
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dibenzocyclooctatetraene based poly-Lewis-acids: flapping hosts for multidentate guests†
Maximilian J. Klingsiek ,
Julian Buth,
Pia C. Trapp,
Andreas Mix,
Jan-Hendrik Lamm,
Beate Neumann,
Hans-Georg Stammler and
Norbert W. Mitzel*
,
Julian Buth,
Pia C. Trapp,
Andreas Mix,
Jan-Hendrik Lamm,
Beate Neumann,
Hans-Georg Stammler and
Norbert W. Mitzel*
Chair of Inorganic and Structural Chemistry, Center for Molecular Materials CM2, Faculty of Chemistry, Bielefeld University, Universitätsstraße 25, Bielefeld 33615, Germany. E-mail: mitzel@uni-bielefeld.de
First published on 26th March 2025
Abstract
A synthetic route to flapping poly-Lewis acids (PLAs) based on dibenzocyclooctatetraene was developed. The reactivity of tetradentate boron and aluminium containing PLAs towards various Lewis bases (LB) including drugs was investigated in detail. By using different monodentate LBs, the different effective Lewis acidities (eLA) of the respective pairings were determined and compared with each other. The reaction with multidentate LBs yielded a range of adducts, which were analysed in solution by NMR methods, including DOSY NMR spectroscopy, and in the solid state by X-ray diffraction. The adducts include specific oligomers, and coordination polymers constructed via B–P, Al–P and Al–N interactions. The solid-state structures of these coordination polymers have been determined. The results of complexation of bidentate LB by the tetradentate PLA show that LB is preferentially chelated in the binding pockets, which does not impede the flapping movement during chelating complexation. This phenomenon can be attributed to a reduction in entropy when this flapping motion is lost. The aluminium containing PLAs were observed to form a complex with two caffeine molecules, exhibiting remarkable preference for oxygen functions. In contrast, the reaction with theobromine resulted in the degradation of the PLAs due to the presence of acidic protons in theobromine, leading to the formation of a dimeric aluminium containing theobromine derivative. These findings demonstrate the interaction of PLAs with drugs. They provide a foundation for the advancement of PLAs with a tuneable orientation of Lewis acidic functions.
Introduction
Poly-Lewis acids (PLAs) are molecules that are usually constructed from a donor-free organic scaffold with several Lewis acidic units. They are the phenomenological counterpart of poly-Lewis bases (e.g. crown ethers and cryptands), which can selectively complex monoatomic cations.1,2 Conversely, PLAs can complex small anions (H−, F−, Cl−, Br−, I−, HO−), in some cases selectively.3–9 They are also used in catalysis,10–14 in the recognition of small molecules15–23 and in the formation of supramolecular structures.20,24–27 The Lewis acid units of such PLAs can contain many different elements including silicon,6,18,28 tin,29 antimony3,8,9 or mercury.27,30 However, group 13 elements (B, Al, Ga, In) are commonly used as they are intrinsically electron deficient and do not necessarily require electron withdrawing substituents to generate high Lewis acidities.15–17,19–23,31–36 The scaffolds of PLAs have to be donor-free and are mostly based on hydrocarbons, often constructed from rigid aromatic systems (sometimes extended by alkynyl units) to ensure a defined distance between the acidic functions,3,19,24,33,34,37 but organosilane-based systems have also been used.20,28,38,39 In recent years, bidentate8,21,28,32,39,40 PLAs have been intensely investigated, but tri-,6,20,24,39 tetra-,3,41 penta-,42 hexa-19,20,38 and octadentate43 systems have also been reported. The flexibility of the PLAs produced varies, with rigid systems for example based on anthracene3,9,33,34 or TBTQ19,24 as well as (semi-)flexible systems based on silanes20,39 or ethane.32 However, the use of scaffolds that allow a controllable flexibility, or better, a controllable orientation of the Lewis acid units, has not been investigated in detail.For such PLAs, dibenzocyclooctatetraene (dbCOT) is an interesting backbone. The dbCOT is a non-aromatic compound that assumes a tub shape at equilibrium and undergoes inversion in solution by overcoming an energy barrier of 51 kJ mol−1.44,45 This property gives PLAs based on the dbCOT framework a certain degree of flexibility. In addition, the dbCOT system can be converted by a two-electron reduction or oxidation into Hückel aromatic compounds with planar structure.46–50 Additionally, a Baird aromatic compound (Baird's rule: triplet molecules with [4n] electrons in a cyclic planar through-conjugated system are particularly stable)51,52 can be generated by excitation of dbCOT-containing compounds, which are also planar until they revert to the ground state and return to the tub shape.53–55 We expect that dbCOT frameworks will provide access to PLAs with a switchable orientation of the Lewis acid functions.
Drover et al. presented an octadentate PLA comprising a nickel atom as a redox-active site, which forms a B–N coordination polymer upon reaction with 4,4′-bipyridine.43 These polymers have potential applications as functional materials, including electrical conductivity, magnetism, and optical applications.43,56–58 This could also be possible with the dbCOT system but without the use of transition metals.
Here we report the synthesis of dibenzocyclooctatetraene-based PLAs and investigate their general reactivity towards various bases. In addition to multidentate phosphanes and N-heterocycles, we also use commonly known biomolecules, such as caffeine, theobromine, nicotine and γ-butyrolactone in the complexation experiments.
Results and discussion
Synthesis of the poly-Lewis acids
The dibenzocyclooctatetraene (dbCOT) backbone was prepared via the one-pot reaction protocol reported by Saito et al.53 starting from 1,2-dibromo-4,5-bis(dibromomethyl)benzene (1) (Scheme 1). Next, the bromo substituents were replaced with trimethylsilyl-protected alkynyl spacers via a Sonogashira–Hagihara coupling to give [Me3SiCC]4-dbCOT (3) after six days in 43% yield. In this coupling, an addition of triphenylphosphane to the reaction mixture turned out to be beneficial in ensuring complete conversion. One potential explanation for this observation is the coordinating effect of the dbCOT system towards Pd,59 which may interfere with the reaction process. The added phosphane coordinates stronger than the dbCOT system, and therefore partially suppresses the formation of Pd·dbCOT species. Reduced or no addition of phosphane afforded the double-alkynylated systems as the main products after 15 days. | ||
| Scheme 1 Synthesis of the four dibenzocyclooctatetraene PLAs 6–9 starting from hexabrominated xylene 1. The colour coding of the B and Al atoms used is consistent with the coding in the solid-state structures. | ||
The deprotection of the trimethylalkynyl units using potassium hydroxide resulted in the formation of the free tetraalkyne 4 with a yield of 90%. In hydrometallation reactions, [HCC]4-dbCOT (4) can be converted into tetradentate PLAs with flexible vinyl spacers. To obtain PLAs with more rigid alkynyl spacers, we employed the frequently used route via tin element exchange. The [Me3SnCC]4-dbCOT (5) required for this exchange was generated by the quantitative reaction of 4 with dimethylaminotrimethylstannane under amine elimination. Single crystals of each of the PLA precursors 1–5 were obtained, which were suitable for determining the solid-state structures by X-ray diffraction (for 1–3 see ESI S168−S170†). In the solid state, the structures of 4 and 5 (Fig. 1) show bond lengths and angles within the typical range for such compounds, with the greatest variance observed in the bending angle (θ), defined as the angle between the base surface and the side wings of the dbCOT unit (Fig. 1). The angle is largely dependent on steric and packing effects. The tetraalkyne 4 has a relatively small bending angle of 37.3(1)° which might be due to attractive intermolecular dispersion interactions between the tetraalkynes 4 in the crystal which contains stacks of molecules by their aryl surfaces. In contrast, the molecules of 5 show no such stacking and the bend angles of 5 (θ = 43.2(1) resp. 43.7(1)°) are comparable to the angle of unsubstituted dbCOT (θ = 43.5(1)°).46 The conversion of tetraalkyne 4 into the vinyl-containing PLAs [Cy2BCHCH]4-dbCOT (6) and [Bis2AlCHCH]4-dbCOT (7) was achieved in quantitative hydrometallation reactions using dicyclohexylborane (HBCy2) or bis-(bis(trimethylsilyl)methyl)alane (HAlBis2) (Scheme 1). A crystal of the free PLA 6 suitable for X-ray diffraction showed three molecules in the asymmetric unit (Fig. 2). The bending angles of the three [Cy2BCHCH]4-dbCOT (6) molecules are between 46.3(2) and 48.6(2)°. The distance between the boron atoms bound to one of the side wings (binding pocket A) is 6.276(5)–7.052(4) Å, whereas the distance across the cavity between the two different side wings (binding pocket B) is 9.360(4)–10.849(5) Å (Fig. 2). The cavity spanned by the diagonal Lewis acid functions (binding pocket C) is 11.800(4)–12.602(6) Å in size. This gives a first impression of the size and flexibility of the binding pockets in which different basic molecules can be complexed.
