
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural variations in the trans-carboxylate/chlorido axis that impact the mode of action of Pt(II) complexes†
David Fabra‡
 a,
Theresa Mendrina‡bc,
Ana I. Matesanza,
Ángeles Medrano§
a,
Rastislav Pitekb,
Isabella Poetschbc,
Walter Bergerb,
Petra Heffeter¶
*b and
Adoración G. Quiroga¶*ad
a,
Theresa Mendrina‡bc,
Ana I. Matesanza,
Ángeles Medrano§
a,
Rastislav Pitekb,
Isabella Poetschbc,
Walter Bergerb,
Petra Heffeter¶
*b and
Adoración G. Quiroga¶*ad
aDepartment of Inorganic Chemistry, School of Sciences, Universidad Autónoma de Madrid (UAM), Madrid, 28049, Spain. E-mail: adoracion.gomez@uam.es
bCenter for Cancer Research and Comprehensive Cancer Center, Medical University of Vienna, Borschkegasse 8a, 1090 Vienna, Austria. E-mail: petra.heffeter@meduniwien.ac.at
cFaculty of Chemistry, Institute of Inorganic Chemistry, University of Vienna, Währinger Strasse 42, 1090 Vienna, Austria
dInstitute for Advance Research in Chemistry UAM, Madrid, 28049, Spain
First published on 12th May 2025
Abstract
The design of trans-platinum(II) complexes marked a significant turning point in the development of unconventional anticancer metallodrugs. Compared to cisplatin, these complexes induce distinctly different cellular responses and are often active against cisplatin-resistant cell lines. In this study, we synthesized and fully characterized two new Pt(II) complexes, introducing one acetate (−OCOCH3) ligand (X) into the trans-PtXX′ axis, where X′ is either acetate or chlorido. We evaluated their cytotoxicity across a panel of malignant (Capan-1, B16, MCF7, HCT-116, CT26 and P31) and non-malignant (HaCaT, HUVEC, BEC, and MCF10A) cell lines, finding that the complex with only one acetate trans to a chlorido group is more active and selective than the complex with two acetates (X = X′). Furthermore, the two complexes differ from cisplatin in their cellular uptake route as well as mode of action by inducing cancer cell death via non-DNA-associated mechanisms.
Introduction
Cisplatin and carboplatin are clinically approved metal drugs used in patients with advanced cancer. In fact, it is often assumed that every second patient receives platinum-based therapy during treatment. Many limitations (such as strong adverse effects and resistance development) are reported for platinum therapy and a lot of effort has been put into the investigation of the underlying molecular reasons. For example, the aquation of cisplatin leads to the formation of hydroxo-bridged oligomers, which may contribute to some of its toxicity. However, the replacement of chlorido ligands by carboxylate groups in carboplatin reduces such toxicity and improves its solubility in water.1 Roberts et al.2 studied the kinetics of the cisplatin and carboplatin aquation reaction, and their subsequent reactions with DNA, both in vitro and in vivo, finding that once the carboplatin chelate ring is broken, a monoadduct is formed and the monodentate carboxylate ligand becomes even more labile. In the case of cisplatin, both chlorido ligands are lost at the same rate.Thus, carboxylates became better leaving groups for platinum drug optimization than chloridos. Since then, we have witnessed a plethora of publications with structural variations that could potentially control the aquation and the reactivity of platinum drugs towards DNA and beyond. As none of these complexes were as good as the drugs already used clinically, research has continued towards new developments, and those researchers proposing rule-breaking ideas have offered the basis for new solutions. One of the most captivating examples was first published by Farrell et al. suggesting potential drugs with the general formula trans-[PtCl2(L)(L′)], where L and L′ are NH3 and/or pyridine and quinoline ligands, leading to non-conventional structural chemotypes with meaningful in vivo activity.3 Their specific activity was ascribed to the formation of ternary adducts (Pt–protein–DNA), based on the high affinity of the trans complex towards sulfur of methionine.4
Many other examples of trans complexes have been published over the years,5 revealing that variations of the non-leaving ligands modulate their activity, but the reactivity trend towards methionine seems to be a common feature.6,7 Within this large trans-Pt(II) series, the use of carboxylate ligands has increased the solubility of prototype drugs that led to greater activity in both in vitro and in vivo tests.8 The mutually weak trans influence of the carboxylate ligands reduces the substitution lability along a trans-PtX2 axis (where X denotes the leaving groups) and results in compounds that are significantly less reactive.
Here, we propose a new half-way type of compound by introducing an acetate (−OCOCH3) ligand (X) in addition to one classic chlorido (Cl−) ligand (X′), foreseeing that the new trans-PtXX′ axis will show mid-term reactivity, higher than that of the bis-acetates but lower than the classic chlorido compounds. Our rational design includes triphenylphosphine (PPh3) as the non-leaving group, which had previously enhanced the cytotoxicity of a series of trans-Pt(II) chlorido complexes. This was ascribed to the use of bulky ligands with hydrophobic properties and higher steric impediments.9 Small aliphatic amines were selected as the trans non-leaving group, with isopropylamine (ipa) being the ligand with the best performance.10–12
Results and discussion
Synthesis of the complexes
The complexes of general formula trans-[PtCl(amine)(OCOCH3)(PPh3)], where amine is ipa, dma (dimethylamine) and ma (methylamine), were named DO1, DO2 and DO3, respectively. These complexes were prepared by the reaction of Ag(OCOCH3) with trans-[PtCl2(amine)(PPh3)] in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. The complex trans-[Pt(ipa)(OCOCH3)2(PPh3)], DO4, was also synthesized by reacting the chlorido-containing trans-[PtCl2(ipa)(PPh3)] precursor9 with the silver salt. However, this procedure afforded mixtures containing DO1 and DO4 (the latter always in a lower proportion) (Fig. 1). Many reaction attempts were performed by varying both the reaction times, up to 24 h, and the solvents, but a similar proportion of DO1 and DO4 was always produced. The best conditions, described in the experimental section, involved purification by column chromatography, which allowed the separation of both complexes. Ag(OCOCH3) reacted with the iodide precursor, trans-[PtI2(ipa)(PPh3)]13 to afford complex DO4 in good yield after a simple recrystallization step, which proved to be the most effective method for isolating DO4.
:1 molar ratio. The complex trans-[Pt(ipa)(OCOCH3)2(PPh3)], DO4, was also synthesized by reacting the chlorido-containing trans-[PtCl2(ipa)(PPh3)] precursor9 with the silver salt. However, this procedure afforded mixtures containing DO1 and DO4 (the latter always in a lower proportion) (Fig. 1). Many reaction attempts were performed by varying both the reaction times, up to 24 h, and the solvents, but a similar proportion of DO1 and DO4 was always produced. The best conditions, described in the experimental section, involved purification by column chromatography, which allowed the separation of both complexes. Ag(OCOCH3) reacted with the iodide precursor, trans-[PtI2(ipa)(PPh3)]13 to afford complex DO4 in good yield after a simple recrystallization step, which proved to be the most effective method for isolating DO4.
 | ||
| Fig. 1 New type of platinum compounds with trans-PtXX′ carboxylate/chlorido axis. Amine = ipa (DO1/DO4), dma (DO2), ma (DO3). | ||
The four complexes were characterized using analytical and spectroscopic techniques. NMR data were assigned with the aid of the coupling constants and chemical shifts of the 31P and 195Pt-NMR spectra (data are shown in ESI Fig. S1–S16†). All data agree with the proposed stoichiometry. The molecular structure was determined by single-crystal X-ray diffraction (SC-XRD) studies of three of the complexes. Fig. 2 shows the selected trans-Pt(II) structure containing ipa, where one acetate ligand is coordinated to the metal ion in the case of DO1 and two acetate ligands in the case of DO4. The molecular structure of DO2, which is isostructural with DO1, is included in the ESI (Fig. S17, Tables S1 and S4†).
