
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective aromatic halogenation by a manganese compound I model: a mimic of chloroperoxidase†
Lina Zhang‡
a,
Steiny Russelisaac Premakumari‡b,
Maggie Ngb,
Jisheng Zhangc,
Yong-Min Lee *a,
Shunichi Fukuzumi*a,
Kyung-Bin Cho*b and
Wonwoo Nam*ade
*a,
Shunichi Fukuzumi*a,
Kyung-Bin Cho*b and
Wonwoo Nam*ade
aDepartment of Chemistry and Nano Science, Ewha Womans University, Seoul 03760, Korea. E-mail: yomlee@ewha.ac.kr; fukuzumi@chem.eng.osaka-u.ac.jp; wwnam@ewha.ac.kr
bDepartment of Chemistry and Research Institute for Materials and Energy Sciences, Jeonbuk National University, Jeonju 54896, Korea. E-mail: workforkyung@jbnu.ac.kr
cInstitute of Green Chemistry and Chemical Technology, School of Chemistry and Chemical Engineering, Jiangsu University, Zhenjiang 212013, P. R. China
dGraduate Program in Innovative Biomaterials Convergence, Ewha Womans University, Seoul 03760, Korea
eCollege of Chemistry and Chemical Engineering, Henan Key Laboratory of Function-Oriented Porous Materials, Luoyang Normal University, Luoyang 471934, P. R. China
First published on 12th May 2025
Abstract
A high-valent manganese(IV)-hydroxo porphyrin π-cation radical complex, [MnIV(OH)(Por˙+)(OTf)]+ (a protonated manganese compound I analogue), was studied in the halogenation of aromatic compounds. By replacing the triflate anion with Cl− or Br−, we were able to halogenate toluene with high selectivity for C(sp2)–H bonds over C(sp3)–H bonds, such as chlorination with Cl− or bromination with Br− in the aromatic ring. We have also examined the halogenation of naphthalene and benzene derivatives with [MnIV(OH)(Por˙+)(X)]+ (X = Cl− and Br−). In all of these reactions, halogenated products were formed dominantly, and the source of the halogens in the products was found to be halides present in the [MnIV(OH)(Por˙+)(X)]+ complexes. In the absence of halides, naphthalene was found to undergo dimerization. Kinetic isotope effect (KIE) experiments on this reaction showed no isotopic effect in the halogenation reactions. DFT calculations on models with the naphthalene substrate supported a mechanism involving an initial (rate-limiting) electron transfer from the substrate to [MnIV(OH)(Por˙+)(OTf)]+, coupled by the attachment of Cl− to the C1 position of the naphthalene radical cation. This mechanism was also supported by the Marcus theory of outer-sphere electron transfer. The so-formed [MnIV(OH)(Por)(OTf)] (a manganese compound II analogue) underwent a hydrogen atom transfer from the C1 position of the substrate to form chlorinated naphthalene and [MnIII(H2O)(Por)(OTf)]. DFT calculations showed that [MnIV(OH)(Por)(OTf)] can also undergo direct OH-transfer to the substrate competitively, leaving open possibilities for side reactions or alternative reactions in a different environment. This study provides a deeper understanding of chloroperoxidase-like reactions.
Introduction
Halogenated aromatic molecules play a crucial role in synthetic pharmaceuticals,1–3 agriculture,4 and organic synthesis.5–9 However, the conventional method of electrophilic halogenation utilizes hazardous halogen gases, such as Cl2 or Br2, or organic chlorination reagents like N-chlorosuccinimide (NCS) and tBuOCl, which are derived from halogen gases and result in significant amounts of organic waste.10–14 Therefore, a more secure, simple, and environmentally friendly approach is needed. In nature, chloroperoxidase is a unique enzyme capable of halogenating aromatic rings C(sp2)–H over C(sp3)–H under low pH conditions.15–21 It also shares similar properties with peroxidases (dehydrogenation or oxygen evolution) and P450s (C–H bond hydroxylation).16b The mechanism of chlorination by chloroperoxidase is believed to involve the generation of FeIV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O porphyrin π-cation radical species,18 referred to as compound I (Cpd I), followed by the oxidation of halides to produce an enzyme-bound haem-FeIII-OX intermediate (where X is a halide).19–21 This intermediate can either react directly with the substrate or generate free hypohalous acid (HOX or OX−).19–21 However, how the intermediate or the free hypohalous acid reacts selectively with substrates remains a topic of debate.
O porphyrin π-cation radical species,18 referred to as compound I (Cpd I), followed by the oxidation of halides to produce an enzyme-bound haem-FeIII-OX intermediate (where X is a halide).19–21 This intermediate can either react directly with the substrate or generate free hypohalous acid (HOX or OX−).19–21 However, how the intermediate or the free hypohalous acid reacts selectively with substrates remains a topic of debate.
Synthetic metalloporphyrin compounds have been widely used in model studies of haem enzymes in order to gain insights into the details of the enzymatic reaction mechanisms. For example, Fujii et al. attempted to mimic the chlorination reactivity of chloroperoxidase using meso-substituted iron porphyrin complexes.22,23 Although an FeIV(O)(TPFPP+˙)(NO)3 (TPFPP = 5,10,15,20-tetrakis(pentafluorophenyl)porphinato dianion) complex was able to oxidize Cl− to form a chloride radical and FeIV–oxo species, only one-electron oxidation of chloride occurred rather than the expected two-electron oxidation of organic compounds.22 Complexes bearing more electron-donating meso substituents were later screened, and none were found to form the desired iron hypochlorite.23b Pursuant to this, several Fe–OCl bearing porphyrins were synthesized directly by ligand exchange of the Fe(OH) moiety with tetrabutylammonium hypochlorite (TBAOCl). Only (TPFPP)Fe–OCl showed an ability to chlorinate substrates like 1,3,5-trimethoxybenzene.23b Additionally, the Fe–OCl compound was observed to undergo heterolytic O–Cl bond cleavage to form Cpd I, an FeIVO species with a porphyrin π-cation radical, and Cl−. However, the reaction mechanism of Cpd I reacting with chloride remains unclear.23
In the case of manganese, manganese porphyrins have been shown to be effective in selectively chlorinating aliphatic C–H bonds with tetrabutylammonium chloride and NaOCl as halogenating agents.24,25 The MnIII porphyrin complex can also catalyse the oxyfluorination of alkanes by iodosylbenzene in the presence of F−.26 It is believed that MnV-dioxo may be the reactive species involved in these halogenation reactions, although evidence to support this mechanism is limited.25,26 However, there has been no report of selective aromatic halogenation using a Mn porphyrin system. In non-haem systems, MnIV–oxo–Sc3+ has been studied for its ability to selectively brominate benzene derivatives through a rate-limiting electron transfer pathway.27 Nevertheless, due to limitations in oxidation potential, bromination can occur only on electron-rich benzene derivatives, making the selective halogenation of toluene or benzene challenging to achieve.27
Recently, a Mn version of Cpd I was synthesized and characterized as most likely being a [MnIV(OH)(TDCPP˙+)(OTf)]+ species (TDCPP = meso-tetrakis(2,6-dichlorophenyl)porphinato dianion), with an extra hydrogen bonding of OTf− to the OH moiety.28 It showed reactivity in C–H bond activation, sulfoxidation, and water oxidation. In the current study, we demonstrate that this Mn-Cpd I species chlorinates aromatic rings selectively, mimicking the behaviour of chloroperoxidase and further improving our understanding of the chloroperoxidase reactivity. A combined experimental and theoretical study for the mechanism of the chlorination of aromatic compounds by the Mn-Cpd I intermediate is discussed in this work. Additionally, we have compared the reactions of Mn-Cpd I, MnIV(O), and MnIII-OIAr (ArIO = iodosylarene) species with aromatic compounds (Scheme 1).
