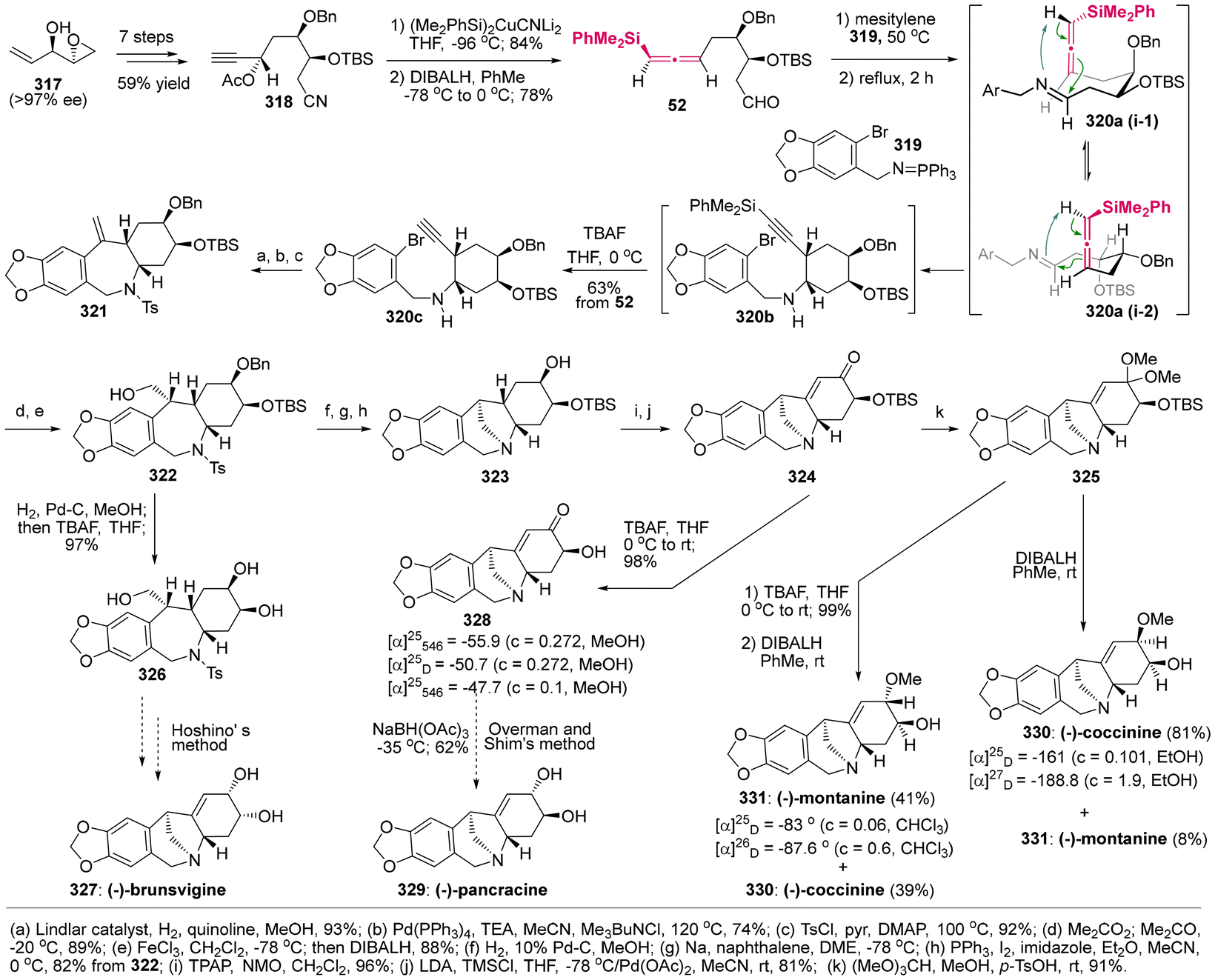
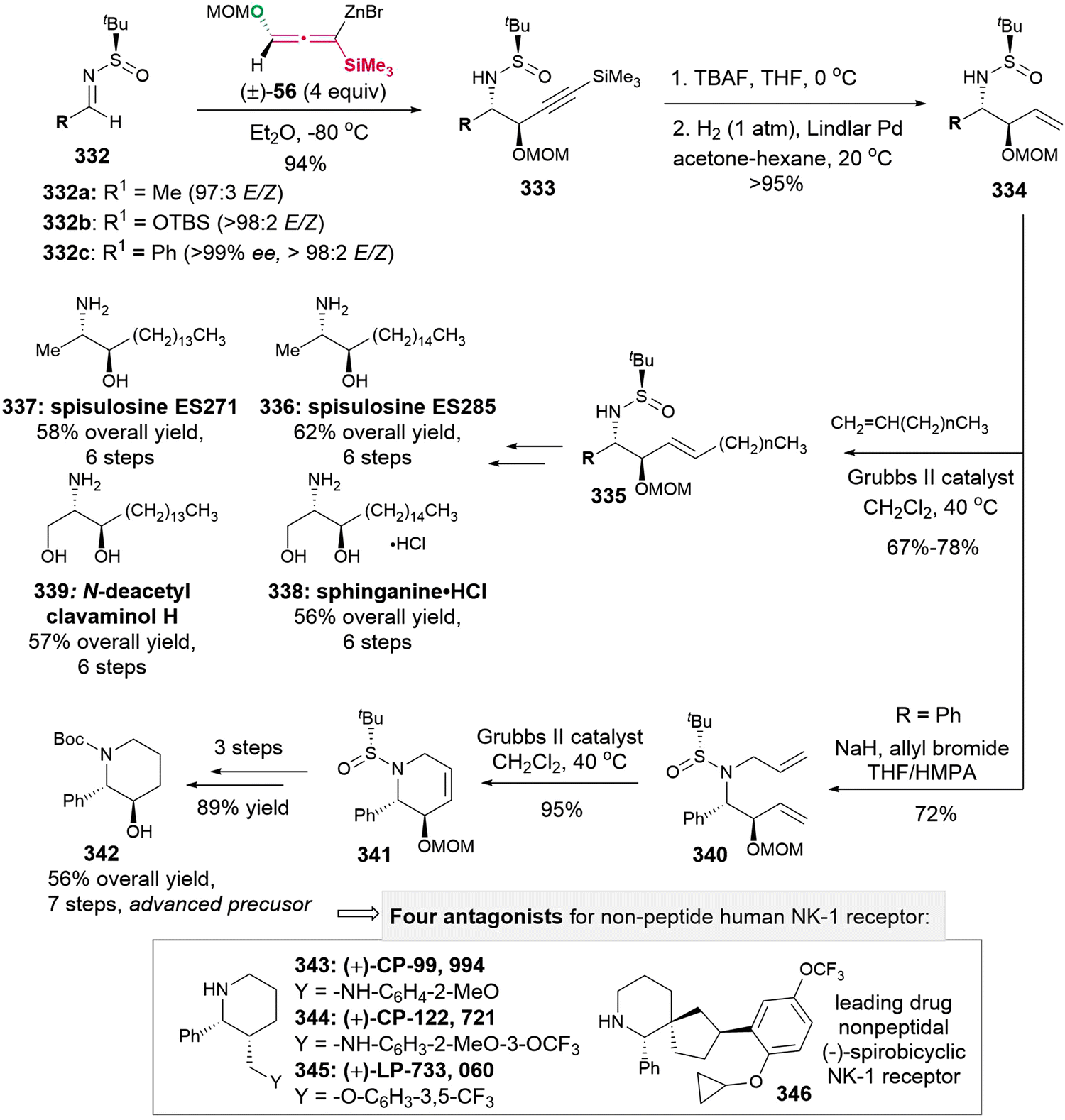
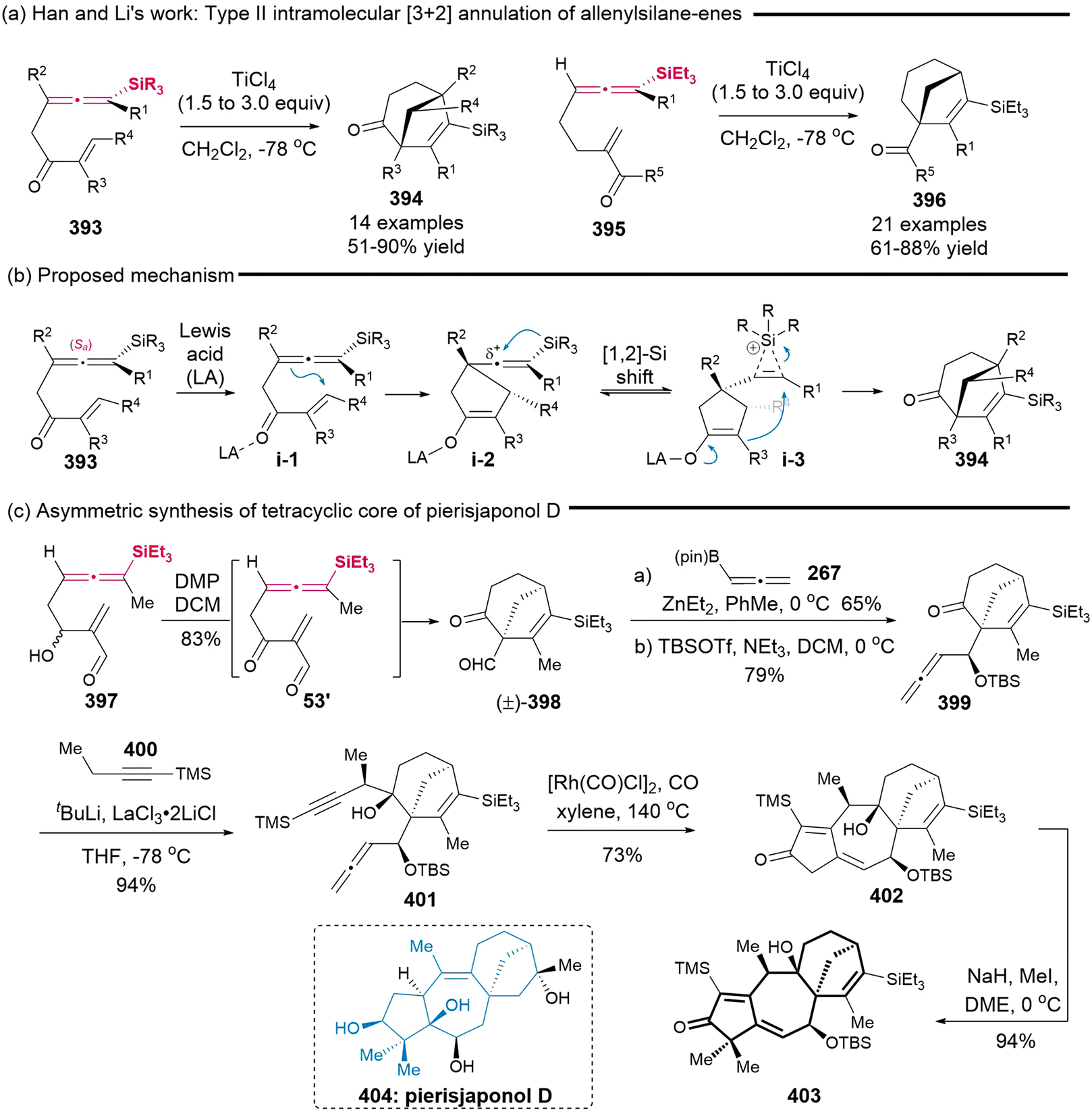
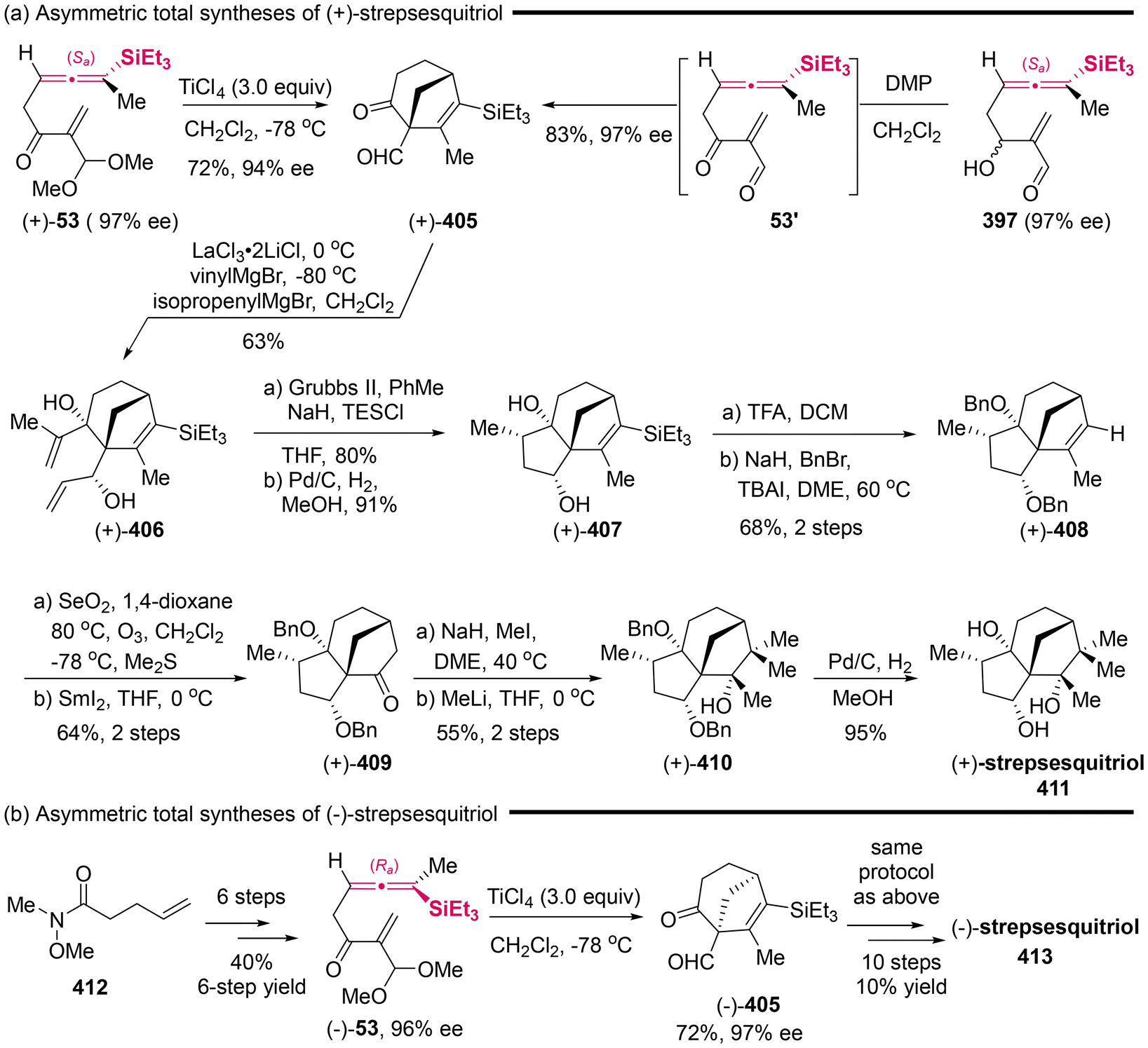
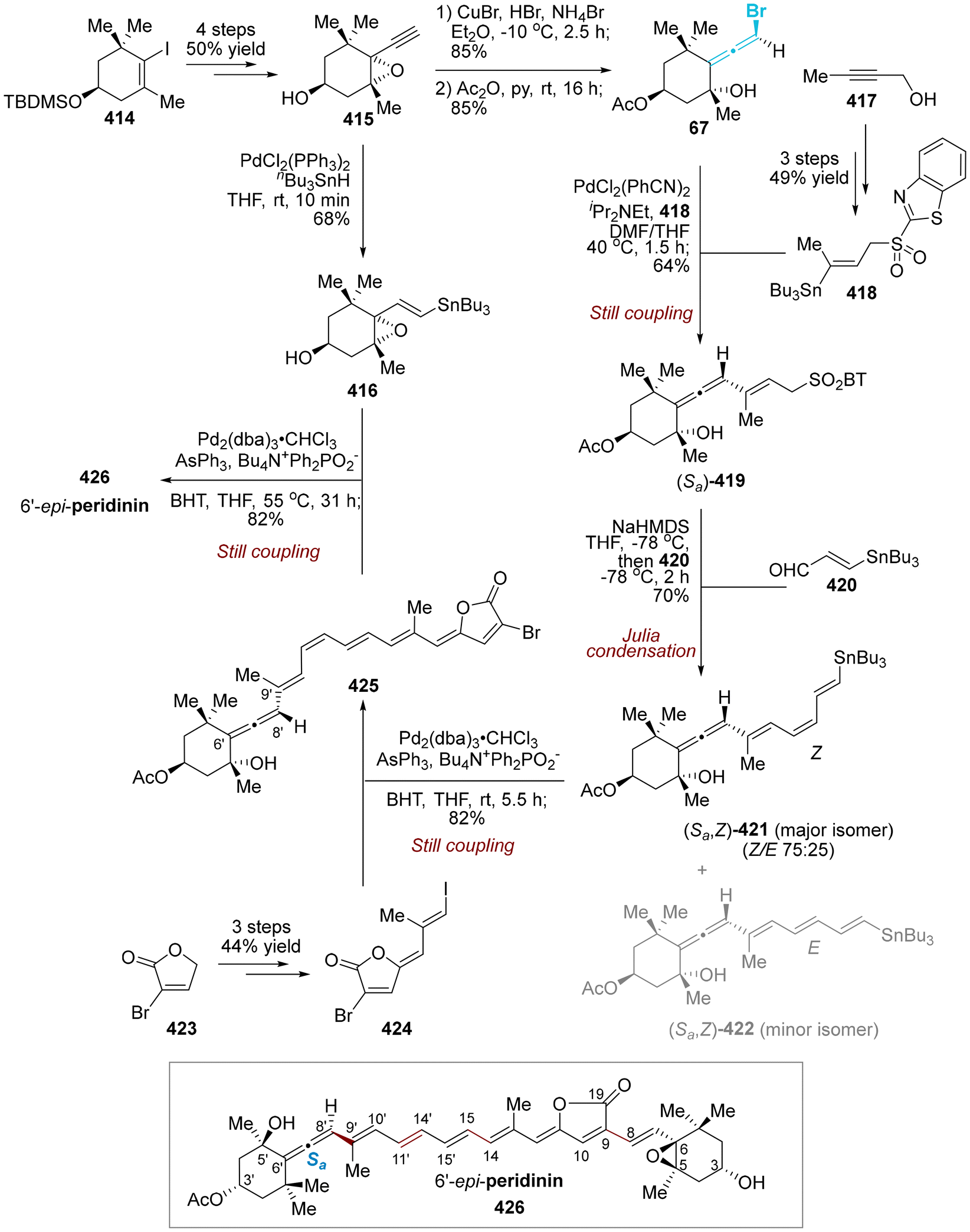
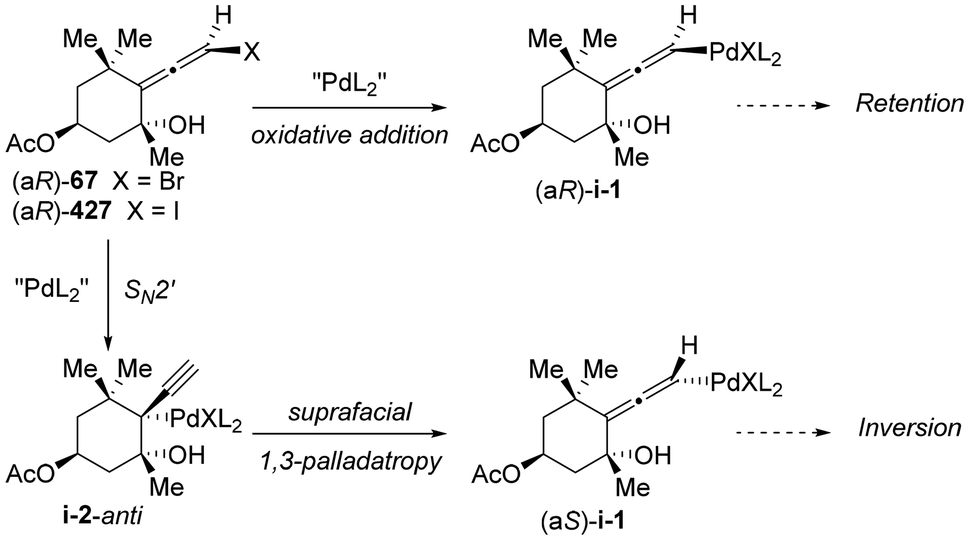
DOI:
10.1039/D4QO02004A
(Review Article)
Org. Chem. Front., 2025, Advance Article
Catalytic asymmetric synthesis and synthetic application of heteroatom-substituted axially chiral allenes
Received
24th October 2024
, Accepted 21st November 2024
First published on 25th November 2024
Abstract
The axially chiral allene framework has garnered significant attention from the synthetic community owing to its wide occurrence and application in natural products, organic synthesis, pharmaceuticals, and materials science. Among the structurally diverse chiral allene compounds, heteroatom-substituted axially chiral allenes represent a large and versatile subclass. Although the