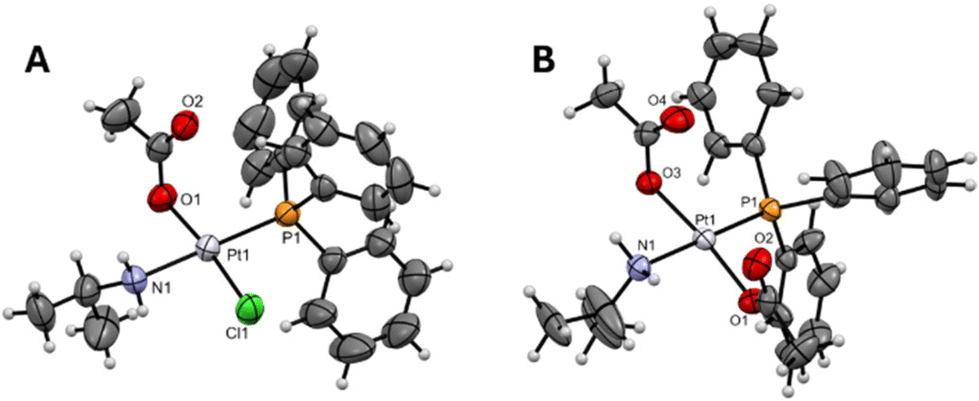 | ||
| Fig. 2 SC-XRD structures of complexes (A) DO1 and (B) DO4. | ||
DO1 and DO4 exhibit slightly distorted square-planar geometry (Tables S1, S2 and S5†). The bond lengths closely resemble those observed in Pt(II) complexes containing PPh3 ligands and aliphatic amines in a trans configuration, including the precursor complex trans-[PtCl2(ipa)(PPh3)].14 The Pt–P bond is shorter than that in complexes with two phosphines in a trans configuration, likely due to the different trans effect produced by the PPh3 ligand with respect to the amine.9,15 Supramolecular interactions have been found for both structures, but these are shown in the ESI† because of their relatively low impact in biological studies (Tables S3 and S5,† respectively).
Preliminary stability and cytotoxic screening of DO1 to DO4
Most of the cis- and trans-Pt(II) drugs are potentially activated via aquation. Notably, speciation of metallic drugs in solution increases in complexity with the use of coordinating solvents such as dimethyl sulfoxide (DMSO) as will be explained in detail in the next section on stability. Therefore, preliminary studies using NMR were performed for all the complexes, and the data are shown in the ESI (Fig. S18 and S19†). The results indicate that DO3 is less stable than DO1 and DO2 in DMSO/D2O mixtures. Experimentally, DO2 shows lower solubility than DO3, which may be correlated to its reduced stability. In contrast, DO1 is not only soluble but also the most stable of the three complexes, as confirmed by NMR analysis. With these new compounds in hand, we performed initial anticancer activity tests in cell cultures using HCT116 cells. The respective IC50 values (after 72 h) for DO1, DO2, and DO3 were 19 ± 1.1 μM, 38 ± 8.4 μM, and 37 ± 3.7 μM. Consequently, we decided to proceed with DO1 and the bis-acetate derivative DO4 for further analysis.Stability studies of complexes DO1 and DO4 in solution
As previously mentioned, the aquation of the complexes with the use of coordinating solvents such as DMSO is not easy to interpret; for example, in some cases DMSO causes certain levels of inactivation,16 while in some others it can act as a transporter17 or as a way to achieve more active species.18 Carboplatin and oxaliplatin solution studies performed by Dyson et al.19 revealed that, although no coordination in full DMSO was observed, the addition of water triggered DMSO coordination and the rate of DMSO adduct formation was dependent on the amount of DMSO. That is why studying the stability of new complexes in solution is a necessary step before studying their potential biological activity. This evaluation must be performed not only in DMSO or water but also in the presence of mixtures of water or water:DMSO and biological buffers.
Our complexes were only slightly soluble in water. For example, to record the 1H-NMR spectra for DO1 in D2O, we needed to use the water suppression method (spectra displayed in Fig. S20†). To prepare the biological stock solutions, it is necessary to reach higher concentrations than those in the D2O NMR sample, so we had to use DMSO. Thus, we studied the stability of DO1 and DO4 in solution with different DMSO:D2O ratios (from 1:0.3 to 1:5, v/v) by monitoring 1H-NMR spectra over 24 h. Both complexes exhibit stability and retain their structural integrity. The NMR spectra of complex DO4 remain virtually unchanged, even at higher water concentrations (Fig. S18C and D†), whereas the spectra of DO1 reveal new minor peaks in the aliphatic and aromatic regions, which are only noticeable after 6 h (Fig. S18A and B†). These signals for new species—free isopropylamine at 0.86 ppm (1.7 mol%) and an additional signal at 0.99 ppm (1.3 mol%)—do not increase over time, and their integration values remain constant after 24 h. Further addition of D2O does not alter this NMR profile (Fig. S18A and B,† peaks marked with a star). Ultimately, only DO1 shows a slightly higher degree of aquation under the test conditions compared to DO4, although it remains significantly more stable than the levels typically reported for cisplatin.20
To prove the low aquation trend in micromolar concentrations and physiological solutions, we used RP-HPLC. The samples evaluated in DMSO (Fig. 3) showed no significant differences in the intensities of the peaks until 24 h, where indicated losses of complex were only 15% and 25% for DO4 and DO1, respectively. The lost proportion in a 1:1 (v/v) mixture of DMSO and 0.9% (w/w) NaCl aqueous solution is slightly greater, but this decrease in stability is a common phenomenon when using high electrolyte concentrations. In general, the compounds maintain their integrity, but DO1 shows slight aquation after 24 h. Normally, we also check the stability of the complexes using UV–visible spectroscopy, but in this case the low absorbance of the bands hampered this study.
 | ||
| Fig. 3 Stability studies using RP-HPLC of DO1 and DO4 in DMSO (light blue and light purple) or DMSO/NaCl 0.9% (1:1) (dark blue and dark purple). Percentage values are given as the ratio of the remaining complex over complex at t = 0. | ||
Overall, it can be concluded that the stability agrees with the data obtained by both NMR and HPLC, indicating that DO1 and DO4 show sufficient stability to proceed with biological studies.
Binding towards N-acetyl-methionine (N-acetyl-met)
Many Pt(II) complexes, including cisplatin, react with several proteins present in biological media.21 There are relevant reports on how some Pt(II) complexes interact with methionine motifs found, for example, in copper transport proteins. In the case of cisplatin, the chlorido ligands are completely replaced by methionine residues, while transplatin retains its ligands and configuration.22 The reaction with N-acetyl-met is a frequently used model to investigate the role of biological S-donor molecules in Pt(II) drug−protein binding.4,8 It has been reported that the reactivity of trans-platinum complexes is significantly enhanced by their previous coordination with S-methionine.6The interaction of N-acetyl-met with DO1 and DO4 was monitored by NMR for 24 h (Fig. 4A). The reaction proceeds at a relatively fast rate for DO1, as a new peak clearly emerges at 9.7 ppm in the 31P-NMR spectrum when the reaction is already at its initial stage (Fig. 4B), becoming the major signal at 6 h. This signal decreases in intensity for another 6 h until it disappears (24 h). 195Pt-NMR chemical shifts are very sensitive to the nature, number, and donor-atom arrangement of the coordinated ligands.23 Consequently, we monitored the same tube by conducting 195Pt-NMR measurements and detected a first new signal at −3359 ppm (deshielded 200 ppm from complex DO1, Fig. 4C) that can be assigned to a [ClNPS(N-acetyl-met)] environment. A second new signal at −4401 ppm appears after 11 h, coexisting for at least 8 h (spectra from 11 h to 19 h), and this can be assigned to an adduct with a [ClPS2] donor set. The 1H-NMR spectrum of the sample clearly shows that the acetate group has been replaced by N-acetyl-met (free AcO− is detected at 1.97 ppm, and the methyl groups of the N-acetyl-met at 1.92 and 2.00 ppm, Fig. S21A†). After 24 h, we observed a signal for a third species in the 31P-NMR spectrum at 11.8 ppm, the poor resolution of which does not improve or change over time.
 | ||
| Fig. 4 (A) Scheme of DO1 interaction with N-acetyl-met in deuterated solvent. (B) Time-dependent 31P-NMR (from t = 6 to 20 h data not shown) of the sample A. (C) Time-dependent 195Pt-NMR of the sample (A). | ||
It is noteworthy that, after many days, visible crystals suitable for single-crystal X-ray diffraction grew in the NMR tube. The molecular structure is depicted in Fig. 5 and the related data are shown in Tables S1 and S6.† The structure of the final product, cis-[Pt2Cl2(PPh3)2(μ-SCH3)2], is the result of a transformation of the third species (δ 195Pt-NMR = –4401 ppm, [ClPS2] donor set), in which N-acetyl-met acted as a bridging ligand, evolving to an S-methyl moiety and releasing isopropylamine. The most important data that we can obtain from this structure are that the Cl− ligand is retained and remains bound to the platinum atom, while the acetate ligand is released first.
 | ||
| Fig. 5 Capped stick representation of the SC-XRD structure for the DO1-N-acetyl-met final product, with the formula cis-[Pt2Cl2(PPh3)2(μ-SCH3)2]. Hydrogen atoms have been omitted to improve clarity. | ||
We tried to monitor the reaction of DO1 with N-acetyl-met by ESI-MS, but the results were not clear. The assay did not result in adequate ion detection, and when using NH4OAc to improve ionization efficiency, we could detect only peaks attributable to fragmentation of the possible adducts (Fig. S21B†). The peaks correspond to species where the Pt(II) is bound to one or two N-acetyl-met and PPh3 ligands. This mass spectrum does not correlate with the purity observed by way of the 1H NMR spectrum, where all the ligands are present and coordinated, except AcO−, which is free (at 1.97 ppm), suggesting the presence of only one pure species.