 | ||
| Scheme 1 Reactions of Mn-porphyrin intermediates with aromatic compounds. | ||
Results and discussion
Aromatic chlorination of 3/Cl−
The Mn porphyrin intermediates were prepared according to the literature procedures.28–30 The names 1/Cl−, 2/Cl−, and 3/Cl− (see Scheme 1 for the chemical structures) referred to below signify intermediates generated with the starting compound containing Cl−, [MnIII(TDCPP)(Cl)]. Later, intermediates 3/Br− and 3/OTf− were also generated using [MnIII(TDCPP)(Br)] and [MnIII(TDCPP)(OTf)] as starting compounds.Typically, the [MnIII(sPhIO)(TDCPP)] species (1/Cl−, sPhIO = 1-(tert-butylsulfonyl)-2-iodosylbenzene) was prepared by adding 5 equiv. of sPhIO to a solution of [MnIII(TDCPP)(Cl)] in CH2Cl2 at −60 °C.29 The temperature was then increased to −10 °C, leading to O–I bond cleavage and the formation of [MnIV(O)(TDCPP)] (2/Cl−).30 When adding 10 equiv. of HOTf to the solution of 2/Cl−, the colour changed directly from yellow to green, indicating the formation of [MnIV(OH)(TDCPP˙+)(OTf)]+ (3/Cl−).28 A comparison of the UV-vis spectra of 1/Cl−, 2/Cl−, and 3/Cl− is shown in Fig. S1 of the ESI.†
1/Cl−, 2/Cl−, and 3/Cl− have been reported as strong oxidants that can activate the C–H bonds of hydrocarbons.28–30 At first, toluene was selected as the substrate probe. The reaction of 1/Cl− with toluene at −60 °C afforded benzaldehyde (∼50% yield), whereas the reaction of 2/Cl− with toluene in CH2Cl2 at −10 °C yielded benzyl chloride (∼45%; Scheme 1 and Fig. S2†). The product analysis indicated that the reactions of 1/Cl− and 2/Cl− occurred at the C(sp3)–H site, which was the same C–H bond activation site depicted in previous studies.29,30 Addition of toluene to a solution containing 3/Cl− at −10 °C led to clean spectral changes with isosbestic points (Fig. 1). The organic product was analysed by GC-MS, revealing that 2-Cl-toluene and 4-Cl-toluene were formed (Scheme 1 and Fig. S2†). Interestingly, this indicates that the reaction of 3/Cl− with toluene took place at the aromatic ring, showing a preference towards a C(sp2)–H site over a C(sp3)–H site.
 | ||
Fig. 1 UV-vis spectral change observed in the reaction of 3/Cl− (0.10 mM, red line) and toluene (0.40 M) in CH2Cl2/CH3CN (v/v 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) at −10 °C. The inset shows the time trace monitored at 478 nm. 3/Cl− (0.10 mM, red line) was generated in situ by the reaction of [MnIII(TDCPP)(Cl)] with PhIO (5 equiv.) and HOTf (10 equiv.). :1) at −10 °C. The inset shows the time trace monitored at 478 nm. 3/Cl− (0.10 mM, red line) was generated in situ by the reaction of [MnIII(TDCPP)(Cl)] with PhIO (5 equiv.) and HOTf (10 equiv.). | ||
To verify the aromatic halogenation reaction by 3/Cl−, we used naphthalene as the substrate. Upon addition of naphthalene to a solution containing 3/Cl− in CH2Cl2, the occurrence of a fast reaction was observed, as confirmed by the UV-vis spectral change (Fig. S3†). The appearance of an absorption band at 478 nm due to the formation of [MnIII(TDCPP)]+ was confirmed by EPR and ESI-MS (Fig. S4†). The product in the oxidation of naphthalene was determined to be 1-Cl-naphthalene (∼95% yield) (Table 1). The reaction with benzene was also confirmed to give ∼40% chlorobenzene as the product (Fig. S3b and S5†). Thus, this once again shows that the C(sp2)–H halogenation in the aromatic ring is the preferred reaction by 3/Cl−.
|
||||
|---|---|---|---|---|
| X− | Solvent | Product (%) | ||
| 1-Cl-Naphthalene | 1-Br-Naphthalene | 1,1′-Binaphthyl | ||
| a Reactions of 3/X− (1.0 mM) with naphthalene (20 mM) were carried out in different solvents at −10 °C. Yields were determined based on the amount of 3/X− used.b Not detected. | ||||
| Cl− | CH2Cl2 | 95(7) | n.d.b | n.d.b |
| Cl− | CH2Br2 | 77(5) | 15(2) | n.d.b |
| Cl− | CH3CN | 90(8) | n.d.b | n.d.b |
| Br− | CH2Br2 | n.d.b | 88(7) | n.d.b |
| Br− | CH2Cl2 | 35(2) | 57(3) | n.d.b |
| OTf− | CH2Cl2 | 30(3) | n.d.b | 15(1) |
| OTf− | CH3CN | n.d.b | n.d.b | 45(3) |

Halogen source in the aromatic halogenation by 3
In order to pinpoint the source of chlorine in the product, the CH2Cl2 solvent was changed to CH2Br2. Analysis of the reaction solution of 3/Cl− and naphthalene in CH2Br2 revealed that the chlorinated product was found to be ∼77% as the major product and only ∼15% of the brominated product was observed (Table 1). The reaction of 3/Cl− with toluene was also carried out in CH2Br2, affording the chlorinated product as the major component (Fig. S2a†). This is distinct from the reaction of 2/Cl− with toluene, in which only benzyl bromide was produced when the solvent was changed to CH2Br2 (Fig. S2b†). The reaction of 3/Cl− with naphthalene was then performed in CH3CN and ∼90% 1-Cl-naphthalene was obtained as the sole product (Table 1). Therefore, it is strongly implied that the halogenating chlorine mainly comes from the Cl− axial ligand of the starting compound.To further validate this observation, [MnIII(TDCPP)(Br)] and [MnIII(TDCPP)(OTf)] were synthesized according to the literature procedures.31 A comparison of the UV-vis spectra of [Mn(TDCPP)(Cl)], [Mn(TDCPP)(Br)], and [Mn(TDCPP)(OTf)] is shown in Fig. S6.† It is worth noting that the UV-vis spectra of 3 with different counter anions from the starting materials are identical to each other. Based on the published EXAFS data,28 3 is six-coordinate and Cl− is ruled out as the axial ligand. Density functional theory (DFT) calculations at that time found that a [MnIV(OH)(TDCPP+˙)(OTf)]+ species with an additional OTf− hydrogen bonding to the OH moiety fitted best with the experimental results, leading us to designate 3 as such a species.
We also used 3/Br− as a catalyst, and the results were very similar to those of 3/Cl−. When the reaction was conducted in CH2Br2, the product was the brominated one. Even when the solvent was changed to CH2Cl2, the brominated products still accounted for the majority (Table 1 and Fig. S2a, S5†). We also studied the reactions of 3/OTf− with various substrates. In the reaction of 3/OTf− with naphthalene in CH2Cl2, only a small amount of halogenated products (30(3)%) was detected, but a non-negligible amount of 1,1′-binaphthyl (15(1)%) was also produced (Table 1). When the reaction was performed in CH3CN, which cannot be a source of halogen, ∼42% of 1,1′-binaphthyl (based on the amount of 3/OTf−) was detected as the sole product (Table 1 and Fig. 2). The formation of 1,1′-binaphthyl is presumably a result of dimerization of two naphthalene radicals, each created by 3/OTf−, but the exact reaction mechanism has yet to be elucidated. The reaction of 3/OTf− with toluene in CH3CN resulted in the formation of benzaldehyde instead, and neither halogenated nor dimer products were detected (Fig. 2). No products were detected in the reaction solution when benzene was used as the substrate under the same reaction conditions (Fig. 2).
 | ||
| Fig. 2 Products with yields in the reaction of 3/OTf− (1.0 mM) with naphthalene (10 mM), toluene (200 mM), and benzene (400 mM) in CH3CN at −10 °C. 3/OTf− was generated by reacting [MnIII(TDCPP)(OTf)] (1.0 mM) with PhIO (5 equiv.) and HOTf (10 equiv.) at −10 °C. | ||
Taking all the results into consideration, we found that the halogen source may come from both the solvent and the starting materials, although the major source is the starting counter anion. In the absence of a halogen source, the reaction of naphthalene leads to dehydrogenation (and/or dimerization), while the reactive site shifts from C(sp2) to C(sp3) with toluene, and no reaction occurs with benzene. These results suggest that the presence of Cl− or Br− assists in the activation of the aromatic ring.