utility of heteroatom-substituted chiral allenes has been extensively explored, the asymmetric synthesis of these valuable chiral frameworks presents a long-standing challenge. Benefitting from the development of asymmetric catalysis, many efficient synthetic methods in enantioselective catalysis and a variety of competent reaction partners have recently emerged. This review primarily summarizes the contributions made to date to the catalytic asymmetric synthesis and synthetic applications of heteroatom-substituted axially chiral allenes.
 Lifei Gan | Lifei Gan was born in Gangxi Province, China. After completing her B.S. in chemistry from Guangxi Normal University in 2018, she received her Ph.D. in Organic Chemistry from Yunnan University in 2024 under the supervision of Prof. Zhihui Shao. Currently, she works as a postdoctoral researcher at Yunnan University and her research interest is focused on asymmetric catalysis. |
 Xuanchen Wan | Xuanchen Wan was born in Zhejiang Province, China. After obtaining his B.S. in Chemistry from the University of Science and Technology of China in 2022, he commenced his Master of Science studies at the Southwest United Graduate School (Yunnan University) in 2013, under the guidance of Prof. Zhihui Shao. His current research is focused on catalytic asymmetric synthesis. |
 Yucheng Pang | Yucheng Pang was born in Heilongjiang Province, China. He obtained his B.S. in Chemistry from Heilongjiang University in 2021. Subsequently, in 2022, he became a member of Prof. Zhihui Shao's research team in the faculty of the School of Chemical Science and Technology at Yunnan University, where he is currently pursuing a Master of Science degree. His research is focused on catalytic asymmetric synthesis. |