The spectra of the reaction of DO4 with N-acetyl-met indicated immediate coordination, as signals for only one new species appear at δ = 6.14 ppm and −2759 ppm, respectively, in the time-dependent 31P- and 195Pt-NMR experiments (Fig. 6A and B). It is noteworthy that there are so far no reports of Pt(II)-phosphine derivatives bound to N-acetyl-met, but only one chemical shift signal in the 195Pt-NMR spectrum is observed, being deshielded by approximately 200 ppm as in the case of DO1. Based on the values reported for trans-Pt(II) complexes containing N-acetyl-met,4 and based on the fact that the 1H-NMR spectra show that the isopropylamine group is still coordinated but not the acetates, we suggest that the adduct contains [Pt(N-acetyl-met-κS)2(ipa)(PPh3)].
 | ||
| Fig. 6 (A) Time-dependent 31P-NMR of DO4 interaction with N-acetyl-met. (B) Time-dependent 195Pt-NMR of the same sample. | ||
Overall, the reactivity of DO4 was not only more rapid than that of DO1 but the new adduct remained unchanged for more than 24 h.
The mass spectra also showed the occurrence of many fragmentations. Fig. S22B† shows the sample ionization, which revealed a major peak at m/z = 838.17, corresponding to a [Pt(S-Met-H)2(PPh3)]+ fragment. However, when using NH4OAc (Fig. S22C†) with the same sample, we detected a peak at m/z = 664.13 indicating that the NH2 fragment of the isopropylamine is retained.
Studies of binding to supercoiled DNA in vitro
The trans-Pt(II) complexes can potentially bind to DNA.24 Consequently, we were interested in how DO1 and DO4 interact with plasmidic DNA in a cell-free in vitro setting. Gel electrophoresis of DNA pBR322 samples is a well-established analytical technique used to assess the possible interactions between metal complexes and DNA. pBR322 is generally found in a supercoiled (SC) state, and its treatment with endonucleases or a damage-inducing agent results in the formation of open-circular (OC) and linear (L) isoforms24 and a change in its migration rate. SC DNA is not only more compact, but it has a lower charge than other states (OC, L, etc.), and migrates faster in the agarose gel matrix. Once the plasmid becomes nicked, the supercoils unwind, which can result in the formation of OC DNA, and this migrates more slowly through the agarose matrix. Metallic complexes can interact with DNA generally by covalent binding or through non-covalent interactions. For instance, covalent binders such as cisplatin slow down the migration of the SC form while increasing the migration rate of the OC form, until they reach a comigration point, which will be used as the positive control.In our experiment, we evaluated the interaction of DO1 and DO4 with a pBR322 plasmid containing only the SC form (Fig. 7) and also with a plasmid that contained the OC and SC forms (Fig. S23†). The interaction of DO1 (lanes 8–12) with the supercoiled plasmid DNA shows a similar pattern to that of cisplatin (used as a positive control in lanes 3–7 in Fig. 7 and lanes 3–6 in Fig. S23†): first, unwinding the SC to the OC form, and later, with higher ratios, both bands comigrate (covalent binding). DO1 produces this effect with lower complex:DNA ratios and the comigration is reached when the SC form collides with the OC form. On the other hand, DO4 (lanes 13–17) also produces unwinding of the SC form, but it does not result in comigration, indicating non-covalent binding. The interaction of the complexes with the plasmid containing two forms (SC and OC, Fig. S23†) shows very similar results for cisplatin and DO1 (lanes 3–6 and 7–10, respectively). However, DO4 (lanes 13–17) does not bind to either OC or SC forms, which can be attributed to its low aquation tendency, as observed by NMR and HPLC.
 | ||
| Fig. 7 Gel electrophoresis. Lane 1 and 18: 1 kb DNA ladder; lane 2: pBR322 control; lanes 3–7: cisplatin at ri: 0.01 to 0.25; lanes 8–12: DO1 at ri: 0.01 to 0.25; lanes 13–17: DO4 at ri: 0.01 to 0.25. CDNA = 0.0625 μg μL−1. ri = complex:DNA (base pair) ratio. | ||
Cytotoxicity of the novel platinum drugs DO1 and DO4
Finally, we became interested in how these differences in reactivity impact the biological behavior of the two new compounds. To this end, as a first step, the antitumoral activities of DO1 and DO4 were evaluated using malignant (Capan-1, B16, MCF7, HCT-116, CT26 and P31) and non-malignant (HaCaT, HUVEC, BEC, and MCF10A) cell lines during 72 h of treatment. In addition, the platinum-resistant subclones HCT-116oxR and P31/cis were included as part of the cell-line panel. In all models tested, both compounds showed a dose-dependent reduction in cell viability in the μM range (Fig. 8). The obtained IC50 values are shown in Table 1 and indicate that, in general, DO1 is more active than DO4 in cancer cell lines (mean IC50 17.5 μM vs. 36.0 μM), with the exception of B16, melanoma, and P31, mesothelioma. Here, in particular, the sensitivity of the human colorectal cancer model HCT-116 with an IC50 value of 8.0 μM to DO1 treatment needs to be highlighted. With respect to drug resistance, no cross-resistance with either cisplatin or oxaliplatin was found, indicating a mechanism of action for both of our novel complexes that is different from that of clinically used Pt(II) drugs. | ||
| Fig. 8 Dose–response curves of DO1 and DO4 in cancerous and non-cancerous cells after 72 h of incubation. Viability was quantified using MTT assays. Red represents cancerous cell lines and green represents non-cancerous cell lines. The mean ± standard deviation (SD) was derived from triplicates of one representative experiment out of at least three repetitions. | ||
| Ratios of the IC50 values of the cisplatin- or oxaliplatin-resistant cell lines compared to the respective control were calculated and are indicated in red. Statistical significance was calculated using an unpaired two-tailed t-test; ns – non-significant, ***p < 0.05. |
|---|
 |
Finally, tests on healthy tissue cultures (revealing mean IC50 values of 30.8 μM and 35.9 μM), indicate a therapeutic window for DO1 (but not DO4), especially against pancreatic, colon and breast cancers (Fig. 8, green vs. red lines).
Cellular uptake
To better understand whether differences in sensitivity were based on intracellular drug levels, drug uptake was evaluated by ICP-MS. In more detail, the total platinum content in the cells was quantified after treatment with complex concentrations of 10 or 30 μM for 1, 3 and 5 h using the CT26 and HCT116 cell lines. Fig. 9A shows that in CT26, both complexes were taken up very quickly in a dose-dependent manner. It is notable that there was a distinct difference in the kinetics between the two complexes, especially at higher drug doses (30 μM): while the maximal platinum levels with DO1 had already been detected at the first time point of 1 h, for DO4 a slower, continuous increase was seen over the 5 h of the test. This indicates that the cellular drug uptake mechanisms differ between the two complexes. This hypothesis was further strengthened by the uptake data generated from the HCT116 cells. Also in this cell model, dose-dependent drug uptake for DO1 was observed. In contrast, the cellular platinum levels did not differ between cells treated with 10 μM or 30 μM DO4 (Fig. 9B). | ||
| Fig. 9 Cellular platinum distribution in (A) CT26 and (B) HCT116 cells after 1, 3 and 5 h of treatment with 10 or 30 μM of DO1 or DO4. (C) Cellular platinum levels in HCT116 cells after 3 h of 30 μM DO1 or DO4 treatment in 37 °C or 4 °C. In all experiments, the platinum levels were measured using ICP-MS. The data are expressed as the mean ± SD, obtained in two different experiments and normalized to cell count. Significance was calculated using two-way ANOVA and Tukey's multiple comparison test (in A) or Sidak's multiple comparison test (in B); ns – non-significant, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. | ||
Consequently, as a next step, an experiment comparing drug uptake at 4 °C (only passive diffusion) and at 37 °C (passive diffusion plus active transport processes) was performed using HCT116 cells (Fig. 9C). Indeed, a temperature-induced decrease in platinum accumulation was observed only in DO4-treated cells, while cellular DO1 levels were similar under both temperature conditions. This strongly suggests that an active transport mechanism is involved for the cellular uptake of DO4, while DO1 enters the cell by passive mechanisms such as diffusion. These data are in good agreement with the rapid and stable binding of DO4 to N-acetyl-met and could indicate an interaction of DO4 with the methionine motifs of copper transport proteins. Notably, since the cellular platinum concentrations of DO1 and DO4 are largely similar, the enhanced sensitivity of the HCT116 cells cannot simply be attributed to elevated intracellular drug levels.
