
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLow-valent tantalum/gold clusters: oxidation, protonation, and C–H activation†
Michela L.
Maiola
and
Joshua A.
Buss
 *
*
Willard Henry Dow Laboratory, Department of Chemistry, University of Michigan, 930 N. University Avenue, Ann Arbor, MI 48109, USA. E-mail: jbuss@umich.edu
First published on 2nd April 2025
Abstract
Gold-based catalysts are topical heterogeneous and molecular species, the chemical diversity of which can be expanded through heterometal doping. Herein, we leverage a carbonyl-free metal–metal salt metathesis protocol to access rare examples of low-valent tantalum/gold multimetallics. The initial reaction between [Ta(naphthalene)3]− and gold(I) synthons affords a trimetallic monohydride cluster (2). Whereas dihydrogen addition to 2 results in deauration en route to a Ta–μH2–Au complex (1), oxidative transformations—either addition of chemical oxidants or cluster protonation—conserve the trimetallic core, even in the absence of a polynucleating ligand. The resultant series of compounds provides experimental anchors for computational interrogation of polarized metal–metal interactions as a function of metal identity, formal oxidation state, and ligand sphere. The electronic structure of these clusters showcases significant Ta–arene covalency, even at higher oxidation states, rationalizing a recalcitrance to undergo ligand substitution. Furthermore, the addition of in situ generated Au+ to 2 results in an arene C–H activation process, highlighting that the naphthalene ligands in these complexes are simultaneously substitutionally inert and prone to functionalization.
Introduction
The synthesis of heterometallic gold clusters flourished in the 1980s,1,2 during which time their relevance to surface catalysis was emphasized as the so-called cluster-surface analogy gained prominence.3,4 Synthetically, many of these species were accessed via addition of Au(I) precursors to existing clusters or via salt-metatheses with polycarbonyl metallates (Fig. 1A).2,5–15 In spite of the diversity of cluster architectures accessed in this way, the thermal reactivity of the resultant species is markedly distinct from that of heterogeneous active sites.16–18 Contrasting the latter, in which highly reactive, coordinatively unsaturated fragments are responsible for substrate binding and activation,19,20 the carbonyl ligands commonly found in low-valent molecular systems stunt their reactivity via electronic attenuation and coordinative saturation.17 Focus therefore shifted to using these platforms to explore the electronic structure of metal–metal bonding and the impact of metal ion identity on bonding modes.2,12 | ||
| Fig. 1 Established synthetic routes to heterometallic gold coordination complexes. Low-valent mixed-metal gold complexes are generally accessed via salt-metatheses with transition metal polycarbonyls (A). High-valent M/Au compounds are routinely prepared via treatment of hydride or alkyl complexes with appropriate gold(I) precursors (B). | ||
Higher valent heterometallic gold fragments, particularly those bearing bridging hydride ligands, have likewise been explored as informers of electronic structure and fundamental reactivity trends relevant to higher nuclearity gold-based nanoparticles and materials.21–36 Distinct from the preparations of low-valent clusters, these bi- and trimetallic compounds are often accessed by addition of aurous precursors bearing labile or strongly Brønsted-basic ligands (Fig. 1B).21,24,26,29,31–36 This strategy is effective for the generation of mid- to high-oxidation state complexes that often feature bridging hydride ligands. Quite recently, Camp and coworkers disclosed a salt-metathesis synthesis of Ta(V)/Au(I) complexes bridged by unsaturated Ta–C bonds, in which bimetallic cooperativity enables unusual ketenyl formation via deoxygenation of CO2.22 There is a scarcity of literature, however, disclosing low-valent early–late heteromultimetallic species, compounds with formal valencies that more accurately mirror a heterogeneous surface.
In heterogeneous catalysis, the multicomponent reactivity of either gold and a non-innocent support or heterometallic gold catalysts is well-established.37,38 These effects are particularly important for imparting gold with atypical reactivity.39–41 Hydrogen spillover—the exchange of surface hydrogen atoms between metals or between gold and a support—is a critical mechanistic phenomenon leveraging bimetallic or metal/support cooperativity.42–47 Both the intrinsic metal–metal interactions and their influence on the electronic structure of bridging hydride ligands is central to understanding this process, a key step toward the rational design and implementation of improved heterometallic catalysts that coordination chemistry is uniquely suited to address.28,48
In line with our group's interest in using well-defined molecular clusters as models of surface active sites,49,50 we recently reported an enabling synthetic methodology for the formation of early–late heterobimetallics.51 Our approach addressed a longstanding challenge in bottom-up mixed-metal cluster synthesis, facilitating a rational, stepwise increase in cluster nuclearity and providing access to a series of bi-, tri-, and tetra-metallic carbene-supported Ta/Cu compounds (Fig. 2A). Herein, we expand the scope of the carbonyl-free metal–metal salt metathesis to include the synthesis of carbene-supported heteromultimetallic gold clusters. Taking advantage of the more accessible redox properties of gold, we demonstrate oxidation and protonation chemistry, subsequently targeting naphthalene substitution (Fig. 2B). Contrasting analogous reaction conditions with copper, the major product of the initial salt metathesis reaction is a TaAu2 trimetallic species featuring unsupported Au–Au and Au–Ta bonds. Redox chemistry, informed by cyclic voltammetry studies, generates two novel Ta/Au clusters. Both single crystal X-ray diffraction (SCXRD) and density functional theory (DFT) studies provide structural and electronic comparisons across Ta/Au trimetallics, highlighting changes in metal–metal bonding as a function of ligation and redox state. This work showcases the versatility of the salt metathesis strategy for rationally accessing rare low-valent mixed-metal complexes absent electronically deactivating, strongly π-acidic carbonyls.
