
Advances in organic room-temperature phosphorescence: design strategies, photophysical mechanisms, and emerging applications
Yujie
Yang
a,
Qianqian
Li
*b and
Zhen
Li
 *bc
*bc
aSchool of Sports Medicine, Wuhan Sports University, Wuhan 430079, China
bHubei Key Lab on Organic and Polymeric Opto-Electronic Materials, Department of Chemistry, Wuhan University, Wuhan 430072, China. E-mail: liqianqian@whu.edu.cn; lizhen@whu.edu.cn
cTaiKang Center for Life and Medical Sciences, Wuhan University, Wuhan 430072, China
First published on 31st January 2025
Abstract
Organic room temperature phosphorescent (RTP) materials have garnered significant interest due to their potential applications in anticounterfeiting, biological imaging, and optoelectronic devices. This Chemistry Frontiers paper comprehensively analyzes the photophysical processes underlying phosphorescence to identify the key factors that facilitate phosphorescence emission. It critically evaluates the intrinsic mechanisms of various construction strategies and explores the relationship between aggregated structures and their properties. Detailed discussions on molecular arrangement, packing modes, and intra/intermolecular interactions are presented, offering systematic design principles. Finally, it outlines the current application areas of organic RTP materials, forecasts future advancements, and proposes performance criteria and strategic design approaches to guide further progress in this field.
 Yujie Yang | Yujie Yang currently holds a position as a lecturer at the School of Sports Medicine, Wuhan Sports University. She received her PhD degree from Tianjin University in 2023. Her current research interest is focused on organic photogenerated radical materials and room-temperature phosphorescent materials and exploring their various applications. |
 Qianqian Li | Qianqian Li received her BSc degree from Hubei University, China, in 2004 and then obtained her PhD degree from Wuhan University in 2009. She is now a full professor at Wuhan University, and her research interests are in the modulation of aggregated structures of optoelectronic functional materials. |
 Zhen Li | Zhen Li received his BSc and PhD degrees from Wuhan University, China, in 1997 and 2002, respectively. He has been a full professor at Wuhan University since 2006 and his research interests are in the development of organic molecules and polymers with new structures and new functions for organic electronics and photonics. |
Introduction
Room-temperature phosphorescence (RTP) is a photoluminescence phenomenon, in which emission persists after removing the excitation source. In 1939, Clapp reported a pivotal finding that tetraphenylmethane, tetraphenylsilane, and their derivatives exhibited a bright blue-green afterglow lasting up to 23 s.1 However, the development of organic RTP materials at that time was still lagging because of unclear internal mechanisms. Also, the intrinsic limitations of organic compounds, including low spin–orbit coupling (SOC) coefficients, and dominant non-radiative decay processes, restricted advancements in long-lived RTP.Fortunately, breakthroughs have emerged since Tang et al. reported the “crystallization-induced phosphorescence” (CIP) strategy to achieve RTP in aggregated states.2 From then on, an increasing number of organic RTP materials have been explored (Fig. 1a) with the dominated green afterglow, thanks to the great efforts of scientists.3–8 The lifetimes have been prolonged to 33 s (505 nm) with modulation of aggregated structures by our group in 2021.9 For full coverage of the whole emission spectra, considerable efforts have been devoted to addressing the challenges with stabilizing the high-energy triplet excited states. An, Liu and Huang et al. have achieved the emission wavelength blue-shifted to 380 nm with deep-blue phosphorescence.10 Generally, with the wavelengths extended to the red/NIR region, the non-radiative decays were usually accelerated by the low energy gaps. Recently, through the host–guest doped strategy, NIR phosphorescence was extended to 732 nm with the lifetime of 102 ms,11 and the highest brightness under physiological conditions (8.21 ± 0.36 × 108 ps−1 cm−2 sr−1) has been achieved using branched-luminogens with strong hydrophobicity.12
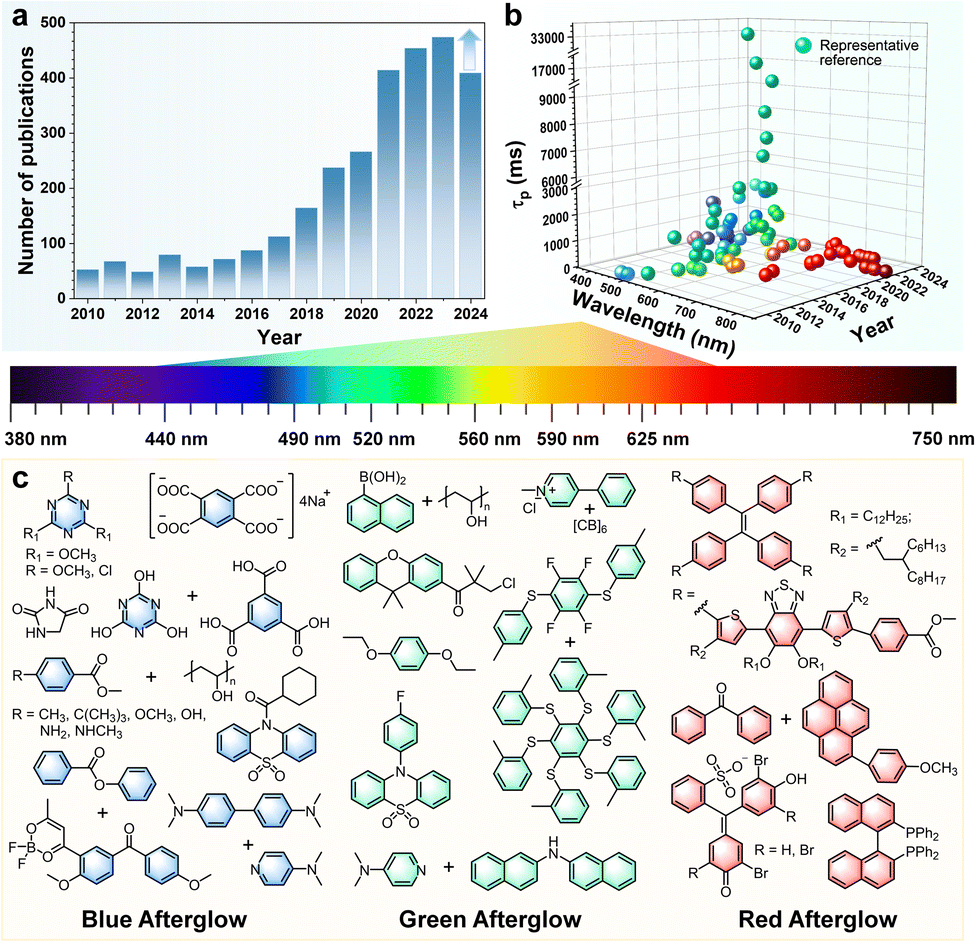 | ||
| Fig. 1 (a) Trends in RTP research from 2010 to 2024 as reflected in scientific publications; (b) the emission wavelengths and lifetimes of phosphorescence materials as reported in the representative literature; and (c) chemical structures of RTP materials with blue, green, and red afterglow as described in the representative literature. | ||
These improved RTP properties show promising potential in various applications, including optoelectronics,13,14 information encryption,4,5 anti-counterfeiting,8,10 and biological imaging,6,9,11,12 which is largely attributed to their structurally tunable nature, low costs, diverse emission spectra, extended luminescence, and excellent biocompatibility. For instance, capitalizing on its excitation-free capability and distinct photophysical properties with large Stokes shifts and extended luminescence lifetimes, phosphorescence bioimaging can effectively shield autofluorescence from biological tissues through time-resolved imaging techniques. The captured afterglow imaging demonstrated high quality and signal-to-noise ratios, representing a substantial advancement in biological imaging techniques.11,12 These properties primarily manifest in aggregated states, where molecular arrangement, packing modes, and inter/intramolecular interactions play a critical role in achieving high-performance organic RTP materials.15–17
In this Chemistry Frontiers paper, we highlight a range of construction strategies proposed to improve the RTP properties by deeply understanding the fundamental relationships between molecular structures, molecular aggregation, and the photophysical properties, and underscore the design and fabrication needs for advancing the field of organic phosphorescence.