DFT calculations
In order to map the exact detailed reaction mechanism, DFT calculations were performed on the reaction (Tables S1–S31†). At the exploratory phase, a simple porphine with OTf− as an axial ligand was used as a model of 3 (Fig. S7†), as this structure has been previously studied and found to fit the experimental results.28 Some results with the Cl− axial ligand are also included in Tables S1–S3,† and by and large, they do not change the conclusions drawn here. The electronic configuration is energetically degenerate between S = 1 (MnIVO porphine β-radical) and S = 2 (MnIVO porphine α-radical),28 as the porphine radical has a weak interaction with the rest of the system. For the reaction itself, however, the S = 2 state consistently showed lower energy barriers, and this is therefore the state we present in this text (for S = 1, see Tables S4–S6†).Looking at the interaction between 3 and the naphthalene substrate, the complexation between naphthalene and 3 yields a reactant complex that is largely devoid of any interactions; thus, the interaction energy is very minor (even repulsive) by up to 0.8 kcal mol−1 (Table S7†). We found, however, two different solutions to the wave function. While the energetically lowest complex did not generate any radical on the naphthalene, a second solution, which was 1.1 kcal mol−1 higher in energy (Table S7†), showed the naphthalene substrate developing a partial radical upon complexation (0.3 in spin; Table S8†). Hence, it hints at a tendency where an electron could be transferred from the substrate to 3 under the right conditions. Performing a C–H abstraction from the C1 carbon of naphthalene to form MnIII–aqua and a naphthalene radical indicated that the barrier was of 37.0 kcal mol−1, with a late Mn(HO)–HC transition state distance of 1.12 Å (Table S9†). Thus, this did not seem to be a feasible pathway. However, a nucleophilic interaction (hydroxylation) where MnOH attacks the C1 and C2 carbon directly resulted in 11.2 and 13.1 kcal mol−1 of the lowest barrier, respectively; each transfer can also occur with initial α- or β-electron transfer, with different barriers (see Table S7;† these barriers are larger with a full model, at least 16.6 kcal mol−1 due to repulsion from hydrogen-bonded OTf−, Table S22† and vide infra). This indicates that an initial naphthalene hydroxylation would be possible. This would seem to be in agreement with KIE studies (KIE = 0.8, vide infra), and it may be the path to the dimerization of the substrate when a halide is not available (vide supra). However, these pathways would actually not be competitive with other pathways found in the presence of a halide (vide infra).
As we know that the substrate is to be chlorinated in this reaction, we added a free chloride anion to the model in the vicinity near the substrate. Surprisingly, there is an immediate electron transfer, reducing the porphine radical on 3 to form a [MnIV(OH)(Por)(OTf)] species (denoted as 2OH-OTf), which would be equivalent to a manganese compound II model. According to the Mulliken spin densities, the source of the reducing electron is both the Cl− ion and the substrate, contributing about half an electron each (Table S5†). Indeed, the interaction energy between Cl− and the rest of this reactant complex (RC) is 12.5 kcal mol−1 (Table S4†). This indicates that the presence of the chloride anion plays a key role in facilitating an electron transfer from the substrate to 3. This in turn indicates that the halogenation-enabling species is in fact the 2OH-OTf species, acting on one electron-deficient naphthalene-chloride complex. To give a context, we have calculated the ionization energy of naphthalene and chloride to be 143.0 kcal mol−1 and 141.4 kcal mol−1, respectively (Table S28†). In contrast, the electron affinity of 3 is calculated to be at 134.3 kcal mol−1 (Table S28†). Thus, under these conditions, an electron transfer from either naphthalene or chloride would be an endothermic reaction by 8.7 kcal mol−1 and 7.1 kcal mol−1, respectively. However, naphthalene and chloride having such close ionization energies hint at the possibility of a resonance structure when complexed together. Indeed, the ionization energy in a complexed model containing both the molecules is 121.6 kcal mol−1, and an electron transfer to 3 would now be exothermic (12.7 kcal mol−1). We propose that this electron transfer is the rate-limiting step, based on experiments, the Marcus theory of electron transfer, and further DFT calculations (vide infra). If true, then the issue of nucleophilic or electrophilic reaction in the ensuing steps would be immaterial, as those non-rate-limiting reaction steps do not affect the KIE value.
In the course of our investigation, we indeed found two theoretically feasible reaction pathways to obtain our halogenated product, one electrophilic pathway (hydrogen atom transfer) and one nucleophilic pathway (Fig. 3). The simplest possible pathway, and therefore also the most likely pathway, is the electrophilic pathway. 2OH-OTf abstracts a hydrogen atom from the C1 position of naphthalene, creating a MnIII–aqua species (denoted as 1H2O-OTf). This results in a very reactive substrate that will bind the nearby Cl half-radical concertedly to C1. The barrier value of this reaction depends slightly on the calculation model used. The exploratory model with porphine yields an energy barrier of 7.1 kcal mol−1 (Table S4†). A larger model, using the full TDCPP structure as a ligand and with an extra hydrogen bonded OTf− (which is the most realistic model tried in this study), results in a barrier value of 9.2 kcal mol−1 (Table S10†). This electrophilic reaction is calculated to be 54.2 kcal mol−1 (larger model: 56.5 kcal mol−1) exothermic. In contrast, a hydrogen atom abstraction from the C2 position results in a barrier of 29.1 kcal mol−1 (larger model). While the energies are slightly different between the simple and larger model (vide infra), the Mulliken spin densities and the geometries are comparable (Tables S11 and S12, compared with Tables S4 and S5†). We have also calculated this reaction with a Mn–oxo species, [MnIVO(Por+˙)(OTf)] (Tables S13–S15†); however, the barrier seems to be slightly higher on this route (13.9 kcal mol−1), and deprotonation of 3 before catalysis therefore seems unnecessary.