 | ||
| Fig. 2 Synthesis of early–late heterometallic hydride complexes via carbonyl-free metal–metal salt metathesis. Our prior disclosure investigated the metathesis, hydride origination, and reductive chemistry of Ta/Cu compositions (A). This work expands the chemistry to gold, showcasing the effect of oxidation and protonation state on metal–metal bonding (B). | ||
Results and discussion
Ta/Au mixed-metal cluster synthesis and reactivity
Building on our previous work, Ellis’ tris(naphthalene) tantalate ([Ta(naph)3]−)52 was treated with one equivalent of cyclic (alkyl)(amino)carbene gold chloride (EtCAACAuCl)53 in dimethoxyethane (DME). Distinct from the analogous reaction with carbene-supported cuprous salts, the initial metathesis produced a mixture of bimetallic (EtCAACAu)H2Ta(naph)2 (1) and trimetallic (EtCAACAu)2HTa(naph)2 (2) complexes in 13% and 30% yield, respectively (Fig. 3A). The incorporation of hydride ligands during the salt metathesis reaction aligns with our findings from the Ta/Cu chemistry, where detailed isotopic labeling experiments showed that the hydride ligands were sourced from both naphthalene and solvent during the formation of the bis(μH) diamond core.54 Compounds 1 and 2 can be separated based on differential solubility, with the former readily dissolving in aliphatic solvents. | ||
| Fig. 3 Synthetic pathway to (A) and solid-state structures of (B) mixed Ta/Au bi- and trimetallic hydride complexes. SCXRD structures are shown with thermal anisotropic displacement ellipsoids at a 50% probability level and non-hydride H atoms omitted for clarity. The hydride H-atoms in 1 were resolved in the Fourier map; their locations were refined freely and their Uiso values were treated with a riding model. Bond metrics for the bimetallic cores are reported in Å. | ||
The bimetallic complex 1, best described as a TaIAuI species, was fully characterized via nuclear magnetic resonance (NMR) spectroscopy and SCXRD. The 1H NMR spectrum of 1 reveals a mixture of both a symmetric (C2v) major species (δμH = −2.78 ppm) and an asymmetric (Cs) minor species (δμH = –1.48 ppm), assigned to coordination isomers with syn- and anti-naphthalene rings, respectively. This structural phenomenon was likewise observed for the copper congeners, in which the proportion of the anti-isomer correlated inversely with the steric profile of the carbene ligand. For 1, the anti-isomer is favored more so than in the isostructural copper analog (34% vs. 22%), consistent with the larger radius of Au. Despite being the minor species in solution, complex 1 preferentially crystalizes in an anti-naphthalene conformation. SCXRD analysis reveals a Ta–Au distance of 2.8536(2) Å, longer than the only previously reported low-valent Ta–Au contact (2.7202(2) Å),55 but decidedly shorter than a recently disclosed alkylidyne-supported Ta–Au separation (2.9462(3) Å).22 The formal shortness ratio (FSR)56 of the Ta–Au contact (1.05) resides on the upper range of a covalent single bond, reflecting the key role the hydrides play in stabilizing the heterobimetallic core (vide infra).
The structure of 2 was likewise interrogated via a combination of solution spectroscopy and SCXRD.57 The 1H NMR spectrum displays broad, but diamagnetic, resonances at 5.83, 4.79 and 3.12 ppm, accounting for sixteen protons of the Ta-bound naphthyl rings. The presence of a single hydride was supported by a broad high-field singlet at −1.79 ppm (THF-d8, 25 °C) with a relative integration of one. The breadth of these signals is attributed to a ring slip isomerization process—interconversion of syn- and anti-conformations—that is intermediate on the NMR timescale at room temperature. Cooling the sample to −80 °C, the hydride singlet sharpens (ΔFWHM = 106 Hz at 25 °C vs. ΔFWHM = 16 Hz at −80 °C), and the three naphthalene peaks split into sixteen distinct signals with relative integrations of one proton each, supporting this hypothesis (Fig. S25†). Notably, the hydride chemical shift (PhMe-d8, −80 °C: δ = −1.29 ppm) is similar to that of the related μH motif in 1anti, which, in combination with the inferred C1 symmetry, suggests that the anti-isomer of 2 is thermodynamically preferred in solution. Even so, trimetallic 2 crystallizes with a syn-naphthalene geometry, featuring both supported (Ta1–Au1: 2.8760(3) Å) and unsupported (Ta1–Au2: 2.8255(3) Å) mixed-metal bonds (Fig. 3B). The hydride ligand in 2 was not discernable in the Fourier map; however, the combined structural and spectroscopic data, particularly the orientation of the naphthyl rings, are consistent with a Ta1–μH–Au1 structure. A more in-depth discussion precluding alternative structures is presented in the ESI.† The naphthyl rings are rotated 60.66(5) degrees relative to the M3 centroid, biasing the cluster edge featuring the unsupported Ta–Au bond. The Au1–Au2 distance of 2.7400(3) Å is significantly shorter than a related heterotrimetallic cluster generated from salt metatheses between Collman's reagent [Fe(CO)4]2− and PPh3AuCl (cf. 2.93 Å)15 and is very similar to the Au–Au distance in a previously reported Au2Nb complex (2.736 Å),35 which bears a formal Au–Au bond. This coinage metal–coinage metal bond is a notable difference in the salt metathesis chemistry of copper and gold. The former generates a trimetallic complex upon reaction with [Ta(naph)3]− with no Cu–Cu interaction, a σ-bound copper aryl, and a second copper center bridged to tantalum by both a μH and μCl (cf.Cu2TaHCl; Fig. 2A). With gold, the trimetallic features a direct Au–Au bond, which we attribute to both the aurophilicity of gold and its more favorable reduction (relative to copper).
Early metal-doped coinage metal surfaces are of significant interest within the context of apolar bond activation. Biener and coworkers recently analyzed H/D exchange on a titanium doped Cu(111) surface through a combined experimental and computational effort.58 This work, and others,45,59–63 purport a mechanism in which the heterometal (i.e. Ti) serves as the site of H2 activation due to its higher affinity to bind H2 as compared to copper, which only weakly interacts with H2. The molecularly adsorbed H-atoms are then proposed to migrate away from the active site and onto the coinage metal (i.e. spillover). Against this backdrop, we were keen to explore H2 reactivity with complexes 1, 1Cu, and 2.
Addition of 1 atm. of H2 gas to a THF solution of complex 2 results in deauration, yielding the bimetallic dihydride 1 (Fig. 3A). This chemistry is reminiscent of the reactivity observed between the EtCAAC-supported copper/tantalum trimetallic, Cu2TaHCl, and H2, despite the significant structural differences described above. Whereas H2 addition (1 atm.) to 1 does not result in conversion to a new species, H/D exchange was observed when treating 1 with D2. Room temperature addition of D2 gas affords a mixed hydride/deuteride diamond core, 1H/D, which, upon heating to 45 °C, is fully deuterated within 24 hours.64 This is in stark contrast to the copper congener, 1Cu, which shows no discernable H/D exchange, even after heating to 65 °C (Fig. S25–27†). The heightened reactivity of the tantalum/gold complexes is attributed to the decreased steric pressure around the metal centers (corroborated by the increased propensity for naphthalene isomerization) and more covalent Ta–μH–Au bonding (vide infra). Both of these features are envisioned to promote D2 interaction with the low-valent Ta center in the tantalum/gold heterobimetallic. Despite the high degree of naphthalene activation, there was no evidence with either H2 or D2 for (partial) reduction of the Ta-bound arene rings.
