complexes of general structure 3 have an adt bridge (as in natural enzymes 1), while complexes such as 4 (propanedithiolate bridge, pdt) and 5 (benzenedithiolate bridge, bdt) have all-carbon chains between the sulfur atoms5 (Fig. 1).
4 and also show an analogous acid–base and redox behavior.5 In mimics 2, the iron centers of the [FeFe] subsite have a formal oxidation state of +1, form a single Fe–Fe bond (∼2.5 Å)6 and complete their coordination sphere with CO ligands. Depending on the substituents on the dithiolate bridgehead, these complexes belong to two different structural classes: complexes of general structure 3 have an adt bridge (as in natural enzymes 1), while complexes such as 4 (propanedithiolate bridge, pdt) and 5 (benzenedithiolate bridge, bdt) have all-carbon chains between the sulfur atoms5 (Fig. 1).
 | ||
| Fig. 1 The schematic representation of a [FeFe]-H2ase active center and the main synthetic mimics. | ||
The hydrogen evolution reaction (HER) in 1 and [FeFe]-hydrogenase mimics is a two-proton/two-electron reduction process, but the mechanistic steps differ depending on the type of mimic (3, 4, 5) and the catalytic conditions.7 The introduction of additional basic centers in pdt- and bdt-[FeFe]-H2ase mimics has been recognized as beneficial for the electrocatalytic properties of these molecules, and this effect has been studied.8 It is known that the HER cycle is facilitated by the initial protonation of an additional basic site8h and is also favored by the presence of basic substituents (i.e. phosphines) that increase the basicity of the metal centers.8d,i In natural [FeFe] and [NiFe]-hydrogenase enzymes, the protonation of one of the cysteinyl thiolate substituents of the [Fe4S4] cluster has been proposed as a key step in the catalytic cycle.8a,k,l Also, in [FeFe]-hydrogenases, it has been demonstrated that the strongly conserved secondary ligand sphere around the (2FeH) cluster plays a major role in preserving a stable cofactor coordination and geometry, favoring the stabilization of the CN-ligands.8m However, while the protonation of the N atom of the adt bridge is generally accepted to be the first step in the HER cycle for adt-[FeFe] mimics, the influence of additional basic centers in the process has been less explored. In this regard, recent work by our group showed that the basic 1,2,3-triazole ring in complex 6 (Fig. 1) can be protonated with a strong acid (H2SO4), and the formation of the triazolium species considerably enhances the electrocatalytic activity of the complex.9
The incorporation of secondary metal ions has emerged as an attractive strategy to tune chemical properties and to observe new reactivity patterns in metal complexes.10,11 In fact, recent attention has been paid to the study of the effect of adding redox-inactive metal ions (i.e. Li+, Na+, K+, Ca2+), as a way to modulate the electrochemical properties of a metal complex.10b,e Inspired by this idea, we devised compounds 7 and 8, which combine two [(μ-adt)Fe2(CO)6] moieties linked by polyethylene glycol (PEG) chains of different lengths, through 1,2,3-triazole rings (Fig. 2). The coordination of 7 and 8 to alkali ions (Na+, K+) must involve the O-PEG and the basic triazole-N3 atoms.12 In these complexes, we should be able to control the participation/inhibition of the triazole rings in cation binding by acid addition, modulating the electrochemical properties. Therefore, the study of the electrochemical response of 7 and 8 in the presence/absence of Na+ or K+ salts in acid media will allow us to determine the contribution of the N-amino-adt versus N3 triazole-protonated species to the HER process.
 | ||
| Fig. 2 The structure of the compounds 7 (n = 3) and 8 (n = 4) studied in this work. | ||
Results and discussion
The synthesis of [FeFe]-H2ase mimetics 7 and 8 was achieved by a cycloaddition reaction between alkynes 9 and 10 and azide 11, catalyzed by Cu(I) (Scheme 1).13 The synthesis of azide 11 was previously described by us.9 Meanwhile, alkynes 914 and 1015 were prepared by reacting the corresponding commercially available diols with propargyl bromide and NaH in THF at room temperature. Compounds 7 and 8 were obtained in 92% and 74% yields, respectively, after column chromatography (SiO2, CH2Cl2/MeOH 95:5) and were characterized by spectroscopic techniques. Significant in the NMR spectra of 7 and 8 are the signals attributable to the CH of the newly formed 1,2,3-triazole rings at 7.96 ppm in the 1H NMR and 120.9 ppm in the 13C{1H} NMR spectra.
 | ||
| Scheme 1 Preparation of the tetrametallic complexes 7 and 8. | ||
Coordination studies with alkali metal cations
The ability of complexes 7 and 8 to coordinate to alkali metal cations was evaluated by 1H-NMR. The polyethylene glycol (PEG) bridges in compounds 7 and 8 are analogous to those of 15-crown-5 and 18-crown-6 ethers, which have cavity diameters of 1.7–2.2 Å and 2.6–3.2 Å, respectively, capable of forming 1:1 coordination complexes with Na+ and K+.16 Coordination studies were performed in solutions of complexes 7 and 8 in CD3CN with increasing concentrations of NaPF6 or KPF6 (0–4 equivalents). The selection of NaPF6 and KPF6 as cation sources was based on their higher solubility under the experimental conditions. In all cases, in the 1H NMR spectra, the signals of the PEG fragments were shifted to lower fields as the cation concentration increased, indicating cation coordination to the oxygen atoms (Fig. 3). The most affected signals were those of the four central methylene protons of the polyether units (singlets at 3.55 ppm and 3.54 ppm, for 7 and 8, respectively, in Fig. 3), which served as references for the subsequent calculations. Fig. 4 shows the plots of Δδ (ppm) versus the amounts of Na+ and K+ for compounds 7 and 8. The plots reveal a steady increase in Δδ values until the stationary region is reached, indicating saturation. Job plots were constructed from the 1H NMR data to determine the stoichiometry of the complexes (Fig. 5), with maxima at χ1 and χ2 = 0.5, suggesting a 1:1 stoichiometry for the metal cation coordination complexes.17
 | ||
| Fig. 3 1H NMR spectra (500 MHz) of complexes 7 (left) and 8 (right) upon the addition of 4 equiv. of NaPF6 (in green) and KPF6 (in red) in CD3CN at 25 °C. The signal at 3.55 ppm (*) was used as reference in the study. | ||
 | ||
| Fig. 4 Representation of Δδ (ppm) vs. equivalents of Na+ and K+ for 7 (left) and 8 (right) at 25 °C. | ||
 | ||
| Fig. 5 Job plots of compounds 7 (left) and 8 (right) with NaPF6 and KPF6 at 25 °C. | ||
Once the stoichiometry of coordination complexes was known, the association constants (Ka, M−1) of the different equilibria were calculated following the reported procedure.17 Experimental Ka values in CH3CN for [7-Na+] and [7-K+] were 6.66 × 102 M−1 and 1.16 × 103 M−1, respectively, while for [8-Na+] and [8-K+], they were 8.49 × 102 M−1 and 1.63 × 103 M−1, respectively. The calculated association constants are about two orders of magnitude higher than those obtained for analogous acyclic polyethers in the presence of Na+ and K+, which highlights the ability of 7 and 8 to bind sodium and potassium and points to the participation of the triazole N3 positions in the process.17,18
Additional support for the structures of the alkali complexes was obtained using DFT calculations at the SMD (CH3CN)-M06/6-31G** level. As shown in Fig. 6, in [7-Na+], the cation is coordinated to seven donor atoms: the five O (PEG) atoms and the two triazole N3 atoms. The Na–O distances fit well with the expected Na–O distance (2.42 Å), and the Na–N distances (2.48 Å and 2.65 Å, respectively) agree with the expected Na–N distance (2.54 Å), which confirms the strong interaction between the sodium ion and the N3 positions of both triazole rings. In turn, in the potassium complex [7-K+], the cation is coordinated to the five O positions of the PEG moiety (K–O average distance of 2.72 Å) and to one of the triazole N3 atoms (K–N distance of 2.83 Å), in a pseudo-crown-6 structure, with K–O and K–N distances in the range of the expected values (K–O 2.78 Å and K–N 2.80 Å, respectively). The remaining triazole ring is positioned above the macrocycle at a distance of K–N3 of 3.08 Å.