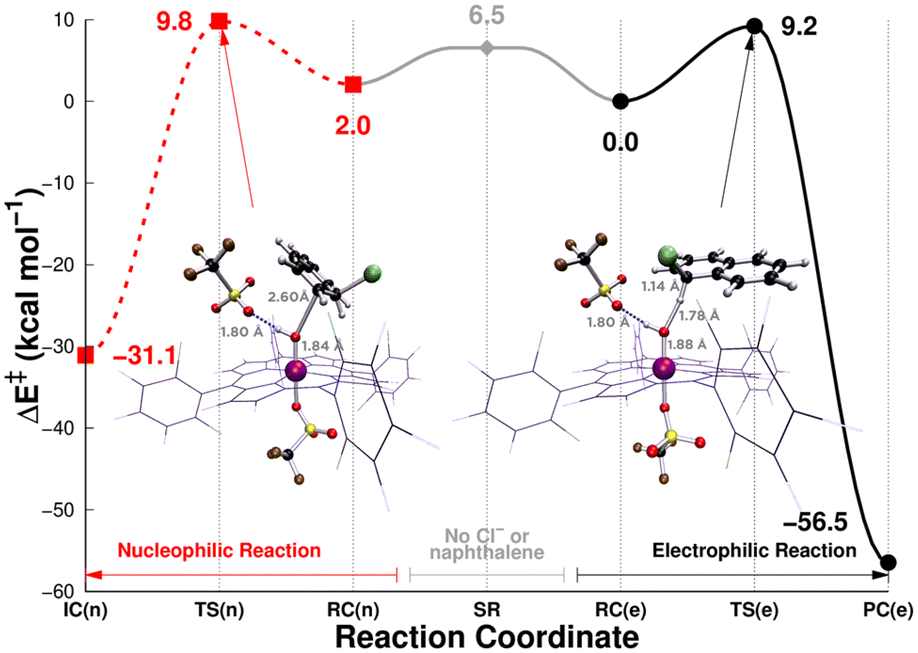 | ||
| Fig. 3 The DFT calculated energetics of the C1 electrophilic pathway (black, solid) and the nucleophilic C2 hydroxylation pathway (red, dashed) in the reaction of [MnIV(OH)(TDCPP+˙)(OTf)]+ (3) with naphthalene. The difference in the energies between the separated reactants SR (centre, grey) and the reactant complexes RC(n) or RC(e) is the complexation energy of 3 (with additional OTf−, which is hydrogen bonded to MnOH) and Cl− and naphthalene. Energies are results of B3LYP/Def2-TZVPP//Def2-SVP calculations on a large model of 3 with TDCPP as a ligand and the additional OTf− present (Table S10†). SR = separated reactants, RC = reactant complex, TS = transition state, PC = product complex; (n) and (e) denote the nucleophilic and electrophilic reactions, respectively. | ||
The second possible pathway involves 2OH-OTf attacking the C1 or C2 position of naphthalene in a nucleophilic reaction. These steps have very small barriers of 7.1 and 4.4 kcal mol−1, respectively (larger models: 12.8 and 9.8 kcal mol−1, respectively) and are fully competitive with the activation barriers of the above-described electrophilic reaction. Thus, the conclusion from theory is that there could be a competition between the electrophilic and nucleophilic pathways. Thus, theory alone cannot distinguish which one is the preferred (initial) pathway. However, despite our best efforts, we could not find a viable pathway after the initial nucleophilic step that was leading to the experimentally detected products.
At this point, the differences between the calculation model sizes warrant an explanation. The important lowest electrophilic and nucleophilic barriers in the porphine model (7.1 and 4.4 kcal mol−1, respectively) do not change much upon replacing porphine with TDCPP (7.2 and 4.6 kcal mol−1, respectively, Table S19†). However, when adding an OTf− to hydrogen-bond to the MnOH moiety, the barriers become 9.2 and 9.8 kcal mol−1, respectively (Table S10†). While the increase for the electrophilic reaction is moderate (2.0 kcal mol−1), it affects the nucleophilic barrier noticeably more (5.2 kcal mol−1). This can probably be attributed to the larger repulsion between OTf− and the substrate, which are closer to each other in the nucleophilic reaction than in the electrophilic one. Thus, we can see that the presence of a hydrogen bond in the MnOH moiety can affect the reaction selectivity of the catalyst through steric repulsion.
While the KIE results would seemingly not support an electrophilic pathway (vide infra), we also have the kinetic information from the experiments. The k2 value of 3/Cl− with naphthalene at −10 °C, which is 2.0(2) M−1 s−1 (Fig. S8a and Table S32;† vide infra), corresponds to the activation free energy of 15.3 kcal mol−1 in barrier, which is obtained from the Eyring equation. This indicates that both the electrophilic and nucleophilic pathways seem to have too low calculated barriers to be compatible with experiments. However, these barrier values are compatible with experiments if we assume that the rate-limiting step is in fact the first electron transfer step from the substrate–Cl complex to 3, with an activation barrier higher than those calculated in the electrophilic and nucleophilic pathways in Fig. 3. To show this, we have performed a kinetics study involving electron transfer and Marcus theory (vide infra). If so, then the ensuing C–H activation step would not generate a KIE value as it is not rate-limiting, and the reaction can go on along the simpler electrophilic pathway. Even though the nucleophilic pathway is theoretically plausible at the initial step, the necessary involvement of further steps to reach the products (when there is a simple electrophilic one-step reaction) makes it a less likely pathway.
Kinetics study
Kinetics data were collected to support the above-described aromatic halogenation mechanism. The first-order rate constants, determined by the first-order fitting for the decay of 3/Cl−, increased linearly with increasing substrate concentration in CH2Cl2 (Fig. S8†). The k2 value of the reaction of 3/Cl− with naphthalene was determined to be 2.0(2) M−1 s−1 in CH2Cl2 at −10 °C and an inverse KIE value of 0.8 was obtained (Fig. S8a†). The inverse KIE value in this case clearly shows that the electrophilic hydrogen abstraction is not the rate-determining step, but that the hybridization change from sp2 to sp3 of the carbon atom of the naphthalene radical cation undergoing nucleophilic attack by Cl− is involved in the rate-determining step.32 This should be compatible with the theoretical results above, where there is an initial electron transfer from the chloride and substrate system and a loose association between those components as a result. The k2 values of the reaction of 3 with toluene and benzene were determined to be 2.5(2) × 10−2 M−1 s−1 and 4.7(4) × 10−3 M−1 s−1, respectively (Fig. S9 and Table S32†). Benzene derivatives and 1-X-naphthalene were also studied, as well as naphthalene with 3/Br− and 3/OTf−, and the k2 values are summarized in Table S32 and Fig. S8–S11.†The rate constants of the reactions of 3/Cl− with different substrates are then evaluated in light of the Marcus theory of outer-sphere electron transfer using eqn (1):
|
ket = Zexp[−(λ/4)(1 + ΔGet/λ)2/(kBT)]
| (1) |
where Z is the frequency factor (normally, 1011 M−1 s−1) that corresponds to kBTK/h (kB is the Boltzmann constant, T is the absolute temperature, K is the formation constant of the precursor complex, and h is the Planck constant), ΔGet is the free energy change of the electron transfer from the substrate to 3/Cl−, and λ is the reorganization energy of the electron transfer.33 The driving force of the electron transfer (−ΔGet) is given by eqn (2):
| −ΔGet = −e(Eox − Ered) | (2) |
 | ||
| Fig. 4 Plot of logk2 for the oxidation of different substrates [(1) 1-Me-naphthalene, (2) naphthalene, (3) 1-Br-naphthalene, (4) 1-NO2-naphthalene, (5) anisole, (6) toluene, and (7) benzene] vs. the driving force of electron transfer [−ΔGet; eqn (2)] by 3/Cl− in CH2Cl2 at −10 °C. | ||
Mechanism of the aromatic halogenation by 3
In the absence of a halogen source, naphthalene gives a dimer as the product. Combined with the fact that the Eox of naphthalene is determined to be 1.75 V vs. SCE, which is only 0.21 V higher than the Ered of 3, and the good fitting in the Marcus plot, an outer-sphere electron transfer from naphthalene to 3 seems to be possible. In particular, the electron transfer should be facilitated in the presence of electron-rich anions such as Cl− or Br−. We therefore propose that as a first step of the reaction mechanism, this electron transfer occurs concertedly from the halide and substrate to 3, and this result is supported by the DFT calculations. Thus, the actual reacting species after this electron transfer stage are [naphthalene + Cl]˙ and [MnIV(OH)(TDCPP)]+. Hydrogen abstraction then occurs to form the chlorinated naphthalene and MnIII(TDCPP) (Scheme 2). In the absence of Cl−, the electron transfer may happen after hydroxylation of the substrate at the C1 or C2 position by 3, as it forms a naphthalene–OH cation with an activation barrier that is within the realms of possibility, according to our DFT calculations (16.6 kcal mol−1, Table S22†). We speculate that this may lead to substrate–substrate dimerization (concomitant with water formation) to form the final product di-naphthalene; however, we have so far not been able to find a viable theoretical pathway for this. | ||