Role of apoptotic cell death in the mode of action of the novel platinum drugs
As a next step, we aimed to better understand the mode of action of DO1 vs. DO4 using live-cell microscopy (Fig. 10A). In order to exclude effects that are mainly based on the difference in intracellular drug levels, these experiments were performed using CT26 cells. In more detail, the cells were treated with drug doses of 30 μM and images were captured every 20 min for a total of 72 h. 0.6% DMSO and cisplatin were included as references. Both DO1 and DO4 were characterized by inducing a rather fast form of cell death within the first 24 h after drug treatment. Moreover, in contrast to cisplatin (Fig. S24†), during the first couple of hours after treatment, the cells started to generate large numbers of vacuoles followed by their detachment and and appearance of cells with apoptotic morphology already after 12 h (Fig. 10B and C). | ||
| Fig. 10 Morphological analysis of the drug-induced effects. (A) Live-cell microscopy of control, DMSO, DO1- and DO4-treated CT26 cells (30 μM) after various time points (20× and additional 1.5× magnification). Red arrows indicate a representative cell. The graphs show the quantification of vacuolized (B) and dead (C) cells of the live-cell microscopy pictures. Bars depict mean ± SD from three representative images. | ||
To acquire more information on the origin of these vacuoles, as a next step, we employed ER-YFP-transfected SW480 cells.27 In this cell model, the behavior of the ER can be easily followed by its green fluorescence. In addition, a Mitotracker stain was used to visualize the mitochondrial morphology. Indeed, these experiments indicated that in the case of both drugs, the vacuoles were derived from the ER (Fig. 11A). Moreover, both compounds distinctly impacted the mitochondria: while DO4 induced mitochondrial fusion, DO1 completely disrupted the mitochondrial integrity resulting in a cytoplasmic localization of the Mitotracker. No such effects could be observed with cisplatin (Fig. S25†). In contrast, the lysosome number and morphology were completely unaltered (Fig. S26†). The mitochondrial damage caused by DO1 was subsequently confirmed by JC-1 staining (Fig. 11B), where ∼30% of the DO1-treated cells exhibited a low mitochondrial potential within 5 h. These effects were distinctly less pronounced with DO4 (2.6% vs. 1.1% under DMSO treatment).
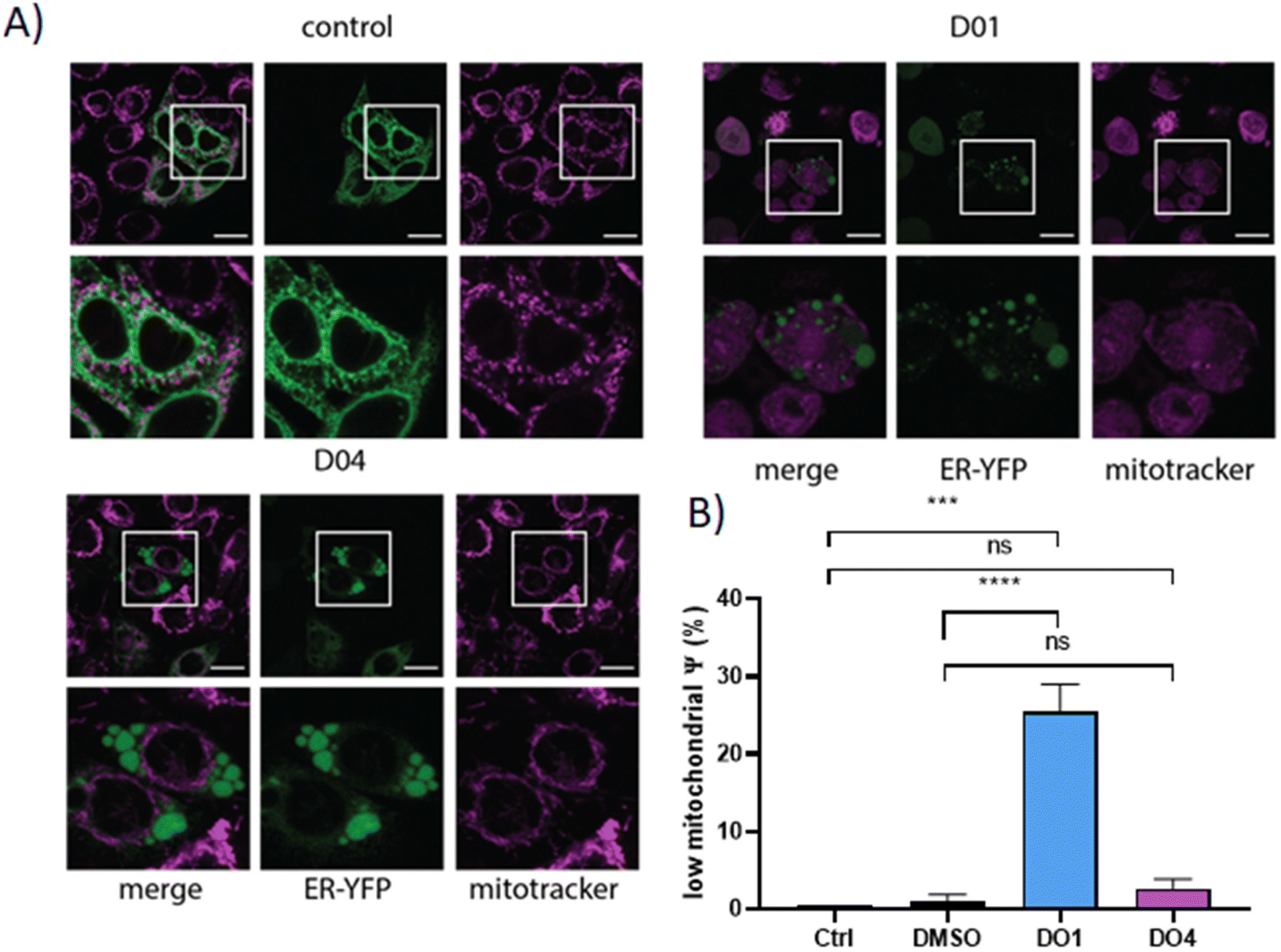 | ||
| Fig. 11 Effects of DO1 and DO4 on ER and mitochondria of ER-targeting YFP-transfected SW480 cells. (A) Representative confocal microscopy images of SW480-ER/YFP cells. Cells were treated with 30 μM DO1 or DO4 and pictures taken after 3 h in the case of DO1 and 6 h in the case of DO4 (63× magnification). ER-YFP localisation is indicated in green, mitochondria were co-stained with 1 μM MitoTracker™ Red CMXRos in pink. (B) Low-mitochondrial membrane potential in CT26 cells after 5 h of incubation with 30 μM DO1 or DO4. Cells were stained with JC-1 followed by flow cytometry. Statistical significance was calculated using one-way ANOVA and Tukey's multiple comparison test; ns – non-significant, ***p < 0.001, ****p < 0.0001. | ||
In becoming interested in the explicit role of apoptotic cell death in the mode of action of the new test drugs, annexin V/PI staining and combination studies using the pan-caspase inhibitor Z-VAD were performed.
As shown in Fig. 12, at 30 μM, DO1 induced rapid cell death within 24 h (Fig. 12A), which could not be prevented by co-incubation with the caspase inhibitor. In contrast, caspase inhibition partly protected CT26 cells from DO4 activity (Fig. 12B). The activity of the two new trans-platinum complexes was subsequently characterized by combining them with different inhibitors. In more detail, the influence of the necroptosis inhibitor necrostatin, the autophagy inhibitor bafilomycin, the protein synthesis inhibitor cycloheximide, the MEK inhibitor UO126 and the thiol-based reductant and radical scavenger N-acetyl cysteine (NAC) was assessed by conducting MTT assays after 24 h.
 | ||
| Fig. 12 Role of apoptotic cell death in the mode of action of DO1 and DO4. (A) Quantification of annexin-V/PI stains of DO1- and DO4-treated CT26 cells (10 μM and 30 μM) for 5 and 24 h in comparison with DMSO and cisplatin-treated cells. Values are measured using flow cytometry. Statistical significance was calculated using two-way ANOVA and Tukey's multiple comparisons test compared to DMSO control (*) or cisplatin (#); *p < 0.05, ****p < 0.0001. (B) Dose–response curves of CT26 cells with DO1 or DO4 in combination with the pan-caspase inhibitor Z-VAD-FMK after 24 h of co-treatment. The proportion of viable cells was quantified by MTT assays. Curves are shown of one representative experiment out of three. Statistical significance was calculated using one-way ANOVA and Tukey's multiple comparisons test; ns – non-significant, *p < 0.05. | ||
Fig. 13A shows that bafilomycin co-treatment had no relevant impact on both compounds (which is in good agreement with the unaltered cellular lysosome composition observed with confocal microscopy). In contrast, cycloheximide protected the CT26 cells especially from DO4 (>2.9-fold increase in IC50) but also from DO1 (1.5-fold increase in IC50) (Fig. 13B).
 | ||
| Fig. 13 Impact of bafilomycin and cycloheximide on the anticancer activity of DO1 and DO4. Dose-dependent activity of DO1 and DO4 in combination with (A) translation inhibitor cycloheximide (CyHx) or (B) the autophagy inhibitor bafilomycin (Bafi) was tested by MTT assay after 24 h of co-incubation. Curves are shown of one representative experiment out of three performed in triplicates. | ||
Interestingly, necrostatin was able to protect CT26 cells from DO4 activity at concentrations up to 25 μM, while it had no relevant impact on DO1 (Fig. S27†). Moreover, co-treatment with NAC protected cells from both DO1 and DO4 activity (Fig. S27†), an effect that was even more pronounced when the experiment was extended to 72 h (Fig. S28†). Finally, co-incubation with UO126 also did not influence either DO1 or DO4 activity (Fig. S27†).