 | ||
| Fig. 1 Molecular structures of 4 and 5 in the crystalline state. Displacement ellipsoids drawn at 40% probability level. Minor occupied disordered atoms and H atoms are omitted for clarity. Selected distances [Å] and angles [°]: 4: C(8)–C(7) 1.324(7), C(7)–C(4) 1.474(7), C(4)–C(5) 1.395(7), C(4)–C(3) 1.408(7), C(3)–C(2) 1.392(7), C(2)–C(1) 1.407(7), C(1)–C(9) 1.435(7), C(9)–C(10) 1.184(8), C(8)–C(7)–C(4) 130.7(5), C(7)–C(4)–C(3) 116.2(4), C(2)–C(1)–C(9) 121.1(5), C(1)–C(9)–C(10) 179.3(6), θ 37.3(1), symmetry code: 1 − x, −y, +z; 5: C(6)–C(5) 1.327(3), C(5)–C(4) 1.480(3), C(4)–C(15) 1.406(3), C(1)–C(17) 1.440(3), C(17)–C(18) 1.203(3), C(18)–Sn(1) 2.113(2), C(6)–C(5)–C(4) 127.5(2), C(1)–C(17)–C(18) 176.4(2), C(17)–C(18)–Sn(1) 171.6(2), θ 43.2(1)/43.7(1). The illustration in the bottom right-hand corner provides an illustration of the meaning of the bending angle θ. | ||
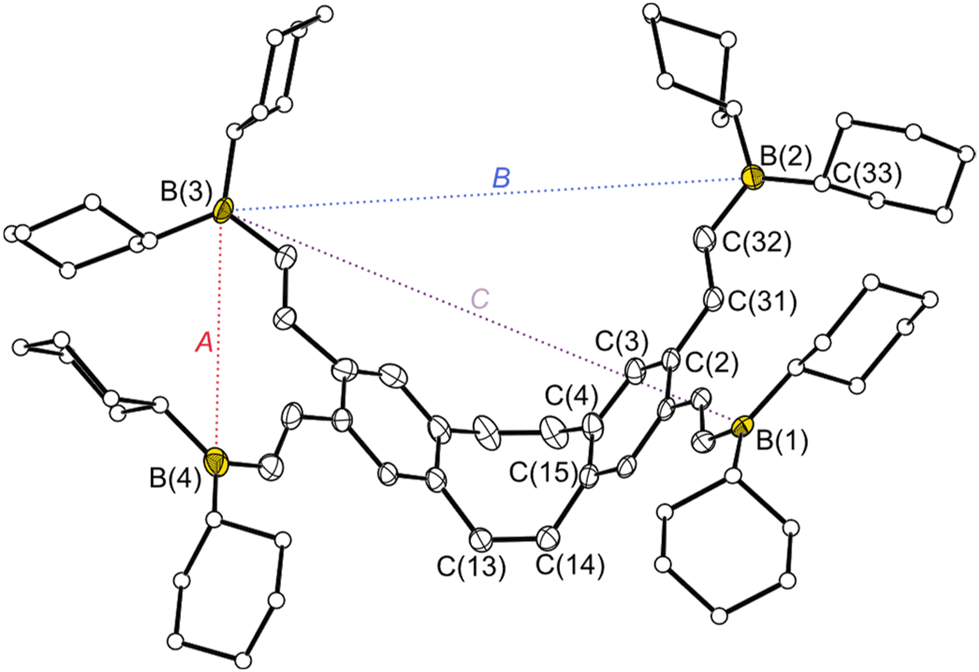 | ||
| Fig. 2 Molecular structure of [Cy2BCHCH]4-dbCOT 6 in the crystalline state. Displacement ellipsoids drawn at the 40% probability level. For reasons of clarity, only one of the three molecular structures is shown, minor occupied disordered atoms and H atoms are omitted and the carbon atoms of the cyclohexyl groups are only shown as spheres. The different binding pockets A, B and C which are spanned by the boron units are marked with dashed coloured lines (red: binding pocket A, blue: binding pocket B, purple: binding pocket C). Selected distances [Å] and angles [°]: B(1)⋯B(2) 6.953(4), B(3)⋯B(4) 6.342(5), B(1)⋯B(4) 9.360(4), B(3)⋯B(2) 10.530(4), B(2)⋯B(4) 12.069(5), B(1)⋯B(3) 11.800(4), C(13)–C(14) 1.337(4), C(14)–C(15) 1.484(3), C(15)–C(4) 1.407(3), C(4)–C(3) 1.401(3), C(3)–C(2) 1.391(4), C(2)–C(31) 1.475(4), C(31)–C(32) 1.345(4), C(32)–B(2) 1.564(4), C(13)–C(14)–C(15) 124.5(2), C(15)–C(4)–C(3) 118.4(2), C(2)–C(31)–C(32) 127.1(2), C(31)–C(32)–B(2) 123.3(3), C(32)–B(2)–C(33) 119.6(2), θ 46.3(2)/48.6(2). | ||
More rigid PLAs with alkynyl groups linking the Lewis acid functions to the dbCOT skeleton were synthesised via tin element exchange reactions starting from 5. The exchange using ClBEt2 and HAlBis2 proceeded without side reactions and afforded 8 and 9 in excellent yields (Scheme 1, HAlBis2 is preferred over ClAlBis2, as it reacts more cleanly and is easier accessible). The reaction with ClBCy2 gave access to a system with four –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–BCy2 units (related to the –CH
C–BCy2 units (related to the –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–BCy2 units in the vinyl system 6), however, in a less clean way and with non-separable decomposition products than the reaction with ClBEt2. Therefore, the ethyl substituents were used instead of the cyclohexyl substituents despite the more limited comparability of the systems 6 and 8. Other exchange reactions were also investigated in more detail as part of this study (see ESI†).
CH–BCy2 units in the vinyl system 6), however, in a less clean way and with non-separable decomposition products than the reaction with ClBEt2. Therefore, the ethyl substituents were used instead of the cyclohexyl substituents despite the more limited comparability of the systems 6 and 8. Other exchange reactions were also investigated in more detail as part of this study (see ESI†).
Insights into the Lewis acidity of the PLAs
The prepared PLAs 6–9 were reacted with a range of monodentate neutral guests containing basic C, N, P or O atoms. For these studies, the PLAs were reacted with an excess of the respective base in C6D6 and analysed by NMR spectroscopy. In addition, the resulting mixture was stripped of all volatile components in a vacuum, dissolved and analysed again by NMR spectroscopy. This showed that some of the interactions are so weak that the Lewis base (LB) can be removed under reduced pressure despite interacting with the PLAs, while in other cases exactly four equivalents of the base remain in the mixture, indicating a stronger interaction.As shown in previous work,60,61 the Lewis acidity of a compound and the interaction between LA and LB depend on a variety of parameters (atom type, Hard and Soft Acids and Bases concept (HSAB), steric requirements, reorganisation energy, London dispersion attraction, Pauli repulsion, etc.).62,63 Furthermore, established spectroscopic test methods such as the Childs64 or Gutmann–Beckett65,66 tests only allow a valid classification of LAs of the same type (e.g. arylboranes67) and only consider the interaction of the LAs with the LB used for the test, which usually contain oxygen.
To estimate the extent to which the PLAs 6–9 interact with LBs, different combinations were analysed by NMR spectroscopy and the strength of the interaction was determined from the shifts of the base signals relative to those of the free base as well as the shift of the 11B NMR signal of PLA 8 and the corresponding adducts 8·4LB. This allowed a semi-quantitative classification of the interactions between LB and LA, below referred to as effective Lewis acidity (eLA), following the work of Greb et al.63 The eLAs were determined by first ranking the interaction of PLA 8 with all tested monodentate LB based on the shift of the 11B resonance of the 8·4LB adducts relative to the shift of 8. These rankings resulted in ratios or factors for the different strengths of the interaction. In the second step, the shifts of selected base signals depending on the interaction with the different PLAs 6–9 were analysed. Again, a sorting was applied from the weakest to the strongest shift relative to the signal of the free LB. The resulting relative shifts for a specific LB were then set in relation to the shift determined in the first step, so that a comparison between the different series was possible (for more details and illustrations see ESI†). In this way we obtained the semi-quantitative classification of the different interactions shown in Fig. 3. A relative eLA of 0 means that no interaction was observed by NMR spectroscopy.
 | ||
| Fig. 3 Semiquantitative classification of the relative effective Lewis acidity (eLA) between the PLAs and the tested monodentate bases. 6 decomposed during the reaction with tBuNC, therefore this interaction could not be ranked. | ||
The coloured areas are based on empirical observations. In the yellow area, the interaction is weak, as indicated by the small shifts of the observed NMR signals, and the bases can be completely removed under reduced pressure. In the orange and red areas, on the other hand, exactly four equivalents of the LB are found after heating to 60 °C under reduced pressure followed by dissolving the residue. In the red area, the shifts are the strongest. When there is an excess of base, it is usually possible to distinguish between the bound and free base by two separate sets of NMR signals. This indicates strong adduct formation which does not allow the free and bound molecules to exchange in solution at room temperature on the time scale of the NMR measurement. For comparison, the orange region shows interactions where no distinction can be made between free and bound base, as rapid exchange occurs in solution due to the weaker interaction.
This study shows that the PLAs 7–9 interact strongly with the strong LB tBuNC, resulting in comparatively high eLA, while decomposition occurs with 6. Furthermore, the alkynyl-containing PLAs 8 and 9 show higher eLA with the nitrogen LB than the vinyl-containing PLAs (6 and 7). It is also evident that the interactions between 9 and the nitrogen LB are the strongest of all the interactions analysed. It could also be seen that there are preferred pairings between the interacting units. For example, the aluminium atoms are more oxophilic than the boron atoms and therefore the Al-containing PLAs 7 and 9 interact more strongly with THF and Et3PO than the boron-containing PLAs 6 and 8, which is reflected in a higher eLA. In contrast, the B–P interaction is more favourable than the Al–P interaction. It is also generally true that 6 has the weakest interaction with all bases except PMe3, which is an indication of the low Lewis acidity of the CHCHBCy2 unit. In general, the PLAs with alkynyl spacers, 8 and 9, show a stronger interaction with the respective base than their counterparts 6 and 7 with their vinyl spacers. This implies a higher Lewis acidity of the Lewis acidic units induced by the alkyne spacer.