| Scheme 2 Proposed halogenation pathway of the naphthalene substrate. At the intermediate stage in this reaction, the Mn catalyst abstracts approximately half an electron from the substrate and the other half from the chloride. | ||
Conclusions
In summary, a high-valent manganese compound I complex {[MnIV(OH)(TDCPP˙+)(OTf)]+} displays high reactivity in aromatic halogenation. This occurs without any observed direct oxidation of Cl− leading to Mn–OCl formation or HOCl release. Product analysis indicates that the halogen source mainly comes from the counter anion of the starting Mn porphyrins. The kinetics data reveal a significant contribution of electron transfer in the rate-limiting step, leading to the proposal of a new reaction mechanism in which the electron transfer occurs in a concerted manner from both Cl− and the substrate to Mn–Cpd I. Moreover, this system exhibits similarities in C–H bond hydroxylation, O2 formation, and aromatic halogenation, as well as dehydrogenation properties to chloroperoxidase. Overall, this study provides valuable insights into the understanding of the mechanism of aromatic halogenation, and we expect to gain more knowledge about this system in the future.Experimental section
Materials
All organic chemicals, obtained from Aldrich Chemical Co. and Tokyo Chemical Industry, were of the maximum purity available and used without further purification unless otherwise indicated. Solvents were dried according to the published procedures and redistilled under an argon atmosphere before use.35 1-(tert-Butylsulfonyl)-2-iodosylbenzene (sPhIO) was synthesized using the literature procedure.36 [MnIII(TDCPP)(Cl)] was purchased from Frontier Scientific Inc. [MnIII(TDCPP)(Br)] and [MnIII(TDCPP)(OTf)] were synthesized by following the literature procedure.28Instrumentation
UV-vis spectra were recorded on a Hewlett Packard 8453 diode array spectrophotometer equipped with a UNISOKU cryostat system (USP-203; UNISOKU, Japan) for low-temperature experiments or on a UNISOKU RSP-601 stopped-flow spectrometer equipped with a MOS-type highly sensitive photodiode array. Electrospray ionization mass (ESI-MS) spectra were collected on an LCQ Fleet ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) by infusing samples directly into the source using a sample syringe. The spray voltage was set at 4.2 kV and the capillary temperature at 120 °C. X-band electron paramagnetic resonance (EPR)37a spectra were recorded at 5 K using an X-band Bruker EMX-plus spectrometer equipped with a dual mode cavity (ER 4116DM). Low temperatures were achieved and controlled with an Oxford Instruments ESR900 liquid He quartz cryostat with an Oxford Instruments ITC503 temperature and gas flow controller. The experimental parameters for EPR measurements were as follows: microwave frequency = 9.647 GHz, microwave power = 1.0 mW, modulation amplitude = 10 G, gain = 1 × 104, modulation frequency = 100 kHz, time constant = 40.96 ms and conversion time = 81.00 ms. Product analysis was performed with a Thermo Finnigan (Austin, Texas, USA) FOCUS DSQ (dual stage quadrupole) mass spectrometer interfaced with a Finnigan FOCUS gas chromatograph (GC-MS).Generation of Mn-porphyrin intermediates
[MnIII(sPhIO)(TDCPP)]+ (1/Cl−) was generated by reacting [MnIII(TDCPP)(Cl)] with 1-(tert-butylsulfonyl)-2-iodosylbenzene (sPhIO, 5 equiv.) in CH2Cl2 at −60 °C.29 The reaction solution was kept at −60 °C for further reactivity studies. A MnIV–oxo porphyrin complex, [MnIV(O)(TDCPP)] (2/Cl−), was generated according to literature procedures. 2/Cl− was generated within 1 min by reacting [MnIII(TDCPP)(Cl)] with 1-(tert-butylsulfonyl)-2-iodosylbenzene (sPhIO, 5 equiv.) in CH2Cl2 at −10 °C.30 A MnIV-hydroxo porphyrin π-cation radical complex, [MnIV(OH)(TDCPP˙+)(OTf)]+ (3/Cl−), was generated by adding triflic acid (CF3SO3H (HOTf); 10 equiv. in 25 μL of CH3CN) to the solution of [MnIV(O)(TDCPP)] at −10 °C.31 Formation of [MnIV(OH)(TDCPP˙+)(OTf)]+ (3/Cl−) was confirmed by monitoring UV-vis spectral changes for the formation of absorption bands at 390 nm and 674 nm due to 3/Cl−, accompanied by the decay of absorption bands at 420 nm and 522 nm due to 2/Cl−.Preparation of [MnIII(TDCPP)(Br)]
In a 100 mL round-bottom flask equipped with a stir bar were placed TDCPP (973 mg, 1 mmol) and Mn(C2H3O2)2·4H2O (865 mg, 5 mmol). Dimethylformamide (50 mL) was added to the flask and the reaction mixture was heated at 153 °C under air until the UV-vis spectrum showed no free TDCPP left. The mixture was cooled to room temperature and 10 mL HBr (48% aqueous) was added to the mixture. The precipitate was filtered and washed with de-ionized water until the filtrate was almost colourless. The product was dried overnight under reduced pressure prior to use. 1.025 g of a dark red solid product was obtained (96% yield). The UV-vis spectrum was consistent with the previous report.31Preparation of [MnIII(TDCPP)(OTf)]
[MnIII(TDCPP)(OTf)] was generated by adding 2.2 mg AgOTf to a solution of [MnIII(TDCPP)(Cl)] in CH3CN and stirred for 5 h until the UV-vis spectra did not change and no precipitates occurred. The resulting solution was filtered, dried and recrystallized to obtain the final product.31Kinetic measurements
Kinetic measurements were performed on a Hewlett Packard 8435 photodiode-array spectrophotometer at −10 °C for C–H bond activation and water oxidation reactions. Reactions were run in a 1.0 mm UV cuvette and followed by monitoring UV-vis spectral changes of reaction solutions. Rate constants were determined under pseudo-first-order conditions (e.g., [substrate]/[Mn intermediate] > 10) by fitting the changes in absorbance at 478 nm due to the MnIII species formed. Naphthalene and benzene derivatives were used in the aromatic halogenation reactions. Sulfoxidation reactions were performed under second-order kinetic conditions, in which the concentrations of the Mn intermediate and substrate are the same, and the k2 value was obtained from the second-order plots in CH2Cl2/CH3CN (v/v 20:1) at −40 °C. Thioanisole derivatives, such as para-X-thioanisoles (X = H, CN, CHO and NO2), have been employed for the oxygen atom transfer reactions. All reactions were run at least in triplicate, and the data reported represent the average of these reactions.