Design strategies
The luminescence mechanism of organic RTP materials is well delineated by the Jablonski diagram (Fig. 2a). Initially, excitons in the lowest singlet excited state (S1) are generated from the ground state (S0) upon excitation. They may engage in intersystem crossing (ISC) to the triplet excited states (Tn) and undergo further internal conversion (IC) to the stable lowest triplet excited state (T1). Phosphorescence can then occur as excitons return to S0 through radiative decay. The quantum mechanics theory suggests that the spin-forbidden transitions from the singlet to triplet excited states, and then from triplet excited states to the ground state, significantly prolong the phosphorescence lifetime to the microsecond or millisecond range or longer. According to the photophysical processes of phosphorescence emission, the ISC process can be efficiently promoted by enhancing the SOC and reducing the energy gap between the singlet and triplet excited states, thereby generating substantial triplet excitons for RTP emission. Furthermore, the ISC process can be facilitated if the transition involves a change of orbital types.18,19 Thus, the preferred singlet–triplet transitions between n–π* and π–π* features enhance RTP emission by promoting the generation of triplet excitons. The n–π* transitions can be induced by incorporating functional groups bearing lone pairs of electrons, including carbonyl, cyano, halogens (F, Cl, and Br), other heteroatoms (S, Se, and P), etc. Meanwhile, it is essential to stabilize the triplet excitons by creating a rigid molecular framework to suppress non-radiative decay and avoid quenching effects exerted by oxygen and water. Drawing on these principles, some efficient strategies have been explored, such as crystallization, doping into rigid matrices, polymerization, supramolecular self-assembly, heavy-atom effect, and deuteration.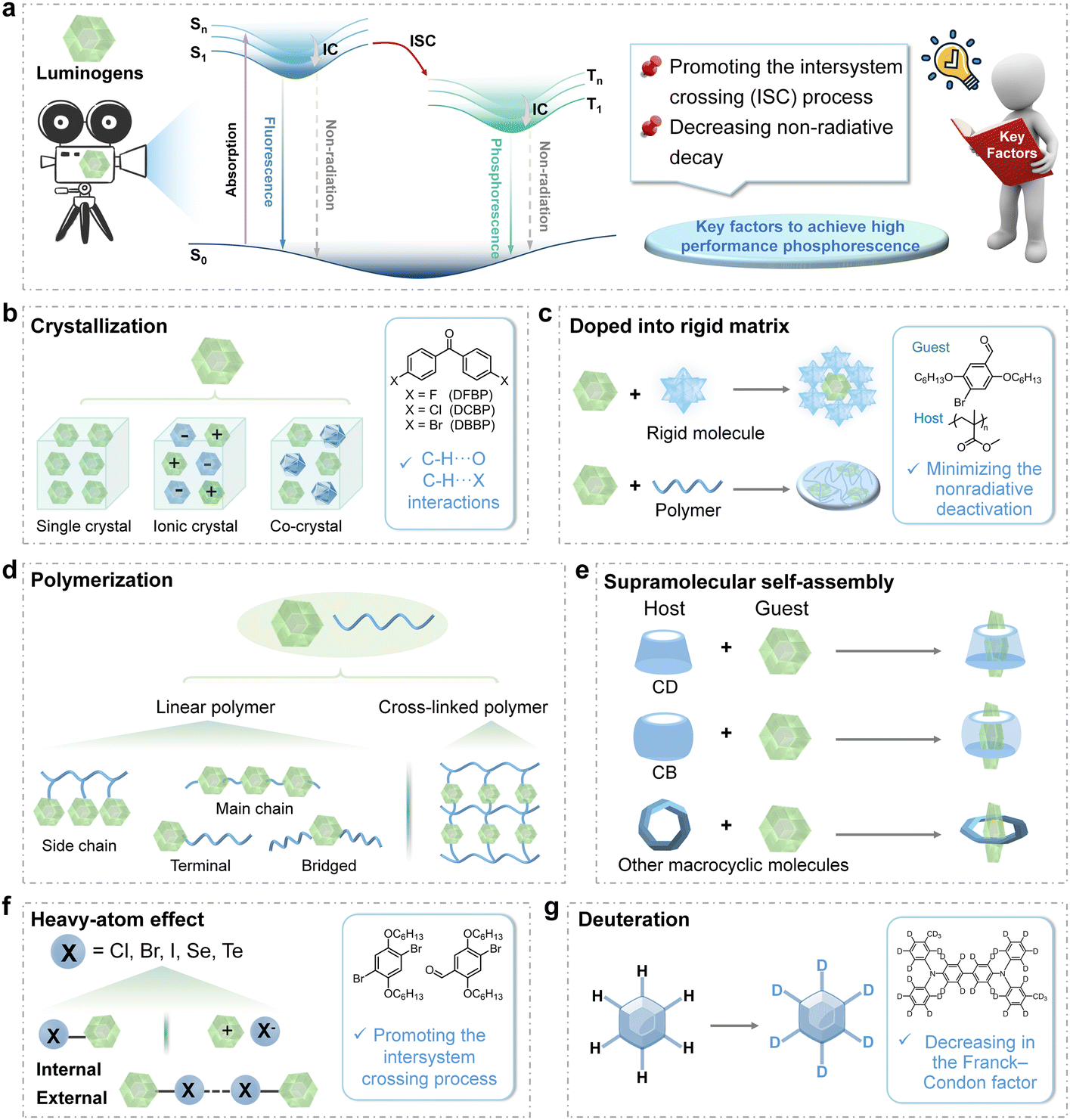 | ||
| Fig. 2 (a) A simplified Jablonski diagram illustrating the photophysical processes involved in room-temperature phosphorescence (RTP) emission, emphasizing the key factors required to achieve effective phosphorescence performance. (b) The crystallization strategy for achieving rigid packing includes the formation of single crystals, ionic crystals, and co-crystals. The inset shows an example involving single crystals to demonstrate phosphorescence emission, highlighting the C–H⋯O and C–H⋯X interactions. (c) The doping strategy is to dope phosphors into the rigid matrix directly. The inset shows an example of doping a phosphor into polymethyl methacrylate to minimize the nonradiative deactivation and achieve efficient RTP. (d) The polymerization strategy to effectively achieve phosphorescence emission with a rigid environment. (e) The supramolecular self-assembly strategy provides a rigid environment to stabilize the triplet excitons using hosts such as cyclodextrins (CDs), cucurbiturils (CBs), and other macrocyclic molecules. (f) The strategy of introducing the heavy-atom effect to facilitate the ISC process through both internal and external heavy-atom effects. The inset shows an example of the materials displaying high-efficiency phosphorescence due to the introduction of bromine atoms. (g) The deuteration strategy to achieve RTP emission. The inset shows an example of deuteration to lower the C–H stretching vibration energy and decrease the Franck–Condon factor. | ||
As to crystal engineering, Sun and Tang et al. made a pivotal advancement by elucidating the principle of CIP in 2010 (Fig. 2b).2 The development of crystallization strategies to stabilize triplet excitons and suppress non-radiative transitions has since become a foundational approach, guiding the rational design of efficient RTP materials.10,13,19–24 Crystalline systems can be classified as pure organic single crystals, ionic crystals, and co-crystals based on their composition. Intermolecular interactions, including halogen bonding, π–π interaction, hydrogen bonding, and ionic bonding, have been employed to improve phosphorescence intensities and lifetimes. In 2018, our group investigated the impact of substituent modifications on π–π interactions and found that stronger π–π interactions led to the stabilization of triplet excitons and an increase in RTP lifetimes from 88 ms to 410 ms.21 Recently, Li, Hu, Tang and Phillips et al. demonstrated that strong hydrogen bonding networks stabilized triplet excited states and suppressed non-radiative transitions within cocrystals.22 The stabilization effect resulted in significantly prolonged phosphorescence, extending the lifetime from 76 ms for uracil (U) to 530 ms for U–MA and 663 ms for U–B after the formation of cocrystals between U and melamine (MA) or boric acid (B). Furthermore, the presence of tight molecular packing with strong intermolecular interactions is a pivotal factor in determining the RTP performance not only in organic aromatic compounds, but also in non-aromatic compounds. For example, in 2018, our group reported that cyanoacetic acid (CAA) crystals displayed persistent RTP with the lifetime of 862 ms, as the result of the strong intermolecular hydrogen bonds.23 Also, the “cluster-triggered phosphorescence” (CTP) has been proposed by Tang and Yuan to explain the RTP properties of various non-aromatic compounds.25