Together, these data suggest that DO1 and DO4 both distinctly differ in their mode of action from other platinum complexes like cisplatin. This is indicated by not only their different in vitro DNA-binding behaviors but also their activities against platinum-resistant cell models. Indeed, DNA damage might not be a central factor in the mode of action of these drugs, since for example the p53 status of the cell line did not have any impact on drug sensitivity. Interestingly, distinct differences between the two new drug complexes were observed. (1) Both drugs differed in their tumor selectivity with DO1 showing promising effects on cancer cells especially HCT116 cells. (2) The cellular uptake of DO1 occurs via passive diffusion, while for DO4 active transport mechanisms are involved. (3) While DO1 is a highly reactive compound causing rapid mitochondrial dysfunction and cell death, DO4 seems less reactive towards the mitochondria. It is noteworthy that these effects did not translate into the full apoptotic signaling cascade (especially in the case of DO1), which could be explained by the rapidness of the cell death induced by the two drugs. Moreover, live-cell imaging showed that both compounds induce ER vacuolization. Subsequent experiments indicated that the mode of action of the drugs does not include autophagy but rather it is dependent on active protein translation (and some necroptosis in the case of DO4). This is also in good agreement with the observed formation of N-acetyl-met adducts in cell-free systems. Ultimately, these data suggest the involvement of other forms of cell death. For example, paraptosis induction appears to be involved, which is a novel form of cell death characterized by mitochondrial and ER swelling, caspase-independent signaling, prevention by NAC cotreatment, and reliance on active protein synthesis.28 However, interestingly, MEK inhibition was not able to prevent DO1/DO4-induced cell death, pointing towards a more complex, multifaceted mode of action, which should be further investigated in future studies. In summarizing, both drugs probably induce anticancer activity via non-DNA-associated mechanisms, which need to be further dissected and characterized in future studies.
Conclusions
Two new trans-Pt(II) complexes of formula trans-[PtCl(OCOCH3)(ipa)(PPh3)], DO1, and trans-[Pt(OCOCH3)2(ipa)(PPh3)], DO4, were synthesized and fully characterized. The compounds showed no significant speciation in solution. DO1 was more active and selective than DO4 against the tested cancer cell lines. Both compounds have different modes of action than cisplatin or oxaliplatin, which is also supported by the observation that no cross-resistance was observed. In addition, compared with DO4, DO1 was taken up differently and produced rapid mitochondrial dysfunction and cell death. Despite DO1's covalent interaction with pBR322, both drugs probably induce anticancer activity via non-DNA-associated mechanisms. Ultimately, the mode of action of the drugs is dependent on active protein translation consistent with the observed formation of N-acetyl-met adducts in cell-free systems.Experimental section
Materials and methods
NMR spectra were recorded at room temperature, using a two-channel 300 MHz Bruker Avance III-HD Nanobay spectrometer equipped with a 5 mm BBO 1H/X probe and Z gradients. CDCl3 and DMSO-d6 were used as solvents (containing 0.05% (v/v) tetramethylsilane (TMS) as a reference). Chemical shift values are given in parts per million (ppm) relative to the residual TMS signals. The following abbreviations were used: s (singlet), bs (broad singlet), d (doublet), and m (multiplet). Elemental analyses were performed on a LECO CHNS-932 elemental analyzer. Mass spectra were recorded using electrospray ionization (ESI) with a Qstar Pulsar I mass spectrometry unit. All of these instruments are located at the Interdepartmental Investigation Service of UAM (SIdI-UAM).Synthesis
trans-[PtCl2(ma)(PPh3)]. Yield: 84%. 1H-NMR (300.13 MHz, acetone-d6), δ (ppm): 7.69 (m, 6H, PPh3), 7.46 (m, 9H, PPh3), 4.02 (bs, 2H, NH2), 2.61 (d, 3H, CH3, JH–H = 4.6 Hz). 13C-NMR (75.47 MHz, acetone-d6), δ (ppm): 134.7 (d, Corto, JC–P = 8.4 Hz), 127.7 (d, Cmeta, JC–P = 12.0 Hz), 130.6 (d, Cpara, JC–P = 3.1 Hz), 14.2 (s, CH3(ipa)). 31P-NMR (121.5 MHz, acetone-d6), δ (ppm): 3.60 (JP–Pt = 3581.2 Hz). 195Pt-NMR (64.53 MHz, acetone-d6; Na2PtCl6), δ (ppm): −3597.6 (d, JPt–P = 3592.1 Hz). Elemental analysis CHN, found: C, 40.60%; H, 3.42%; N, 2.41%. Calculated for C19H20Cl2NPPt: C, 40.80%; H, 3.60%; N: 2.50%.
trans-[PtCl(ipa)(OCOCH3)(PPh3)] (DO1). A mixture of trans-[PtCl2(ipa)(PPh3)] (106.0 mg, 0.18 mmol) in 3 mL of CHCl3 and silver acetate (63.29 mg, 0.38 mmol) in 4 mL of MeOH was stirred at 40 °C in the dark for 1 day. The reaction mixture was then filtered through Celite and concentrated until dryness. The crude product was dissolved in a minimum amount of CH2Cl2, and purified by column chromatography using silica gel, with CH2Cl2 and CH2Cl2
:AcOEt 2:1 as eluents. The final product was eluted last and concentrated to dryness, yielding a beige solid. Yield: 37%. 1H-NMR (300.13 MHz, CDCl3), δ (ppm): 7.71 (m, 6H, PPh3), 7.42 (m, 9H, PPh3), 4.09 (bs, 2H, NH2), 3.32 (m, 1H, CH(ipa), JH–H = 6.4 Hz), 1.45 (s, 3H, CH3(OAc)), 1.37 (d, 6H, CH3(ipa), JH–H = 7 Hz). 13C-NMR (75.47 MHz, CDCl3), δ (ppm): 178.6 (s, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(OAc)) 134.5 (d, Corto, JC–P = 10.8 Hz), 128.2 (d, Cmeta, JC–P = 11.0 Hz), 130.8 (d, Cpara, JC–P = 2.4 Hz), 46.7 (s, CH(ipa)), 24.5 (s, CH3(ipa)), 22.5 (s, CH3(OAc)). 31P-NMR (121.5 MHz, CDCl3), δ (ppm): 4.03 (JP–Pt = 3825.3 Hz). 195Pt-NMR (64.53 MHz, CDCl3; Na2PtCl6), δ (ppm): −3130.0 (d, JPt–P = 3831.3 Hz). ESI+-MS (m/z): 612.1 [M + H]+. Elemental analysis CHN, found: C, 41.92%; H, 4.38%; N, 2.16%. Calculated for C23H27ClNO2PPt·0.5CH2Cl2: C, 42.08%; H, 4.13%; N: 2.09%.