 Yuqi Zou | Yuqi Zou was born in Chongqing, China, in 2005. In 2024, he commenced his studies at the School of Pharmacy at Yunnan University, where he is pursuing a major in pharmacy. He is presently an undergraduate student engaged in the study of asymmetric synthesis chemistry. |
 Yu-Hua Deng | Yu-Hua Deng obtained her Master of Science degree in Organic Chemistry from China West Normal University in 2013. Subsequently, she pursued her doctoral studies at the Lanzhou Institute of Chemical Physics, Chinese Academy of Sciences, under the guidance of Prof. Chun-An Fan and Hanmin Huang, and was awarded her Ph.D. in 2017. Currently, she serves as Associate Researcher in Professor Zhihui Shao's team at Yunnan University. Her research interests are primarily centered on the advancement of novel methodologies for organic synthesis. |
 Zhihui Shao | Prof. Zhihui Shao received his Ph.D. from Lanzhou University in 2004 under the supervision of Professors Yongqiang Tu and Hongbin Zhang. He then carried out his postdoctoral studies under the guidance of Prof. Stephen Hanessian at the University of Montreal (2004–2006) and Prof. Albert S. C. Chan at the Hong Kong Polytechnic University (2006–2007). He began his independent academic research in the faculty of the School of Chemical Science and Technology at Yunnan University. His current research interests include asymmetric catalysis and total synthesis. |
1. Introduction
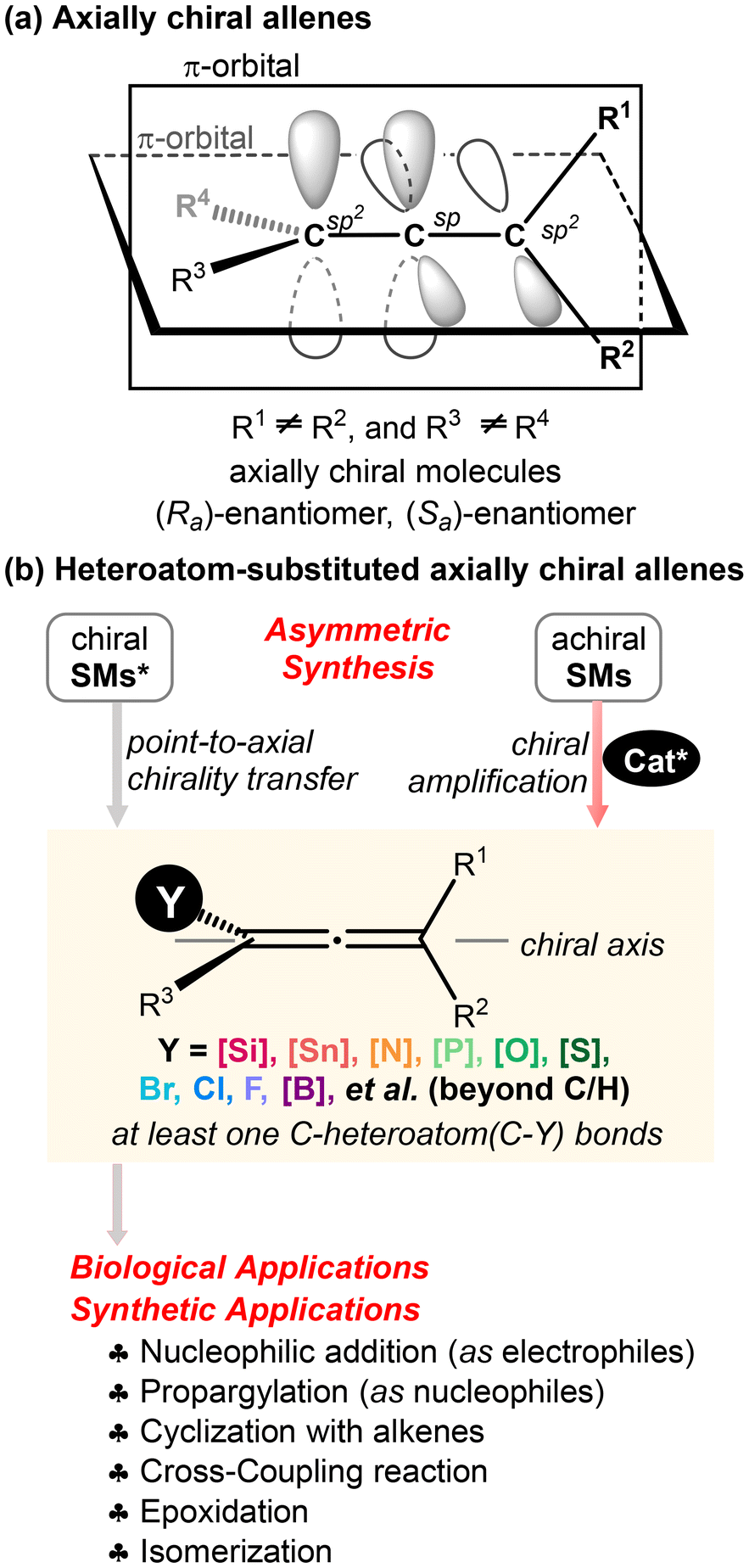
Allenes1 represent a versatile class of unsaturated compounds characterized by their unique cumulative double bonds or the 1,2-propadiene structural moiety. The distinct molecular structure of allenes was elucidated in 1954—67 years after their initial synthesis. The unique structure of allenes endows them with a richer and more distinctive array of chemical properties, compared to unsaturated conjugated alkenes and alkynes. Besides their occurrence in significant