This experimental trend was also confirmed by calculated ion affinities (Table 1) of models representing the individual LA functions contained in the PLAs. The hydride (HIA) and fluoride ion affinities (FIA) of the model compounds PhCHCHBCy2, PhCCBCy2, PhCHCHAlBis2 and PhCCAlBis2 were calculated using established methods68,69 and allow the analysis of the intrinsic Lewis acidities (iLA).63 The calculated values also show an increased acidity of the alkynyl systems compared to the vinyl systems. Furthermore, the AlBis2 containing systems have higher ion affinities than the BCy2 containing systems. Note that the ionic affinities of PhCCBCy2 were calculated instead of the PhCCBEt2 units used to improve the comparability of the calculated values. The apparently higher Lewis acidity of the alkynyl-containing systems can be explained by the higher group electronegativity of the alkynyl compared to the vinyl group,70 but other effects such as lower repulsion or reorganisation energy during adduct formation may also play a role for the eLA. In some cases, the reactivity studies with the monodentate guests yielded single crystals suitable for structure determination by X-ray diffraction. All solid state structures are in agreement with the NMR spectroscopic results and show the formation of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 adducts (host:guest). The bond lengths and angles of 6·4PMe3, 7·4Py, 9·4Py and 9·4PMe3 (see below) are in the typical range for such compounds as well as for the structures of 7·4THF, 7·4CNtBu, 7·4OPEt3, 8·4PMe3, 8·4NMe3 and 9·4OPEt3, which are described in the ESI (Fig. S171–S176†).
:4 adducts (host:guest). The bond lengths and angles of 6·4PMe3, 7·4Py, 9·4Py and 9·4PMe3 (see below) are in the typical range for such compounds as well as for the structures of 7·4THF, 7·4CNtBu, 7·4OPEt3, 8·4PMe3, 8·4NMe3 and 9·4OPEt3, which are described in the ESI (Fig. S171–S176†).
| Lewis acid function | HIA/kJ mol−1 | FIA/kJ mol−1 |
|---|---|---|
| PhCHCHBCy2 | 307 | 308 |
| PhCCBCy2 | 323 | 314 |
| PhCHCHAlBis2 | 347 | 434 |
| PhCCAlBis2 | 355 | 439 |
Interaction of the PLAs with multidentate Lewis-bases
The PLAs were reacted with multidentate LB (Schemes 2–4) to investigate the complexation behaviour of the PLAs in more detail. The NMR spectroscopic studies are consistent with the eLA study described above, so that the shift of the NMR signals after complexation of multidentate LB is comparable to the shifts occupied by the complexation of comparable monodentate LB. In addition, some of the adducts formed were analysed by single crystal X-ray diffraction experiments as well as in solution by diffusion ordered spectroscopy. | ||
| Scheme 2 Complexation of the (multidentate) phosphanes: PMe3, BisPhos and Bis[BisPhos] by the PLAs 6–9. | ||
 | ||
| Scheme 3 Complexation of the (multidentate) N-heterocyclic ligands: pyridine (Py), bisimidazolylmethane (N-1), bistriazolylethane (N-2) and bistriazolylbutane (N-4) by the PLAs 6–9. | ||
 | ||
| Scheme 4 Complexation of the drugs: γ-butyrolactone (GBL), nicotine (Nico), theobromine and caffeine (Caff) by the PLAs 6–9. R = –CCH (4), –CHCH2 (11). | ||
Interaction of the PLAs with multidentate phosphanes
Based on the determined eLA of 6 with PMe3, this PLA was reacted with bi- (BisPhos) and tetradentate (Bis[BisPhos]) phosphane bases (Scheme 2). NMR spectra show the reaction of 6 with BisPhos to afford an adduct and are consistent with the shifts of the related 6·4PMe3. However, the interaction with BisPhos results in a slightly stronger shift of the 11B resonance (6·4PMe3: Δδ(11B) = δ(11B)6 − δ(11B)adduct = 83 ppm, 6·2BisPhos: Δδ(11B) = 88 ppm) and 31P signals (6·4PMe3: Δδ(31P) = 43 ppm, 6·2BisPhos: Δδ(31P) = 46 ppm), indicating a stronger interaction.In contrast, the reaction with the ethyl-bridged BisPhos dimer (Bis[BisPhos]) resulted in a non-selective adduct formation and an adduct mixture, observed by several sets of signals. The determined solid state structures of 6·4PMe3 and the BisPhos adduct [6·2BisPhos]2 are shown in Fig. 4. 6·4PMe3 shows the B:P adduct in the ratio 1:1. As expected, BisPhos acts as a bridge between the boron atoms, but surprisingly both intramolecularly in the long binding pocket B (Fig. 2) and intermolecularly. In [6·2BisPhos]2 a 2:4 adduct has formed between 6 and BisPhos in the solid state, with two BisPhos units being chelated and two BisPhos units bridging connecting two such [6·BisPhos] units. The bond lengths and angles of the dbCOT unit are comparable to those of the free PLA 6, although the bending angles in 6·4PMe3 (42.7(2)° and 42.9(2)°) are smaller than in 6 (46.3(2)° and 48.6(2)°), whereas the dimer [6·2BisPhos]2 has comparable bending angles of 46.6(1)° and 48.3(1)°. This is due to the additional repulsion caused by the complexed PMe3 molecules pointing into the cavity. This repulsion also leads to larger B⋯B distances in the binding pocket A and results from the flexible vinyl units being aligned as far apart as possible. In comparison, [6·2BisPhos]2, with its flexible vinyl spacer, allows to bring B(1) and B(2) closer together (9.193(3) Å) and can thus complex a BisPhos molecule in the binding pocket B. The distance in C (11.594(3)–12.405(3) Å) also seems far too large to be bridged by BisPhos. The BisPhos molecule also adapts to cavity B due to its flexibility: BisPhos in binding pocket B has a stretched P(1)⋯P(2) distance of 6.371(1) Å, while the connecting BisPhos has a P(3)⋯P(4) distance of 5.140(1) Å. This demonstrates the affinity of both molecules, PLA and LB, to form the 2:4 adduct.
 | ||
| Fig. 4 Molecular structure of 6·4PMe3 and [6·2BisPhos]2 in the crystalline state. Displacement ellipsoids drawn at 40% probability level. For reasons of clarity, minor occupied disordered atoms and H atoms are omitted and the carbon atoms of the cyclohexyl groups are only shown as spheres or are omitted. Selected distances [Å] and angles [°]: 6·4PMe3: B(1)⋯B(3) 12.795(5), B(4)⋯B(2) 13.090(5), B(1)⋯B(2) 10.689(2), B(3)⋯B(4) 10.680(2), B(1)⋯B(4) 7.380(2), B(3)⋯B(2) 7.234(1), B(1)–P(1A) 2.076(10), B(2)–P(2) 2.003(3), B(3)–P(3) 2.000(4), B(4)–P(4) 2.002(3), B(1)–C(18) 1.607(5), C(17)–C(18) 1.324(5), C(1)–C(17) 1.468(4), C(1)–C(17)–C(18) 127.5(3), C(17)–C(18)–B(1) 131.9(3), C(18)–B(1)–P(1A) 101.9(3), θ 42.7(2)/42.9(2); [6·2BisPhos]2: B(1)⋯B(3) 11.594(3), B(2)⋯B(4) 12.405(3),B(1)⋯B(2) 9.193(3), B(3)⋯B(4) 9.957(13), B(1)⋯B(4) 7.404(3), B(3)⋯B(2) 7.155(3), P(1)⋯P(2) 6.371(1), P(3)⋯P(4) 5.140(1), B(1)–P(1) 2.034(2), B(2)–P(2) 2.046(2), B(3)–P(3) 2.029(2), B(4)–P(4) 2.034(2), B(1)–C(18) 1.617(3), C(17)–C(18) 1.335(3), C(1)–C(17) 1.482(2), P(1)–C(75) 1.823(2), C(75)–Si(1A) 1.894(2), Si(1A)–C(78) 1.900(2), P(3)–C(83) 1.812(2), C(83)–Si(2) 1.894(2), Si(2)–C(86) 1.896(2), C(1)–C(17)–C(18) 125.5(3), C(17)–C(18)–B(1) 129.7(2), C(18)–B(1)–P(1) 96.9(1), Si(1A)–C(75)–P(1) 123.9(1), C(78)–Si(1A)–C(75) 102.3(1), P(3)–C(83)–Si(2) 124.9(1), C(83)–Si(2)–C(86)114.7(1), θ 46.6(1)/48.3(1), symmetry code: 1 − x, 1 − y, 1 − z. | ||
To investigate whether the dimer was already formed in solution or only during crystallisation, a CDCl3 solution of 6 with two equivalents of BisPhos was studied by diffusion ordered spectroscopy (DOSY). From the measured diffusion coefficients, we calculated the hydrodynamic radii and the hydrodynamic volumes (VH) of the individual components and the adduct formed (Table 2). They allow us to specify the stoichiometry of the adduct and whether one or more different adducts have been formed. The DOSY measurements indicate that the dimer [6·2BisPhos]2 is already present in solution and that neither a 1:2 adduct nor larger oligomers have been formed. The VH determined for [6·2BisPhos]2 is equal to the sum of the volumes of the individual compounds (2·VH,6 + 4·VH,BisPhos = 5904 Å3) and also agrees with the volume determined from the solid state structure (VXRD). This result suggests that the formation of a 1:2 adduct with both BisPhos complexed in B is unfavourable due to the repulsion between the SiMe2 units. In contrast, the formation of the dimer appears to be favoured over the complexation of the two LBs in A to form a 1:2 adduct.
| Compound | VvdW/Å3 | VH/Å3 | VXRD/Å3 |
|---|---|---|---|
| BisPhos | 257 | 262 | — |
| 6 | 1401 | 2428 | 1655 |
| [6·2BisPhos]2 | 3783 | 5858 | 5541 |
From the NMR spectroscopic investigations we can conclude that PLA 8 interacts with PMe3 and BisPhos under formation of the adducts 8·4PMe3 and 8·2BisPhos but forms an adduct mixture with Bis[BisPhos].
For 8·4PMe3 and 8·2BisPhos there is only one characteristically shifted signal in the 31P and 11B NMR spectra. In contrast, the reaction with Bis[BisPhos] gives a mixture of products that shows many partially overlapping signals in the NMR spectra. These signals are in the same chemical shift range as the signals of the other two adducts, indicating the formation of a mixture of different adducts.