Oxidation versus protonation
Toward our goal of substituting the naphthalene rings for surface-catalysis-relevant small molecules,52,65 we hypothesized that a more oxidized Ta center would better facilitate naphthalene exchange and pursued cyclic voltammetry (CV) studies of both 1 and 2. The CV of 1 showed an apparent quasi-reversible two-electron oxidative feature at −0.66 V coupled with re-reductions at −0.83 V and −2.33 V (vs. Fc/Fc+; Fig. S31†). The electrochemistry of 2 displayed two overlapping quasi-reversible oxidations at −1.18 V and −1.10 V (vs. Fc/Fc+; Fig. 4). Square wave voltammetry (SWV) better resolved these CV features, clearly displaying two events; however, the second oxidation event passes attenuated current as compared to the first, consistent with a chemical change occurring on the timescale of the electrochemical measurement.66 The reductive sweep of the SWV is symmetric, with features at −1.10 V and −1.18 V (vs. Fc/Fc+), suggesting no chemical changes occur during reduction. Encouraged by the apparent reversibility of the oxidation features for 2, we explored chemical oxidations of this cluster with ferrocenium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (FcBArF).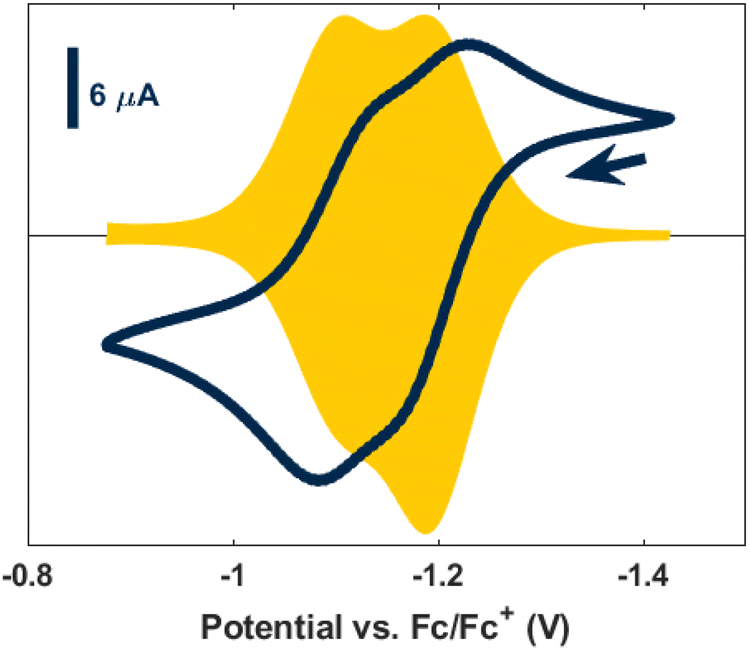 | ||
| Fig. 4 Cyclic voltammogram (blue) and square wave voltammogram (yellow) of 2. The tail of the arrow designates the open-circuit potential. The arrowhead indicates the direction of the scan. | ||
To take advantage of diamagnetic 1H NMR handles, a two-electron oxidation was targeted first. The addition of two equivalents of FcBArF to 2 in thawing DME resulted in formation of an oily, orange solid. 1H NMR spectroscopy revealed chemically inequivalent naphthalene rings with a total of eight naphthyl proton peaks, each with a relative integration of two, ranging from 7.27 to 3.25 ppm. Both the EtCAAC ligand and BArF anion presented a relative integration of two, consistent with an intact, doubly oxidized trimetallic complex, [(EtCAACAu)2HTa(naph)2]2+ (3, 72% yield, Scheme 1). Upon oxidation to a putative TaIAuI2 trimetallic, the hydride shifts from a broad peak at −1.79 ppm (2) to a sharp singlet at +1.45 ppm (3). Although this complex has thus far eluded crystallization, the proposed formulation is supported by a full suite of homo- and hetero-nuclear 2D NMR spectroscopies (see ESI† for additional discussion).
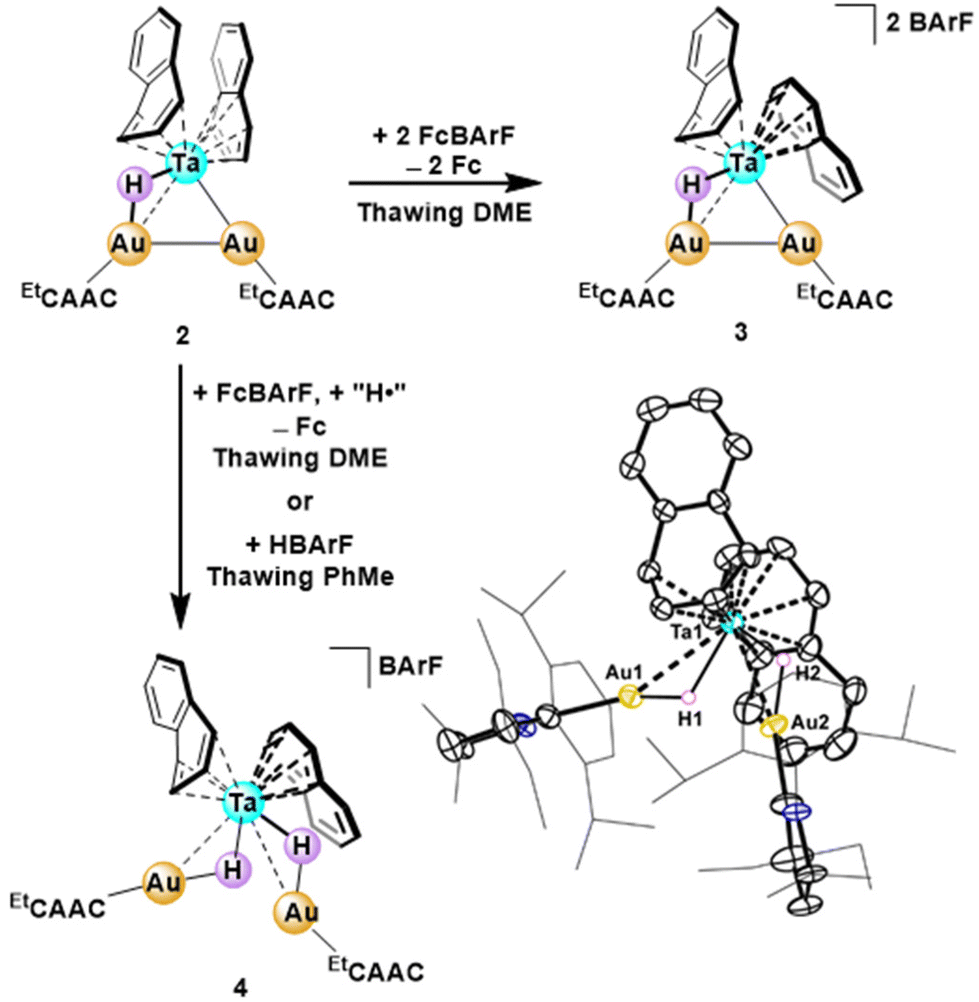 | ||
| Scheme 1 Either oxidation or protonation of complex 2 affords intact cluster architectures in distinct redox states. The solid-state structure of 4 is shown with thermal anisotropic displacement ellipsoids displayed at the 50% probability level and both non-hydride H-atoms and counterions are omitted for clarity. The hydride H-atoms were resolved in the Fourier map; their locations were refined freely and their Uiso values were treated with a riding model. | ||
A single-electron oxidation of 2 was targeted via the addition of one equivalent of FcBArF in thawing DME. Surprisingly, the resultant red-brown product remained diamagnetic; two high-field hydride resonances (−4.33 and −5.33 ppm, each with a relative integration of one proton) were observed in the 1H NMR spectrum. These data are consistent with a one-electron oxidation of 2, followed by a hydrogen atom abstraction by the open-shell cluster, yielding a formally TaIAuI2 trimetallic dihydride complex [(EtCAACAu)2H2Ta(naph)2]+ (4, 36% yield, Scheme 1).67 Hydrogen atom transfer to the singly oxidized trimetallic is in agreement with the observed electrochemical/chemical behavior in both the SWV and CV of 2 (vide supra). To further corroborate the structural identity of 4, we targeted an independent synthesis through protonation of 2 with Brookhart's acid (HBArF).68 Addition of one equivalent of HBArF to 2 in thawing toluene produced the same spectral features as the single electron oxidation, further supporting a dihydride cation assignment for 4.