O(OAc)) 134.5 (d, Corto, JC–P = 10.8 Hz), 128.2 (d, Cmeta, JC–P = 11.0 Hz), 130.8 (d, Cpara, JC–P = 2.4 Hz), 46.7 (s, CH(ipa)), 24.5 (s, CH3(ipa)), 22.5 (s, CH3(OAc)). 31P-NMR (121.5 MHz, CDCl3), δ (ppm): 4.03 (JP–Pt = 3825.3 Hz). 195Pt-NMR (64.53 MHz, CDCl3; Na2PtCl6), δ (ppm): −3130.0 (d, JPt–P = 3831.3 Hz). ESI+-MS (m/z): 612.1 [M + H]+. Elemental analysis CHN, found: C, 41.92%; H, 4.38%; N, 2.16%. Calculated for C23H27ClNO2PPt·0.5CH2Cl2: C, 42.08%; H, 4.13%; N: 2.09%. 
trans-[PtCl(amine)(OCOCH3)(PPh3)] [amine = dma (DO2) or ma (DO3)]. A solution of 2 equivalents of silver acetate in acetone–water was added to a solution of the respective complex trans-[PtCl2(amine)(PPh3)] in acetone. After stirring at the reflux temperature for 6 h protected from light, the mixture was cooled to room temperature and filtered through Celite. The filtrate was concentrated until dryness. The resulting crude product was dissolved in a minimum amount of chloroform, a layer of hexane was added, and the mixture was left at low temperature overnight. The resulting beige precipitate was filtered and dried under vacuum. Several precipitations are needed in order to purify the complexes.
trans-[PtCl(dma)(OCOCH3)(PPh3)] (DO2). Yield: 64%. 1H-NMR (300.13 MHz, CDCl3), δ (ppm): 7.74 (m, 6H, Horto), 7.43 (m, 9H), 2.71 (d, 6H, CH3(dma), JH–H = 7.0 Hz), 4.35 (bs, 1H, NH(dma)), 1.38 (s, 3H, CH3(OAc)). 13C-NMR (75.47 MHz, CDCl3), δ (ppm): 177.8 (s, C
O(OAc)) 134.6 (d, Corto, JC–P = 10.4 Hz), 128.2 (d, Cmeta, JC–P = 11.2 Hz), 130.7 (d, Cpara, JC–P = 2.1 Hz), 40.7 (s, CH3(dma)), 21.7 (s, CH3(OAc)). 31P-NMR (121.5 MHz, CDCl3), δ (ppm): 3.50 (JP–Pt = 3725.3 Hz). 195Pt-NMR (64.53 MHz, CDCl3; Na2PtCl6), δ (ppm): −3195.8 (d, JPt–P = 3725.3 Hz). ESI+-MS (m/z): 598.1 [M]+. Elemental analysis CHN, found: C, 36.49%; H, 3.63%; N, 1.79%. Calculated for C22H25NClO2PPt·1.65CHCl3·0.15C6H14: C, 36.55%; H, 3.59%; N, 1.74%. 
trans-[PtCl(ma)(OCOCH3)(PPh3)] (DO3). Yield: 58%. 1H-NMR (300.13 MHz, CDCl3), δ (ppm): 7.74 (m, 6H, Horto), 7.43 (m, 9H), 4.35 (bs, 1H, NH2(ma)), 2.71 (d, 6H, CH3(ma), JH–H = 6.9 Hz), 1.38 (s, 3H, CH3(OAc)). 13C-NMR (75.47 MHz, CDCl3), δ (ppm): 178.6 (s, C
O(OAc)) 134.6 (d, Corto, JC–P = 10.1 Hz), 128.1 (d, Cmeta, JC–P = 11.0 Hz), 130.8 (d, Cpara, JC–P = 2.2 Hz), 30.6 (s, CH3(ma)), 22.4 (s, CH3(OAc)). 31P-NMR (121.5 MHz, CDCl3), δ (ppm): 3.93 (JP–Pt = 2289.8 Hz). 195Pt-NMR (64.53 MHz, CDCl3; Na2PtCl6), δ (ppm): −3152.2 (d, JPt–P = 2299.9 Hz). ESI+-MS (m/z): 526.1 [M − Cl]+. Elemental analysis CHN, found: C, 44.49%; H, 4.47%; N, 1.28%. Calculated for C21H23NClO2PPt·0.9C3H6O·0.05CHCl3: C, 44.88%; H, 4.10%; N, 2.08%. 
trans-[Pt(ipa)(OCOCH3)2(PPh3)] (DO4). A solution of silver acetate (53.7 mg, 0.322 mmol) in 5 mL of MeOH was added to a solution of trans-[PtI2(ipa)(PPh3)] (118 mg, 0.153 mmol) in 5 mL of acetone. The mixture was stirred and heated at 40 °C for 1 day in darkness. The reaction was cooled down to room temperature and was filtered with Celite. The solvent was removed under reduced pressure and the crude product was then recrystallized in CHCl3. The resulting solid was dissolved again in CHCl3 and the same amount of hexane was slowly added and the solution left until the precipitation of a white solid. Yield: 57%. 1H-NMR (300.13 MHz, CDCl3), δ (ppm): 7.69 (m, 6H, PPh3), 7.42 (m, 9H, PPh3), 5.17 (bs, 2H, NH2), 3.20 (m, 1H, CH(ipa), JH–H = 6.5 Hz), 1.46 (s, 6H, CH3(OAc)), 1.34 (d, 6H, CH3(ipa), JH–H = 7 Hz). 13C-NMR (75.47 MHz, CDCl3), δ (ppm): 179.3 (s, C
O(OAc)), 134.4 (d, Corto, J = 10.9 Hz), 130.9 (d, Cpara, J = 2.4 Hz), 128.4 (d, Cmeta, J = 11.1 Hz), 127.7 (s, Cipso), 46.6 (s, CH(ipa)), 24.4 (s, CH3(CO)), 22.6 (s, CH3(ipa)). 31P-NMR (121.5 MHz, CDCl3), δ (ppm): 3.98 (JP–Pt = 4006.4 Hz). 195Pt-NMR (64.53 MHz, CDCl3; Na2PtCl6), δ (ppm): −2550.0 (d, JPt–P = 4021.8 Hz). ESI+-MS (m/z): 635.2 [M + H]+, 657.2 [M + Na]+. Elemental analysis CHN, found: C, 47.05%; H, 5.06%; N, 2.15%. Calculated for C25H30NO4PPt: C, 46.85%; H, 5.14%; N, 2.10%. 
Stability assays
The stability of the trans-Pt(II) complexes DO1 to DO4 was first determined using NMR in DMSO and DMSO/D2O to select the best candidate for biological activity. Once DO1 and DO4 were chosen, the stability was further evaluated by HPLC analysis using DMSO and saline solution (0.9% NaCl) as solvents. Stock solutions of the complexes were freshly prepared in DMSO (5 mM) and diluted to 100 μM using the appropriate solvent. The samples were then incubated at 37 °C and stirred at a mixing frequency of 300 rpm. 20 μL aliquots were taken at different time points (0, 1, 2, 4, 6 and 24 h) and analyzed by RP-HPLC on an Agilent 1200 system using a Zorbax Eclipse Plus C18 column (4.6 × 100 mm, 3.5 μm): flow rate, 1 mL min−1; detection, UV 254 nm; gradient solvent system A/B (acetonitrile/water), initial 30% A + 70% B; 10 min linear gradient to 70% A + 30% B; 5 min linear gradient to 100% A. The disappearance of the compound over time was expressed as the remaining percentage in comparison with the initial amount.Interaction with N-acetyl-met
N-acetyl-methionine (N-acetyl-met) was purchased from VWR. The interaction of complexes DO1 and DO4 with N-acetyl-met was assessed by NMR and ESI+-MS. In all cases, the samples were prepared by dissolving the complexes in a 5:2 acetone:water mixture (final concentration 8.2 mM), with an excess amount of N-acetyl-met (5:1 molar ratio). For the NMR studies, deuterated solvents were used, and the respective 1H- and 31P-NMR spectra were recorded at various times, up to 24 h. The 195Pt-NMR spectra were acquired every 4 h using 2 h of acquisition time, up to 24 h. For the ESI+-MS experiments, protic solvents were used, and aliquots of the reaction were taken at 0, 3, 6 and 24 h after incubation at 37 °C while stirring at a mixing frequency of 300 rpm. 50 μL of each aliquot was then diluted 20-fold using a mixture of acetone/water or acetone/ammonium acetate 2 mM aqueous buffer, and the ESI+-MS spectra were recorded.
Crystallography
Single crystals of complexes DO1, DO2 and DO4, suitable for X-ray diffraction analysis (SC-XRD), were grown from a chloroform solution in the case of DO1 and DO2 and from a freshly filtered acetone/water solution for DO4. Single crystals from the reaction of DO1 with N-acetyl-met were grown in the NMR sample in acetone-d6/D2O solution. Data were collected on a Bruker X8 APEX II CCD (located at SIdI). Table S1† contains crystal data and details of the refinement of the crystal structures. For both compounds, the software package SHELXTL was used for space group determination, structure solution, and refinement (SHELTXTL-NTversion 6.12, Structure Determination Package, Bruker–Nonius XS, Madison, Wisconsin, USA, 1997–2001). The structures were solved by direct methods, completed with difference Fourier syntheses, and refined with anisotropic displacement parameters.CCDC IDs 2401350, 2447565, 2401352 and 2401353† contain the supplementary crystallographic data for compounds DO1, DO2, DO4, and cis-[Pt2Cl2(PPh3)2(μ-SCH3)2], respectively.