Product analysis
Organic product analysis was performed using GC, GC-MS and NMR37b spectroscopic measurements. For example, the reaction solution was filtered through a basic silica gel plug, and then an internal standard (decane) was added into the reaction solution, followed by analysing the resulting solution with GC and GC-MS. Products were identified by comparing them with authentic samples. Product yields were determined by comparing the peak areas of the reaction solutions with that of decane as an internal standard. The product yields were calculated based on the concentration of the Mn intermediates.Electrochemical measurements
Electrochemical measurements were performed on a CH Instruments (CHI630B) electrochemical analyser (CH Instruments, Inc.) under an Ar atmosphere in deaerated CH3CN containing 0.10 M Bu4NPF6 (TBAPF6) as a supporting electrolyte. A conventional three-electrode cell was used with a platinum working electrode (surface area of 0.30 mm2), a platinum wire as a counter electrode, and an Ag/Ag+ electrode as a reference electrode. The measured potentials were recorded with respect to an Ag/Ag+ (0.010 M) reference electrode. All potentials (vs. Ag/Ag+) were converted to values vs. SCE by adding 0.29 V. One-electron oxidation potentials of substrates were determined using second-harmonic alternating current voltammetry (SHACV) techniques at 25 °C. The one-electron oxidation potential of [MnIV(O)(TDCPP)] and the one-electron reduction potential of [MnIV(OH)(TDCPP˙+)(OTf)]+ were determined by cyclic voltammetry at −40 °C.Density functional theory calculations
Density functional theory38 calculations were carried out using the Gaussian 16 software.39 Geometry optimizations and frequency calculations were performed with the unrestricted B3LYP functional40 using the Def2-SVP basis set,41 followed by a single-point calculation with the Def2-TZVPP basis set41 to obtain more accurate energy values. Frequency calculations on the optimized geometries were performed to confirm the nature of the stationary points, and intrinsic reaction coordinate (IRC) calculations have been performed to ensure that the reactants and products really connect with the transition states. The conductor-like polarizable continuum model (CPCM)42 with a UFF cavity, per G16 default, was used to include the solvent (dichloromethane) effects in all the calculations. Gibbs free energies, including vibrational energy and entropy at 298 K, zero-point vibrational energy, dispersion43 and complexation energy due to a change in the reference state at 298 K44 were calculated and are presented in the ESI;† however, we usually do not consider this more accurate than the electronic energies. This is because we believe that the B3LYP parametrization and the CPCM solvent parametrization already consider these effects in small models such as ours,45 and quite frequently, we see unreasonable energies vis-à-vis experiments if using the calculated Gibbs free energy. Therefore, the energies discussed in the text are thus the electronic energies at the B3LYP/Def2-TZVPP//Def2-SVP level, modified by the CPCM solvent scheme only, which in our repeated experience shows good agreement with experiments, as it does in the current case.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NRF of Korea (NRF-2021R1A3B1076539 and NRF-2023K2A9A2A11098996 to W. N., NRF-2023R1A2C1007668 to Y.-M. L. and RS-2021-NR058689 to K.-B. C.), the MoE G-LAMP Program (No. RS-2024-00443714 to K.-B. C), and the Henan Center for Outstanding Overseas Scientists, China (GZS2024020 to W. N.).References
- W.-Y. Fang, L. Ravindar, K. P. Rakesh, H. M. Manukumar, C. S. Shantharam, N. S. Alharbi and H.-L. Qin, Synthetic approaches and pharmaceutical applications of chloro-containing molecules for drug discovery: A critical review, Eur. J. Med. Chem., 2019, 173, 117–153 CrossRef CAS PubMed.
- A. Jain, L. S. Duvvuri, S. Farah, N. Beyth, A. J. Domb and W. Khan, Antimicrobial polymers, Adv. Healthcare Mater., 2014, 3, 1969–1985 CrossRef CAS PubMed.
- R. D. Birkenmeyer, S. J. Kroll, C. Lewis, K. F. Stern and G. E. Zurenko, Synthesis and antimicrobial activity of clindamycin analogues: pirlimycin, a potent antibacterial agent, J. Med. Chem., 1984, 27, 216–223 CrossRef CAS PubMed.
- D. Xu, J. Guan, X. Xu, S. Gong and H. Xu, A new method for the synthesis of oxadiazine insecticide indoxacarb, J. Heterocycl. Chem., 2016, 53, 1469–1473 CrossRef CAS.
- M. L. Tang and Z. Bao, Halogenated materials as organic semiconductors, Chem. Mater., 2011, 23, 446–455 CrossRef CAS.
- (a) A. Suzuki, Cross-coupling reactions of organoboranes: an easy way to construct C-C bonds (Nobel lecture), Angew. Chem., Int. Ed., 2011, 30, 6722–6737 CrossRef PubMed; (b) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Palladium-catalyzed cross-coupling: a historical contextual perspective to the 2010 Nobel prize, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef CAS PubMed; (c) V. V. Grushin and H. Alper, Transformations of chloroarenes, catalyzed by transition-metal complexes, Chem. Rev., 1994, 94, 1047–1062 CrossRef CAS.
- (a) A. Biffis, P. Centomo, A. Del Zotto and M. Zecca, Pd metal catalysts for cross-couplings and related reactions in the 21st century: a critical review, Chem. Rev., 2018, 118, 2249–2295 CrossRef CAS PubMed; (b) L.-C. Campeau and N. Hazari, Cross-coupling and related reactions: connecting past success to the development of new reactions for the future, Organometallics, 2019, 38, 3–35 CrossRef CAS PubMed.
- H. Li, C. C. C. Johansson Seechurn and T. J. Colacot, Development of preformed Pd catalysts for cross-coupling reactions, beyond the 2010 Nobel prize, ACS Catal., 2012, 2, 1147–1164 CrossRef CAS.
- F. Sabuzi, G. Pomarico, B. Floris, F. Valentini, P. Galloni and V. Conte, Sustainable bromination of organic compounds: a critical review, Coord. Chem. Rev., 2019, 385, 100–136 CrossRef CAS.
- W. Lorpaiboon and P. Bovonsombat, Halogen bond-induced electrophilic aromatic halogenations, Org. Biomol. Chem., 2021, 19, 7518–7534 RSC.
- T. Hering and B. König, Photocatalytic activation of N-chloro compounds for the chlorination of arenes, Tetrahedron, 2016, 72, 7821–7825 CrossRef CAS.
- X. Xiong and Y.-Y. Yeung, Highly ortho-selective chlorination of anilines using a secondary ammonium salt organocatalyst, Angew. Chem., Int. Ed., 2016, 55, 16101–16105 CrossRef CAS PubMed.
- Y. Zhang, K. Shibatomi and H. Yamamoto, Lewis acid catalyzed highly selective halogenation of aromatic compounds, Synlett, 2005, 18, 2837–2842 Search PubMed.
- G. K. S. Prakash, T. Mathew, D. Hoole, P. M. Esteves, Q. Wang, G. Rasul and G. A. Olah, N-halosuccinimide/BF3-H2O, efficient eloctrophilic halogenating systems for aromatics, J. Am. Chem. Soc., 2004, 126, 15770–15776 CrossRef CAS PubMed.
- S. Engbers, R. Hage and J. E. M. N. Klein, Toward environmentally benign electrophilic chlorinations: from chloroperoxidase to bioinspired isoporphyrins, Inorg. Chem., 2022, 61, 8105–8111 CrossRef CAS PubMed.
- (a) D. R. Morris and L. P. Hager, Chloroperoxidase: I. Isolation and properties of the crystalline glycoprotein, J. Biol. Chem., 1966, 241, 1763–1768 CrossRef CAS PubMed; (b) M. Sundaramoorthy, J. Terner and T. L. Poulos, The crystal structure of chloroperoxidase: a heme peroxidase-cytochrome P450 functional hybrid, Structure, 1995, 3, 1367–1378 CrossRef CAS PubMed; (c) P. M. Champion, E. Muenck, P. G. Debrunner, P. F. Hollenberg and L. P. Hager, Mössbauer investigations of chloroperoxidase and its halide complexes, Biochemistry, 1973, 12, 426–435 CrossRef CAS PubMed; (d) G. E. Krejcarek, R. G. Bryant, R. J. Smith and L. P. Hager, Broad-line nuclear magnetic resonance studies of chloroperoxidase, Biochemistry, 1976, 15, 2508–2511 CrossRef CAS PubMed; (e) P. F. Hollenberg, L. P. Hager, W. E. Blumberg and J. Peisach, An electron paramagnetic resonance study of the high and low spin forms of chloroperoxidase, J. Biol. Chem., 1980, 255, 4801–4807 CrossRef CAS PubMed; (f) M. Sono, J. H. Dawson and L. P. Hager, Ligand and halide binding properties of chloroperoxidase: peroxidase-type active site heme environment with cytochrome P-450 type endogenous axial ligand and spectroscopic properties, Biochemistry, 1986, 25, 347–356 CrossRef CAS PubMed; (g) R. Vázquez-Duhalt, M. Ayala and F. J. Márquez-Rocha, Biocatalytic chlorination of aromatic hydrocarbons by chloroperoxidase of Caldariomyces fumago, Phytochemistry, 2001, 58, 929–933 CrossRef PubMed; (h) R. D. Libby, N. S. Rotberg, J. T. Emerson, T. C. White, G. M. Yen, S. H. Friedman, N. S. Sun and R. Goldowski, The chloride-activated peroxidation of catechol as a mechanistic probe of chloroperoxidase reactions, J. Biol. Chem., 1989, 264, 15284–15292 CrossRef CAS PubMed; (i) K. Murali Manoj, Chlorinations catalyzed by chloroperoxidase occur via diffusible intermediates) and, the reaction components play multiple roles in the overall process, Biochim. Biophys. Acta, Biomembr., 2006, 1764, 1325–1339 CrossRef PubMed; (j) K. M. Manoj and L. P. Hager, Chloroperoxidase, a Janus enzyme, Biochemistry, 2008, 47, 2997–3003 CrossRef CAS PubMed; (k) R. D. Libby, J. A. Thomas, L. W. Kaiser and L. P. Hager, Chloroperoxidase halogenation reactions: chemical versus enzymic halogenating intermediates, J. Biol. Chem., 1982, 257, 5030–5037 CrossRef CAS PubMed.