The structure of 4 was verified through a SCXRD study, in which both hydride ligands were resolved in the Fourier map and refined freely (Scheme 1). Contrasting the core of 2, 4 features a significantly elongated Au–Au distance of 3.944 Å, expanding to provide room for the second μH ligand. The hydride-supported Ta–Au distances also lengthen upon oxidation to an average of 2.9713(4) Å. However, the average Ta–arene bond distances in 4 (Ta–Cave = 2.366 Å and 2.376 Å, for the η4 and η6 naphthyl groups, respectively) are very similar to 1 and 2 (2.348(4)/2.373(4) Å and 2.352(5)/2.411(5) Å for the η4/η6 Ta–Cave distances in 1 and 2, respectively). Correspondingly, naphthalene exchange reactions with myriad substrates across the four multimetallic complexes (1–4) have thus far proven unsuccessful, either showing no reaction, resulting in intractable decomposition, or forming the bimetallic complex, 1 (Table S1†).
Density functional theory calculations
Whereas oxidation did not render the Ta-bound naphthyl groups labile, the heteromultimetallic series provides a means of exploring the effects of metal identity, redox state, and hydride content on metal–metal bonding. We conducted density functional theory calculations for complexes 1, 1Cu, 2, 3, and 4. The full structures of 1 and 1Cu were optimized at the PBE0-D3(0)/def2-TZVPD level of theory for Ta and Au/Cu, and at the PBE0-D3(0)/def2-SVPD level of theory for all other atoms.69–72 The full structures of 2, 3, and 4 were optimized at the PBE0-D3(0)/def2-SVPD level of theory. In all cases an effective core potential was applied to Ta and Au. Salient DFT findings are summarized in Fig. 5. | ||
| Fig. 5 Calculated HOMOs, Wiberg bond indices, and natural population analysis charges for 1 and 1Cu (A). Select frontier molecular orbitals, as well as select optimized bond metrics, for 2, 3, and 4 (B). QTAIM bond critical points are represented by red circles. Orbital isosurfaces are presented at a 0.04 e Å−3 level and energies, relative to the HOMO, are provided parenthetically. | ||
The Wiberg bond indices (WBI)73 for 1 and 1Cu both show Ta-centered M–H bonding, however, the bridging hydrides are more symmetric in the Ta/Au bimetallic. Concomitantly, Natural Population Analysis (NPA) charges and the Ta–M′ WBIs are consistent with greater covalency for the heavier metal pairing. Quantum Theory of Atoms-In-Molecules (QTAIM) analysis74 shows no Ta–M′ bond critical points, suggesting the bimetallic is supported by 3c2e bonding through the μ2-hydride. Analysis of the frontier molecular orbitals, which, in spite of the different naphthalene orientations are nearly identical for both the gold and copper congeners, lends additional veracity to the supported nature of the bimetallic core. The HOMOs for each 1 and 1Cu have Ta 5dz2 parentage and reflect δ-backbonding into both of the naphthyl rings. Similar Ta–arene interactions dominate the lower energy frontier molecular orbitals (Fig. S35†).
The optimized geometries of 2, 3, and 4 were used to inform electronic structure comparisons across the trimetallics, as well as support the locations of the hydride ligands inferred from the SCXRD data. Comparing the structure of 2 to the two-electron oxidized 3 reveals an elongation of the Au–Au and supported Ta–Au distance of approximately 0.1 Å each. Despite this longer Au–Au distance, QTAIM analysis still prescribes a bond critical point between the Au centers. The unsupported Ta–Au bond remains quite similar in both 2 and 3 and likewise displays a bond critical point between the metals. For 2, the HOMO−2 captures this direct Ta–Au bond, in which a Ta 5d orbital engages in σ-bonding to the unsupported Au-center (and the μH on the opposite side of the cluster). The Au–Au bonding interaction is likewise clear in the orbital analysis (HOMO−7; Fig. S36†) of 2. The Ta–Au interaction is present in the HOMO−1 of the oxidized cluster, 3. In agreement with the weaker Au–Au contact intuited from the bond metrics, no significant orbital overlap is observed between the coinage metals. Consistent with the demonstrated recalcitrance to undergo substitution (vide infra), the HOMOs of both 2 and 3 are dominated by Ta–arene backbonding.
The dihydride complex, 4, is the most structurally distinct trimetallic. Upon protonation, the Au–Au distance increases dramatically (3.880 Å) and the Ta–Au bonds, now both featuring 3c2e hydride bridges, are approximately symmetric. There are no formal M–M bonds in this cluster; QTAIM demonstrates bond critical points exclusively located between M–H interactions. The degradation of direct M–M bonding in 4 is postulated to correlate with experimentally observed instability. Complex 4 decomposes as a solid at room temperature after approximately one week and is only stable in solution at room temperature on the timescale of a few hours. This thermal decomposition leads to intractable mixtures.