 | ||
| Fig. 6 Calculated structures of complexes [7-Na+] (top) and [7-K+] (bottom) (SMD (CH3CN)-M06/6-31G** level). Hydrogen atoms have been omitted for clarity. Bond distances in Å. Na–N and K–N distances are shown in red. | ||
The computed structures of [8-Na+] and [8-K+] are depicted in Fig. 7. In both cases, the N3 positions of the triazole fragments are deeply involved in the coordination to the alkali cations together with the PEG fragments. For complex [8-Na+], the average distance Na–O is 2.68 Å and the Na–N3 distances are 2.49 Å and 2.68 Å, respectively. In turn, for complex [8-K+], the average K–O distance is 2.82 Å and the Na–N3 distances are 2.89 Å and 2.85 Å, all within the range of the literature data.19
 | ||
| Fig. 7 Calculated structures of complexes [8-Na+] (top) and [8-K+] (bottom) (SMD (CH3CN)-M06/6-31G** level). Hydrogen atoms have been omitted for clarity. Bond distances in Å. Na–N and K–N distances are shown in red. | ||
Electrochemistry
The electrochemical and electrocatalytic properties of compounds 7 and 8 and their Na+ and K+ coordination complexes were explored. Compounds 7 and 8 exhibited similar electrochemical behavior. Both showed a reduction wave at Epc = −1.57 V attributed to the [FeIFeI]/[Fe0FeI] process in the [(μ-adt)Fe2(CO)6] moieties, together with an irreversible second reduction wave at Epc = −2.18 V, corresponding to the [Fe0FeI]/[Fe0Fe0] process in the same fragment (Fig. 8).20 | ||
| Fig. 8 Cyclic voltammograms of complexes 7 and 8 in a 10−3 M MeCN solution containing [(nBu)4N]PF6 10−1 M as the supporting electrolyte. Voltammograms were registered at 25 °C using a Pt counter electrode and a glassy carbon working electrode. Potentials are given in V vs. Fc+/Fc. Scan rate: 100 mVs. | ||
In the presence of a weak acid (AcOH), 7 and 8 display the expected behavior for a [(μ-adt)Fe2(CO)6] derivative.21 The band at −1.56 V does not change, but the intensity of the second reduction wave at −2.18 V steadily increases with the successive additions of acid (Fig. 9, top). Upon the addition of the stronger trifluoroacetic acid (TFA, pKMeCNa ≈ 12.6), there is a clear electrocatalytic response of the wave at −1.56 V (Fig. 9, bottom). However, at high concentrations of acid (6–10 equiv.), the catalytic current (icat) at −2.18 V also increases. In agreement with our previous results, the electrocatalytic behavior observed for 7 and 8 in TFA must be due to the contribution of the species protonated in both the triazole ring and the adt-amino groups.8 The icat/ip plots vs. increasing acid concentrations were used to evaluate the electrocatalytic properties of 7 and 8 in the reduction of protons from TFA (ip is the peak current in the absence of acid, Fig. 10). The data revealed that 8 displayed a higher (about 15% more) electrocatalytic activity than 7 with AcOH, but their performance was similar with TFA.
 | ||
| Fig. 9 CVs of compounds 7 (left) and 8 (right) with increasing amounts of AcOH (top) and TFA (bottom) obtained from 10−3 M solutions in MeCN containing [(nBu)4N]PF6 10−1 M as the supporting electrolyte, recorded at 25 °C. Counter-electrode: Pt; working electrode: glassy carbon; potentials given in V vs. Fc+/Fc; scan rate: 100 mV s−1. | ||
 | ||
| Fig. 10 Representation of icat/ip of 7 and 8 vs. equivalents of AcOH and TFA. | ||
To determine the effect of cation binding on the process, the cyclic voltammograms (CVs) of 7 and 8 were recorded in the presence of NaPF6 or KPF6 (5 equiv.) (Fig. 11). Compared to the CVs of 7 and 8, the current intensity of the first reduction wave increased for both [7-Na+] and [8-Na+] but slightly decreased for the potassium complexes. However, the second reduction wave that appeared at −2.18 V in the CV of the uncoordinated compounds was significantly altered by the nature of the cation. The CV of complex [7-Na+] displayed a crossover upon reversing the potential scan direction, likely due to the deposition of insoluble material on the working electrode. In contrast, complex [8-Na+] showed no such deposition. This displacement has been observed previously in different transition metal complexes bearing crown ether moieties.22
 | ||
| Fig. 11 CVs of compounds [7-Na+], [7-K+], [8-Na+] and [8-K+] obtained from 10−3 M solutions in MeCN containing [(nBu)4N]PF6 10−1 M as the supporting electrolyte, recorded at 25 °C. Counter-electrode: Pt; working electrode: glassy carbon; potentials given in V vs. Fc+/Fc; scan rate: 100 mV s−1. | ||