natural products, pharmaceutical molecules,2 and advanced functional materials,3 allenes have emerged as versatile synthetic building blocks in organic synthesis.4 The chirality of allenes is axial, arising from the perpendicular geometry of two π-bond orbitals and two pairs of substituents (Scheme 1a).5 When each terminal carbon C(sp2) of allenes is bonded to two different substituents, the resulting allenes lack a plane of symmetry, rendering them chiral. Axially chiral allenes have even been proven to be privileged chiral ligands in asymmetric catalysis.6
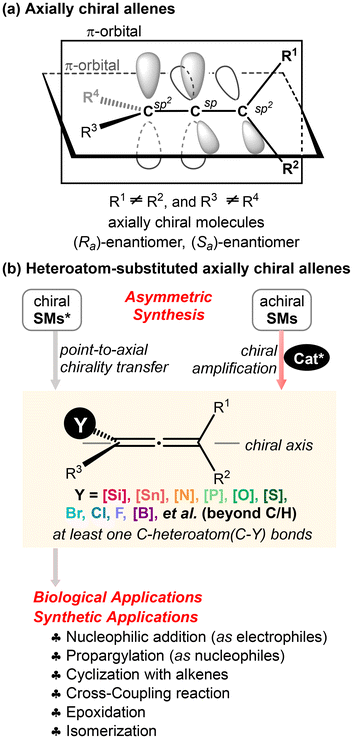 |
| | Scheme 1 Axially chiral allenes and heteroatom-substituted axially chiral allenes. | |
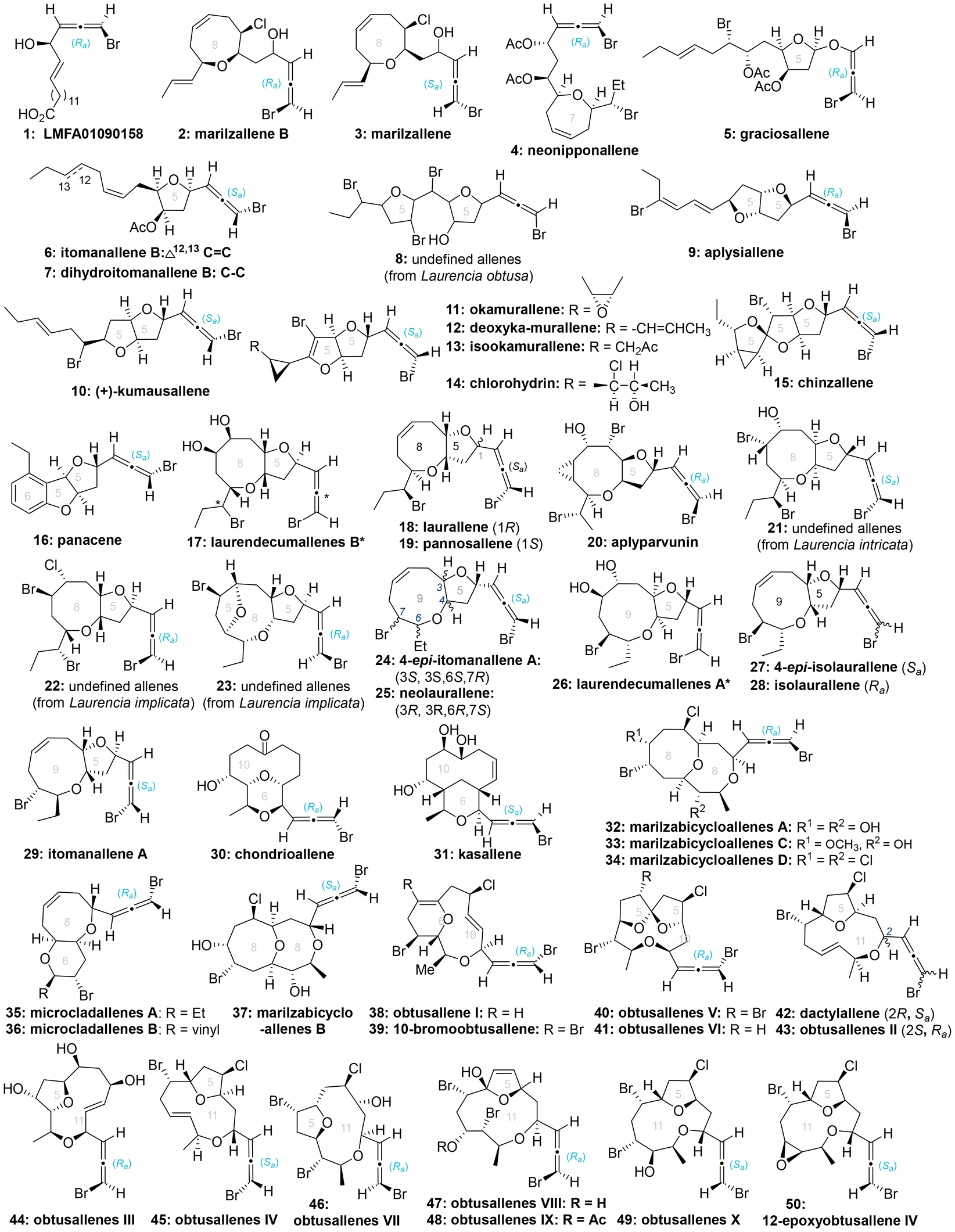
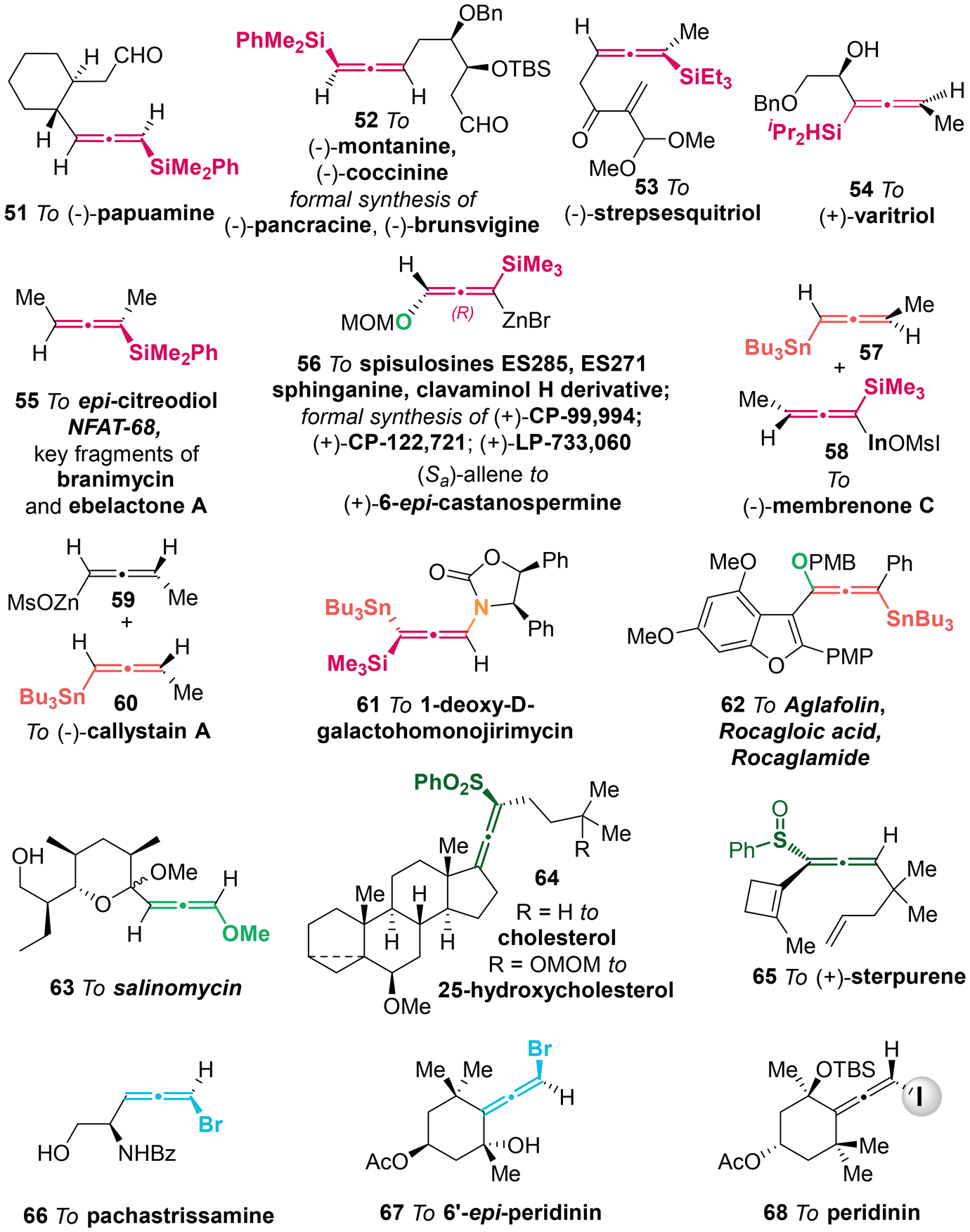
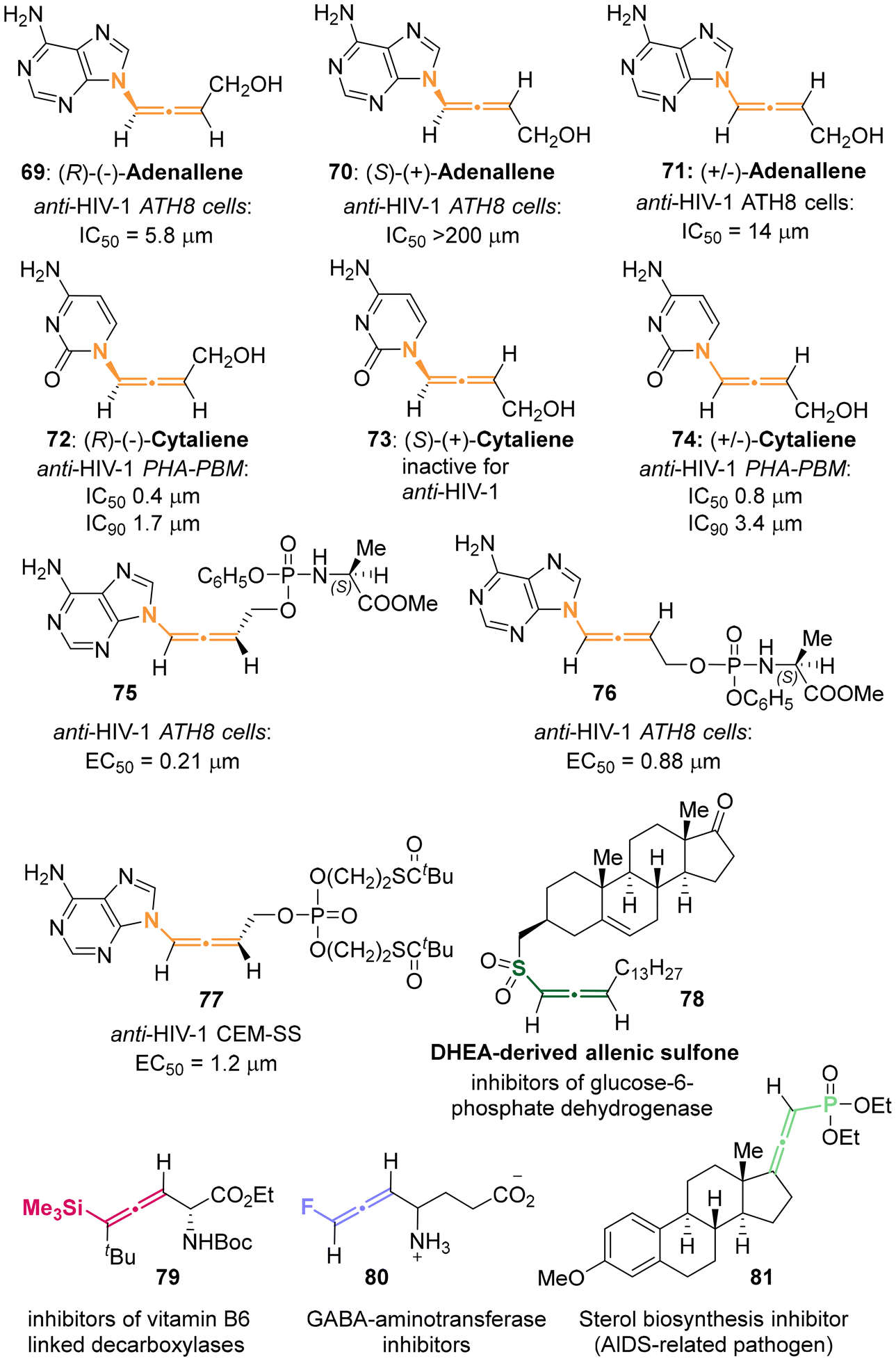
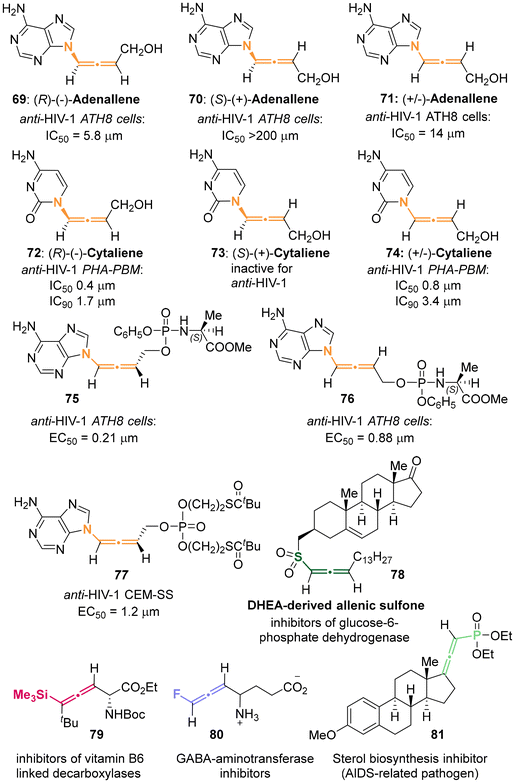