The average Ta–arene bond distances for the optimized structures were compared across the five complexes (Table S2†). In agreement with the SCXRD data, no significant difference between Ta–arene bond distances is observed, suggesting that coinage metal identity (Cu vs. Au) and cluster charge do not significantly impact Ta–arene bond strength. The HOMOs for all five compounds show significant Ta–arene bonding character, as do lower energy frontier molecular orbitals (vide supra). These calculations support the robustness of the Ta–arene interactions, even in the more oxidized structures, as inferred experimentally from the unsuccessful naphthalene exchange experiments.
Arene C–H activation
In an effort to leverage aurophilicity to increase cluster nuclearity,35,75 trimetallic 2 was treated with one equivalent of both EtCAACAuCl and NaBArF (a net addition of carbene-supported Au+), affording a dark red solid in 89% yield. The 1H NMR spectrum of this product was consistent with a C1 symmetric diamagnet featuring two hydrides (δ = −3.16 and −5.89 ppm), three carbenes, and one BArF counter anion. SCXRD revealed the complex as the C–H activated cluster [(EtCAACAu)3H2Ta(C10H8)(C10H7)]+, 5, in which the second hydride is sourced from auration of one of the Ta-bound naphthyl rings (Scheme 2). This result highlights the strong Ta–arene interaction, which renders the naphthyl ring prone to functionalization, and recalcitrant to substitution. Moreover, the coinage metal σ-aryl adduct is reminiscent of our previously reported Ta/Cu trimetallic, Cu2TaHCl (cf.Fig. 2A), in which one copper center activates a naphthalene C–H bond during the initial salt metathesis reaction, and may follow a similar mechanism to related heterobimetallic arene C–H activations reported in the literature.51,76,77 A comparison of the SCXRD structures of 4 and 5, which both feature TaAu2H2 cores, shows little difference in the average Ta–Au distances (2.98 vs. 3.06 Å for 4 and 5, respectively), suggesting that arene substitution has minimal effect on the M–M bonding in the core. A significant discrepancy between the complexes, however, is their solution stability; 5 is stable in THF solution at 40 °C for at least 24 hours. Adding H2 (1 atm.) at room temperature, however, does impose a transformation, converting 5 to the bimetallic dihydride complex 1, with concomitant loss of two equivalents of gold. This transformation, though unproductive in terms of accessing higher nuclearity mixed-metal compounds, is analogous to what was observed for closely related Cu2TaHCl. Holistically, the strength of the Ta–arene backbonding renders these ligands simultaneously inert to substitution and prone to functionalization, complicating further cluster evolution from these well-defined heteromultimetallic platforms. | ||
| Scheme 2 Synthesis and H2 reactivity of tetranuclear complex 5. The solid-state structure of 5 is provided with thermal anisotropic displacement ellipsoids displayed at the 50% probability level. BArF anion and non-hydride H-atoms are omitted for clarity. The hydride H-atoms were resolved in the Fourier map; their locations were refined freely and their Uiso values were treated with a riding model. | ||
Conclusions
In summary, we have successfully demonstrated the versatility of arene-supported metalates in metal–metal salt metatheses through the synthesis of rare low-valent Ta/Au heteromultimetallic clusters. Contrasting analogous reactions with copper, carbene-supported gold(I) synthons generate a trimetallic cluster, 2, upon reaction with [Ta(naph)3]−. Both SCXRD data and electronic structure calculations corroborate both supported (μH) and unsupported Ta–Au bonding in the trimetallic core. Cluster 2 can activate H2, but dissociates gold in the process, generating a bimetallic species featuring a Ta–(μH)2–Au core. This compound, adds to the scarce examples of Ta/Au mixed metal complexes, and demonstrates notable structural and reactivity differences in comparison to related Cu analogs. Furthermore, despite the absence of a chelating ancillary ligand framework, trimetallic 2 proves robust, remaining intact through CV-informed chemical oxidation and protonation reactions. Redox chemistry and cluster protonation perturb the Ta–Au bonding, but ultimately seem to have little effect on the lability of the Ta–arene interactions. Further attempts to increase cluster nuclearity highlight the activated nature of the Ta-bound arene rings, resulting in C–H auration en route to the tetranuclear dihydride compound 5. Towards the goal of installing labile ligands on the apical metal center, studies to expand our carbonyl-free salt metathesis protocols to homoleptic polyhydrido metalates are underway in our laboratory.Author contributions
M. L. M. performed all experiments and DFT calculations, drafted the manuscript, and prepared the ESI.† J. A. B. directed the project, aided in SCXRD refinement, and revised the manuscript/ESI.† Both authors have given approval of the final version of this manuscript.Data availability
Crystallographic data are available from the Cambridge Structural Database under refcodes 2409547 (1anti), 2409548 (2), 2409549 (4), 2409550 (5), 2432513 (2SIMes), and 2432514 (LAu(μH)AuL+).† Full synthetic and computational details, including preparative procedures and spectroscopic data for compound characterization, are available in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the University of Michigan and the NSF (Graduate Research Fellowship to M. L. M.—DGE-2241144; XRD Instrumentation—CHE-0840456). We thank Dr Eugenio Alvarado for NMR Spectroscopy expertise and Dr Fengrui Qu and Jeff Kampf are gratefully acknowledged for assistance with SCXRD. Roy Wentz aided in the design and fabrication of glassware that made this work possible.References
-
M. I. Bruce, B. K. Nicholson, O. B. Shawkataly, J. R. Shapley and T. Henly, Synthesis of Gold-Containing Mixed-Metal Cluster Complexes, in Inorganic Syntheses, John Wiley and Sons, 1989, Vol. 26, p 324 Search PubMed
.