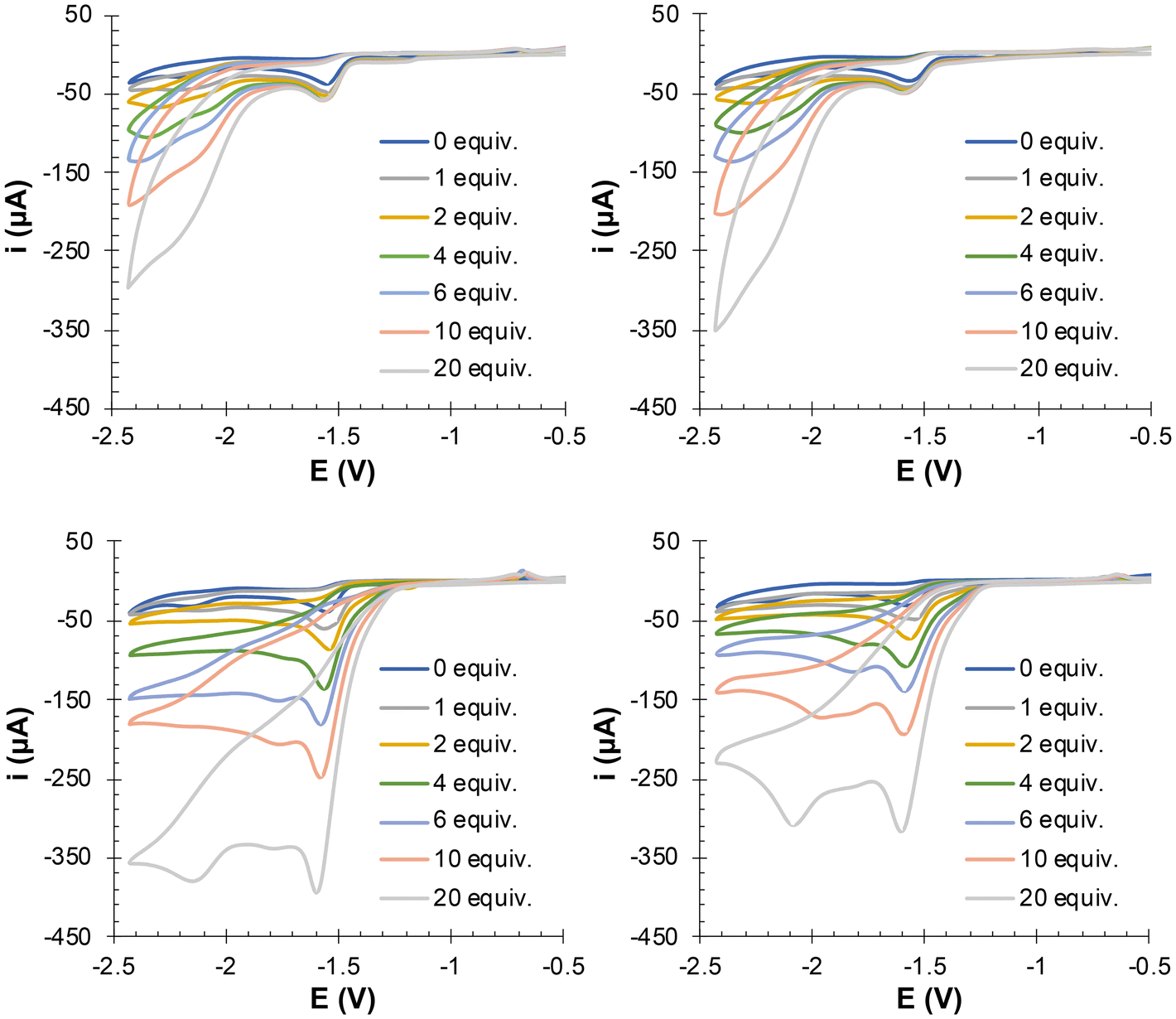
The electrocatalytic activity of 7 and 8 in the presence of 5 equiv. of NaPF6 or KPF6 and with increasing amounts of TFA (1–20 equiv.) was next addressed (Fig. 12). The icat/ip plots and the turnover frequency (TOF s−1) values for the [FeIFeI]/[FeIFe0] reduction wave, with 20 equiv. of TFA, were used to evaluate the electrocatalytic activity of the complexes (Fig. 13 and Table 1). The calculated overpotential was 0.68 V. The results revealed that, in TFA, the electrocatalytic activity of 7 and 8 in the presence of NaPF6 was considerably diminished, as indicated by the 50% reduction in the TOF values. As compounds 7 and 8 could be protonated in both the triazole rings and the adt-amino groups,9 the reduction in the TOF values suggests that the contribution of the triazolium species to the electrocatalytic process is inhibited with NaPF6. Undoubtedly, despite the addition of acid, complexes [7-Na+] and [8-Na+] are formed in the medium, with the N3 positions of the triazole rings involved in the Na+ binding.
 | ||
| Fig. 12 CVs of (a) complex 7 with TFA + 5 equiv. of NaPF6, (b) complex 8 with TFA + 5 equiv. of NaPF6, (c) complex 7 with TFA + 5 equiv. of KPF6, and (d) complex 8 with TFA + 5 equiv. of KPF6, obtained from 10−3 M solutions in MeCN containing [(nBu)4N]PF6 10−1 M as the supporting electrolyte, recorded at 25 °C. Counter-electrode: Pt; working electrode: glassy carbon; potentials given in V vs. Fc+/Fc; scan rate: 100 mV s−1. | ||
 | ||
| Fig. 13 Representation of icat/ip of complexes 7 and 8 with 5 equivalents of NaPF6 and KPF6 vs. equivalents of TFA. | ||
These assertions were confirmed by an 1H NMR study carried out with complex 8. The spectra in Fig. 14 show the protonation of the triazole rings in compound 8 as the amount of TFA increased in solution (0–10 eq.) The formation of the triazolium salt causes the broadening and the significant downfield shift of the H5 signal of the triazole due to a rapid proton exchange between the protonated centers in the triazole-PEG region.9 The signals corresponding to the [(μ-adt)Fe2(CO)6] fragment (7.70, 6.96 and 4.45 ppm) are hardly affected by the addition of acid, which suggests that protonation is specific to the triazole moiety. When the titration with TFA was done in the presence of NaPF6, the 1H NMR spectra of 8 changed significantly (Fig. 15). Unlike the free complex, the signals corresponding to the triazole fragment remain sharp and barely shift as the TFA concentration increases. This indicates that the triazole-PEG region does not undergo rapid proton exchange when Na+ is present, supporting the formation of the coordination complex [8-Na+] in the acid media.
 | ||
| Fig. 14 1H NMR spectra (300 MHz) of compound 8 with TFA (0–10 equiv.) in CD3CN at 25 °C. Arrows indicate signals corresponding to the triazole-PEG fragment. | ||
 | ||
| Fig. 15 1H NMR spectra (300 MHz) of compound 8 with 5 equiv. of NaPF6 and TFA (0–10 equiv.) in CD3CN at 25 °C. | ||
The electrochemical experiments of 7 and 8 with the potassium salt in the presence of TFA had a different outcome. Data for compound 7 in Fig. 12 and 13 and Table 1 indicate a similar electrocatalytic response in the presence or absence of KPF6 (TOF values of 20.4 s−1 and 19.9 s−1, respectively). However, the addition of the potassium salt in TFA enhances the electrocatalytic activity of 8 (TOF values of 21.0 s−1 versus 37.9 s−1 with KPF6). These experimental data suggest that the addition of KPF6 does not hamper the protonation of the triazole rings of 7 and 8. Obviously, in the presence of KPF6, the triazole N3-positions are not involved in the coordination of K+ in acid media. The TOF values in these cases should reflect the contributions of the species protonated in both the triazole rings and the adt-amino groups to the HER process.
The 1H NMR spectroscopy study of complex 8 with KPF6 in the presence of increasing amounts of TFA (0–10 eq.) supported these claims. As shown in Fig. 16, the signal of the H5 proton of the triazole ring broadens and experiences a significant downfield shift as the amount of TFA increases in solution. The process is almost identical to that observed in Fig. 14 for complex 8 in the presence of increasing amounts of TFA.