8·4PMe3 was analysed by X-ray diffraction experiments (ESI S174†), but it was not possible to obtain suitable single crystals of 8·2BisPhos, so it is not possible to say with any certainty whether the BisPhos is complexed in the A or in B cavity of the host. However, due to the large B⋯B distances in the pocket B (12.145(11) Å and 11.969(11) Å) compared to the distance in A (6.269(11) Å and 6.643(11) Å) in 8·4PMe3, the complexation of BisPhos by 8 in binding pocket B seems unlikely. We assume that BisPhos is complexed in binding pocket A, analogous to the aluminium homologue 9·2BisPhos described below.
Despite the relatively weak interaction of the Al-PLAs (7, 9) with PMe3, they were also reacted with BisPhos and Bis[BisPhos]. In these experiments, 7 shows very weak to no interaction with the tested phosphanes, which is expressed in very small to no shifts of the 1H, 29Si and 31P NMR signals compared to the signals of the individual components. These small shifts indicate that the reaction equilibrium is tilted towards the free species rather than the adduct. It was not possible to obtain crystals suitable for solid-state structure determination from these mixtures.
The phosphanes interacted preferentially with the stronger PLA 9, but the extremely broadened signals in the 31P and 1H NMR spectra (see ESI S105–S114†) indicated a highly dynamic behaviour. Nevertheless, the adducts 9·2BisPhos and [9·Bis[BisPhos]]n crystallised from the solutions (Fig. 5). The complexation of the BisPhos molecules in cavity A is consistent with a known BisPhos adduct19 and comparable in terms of bond lengths and angles. The formation of a polymer chain through the interaction with Bis[BisPhos] could be attributed to the highly dynamic behaviour in solution, whereby the uniform coordination polymer can build up in solution until it crystallises. The fact that two of the phosphorus atoms of Bis[BisPhos] are attached intermolecularly, forming a polymer chain instead of a 1:1 adduct, could be due to a too short distance between the P atoms in the guest molecules. The intramolecular Al⋯Al distance between the opposing aluminium atoms is 10.884(1)/11.000(1) Å and the intermolecular distance (bridged by Bis[BisPhos]) is 9.986(1)/10.082(1) Å. Furthermore, the bending angles of [9·Bis[BisPhos]]n of 46.7(1)° and 46.1(1)° are already wider than in the other adducts (9·4PMe3: 43.7(2)°, 9·2BisPhos: 43.4(2)° and 42.9(2)°), which therefore also have larger Al⋯Al distances. This finding could indicate that further folding of the system, which reduces the Al⋯Al distance and increases the bending angle, appears to be less energetically favourable than the formation of a polymer.
 | ||
| Fig. 5 Molecular structure of 9·4PMe3, 9·2BisPhos and a section from the polymer [9·Bis[BisPhos]]n in the crystalline state. Displacement ellipsoids drawn at 40% probability level. Minor occupied disordered atoms, methyl groups and H atoms are omitted for clearly. Selected distances [Å] and angles [°]: 9·4PMe3: Al(1)⋯Al(1′) 14.034(3), Al(2)⋯Al(2′) 14.034(3), Al(1)⋯Al(2) 8.127(2), Al(1)⋯Al(2′) 11.386(2), Al(1)–P(1A) 2.485(2), Al(2)–P(2) 2.528(2), Al(2)–C(26) 1.972(5), C(26)–C(25) 1.208(6), C(25)–C(8) 1.440(6), C(8)–C(25)–C(26) 171.6(4), C(25)–C(26)–Al(2) 168.8(4), C(26)–Al(2)–P(2) 88.3(1), θ 43.7(1), symmetry code: 1/2 − x, 3/2 − y, +z; 9·2BisPhos: Al(1)⋯Al(3) 14.162(1), Al(2)⋯Al(4) 13.343(1), Al(1)⋯Al(2) 11.217(1), Al(3)⋯Al(4) 11.176(1), Al(1)⋯Al(4) 8.084(1), Al(2)⋯Al(3) 7.916(1), P(1)⋯P(2) 5.696(1), P(3)⋯P(4) 5.776(1), Al(1)–P(1) 2.527(1), Al(2)–P(3) 2.540(1), Al(3)–P(4) 2.509(1), Al(4)–P(2) 2.495(1), Al(1)–C(18) 1.975(3), C(18)–C(17) 1.212(4), C(17)–C(1) 1.444(3), P(1)–C(83) 1.817(3), C(83)–Si(17) 1.889(3), Si(17)–C(86) 1.890, C(86)–P(2) 1.813(3), C(1)–C(17)–C(18) 169.3(3), C(17)–C(18)–Al(1) 176.6(2), C(18)–Al(1)–P(1) 95.6(1), P(1)–C(83)–Si(17) 124.2(2), C(83)–Si(17)–C(86) 103.4(1), θ 42.9(2)/43.4(2); [9·Bis[BisPhos]]n: Al(1)⋯Al(2) 7.691(1), Al(3)⋯Al(4) 7.983(1), Al(1)⋯Al(4) 11.000(1), Al(2)⋯Al(3) 10.884(1), Al(1)⋯Al(2′) 10.082(1), Al(3)⋯Al(4′) 9.986(1), P(1)⋯P(2) 5.467(1), P(3)⋯P(4) 5.498(1), P(3)⋯P(4′) 8.256(1), P(1)⋯P(2′) 8.204(1), Al(1)–P(1) 2.497(1), Al(2)–P(2) 2.518(1), Al(3)–P(3) 2.512(1), Al(4)–P(4) 2.508(1), C(9)–C(49) 1.439(2), C(49)–C(50) 1.213(2), C(50)–Al(3) 1.971(2), C(9)–C(49)–C(50) 168.6(2), C(49)–C(50)–Al(3) 169.2(1), C(50)–Al(3)–P(3) 91.2(1), θ 46.0(1)/46.6(1), symmetry codes: ‘ −x, 1 − y, 1 − z; “1 − x, 2 − y, −z. | ||
The complexation experiments carried out with phosphanes show that the boron-containing PLAs 6 and 8 interact stronger with the phosphorus atoms than the Al-containing PLAs 7 and 9. Furthermore, when comparing the interactions of the two Al-containing PLAs with the LBs, it is apparent that stronger interactions occur due to the alkyne functions rather than the vinyl functions and this is in line with the reactivity study described above. In addition, the solid state structures obtained show that BisPhos and Bis[BisPhos] are chelated in the rigid binding pocket A of 9.
Interaction of the PLAs with multidentate N-heterocycle ligands
Due to the high eLA of all PLA towards pyridine (Py), bidentate N-heterocyclic LBs of different sizes, bisimidazolylmethane (N-1), bistriazolylethane (N-2) and bistriazolylbutane (N-4) (Scheme 3) were used to investigate the complexation modes concerning the different binding pockets. To find out what size a guest must have to be complexed via the cavity (binding pocket B), similar to [6·2BisPhos]2, and whether it is possible to complex two such N-LBs in this binding pocket.NMR studies show that 6 reacts selectively with pyridine to form the 6·4Py adduct. A mixture of different adducts is formed with the multidentate nitrogen bases N-1, N-2 and N-4. The interaction can be seen from the 11B NMR signal, which undergoes a high field shift after the addition of the N-heterocyclic ligands. The signal is now at chemical shifts comparable to those of 6·4Py, indicating tetra- coordination at the boron atoms. However, there is a strong broadening of the signal and also several broad, partially overlapping signals in the 1H NMR spectra, making the formation of an adduct mixture realistic. The more rigid boron containing PLA 8 reacted similarly with N-1 and N-4, forming an adduct mixture. However, the reaction with N-2 did not lead to the formation of a mixture, so the 1H NMR spectra show a shifted signal set for 8 and N-2, indicating the formation of a specific adduct. As no crystals suitable for X-ray diffraction measurements could be obtained, it is not possible to determine the connectivity within the adduct in the solid-state and to decide in which binding pocket N-2 is bound and whether it is a monomeric or oligomeric species. However, a potential explanation for this observation is that N-2 has an appropriate B⋯B distance for one of the two binding pockets (8·4PMe3†: A 6.295(11)/6.643(11), B 11.969(11)/12.147(11) Å; 8·4NMe3†: A 6.020(7)/6.585(4), B 12.646(4)/12.564(9) Å). We suggest that N-1 has an insufficient distance between the nitrogen domains to be captured in a chelating manner in one of the cavities. Similarly, N-4 is too large for A but too small for B, whereas N-2 is likely to fit well within cavity A in terms of size. This could explain the formation of adduct mixtures using N-1 and N-4.
In preliminary test reactions, the aluminium-containing PLAs (7, 9) formed distinct adducts with the multidentate N-heterocycle ligands that crystallised rapidly from C6D6. Therefore, preparative scale complexation reactions were carried out in polar solvents (o-difluorobenzene, THF) and these solvents were also used for spectroscopic analysis after crystallisation. The formation of adducts in THF is in fact exchange experiments, as two THF molecules are displaced for each complexed nitrogen-LB. Furthermore, despite the presence of a large excess of THF, we observed no exchange of the guest. This is due to the higher eLA of the pairs of the aluminium-PLAs and the nitrogen-LBs (see Fig. 1). In addition, a chelate-like effect may also play a role in ensuring that the bidentate LBs are not displaced by THF. The NMR spectroscopic studies show the expected significant shifts in each case, and the signals of the free LB are also observed when an excess of LB is used. The NMR spectra thus indicate the clean formation of stable 1:2 adducts (7·2N-1, 7·2N-2, 7·2N-4, 9·2N-1, 9·2N-2). Only for the reaction of 9 with bistriazolylbutane (N-4) the number of available signal sets suggest the formation of an adduct in which the two base molecules are each complexed in different ways or a mixture of different adducts was formed (NMR spectra see ESI S128–S130†).