- P. Braunstein and J. Rose, Gold in Bimetallic Molecular Clusters, Gold Bull., 1985, 18, 17 CrossRef CAS
- E. L. Muetterties, Molecular Metal Clusters, Science, 1977, 196, 839 CrossRef CAS PubMed
- E. L. Muetterties, T. N. Rhodin, E. Band, C. F. Brucker and W. R. Pretzer, Clusters and Surfaces, Chem. Rev., 1979, 79, 91 CrossRef CAS
- B. Berti, M. Bortoluzzi, C. Cesari, C. Femoni, M. C. Iapalucci, R. Mazzoni and S. Zacchini, A Comparative Experimental and Computational Study of Heterometallic Fe-M (M = Cu, Ag, Au) Carbonyl Clusters Containing N-Heterocyclic Carbene Ligands, Eur. J. Inorg. Chem., 2020, 2020, 2191 CrossRef CAS
- I. Ciabatti, C. Femoni, M. C. Iapalucci, S. Ruggieri and S. Zacchini, The role of gold in transition metal carbonyl clusters, Coord. Chem. Rev., 2018, 355, 27 CrossRef CAS
- O. Lopez-Acevedo, J. Rintala, S. Virtanen, C. Femoni, C. Tiozzo, H. Grönbeck, M. Pettersson and H. Häkkinen, Characterization of Iron−Carbonyl-Protected Gold Clusters, J. Am. Chem. Soc., 2009, 131, 12573 CrossRef CAS PubMed
- C. Femoni, M. C. Iapalucci, G. Longoni, C. Tiozzo and S. Zacchini, An Organometallic Approach to Gold Nanoparticles: Synthesis and X-Ray Structure of CO-Protected Au21Fe10, Au22Fe12, Au28Fe14, and Au34Fe14 Clusters, Angew. Chem., Int. Ed., 2008, 47, 6666 CrossRef CAS PubMed
- N. T. Tran, D. R. Powell and L. F. Dahl, Generation of AuPd22/Au2Pd21 Analogues of the High-Nuclearity Pd23(CO)20(PEt3)10 Cluster Containing 19-atom Centered Hexacapped-Cuboctahedral (ν2-octahedral) Metal Fragment: Structural-to-Synthesis Approach Concerning Formation of Au2Pd21(CO)20(PEt3)10, Dalton Trans., 2004, 209 RSC
- N. T. Tran, D. R. Powell and L. F. Dahl, Nanosized Au2Pd41(CO)27(PEt3)15 Containing Two Geometrically Unprecedented 13-coordinated Au-centered (μ13-Au)Pd13 Polyhedra Connected by Triangular Face-sharing and Three Interpenetrating 12-coordinated Pd-centered (μ12-Pd)Au2Pd10 Icosahedra: Geometrical Change in Centered Polyhedra Induced by Au/Pd Electronegativity-Mismatch, Dalton Trans., 2004, 217 RSC
- N. T. Tran, M. Kawano, D. R. Powell, R. K. Hayashi, C. F. Campana and L. F. Dahl, Isostructural [Au6Pd6(Pd6-xNix)Ni20(CO)44]6− and [Au6Ni32(CO)44]6− Clusters Containing Corresponding Nonstoichiometric Au6Pd6(Pd6-xNix)Ni20 and Stoichiometric Au6Ni32 Nanosized Cores: Substitutional Pd/Ni Crystal Disorder (Coloring Problem) at Only Six Specific Nonadjacent Pseudoequivalent Metal Sites in the 38-Atom Trimetallic Close-Packed Framework, J. Am. Chem. Soc., 1999, 121, 5945 CrossRef CAS
-
D. M. P. Mingos and M. J. Watson, Heteronuclear Gold Cluster Compounds, in Advances in Inorganic Chemistry, ed. A. G. Sykes, Academic Press, 1992, vol. 39, p. 327 Search PubMed
- A. J. Whoolery and L. F. Dahl, Synthesis and structural-bonding analysis of the [Au6Ni12(CO)24]2− dianion containing an unprecedented 18-vertex cubic Td metal core composed of five face-fused octahedra: the first example of a discrete gold/nickel bimetallic-bonded species, J. Am. Chem. Soc., 1991, 113, 6683 CrossRef CAS
- J. E. Ellis, Highly reduced organometallics. 5. Synthesis, properties, and the molecular structure of (Ph3PAu)3V(CO)5, a gold-vanadium cluster, J. Am. Chem. Soc., 1981, 103, 6106 CrossRef CAS
- C. E. Coffey, J. Lewis and R. S. Nyholm, 339. Metal–metal bonds. Part I. Compounds of gold(0) with the carbonyls of manganese, iron, and cobalt, J. Chem. Soc., 1964, 1741 RSC
- B. C. Gates, Extending the Metal Cluster–Metal Surface Analogy, Angew. Chem., Int. Ed. Engl., 1993, 32, 228 CrossRef
-
G. Ertl, Chapter 11 Relations Between Metal Clusters and Metal Surfaces, in Studies in Surface Science and Catalysis, ed. B. C. Gates, L. Guczi and H. Knozinger, Elsevier, 1986, vol. 29, p. 577 Search PubMed
- M. Moskovits, Metal cluster complexes and heterogeneous catalysis - a heterodox view, Acc. Chem. Res., 1979, 12, 229 CrossRef CAS
- H. S. Taylor and E. F. Armstrong, A theory of the catalytic surface, Proc. R. Soc. London, 1997, 108, 105 Search PubMed
- G. A. Somorjai, Surface Science and Catalysis, Science, 1985, 227, 902 CrossRef CAS PubMed
- M. Landrini, M. Navarro, J. Campos and L. Rocchigiani, Enhanced reactivity of cationic Au(μ-H)2MCp2 complexes (M = Mo and W) enabled by bulky tris-biaryl phosphines, Dalton Trans., 2025, 54, 898 RSC
- A. Lachguar, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, π-Bonding of Group 11 Metals to a Tantalum Alkylidyne Alkyl Complex Promotes Unusual Tautomerism to Bis-alkylidene and CO2 to Ketenyl Transformation, J. Am. Chem. Soc., 2024, 146, 18306 CrossRef CAS PubMed
- M. Landrini, R. Patel, J. Tyrrell-Thrower, A. Macchioni, D. L. Hughes, L. Tensi, P. Hrobárik and L. Rocchigiani, Exploring Ligand Effects on Structure, Bonding, and Photolytic Hydride Transfer of Cationic Gold(I) Bridging Hydride Complexes of Molybdocene and Tungstenocene, Inorg. Chem., 2024, 63, 13525 CrossRef CAS PubMed
- L. Rocchigiani, W. T. Klooster, S. J. Coles, D. L. Hughes, P. Hrobárik and M. Bochmann, Hydride Transfer to Gold: Yes or No? Exploring the Unexpected Versatility of Au⋯H−M Bonding in Heterobimetallic Dihydrides, Chem. – Eur. J., 2020, 26, 8267 CrossRef CAS PubMed
- N. P. Mankad, Diverse bimetallic mechanisms emerging from transition metal Lewis acid/base pairs: development of co-catalysis with metal carbenes and metal carbonyl anions, Chem. Commun., 2018, 54, 1291 RSC
- A. Hicken, A. J. P. White and M. R. Crimmin, Selective Reduction of CO2 to a Formate Equivalent with Heterobimetallic Gold–Copper Hydride Complexes, Angew. Chem., Int. Ed., 2017, 56, 15127 CrossRef CAS PubMed
- M. K. Karunananda, S. R. Parmelee, G. W. Waldhart and N. P. Mankad, Experimental and Computational Characterization of the Transition State for C–X Bimetallic Oxidative Addition at a Cu–Fe Reaction Center, Organometallics, 2015, 34, 3857 CrossRef CAS
- P. Buchwalter, J. Rosé and P. Braunstein, Multimetallic Catalysis Based on Heterometallic Complexes and Clusters, Chem. Rev., 2015, 115, 28 CrossRef CAS PubMed