For the doping strategy, the crystalline samples as hosts can create a rigid environment for guest molecules to suppress non-radiative transitions of triplet excitons (Fig. 2c).11,26 For example, 4,4′-dibromobiphenyl crystals establish a rigid environment through intermolecular C–Br⋯Br–C, C–H⋯π, and C–H⋯Br interactions, effectively restricting the molecular motions of guests.26 The doping strategy is an effective and simple strategy to achieve high-efficiency RTP materials. Also, polymers have been employed as effective rigid matrices to promote RTP properties for creating a rigid environment through molecular chain entanglement and interactions between functional groups.8,27,28 In 2013, a significant advancement in amorphous metal-free organic phosphorescence was achieved by doping phosphors into a glassy polymethyl methacrylate (PMMA) matrix.27 It was demonstrated that the polymer matrix could effectively restrict molecular motions and reduce triplet deactivation. Similarly, polyvinyl alcohol (PVA), bearing hydroxyl units with high density, can form hydrogen bonding with guest molecules bearing active hydrogens, promoting RTP properties by the strong restriction effect and excellent oxygen barrier properties.29–31 Also in 2022, our group achieved ultralong RTP with the lifetime of 2.43 s by promoting the hydrogen bonding between arylboronic acids and PVA after the removal of H2O.29
Polymerization has been utilized to further strengthen the rigidity of systems by forming covalent bonds between polymer chains and luminogens, which can further suppress non-radiative transitions (Fig. 2d).7,32–34 The tunable luminogens in various regions of linear polymers—such as in the side chains, main chain, or at the terminal and bridge positions—enable the modulation of both emission wavelengths and lifetimes of phosphorescence emission. In 2023, Ma, Huang, and Zhao et al. prepared a series of RTP polymers by integrating phosphorescence moieties into the polymer backbone.33 The P1 polymer demonstrated color-tunable phosphorescence ranging from 474 to 506 nm when excited at 300 nm and 360 nm, respectively, along with lifetimes of approximately 1050 ms (at 474 nm) and 261 ms (at 507 nm). The excitation wavelength-dependent RTP resulted from the various molecular conformations stabilized by the polymer chains, indicating the potential for achieving multi-color emitting RTP in aggregated states by altering the molecular conformations of monomers. Moreover, the restriction of molecular rotations can be further strengthened through cross-linking between luminogens and polymer chains.35,36 For instance, in 2015, Kim et al. demonstrated that cross-linking between phosphors and polymer matrices could significantly improve phosphorescence properties.35 These covalent bonds not only established a rigid environment that suppressed non-radiative decay and optimized the luminescence properties, but also highlighted the significance of modulating molecular motions in aggregated states to influence non-radiative decay pathways. The cross-linked system exhibited a high phosphorescence quantum yield, which was 2.5 times higher (13%) than that of the blend system (5%). In comparison with the crystallization strategy, the strategies involving amorphous polymers, which can be fabricated as thin films for practical applications, demonstrated better processability and flexibility, and various organic luminogens can be incorporated by simple doping or covalent linkage to realize the RTP emission by the rigid environment constructed using a polymer matrix. For the crystallization strategy, more requirements are needed to form the organic crystals/co-crystals, which are related to the rigidity of molecular structures, the crystal growth technologies, the compatibility of multiple components, etc.
Supramolecular assemblies can also be utilized to effectively restrict the vibrational motions of RTP luminogens through non-covalent interactions, offering precise control over molecular orientation and packing. Host molecules such as cyclodextrin (CD), cucurbituril (CB), and other macrocyclic compounds have been selected to support the guest molecules as emissive sources (Fig. 2e).37–40 As early as 1982, Turro et al. reported a host–guest assembly-based RTP material in a nitrogen-purged aqueous solution by doping 1-bromonaphthalene or 1-chloronaphthalene into β-CD.41 This pioneering work paved the way for development of supramolecular assembly-induced RTP systems. CBs also serve as ideal host materials due to their strong binding affinity for positively charged guests, and the multiple carbonyl groups can facilitate intermolecular interactions, such as C–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C interactions, to promote phosphorescence emission. Accordingly, a high phosphorescence quantum yield of 81.2% with the lifetime of 5.40 ms has been achieved by grinding CB[6] with the pyridinium derivative PYCl, while that of PYCl was only 2.6% with the lifetime of 5.76 μs.39 This was mainly attributed to the strict encapsulation by host–guest interactions, which helped to suppress non-radiative relaxation and promote ISC. This principle of supramolecular self-assembly presents a convenient method for enhancing RTP performance, where the binding constant between host macrocyclic molecules and phosphor guests is a critical factor. Moreover, this strategy effectively addresses the challenge of poor water solubility of organic materials, thereby broadening their potential applications in biological fields. Most recently, the quantum yield of 18.5% was achieved using aqueous organic red/NIR RTP probes in a highly viscous solution.40 It was accomplished through the supramolecular assembly of CB[8] with pyridinium derivatives, which were successfully employed in two-photon phosphorescence imaging of lysosomal viscosity and time-resolved in vivo imaging for lipopolysaccharide-induced inflammation diagnosis.