To verify whether the adducts are stable in solution at room temperature and not subject to dynamic exchange, which is already indicated by the presence of signal sets of the free base when an excess is used, DOSY measurements were carried out. Therefore, 7·4Py was analysed in the presence of an excess of pyridine at 20 °C and at 50 °C in DOSY measurements. 7·4Py was chosen as a model because 7 is the aluminium-PLA with the lower Lewis acidity compared to 9, according to the above study, and pyridine was chosen as a weaker base representative of the imidazolyl and triazolyl units used in N-1, N-2 and N-4, so that all the stronger interacting adducts reported below are also expected to exist non-dissociated in solution. The DOSY measurements show that 7·4Py is a stable adduct at room temperature and even at 50 °C only a small exchange of the complexed pyridines with the free ones can be observed (see ESI†). It can therefore be assumed that all the solid-state structures obtained in Fig. 6 correspond to the adducts present in solution at room temperature. With the exception of 7·2N-1, the binding pockets in which the nitrogen-LB were complexed could be seen from the solid state structure determined by X-ray diffraction of suitable single crystals (Fig. 6). The vinyl-containing PLA 7 complexes all nitrogen-LB (N-1, N-2, N-4) in its binding pocket A. Due to the vinyl units and the associated flexibility, the binding pockets can vary in size and thus adapt to the guest molecule. This results in different Al⋯Al distances: the rather large base N-4 expands the binding pocket A up to 8.332(5) Å, whereas the adduct 7·2N-2 with the smaller guest N-2 only shows a distance of 7.642(5)/7.669(5) Å.
 | ||
| Fig. 6 Molecular structure of 7·4Py, 9·4Py, 9·2N-1 with one N-1 as cocrystal, 7·2N-2, 9·2N-2, 7·2N-4 and a section from the polymer [9·2N-4]n in the crystalline state. Displacement ellipsoids drawn at 40% probability level. Minor occupied disordered atom sites, methyl groups and H atoms are omitted for clarity. | ||
The rigid Al-PLA 9 binds N-1 and N-2 in the binding pocket A, where by the solid-state structure of 9·2N-1 shows the formation of a co-crystal with a third N-1 molecule located in the cavity of the dbCOT unit. No interaction between the basic units and the Lewis-acid units was observed. The third N-1 is found as a non-coordinating molecule in the cavity and fills the void that exists within the dbCOT unit. The Al⋯Al distance in A of the adducts with 9 shows no big variance in the complexation of the bidentate bases due to the rigid alkyne spacer (Table 3). Given its dimensions (d(N⋯N): 8.709(2) Å), N-4 does not appear to align with the rigid binding pocket A, which can be widened to a maximum of 8.084(1) Å (9·2BisPhos) in the solid-state structures determined. Therefore, N-4 is thus complexed into the larger binding pocket B, but similar to [6·2BisPhos]2, only one of the two binding pockets is occupied. The second N-4 molecule then forms a link between several such units, thereby resulting in the formation of the Al–N coordination polymer [9·2N-4]n. The complexation of the two N-4 molecules in different ways results in the chemical inequivalence of the hydrogen atoms of the dbCOT system, thereby generating three signals in the 1H-NMR spectrum, in contrast to the usual two of other systems. Moreover, the signal of the NCH2 group, which was a single signal before the interaction, is now observed as four distinct signals. This finding is in agreement with the results obtained from the solid-state structure analysis. Both PLAs 7 and 9 are widened by the interaction with N-4, resulting in relatively small bending angles θ in the adducts (Table 3). For [9·2N-4]n, this can be explained by the fact that N-4 exerts a pushing force on PLA system 9 due to its intermolecular complexation in binding pocket B. In all adducts, the Al–N distances are almost identical and are a maximum of 0.08 Å above the sum of the covalent radii (1.97 Å).71 The results show that both PLAs facilitate the complexation of bidentate LB in binding pocket A. Complexation of a guest in cavity B was only observed for the combination of 9 and N-4. It may be reasonably expected that N-1 will also complex in the binding pocket A of the two aluminium containing PLS 7 and 9, due to the size and lower flexibility of N-1 compared to N-2 and N-4. Nevertheless, the more flexible and slightly larger N-2 is capable of binding two aluminium atoms with an Al⋯Al distance of 11.610(2) Å, as evidenced by 2Bis2AlCl·N-2 (for more details see ESI†). As can be observed from the distances listed in Table 3, N-2 tends to fit into cavity B of 7 or 9. Moreover, the notable compression of N-4 to facilitate its positioning within pocket A in adduct 7·2N-4 is somewhat unexpected. However, complexation within pocket B of 7 represents a potential alternative that does not necessitate the compression of N-4.
| d(A[Al⋯Al])/Å | d(B[Al⋯Al])/Å | d(Al–N)/Å | d(N⋯N)/Å | θ/° | |
|---|---|---|---|---|---|
| 7·4Py | 7.923(1) | 11.521(1) | 2.010(3)/2.017(2) | — | 42.2(1) |
| 7·2N-2 | 7.642(5)/7.669(5) | 11.000(5)/11.218(5) | 1.989(9)/2.051(12) | 5.445(17)/5.519(14) | 44.2(9)/45.5(7) |
| 7·2N-4 | 7.987(5)/8.332(5) | 11.402(7)/12.007(7) | 1.973(10)/2.012(11) | 5.811(15)/6.300(15) | 39.5(9)/40.1(9) |
| 9·4Py | 7.625(1)/7.627(1) | 11.451(1)/11.493(1) | 2.012(2)/2.035(2) | — | 44.5(1)/44.8(1) |
| 9·2N-1 | 7.213(1)/7.310(1) | 11.822(1)/13.588(1) | 1.974(3)/1.998(2) | 5.392(3)/5.444(4) | 41.6(2)/42.4(2) |
| 9·2N-2 | 7.266(1) | 11.282(1) | 2.000(1)/2.009(1) | 5.119(1) | 44.8(1) |
| [9·2N-4]n | 7.240(2)/7.257(2) | 13.107(2)/14.041(2) | 1.984(5)/1.994(4) | 8.848(8)/9.959(6) | 34.6(4)/34.7(5) |
| N-2 | — | — | — | 7.964(1) | — |
| 2Bis2AlCl·N-2 | — | 11.610(2) | — | 7.885(7) | — |
| N-4 | — | — | — | 8.709(2) | — |
One potential explanation for observed preference for bidentate Lewis-bases, whether nitrogen- or phosphorus-containing, to complex in pocket A, despite their size being compatible with pocket B, could be that the complexation of a guest in B or in C would no longer allow the molecule to flap, which is associated with a reduction in entropy.
Nevertheless, the [9·2N-4]n data indicate that when the N⋯N distances are sufficiently large, binding pocket A is no longer the preferred location for complexation, and intermolecular linkages also occur with greater frequency.
Interaction with multidentate drug molecules
We have also studied the complexation of some drug molecules (Scheme 4). These molecules typically possess multiple base functions, thereby prompting the inquiry as to which of the sites are preferred by the specific PLA. In addition, neurotransmitters interact with the receptors in the body in a comparable manner, subsequently triggering biochemical processes, so broadly defined, these are also host–guest interactions.The drugs caffeine (Caff), theobromine and γ-butyrolactone (GBL) found in coffee,72 and nicotine (Nico) as component of tobacco, were tested. GBL showed no interaction with the boron PLAs 6 and 8, which is reflected in non-shifted signals in the NMR spectra, and is consistent with previous results in which both showed weak to no interactions with oxygen-containing LBs (see Fig. 1). In contrast, the Al-PLAs 7 and 9 reacted with GBL, with 7 resulting in a non-selective reaction leading to degradation of the PLA. In contrast, 9 selectively formed a 1:4 adduct with GBL. This follows from the NMR spectra which show a shift comparable to 9·4THF and 9·4OPEt3 and no further signals indicating decomposition.
The bidentate base nicotine, comprising a pyridine and a pyrrolidine unit, may potentially form a 1:2 adduct analogous to N-1 and N-2. However, the NMR spectra obtained show that the reaction of the aluminium-containing PLAs 7 and 9 with two equivalents of nicotine resulted in both cases in the formation of adduct mixtures (1:1, 1:2, 1:3 and 1:4 adducts), which were converted to defined 1:4 adducts by the addition of two further equivalents. This shows that the interaction of the aluminium-PLAs with the pyridyl moiety is strongly favoured over the interaction with the pyrrolidine moiety so that the aluminium-PLAs 7 and 9 seek saturation with nicotine and form 1:4 adducts.
In contrast, PLA 8, which exhibited comparable interactions with NMe3 and pyridine (Fig. 1), formed an insoluble solid when two equivalents of nicotine were added. This solid was not amenable to detailed analysis, and the supernatant liquid contained neither nicotine nor 8. It was not possible to determine whether a polymer, a 1:2 adduct or a mixture of different adducts was formed, but a polymer is most likely. As 6 did not interact with NMe3 in the previous experiments, it was not reacted with nicotine, as only the formation of a 1:4 adduct is possible anyway.
Further complexation experiments were conducted with theobromine and caffeine, which represent more challenging structures. These molecules possess multiple base functions, and complexation experiments can therefore elucidate the preference for some over others. The weak PLA 6 showed no interaction with these xanthine derivatives and 8 showed only a weak interaction with caffeine, as evidenced by a slight shift in the PLA NMR signal (Δδ 11B = 17 ppm).