- A. Albinati and L. M. Venanzi, Transition metal hydrides as ligands, Coord. Chem. Rev., 2000, 200–202, 687 CrossRef CAS
- L. H. Pignolet, M. A. Aubart, K. L. Craighead, R. A. T. Gould, D. A. Krogstad and J. S. Wiley, Phosphine-stabilized, platinum-gold and palladium-gold cluster compounds and applications in catalysis, Coord. Chem. Rev., 1995, 143, 219 CrossRef CAS
- A. Antiñolo, F. A. Jalón, A. Otero, M. Fajardo, B. Chaudret, F. Lahoz and J. A. López, Reactivity of ruthenium and niobium trihydrides with gold fragments. Crystal structure of the hexanuclear raft cluster [Au3Nb3(μ-H)6(η-C5H4SiMe3)6], J. Chem. Soc., Dalton Trans., 1991, 1861 RSC
- B. D. Alexander, M. P. Gomez-Sal, P. R. Gannon, C. A. Blaine, P. D. Boyle, A. M. Mueting and L. H. Pignolet, Heterobimetallic gold-osmium and gold-ruthenium hydrido complexes. X-ray crystal and molecular structures of [Au2Os(H)3(PPh3)5]PF6 and [AuRu(H)2(CO)(PPh3)4]PF6, Inorg. Chem., 1988, 27, 3301 CrossRef CAS
- B. D. Alexander, B. J. Johnson, S. M. Johnson, P. D. Boyle, N. C. Kann, A. M. Mueting and L. H. Pignolet, Heterobimetallic gold-iridium, silver-iridium, and gold-ruthenium bis(μ-hydrido) complexes. X-ray crystal and molecular structures of [AuRu(H)2(dppm)2(PPh3)]PF6 and [Ir(H)2(bpy)(PPh3)2]PF6, Inorg. Chem., 1987, 26, 3506 CrossRef CAS
- B. R. Sutherland, K. Folting, W. E. Streib, D. M. Ho, J. C. Huffman and K. G. Caulton, Synthetic and mechanistic features of alcohol elimination between gold alkoxides
and rhenium polyhydrides. Metal polyhedron reconstruction upon protonation, J. Am. Chem. Soc., 1987, 109, 3489 CrossRef CAS
- M. Fajardo, M. P. Gómez-Sal, P. Royo, S. Martínez Carrera and S. García Blanco, Synthesis and structural characterisation of the mixed-metal cluster cation [Nb(η5-C5H4R)2{AuP(C6H5)3}2]+ with R = H or Si(CH3)3, J. Organomet. Chem., 1986, 312, C44 CrossRef CAS
- H. Lehner, D. Matt, P. S. Pregosin, L. M. Venanzi and A. Albinati, Stable gold hydride complexes, J. Am. Chem. Soc., 1982, 104, 6825 CrossRef CAS
- M. A. Gawish, Q. A. Drmosh and S. A. Onaizi, Single Atom Catalysts: An Overview of the Coordination and Interactions with Metallic Supports, Chem. Rec., 2022, 22, e202100328 CrossRef CAS PubMed
- X. Y. Liu, A. Wang, T. Zhang and C.-Y. Mou, Catalysis by gold: New insights into the support effect, Nano Today, 2013, 8, 403 CrossRef CAS
- J. A. Rodriguez, S. D. Senanayake, D. Stacchiola, P. Liu and J. Hrbek, The Activation of Gold and the Water–Gas Shift Reaction: Insights from Studies with Model Catalysts, Acc. Chem. Res., 2014, 47, 773 CrossRef CAS PubMed
- B. Hammer and J. K. Norskov, Why gold is the noblest of all the metals, Nature, 1995, 376, 238 CrossRef CAS
- J. Schwank, Gold in bimetallic catalysts, Gold Bull., 1985, 18, 2 CrossRef CAS
- A. Mahdavi-Shakib, T. N. Whittaker, T. Y. Yun, K. B. Sravan Kumar, L. C. Rich, S. Wang, R. M. Rioux, L. C. Grabow and B. D. Chandler, The role of surface hydroxyls in the entropy-driven adsorption and spillover of H2 on Au/TiO2 catalysts, Nat. Catal., 2023, 6, 710 CrossRef CAS
- R. Guo, X. Xu, Y. Xia, W. Huang, Z. Li and B. Teng, , Insights into electrocatalytic hydrogen evolution reaction in acidic medium at in situ dispersed Pt atoms on nanoporous gold films, J. Catal., 2018, 368, 379 CrossRef CAS
- J. Cornejo-Romero, A. Solis-Garcia, S. M. Vega-Diaz and J. C. Fierro-Gonzalez, Reverse hydrogen spillover during ethanol dehydrogenation on TiO2-supported gold catalysts, Mol. Catal., 2017, 433, 391 CrossRef CAS
- P. Schlexer and G. Pacchioni, Anchoring Small Au Clusters on the Dehydroxylated and Hydroxylated SiO2 α-Quartz (001) Surface via Ti-Alloying, J. Phys. Chem. C, 2017, 121, 14717 CrossRef CAS
- D. Nabaho, J. W. Niemantsverdriet, M. Claeys and E. v. Steen, Hydrogen spillover in the Fischer–Tropsch synthesis: An analysis of gold as a promoter for cobalt–alumina catalysts, Catal. Today, 2016, 275, 27 CrossRef CAS
- S. S. E. Collins, M. Cittadini, C. Pecharromán, A. Martucci and P. Mulvaney, Hydrogen Spillover between Single Gold Nanorods and Metal Oxide Supports: A Surface Plasmon Spectroscopy Study, ACS Nano, 2015, 9, 7846 CrossRef CAS PubMed
- J. Campos, Bimetallic cooperation across the periodic table, Nat. Rev. Chem., 2020, 4, 696 CrossRef CAS PubMed
- A. W. Beamer and J. A. Buss, Surface-like NOx Reduction at an Atomically-Precise Tricopper Cluster, Angew. Chem., Int. Ed., 2025, e202424772 CAS
- A. W. Beamer and J. A. Buss, Synthesis, Structural Characterization, and CO2 Reactivity of a Constitutionally Analogous Series of Tricopper Mono-, Di-, and Trihydrides, J. Am. Chem. Soc., 2023, 145, 12911 CrossRef CAS PubMed
- M. L. Maiola and J. A. Buss, Accessing Ta/Cu Architectures via Metal-Metal Salt Metatheses: Heterobimetallic C−H Bond Activation Affords μ-Hydrides, Angew. Chem., Int. Ed., 2023, 62, e202311721 CrossRef CAS PubMed
- W. W. Brennessel, J. E. Ellis, M. K. Pomije, V. J. Sussman, E. Urnezius and V. G. Young, Tris(η4-naphthalene)- and Tris(1–4-η4-anthracene)tantalate(1−): First Homoleptic Arene Complexes of Anionic Tantalum, J. Am. Chem. Soc., 2002, 124, 10258 CrossRef CAS PubMed
- A. S. Romanov, C. R. Becker, C. E. James, D. Di, D. Credgington, M. Linnolahti and M. Bochmann, Copper and Gold Cyclic (Alkyl)(amino)carbene Complexes with Sub-Microsecond Photoemissions: Structure and Substituent Effects on Redox and Luminescent Properties, Chem. – Eur. J., 2017, 23, 4625 CrossRef CAS PubMed
- In our previously reported Cu/Ta system (see ref. 51), we found that the reducing reaction conditions promoted C–H activation of the naphthalene rings and HAT from solvent, resulting in the incorporation of hydride ligands. H/D scrambling in the gold system prevents such precise isotopic labeling experiments; reactions from either [Ta(naph-d8)3]− or those run in THF-d8 result in partial deuterium incorporation in 2. In light of the multi-step reaction sequence involved in generating 2, the low reaction yield, and the scrambling observed in attempts to label the hydride, we hesitate to speculate as to the exact balance of this reaction.