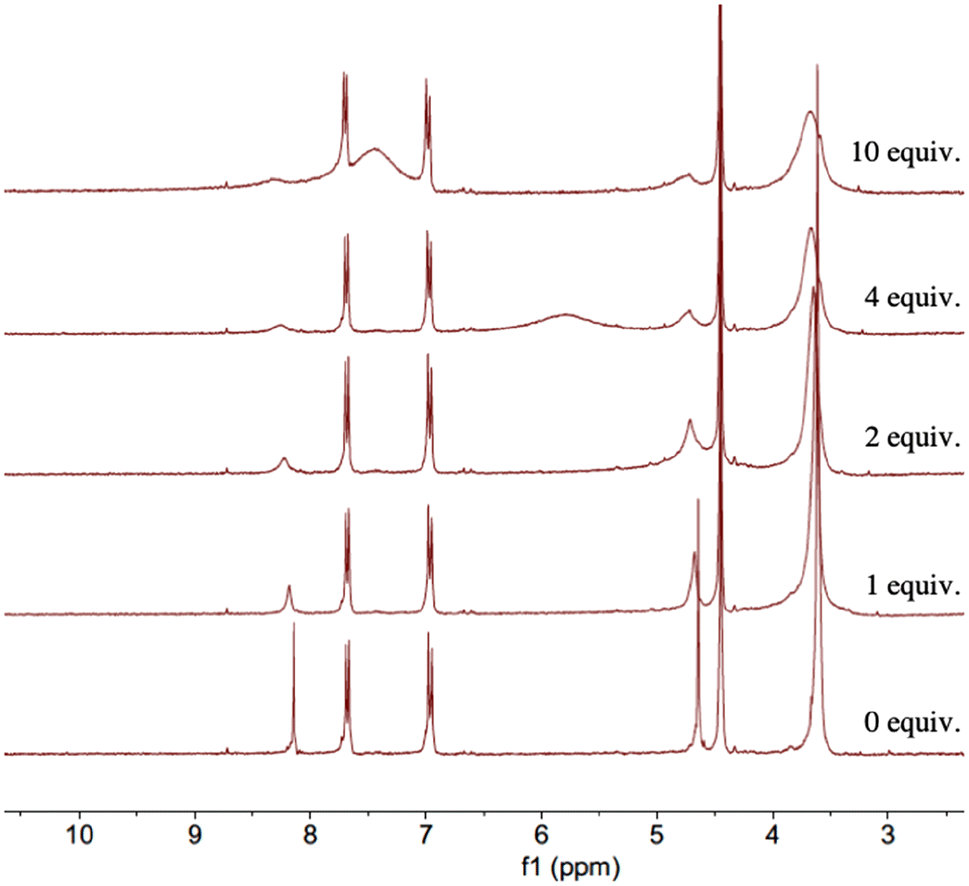 | ||
| Fig. 16 1H NMR spectra (300 MHz) of compound 8 with 5 equiv. of KPF6 and TFA (0–10 equiv.) in CD3CN at 25 °C. | ||
Additional insights into the coordinating behavior of complex 8 in trifluoroacetic acid media were obtained by determining its complexation constants using the same methodology applied under neutral conditions, in the presence of 5 equivalents of TFA (see above and the ESI‡) and NaPF6 and KPF6. The data from Job's plots show that the ratio of ligand 8 to the alkali cation was 1:1 in both cases (Fig. S69‡). The association constants (Ka) in MeCN for [8-Na++TFA] and [8-K++TFA] were found to be 4.04 × 102 M−1 and 1.26 × 103 M−1, respectively. As anticipated, these values were lower than those obtained under neutral conditions (6.66 × 102 M−1 and 1.63 × 103 M−1 for [8-Na+] and [8-K+], respectively), since, under acidic conditions, the 1,2,3-triazole moiety is expected to be protonated, rendering it unavailable for coordination with the alkali metal cations.
The electrocatalytic and NMR experiments demonstrate the fundamental role of the alkali ions in modulating the electrocatalytic response of 7 and 8 in acid medium. With NaPF6/TFA, the triazole units are engaged in the coordination with Na+ and are not able to contribute to the HER process. The experimental TOF values of 9.0 s−1 for 7 and 9.2 s−1 for 8 in the presence of NaPF6/TFA essentially show the contribution of the species protonated in the amino adt groups, whereas with KPF6/TFA, the triazole units in 7 and 8 are protonated and can participate in the reaction.
It is well established that the protonated amine plays a fundamental role in the mechanism of hydrogen generation from [(μ-adt)Fe2(CO)6] mimics, serving as a readily available source of protons for the neighboring Fe atom.5b,7f,21a Our results provide new insights into the contribution of N3-protonated 1,2,3-triazolium salts, which significantly enhance the electrocatalytic activity of [(μ-adt)Fe2(CO)6] mimics in strong acidic media. While the protonated amine serves as a direct proton donor for the Fe atom, the N3-protonated 1,2,3-triazolium salts would provide an additional reservoir of protons, enhancing the overall catalytic efficiency. Although the potential role of the 1,2,3-triazole moieties as electron reservoirs could also be considered, recent studies on [FeFe]H2ase mimics with alkyl 1,2,3-triazolium salts as ligands exclude the triazolium ring as an additional redox-active site in the hydrogen evolution reaction (HER) process.8e,f,9
The observed increase in the turnover frequency (TOF) for compound 8 in trifluoroacetic acid (TFA) with KPF6 (37.9 s−1) compared to the TOF for compound 8 without the potassium salt (21.0 s−1) remains unclear. Experimental evidence indicates that the triazole rings are protonated in both cases and can contribute to the HER process together with the protonated amino adt groups. A computational study would shed some light on this question. The SMD (CH3CN)-M06/6-31G** computed structure of 8, protonated in both the triazole and [(μ-adt)Fe2(CO)6] amino groups [8H4-K], reveals the six PEG oxygen atoms coordinated to K+ in a very regular pseudo-18-crown-6 structure (Fig. 17). The average K–O distance in the hexacoordinated potassium complex is 2.77 Å, in excellent agreement with the expected K–O distance (2.78 Å), calculated as the sum of the oxygen van der Waals radius (1.40 Å) and the effective ionic radius of K+ (1.38 Å).23 The incorporation of alkali metal ions is known to enhance the electrocatalytic activity of metal complexes,10,11 and the formation of the coordination complex [8H4-K] could account for the increased TOF observed for compound 8 in TFA in the presence of KPF6. The fact that the TOF values for complex 7 in TFA are almost identical in the presence/absence of KPF6 (20.4 s−1 and 19.9 s−1, respectively) supports the importance of the six-oxygen coordination environment for the K+ in [8H4-K], likely allowing greater catalytic performance. These findings underscore the critical role of ligand design and metal-ion coordination in optimizing the performance of hydrogenase mimics for electrocatalytic applications.
 | ||
| Fig. 17 Computed SMD (CH3CN)-M06/6-31G** structure of [8H4-K]. The hydrogen atoms of the PEG linker have been omitted for clarity. Bond distances are in Å. | ||
Conclusions
[FeFe]H2ase mimics 7 and 8, which feature two [(μ-adt)Fe2(CO)6] moieties and two 1,2,3-triazole rings linked through PEG chains of different lengths, have been prepared to study the effect of additional basic centers on the electrocatalytic properties of [(μ-adt)Fe2(CO)6] derivatives in acidic media. Compounds 7 and 8 are capable of binding alkali ions (Na+, K+) to form 1:1 coordination complexes, in which the oxygen atoms of the PEG moiety and the N3 positions of the triazole rings are involved. Through 1H NMR titration experiments and electrochemical studies of 7 and 8 in the presence or absence of NaPF6 or KPF6, we determined the relative contributions of N-adt amino versus N3 triazole protonated species to the HER process in TFA. The addition of Na+ ions inhibits the protonation of the triazole rings in both 7 and 8, and their electrochemical response is primarily attributed to the species protonated at the amino adt groups. However, with K+, the triazolium salts are formed in TFA, and the electrocatalytic behavior of 7 and 8 reflects the contributions from species protonated at both the triazole and amino adt groups.
A computational study of the protonated complex 8 with KPF6 in the presence of TFA could explain the increase in the turnover frequency (TOF) observed for this complex (37.9 s−1) compared to the TOF calculated in the absence of the potassium salt (21.0 s−1). The computed complex [8H4-K], protonated in both the triazole and amino [(μ-adt)Fe2(CO)6] groups, is still able to coordinate the K+ ion through the six O-PEG atoms, in a very regular pseudo-18-crown-6 structure. Since the incorporation of alkali metal ions is known to enhance the electrocatalytic activity of metal complexes, the formation of complex [8H4-K] could account for the increased TOF observed for compound 8 in TFA in the presence of KPF6. These findings highlight the intricate interplay between ligand design, ion-binding fragments and their effects on catalytic behavior in acidic media. Future studies are underway to explore the incorporation of other ion-binding fragments and investigate their impact on the HER process, with the aim of further optimizing the performance of hydrogenase mimics for electrocatalytic applications.