- J. Niu and G. Yu, Molecular structural characteristics governing biocatalytic chlorination of PAHs by chloroperoxidase from caldariomyces fumago, SAR QSAR Environ. Res., 2004, 15, 159–167 CrossRef CAS PubMed.
- (a) R. Rutter, L. P. Hager, H. Dhonau, M. Hendrich, M. Valentine and P. Debrunner, Chloroperoxidase compound I: electron paramagnetic resonance and mössbauer studies, Biochemistry, 1984, 23, 6809–6816 CrossRef CAS PubMed; (b) C. M. Hosten, A. M. Sullivan, V. Palaniappan, M. M. Fitzgerald and J. Terner, Resonance Raman spectroscopy of the catalytic intermediates and derivatives of chloroperoxidase from caldariomyces fumago, J. Biol. Chem., 1994, 269, 13966–13978 CrossRef CAS PubMed; (c) K. L. Stone, R. K. Behan and M. T. Green, X-ray absorption spectroscopy of chloroperoxidase compound I: Insight into the reactive intermediate of P450 chemistry, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16563–16565 CrossRef CAS PubMed.
- (a) J. A. Thomas, D. R. Morris and L. P. Hager, Chloroperoxidase: VIII. Formation of peroxide and halide complexes and their relation to the mechanism of the halogenation reaction, J. Biol. Chem., 1970, 245, 3135–3142 CrossRef CAS PubMed; (b) H. B. Dunford, A.-M. Lambeir, M. A. Kashem and M. Pickard, On the mechanism of chlorination by chloroperoxidase, Arch. Biochem. Biophys., 1987, 252, 292–302 CrossRef CAS PubMed; (c) R. D. Libby, A. L. Shedd, A. K. Phipps, T. M. Beachy and S. M. Gerstberger, Defining the involvement of HOCl or Cl2 as enzyme-generated intermediates in chloroperoxidase-catalyzed reactions, J. Biol. Chem., 1992, 267, 1769–1775 CrossRef CAS PubMed; (d) R. D. Libby, T. M. Beachy and A. K. Phipps, Quantitating direct chlorine transfer from enzyme to substrate in chloroperoxidase-catalyzed reactions, J. Biol. Chem., 1996, 271, 21820–21827 CrossRef CAS PubMed.
- (a) M. Sundaramoorthy, J. Terner and T. L. Poulos, Stereochemistry of the chloroperoxidase active site: crystallographic and molecular-modeling studies, Chem. Biol., 1998, 5, 461–473 CrossRef CAS PubMed; (b) A. Timmins and S. P. De Visser, Enzymatic halogenases and haloperoxidases: computational studies on mechanism and function, in Advances in Protein Chemistry and Structural Biology, ed. T. Karabencheva-Christova, Academic Press, 2015, vol. 100, pp. 113–151 Search PubMed; (c) F. S. Brown and L. P. Hager, Chloroperoxidase: IV. Evidence of an ionic electrophilic substitution mechanism, J. Am. Chem. Soc., 1967, 89, 719–720 CrossRef CAS PubMed.
- (a) H.-A. Wagenknecht and W.-D. Woggon, Identification of intermediates in the catalytic cycle of chloroperoxidase, Chem. Biol., 1997, 4, 367–372 CrossRef CAS PubMed; (b) H.-A. Wagenknecht and W.-D. Woggon, New active-site analogues of chloroperoxidase - syntheses and catalytic reactions, Angew. Chem., Int. Ed. Engl., 1997, 36, 390–392 CrossRef CAS; (c) H.-A. Wagenknecht, C. Claude and W.-D. Woggon, New enzyme models of chloroperoxidase: Improved stability and catalytic efficiency of iron porphyrinates containing a thiolato ligand, Helv. Chim. Acta, 1998, 81, 1506–1520 CrossRef CAS; (d) W.-D. Woggon, H.-A. Wagenknecht and C. Claude, Synthetic active site analogues of heme−thiolate proteins. Characterization and identification of intermediates of the catalytic cycles of cytochrome p450cam and chloroperoxidase, J. Inorg. Biochem., 2001, 83, 289–300 CrossRef CAS PubMed; (e) S. M. Maddox, C. J. Nalbandian, D. E. Smith and J. L. Gustafson, A practical Lewis base catalyzed electrophilic chlorination of arenes and heterocycles, Org. Lett., 2015, 17, 1042–1045 CrossRef CAS PubMed.
- Z. Cong, T. Kurahashi and H. Fujii, Oxidation of chloride and subsequent chlorination of organic compounds by oxoiron(IV) porphyrin π-cation radicals, Angew. Chem., Int. Ed., 2011, 50, 9935–9939 CrossRef CAS PubMed.
- (a) Z. Cong, S. Yanagisawa, T. Kurahashi, T. Ogura, S. Nakashima and H. Fujii, Synthesis, characterization, and reactivity of hypochloritoiron(III) porphyrin complexes, J. Am. Chem. Soc., 2012, 134, 20617–20620 CrossRef CAS PubMed; (b) S. Yokota and H. Fujii, Critical factors in determining the heterolytic versus homolytic bond cleavage of terminal oxidants by iron(III) porphyrin complexes, J. Am. Chem. Soc., 2018, 140, 5127–5137 CrossRef CAS PubMed.
- J. P. Biswas, S. Guin and D. Maiti, Highvalent 3d metal-oxo mediated C-H halogenation: biomimetic approaches, Coord. Chem. Rev., 2020, 408, 213174 CrossRef CAS.
- (a) W. Liu and J. T. Groves, Manganese porphyrins catalyze selective C-H bond halogenations, J. Am. Chem. Soc., 2010, 132, 12847–12849 CrossRef CAS PubMed; (b) W. Liu and J. T. Groves, Manganese catalyzed C-H halogenation, Acc. Chem. Res., 2015, 48, 1727–1735 CrossRef CAS PubMed.
- W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Oxidative aliphatic C-H fluorination with fluoride ion catalyzed by a manganese porphyrin, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed.
- N. Sharma, Y.-M. Lee, X.-X. Li, W. Nam and S. Fukuzumi, Regioselective oxybromination of benzene and its derivatives by bromide anion with a mononuclear nonheme Mn(IV)-oxo complex, Inorg. Chem., 2019, 58, 14299–14303 CrossRef CAS PubMed.
- L. Zhang, M. S. Seo, Y. Choi, R. Ezhov, O. Maximova, D. D. Malik, M. Ng, Y.-M. Lee, R. Sarangi, Y. N. Pushkar, K.-B. Cho and W. Nam, A manganese compound I model with a high reactivity in the oxidation of organic substrates and water, J. Am. Chem. Soc., 2023, 145, 8319–8325 CrossRef CAS PubMed.