- K. Chakarawet, Z. W. Davis-Gilbert, S. R. Harstad, V. G. Young Jr, J. R. Long and J. E. Ellis, Ta(CNDipp)6: An Isocyanide Analogue of Hexacarbonyltantalum(0), Angew. Chem., Int. Ed., 2017, 56, 10577 CrossRef CAS PubMed
-
F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds Between Metal Atoms. 3rd ed, Springer, New York, 2005 Search PubMed
- The formal oxidation state assignments in cluster 2 are ambiguous. We favor Ta−IAuIAuI; however, another reasonable assignment invokes a antiferromagnetically coupled Ta0AuIAu0 core, where the oxidized Au center bears the bridging hydride. Efforts to inform the formal oxidation state at Ta via Ta–arene bond metrics were unsuccessful (see Tables S2 & S4; additional discussion is presented in the ESI†).
- J. D. Lee, Z. Qi, A. C. Foucher, H. T. Ngan, K. Dennis, J. Cui, I. I. Sadykov, E. J. Crumlin, P. Sautet, E. A. Stach, C. M. Friend, R. J. Madix and J. Biener, Facilitating Hydrogen Dissociation over Dilute Nanoporous Ti–Cu Catalysts, J. Am. Chem. Soc., 2022, 144, 16778 CrossRef CAS PubMed
- R. Réocreux, E. C. H. Sykes, A. Michaelides and M. Stamatakis, Stick or Spill? Scaling Relationships for the Binding Energies of Adsorbates on Single-Atom Alloy Catalysts, J. Phys. Chem. Lett., 2022, 13, 7314 CrossRef PubMed
- J. E. S. van der Hoeven, H. T. Ngan, A. Taylor, N. M. Eagan, J. Aizenberg, P. Sautet, R. J. Madix and C. M. Friend, Entropic Control of HD Exchange Rates over Dilute Pd-in-Au Alloy Nanoparticle Catalysts, ACS Catal., 2021, 11, 6971 CrossRef CAS
- F. R. Lucci, M. D. Marcinkowski, T. J. Lawton and E. C. H. Sykes, H2 Activation and Spillover on Catalytically Relevant Pt–Cu Single Atom Alloys, J. Phys. Chem. C, 2015, 119, 24351 CrossRef CAS
- F. R. Lucci, J. Liu, M. D. Marcinkowski, M. Yang, L. F. Allard, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Selective hydrogenation of 1,3-butadiene on platinum–copper alloys at the single-atom limit, Nat. Commun., 2015, 6, 8550 CrossRef PubMed
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Isolated Metal Atom Geometries as a Strategy for Selective Heterogeneous Hydrogenations, Science, 2012, 335, 1209 CrossRef CAS PubMed
- Partial decomposition to unknown hydride/deuteride containing byproducts is observed concomitantly with the conversion of 1H to 1D; however, 1H NMR signals corresponding to the carbene ligand of 1 support it is the major species in solution following treatment of 1H with D2 gas (see Fig. S27).†.
- V. J. Sussman and J. E. Ellis, From Storable Sources of Atomic Nb− and Ta− Ions to Isolable Anionic Tris(1,3-butadiene)metal Complexes: [M(η4-C4H6)3]−, M=Nb, Ta, Angew. Chem., Int. Ed., 2008, 47, 484 CrossRef CAS PubMed
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, A Practical Beginner's Guide to Cyclic Voltammetry, J. Chem. Educ., 2018, 95, 197 CrossRef CAS
- We have repeated the single electron oxidation of 2 in DME-d10; however, no deuterium is incorporated into the product, 4.
- M. Brookhart, B. Grant and A. F. Volpe, Jr., [(3,5-(CF3)2C6H3)4B]−[H(OEt2)2]+: a convenient reagent for generation and stabilization of cationic, highly electrophilic organometallic complexes, Organometallics, 1992, 11, 3920 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS
- D. Andrae, U. Huermann, M. Dolg, H. Stoll and H. L. Preu, Energy-adjustedab initio pseudopotentials for the second and third row transition elements, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS
- K. B. Wiberg, Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane, Tetrahedron, 1968, 24, 1083 CrossRef CAS
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580 CrossRef CAS PubMed
- E. Y. Tsui, P. Müller and J. P. Sadighi, Reactions of a Stable Monomeric Gold(I) Hydride Complex, Angew. Chem., Int. Ed., 2008, 47, 8937 CrossRef CAS PubMed
- I. Del Rosal, S. Lassalle, C. Dinoi, C. Thieuleux, L. Maron and C. Camp, Mechanistic investigations via DFT support the cooperative heterobimetallic C–H and O–H bond activation across Ta=Ir multiple bonds, Dalton Trans., 2021, 50, 504 RSC
- M. G. Alférez, J. J. Moreno, N. Hidalgo and J. Campos, Reversible Hydride Migration from C5Me5 to RhI Revealed by a Cooperative Bimetallic Approach, Angew. Chem., Int. Ed., 2020, 59, 20863 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic and computational details; spectral data. CCDC 2409547–2409550, 2432513 and 2432514. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5qi00334b |
| This journal is © the Partner Organisations 2025 |