Experimental section
General
Unless otherwise stated, all the reactions were carried out under an Ar atmosphere using anhydrous solvents. Alkynes 914 and 1015 and azide 119 were prepared according to reported protocols. 1H and 13C{1H} NMR spectra were recorded at ambient temperature in CDCl3 using Bruker 500 or 300 MHz spectrometers. Chemical shifts are expressed in part per million and are referenced to residual solvent peaks. ESI-HRMS was performed using an Agilent 6500 accurate mass spectrometer with a Q-TOF analyser. Cyclic voltammograms were recorded using a Metrohm PGSTAT302N potentiostat model. In all cases, the solutions were degassed with an argon flow and sonicated prior to electrochemical analysis. For cyclic voltammograms, a 3 mm glassy-carbon working electrode, an Ag/AgCl 3 M reference electrode, and a 2 mm Pt-wire counter electrode were used. All measurements were performed at room temperature and under an argon atmosphere in CH3CN solutions containing 10−1 M [nBu4]PF6 as the supporting electrolyte, with analyte concentrations of 10−3 M.
General procedure for the CuAAC reactions
In a round-bottom flask, 1 equiv. of the dialkyne, 2 equiv. of azide 11, and a MeCN/THF mixture in a 1:1 ratio were added. The resulting suspension was bubbled with Ar for 10 minutes. Subsequently, 2 equiv. of CuI and 2 equiv. of DIPEA were added. The reaction mixture was stirred at room temperature for 72 h. The solvent was evaporated under reduced pressure, and the obtained solid was purified by column chromatography (SiO2, CH2Cl2/MeOH 95:5).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O 2073, 2031 and 1992 cm−1. HRMS-ESI m/z: calcd for C42H39Fe4N8O17S4 [M + H]+: 1278.8714; found [M + H]+: 1278.8724.O 2073, 2030 and 1991 cm−1. HRMS-ESI m/z: calcd for C44H43Fe4N8O18S4 [M + H]+: 1322.8976; found [M + H]+: 1322.8986.
O 2073, 2031 and 1992 cm−1. HRMS-ESI m/z: calcd for C42H39Fe4N8O17S4 [M + H]+: 1278.8714; found [M + H]+: 1278.8724.O 2073, 2030 and 1991 cm−1. HRMS-ESI m/z: calcd for C44H43Fe4N8O18S4 [M + H]+: 1322.8976; found [M + H]+: 1322.8986.Computational details
Calculations were performed at the DFT level using the M06 functional24 with an ultrafine integration grid25 as implemented in Gaussian 16.26 Fe, S, and K atoms were described using the scalar relativistic Stuttgart–Dresden SDD pseudopotential27 and its associated double-ζ basis set complemented with a set of d- and f-polarization functions.28 The 6-31G** basis set was used for the H, C, N, and O atoms.29 All structures were fully optimized in acetonitrile solvent using the SMD continuum model.24,30Author contributions
Miguel A. Sierra (MAS) designed the work. MAS and Alba Collado (AC) directed the research and interpreted the results. Elena Sáez recorded all the NMR experiments and titrations. Alejandro Torres and Sergio Aguado executed the experimental part and interpreted the experimental results. Mar Gómez-Gallego executed the DFT calculations and was involved in the electrochemical studies. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.‡Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support for this work from the Ministerio de Ciencia, Innovación y Universidades (Grants PID2022-139177OB-I00 and RED2022-134287-T) is acknowledged. AT was the recipient of an FPU fellowship (FPU16/02062).References
- (a) J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Structure/function relationships of [NiFe]- and [FeFe]-hydrogenases, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS PubMed; (b) P. M. Vignais and B. Billoud, Occurrence, classification and biological function of hydrogenases: an overview, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS PubMed; (c) W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Hydrogenases, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- (a) C. Esmieu, M. Guo, H. J. Redman, M. Lundberg and G. Berggren, Synthesis of a miniaturized [FeFe] hydrogenase model system, Dalton Trans., 2019, 48, 2280–2284 RSC; (b) M. Senger, S. Mebs, J. Duan, O. Shulenina, K. Laun, L. Kertess, F. Wittkamp, U.-P. Apfel, T. Happe, M. Winkler, M. Haumann and S. T. Stripp, Protonation/reduction dynamics at the [4Fe-4S] cluster of the hydrogen-forming cofactor in [FeFe]-hydrogenases, Phys. Chem. Chem. Phys., 2018, 20, 3128–3140 RSC; (c) P. Rodríguez-Maciá, L. Kertess, J. Burnik, J. A. Birrell, E. Hofmann, W. Lubitz, T. Happe and O. Rüdiger, His-ligation to the [4Fe-4S] subcluster tunes the catalytic bias of [FeFe] hydrogenase, J. Am. Chem. Soc., 2019, 141, 472–481 CrossRef PubMed; (d) A. J. Pierik, M. Hulstein, W. R. Hagen and S. P. Albracht, A Low-Spin Iron with CN and CO as Intrinsic Ligands Forms the Core of the Active Site in [Fe]-Hydrogenases, Eur. J. Biochem., 1998, 258, 572–578 CrossRef CAS PubMed; (e) A. L. De Lacey, C. Stadler, C. Cavazza, E. C. Hatchikian and V. M. Fernandez, FTIR Characterization of the Active Site of the Fe-Hydrogenase from Desulfovibrio Desulfuricans, J. Am. Chem. Soc., 2000, 122, 11232–11233 CrossRef CAS; (f) Y. Nicolet, A. L. De Lacey, X. Vernède, V. M. Fernandez, E. C. Hatchikian and J. C. Fontecilla-Camps, Crystallographic and FTIR Spectroscopic Evidence of Changes in Fe Coordination upon Reduction of the Active Site of the Fe-Only Hydrogenase from Desulfovibrio Desulfuricans, J. Am. Chem. Soc., 2001, 123, 1596–1601 CrossRef CAS PubMed.
- (a) M. T. Lachmann, Z. Duan, P. Rodríguez-Macía and J. A. Birrell, The missing pieces in the catalytic cycle of [FeFe]hydrogenases, Chem. Sci., 2024, 15, 14062–14080 RSC; (b) J. A. Birrell, P. Rodríguez-Macía, E. J. Reijerse, M. A. Martini and W. Lubitz, The catalytic cycle of [FeFe] hydrogenase: A tale of two sites, Coord. Chem. Rev., 2021, 449, e214191 Search PubMed; (c) M. Senger, V. Eichmann, K. Laun, J. Duan, F. Wittkamp, G. Knör, U.-P. Apfel, T. Happe, M. Winkler, J. Heberle and S. T. Stripp, How [FeFe]-Hydrogenase Facilitates Bidirectional Proton Transfer?, J. Am. Chem. Soc., 2019, 141, 17394–17403 CrossRef CAS PubMed; (d) J. Duan, S. Mebs, K. Laun, F. Wittkamp, J. Heberle, E. Hofmann, U.-P. Apfel, M. Winkler, M. Senger, M. Haumann and S. T. Stripp, Geometry of the Catalytic Active Site in [FeFe]-Hydrogenase Is Determined by Hydrogen Bonding and Proton Transfer, ACS Catal., 2019, 9, 9140–9149 CrossRef CAS; (e) J. Duan, M. Senger, J. Esselborn, V. Engelbrecht, F. Wittkamp, U.-P. Apfel, E. Hofmann, S. Y. Stripp, T. Happe and M. Winkler, Crystallographic and spectroscopic assignment of the proton transfer pathway in [FeFe]-hydrogenases, Nat. Commun., 2018, 9, 4726–4737 Search PubMed; (f) A. J. Cornish, K. Gärtner, H. Yang, J. W. Peters and E. L. Hegg, Mechanism of proton transfer in [FeFe]-hydrogenase from Clostridium pasteurianum, J. Biol. Chem., 2011, 286, 38341–38347 CrossRef CAS; (g) H. Long, P. W. King and C. H. Chang, Proton transport in Clostridium pasteurianum [FeFe] hydrogenase I: a computational study, J. Phys. Chem. B, 2014, 118, 890–900 CrossRef CAS; (h) G. Hong, A. J. Cornish, E. L. Hegg and R. Pachter, On understanding proton transfer to the biocatalytic [Fe-Fe]H sub-cluster in [Fe-Fe]H2ases: QM/MM MD simulations, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 510–517 CrossRef CAS.