- (a) M. Guo, Y.-M. Lee, M. S. Seo, Y.-J. Kwon, X.-X. Li, T. Ohta, W.-S. Kim, R. Sarangi, S. Fukuzumi and W. Nam, Mn(III)-iodosylarene porphyrins as an active oxidant in oxidation reactions: synthesis, characterization, and reactivity studies, Inorg. Chem., 2018, 57, 10232–10240 CrossRef CAS PubMed; (b) L. Zhang, Y.-M. Lee, M. Guo, S. Fukuzumi and W. Nam, Unprecedented reactivities of highly reactive manganese(III)-iodosylarene porphyrins in oxidation reactions, J. Am. Chem. Soc., 2020, 142, 19879–19884 CrossRef CAS PubMed.
- (a) M. Guo, M. S. Seo, Y.-M. Lee, S. Fukuzumi and W. Nam, Highly reactive manganese(IV)-oxo porphyrins showing temperature-dependent reversed electronic effect in C-H bond activation reactions, J. Am. Chem. Soc., 2019, 141, 12187–12191 CrossRef CAS PubMed; (b) M. Guo, J. Zhang, L. Zhang, Y.-M. Lee, S. Fukuzumi and W. Nam, Enthalpy-entropy compensation effect in oxidation reactions by manganese(IV)-oxo porphyrins and nonheme iron(IV)-oxo models, J. Am. Chem. Soc., 2021, 143, 18559–18570 CrossRef CAS PubMed.
- (a) A. D. Adler, F. R. Longo, F. Kampas and J. Kim, On the preparation of metalloporphyrins, J. Inorg. Nucl. Chem., 1970, 32, 2443–2445 CrossRef CAS; (b) G. Li, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, Selective C-H halogenation with a highly fluorinated manganese porphyrin, Angew. Chem., Int. Ed., 2018, 57, 1251–1255 CrossRef CAS PubMed; (c) R. R. Gaughan, D. F. Shriver and L. J. Boucher, Resonance Raman spectra of manganese(III) tetraphenylporphin halides, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 433–436 CrossRef CAS PubMed; (d) G. W. Farley, M. A. Siegler and D. P. Goldberg, Halogen transfer to carbon radicals by high-valent iron chloride and iron fluoride corroles, Inorg. Chem., 2021, 60, 17288–17302 CrossRef CAS PubMed.
- (a) V. D. Parker, Y. T. Chao and G. Zheng, Dynamics of proton transfer from radical cations. Addition−elimination or direct proton transfer?, J. Am. Chem. Soc., 1997, 119, 11390–11394 CrossRef CAS; (b) H. Slebocka-Tilk, A. Neverov, S. Motallebi, R. S. Brown, O. Donini, J. L. Gainsforth and M. Klobukowski, Electrophilic bromination of specifically deuterated cyclohexenes: A combined experimental and theoretical investigation, J. Am. Chem. Soc., 1998, 120, 2578–2585 CrossRef CAS; (c) S. Bhosale, D. P. Sonune, U. V. Prasad and D. Bhuniya, Inverse kinetic isotope effect in MagtrieveTM mediated oxidation or deoximation of benzaldoxime: mechanistic implication, Tetrahedron Lett., 2012, 53, 1794–1797 CrossRef CAS; (d) S. Fukuzumi, Y.-M. Lee and W. Nam, Deuterium kinetic isotope effects as redox mechanistic criterions, Bull. Korean Chem. Soc., 2021, 42, 1558–1568 CrossRef CAS.
- (a) S. Bang, S. Park, Y.-M. Lee, S. Hong, K.-B. Cho and W. Nam, Demonstration of the heterolytic O-O bond cleavage of putative nonheme iron(II)-OOH(R) complexes for fenton and enzymatc reactions, Angew. Chem., Int. Ed., 2014, 53, 7843–7847 CrossRef CAS PubMed; (b) J. Chen, H. Yoon, Y.-M. Lee, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Tuning the reactivity of mononuclear nonheme manganese(IV)-oxo complexes by triflic acid, Chem. Sci., 2015, 6, 3624–3632 RSC.
- (a) S.-S. Xue, X.-X. Li, Y.-M. Lee, M. S. Seo, Y. Kim, S. Yanagisawa, M. Kubo, Y.-K. Jeon, W.-S. Kim, R. Sarangi, S. H. Kim, S. Fukuzumi and W. Nam, Enhanced redox reactivity of a nonheme iron(V)–oxo complex binding proton, J. Am. Chem. Soc., 2020, 142, 15305–15319 CrossRef CAS PubMed; (b) Y.-M. Lee, S. Kim, K. Ohkubo, K.-H. Kim, W. Nam and S. Fukuzumi, Unified mechanism of oxygen atom transfer and hydrogen atom transfer reactions with a triflic acid-bound nonheme manganese(IV)–oxo complex via outer-sphere electron transfer, J. Am. Chem. Soc., 2019, 141, 2614–2622 CrossRef CAS PubMed; (c) J. Park, Y. Morimoto, Y.-M. Lee, W. Nam and S. Fukuzumi, Unified view of oxidative C–H bond cleavage and sulfoxidation by a nonheme iron(IV)–oxo complex via Lewis acid-promoted electron transfer, Inorg. Chem., 2014, 53, 3618–3628 CrossRef CAS PubMed; (d) N. Sharma, Y.-M. Lee, X.-X. Li, W. Nam and S. Fukuzumi, Singly unified driving force dependence of outer-sphere electron-transfer pathways of nonheme manganese(IV)−oxo complexes in the absence and presence of Lewis acids, Inorg. Chem., 2019, 58, 13761–13765 CrossRef CAS PubMed; (e) J. Jung, S. Kim, Y.-M. Lee, W. Nam and S. Fukuzumi, Switchover of the mechanism between electron transfer and hydrogen-atom transfer for a protonated manganese(IV)–oxo complex by changing only the reaction temperature, Angew. Chem., Int. Ed., 2016, 55, 7450–7454 CrossRef CAS PubMed.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Pergamon Press, Oxford, 6th edn, 2009 Search PubMed.
- M. Guo, H. Dong, J. Li, B. Cheng, Y.-Q. Huang, Y.-Q. Feng and A. Lei, Spectroscopic observation of iodosylarene metalloporphyrin adducts and manganese(V)-oxo porphyrin species in a cytochrome P450 analogue, Nat. Commun., 2012, 3, 1190 CrossRef PubMed.
- (a) M. Ju, J. Kim and J. Shin, EPR spectroscopy: A versatile tool for exploring transition metal complexes in organometallic and bioinorganic chemistry, Bull. Korean Chem. Soc., 2024, 45, 835–862 CrossRef CAS; (b) J. Shin, M. H. Lim and J. Han, NMR spectroscopic investigations of transition metal complexes in organometallic and bioinorganic chemistry, Bull. Korean Chem. Soc., 2024, 45, 593–613 CrossRef CAS.
- W. Kohn and L. J. Sham, Self-consistent equations including exchange and correlation effects, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- (a) A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A:At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) A. D. Becke, Density-functional thermochemistry. I. The effect of the exchange-only gradient correction, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS; (c) A. D. Becke, A new mixing of Hartree-Fock and local density-functional theories, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS; (d) C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed; (e) S. H. Vosko, L. Wilk and M. Nusair, Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- (a) F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC; (b) F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- (a) V. Barone and M. Cossi, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- (a) D. G. Truhlar, C. J. Cramer, A. Lewis and J. A. Bumpus, Molecular modeling of environmentally important processes: reduction potentials, J. Chem. Educ., 2004, 81, 596 CrossRef; (b) P. Winget, C. J. Cramer and D. G. Truhlar, Computation of equilibrium oxidation and reduction potentials for reversible and dissociative electron-transfer reactions in solution, Theor. Chem. Acc., 2004, 112, 217–227 Search PubMed.
- J. Ho, A. Klamt and M. L. Coote, Comment on the correct use of continuum solvent models, J. Phys. Chem. A, 2010, 114, 13442–13444 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Kinetic data, spectroscopic data, cyclic voltammograms, product yields, energies, Mulliken spin density distribution, geometries and calculated coordinates. See DOI: https://doi.org/10.1039/d5qi00807g |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |