
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Restricting intramolecular motion converts non-fluorescent semicroconaine dyes into turn-on aggregation-induced emission probes†
Rebecca Stradaa,
David Dunlop ce,
Michal Vorbab,
Amar Rajf,
Büşra Buse Tütüncücd,
Pasi Myllyperkiöf,
Tomáš Slaninac,
Tatu Kumpulainenf and
Peter Šebej*a
ce,
Michal Vorbab,
Amar Rajf,
Büşra Buse Tütüncücd,
Pasi Myllyperkiöf,
Tomáš Slaninac,
Tatu Kumpulainenf and
Peter Šebej*a
aRECETOX, Faculty of Science, Masaryk University, Kamenice 5, Brno, 625 00, Czech Republic. E-mail: sebej@recetox.muni.cz
bDepartment of Chemistry, Faculty of Science, Masaryk University, Kamenice 5, Brno, 625 00, Czech Republic
cInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo náměstí 542/2, Prague 6, 160 00, Czech Republic
dDepartment of Organic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, Prague 2, 128 40, Czech Republic
eDepartment of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, Prague 2, 128 40, Czech Republic
fDepartment of Chemistry/Nanoscience Center, Faculty of Mathematics and Science, University of Jyväskylä, Survontie 9B, Jyväskylä, FI-40014, Finland
First published on 28th March 2025
Abstract
Bucking the trend of all other polymethines, semicroconaines are virtually non-fluorescent (Φf < 0.1%) dyes. But their fluorescence could be increased through external physicochemical factors, thereby converting them into turn-on fluorescent probes. To test this hypothesis, we analyzed excited-state dynamics of a small library of semicroconaines bearing various auxochrome substituents to determine how they lose excited-state energy. Using a combined experimental/quantum-chemical approach, we found that isomerization of their methine bridge leads to non-radiative S1–S0 relaxation through a conical intersection. This relaxation pattern was consistently identified in semicroconaines with various auxochrome substituents (–F, –I, –OCH3, –SO3H and –NO2). Just as consistently, their fluorescence in solution significantly increased when increasing solvent viscosity and inducing complexation with two macromolecules, namely glycoluril dimer and DNA. Therefore, semicroconaine dyes display turn-on aggregation-induced emission, a mechanism that may be exploited for macromolecular sensing under physiological and pathological conditions.
Introduction
Commonly used in imaging and diagnostics, polymethines encompass a large class of small molecule fluorophores.1–3 In their simplest form, polymethine dyes consist of a conjugated carbon chain terminated by electron-donating and -withdrawing groups (Fig. 1). These groups can be rationally designed to modulate the photophysical and physicochemical properties of these dyes and to increase their stability.4–6 Case in point, polymethines with conjugated cyclic moieties, such as squarate subunits embedded in squaraines, often replace archetypal linear polymethines.7 These polymethines show enhanced photophysical properties, particularly their fluorescence quantum yield, because the planar conjugated subunit rigidifies their otherwise flexible carbon chain.8,9 Another rigidifying subunit is croconate, formally a pentamer of carbon monoxide and a higher analog of squarate. Polymethines with embedded croconate are known as croconaines and semicroconaines (Fig. 1). | ||
| Fig. 1 General polymethine, squaraine and croconaine (top left) structures and the most common non-radiative deexcitation pathways (top right; TICT stands for twisted intramolecular charge transfer). As with HBDI (bottom left), confining the rotation of semicroconaines 2 (bottom right) may increase their fluorescence substantially. | ||
(Semi-)croconaines are typically synthesized through a one-step condensation reaction between an electron-rich aromatic heterocycle and croconic acid.10 Also leveraging the well-known reactivity of squaric acid, alternative synthetic procedures include carbodiimide activation,11 microwave-assisted,12 green-chemistry approaches with choline chloride/urea mixtures as solvent and catalyst,13 and solvent-free methods.14 Independently of their synthetic procedure, semicroconaines share the high stability and absorptivity of squaraines.15 But under the same conditions, semicroconaines show fluorescence emission quantum yields approximately 2 orders of magnitude lower than those of polymethines and other squaraines.7 Consequently, (semi-)croconaines can be applied as absorption-change-based chemometers,16 photothermal therapeutic agents17 and in photoacoustic imaging,18 but not as fluorescence probes for imaging, in contrast to squaraines7 and linear polymethines. Moreover, the reasons for the low fluorescence quantum yields of semicroconaines are still unknown, raising major questions.”How do semicroconaines lose their excited-state energy? Can we prevent their non-radiative loss of excitation energy?”
Broadly speaking, non-radiative loss of excitation energy can proceed through several pathways, including thermal dissipation, energy or electron transfer, intersystem crossing, excited-state chemical reaction and structural reorganization.19 Structural reorganization is the most prevalent mechanism in polymethines, whether involving intramolecular motion (deformation and rotation),20,21 double-bond isomerization,22,23 twisted intramolecular charge transfer (TICT),24 or reversible intra- and intermolecular proton transfer.25 Regardless of mechanism, structural reorganization can be controlled by restricting the molecular geometry (e.g., through aggregation or host–guest interactions) to promote radiative deexcitation pathways. The resulting phenomenon is known as aggregation-induced emission (AIE).26,27
As a paradigmatic example of an AIE probe, hydroxybenzylidene-1,2-dimethylimidazolinone (HBDI) shows a significant increase in fluorescence quantum yield upon geometric restriction.28 Most notably, HBDI encapsulated into the β-barrel cavity of green fluorescent protein (GFP) displays a ≥100-fold higher fluorescence quantum yield (Φf ∼ 1) than the free fluorophore in solution (with Φf < 0.01)29 because its methine bridge is no longer able to undergo excited-state isomerization – a major, nonradiative deexcitation pathway of HBDI30 (Fig. 1). In other words, fluorescence emission becomes the main deexcitation pathway when the nonradiative deexcitation pathway is hindered by geometric restriction.
Geometric restriction can also be induced by encapsulation into other macromolecules, such as “octaacids”,28,31 by DNA-intercalation,32,33 or by increasing the viscosity of the solvent.34,35 Based on their structural similarity to HBDI, we hypothesized that semicroconaines could also be converted into fluorescent probes by geometric restriction. However, their underlying mechanism of non-fluorescent deexcitation remains unknown. Therefore, further research must be conducted to investigate the photophysics and deexcitation processes of semicroconaines.
Using a combined spectroscopic and quantum-chemical approach, this study aims at leveraging geometric restriction to improve semicroconaine fluorescence. For this purpose, we prepared and characterized a small library of indolenine-semicroconaines bearing various auxochrome substituents (–F, –I, –NO2, –OCH3, and –SO3H) to assess whether methine bridge isomerization is the main deexcitation pathway in semicroconaines. Subsequently, we (1) increased the viscosity of the solvent medium, and (2) promoted interactions with various macromolecules to prevent non-radiative deexcitation pathways. The ultimate goal of this research was to convert semicroconaines into turn-on AIE probes.
Results and discussion
We synthesized a library of semicroconaine dyes (2a–2f) in 10–50% overall yield by a one-pot condensation of a series of indolenines (1a–1f) with croconic acid under conditions optimized by Capozzi et al. (water/acetone, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, (v/v)) (see ESI,† for more details) (Scheme 1).15 All dyes 2 were relatively chemically stable in methanol and aqueous (including buffered) solutions in the dark (Table S8, ESI†). In both MeOH and PBS, 2 was also photochemically stable upon irradiation with a λexc = 572 nm LED (see ESI,† for more details).
:1, (v/v)) (see ESI,† for more details) (Scheme 1).15 All dyes 2 were relatively chemically stable in methanol and aqueous (including buffered) solutions in the dark (Table S8, ESI†). In both MeOH and PBS, 2 was also photochemically stable upon irradiation with a λexc = 572 nm LED (see ESI,† for more details).
 | ||
| Scheme 1 Synthesis of semicroconaines 2a–2f. | ||
The fluorescence quantum yield of semicroconaine dyes increases with solvent viscosity
To assess solvent effects on semicroconaines dyes, we analyzed the spectroscopic properties of 2a–2f in solvents (chloroform, DMSO, acetonitrile, tetrahydrofuran, methanol and PBS; Table 1 and Tables S6, S7, ESI†) with varying (i) polarity, (ii) H-donor/H-acceptor ability, and (iii) viscosity by steady-state, UV-vis spectroscopy. In all compounds, absorption maximum was observed in the 560–570 nm range, with weak (Φf < 0.1%) or below-the-limit-of-quantification (LoQ) emission in the 600–630 nm range. The full list of spectroscopic properties of the dyes is presented in Table 1 for methanol solutions and in Table S6 for other solvents, with more details in ESI.†| Cmpdb | λmaxabs/nm | εmax/mol−1 dm3 cm−1 | λmaxem/nm | Δṽ/cm−1c |
Φf/% | εΦd |
|---|---|---|---|---|---|---|
| a Methanol solutions: A(λmax) ≅ 1 (c(dye) = 1–2 × 10−3 mol dm−3) was adjusted for absorption spectroscopy, and A(λmax) < 0.1 (c(dye) < 8 × 10−6 mol dm−3) was used for emission spectroscopy and fluorescence quantum-yield measurements; all experiments were performed at 22 °C.b Structures are shown in Scheme 1.c Stokes shift.d εΦ/mol−1 dm3 cm−1; fluorophore brightness (the product of the molar absorption coefficient at λmaxem and the corresponding fluorescence quantum yield).e Data from the literature.15f Below the limit of quantification (LoQ).g Not measurable. | ||||||
| 2a | 565 (564)e | 13000 |
600 (631)e | 1032 | 0.01 ± 0.01 | 1.3 |
| 2b | 564 (565)e | 17200 |
602 (605)e | 1119 | Below LoQf | n.m.g |
| 2c | 564 | 19900 |
606 | 1229 | 0.04 ± 0.01 | 8.0 |
| 2d | 563 | 32800 |
599 | 1068 | 0.01 ± 0.01 | 3.3 |
| 2e | 571 (568)e | 1600 | 649 (640)e | 2105 | Below LoQf | n.m.g |
| 2f | 564 | 49000 |
607 | 1255 | 0.09 ± 0.01 | 44.1 |
Stokes shifts fell within the 900–2100 cm−1 range, close to that of analogous pentamethines (≤900 cm−1),36,37 with only minor geometric differences between absorbing and emitting species. This interpretation was supported by TD-DFT calculations, which show that S0–S1 transitions of 2a–2f have a π–π* character with only a minor charge transfer character (except for 2d; ESI,† Chapter S11.3). Moreover, no significant relationship between absorption/emission band maxima and electronic effects of the R-substituent was found in any of the semicroconaines dyes analyzed in this study (Scheme 1; quantified using the Hammett constant).
No significant correlation was found between the photophysical properties of 2a–2d and solvent polarity or hydrogen-bonding ability either (Table S6, ESI†). However, we did observe significant changes in emission band shapes and fluorescence quantum yields as a function of solvent viscosity (Table S5, ESI†). To better characterize this effect, we measured UV-vis, transient absorption and emission spectra in carefully selected binary DMSO/glycerol solvent mixtures with constant polarity across a wide range of glycerol molar fractions and, hence, viscosity values (from η = 2.2 cP to η = 100 cP).38
The hydrogen-donating ability of the solvent increased with the molar fraction of glycerol, potentially stabilizing negatively charged species such as the nitro group in the excited state. However, the absorption spectra of 2a–2d did not vary significantly with the glycerol molar fraction (Fig. S52, S54, S56, S58, ESI†), suggesting that hydrogen bonding between the solvent and semicroconaines does not play a major role. The minor changes observed in the emission spectra of 2a–2c in both low (DMSO, η = 2.2 cP) and high (DMSO/glycerol, x(gly) = 0.615, η = 100 cP) viscosity media indicate that solvent viscosity has only a weak effect on the radiation deactivation pathway. This inference is supported by the small (∼2-fold increase) change in the Φf of 2a–2c (Table S5, ESI†).
In contrast to 2a–2c, the emission band of 2d is significantly broadened, with a shoulder at λ = 730 nm (Fig. 2) in low-viscosity medium (DMSO, η = 2.2 cP), but not in high-viscosity medium (DMSO/glycerol, x(gly) = 0.615, η = 100 cP). This shoulder in the emission spectra of 2d in low-viscosity medium may be explained by the known effect of its nitro group, which can rotate out of plane in the excited state.39,40 So in the low-viscosity medium (DMSO), the locally excited singlet state (1LE) relaxes by geometrical reorganization of the nitro group into a twisted charge-transfer state with a weak, red-shifted emission (λ ∼ 730 nm).41,42 As a result, the partial charges on electron-donating and -withdrawing moieties of these semicroconaines can be affected by the viscosity and hydrogen-bonding ability of the solvent, respectively.
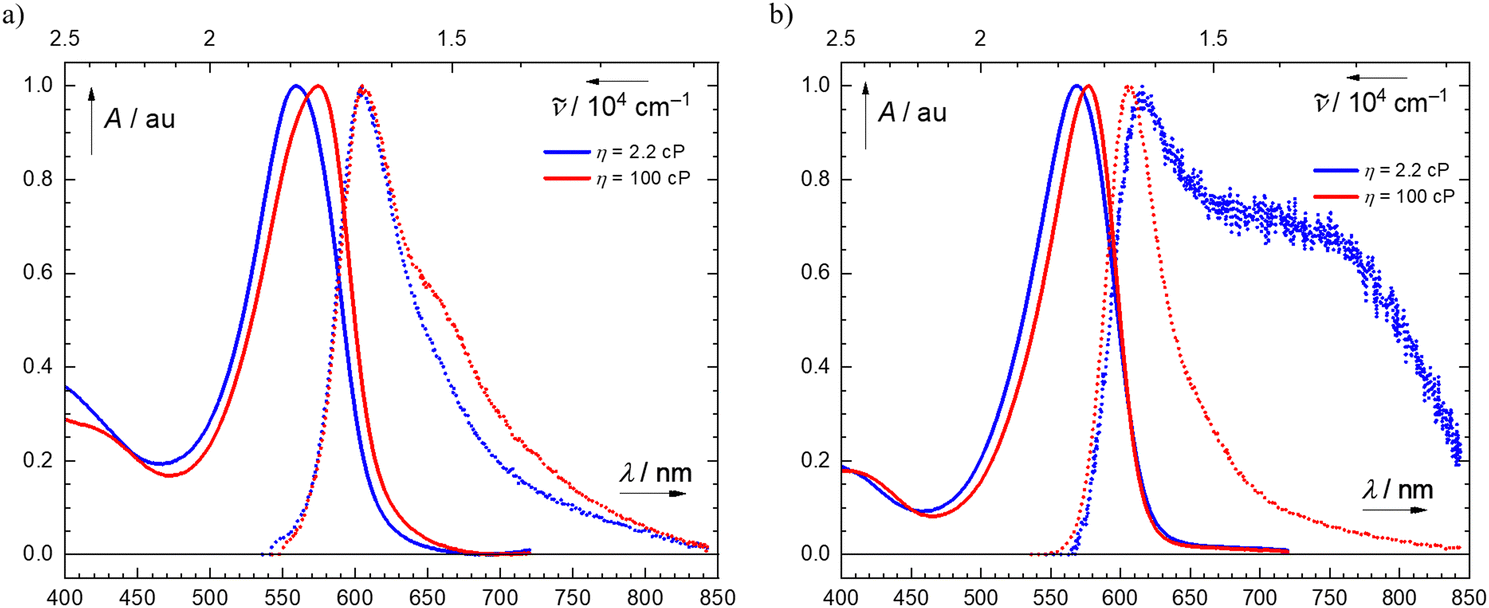 | ||
| Fig. 2 Normalized steady-state absorption (solid line) and emission (dotted line) spectra of (a) 2a in DMSO (η = 2.2 cP, blue) and in DMSO/glycerol (η = 100 cP, red) (left panel); and (b) 2d in DMSO (η = 2.2 cP, blue) and in DMSO/glycerol (η = 100 cP, red) (right panel). | ||
Based on this evidence, in low-viscosity media, 2d undergoes a large geometry change in the excited state. In line with these findings, the fluorescence emission quantum yield of 2d increased ∼7-fold from 0.08 to 0.53% (Table 2 and Table S5, ESI†), also implying that the nonradiative deactivation pathway changes with the increase in solvent viscosity. This viscosity effect may be attributed to donor–acceptor interactions between the electron-rich indolenine moiety and the electron-withdrawing nitro group of 2d.
| Observed by: | A(S1) → B(S1) → C | |||||
|---|---|---|---|---|---|---|
| fs-TA, FLUPS | fs-TA, FLUPS | Ifl | fs-TA, ns-TA | ns-TA | ||
| Monitoring wavelengths: | λabs(max) ∼565–590 nm | λabs(max) ∼565–585 nm | λabs(max) ∼460 nm | λabs(max) ∼640 nm | ||
| λem(max) ∼ 610 nm | λem(max) ∼610 nm | |||||
| Cmpd. | η/cPa | τA/psb,e | τB/psc,e | Φf/% | τC/nsd | τC′/nsd |
| a For more details on the solvent mixtures composition, please refer to the ESI (Table S4).b For more details on spectral component A, and its properties, please refer to ESI (Table S10).c For more details on spectral component B, and its properties, please refer to ESI (Table S10).d For more details on spectral component C, and its properties, please refer to ESI (Tables S10 and S11).e The spectral components were observed by both TA and FLUPS, but the respective τ values were measured by FLUPS (Table S10).f Measured in methanol (Fig. S117, S119 and S120, ESI).g Not observed.h In degassed media, the observed lifetimes were τC = 138 ± 5 μs (Fig. S142, ESI) and τC’ = 140 ± 7 μs (Fig. S143, ESI).i In degassed media, the observed lifetimes were τC = 113 ± 3 μs (Fig. S149, ESI) and τC′ = 111 ± 5 μs (Fig. S151, ESI). | ||||||
| 2a | 2.2 | 1.9 ± 0.1 | 49.9 ± 0.2 | 0.32 | 212 ± 20f | n.o.f,g |
| 100 | 4.2 ± 0.1 | 47.8 ± 0.2 | 0.53 | 10600 ± 100 |
n.o.g | |
| 2d | 2.2 | 2.8 ± 0.1 | 12.7 ± 0.2 | 0.08 | 830 ± 50h | 864 ± 30h |
| 100 | 5.6 ± 0.2 | 45.5 ± 0.3 | 0.53 | 13000 ± 2000i |
13500 ± 700i |
|
High viscosity media impair the non-radiative S1-deexcitation of semicroconaines
We further investigated the deexcitation pathways of 2a–2d by time-resolved absorption and emission spectroscopy. More specifically, we used femto- and nanosecond transient absorption (fs- and ns-TA) and fluorescence up-conversion spectroscopy (FLUPS) to shed light on excited-state dynamics in S1, in three DMSO/glycerol solvent mixtures (η = 2.2, 50 and 100 cP) (see ESI,† for more details). In fs-TA, the samples were excited to S1 by a 40–45-fs laser pulse centralized around 540 nm (close to the absorption maxima of 2) before recording absorption spectra in the 490–750 nm range using a continuum white-light in times up to 1.2 ns. The resulting spectra were interpreted by global lifetime analysis using a sequential kinetics model (see ESI,† for more details). All compounds shared similar spectral features in fs-TA, as shown by the negative signals of ground state bleach (GSB) and stimulated emission (SE) in the region of λ ∼ 500–700 nm (Fig. 3). Our global lifetime analysis revealed three components, corresponding to the sequential A → B → C kinetic model. | ||
| Fig. 3 Femtosecond transient absorption (fs-TA) spectra of 2a (top row) and 2d (bottom row) in either DMSO (η = 2.2 cP) (a) and (e) or DMSO/glycerol mixture (η = 100 cP) (c) and (g), alongside the respective deconvoluted spectra of components A (red solid line), B (green solid line) and C (grey dashed line, end spectrum) (b), (d), (f) and (h). The steady-state absorption (dashed grey line) and fluorescence emission (full grey line) spectra normalized to match the spectral shape of fs-TA spectra are superimposed for direct comparison. | ||
As shown by fs-TA, after excitation, the initially excited hot S1 species undergoes ultra-fast vibrational cooling (spectral component A, τA ∼ 3–9 ps) to a relaxed S1 state (spectral component B, τB ∼ 15–55 ps) before rapidly returning to the ground state, either directly or through C. The time constant of the initial relaxation process significantly increases with viscosity, suggesting that the relaxation involves a significant structural change. FLUPS also showed two spectral components, with λem ∼ 610 nm, corresponding to A and B based on their matching lifetimes (see Table S10 for details, ESI†). As such, both A and B are emissive components, whereas C undergoes only non-radiative decay.
C is the last spectral component shared by all studied compounds (2a–2d). This component decays on a much longer timescale (τC ≫ 2 ns) than that achievable by fs-TA. Moreover, the lifetime of C is highly sensitive to oxygen, with ∼100-fold longer lifetimes in the absence of oxygen. For example, for 2d, the lifetime of C is ∼0.8 μs in aerated DMSO vs. ∼240 μs in deoxygenated DMSO (ns-TA, Table 2 and Table S11, ESI†). Corroborating the observed quenching of C by the native, triplet oxygen, we detected sensitized singlet oxygen in the presence of excited 2, by spectrofluorimetry. The singlet oxygen was sensitized at quantifiable concentrations for 2c (ΦΔ = 0.018 ± 0.001 in methanol, see ESI,† for details). At these concentrations, intersystem crossing (ISC) is enhanced by the iodine atom of 2c. Thus, we identified C as a triplet T1 state formed by S1–T1 ISC.
Although the spectral features of 2a–2c did not significantly vary, 2d in DMSO displayed positive transient absorption signals, with λmax ∼ 640–680 nm on both ps and ns timescales (Fig. 3). Both steady-state and FLUPS measurements indicated that the initially excited hot S1 species of 2d (A) in DMSO yields a relaxed S1 state with charge-transfer character (B′). The B′ species of 2d (i) has ∼4-fold shorter lifetime than that of 2a, and (ii) its emission is red-shifted (∼730 nm).43
This clear difference in relaxed singlet state was confirmed by transient spectroscopy, given the positive absorption signal at ∼650 nm on a ps timescale. Further ns measurements highlighted the relaxation of B′ into a triplet state C′ with charge transfer character and an oxygen-sensitive lifetime. As with B′, C′ was detected by the positive transient absorption signal at ∼640 nm on a μs timescale.
Semicroconaine deexcitation involves three competing pathways
In examining the relationship between the lifetimes of the three spectral components (A–C) and solvent viscosity, we distinguished relationships found in 2a–2c from those found in 2d considering the results described above (Fig. 3 and Table 2). In 2a–2c, the lifetime of A was ∼2 times higher in high-than in low-viscosity medium. This increase in lifetime mirrored the increase in fluorescence emission quantum yield with solvent viscosity (Table S6, ESI†). By contrast, the lifetime of B remained unchanged when increasing the viscosity of the medium, failing to match the significant increase in the lifetime of A even though the rate of overall S1–S0 internal conversion was affected by viscosity. Accordingly, the “hot” S1 state A likely undergoes nonradiative deexcitation to S0 more efficiently than the equilibrium S1 state B through an orthogonal, viscosity-dependent deexcitation pathway facilitated by excited-state isomerization. Because fast relaxation from A to B depends on viscosity, the initially excited population should not be located at the equilibrium geometry of the S1 state (Scheme 2). | ||
| Scheme 2 Model of excited-state dynamics of semicroconaine, representing absorption from the ground state S0 to the excited-state S1 (hν) and fluorescence emission (kf) (straight black arrows) and non-radiative processes, such as internal conversion (IC, kIC) and intersystem crossing (ISC, kISC); other processes are omitted for clarity. The spectral components are assigned to the respective states in the corresponding capital letters, in italics (A, B, and C). | ||
In 2d, A follows the same trend, but the lifetime of B is ∼ 4 times longer in high- than in low-viscosity medium (Table 2 and Table S10, ESI†). This increase in lifetime results from the charge-transfer character of the B′ state in which the nitro group acts as an electron-accepting moiety. The formation of the charge-transfer triplet C′ confirms that the nitro group plays a key role in excited-state electron distribution. Associated with a large structural reorganization, these changes in the electronic coupling of the nitro group in the excited state are hindered by high viscosity media. Based on the combined results from (i) steady-state and (ii) time-resolved spectroscopy and from (iii) the relationship between spectral component lifetimes and solvent viscosity, we derived a model for semicroconaine deexcitation involving the following competing pathways:
(1) An intramolecular deexcitation pathway facilitated by isomerization, with a major role in 2a–2d;
(2) S1–T1 ISC, with a minor role in 2a–2d;
(3) NO2 group coupling/decoupling (charge transfer in B′ and C′), with a major role in 2d in low viscosity environments.
S1-Deexcitation proceeds through excited-state methine bridge isomerization
ISC and the –NO2 effect are well-known non-radiative deexcitation mechanisms, but the third pathway is not, despite being common to all semicroconaines analyzed in this study. To identify this deexcitation mechanism on a monomolecular level, we performed quantum-chemical calculations. Building upon ground-state isomer and TD-DFT energies previously reported by Capozzi et al.,15 we used 2a as the model compound to describe excited-state dynamics. Based on the restricted Kohn–Sham density functional theory (in the ground state), we optimized the geometry of plausible conformers/isomers of 2a, calculated their corresponding (climbing-image nudged-elastic band) minimum-energy isomerization pathways and optimized their respective transition states (Fig. 4). Based on the time-dependent density functional theory, we then calculated the unrelaxed energy profile of the isomerization pathway in the first singlet excited state. Wherever the excited-state isomerization pathway approached the ground-state isomerization pathway, we used the closest available geometry as the starting geometry for S1–S0 conical-intersection optimization (details are provided in ESI†). | ||
| Fig. 4 Top row: schemes of the global minimum energy geometry of 2a and geometries of the isomerization products. Bottom row: plots of nudged-elastic band pathways (dotted line) calculated at the B3LYP/def2-TZVP level; corrected pathway towards the analytically optimized transition state (full red line) and S1 energy profile of the unrelaxed pathway calculated at the TD-B3LYP/def2-TZVP level for each image on the isomerization trajectory. All NEB pathways share the “Common Reactant Geometry” in Step = 1, which is also used to reference the energy at 0.00 kcal mol−1. In each plot, product geometry is denoted as a, b and c (with an * in S1). | ||
In line with the literature,15 our conformational analysis using the optimized geometry of 2a identified the starting geometry as the most stable conformer (Fig. 4, top row, left panel). All plausible geometry changes were then considered. Thus, we optimized the products of (a) intramolecular proton transfer, and (b) indolenine– and (c) croconate– moiety rotation (Fig. 4, top row).
Using these geometries, we applied the protocol described above (Fig. 4, bottom row). The results showed that proton transfer (a) is only subtly disfavored in the ground state and that a may be present in a non-negligible equilibrium concentration under experimental conditions given the particularly low reaction barrier (ΔETS = 2.0 kcal mol−1; Table S21, ESI†). In the excited state, conversely, proton transfer is a highly disfavored, barrierless, uphill reaction, so a is likely readily converted into the common reactant geometry in S1. The S1 and S0 potential energy surfaces are well separated, with no crossing. Indolenine-moiety rotation (b) is disfavored in the ground state (ΔETS = 24.6 kcal mol−1; Table S21, ESI†) and remains so in S1 even though the energy profile is shallower (Fig. 4, bottom panel). The S1 and S0 potential energy surfaces are well separated, with no crossing. Croconate-moiety rotation (c) is strongly disfavored in the ground state and has a very high reaction barrier (ΔETS = 42.0 kcal mol−1; Table S21, ESI†). But in the excited state, the transition geometry represents a local minimum on the S1 potential energy surface and closely approaches the S0 potential energy surface (Fig. 4, bottom row, right, step 5). Using this transition state geometry, we optimized the S1–S0 conical intersection geometry (Fig. S171–S175, ESI†) and found that the optimized transition state geometry and the S1–S0 conical intersection geometry are virtually identical. Therefore, croconate moiety rotation c is the suspected deexcitation pathway.
Based on these findings, excited-state intramolecular proton transfer (ESIPT) may contribute to subtle changes in band shape between A and B as the ground-state population of the proton-transfer a is readily and exclusively converted into the excited-state population of the global minimum energy geometry. But while indolenine-moiety rotation does not contribute to the photophysical properties of semicroconaines 2 whatsoever, croconate moiety rotation provides the main non-radiative deexcitation pathway through an S1–S0 conical intersection. For this reason, hampering this deexcitation pathway of semicroconaines by geometric restriction may convert them into “turn-on” AIE probes.
Host–guest complexation with macromolecules hinders excited-state isomerization of semicroconaines, “turning on” their fluorescence
To confirm that semicroconaines can be used as “turn-on” AIE probes, we induced the complexation of 2a with an open (acyclic) glycoluril dimer-based clip (bis-o-xylylene-capped) (GU)244 and double-stranded DNA (dsDNA) and measured its steady-state fluorescence. When adding (GU)2 to a solution of 2a in water (with PBS), we observed an increase in fluorescence intensity (λexc = 555 nm) and, concurrently, a bathochromic shift of the fluorescence maximum. At low concentrations of (GU)2 (up to 1 eq.), fluorescence intensity increased only marginally, but with an excess 100 eq. of (GU)2, fluorescence intensity increased up to 30-fold (Fig. 5a) (see ESI,† for details). | ||
| Fig. 5 Encapsulation “turns on” semicroconaine fluorescence: (a) steady-state emission spectra (upon excitation at λexc = 555 nm) of 2a (c = 1.2 × 10−5 mol dm−3) in PBS alone (black solid line) and with 0.1 eq. and 1 eq. (blue solid line) or 10 eq. and 100 eq. (red solid line) of glycoluril dimer-based clip (GU)2. Inset: (GU)2 structure;44 (b) steady-state emission spectra (upon excitation at λexc = 460 nm) of thiazole orange (TO, c = 1 × 10−5 mol dm−3) and 2a (black dotted line); TO (c = 1 × 10−5 mol dm−3) and dsDNA (0.1 eq.) in PBS (black solid line); 2a (c = 1 × 10−5 mol dm−3) (red dotted line; asterisk denotes a scatter artefact signal, which was subtracted to calculate the turn-on value, see ESI,† for more details); and 2a (c = 1.0 × 10−5 mol dm−3) and dsDNA (0.1 eq.) (black solid line). All spectra were recorded in PBS. The TO and 2a spectra without dsDNA were normalized to each other, and the spectra of the dyes intercalated with dsDNA were scaled to keep the ratio of Iem(dye)/Iem(dye + DNA). Inset: TO structure. Note: arrows indicate changes in the emission spectrum band upon adding (GU)2 (panel a) or dsDNA (panel b). | ||
In contrast to (GU)2, even a substochiometric amount of dsDNA (0.1 eq.) led to a 5.1-fold increase in the fluorescence intensity of 2a (λexc = 460 nm) and to a hypsochromic shift of the fluorescence maximum (Fig. 5b). For comparison, this increase is significantly higher than that of thiazole orange (TO), a commercially available DNA-intercalation probe, under the same conditions (1.9-fold increase at λexc = 460 nm). The higher affinity of 2a to dsDNA may be explained by the zwitterionic character of the dye. As a zwitterionic molecule, 2a interacts more favorably with the polar dsDNA molecule than with the lipophilic cavity of (GU)2. Furthermore, 2a also carries multiple H-bond donor and acceptor functional groups, which increase its propensity for dsDNA intercalation. Supporting this explanation, the fluorescence maximum shifts were bathochromic with (GU)2 and hypsochromic with dsDNA, suggesting two different interaction mechanisms. In both, geometric restriction of 2a hinders its major non-radiative deexcitation pathway (excited-state methine bridge isomerization), thereby increasing its fluorescence intensity.
Conclusions
In semicroconaines, the main deexcitation pathway involves excited-state isomerization of the croconate moiety. Increasing solvent viscosity or promoting complexation with (GU)2 and dsDNA inhibits semicroconaine deexcitation, intensifying their fluorescence emission. This increase in fluorescence emission induced by geometric restriction mirrors the effect of GFP on its fluorophore (HBDI). Therefore, virtually non-fluorescent semicroconaine dyes can be converted into AIE probes for (bio)macromolecular sensing.Author contributions
R. S. and M. V. synthesized compounds and measured steady-state spectroscopy. R. S. and A. R. measured time-resolved spectroscopy and processed data. D. D. performed the computational study. B. B. T. performed the DNA intercalation study with inputs from D. D. P. Š. performed photochemical experiments. P. M. built the fs-TA setup, excitation sources and provided assistance during the fs-experiments. T. S. supervised D. D. and B. B. T., T. K. supervised A. R., P. Š. supervised R. S. and M. V. The original idea was conceived by P. Š. and further shaped with input from R. S., D. D., T. S. and T. K. Funding was acquired by P. Š., T. S., and T. K. The manuscript was written by P. Š. with inputs from R. S., D. D. and T. S., and the final version was approved by all authors.Data availability
Photophysical properties of select chromophores, NMR, UV/vis absorption and fluorescence emission spectra, stability determination experiments, time-resolved absorption and fluorescence emission spectral data, details on quantum chemical calculations and coordinates, and additional information on the structure-properties relationships are available in the ESI.†Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
This study was funded by the Czech Science Foundation (GJ20-30004Y, P. Š.) and supported from the European Union’s Horizon 2020 research and innovation program under grant agreement No. 857560 (CETOCOEN Excellence). This publication reflects only the author's view, and the European Commission is not responsible for any use that may be made of the information it contains. The authors thank the MEYS INTER-COST project No. LTC20076 (T. S.), CETOCOEN EXCELLENCE (CZ.02.1.01/0.0/0.0/17_043/0009632) and the RECETOX research infrastructure (LM2023069) (P. Š.). R. S. thanks RECETOX and University of Jyväskylä for supporting her stay in Jyväskylä. The authors also express their gratitude to Marek Martínek for his aid with photochemical experiments and singlet oxygen measurements, to Lukáš Maier and Miroslava Bittová for their help with the NMR and mass spectrometry measurements, to Luboš Jílek for his assistance with time-resolved spectroscopic methods and to Eliška Klimešová and Jana Bruzlová (all from Masaryk University) for their help with the synthetic work. We thank Vladimír Šindelář (Masaryk University) for providing samples of cyclic and open cucurbiturils. The authors thank Dr. Carlos V. Melo (Charles University) for editing the manuscript.References
- L. D. Lavis and R. T. Raines, Bright Building Blocks for Chemical Biology, ACS Chem. Biol., 2014, 9(4), 855–866, DOI:10.1021/cb500078u.
- R. Strack, Organic Dyes for Live Imaging, Nat. Methods, 2021, 18(1), 30, DOI:10.1038/s41592-020-01032-z.
- A. Martin and P. Rivera-Fuentes, A General Strategy to Develop Fluorogenic Polymethine Dyes for Bioimaging, Nat. Chem., 2024, 16(1), 28–35, DOI:10.1038/s41557-023-01367-y.
- Q. Zheng and L. D. Lavis, Development of Photostable Fluorophores for Molecular Imaging, Curr. Opin. Chem. Biol., 2017, 39, 32–38, DOI:10.1016/j.cbpa.2017.04.017.
- Y. Fu and N. S. Finney, Small-Molecule Fluorescent Probes and Their Design, RSC Adv., 2018, 8(51), 29051–29061, 10.1039/C8RA02297F.
- T. Terai and T. Nagano, Small-Molecule Fluorophores and Fluorescent Probes for Bioimaging, Pflugers Arch – Eur. J. Physiol., 2013, 465(3), 347–359, DOI:10.1007/s00424-013-1234-z.
- K. Ilina, W. M. MacCuaig, M. Laramie, J. N. Jeouty, L. R. McNally and M. Henary, Squaraine Dyes: Molecular Design for Different Applications and Remaining Challenges, Bioconjugate Chem., 2020, 31(2), 194–213, DOI:10.1021/acs.bioconjchem.9b00482.
- A. P. Gorka, R. R. Nani and M. J. Schnermann, Harnessing Cyanine Reactivity for Optical Imaging and Drug Delivery, Acc. Chem. Res., 2018, 51(12), 3226–3235, DOI:10.1021/acs.accounts.8b00384.
- M. S. Michie, R. Götz, C. Franke, M. Bowler, N. Kumari, V. Magidson, M. Levitus, J. Loncarek, M. Sauer and M. J. Schnermann, Cyanine Conformational Restraint in the Far-Red Range, J. Am. Chem. Soc., 2017, 139(36), 12406–12409, DOI:10.1021/jacs.7b07272.
- S. Lei, Y. Zhang, N. T. Blum, P. Huang and J. Lin, Recent Advances in Croconaine Dyes for Bioimaging and Theranostics, Bioconjugate Chem., 2020, 31(9), 2072–2084, DOI:10.1021/acs.bioconjchem.0c00356.
- M. H. Sleiman and S. Ladame, Synthesis of Squaraine Dyes under Mild Conditions: Applications for Labelling and Sensing of Biomolecules, Chem. Commun., 2014, 50(40), 5288–5290, 10.1039/C3CC47894G.
- N. Barbero, C. Magistris, J. Park, D. Saccone, P. Quagliotto, R. Buscaino, C. Medana, C. Barolo and G. Viscardi, Microwave-Assisted Synthesis of Near-Infrared Fluorescent Indole-Based Squaraines, Org. Lett., 2015, 17(13), 3306–3309, DOI:10.1021/acs.orglett.5b01453.
- S. Khopkar, S. Deshpande and G. Shankarling, Greener Protocol for the Synthesis of NIR Fluorescent Indolenine-Based Symmetrical Squaraine Colorants, ACS Sustainable Chem. Eng., 2018, 6(8), 10798–10805, DOI:10.1021/acssuschemeng.8b02095.
- N. Zappimbulso, M. A. M. Capozzi, A. Porcheddu, G. M. Farinola and A. Punzi, Solvent-Free Reactions for the Synthesis of Indolenine-Based Squaraines and Croconaines: Comparison of Thermal Heating, Mechanochemical Milling, and IR Irradiation, ChemSusChem, 2021, 14(5), 1363–1369, DOI:10.1002/cssc.202002763.
- M. A. M. Capozzi, A. Punzi, F. Babudri, R. Musio and G. M. Farinola, Synthesis and Computational Study of Semicroconaines and Nonsymmetric Croconaines, J. Org. Chem., 2018, 83(23), 14396–14405, DOI:10.1021/acs.joc.8b01930.
- S. Zhang, C. Zhou, M. Zhang, Y. Zhao, B. Yuan, B. Yang and H. Li, Solvent-Driven Micro/Nanostructures of Squaraine and Croconaine Dyes Based on Quinoxalinone for Visual Detection of Nerve Agent Simulants, Dyes Pigm., 2023, 208, 110824, DOI:10.1016/j.dyepig.2022.110824.
- Y. Dong, H. Wang, X. Zhang, Y. Ding, Y. Zou, J. Wang, S.-C. Zhao and Z. Li, Croconaine-Based NIR-II Fluorescence Imaging-Guided Tumor Photothermal Therapy Induces Long-Term Antitumor Immune Memory, J. Nanobiotechnol., 2024, 22(1), 481, DOI:10.1186/s12951-024-02695-y.
- Y. Tang, W. Li, X. Zhou, N. Hao, X. Zhou, Z. Zhong, Y. Jian, S. Wei, X. Zheng and L. Yang, NIR Diradical-Featured Croconaine Nanoagent for Cancer Photoacoustic Imaging and Photothermal Therapy, Chem. – Eur. J., 2025, 31(4), e202403372, DOI:10.1002/chem.202403372.
- A. Sanchez-Galvez, P. Hunt, M. A. Robb, M. Olivucci, T. Vreven and H. B. Schlegel, Ultrafast Radiationless Deactivation of Organic Dyes: Evidence for a Two-State Two-Mode Pathway in Polymethine Cyanines, J. Am. Chem. Soc., 2000, 122(12), 2911–2924, DOI:10.1021/ja993985x.
- L. Dähne and G. Reck, Deformation of Polymethine Structures by Intermolecular Interactions, Angew. Chem., Int. Ed. Engl., 1995, 34(6), 690–692, DOI:10.1002/anie.199506901.
- A. D. Kachkovski and O. O. Zhukova, Electronic Properties of Polymethine Systems. 6. Bond Order and Bond Length Changes upon Excitation, Dyes Pigm., 2004, 63(3), 323–332, DOI:10.1016/j.dyepig.2004.01.006.
- A. Weigel, M. Pfaffe, M. Sajadi, R. Mahrwald, R. Improta, V. Barone, D. Polli, G. Cerullo, N. P. Ernsting and F. Santoro, Barrierless Photoisomerisation of the “Simplest Cyanine”: Joining Computational and Femtosecond Optical Spectroscopies to Trace the Full Reaction Path, Phys. Chem. Chem. Phys., 2012, 14(38), 13350–13364, 10.1039/C2CP41522D.
- C. Dugave and L. Demange, Cis−Trans Isomerization of Organic Molecules and Biomolecules: Implications and Applications, Chem. Rev., 2003, 103(7), 2475–2532, DOI:10.1021/cr0104375.
- C. Wang, W. Chi, Q. Qiao, D. Tan, Z. Xu and X. Liu, Twisted Intramolecular Charge Transfer (TICT) and Twists beyond TICT: From Mechanisms to Rational Designs of Bright and Sensitive Fluorophores, Chem. Soc. Rev., 2021, 50(22), 12656–12678, 10.1039/D1CS00239B.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Ultrafast Elementary Photochemical Processes of Organic Molecules in Liquid Solution, Chem. Rev., 2017, 117(16), 10826–10939, DOI:10.1021/acs.chemrev.6b00491.
- D. Su, C. L. Teoh, L. Wang, X. Liu and Y.-T. Chang, Motion-Induced Change in Emission (MICE) for Developing Fluorescent Probes, Chem. Soc. Rev., 2017, 46(16), 4833–4844, 10.1039/C7CS00018A.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Aggregation-Induced Emission: Together We Shine, United We Soar, Chem. Rev., 2015, 115(21), 11718–11940, DOI:10.1021/acs.chemrev.5b00263.
- A. Baldridge, S. R. Samanta, N. Jayaraj, V. Ramamurthy and L. M. Tolbert, Activation of Fluorescent Protein Chromophores by Encapsulation, J. Am. Chem. Soc., 2010, 132(5), 1498–1499, DOI:10.1021/ja908870k.
- A. Follenius-Wund, M. Bourotte, M. Schmitt, F. Iyice, H. Lami, J.-J. Bourguignon, J. Haiech and C. Pigault, Fluorescent Derivatives of the GFP Chromophore Give a New Insight into the GFP Fluorescence Process, Biophys. J., 2003, 85(3), 1839–1850, DOI:10.1016/S0006-3495(03)74612-8.
- D. Mandal, T. Tahara and S. R. Meech, Excited-State Dynamics in the Green Fluorescent Protein Chromophore, J. Phys. Chem. B, 2004, 108(3), 1102–1108, DOI:10.1021/jp035816b.
- A. Baldridge, S. R. Samanta, N. Jayaraj, V. Ramamurthy and L. M. Tolbert, Steric and Electronic Effects in Capsule-Confined Green Fluorescent Protein Chromophores, J. Am. Chem. Soc., 2011, 133(4), 712–715, DOI:10.1021/ja1094606.
- H. Ihmels and D. Otto, Intercalation of Organic Dye Molecules into Double-Stranded DNA – GeneralPrinciples and Recent Developments, in Supermolecular Dye Chemistry, ed. F. Würthner, Springer, Berlin, Heidelberg, 2005, pp. 161–204 DOI:10.1007/b135804.
- J. M. Alaranta, K.-N. Truong, M. F. Matus, S. A. Malola, K. T. Rissanen, S. S. Shroff, V. S. Marjomäki, H. J. Häkkinen and T. M. Lahtinen, Optimizing the SYBR Green Related Cyanine Dye Structure to Aim for Brighter Nucleic Acid Visualization, Dyes Pigm., 2023, 208, 110844, DOI:10.1016/j.dyepig.2022.110844.
- C. Ma, W. Sun, L. Xu, Y. Qian, J. Dai, G. Zhong, Y. Hou, J. Liu and B. Shen, A Minireview of Viscosity-Sensitive Fluorescent Probes: Design and Biological Applications, J. Mater. Chem. B, 2020, 8(42), 9642–9651, 10.1039/D0TB01146K.
- S. Upadhyayula, V. Nuñez, E. M. Espinoza, J. M. Larsen, D. Bao, D. Shi, J. T. Mac, B. Anvari and V. I. Vullev, Photoinduced Dynamics of a Cyanine Dye: Parallel Pathways of Non-Radiative Deactivation Involving Multiple Excited-State Twisted Transients, Chem. Sci., 2015, 6(4), 2237–2251, 10.1039/C4SC02881C.
- B. Ciubini, S. Visentin, L. Serpe, R. Canaparo, A. Fin and N. Barbero, Design and Synthesis of Symmetrical Pentamethine Cyanine Dyes as NIR Photosensitizers for PDT, Dyes Pigm., 2019, 160, 806–813, DOI:10.1016/j.dyepig.2018.09.009.
- E. A. Savicheva, M. L. Bossi, V. N. Belov and S. W. Hell, Introduction of the Functional Amino Group at the Meso Position of Cy3 and Cy5 Dyes: Synthesis, Stability, Spectra and Photolysis of 4-Amino-1-diazo-2-butanone Derivatives, ChemPhotoChem, 2023, 7(1), e202200222, DOI:10.1002/cptc.202200222.
- G. Angulo, M. Brucka, M. Gerecke, G. Grampp, D. Jeannerat, J. Milkiewicz, Y. Mitrev, C. Radzewicz, A. Rosspeintner, E. Vauthey and P. Wnuk, Characterization of Dimethylsulfoxide/Glycerol Mixtures: A Binary Solvent System for the Study of “Friction-Dependent” Chemical Reactivity, Phys. Chem. Chem. Phys., 2016, 18(27), 18460–18469, 10.1039/C6CP02997C.
- J. A. Mondal, M. Sarkar, A. Samanta, H. N. Ghosh and D. K. Palit, Charge-Transfer-Induced Twisting of the Nitro Group, J. Phys. Chem. A, 2007, 111(28), 6122–6126, DOI:10.1021/jp0737193.
- S. K. Panja, N. Dwivedi and S. Saha, Tuning the Intramolecular Charge Transfer (ICT) Process in Push–Pull Systems: Effect of Nitro Groups, RSC Adv., 2016, 6(107), 105786–105794, 10.1039/C6RA17521J.
- M.-C. Chen, D.-G. Chen and P.-T. Chou, Fluorescent Chromophores Containing the Nitro Group: Relatively Unexplored Emissive Properties, ChemPlusChem, 2021, 86(1), 11–27, DOI:10.1002/cplu.202000592.
- W. Rodríguez-Córdoba, L. Gutiérrez-Arzaluz, F. Cortés-Guzmán and J. Peon, Excited State Dynamics and Photochemistry of Nitroaromatic Compounds, Chem. Commun., 2021, 57(92), 12218–12235, 10.1039/D1CC04999B.
- Note that the hot S1 species A has also a charge-transfer character because the red-shifted emission as well as red-absorption peak (680 nm) is there from the very beginning (both FLUPS and fs-TA show a signal in this region already at hundreds of femtoseconds).
- L. Gilberg, B. Zhang, P. Y. Zavalij, V. Sindelar and L. Isaacs, Acyclic Cucurbit[n]Uril-Type Molecular Containers: Influence of Glycoluril Oligomer Length on Their Function as Solubilizing Agents, Org. Biomol. Chem., 2015, 13(13), 4041–4050, 10.1039/C5OB00184F.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5qm00030k |
| This journal is © the Partner Organisations 2025 |