Electrophoresis assays
The DNA plasmid pBR322 was purchased from Gencust (supercoiled and open circular forms) or from Fisher Scientific (supercoiled form only), both at a concentration of 0.5 μg μL−1 in phosphate buffer (50 mM; pH 7.4). Stock solutions of the complexes were prepared in DMSO (5 mM) and then diluted with water to the desired concentration, expressed as ri = complex:DNA (base pair) ratio. The ri value used is from 0.01 to 0.20, in a total volume of 20 μL. After 24 h of incubation at 37 °C, 2 μL of a loading dye buffer was added, and the mobility of the complex-treated samples was analyzed by agarose gel electrophoresis (1.2% w/w) at 70 V in Tris/acetate/EDTA buffer 1× for 120 min. The gel was then stained using 2.5 μg mL−1 ethidium bromide aqueous solution for 20 min, and DNA bands were visualized with a UV-transilluminator UVITEC Cambridge UVIDOC HD2 instrument.
Cell culture
The cell lines, their sources, and specific growth medium are summarized in Table S7.† All media were supplemented with 10% fetal calf serum (FCS, PAA, Linz, Austria). The generation of the oxaliplatin-resistant HCT116 cell line by continuous exposure to oxaliplatin was based on a previously published report.29 The cells were selected every week with 10 μM oxaliplatin for 72 h. Human HCT-116 and HCT-116oxR were supplemented with 10% FCS and 1% L-glutamine (Sigma-Aldrich, St Louis, MO, USA). p31/cisR cells were selected every week with 4 μM cisplatin. All cells were regularly checked for Mycoplasma contamination. All cells were cultured under a humidified 5% CO2 atmosphere at 37 °C.Cytotoxicity assays
Cells were seeded at a density of 2000–20000 cells per well in 96-well plates according to the proliferation rate of the respective cell line and allowed to recover for 24 h. Cells were then treated with the indicated concentrations of DO1 or DO4 for 24 h or 72 h. For the combination assays, cells were also treated with cycloheximide (Merck, Darmstadt, Germany), Z-VAD-FMK (MedchemExpress, New Jersey, USA), or bafilomycin A1 (Santa Cruz, California, USA) in broad concentration ranges. The cell viability was subsequently measured by the MTT-based vitality assay (EZ4U; Biomedica, Vienna, Austria), following the manufacturer's recommendations. Full dose–response curves were generated using GraphPad Prism software (version 8.0.1) to calculate IC50 values (drug concentrations at 50% reduced cell viability compared to control).
ICP-MS measurements
HCT-116 and CT26 cells were seeded at 5 × 105 cells per well in 6-well plates and allowed to recover for 24 h. The cells were then exposed to DO1 or DO4 at 10 or 30 μM concentration for 1, 3 or 5 h under normal cell culture conditions. Cells were subsequently trypsinized, counted and washed twice with phosphate-buffered saline (PBS) and finally lysed at room temperature in 100 μL of HNO3 (>69%, Rotipuran Supra, Carl Roth, Karlsruhe, Germany) for 1 h. The lysate was then diluted to 6 mL using ultrapure water and the Pt concentration was determined by ICP-MS and normalized to the cell count.Platinum concentrations were measured using an Agilent 7800 ICP-QMS instrument (Agilent Technologies, Tokyo, Japan), equipped with an Agilent SPS 4 autosampler and a MicroMist nebulizer at a sample uptake rate of approximately 0.2 mL min−1. Data evaluation was performed using the Agilent MassHunter software package (Workstation Software, Version C.01.04, 2018). Elemental standard solutions were purchased from Labkings (Hilversun, The Netherlands).
Live-cell microscopy
CT26 cells were seeded at a density of 3 × 105 cells per mL in a μ slide (8-well glass bottom, ibidi GmbH, Gräfelfing, Germany) and allowed to recover for 24 h. The cells were then treated with complexes DO1 and DO4 at 30 μM concentration for 72 h and one image per well (bright field) was captured every 20 min using a Nikon Eclipse Ti with a 20× (Super Plan Fluor NA 0.45 Ph1) objective in an OkoLAB incubation box (5% CO2, 37 °C, passive humidifier) with a PCS sCMOS 4.2 MPxl monochrome camera.Confocal fluorescence microscopy
SW480ER-YFP cells, generated according to a previous study30 were seeded at a density of 3.7 × 105 cells in a μ slide (8-well glass bottom, ibidi GmbH, Gräfelfing, Germany) and allowed to recover for 24 h. The cells were then treated with cisplatin, DO1 and DO4 at 30 μM concentration. After 30 min, 1 μM Mitotracker was added. Confocal microscopy was performed using a Zeiss LSM 700 (Carl Zeiss AG), equipped with 405, 488, 555 and 639 nm solid state laser diodes, using a 63× Plan-Aochromat NA 1.4 Oil DIC. For each condition, one representative picture out of 5 is shown.JC-1 staining
CT26 cells were seeded at a concentration of 5 × 105 cells per mL into 12-well plates and allowed to settle overnight. Then, the cells were treated with the compounds DO1 or DO4 at 30 μM concentration for 5 h, and the cells were trypsinized and collected. Cells were washed with PBS and their ΔΨm mitochondrial membrane potential was determined by FACS analysis using 200 μL of 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1, Enzo Life Sciences, New York, NY, USA, 1 mg mL−1) solution (1:100 diluted with medium, final concentration 10 μg mL−1) and incubated in the dark for 15 min at 37 °C. Afterwards, the cells were washed, resuspended in PBS, and analyzed at 530 and 605 nm using flow cytometry (LSRFortessa™ X-20 Cell Analyzer, BD Biosciences, Franklin Lakes, NJ, USA). Analysis was performed using FlowJo software.
Cell death analysis
CT26 cells were seeded at a density of 2.5 × 105 cells per mL on 12-well plates and allowed to settle for 24 h. The cells were then treated with complexes DO1 and DO4 (10 and 30 μM concentration) for 5 or 24 h, using cisplatin (5 μM) as a reference. The samples were trypsinized and centrifuged at 300g for 5 min. A dead cell control was included by incubating cells on a heating block (60 °C) for 30 min. The cells were stained with annexin-V (1:50) and propidium iodide (1:50) in annexin-V-binding buffer (ABB: 10 mM HEPES, 140 mM NaCl, 2.5 mM CaCl2) at room temperature in the dark for 15 min. Afterwards, the samples were diluted using 200 μL of ABB and directly measured at 530 and 610 nm using flow cytometry (LSRFortessa™ X-20 Cell Analyzer, BD Biosciences, Franklin Lakes, NJ, USA) and analyzed using FlowJo software.
Author contributions
Conceptualization: DF, TM, WB, PH, and AGQ; data curation: DF, TM, and AIM; formal analysis: DF, TM, RP, and IP; funding acquisition: WB, PH, and AGQ; investigation: DF, TM, RP, IP, PH, and AGQ; methodology: DF, TM, AIM, RP, IP, PH, and AGO; project administration: DF, TM, AIM, RP, IP, PH, and AGO; resources: WB, PH, and AGO; supervision: TM, WB, PH, and AGQ; validation: all authors; visualization: DF, TM, AIM, RP, IP, PH, and AGQ; writing – original draft: DF, TM, PH, and AGQ; writing – review and editing: all authors.Data availability
Data are available upon request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr. A. Álvarez-Valdés for their early contribution to this work. This research was funded by CTQ2015-68779-R and PID2019-106220RB-I00, granted by Agencia Estatal de Investigación from the Spanish Ministerio de Ciencia e Innovación and Ministerio de Economía y Competitividad. All biological studies were funded by the Austrian Research Service (FWF) project FG3 and P37111. Theresa Mendrina was funded via the Obermann-Mahlke Stiftung.References
- J. C. Dabrowiak, Metals in Medicine, John Wiley & Sons, Ltd, 2009, pp. 109–147 Search PubMed.
- R. J. Knox, F. Friedlos, D. A. Lydall and J. J. Roberts, Mechanism of cytotoxicity of anticancer platinum drugs: evidence that cis-diamminedichloroplatinum(II) and cis-diammine-(1,1-cyclobutanedicarboxylato)platinum(II) differ only in the kinetics of their interaction with DNA, Cancer Res., 1986, 46, 1972–1979 Search PubMed.
- N. Farrell, L. R. Kelland, J. D. Roberts and M. Van Beusichem, Activation of the trans Geometry in Platinum Antitumor Complexes: A Survey of the Cytotoxicity of trans Complexes Containing Planar Ligands in Murine L1210 and Human Tumor Panels and Studies on Their Mechanism of Action1, Cancer Res., 1992, 52, 5065–5072 Search PubMed.
- U. Bierbach and N. Farrell, Structural and reactivity studies on the ternary system guanine/methionine/trans-[PtCl2(NH3)L] (L=NH3, quinoline): implications for the mechanism of action of nonclassical trans-platinum antitumor complexes, JBIC, J. Biol. Inorg. Chem., 1998, 3, 570–580 CrossRef.
- U. Kalinowska-Lis, J. Ochocki and K. Matlawska-Wasowska, Trans geometry in platinum antitumor complexes, Coord. Chem. Rev., 2008, 252, 1328–1345 CrossRef.
- C. Li, Z. Li, E. Sletten, F. Arnesano, M. Losacco, G. Natile and Y. Liu, Methionine Can Favor DNA Platination by trans-Coordinated Platinum Antitumor Drugs, Angew. Chem., Int. Ed., 2009, 48, 8497–8500 CrossRef PubMed.
- L. Cubo, A. Casini, C. Gabbiani, G. Mastrobuoni, L. Messori, J. Jiménez-Barbero, C. Navarro-Ranninger and A. G. Quiroga, Solution Behaviour and Biomolecular Interactions of Two Cytotoxic trans-Platinum(II) Complexes Bearing Aliphatic Amine Ligands, Chem. – Eur. J., 2009, 15, 9139–9146 CrossRef CAS PubMed.
- V. Menon, S. J. Katner, D. E. Lee, E. J. Peterson, J. E. Koblinski and N. P. Farrell, Antitumor active trans-platinum complexes through metabolic stability and enhanced cellular accumulation, J. Inorg. Biochem., 2024, 252, 112475 CrossRef CAS PubMed.
- F. J. Ramos-Lima, A. G. Quiroga, B. García-Serrelde, F. Blanco, A. Carnero and C. Navarro-Ranninger, New trans-Platinum Drugs with Phosphines and Amines as Carrier Ligands Induce Apoptosis in Tumor Cells Resistant to Cisplatin, J. Med. Chem., 2007, 50, 2194–2199 CrossRef PubMed.
- A. G. Quiroga, Understanding trans platinum complexes as potential antitumor drugs beyond targeting DNA, J. Inorg. Biochem., 2012, 114, 106–112 CrossRef PubMed.
- F. A. Rico, M. Derogar, L. Cubo and A. G. Quiroga, Synthetic routes and chemical structural analysis for guiding the strategies on new Pt(ii) metallodrug design, Dalton Trans., 2024, 53, 14949–14960 RSC.
- C. M. Santos, S. Cabrera, C. Ríos-Luci, J. M. Padrón, I. L. Solera, A. G. Quiroga, M. A. Medrano, C. Navarro-Ranninger and J. Alemán, Novel clioquinol and its analogous platinum complexes: importance, role of the halogen substitution and the hydroxyl group of the ligand, Dalton Trans., 2013, 42, 13343–13348 RSC.
- A. Medrano, S. M. Dennis, A. Alvarez-Valdés, J. Perles, T. M. Mason and A. G. Quiroga, Synthesis, cytotoxicity, DNA interaction and cell cycle studies of trans-diiodophosphine Pt(II) complexes, Dalton Trans., 2015, 44, 3557–3562 RSC.
- F. J. Ramos-Lima, A. G. Quiroga, J. M. Pérez, M. Font-Bardía, X. Solans and C. Navarro-Ranninger, Synthesis and Characterization of New Transplatinum Complexes Containing Phosphane Groups − Cytotoxic Studies in Cisplatin-Resistant Cells, Eur. J. Inorg. Chem., 2003, 2003, 1591–1598 CrossRef.
- G. Guerrero, P. H. Mutin, F. Dahan and A. Vioux, X-ray crystal structures of novel platinum(II) and palladium(II) complexes of dialkyl phosphonated phosphines, J. Organomet. Chem., 2002, 649, 113–120 CrossRef.
- M. D. Hall, K. A. Telma, K.-E. Chang, T. D. Lee, J. P. Madigan, J. R. Lloyd, I. S. Goldlust, J. D. Hoeschele and M. M. Gottesman, Say No to DMSO: Dimethylsulfoxide Inactivates Cisplatin, Carboplatin, and Other Platinum Complexes, Cancer Res., 2014, 74, 3913–3922 CrossRef PubMed.
- M. Patra, T. Joshi, V. Pierroz, K. Ingram, M. Kaiser, S. Ferrari, B. Spingler, J. Keiser and G. Gasser, DMSO-Mediated Ligand Dissociation: Renaissance for Biological Activity of N-Heterocyclic-[Ru(η6-arene)Cl2] Drug Candidates, Chem. – Eur. J., 2013, 19, 14768–14772 CrossRef PubMed.
- T. Hidalgo, D. Fabra, R. Allende, A. I. Matesanz, P. Horcajada, T. Biver and A. G. Quiroga, Two novel Pd thiosemicarbazone complexes as efficient and selective antitumoral drugs, Inorg. Chem. Front., 2023, 10, 1986–1998 Search PubMed.
- H. P. Varbanov, D. Ortiz, D. Höfer, L. Menin, M. S. Galanski, B. K. Keppler and P. J. Dyson, Oxaliplatin reacts with DMSO only in the presence of water, Dalton Trans., 2017, 46, 8929–8932 Search PubMed.
- S. J. Berners-Price, T. A. Frenkiel, U. Frey, J. D. Ranford and P. J. Sadler, Hydrolysis products of cisplatin: pKa determinations via [1H, 15N] NMR spectroscopy, J. Chem. Soc., Chem. Commun., 1992, 789–791 Search PubMed.
- S. Ishida, J. Lee, D. J. Thiele and I. Herskowitz, Uptake of the anticancer drug cisplatin mediated by the copper transporter Ctr1 in yeast and mammals, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14298–14302 CrossRef PubMed.
- F. Arnesano, S. Scintilla and G. Natile, Interaction between Platinum Complexes and a Methionine Motif Found in Copper Transport Proteins, Angew. Chem., Int. Ed., 2007, 46, 9062–9064 CrossRef PubMed.
- S. J. Berners-Price, L. Ronconi and P. J. Sadler, Insights into the mechanism of action of platinum anticancer drugs from multinuclear NMR spectroscopy, Prog. Nucl. Magn. Reson. Spectrosc., 2006, 49, 65–98 CrossRef.
- A. Kellett, Z. Molphy, C. Slator, V. McKee and N. P. Farrell, Molecular methods for assessment of non-covalent metallodrug–DNA interactions, Chem. Soc. Rev., 2019, 48, 971–988 RSC.
- T. Mendrina, I. Poetsch, H. Schueffl, D. Baier, C. Pirker, A. Ries, B. K. Keppler, C. R. Kowol, D. Gibson, M. Grusch, W. Berger and P. Heffeter, Influence of the Fatty Acid Metabolism on the Mode of Action of a Cisplatin(IV) Complex with Phenylbutyrate as Axial Ligands, Pharmaceutics, 2023, 15, 677 CrossRef CAS.
- P. Fronik, I. Poetsch, A. Kastner, T. Mendrina, S. Hager, K. Hohenwallner, H. Schueffl, D. Herndler-Brandstetter, G. Koellensperger, E. Rampler, J. Kopecka, C. Riganti, W. Berger, B. K. Keppler, P. Heffeter and C. R. Kowol, Structure–Activity Relationships of Triple-Action Platinum(IV) Prodrugs with Albumin-Binding Properties and Immunomodulating Ligands, J. Med. Chem., 2021, 64, 12132–12151 CrossRef CAS PubMed.
- M. Song, M. Cui and K. Liu, Therapeutic strategies to overcome cisplatin resistance in ovarian cancer, Eur. J. Med. Chem., 2022, 232, 114205 CrossRef CAS.
- S. Hanson, A. Dharan, Jinsha P. V., B. G. Nair, R. Kar and N. Mishra, Paraptosis: a unique cell death mode for targeting cancer, Front. Pharmacol., 2023, 14, 1159409, DOI:10.3389/fphar.2023.1159409.
- U. Jungwirth, D. N. Xanthos, J. Gojo, A. K. Bytzek, W. Körner, P. Heffeter, S. A. Abramkin, M. A. Jakupec, C. G. Hartinger, U. Windberger, M. Galanski, B. K. Keppler and W. Berger, Anticancer Activity of Methyl-Substituted Oxaliplatin Analogs, Mol. Pharmacol., 2012, 81, 719–728 CrossRef CAS.
- S. Hager, K. Korbula, B. Bielec, M. Grusch, C. Pirker, M. Schosserer, L. Liendl, M. Lang, J. Grillari, K. Nowikovsky, V. F. S. Pape, T. Mohr, G. Szakács, B. K. Keppler, W. Berger, C. R. Kowol and P. Heffeter, The thiosemicarbazone Me2NNMe2 induces paraptosis by disrupting the ER thiol redox homeostasis based on protein disulfide isomerase inhibition, Cell Death Dis., 2018, 9, 1–17 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental section, figures and tables. CCDC 2401350, 2447565, 2401352 and 2401353. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5qi00674k |
| ‡ Same contribution. |
| § Currently in industry. |
| ¶ Shared position. |
| This journal is © the Partner Organisations 2025 |