In contrast, the aluminium-containing systems 7 and 9 exhibited a discernible interaction with both theobromine and caffeine. However, the reactions with theobromine do not result in its complexation, but in a selective substitution of the AlBis2 units of PLAs 7 and 9 by hydrogen, thereby forming the tetravinyl- (11) or tetralkynyl-dbCOT (4), respectively. This Bis2Al-hydrogen exchange is the result of the reaction between the enol tautomer of theobromine and the aluminium-containing PLAs 7 and 9. In both reactions, the acidic hydrogen atom of theobromine is exchanged for Bis2Al, leading to the formation of the Bis2Al–O–purine dimer 10 (Fig. 7) as a further product, in addition to hydrocarbons 4 or 11. The obtained solid-state structure of 10 shows the formation of a dimer with a planar 12-membered ring (root mean square deviation 0.045 Å). The Al–O distances of 1.811(1) Å are in the range of other comparable adducts (1.801–1.846 Å),73 whereas the Al–O distance of 1.788(1) Å is notably short and lies below the sum of the covalence radii of a Al–O single bond (1.89 Å) but just above those of a covalent AlO double bond (1.70 Å).71 A comparison with theobromine also shows that the C–O bonds in 10 are 0.034/0.124 Å longer and the bonds C(16)–N(1) and C(15)–N(1) are 0.06/0.11 Å shorter. This indicates the formation of an aromatic pericyclic system in 10.74
 | ||
| Fig. 7 Molecular structure of 10 in the crystalline state. Only the structure of the ↓↑ isomer is shown as examples from the both (↑↑, ↓↑) isomers. Displacement ellipsoids drawn at 40% probability level. Minor occupied disordered atoms and methyl groups of the bis ligands are omitted for clarity. Selected distances [Å] and angles [°]: Al(1)–O(1) 1.811(1), O(1)–C(15) 1.285(2), C(15)–N(1) 1.332(2), N(1)–C(16) 1.331(2), C(16)–O(2) 1.279(2), O(2)–Al(1′) 1.788(1), O(2′)–Al(1)–O(1) 116.1(1), Al(1)–O(1)–C(15) 144.1(1), O(1)–C(15)–N(1) 121.0(1), N(1)–C(16)–O(2) 121.2(1), C(16)–O(2)–Al(1′) 158.3(1), symmetry code ‘: 3/2 − x, 1/2 − y, 3/2 − z. | ||
Caffeine contains a methyl group in place of the acidic hydrogen atom in theobromine, which prevents it from forming the aforementioned tautomer. The absence of a protic function, prevents the cleavage of the Al-PLAs and consequently adduct formation occurs with 7 and 9. The interaction of caffeine with 7 appears to be dynamic in solution, such that the use of an excess of the compound precludes the possibility of distinguishing between free and bound caffeine in the NMR spectra (ESI S150†).
In contrast, 9 reacts with caffeine to afford the 1:2 adduct 9·2Caff. The addition of only one equivalent of caffeine also leads to 9·2Caff and leaves an equivalent amount of PLA free in the solution, which can subsequently be converted to the 1:2 adduct by addition of further caffeine (Fig. 8). An excess of caffeine shows no further interaction with the adduct 9·2Caff as detected by unshifted NMR signal of caffeine. The 1H NMR spectra obtained of 9·2Caff shows splitting of the signals of the PLA 9 into several signal sets, resulting from the complexation of the asymmetric base, which makes the hydrogen atoms of the PLA chemically inequivalent. However, a 1:1 mixture of two adduct types is formed, so that the two caffeine molecules in the 1:2 adduct can be oriented either the same way (↑↑) or differently (↓↑). This can be seen in the alignment of the N–CH3 groups. Therefore, signals 1 and 2 of dbCOT in the 1H-NMR spectrum, which were previously each represented by a singlet in the spectrum of 9, split into two sets of signals. This is due to the chemical inequivalence caused by the asymmetric guest. Furthermore, the formation of a 1:1 mixture of the two different isomers upon complexation results in a total of four sets of signals for the dbCOT signals (signals 1 and 2 in Fig. 8) due to the positions of the N–CH3 groups.
 | ||
| Fig. 8 (a) Stacked section of the 1H-NMR spectra of 9, caffeine and 9·2Caff and mixtures of these compounds. For more details see ESI S158–S159.† (b) Molecular structure of the ↑↑ and ↓↑ isomers of 9·2Caff in the crystalline state. Displacement ellipsoids drawn at the 40% probability level. The isomers are present in the crystal as superimposed disordered structures. For reasons of clarity, the isomers are shown as separate structures, and methyl groups of bis and H atoms are omitted. Selected distances [Å] and angles [°]: Al(1A)⋯Al(1B) 6.844(1), Al(1A)–O(2) 1.912(1), Al(1B)–O(1) 1.917(1), C(1)–C(5) 1.436(4), C(5)–C(6) 1.215(4), C(6)–Al(1A) 1.928(6), C(6′)–Al(1B‘) 2.014(7), O(2)–C(22) 1.260(10), C(22)–N(1) 1.382(11), N(1)–C(21) 1.379(11), C(21)–O(1) 1.263(11), C(21)–N(2) 1.330(12), C(1)–C(5)–C(6) 177.0(3), C(5)–C(6)–Al(1A) 176.7(3), C(5′)–C(6′)–Al(1B′) 168.3(3), C(6)–Al(1A)–O(2) 107.20(1), C(6′)–Al(1B′)–O(1′) 98.73(1), Al(1A)–(O2)–C(22) 148.71(1), Al(1B)–O(1)–C(21) 142.04(1), C(22)–N(1)–C(21) 125.1(8), θ 43.0(1); binding pocket B: for ↑↑ Al(1A)⋯Al(1A′) 12.734(10), Al(1B)⋯Al(1B′) 12.213(1); for ↓↑ Al(1A)⋯Al(1B′) 12.479(3). | ||
These different isomers were also found in crystal and detected by X-ray diffraction; they are contained as disordered and overlaid molecules in the same ratio in the same crystal (Fig. 8). The caffeine molecule is complexed in the binding pocket A of PLA 9, and the aluminium atoms of 9 approach another by 6.845(6) Å, which is the smallest distance determined so far for its adducts. The fact that the Al–O bonds are longer than the sum of the covalence radii of 1.89 Å (ref. 71) is consistent with their dative character. However, their lengths of 1.91 Å are up to 0.13 Å longer than those in 10 and 9·4OPEt3† and this is probably due to the fixed distance between the Al atoms in the binding pocket A due to the alkynyl spacers, which prevent any further approach of the Al atoms. These results show that 9 can selectively bind two caffeine via their oxygen atoms due to the favourable orientation of the aluminium atoms within the binding pocket A and the high oxophilicity of aluminium. There is no interaction with any of the other basic positions, such as the very Lewis basic imidazole function. Based on the previous reactivity study (Fig. 1), it is remarkable that no Al–N interaction is observed. The study showed that 9 interacts particularly strongly with nitrogen bases and should therefore interact more strongly with nitrogen than with oxygen-containing functions. However, the fact that the aluminium functions interact preferentially with the oxygen atoms of caffeine is likely due to their more suitable distance for chelate binding. Thus, the orientation of the two O atoms allows the complexation of caffeine by two Al functions, whereas the supposedly stronger interaction with the imidazole nitrogen would not allow such cooperative complexation of caffeine. Steric effects could make the interaction with the nitrogen atoms of caffeine more difficult.
This shows the complexity of the definition of Lewis acidity of PLAs, which cannot be reduced to a sum of the individual Lewis acid functions involved. At least when they interact with multidentate bases, the cooperative effects between the Lewis acid units seem to have a significant influence on the reactivity.
Conclusions
Based on dibenzocyclooctatetraene, a set of tetradentate PLAs were synthesised and their reactivity towards a wide range of bases was tested. Different reactivities of the PLAs were identified due to their different flexibility and Lewis acidity. The boron PLAs 6 and 8 showed strong interactions with phosphane bases, resulting in the formation of stable adducts such as [6·2BisPhos]2 with two kinds of BisPhos coordination: intramolecular and intermolecular concerning the PLA 6. DOSY NMR experiments have shown this adduct to exist both in solution and the solid state. In contrast, the aluminium-containing PLAs 7 and 9 interacted weakly with the phosphanes and showed dynamic exchange in solution. Nevertheless, it was possible to determine the structure in the solid state of the adducts of PLA 9 with phosphanes. To our knowledge, [9·Bis[BisPhos]]n is the first Al–P coordination polymer whose solid-state structure has been determined. In addition, the polymer [9·2N-4]n was prepared by reaction with bistriazolylbutane (N-4) and analysed in the solid state. Moreover, two molecules of caffeine were complexed over the two O atoms containing in caffeine with the four Al-units of 9 to form the remarkably stable adduct 9·2Caff, demonstrating the interaction of a PLA with a biomolecule. This adduct, as well as the other adducts obtained, demonstrates the cooperative interaction between the Lewis acidic functions to complex the multidentate Lewis bases. The complexation experiments performed also show a preference for the complexation of bidentate guests in a way that does not significantly restrict the flapping movement, which may be due to the preservation of this degree of freedom and thus to a gain in entropy.Due to the dibenzocyclooctatetraene units, this new class of poly-Lewis acids and the coordination polymers have redox-active, but also photochemically switchable units and can therefore be used as functional materials in the fields of electrical conductivity, magnetism and optics. Investigations in this area are currently being carried out in our laboratories and initial experiments have shown that the PLAs are redox-switchable, which is associated with a change in the orientation of the Lewis acid functions, their mutual distance and their acidity.
Author contributions
M. J. Klingsiek: investigation, methodology, validation, visualisation, writing (original draft), J. Buth: investigation (supporting synthesis), P. C. Trapp: investigation (DFT), A. Mix: investigation (DOSY-NMR), J.-H. Lamm, B. Neumann and H.-G. Stammler: investigation (SCXRD), N. W. Mitzel: funding acquisition, project administration, supervision, writing, reviewing and editing.Data availability
The data that supports the findings of this study are available in the ESI† of this article. CCDC 2404643–2404671† contain the supplementary crystallographic data for this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Marco Wißbrock and Dr Andreas Mix for recording NMR spectra, Barbara Teichner for performing elemental analyses, Johanna Buddeberg, Delia Hageman, Selina Langen and Verena Orth for support with syntheses, Julian Buth for his flawless Bis2AlH, Calvin Höffelmeyer for the crystallisation of N-2 and Dr Maurice Franke for fruitful discussions. This work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) grant MI 477/39-1, project no. 424957011. We thank the Regional Computing Centre of the University of Cologne (RRZK) for providing computing time and support on the DFG-funded (Funding number: INST 216/512/1FUGG) HPC system CHEOPS.References
- B. Dietrich and J. M. Lehn, Activation anionique a l'aide de cryptates I milieux fortement basiques, Tetrahedron Lett., 1973, 14, 1225–1228 Search PubMed.
- C. J. Pedersen and H. K. Frensdorff, Makrocyclische Polyäther und ihre Komplexe, Angew. Chem., 1972, 84, 16–26 Search PubMed.
- J. L. Beckmann, J. Krieft, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, Poly-pnictogen bonding: trapping halide ions by a tetradentate antimony Lewis acid, Chem. Sci., 2023, 14, 13551–13559 CAS.
- H. E. Katz, Hydride sponge: 1,8-naphthalenediylbis(dimethylborane), J. Am. Chem. Soc., 1985, 107, 1420–1421 CAS.
- H. E. Katz, Hydride sponge: complexation of 1,8-naphthalenediylbis(dimethylborane) with hydride, fluoride, and hydroxide, J. Org. Chem., 1985, 50, 5027–5032 CAS.
- D. Brondani, F. H. Carré, R. J. P. Corriu, J. J. E. Moreau and M. Wong Chi Man, Synthesis and Dynamic Behavior of the Heptafluorotrisilacyclohexane Anion: A New Fluxional Silicate with Rapid Intramolecular Exchange of Fluoride Ligand, Angew. Chem., Int. Ed. Engl., 1996, 35, 324–326 CAS.
- M. Schulte, M. Schürmann and K. Jurkschat, [cyclo-CH2Sn(Cl2)CH2Si(Me2)2O]: Synthesis and Complexation Behaviour of a Novel, Cyclic, Bidentate Lewis Acid and Its Conversion into a Tin-Containing Fluorosilane with Intermolecular Si−F⋯Sn Bridges, Chem. – Eur. J., 2001, 7, 347–355 CrossRef CAS PubMed.
- C.-H. Chen and F. P. Gabbaï, Fluoride Anion Complexation by a Triptycene-Based Distiborane: Taking Advantage of a Weak but Observable C−H⋯F Interaction, Angew. Chem., Int. Ed., 2017, 56, 1799–1804 CrossRef CAS PubMed.
- J. L. Beckmann, J. Krieft, Y. V. Vishnevskiy, B. Neumann, H.-G. Stammler and N. W. Mitzel, A Bidentate Antimony Pnictogen Bonding Host System, Angew. Chem., Int. Ed., 2023, 62, e202310439 CrossRef CAS PubMed.
- G.-W. Yang, Y.-Y. Zhang and G.-P. Wu, Modular Organoboron Catalysts Enable Transformations with Unprecedented Reactivity, Acc. Chem. Res., 2021, 54, 4434–4448 CrossRef CAS PubMed.
- S. N. Kessler, M. Neuburger and H. A. Wegner, Bidentate Lewis Acids for the Activation of 1,2-Diazines – A New Mode of Catalysis, Eur. J. Org. Chem., 2011, 3238–3245 CrossRef CAS.
- S. N. Kessler, M. Neuburger and H. A. Wegner, Domino Inverse Electron-Demand Diels–Alder/Cyclopropanation Reaction of Diazines Catalyzed by a Bidentate Lewis Acid, J. Am. Chem. Soc., 2012, 134, 17885–17888 CrossRef CAS PubMed.
- S. P. Lewis, L. D. Henderson, B. D. Chandler, M. Parvez, W. E. Piers and S. Collins, Aqueous Suspension Polymerization of Isobutene Initiated by 1,2-C6F4[B(C6F5)2]2, J. Am. Chem. Soc., 2005, 127, 46–47 CrossRef CAS PubMed.
- L. Schweighauser, I. Bodoky, S. N. Kessler, D. Häussinger, C. Donsbach and H. A. Wegner, Bidentate Lewis Acid Catalyzed Domino Diels–Alder Reaction of Phthalazine for the Synthesis of Bridged Oligocyclic Tetrahydronaphthalenes, Org. Lett., 2016, 18, 1330–1333 CrossRef CAS PubMed.
- W. Uhl and M. Matar, Syntheses of polyaluminium compounds by hydroalumination reactions, chelating Lewis acids possessing two and four coordinatively unsaturated aluminium atoms, J. Organomet. Chem., 2002, 664, 110–115 CrossRef CAS.
- J. L. Beckmann, B. Neumann, H. Stammler and N. W. Mitzel, Selectivity in Adduct Formation of a Bidentate Boron Lewis Acid, Chem. – Eur. J., 2024, 30, e202400081 CrossRef CAS PubMed.
- N. Aders, P. C. Trapp, J.-H. Lamm, J. L. Beckmann, B. Neumann, H.-G. Stammler and N. W. Mitzel, Hydroalumination of 1,8-Diethynylanthracenes–Al-based Bis-Lewis-Acids and their Isomerization and Complexation Behavior, Organometallics, 2022, 41, 3600–3611 CrossRef CAS.
- F. Schäfer, A. Mix, N. Cati, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Host-guest chemistry of a bidentate silyl-triflate bis-Lewis acid – complex complexation behaviour unravelled by diffusion NMR spectroscopy, Dalton Trans., 2022, 51, 7164–7173 RSC.
- M. Franke, M. J. Klingsiek, J. Buth, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Hexadentate poly-Lewis acids based on the bowl-shaped tribenzotriquinacene, Dalton Trans., 2024, 53, 7752–7762 Search PubMed.
- M. J. Klingsiek, M. Franke, S. M. Walkenhorst, T. Röhr, J. Buth, A. Mix, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Tri– and hexadentate poly–Lewis acids – attempts to unravel the complex dynamics of cage and capsule formation in solution, Eur. J. Inorg. Chem., 2024, e202400445 CrossRef CAS.
- H. E. Katz, 1,8-Anthracenediethynylbis(catechol boronate): a bidentate Lewis acid on a novel framework, J. Org. Chem., 1989, 54, 2179–2183 CrossRef CAS.
- W. Uhl, F. Hannemann, W. Saak and R. Wartchow, A Methylene-Bridged Dialuminium Compound as a Chelating Lewis Acid: Complexation of Nitrite and Nitrate Anions by R2Al–CH2–AlR2[R = CH(SiMe3)2], Eur. J. Inorg. Chem., 1998, 921–926 CrossRef CAS.
- W. Uhl and F. Hannemann, A methylene bridged dialuminium compound as a chelating Lewis acid—complexation of azide and acetate anions by R2Al–CH2–AlR2 [R=CH(SiMe3)2], J. Organomet. Chem., 1999, 579, 18–23 CrossRef CAS.
- M. Franke, M. J. Klingsiek, J. Buth, A. Mix, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Tridentate Lewis Acids Based on Tribenzotriquinacene Chalices, Chem. – Eur. J., 2024, 30, e202401072 CrossRef CAS PubMed.
- A. Dhara and F. Beuerle, Reversible Assembly of a Supramolecular Cage Linked by Boron–Nitrogen Dative Bonds, Chem. – Eur. J., 2015, 21, 17391–17396 CrossRef CAS PubMed.
- B. Icli, E. Sheepwash, T. Riis-Johannessen, K. Schenk, Y. Filinchuk, R. Scopelliti and K. Severin, Dative boron–nitrogen bonds in structural supramolecular chemistry: multicomponent assembly of prismatic organic cages, Chem. Sci., 2011, 2, 1719 RSC.
- M. Fleischmann, J. S. Jones, G. Balázs, F. P. Gabbaï and M. Scheer, Supramolecular adducts based on weak interactions between the trimeric Lewis acid complex (perfluoro-ortho-phenylene)mercury and polypnictogen complexes, Dalton Trans., 2016, 45, 13742–13749 RSC.
- K. Tamao, T. Hayashi, Y. Ito and M. Shiro, Pentacoordinate anionic bis(siliconates) containing a fluorine bridge between two silicon atoms. Synthesis, solid-state structures, and dynamic behavior in solution, Organometallics, 1992, 11, 2099–2114 CAS.
- A. S. Wendji, C. Dietz, S. Kühn, M. Lutter, D. Schollmeyer, W. Hiller and K. Jurkschat, Silicon– and Tin–Containing Open–Chain and Eight–Membered–Ring Compounds as Bicentric Lewis Acids toward Anions, Chem. – Eur. J., 2016, 22, 404–416 CrossRef CAS PubMed.
- M. Fleischmann, J. S. Jones, F. P. Gabbaï and M. Scheer, A comparative study of the coordination behavior of cyclo,-P5and cyclo-As5 ligand complexes towards the trinuclear Lewis acid complex (perfluoro-ortho-phenylene)mercury, Chem. Sci., 2015, 6, 132–139 CAS.
- W. Uhl, A. Hepp, H. Westenberg, S. Zemke, E.-U. Würthwein and J. Hellmann, Hydroalumination and Hydrogallation of 1,2-Bis(trimethylsilylethynyl)benzene: Formation of Molecular Capsules and C−C Bond Activation, Organometallics, 2010, 29, 1406–1412 CAS.
- D. F. Shriver and M. J. Biallas, Observation of the Chelate Effect with a Bidentate Lewis Acid, F2BCH2CH2BF2, J. Am. Chem. Soc., 1967, 89, 1078–1081 CAS.
- P. Niermeier, S. Blomeyer, Y. K. J. Bejaoui, J. L. Beckmann, B. Neumann, H.-G. Stammler and N. W. Mitzel, Bidentate Boron Lewis Acids: Selectivity in Host–Guest Complex Formation, Angew. Chem., Int. Ed., 2019, 58, 1965–1969 CrossRef CAS PubMed.
- J. Chmiel, B. Neumann, H.-G. Stammler and N. W. Mitzel, Dialkylaluminium-, -Gallium-, and -Indium-Based Poly-Lewis Acids with a 1,8-Diethynylanthracene Backbone, Chem. – Eur. J., 2010, 16, 11906–11914 CrossRef CAS PubMed.
- J. Rudlof, N. Aders, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Bidentate Lewis Acids Derived from o-Diethynylbenzene with Group 13 and 14 Functions, Chem. Open, 2021, 10, 1020–1027 CAS.
- W. Uhl, H. R. Bock, F. Breher, M. Claesener, S. Haddadpour, B. Jasper and A. Hepp, Hydrogallation of Trimethylsilylethynylbenzenes: Generation of Potential Di- and Tripodal Chelating Lewis Acids, Organometallics, 2007, 26, 2363–2369 CrossRef CAS.
- J. L. Beckmann, B. Neumann, H.-G. Stammler, J.-H. Lamm and N. W. Mitzel, Bidentate boron Lewis acids: synthesis by tin boron exchange reaction and host–guest complex formation, Dalton Trans., 2024, 53, 7958–7964 RSC.
- F. Schäfer, B. Neumann, H.-G. Stammler and N. W. Mitzel, Hexadentate Poly-Lewis Acids Based on 1,3,5-Trisilacyclohexane, Eur. J. Inorg. Chem., 2021, 3083–3090 CrossRef.
- F. Schäfer, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Silicon-Bridged Bi- and Tridentate Lewis Acidic Host Systems, Eur. J. Inorg. Chem., 2021, 3265–3271 CrossRef.
- H. Schulz, G. Gabbert, H. Pritzkow and W. Siebert, Synthesen und Struktur von Diborylacetylenen, Chem. Ber., 1993, 126, 1593–1595 CrossRef CAS.
- P. Niermeier, K. A. M. Maibom, J.-H. Lamm, B. Neumann, H.-G. Stammler and N. W. Mitzel, Hydrogen-bond-induced selectivity of a head-to-head photo-dimerisation of dialkynylanthracene – access to tetradentate Lewis acids, Chem. Sci., 2021, 12, 7943–7952 RSC.
- U. H. Strasser, B. Neumann, H.-G. Stammler, R. J. F. Berger and N. W. Mitzel, Gilded Chalices: Tetra-aurated Calix[4]arenes, Z. Naturforsch., B:J. Chem. Sci., 2014, 69, 1061–1072 CrossRef CAS.
- M. W. Drover, M. C. Dufour, L. A. Lesperance-Nantau, R. P. Noriega, K. Levin and R. W. Schurko, Octaboraneyl Complexes of Nickel: Monomers for Redox-Active Coordination Polymers, Chem. – Eur. J., 2020, 26, 11180–11186 CrossRef CAS PubMed.
- A. A. Kroeger and A. Karton, Graphene–induced planarization of cyclooctatetraene derivatives, J. Comput. Chem., 2022, 43, 96–105 CrossRef CAS PubMed.
- G. H. Senkler, D. Gust, P. K. Riccobono and K. Mislow, Ring inversion in di- and tetrabenzocyclootatetraenes, J. Am. Chem. Soc., 1972, 94, 8626–8627 CrossRef CAS.
- Y. Zhu, Z. Zhou, Z. Wei and M. A. Petrukhina, Two-Fold Reduction of Dibenzo[a,e]cyclooctatetraene with Group 1 Metals: From Lithium to Cesium, Organometallics, 2020, 39, 4688–4695 CrossRef CAS.
- G. A. Olah, J. S. Staral, G. Liang, L. A. Paquette, W. P. Melega and M. J. Carmody, Novel aromatic systems. 8. Cyclooctatetraene dications, J. Am. Chem. Soc., 1977, 99, 3349–3355 CrossRef CAS.
- T. J. Katz, M. Yoshida and L. C. Siew, The sym-Dibenzcyclooctatetraene Anion Radical and Dianion, J. Am. Chem. Soc., 1965, 87, 4516–4520 CrossRef CAS.
- L. A. Paquette, G. D. Ewing, S. Traynor and J. M. Gardlik, Dicyclooctatetraeno[1,2:4,5]benzene dianion and tetraanion. Experimental assessment of extended paratropic vs. restricted diatropic π-electron delocalization, J. Am. Chem. Soc., 1977, 99, 6115–6117 CrossRef CAS.
- H. Kojima, A. J. Bard, H. N. C. Wong and F. Sondheimer, Electrochemical reduction of sym-dibenzocyclooctatetraene, sym-dibenzo-1,5-cyclooctadiene-3,7-diyne, and sym-dibenzo-1,3,5-cyclooctatrien-7-yne, J. Am. Chem. Soc., 1976, 98, 5560–5565 CrossRef CAS.
- N. C. Baird, Quantum organic photochemistry. II. Resonance and aromaticity in the lowest 3ππ* state of cyclic hydrocarbons, J. Am. Chem. Soc., 1972, 94, 4941–4948 CrossRef CAS.
- H. Ottosson, Exciting excited-state aromaticity, Nat. Chem., 2012, 4, 969–971 CrossRef CAS PubMed.
- R. Kimura, H. Kuramochi, P. Liu, T. Yamakado, A. Osuka, T. Tahara and S. Saito, Flapping Peryleneimide as a Fluorogenic Dye with High Photostability and Strong Visible–Light Absorption, Angew. Chem., Int. Ed., 2020, 59, 16430–16435 CrossRef CAS PubMed.
- C. Yuan, S. Saito, C. Camacho, S. Irle, I. Hisaki and S. Yamaguchi, A π-Conjugated System with Flexibility and Rigidity That Shows Environment-Dependent RGB Luminescence, J. Am. Chem. Soc., 2013, 135, 8842–8845 CrossRef CAS PubMed.
- T. Yamakado, S. Takahashi, K. Watanabe, Y. Matsumoto, A. Osuka and S. Saito, Conformational Planarization versus Singlet Fission: Distinct Excited-State Dynamics of Cyclooctatetraene-Fused Acene Dimers, Angew. Chem., Int. Ed., 2018, 57, 5438–5443 CrossRef CAS PubMed.
- E. Sheepwash, V. Krampl, R. Scopelliti, O. Sereda, A. Neels and K. Severin, Molecular Networks Based on Dative Boron–Nitrogen Bonds, Angew. Chem., 2011, 123, 3090–3093 CrossRef.
- M. Fontani, F. Peters, W. Scherer, W. Wachter, M. Wagner and P. Zanello, Adducts of Ferrocenylboranes and Pyridine Bases: Generation of Charge-Transfer Complexes and Reversible Coordination Polymers, Eur. J. Inorg. Chem., 1998, 1453–1465 CrossRef CAS.
- C. Janiak, Engineering coordination polymers towards applications, Dalton Trans., 2003, 2781 RSC.
- G. A. Lane, W. E. Geiger and N. G. Connelly, Palladium(I).pi.-radicals. Electrochemical preparation and study of their reaction pathways, J. Am. Chem. Soc., 1987, 109, 402–407 CrossRef CAS.
- S. Kobayashi, T. Busujima and S. Nagayama, A Novel Classification of Lewis Acids on the Basis of Activity and Selectivity, Chem. – Eur. J., 2000, 6, 3491–3494 CrossRef CAS PubMed.
- D. P. N. Satchell and R. S. Satchell, Quantitative aspects of Lewis acidity, Q. Rev., Chem. Soc., 1971, 25, 171 RSC.
- P. Erdmann and L. Greb, What Distinguishes the Strength and the Effect of a Lewis Acid: Analysis of the Gutmann–Beckett Method, Angew. Chem., Int. Ed., 2022, 61, e202114550 CrossRef CAS PubMed.
- L. Greb, Lewis Superacids: Classifications, Candidates, and Applications, Chem. – Eur. J., 2018, 24, 17881–17896 CrossRef CAS PubMed.
- R. F. Childs, D. L. Mulholland and A. Nixon, Lewis acid adducts of α,β-unsaturated carbonyl and nitrile compounds. A calorimetric study, Can. J. Chem., 1982, 60, 809–812 CrossRef CAS.
- M. A. Beckett, G. C. Strickland, J. R. Holland and K. Sukumar Varma, A convenient n.m.r. method for the measurement of Lewis acidity at boron centres: correlation of reaction rates of Lewis acid initiated epoxide polymerizations with Lewis acidity, Polymer, 1996, 37, 4629–4631 CrossRef CAS.
- U. Mayer, V. Gutmann and W. Gerger, The acceptor number - A quantitative empirical parameter for the electrophilic properties of solvents, Monatsh. Chem., 1975, 106, 1235–1257 CrossRef CAS.
- I. B. Sivaev and V. I. Bregadze, Lewis acidity of boron compounds, Coord. Chem. Rev., 2014, 270–271, 75–88 CrossRef CAS.
- P. Erdmann and L. Greb, Multidimensional Lewis Acidity: A Consistent Data Set of Chloride, Hydride, Methide, Water and Ammonia Affinities for 183 p-Block Element Lewis Acids, ChemPhysChem, 2021, 22, 935–943 CrossRef CAS PubMed.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, An Extensive Set of Accurate Fluoride Ion Affinities for p-Block Element Lewis Acids and Basic Design Principles for Strong Fluoride Ion Acceptors, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed.
- J. E. Huheey, The Electronegativity of Groups, J. Phys. Chem., 1965, 69, 3284–3291 CrossRef CAS.
- P. Pyykkö and M. Atsumi, Molecular Single-Bond Covalent Radii for Elements 1–118, Chem. – Eur. J., 2009, 15, 186–197 CrossRef PubMed.
- D. R. Seninde and E. Chambers, Coffee Flavor: A Review, Beverages, 2020, 6, 44 CrossRef CAS.
- K. Martinewski, T. Holtrichter-Rößmann, C. Rösener, A. Hepp, E.-U. Würthwein and W. Uhl, Cooperative Activation of Isocyanates by Al–N-Based Active Lewis Pairs and the Generation of a C5 Chain by Simultaneous Formation of Two C–C Bonds, Chem. – Eur. J., 2017, 23, 6129–6141 CrossRef CAS PubMed.
- CCDC 753171 and 658887.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic protocols, spectra, details on calculations, crystallographic data. CCDC 2404643–2404671. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5qi00823a |
| This journal is © the Partner Organisations 2025 |