C interactions, to promote phosphorescence emission. Accordingly, a high phosphorescence quantum yield of 81.2% with the lifetime of 5.40 ms has been achieved by grinding CB[6] with the pyridinium derivative PYCl, while that of PYCl was only 2.6% with the lifetime of 5.76 μs.39 This was mainly attributed to the strict encapsulation by host–guest interactions, which helped to suppress non-radiative relaxation and promote ISC. This principle of supramolecular self-assembly presents a convenient method for enhancing RTP performance, where the binding constant between host macrocyclic molecules and phosphor guests is a critical factor. Moreover, this strategy effectively addresses the challenge of poor water solubility of organic materials, thereby broadening their potential applications in biological fields. Most recently, the quantum yield of 18.5% was achieved using aqueous organic red/NIR RTP probes in a highly viscous solution.40 It was accomplished through the supramolecular assembly of CB[8] with pyridinium derivatives, which were successfully employed in two-photon phosphorescence imaging of lysosomal viscosity and time-resolved in vivo imaging for lipopolysaccharide-induced inflammation diagnosis.
Heavy atoms, such as bromine (Br), iodine (I), selenium (Se), and tellurium (Te), significantly enhance the SOC between singlet and triplet excited states, due to the high nuclear charge, thus leading to a fast phosphorescence radiative decay rate (kp) and boosting the phosphorescence emission by facilitated ISC processes (Fig. 2f).42–44 The heavy atom effect is broadly classified into two types based on the nature of bonding: the internal heavy atom effect, which is mediated through covalent bonds, and the external heavy atom effect, facilitated by non-covalent interactions such as ionic or halogen bonding. In 2011, Kim et al. introduced the key design principles that enabled the development of bright RTP materials by incorporating heavy atoms.42 The presence of halogen bonding in these crystals directly enhanced ISC and minimized vibrational losses, thereby activating phosphorescence emission. As a result, the high RTP quantum yield of up to 55% was achieved. Particularly, the external heavy atom effect with multiple non-covalent interactions in aggregated states has garnered significant scientific interest in inducing phosphorescence emission without necessitating complex and multi-step chemical synthesis.45,46 In 2021, Ma et al. developed a general strategy for amorphous RTP materials by doping fluorescent dyes into a polymer matrix with bromide ions (PAB).45 Compared to the polymer with chloride ions, the phosphorescence emission intensities in PAB greatly enhanced for the external heavy atom effect, while the lifetimes decreased by an order of magnitude due to the increase of kp.
Deuteration can also enhance the spin prohibition factor by inducing spin configurational and nuclear alterations during the transition from the S1 to T1 state, and thus facilitating the ISC process.47–49 Moreover, deuteration can result in reduced vibrational frequencies due to the greater mass, which consequently diminishes the rate of non-radiative decay processes and prolongs the triplet lifetime for RTP emission (Fig. 2g). The underlying principle of deuteration lies in the differences in vibrational energy levels between hydrogen and deuterium. It has been demonstrated by Hirata et al. in 2014 that the selective deuteration of aromatic hydrocarbons significantly lowered the C–H stretching vibration energy, as described by the Franck–Condon principle, thereby extending the RTP lifetimes.47 Combined with the increased ISC rates by deuteration, the high RTP quantum yield of 6.2% with an extended RTP lifetime of up to 1.36 s was achieved. Recently, researchers have explored the use of deuterated reagents or deuterated small molecules to enhance phosphorescence emissions in the solid state, achieving higher efficiency and longer phosphorescence lifetime. For instance, the increased phosphorescence quantum yields from 1.70% to 27.04% have been obtained by our group through the deuteration process of the OH group in phenothiazine derivatives, due to the lower vibrational frequencies of O–D bonds.49 Furthermore, the integration of deuteration strategies with supramolecular assembly or host–guest interactions holds promise for the development of highly efficient and versatile phosphorescent systems.
Thus, the design strategies and intrinsic mechanisms of organic RTP materials have been progressively refined over the years. Moreover, the energy transfer mechanisms to facilitate the development of phosphorescent materials are mainly Förster resonance energy transfer (FRET) and Dexter energy transfer (DET). FRET is a non-radiative energy transfer process that occurs through the dipole–dipole interactions between molecules, resulting in the transfer of energy from the excited state of the donor to acceptor.50 The generation of resonance energy transfer relied on the extent of spectral overlap. The emission spectrum of the donor should overlap with the absorption spectrum of the acceptor. It has also been proven as an effective design strategy for constructing near-infrared (NIR) afterglow materials with high brightness. For instance, when FRET occurs from the excited triplet states of organic RTP materials with visible afterglow to the excited single states of the NIR emissive materials with matched properties, NIR afterglow can be realized, which is hard to achieve by the single components of organic RTP materials.
DET occurs through exchange interaction rather than dipole–dipole coupling. This mechanism is often utilized in systems involving triplet–triplet energy transfer (TTET), which enhances the triplet exciton population and therefore increases phosphorescence emission. It demonstrates the strong dependence of Dexter couplings on the electronic structure of the donor and acceptor and becomes more efficient when the donor and acceptor are in close proximity (typically less than 1 nm), with the donor and acceptor molecules having similar electronic states. For example, once efficient TTET has been observed between phthalimide and 1,8-naphthalimide derivatives, the RTP lifetime can largely enhance by ∼100 ms compared to those without TTET.51 Such advances in RTP materials hold significant promise for applications across various fields, including optoelectronics, anticounterfeiting, sensing, and bioimaging.
Applications and outlook
Compared to fluorescent materials, organic RTP materials are distinguished by their prolonged emission lifetimes in aggregated states, which render them highly attractive for applications in anticounterfeiting8,10,21 and bioimaging.6,7,11,12,20 In the field of anticounterfeiting, organic RTP materials can be strategically employed to develop time-dependent security features that encode, conceal, and reveal information over predetermined durations. As depicted in Fig. 3a, organic RTP materials are employed as inks to print patterns, and the information can be further encrypted by phosphorescence modes, while fluorescence signals act as false information.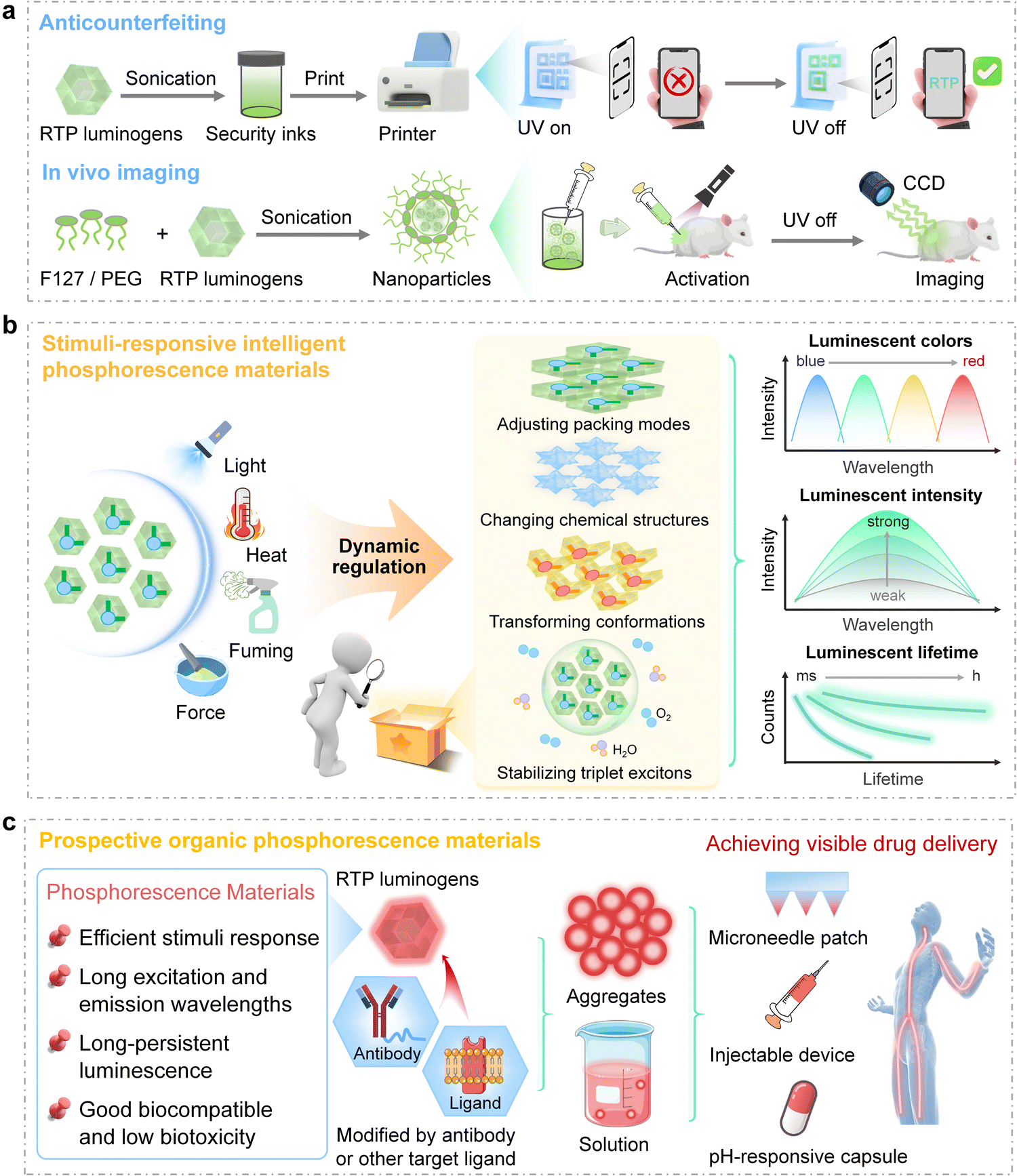 | ||
| Fig. 3 (a) Schematic diagram of the utilization of phosphorescent materials in anticounterfeiting and in vivo imaging applications. (b) The characteristics of stimuli-responsive intelligent phosphorescent materials and the underlying mechanism of their responsiveness, which results in observable phenomena such as color shifts, intensity variations, and changes in lifetime. (c) Schematic diagram of the utilization of stimuli-responsive intelligent phosphorescent materials for future clinical applications, highlighting the necessary advancements and integration strategies required to realize their full potential in drug delivery systems. | ||
Besides, triplet excitons, which are sensitive to molecular aggregates and environments, offer a fascinating opportunity for the development of stimuli-responsive phosphorescent materials, and the stimuli include temperature, force, light, pH, or the presence of certain species (Fig. 3b).52–55 This tunability is achieved by facilitating or hindering the ISC or by altering the microenvironment surrounding the phosphorescent centres, which is always associated with the modulation of aggregated structures. For example, photo-induced phosphorescence of single organic crystals, as first reported by our group, could be attributed to enhanced π–π interactions,21 while force-induced phosphorescence could be related to the variations in molecular conformations.54 Wu, Yu, and Tang et al. have explored a photo-thermo-induced phosphorescence mechanism, which arises from the molecular rearrangements following photoirradiation.56 Additionally, the chemical structures of organic RTP materials can also be changed upon acid/base exposure, ion addition, or photochemical reactions, allowing precise control over RTP emission intensity and color.57 Consequently, these structural and environmental modifications result in dynamic changes in the phosphorescence properties of the materials, such as color shifts, intensity variations, changes in lifetime, and modifications in quantum yield.
There is a growing interest in developing stimuli-responsive phosphorescence materials due to their remarkable reversible sensitivity, rapid response, and ease of control. These materials facilitate the creation of multi-level anti-counterfeiting measures that are challenging for counterfeiters to reproduce.58–60 For instance, temperature-dependent RTP materials with time-resolved RTP emission can be incorporated into labels, enabling genuine products to display distinct time-dependent phosphorescence characteristics, such as specific colors and intensities with different patterns at various temperatures. Also, stimuli-responsive phosphorescent materials can be used to monitor the low concentration of oxygen and humidity due to their quenching effect on excited triplet states.
When organic RTP materials are fabricated into nanoparticles, which are then injected into the dorsal region of animals like mice, the long-lasting phosphorescence emission significantly reduces background noise with fluorescence emission, thereby enhancing high-resolution imaging in vivo.6,9,11,12 Through utilizing phosphorescent materials with specific responses in physiological environments, researchers can achieve highly controlled bioimaging in both spatial and temporal dimensions. Moreover, the development of efficient near-infrared (NIR) RTP materials has opened new avenues for deep tissue imaging. Recently, four-branched luminogens with strong electronic pull–push properties have been reported by our group with ultrahigh afterglow brightness in the red/NIR region, mainly due to the shielding effect against oxygen and water and strong intermolecular interactions of branched structures. They enabled tissue penetration up to the depth of 35 mm, and in vivo phosphorescence imaging of organs with high signal-to-background ratios has been achieved, marking a significant advancement in bioimaging technologies.12
Moreover, organic RTP materials hold the potential for non-invasive monitoring of biological processes in response to physiological changes, such as oxidative stress or pH variations (Fig. 3c). They can be delivered into the body through various methods, such as pH-responsive encapsulation, injectable devices, or microneedle patches. Ultimately, employing time-gated technology enhances the visualization of drug release and enables precise detection and tracking of tumors or other biomarkers without interference from background signals. The modulation of aggregated structures by rational molecular design offers effective guidance for balancing triplet exciton stabilization with responsiveness to stimuli. To increase specificity for disease areas or biomarkers, targeted ligands such as antibodies or peptides can be introduced, guiding the phosphorescent probes to selectively bind to target cells or tissues and thereby amplify the signal within the intended area.
In addition, organic long-persistent luminescence (LPL) materials with charge-separation mechanisms and thermally-activated delayed fluorescence (TADF) emitters are other types of organic luminescent materials with long lifetimes.61–64 For LPL involving charge-separation mechanisms, photoexcitation leads to the generation of charge-separated states (typically electron–hole pairs). These charge carriers are trapped in deep traps or defect states of organic materials. Over time, the trapped charges recombine to release energy in the form of luminescence, which can last for seconds to hours. While the mechanisms underlying RTP have been extensively studied, the trapping and recombination process of charge carriers in defect states of LPL materials remains less well understood. Further research is required to provide universal design principles for the development of efficient LPL materials. TADF involves emission from a singlet excited state with a delayed component, which relies upon reducing the energy gap between singlet and triplet states, so that thermal energy can promote reverse intersystem crossing from the triplet to singlet state. Until now, many highly efficient OLED devices have been fabricated using TADF emitters.
In conclusion, extensive scientific efforts have driven significant advancements in organic RTP materials, including broadening of emission wavelengths from deep blue to NIR, an enhancement of lifetimes from microseconds to seconds, and efficiencies reaching up to 96.5%. The wavelengths and lifetimes of RTP materials are determined by a complex interplay of molecular configuration, electronic structure, and environmental factors. Generally, the emission wavelengths are primarily controlled by the electronic structures of organic luminogens and the possible electronic coupling at aggregated states, which can determine the energy levels of excited triplet states. The RTP lifetimes are strongly related to the stabilization of excited triplet states, which can be affected by the SOC of singlet and triplet states and the rigidity of the environment. Generally, strong SOC can promote the intersystem crossing but usually shorten the RTP lifetimes for the facilitated decay processes. In rigid environments, such as crystalline solids or polymer matrices, the non-radiative transitions as the competitive process can be suppressed largely, which is beneficial for the extension of RTP lifetimes.
The development of RTP materials represents a substantial leap toward their applications across diverse fields. However, with the aim to meet the requirements for practical applications, further enhancement of RTP sensitivity and responsiveness is essential. A deeper investigation into the “molecular uniting set identified characteristic (MUSIC)” concept, as proposed by our group, will offer a strategic framework for optimizing molecular design.13–15 Continued exploration of the MUSIC-guided molecular design and other innovative approaches will be crucial for advancing RTP materials toward real-world applications, ultimately bridging the gap between laboratory research and practical implementation.
Author contributions
Y. Y. prepared the manuscript under the supervision of Q. L. and Z. L. All authors contributed to the general discussion.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22235006 and 22405200) and the Foundation of Hubei Scientific Committee (2022BAA015, 2024AFA021 and 2022EHB010).Notes and references
- D. B. Clapp, The phosphorescence of tetraphenylmethane and certain related substances, J. Am. Chem. Soc., 1939, 61, 523–524 CrossRef CAS
.
- W. Yuan, X. Shen, H. Zhao, J. W. Y. Lam, L. Tang, P. Lu, C. Wang, Y. Liu, Z. Wang, Q. Zheng, J. Z. Sun, Y. Ma and B. Z. Tang, Crystallization-induced phosphorescence of pure organic luminogens at room temperature, J. Phys. Chem. C, 2010, 114, 6090–6099 CrossRef CAS
- W. Zhao, Z. He and B. Z. Tang, Room-temperature phosphorescence from organic aggregates, Nat. Rev. Mater., 2020, 5, 869–885 CrossRef CAS
- Y. Gao, J. Lu, Q. Liao, S. Li, Q. Li and Z. Li, Thermal annealing promoted room temperature phosphorescence: Motion models and internal mechanism, Natl. Sci. Rev., 2023, 10, nwad239 CrossRef CAS PubMed
- Z. Chen, X. Chen, D. Ma, Z. Mao, J. Zhao and Z. Chi, Synergetic conformational regulations in ground and excited states for realizing stimulus-responsive and wide-tuning room temperature phosphorescence, J. Am. Chem. Soc., 2023, 145, 16748–16759 CrossRef CAS PubMed
- K. Chang, L. Xiao, Y. Fan, J. Gu, Y. Wang, J. Yang, M. Chen, Y. Zhang, Q. Li and Z. Li, Lighting up metastasis process before formation of secondary tumor by phosphorescence imaging, Sci. Adv., 2023, 9, eadf6757 CrossRef CAS PubMed
- L. Gu, H. Wu, H. Ma, W. Ye, W. Jia, H. Wang, H. Chen, N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Color-tunable ultralong organic room temperature phosphorescence from a multicomponent copolymer, Nat. Commun., 2020, 11, 944 CrossRef CAS PubMed
- Y. Yang, J. Wang, D. Li, J. Yang, M. Fang and Z. Li, Tunable photoresponsive behaviors based on triphenylamine derivatives: The pivotal role of π-conjugated structure and corresponding application, Adv. Mater., 2021, 33, 2104002 CrossRef CAS PubMed
- Y. Wang, H. Gao, J. Yang, M. Fang, D. Ding, B. Z. Tang and Z. Li, High performance of simple organic phosphorescence host-guest materials and their application in time resolved bioimaging, Adv. Mater., 2021, 33, 2007811 CrossRef CAS PubMed
- L. Gu, H. Shi, L. Bian, M. Gu, K. Ling, X. Wang, H. Ma, S. Cai, W. Ning, L. Fu, H. Wang, S. Wang, Y. Gao, W. Yao, F. Huo, Y. Tao, Z. An, X. Liu and W. Huang, Colour-tunable ultra-long organic phosphorescence of a single-component molecular crystal, Nat. Photonics, 2019, 13, 406–411 CrossRef CAS
- F. Xiao, H. Gao, Y. Lei, W. Dai, M. Liu, X. Zheng, Z. Cai, X. Huang, H. Wu and D. Ding, Guest-host doped strategy for constructing ultralong-lifetime near-infrared organic phosphorescence materials for bioimaging, Nat. Commun., 2022, 13, 186 CrossRef CAS PubMed
- J. Gu, W. Yuan, K. Chang, C. Zhong, Y. Yuan, J. Li, Y. Zhang, T. Deng, Y. Fan, L. Yuan, S. Liu, Y. Xu, S. Ling, C. Li, Z. Zhao, Q. Li, Z. Li and B. Z. Tang, Organic materials with ultrabright phosphorescence at room temperature under physiological conditions for bioimaging, Angew.
Chem., Int. Ed., 2024, 64, e202415637 CrossRef PubMed
- W. Ye, H. Ma, H. Shi, H. Wang, A. Lv, L. Bian, M. Zhang, C. Ma, K. Ling, M. Gu, Y. Mao, X. Yao, C. Gao, K. Shen, W. Jia, J. Zhi, S. Cai, Z. Song, J. Li, Y. Zhang, S. Lu, K. Liu, C. Dong, Q. Wang and W. Huang, Confining isolated chromophores for highly efficient blue phosphorescence, Nat. Mater., 2021, 20, 1539–1544 CrossRef CAS PubMed
- Z. Xu, Z. Wang, W. Yao, Y. Gao, Y. Li, H. Shi, W. Huang and Z. An, Supercooled liquids with dynamic room temperature phosphorescence using terminal hydroxyl engineering, Angew. Chem., Int. Ed., 2023, 62, e202301564 CrossRef CAS PubMed
- Q. Liao, Q. Li and Z. Li, The key role of molecular packing in luminescence property: From adjacent molecules to molecular aggregates, Adv. Mater., 2023, 2306617 CrossRef PubMed
- J. Gu, Z. Li and Q. Li, From single molecule to molecular aggregation science, Coord. Chem. Rev., 2023, 475, 214872 CrossRef CAS
- Q. Li and Z. Li, Molecular packing: Another key point for the performance of organic and polymeric optoelectronic materials, Acc. Chem. Res., 2020, 53, 962–973 CrossRef CAS PubMed
- S. Lower and M. El-Sayed, The triplet state and molecular electronic processes in organic molecules, Chem. Rev., 1966, 66, 199–241 CrossRef CAS
- C. M. Marian, Spin–orbit coupling and intersystem crossing in molecules, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 187–203 CAS
- Z. Cong, M. Han, Y. Fan, Y. Fan, K. Chang, L. Xiao, Y. Zhang, X. Zhen, Q. Li and Z. Li, Ultralong blue room-temperature phosphorescence by cycloalkyl engineering, Mater. Chem. Front., 2022, 6, 1606–1614 RSC
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng, K. Pu and Z. Li, The influence of the molecular packing on the room temperature phosphorescence of purely organic luminogens, Nat. Commun., 2018, 9, 840 CrossRef PubMed
- W. Xu, G. Huang, Z. Yang, Z. Deng, C. Zhou, J. Li, M. Li, T. Hu, B. Z. Tang and D. L. Phillips, Nucleic-acid-base photofunctional cocrystal for information security and antimicrobial applications, Nat. Commun., 2024, 15, 2561 CrossRef CAS PubMed
- M. Fang, J. Yang, X. Xiang, Y. Xie, Y. Dong, Q. Peng, Q. Li and Z. Li, Unexpected room-temperature phosphorescence from a non-aromatic, low molecular weight, pure organic molecule through the intermolecular hydrogen bond, Mater. Chem. Front., 2018, 2, 2124–2129 RSC
- W. Guo, Y. Chen, C. Tung and L. Wu, Ultralong room-temperature phosphorescence of silicon-based pure organic crystal for oxygen sensing, CCS Chem., 2022, 4, 1007–1015 CrossRef CAS
- T. Yang, Y. Li, Z. Zhao and W. Yuan, Clustering-triggered phosphorescence of nonconventional luminophores, Sci. China:Chem., 2023, 66, 367–387 CrossRef CAS
- J. Wei, B. Liang, R. Duan, Z. Cheng, C. Li, T. Zhou, Y. Yi and Y. Wang, Induction of strong long-lived room-temperature phosphorescence of N-phenyl-2naphthylamine molecules by confinement in a crystalline dibromobiphenyl matrix, Angew. Chem., Int. Ed., 2016, 55, 15589–15593 CrossRef CAS PubMed
- D. Lee, O. Bolton, B. C. Kim, J. H. Youk, S. Takayama and J. Kim, Room temperature phosphorescence of metal-free organic materials in amorphous polymer matrices, J. Am. Chem. Soc., 2013, 135, 6325–6329 CrossRef CAS
- L. Zhou, J. Song, Z. He, Y. Liu, P. Jiang, T. Li and X. Ma, Achieving efficient dark blue room-temperature phosphorescence with ultra-wide range tunable-lifetime, Angew. Chem., Int. Ed., 2024, 63, e202403773 CrossRef CAS
- D. Li, Y. Yang, J. Yang, M. Fang, B. Z. Tang and Z. Li, Completely aqueous processable stimulus responsive organic room temperature phosphorescence materials with tunable afterglow color, Nat. Commun., 2022, 13, 347 CrossRef CAS PubMed
- D. Li, J. Yang, M. Fang, B. Z. Tang and Z. Li, Stimulus-responsive room temperature phosphorescence materials with full-color tenability from pure organic amorphous polymers, Sci. Adv., 2022, 8, eabl8392 CrossRef CAS PubMed
- P. She, J. Duan, F. Li, Y. Zhou, Y. Qin, J. Wei, S. Liu, Y. Ma and Q. Zhao, Exploring electronic coupling effects in amorphous polymers for multi-color persistent room-temperature phosphorescence, Sci. China Mater., 2023, 66, 4749–4755 CrossRef CAS
- H. Sun, X. Wei, Y. He, Y. Xiao, Y. Wu, Z. Xie and T. Yu, Regulation of various photo-active UOPs in a polymer matrix by tuning intermolecular charge transfer, Mater. Chem. Front., 2020, 4, 386–399 RSC
- J. Wei, C. Liu, J. Duan, A. Shao, J. Li, J. Li, W. Gu, Z. Li, S. Liu, Y. Ma, W. Huang and Q. Zhao, Conformation-dependent dynamic organic phosphorescence through thermal energy driven molecular rotations, Nat. Commun., 2023, 14, 627 CrossRef CAS PubMed
- X. Zhang, J. You, J. Zhang, C. Yin, Y. Wang, R. Li and J. Zhang, Stimuli-responsive organic ultralong phosphorescent materials with complete biodegradability for sustainable information encryption, CCS Chem., 2023, 5, 2140–2151 CrossRef CAS
- M. S. Kwon, Y. Yu, C. Coburn, A. W. Phillips, K. Chung, A. Shanker, J. Jung, G. Kim, K. Pipe, S. R. Forrest, J. H. Youk, J. Gierschner and J. Kim, Suppressing molecular motions for enhanced room-temperature phosphorescence of metal-free organic materials, Nat. Commun., 2015, 6, 8947 CrossRef PubMed
- Y. Niu, Y. Guan, C. Long, C. Ren, J. Lu, C. Jin, P. Wang, X. Fan and H. Xie, A universal strategy for achieving dual cross-linked networks to obtain ultralong polymeric room temperature phosphorescence, Sci. China:Chem., 2023, 66, 1161–1168 CrossRef CAS
- X. Ma and Y. Liu, Supramolecular purely organic room-temperature phosphorescence, Acc. Chem. Res., 2021, 54, 3403–3414 CrossRef CAS PubMed
- H. Chen, X. Ma, S. Wu and H. Tian, A rapidly self-healing supramolecular polymer hydrogel with photostimulated room-temperature phosphorescence responsiveness, Angew. Chem., Int. Ed., 2014, 53, 14149–14152 CrossRef CAS
- Z. Zhang, Y. Chen and L. Yu, Efficient room-temperature phosphorescence of a solid-state supramolecule enhanced by cucurbit[6]uril, Angew. Chem., Int. Ed., 2019, 58, 6028–6032 CrossRef CAS PubMed
- Y. Li, Z. Wu, Z. Huang, C. Yin, H. Tian and X. Ma, Activatable red/near-infrared aqueous organic phosphorescence probes for improved time-resolved bioimaging, Natl. Sci. Rev., 2024, nwae383 Search PubMed
- N. J. Turro, J. D. Bolt, Y. Kurodat and I. Tabushi, A study of the kinetics of inclusion of halo naphthalenes with β-cyclodextrin via time correlated phosphorescence, Photochem. Photobiol., 1982, 35, 69–72 CrossRef CAS
- O. Bolton, K. Lee, H. Kim, K. Y. Lin and J. Kim, Activating efficient phosphorescence from purely organic materials by crystal design, Nat. Chem., 2011, 3, 205–210 CrossRef CAS PubMed
- Y. He, J. Wang, Q. Li, S. Qu, C. Zhou, C. Yin, H. Ma, H. Shi, Z. Meng and Z. An, Highly efficient room-temperature phosphorescence promoted via intramolecular-space heavy-atom effect, Adv. Opt. Mater., 2023, 11, 2201641 CrossRef CAS
- Y. Su, S. Z. F. Phua, Y. Li, X. Zhou, D. Jana, G. Liu, W. Q. Lim, W. K. Ong, C. Yang and Y. Zhao, Ultralong room temperature phosphorescence from amorphous organic materials toward confidential information encryption and decryption, Sci. Adv., 2018, 4, eaas9732 CrossRef PubMed
- Z. Yan, X. Lin, S. Sun, X. Ma and H. Tian, Activating room-temperature phosphorescence of organic luminophores via external heavy-atom effect and rigidity of ionic polymer matrix, Angew. Chem., Int. Ed., 2021, 60, 19735–19739 CrossRef CAS PubMed
- J. Wang, X. Gu, H. Ma, Q. Peng, X. Huang, X. Zheng, S. H. P. Sung, G. Shan, J. W. Y. Lam, Z. Shuai and B. Z. Tang, A facile strategy for realizing room temperature phosphorescence and single molecule white light emission, Nat. Commun., 2018, 9, 2963 CrossRef PubMed
- S. Hirata, K. Totani, T. Watanabe, H. Kaji and M. Vacha, Relationship between room temperature phosphorescence and deuteration position in a purely aromatic compound, Chem. Phys. Lett., 2014, 591, 119–125 CrossRef CAS
- H. Mieno, R. Kabe, N. Notsuka, M. D. Allendorf and C. Adachi, Long-lived room-temperature phosphorescence of coronene in zeolitic imidazolate framework ZIF-8, Adv. Opt. Mater., 2016, 4, 1015–1021 CrossRef CAS
- X. Liu, Q. Liao, J. Yang, Z. Li and Q. Li, Unveiling one-to-one correspondence between excited triplet states and determinate interactions by temperature-controllable blue-green-yellow afterglow, Angew. Chem., Int. Ed., 2023, 62, e202302792 CrossRef CAS PubMed
- Y. Wang, J. Yang, M. Fang, Y. Yu, B. Zou, L. Wang, Y. Tian, J. Cheng, B. Z. Tang and Z. Li, Förster resonance energy transfer: An efficient way to develop stimulus-responsive room-temperature phosphorescence materials and the applications, Matter, 2020, 2, 449–463 CrossRef
- J. Wang, Y. Yang, X. Sun, X. Li, L. Zhang and Z. Li, Management of triplet excitons transition: Fine regulation of Förster and dexter energy transfer simultaneously, Light: Sci. Appl., 2024, 13, 35 CrossRef CAS PubMed
- Q. Zhou, C. Yang and Y. Zhao, Dynamic organic room-temperature phosphorescent systems, Chem, 2023, 9, 2446–2480 CAS
- J. Yang, M. Fang and Z. Li, Stimulus-responsive room temperature phosphorescence materials: Internal mechanism, design strategy, and potential application, Acc. Mater. Res., 2021, 2, 644–654 CrossRef CAS
- J. Ren, Y. Wang, Y. Tian, Z. Liu, X. Xiao, J. Yang, M. Fang and Z. Li, Force-induced turn-on persistent room-temperature phosphorescence in purely organic luminogen, Angew. Chem., Int. Ed., 2021, 60, 12335–12340 CrossRef CAS PubMed
- J. Wang, C. Wang, Y. Gong, Q. Liao, M. Han, T. Jiang, Q. Dang, Y. Li, Q. Li and Z. Li, Bromine-substituted fluorene: Molecular structure, π–π interactions, room-temperature phosphorescence, and tricolor triboluminescence, Angew. Chem., Int. Ed., 2018, 57, 16821–16826 CrossRef CAS PubMed
- X. Liu, W. Zhao, Y. Wu, Z. Meng, Z. He, X. Qi, Y. Ren, Z. Yu and B. Z. Tang, Photo-thermo-induced room-temperature phosphorescence through solid-state molecular motion, Nat. Commun., 2022, 13, 3887 CrossRef CAS PubMed
- Y. Yang, A. Li, Y. Yang, J. Wang, Y. Chen, K. Yang, B. Z. Tang and Z. Li, Multi-stimulus room temperature phosphorescent polymers sensitive to light and acid cyclically with energy transfer, Angew. Chem., Int. Ed., 2023, 62, e202308848 CrossRef CAS PubMed
- G. Ye, Y. Yang, W. Yuan, J. Gu, S. Li, Q. Li and Z. Li, Multistimuli-responsive room-temperature phosphorescence: Adjustable electrostatic interactions and the corresponding accurate molecular packing, ACS Mater. Lett., 2024, 6, 4639–4648 CrossRef CAS
- S. Li, Y. Xie, A. Li, X. Li, W. Che, J. Wang, H. Shi and Z. Li, Different molecular conformation and packing determining mechanochromism and room-temperature phosphorescence, Sci. China Mater., 2021, 64, 2813–2823 CrossRef CAS
- X. Ma, T. Ye, H. Yang, G. Ling, D. Wang, J. Li, P. Gu, L. Xu, T. Sheng, F. Lin, R. Shen and Q. Zhang, Dynamic color-tunable ultra-long room temperature phosphorescence polymers with photo-chromism and water-stimuli response for multilevel anti-counterfeiting, Aggregate, 2024, 5, e579 CrossRef CAS
- N. Xue, H. Zhou, Y. Han, M. Li, H. Lu and C. Chen, A general supramolecular strategy for fabricating full-color-tunable thermally activated delayed fluorescence materials, Nat. Commun., 2024, 15, 1425 CrossRef CAS PubMed
- W. Ma, Y. Su, Q. Zhang, C. Deng, L. Pasquali, W. Zhu, Y. Tian, P. Ran, Z. Chen, G. Yang, G. Liang, T. Liu, H. Zhu, P. Huang, H. Zhong, K. Wang, S. Peng, J. Xia, H. Liu, X. Liu and Y. M. Yang, Thermally activated delayed fluorescence (TADF) organic molecules for efficient X-ray scintillation and imaging, Nat. Mater., 2022, 21, 210–216 CrossRef CAS PubMed
- R. Kabe and C. Adachi, Organic long persistent luminescence, Nature, 2017, 550, 384–387 CrossRef CAS PubMed
- K. Jinnai, R. Kabe, Z. Lin and C. Adachi, Organic long-persistent luminescence stimulated by visible light in p-type systems based on organic photoredox catalyst dopants, Nat. Mater., 2022, 21, 338–344 CrossRef CAS PubMed
| This journal is © the Partner Organisations 2025 |