- H. Reihlen, A. Gruhl, V. Hessling and G. Über, den photochemischen und oxydativen abbau von carbonylen, Justus Liebigs Ann. Chem., 1929, 472, 268–287 CrossRef CAS.
- (a) Y. Li and T. B. Rauchfuss, Synthesis of diiron(I) dithiolato carbonyl complexes, Chem. Rev., 2016, 116, 7043–7077 CrossRef CAS; (b) D. Schilter, J. M. Camara, M. T. Huynh, S. Hammes-Schiffer and T. B. Rauchfuss, Hydrogenase enzymes and their synthetic models: the role of metal hydrides, Chem. Rev., 2016, 116, 8693–8749 CrossRef CAS.
- M. Y. Darensbourg, E. J. Lyon, X. Zhao and I. P. Georgakaki, The Organometallic Active Site of [Fe]Hydrogenase: Models and Entatic States, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3683–3688 Search PubMed.
- See for example: (a) R. Mejia-Rodriguez, D. Chong, J. H. Reibenspies, M. P. Soriaga and M. Y. Darensbourg, The Hydrophilic Phosphatriazaadamantane Ligand in the Development of H2 Production Electrocatalysts: Iron Hydrogenase Model Complexes, J. Am. Chem. Soc., 2004, 126, 12004–12014 CrossRef CAS; (b) T. Liu, M. Wang, Z. Shi, H. Cui, W. Dong, J. Chen, B. Åkermark and L. Sun, Synthesis, Structures and Electrochemical Properties of Nitro- and Amino- Functionalized Diiron Azadithiolates as Active Site Models of Fe-Only Hydrogenases, Chem. – Eur. J., 2004, 10, 4474–4479 CrossRef CAS PubMed; (c) J. F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, Catalysis of the Electrochemical H2 evolution by Di-Iron Sub-Site Models, Coord. Chem. Rev., 2005, 249, 1664–1676 CrossRef CAS; (d) G. A. N. Felton, A. K. Vannucci, J. Chen, L. T. Lockett, N. Okumura, B. J. Petro, U. I. Zakai, D. H. Evans, R. S. Glass and D. L. Lichtenberger, Hydrogen Generation from Weak Acids: Electrochemical and Computational Studies of a Diiron Hydrogenase Mimic, J. Am. Chem. Soc., 2007, 129, 12521–12530 CrossRef CAS PubMed; (e) R. Zaffaroni, T. B. Rauchfuss, D. L. Gray, L. De Gioia and G. Zampella, Terminal vs Bridging Hydrides of Diiron Dithiolates: Protonation of Fe2(dithiolate)(CO)2(PMe3)4, J. Am. Chem. Soc., 2012, 134, 19260–19269 CrossRef CAS PubMed; (f) M. T. Huynh, W. Wang, T. B. Rauchfuss and S. Hammes-Schiffer, Computational Investigation of [FeFe]-Hydrogenase Models: Characterization of Singly and Doubly Protonated Intermediates and Mechanistic Insights, Inorg. Chem., 2014, 53, 10301–10311 CrossRef CAS PubMed; (g) A. Jablonskyte, L. R. Webster, T. R. Simmons, J. A. Wright and C. J. Pickett, Electronic Control of the Protonation Rates of Fe–Fe Bonds, J. Am. Chem. Soc., 2014, 136, 13038–13044 CrossRef CAS.
- For the synthesis of [FeFe]-H2ase mimics bearing pendant basic centers, see: (a) J. T. Kleinhaus, F. Wittkamp, S. Yadav, D. Siegmund and U.-P. Apfel, [FeFe]-Hydrogenases: maturation and reactivity of enzymatic systems and overview of biomimetic models, Chem. Soc. Rev., 2021, 50, 1668–1784 RSC; (b) G. Hogarth, An unexpected leading role for [Fe2(CO)6(μ-pdt)] in our understanding of [FeFe]-H2ases and the search for clean hydrogen production, Coord. Chem. Rev., 2023, 490, e215174 CrossRef See also ref. 5a. Examples of the study of the effect of the additional basic centers on the electrocatalysis of these mimics: (c) I. O. Shotonwa, O. Ejeromedoghene, A. O. Adesoji, Y. A. Alli, C. Akinremi and S. Adewuyi, Electrochemistry, electrocatalysis, and mechanistic details into hydrogen evolution pathways of hexacoordinated iron scaffolds in hydrogenase mimics, J. Electroanal. Chem., 2023, 938, e117446 CrossRef; (d) G. R. F. Orton, S. Belazregue, J. K. Cockcroft, F. Hartl and G. Hogarth, Biomimics of [FeFe]-hydrogenases with a pendant amine: Diphosphine complexes [Fe2(CO)4{μ-S(CH2)nS}{κ2-(Ph2PCH2)2NR}] (n = 2, 3; R = Me, Bn) towards H2 oxidation catalysts, J. Organomet. Chem., 2023, 991, 122673 CrossRef CAS; (e) A. Mele, F. Arrigoni, C. Elleouet, F. Y. Pétillon, P. Schollhammer and G. Zampella, Insights into Triazolylidene Ligands Behaviour at a Di-Iron Site Related to [FeFe]-Hydrogenases, Molecules, 2022, 27, 4700 CrossRef CAS; (f) A. Mele, S. Bertini, M. Albrecht, C. Elleouet, F. Y. Pétillon and P. A. Schollhammer, Diiron Hydrogenase Mimic Featuring a 1,2,3-Triazolylidene, Chimia, 2020, 74, 499–503 CrossRef CAS PubMed; (g) R. Zaffaroni, W. I. Dzik, R. J. Detz, J. I. van der Vlugt and J. N. H. Reek, Proton Relay Effects in Pyridyl-Appended Hydrogenase Mimics, Eur. J. Inorg. Chem., 2019, 2498–2509 CrossRef CAS; (h) R. Becker, S. Amirjalayer, P. Li, S. Woutersen and J. N. Reek, An iron-iron hydrogenase mimic with appended electron reservoir for efficient proton reduction in aqueous media, Sci. Adv., 2016, 2, e1501014 CrossRef PubMed; (i) N. Wang, M. Wang, J. H. Liu, K. Jin, L. Chen and L. C. Sun, Preparation, Facile Deprotonation and Rapid H/D Exchange of the μ-Hydride Diiron Model Complexes of the [FeFe]-Hydrogenase Containing a Pendant Amine in a Chelating Diphosphine Ligand, Inorg. Chem., 2009, 48, 11551–11558 CrossRef CAS; (j) N. Wen, F.-F. Xu, R.-P. Chen and S.-W. Du, Reversible carbonylation of [2Fe2S] model complexes with pendant quinoline or pyridine arms, J. Organomet. Chem., 2014, 756, 61–67 CrossRef CAS; (k) J. A. Birrell, P. Rodríguez-Maciá, E. J. Reijerse, M. A. Martini and W. Lubitz, The catalytic cycle of [FeFe] hydrogenase: A tale of two sites, Coord. Chem. Rev., 2021, 449, 214191 CrossRef CAS; (l) L. Greene, G. E. Vansuch, B. C. Chica, M. W. W. Adams and R. Brian Dyer, Applications of Photogating and Time Resolved Spectroscopy toMechanistic Studies of Hydrogenases, Acc. Chem. Res., 2017, 50, 2718–2726 CrossRef; (m) O. Lampret, A. Adamska-Venkatesh, H. Konegger, F. Wittkamp, U.-P. Apfel, E. J. Reijerse, W. Lubitz, O. Rüdiger, T. Happe and M. Winkler, Interplay between CN- Ligands and the Secondary Coordination Sphere of the H-Cluster in [FeFe]-Hydrogenases, J. Am. Chem. Soc., 2017, 139, 18222–18230 CrossRef CAS PubMed.
- A. D. Merinero, A. Collado, L. Casarrubios, M. Gómez-Gallego, C. Ramírez de Arellano, A. Caballero, F. Zapata and M. A. Sierra, Triazole-Containing [FeFe]-Hydrogenase Mimics: Synthesis and Electrocatalytic Behavior, Inorg. Chem., 2019, 58, 16267–16278 CrossRef CAS PubMed.
- See, among others: (a) M. S. Somachandra, B. Averkiev and P. E. Sues, Unsymmetric Co-Facial “Salixpyrrole” Hydrogen Evolution Catalysts: Two Metals are Better than One, Inorg. Chem., 2024, 63, 13346–11335 CrossRef CAS; (b) R. R. Golwankar, A. Kumar, V. W. Day and J. D. Blakemore, Revealing the Influence of Diverse Secondary Metal Cations on Redox-Active Palladium Complexes, Chem. – Eur. J., 2022, 28, e202200344 CrossRef CAS; (c) H. Shao, S. K. Muduli, P. D. Tran and H. S. Soo, Enhancing electrocatalytic hydrogen evolution by nickel salicylaldimine complexes with alkali metal cations in aqueous media, Chem. Commun., 2016, 52, 294–2951 Search PubMed; (d) D. C. Lacy, Y. J. Park, J. W. Ziller, J. Yano and A. S. Borovik, Assembly and Properties of Heterobimetallic CoII/III/CaII Complexes with Aquo and Hydroxo Ligands, J. Am. Chem. Soc., 2012, 134, 17526–17535 CrossRef CAS PubMed; (e) Y. J. Park, J. W. Ziller and A. S. Borovik, The Effects of Redox-Inactive Metal Ions on the Activation of Dioxygen: Isolation and Characterization of a Heterobimetallic Complex Containing a MnIII–(μ-OH)–CaII Core, J. Am. Chem. Soc., 2011, 133, 9258–9261 CrossRef CAS; (f) S. Haaf, E. Kaifer, H. Wadepohl and H.-J. Himmel, Use of Crown Ether Functions as Secondary Coordination Spheres for the Manipulation of Ligand–Metal Intramolecular Electron Transfer in Copper–Guanidine Complexes, Chem. – Eur. J., 2021, 27, 959–970 CrossRef CAS PubMed; (g) S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Redox-inactive metal ions modulate the reactivity and oxygen release of mononuclear non-heme iron(III)–peroxo complexes, Nat. Chem., 2014, 6, 934–940 CrossRef CAS PubMed.
- (a) A. Torres, A. Collado, M. Gómez-Gallego, C. Ramírez de Arellano and M. A. Sierra, Heteropolymetallic [FeFe]-Hydrogenase Mimics: Synthesis and Electrochemical Properties, Inorg. Chem., 2023, 62, 3409–3419 CrossRef CAS PubMed; (b) S. Aguado, M. Gómez-Gallego, L. Casarrubios and M. A. Sierra, Electrocatalytic HER Performance of [FeFe]-Hydrogenase Mimics Bearing M-salen Moieties (M=Zn, Ni, Fe, Mn), Chem. – Eur. J., 2025, e202403721 CrossRef CAS PubMed.
- (a) J. M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, Wiley-VCH, Weinheim, 1995 CrossRef; (b) R. M. Izatt, K. Pawlak, J. S. Bradsshaw and R. L. Bruening, Thermodynamic and Kinetic Data for Macrocycle Interaction with Cations, Anions, and Neutral Molecules, Chem. Rev., 1995, 95, 2529–2586 CrossRef CAS; (c) R. Peng, Y. Xu and Q. Cao, Recent advances in click-derived macrocycles for ions recognition, Chin. Chem. Lett., 2018, 29, 1465–1474 Search PubMed; (d) J. W. Steed, First- and second-sphere coordination chemistry of alkali metal crown ether complexes, Coord. Chem. Rev., 2001, 215, 171–221 CrossRef CAS; (e) N. A. Itsikson, G. L. Rusinov, D. G. Beresnev and O. L. Chupakhin, Modification of macrocyclic compounds by Azaheterocycles, Heterocycles, 2003, 61, 593–637 Search PubMed.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. A. Sharpless, Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 Search PubMed; (b) C. W. Torne, C. Christensen and M. Meldal, Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides, J. Org. Chem., 2002, 67, 3057–3064 CrossRef PubMed; (c) H. C. Kolb, M. G. Finn and K. B. Sharpless, Click Chemistry: Diverse Chemical Function from a Few Good Reactions, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 Search PubMed; (d) M. Meldal and C. W. Torne, Cu-Catalyzed Azide-Alkyne Cycloaddition, Chem. Rev., 2008, 108, 2952–3015 CrossRef CAS PubMed.
- T. Koopmans, T. M. Wood, P. ‘t Hart, L. H. J. Kleijn, A. P. A. Hendrickx, R. J. L. Willems, E. Breukink and N. I. Martin, Semisynthetic lipopeptides derived from nisin display antibacterial activity and lipid II binding on par with that of the parent compound, J. Am. Chem. Soc., 2015, 137, 9382–9389 CrossRef CAS.
- P. J. Jervis, M. Moulis, J.-P. Jukes, H. Ghadbane, L. R. Cox, V. Cerundolo and G. S. Besra, Towards multivalent CD1d ligands: synthesis and biological activity of homodimeric α-galactosyl ceramide analogues, Carbohydr. Res., 2012, 356, 152–162 CrossRef CAS PubMed.
- C. J. Pedersen, Cyclic polyethers and their complexes with metal salts, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- T. Terashima, M. Kawabe, Y. Miyabara, H. Yoda and M. Sawamoto, Polymeric pseudo-crown ether for cation recognition via cation template-assisted cyclopolymerization, Nat. Commun., 2013, 4, 2321 CrossRef PubMed.
- The reported values (logKa) of the association constants of crown ethers with Na+ and K+ are deeply influenced by the solvent. See:
(a) H. K. Frensdorff, Stability constants of cyclic polyether complexes with univalent cations, J. Am. Chem. Soc., 1971, 93, 600–606 CrossRef CAS;
(b) D. M. Dishong and G. W. Gokel, Crown Cation Complex Effects. 16. Solvent Dependence of the 15-Crown-5 and 18-Crown-6 Equilibria with Sodium Cation, J. Org. Chem., 1982, 47, 147–148 CrossRef CAS;
(c) J. D. Lin and A. I. Popov, Nuclear Magnetic Resonance Studies of Some Sodium Ion Complexes with Crown Ethers and Cryptands in Various Solvents, J. Am. Chem. Soc., 1981, 103, 3173–3111 CrossRef;
(d) I. M. Kolthoff and M. K. Chantooni Jr., Transfer Activity Coefficients in Various Solvents of Severaln Univalent Cations Complexed with Dibenzo-18-crown-6, Anal. Chem., 1980, 52, 1039–1044 CrossRef CAS;
(e) I. Inoue, Y. Liu, L.-H. Tong, M. Ouchi and T. Hakushi, Complexation Thermodynamics of Crown Ethers. Part 3. 12-Crown-4 to 36-Crown-12: from Rigid to Flexible Ligand, J. Chem. Soc., Perkin Trans. 2, 1993, 1947–1950 RSC.
- L. Fabbrizi, The experimental average Na-O distance in crown ethers is 2.47–2.54 Å. The average K-O distance is 2.80 Å, in Cryptands and Cryptates, World Scientific Publishing Europe Ltd., 2018 Search PubMed.
- (a) Z. Xiao, Z. Wei, L. Long, Y. Wang, D. J. Evans and X. Liu, Diiron Carbonyl Complexes Possessing a Fe(II)Fe(II) Core: Synthesis, Characterization and electrochemical Investigation, Dalton Trans., 2011, 40, 4291–4299 RSC; (b) S. Dey, A. Rana, S. G. Dey and A. Dey, Electrochemical Hydrogen Production in Acidic Water by an Azadithiolate Bridged Synthetic Hydrogenase Mimic: Role of Aqueous Solvation in Lowering Overpotential, ACS Catal., 2013, 3, 429–436 CrossRef CAS; (c) A. Rana, P. K. Das, B. Mondal, S. Dey, D. Crouthers and A. Dey, Investigation of Bridgehead Effects on Reduction Potential in Alkyl and Aryl Azadithiolate-Bridged (μ-SCH2XCH2S) [Fe(CO)3]2 Synthetic Analogues of [FeFe]-H2ase Active Site, Eur. J. Inorg. Chem., 2018, 3633–3643 CrossRef CAS; (d) D. Chong, I. P. Georgakaki, R. Mejia-Rodriguez, J. Sanabria-Chinchilla, M. P. Soriaga and M. Y. Darensbourg, Electrocatalysis of Hydrogen Production by Active Site Analogues of the Iron Hydrogenase Enzyme: Structure/Function Relationships, J. Chem. Soc., Dalton Trans., 2003, 4158–4163 RSC.
- (a) J.-F. Capon, S. Ezzaher, F. Gloaguen, F. Y. Pétillon, P. Schollhammer and J. Talarmin, Electrochemical insights into the mechanisms of proton reduction by [Fe2(CO)6{μ-SCH2N(R)CH2S}] complexes related to the [2Fe]H subsite of [FeFe]hydrogenase, Chem. – Eur. J., 2008, 14, 1954–1964 CrossRef CAS PubMed; (b) F. Gloaguen, J. D. Lawrence, T. B. Rauchfuss, M. Bénard and M. Rohmer, Bimetallic Carbonyl Thiolates as Functional Models for Fe-Only Hydrogenases, Inorg. Chem., 2002, 41, 6573–6582 CrossRef CAS PubMed; (c) J.-F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, Electrochemical Proton Reduction by Thiolate-Bridged Hexacarbonyldiiron Clusters, J. Electroanal. Chem., 2004, 566, 241–247 CrossRef CAS; (d) J.-F. Capon, F. Gloaguen, P. Schollhammer and J. Talarmin, Activation of Proton by the Two-Electron Reduction of a Diiron Organometallic Complex, J. Electroanal. Chem., 2006, 595, 47–52 CrossRef CAS; (e) S. J. Borg, T. Behrsing, S. P. Best, M. Razavet, X. Liu and C. J. Pickett, Electron Transfer at a Dithiolate-Bridged Diiron Assembly: Electrocatalytic Hydrogen Evolution, J. Am. Chem. Soc., 2004, 126, 16988–16999 CrossRef CAS PubMed.
- (a) N. D. Lowe and C. D. Garner, Transition-metal Complexes of Crown Ether Benzodithiolenes, J. Chem. Soc. Dalton Trans., 1993, 2197–2207 RSC; (b) N. D. Lowe and C. D. Garner, Transition-metal Complexes of Crown Ether Benzodithiolenes. Part 2. The Effects of Alkali-metal Cation Binding, J. Chem. Soc., Dalton Trans., 1993, 3333–3340 RSC; (c) H. M. Nguyen, H. W. T. Morgan, T. Chantarojsiri, T. A. Kerr, J. Y. Yang, A. N. Alexandrova and N. G. Léonard, Charge and Solvent Effects on the Redox Behavior of Vanadyl Salen-Crown Complexes, J. Phys. Chem. A, 2023, 127, 5324–5334 CrossRef CAS PubMed; (d) J. L. Alvarez-Hernandez, X. Zhang, K. Cui, A. P. Deziel, S. Hammes-Schiffer, N. Hazari, N. Piekut and M. Zhong, Long-range electrostatic effects from intramolecular Lewis acid binding influence the redox properties of cobalt–porphyrin complexes, Chem. Sci., 2024, 15, 6800–6815 RSC.
- R. D. Shannon, Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chaleogenides, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- Y. Zhao and D. G. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- (a) Y. Zhao and D. G. Truhlar, Density functionals with broad applicability in Chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed; (b) Y. Zhao and D. G. Truhlar, Applications and validations of the Minnesota density functionals, Chem. Phys. Lett., 2011, 502, 1–13 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- U. Andrae, M. Häußermann, H. Dolg, H. Stoll and H. Preuß, Energy-adjusted ab initio pseudopotentials for the second and third row transition elements, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef.
- (a) A. W. Ehlers, M. Böhme, S. Dapprich, A. Gobbi, A. Höllwarth, V. Jonas, K. F. Köhler, R. Stegmann, A. Veldkamp and G. Frenking, A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc-Cu, Y-Ag and La-Au, Chem. Phys. Lett., 1993, 208, 111 CrossRef CAS; (b) A. Höllwarth, M. Biihme, S. Dapprich, A. W. Ehlers, A. Gobbi, V. Jonas, K. F. Kijhler, R. Stegmann, A. Veldkamp and G. Frenking, A set of d-polarization functions for pseudo-potential basis sets of the main group elements Al-Bi and f-type polarization functions for Zn, Cd, Hg, Chem. Phys. Lett., 1993, 208, 237–240 CrossRef.
- (a) W. J. Hehre, R. J. Ditchfield and A. Pople, Self-Consistent molecular orbital methods. XII. Further extensions of Gaussian-Type basis sets for use in molecular orbital studies of organic molecules, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. J. DeFrees and A. Pople, Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Fernando P. Cossío (EHU-UPV-Donostia, San Sebastián) on the occasion of his 65th birthday. |
| ‡ Electronic supplementary information (ESI) available: NMR and IR spectra of compounds 7 and 8, 1H NMR experiments with NaPF6 and KPF6 and computational details. See DOI: https://doi.org/10.1039/d5qi00170f |
| This journal is © the Partner Organisations 2025 |