Heteroatom-substituted axially chiral allenes (HACAs) are compounds that contain one or more heteroatoms bonded to their terminal carbon (Scheme 1b). The tunable reactivity and synthetic versatility of carbon–heteroatom bonds make heteroatom-substituted axially chiral allenes versatile platforms for various applications. Over 50 natural products featuring heteroatom-brominated allenes have been isolated from marine algae (Schemes 2 and 3).7 A distinguishing feature of these marine natural products is the presence of different complex cyclic ether groups attached to a chiral brominated allene motif. Furthermore, complementary diastereoisomers with axial chirality have been observed in nature. In many cases, the unique structures of these natural compounds have been confirmed by X-ray analysis or synthetic methods.8 Furthermore, various structurally simple heteroatom-substituted axially chiral allenes have been proven to be significant as essential synthetic precursors for the development of complex pharmaceutical molecules and natural products (Scheme 4). Many natural and synthetic heteroatom-substituted axially chiral allenes have demonstrated promising biological activities (Scheme 5). For example, adenalene (69–71), cytaliene (72–74) and their derivatives (75–77), which represent N-allenic nucleoside analogues 69–77, exhibit potential therapeutic value for the treatment of AIDS disease.
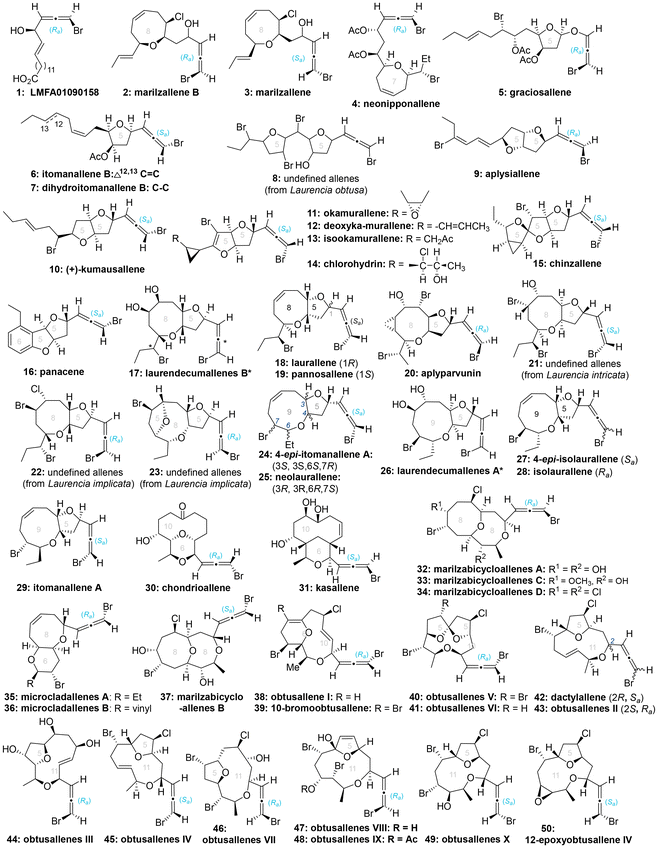 |
| | Scheme 2 Representative natural products bearing heteroatom-substituted axially chiral allenes. | |
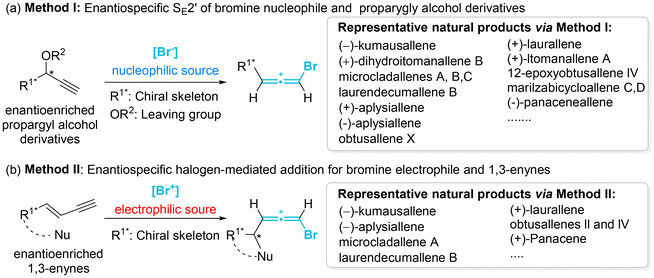 |
| | Scheme 3 Two main methods for the synthesis of natural axially chiral brominated allenes. | |
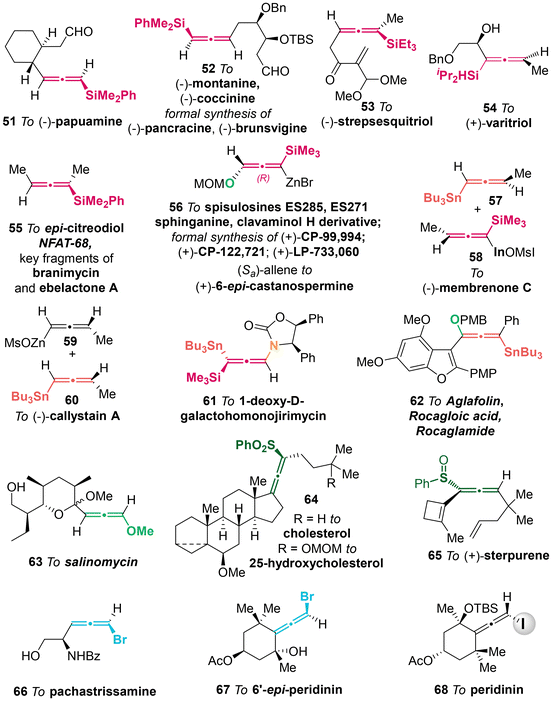 |
| | Scheme 4 Representative synthetic applications involving heteroatom-substituted allenes. | |
 |
| | Scheme 5 Representative bio-active synthetic molecules. | |
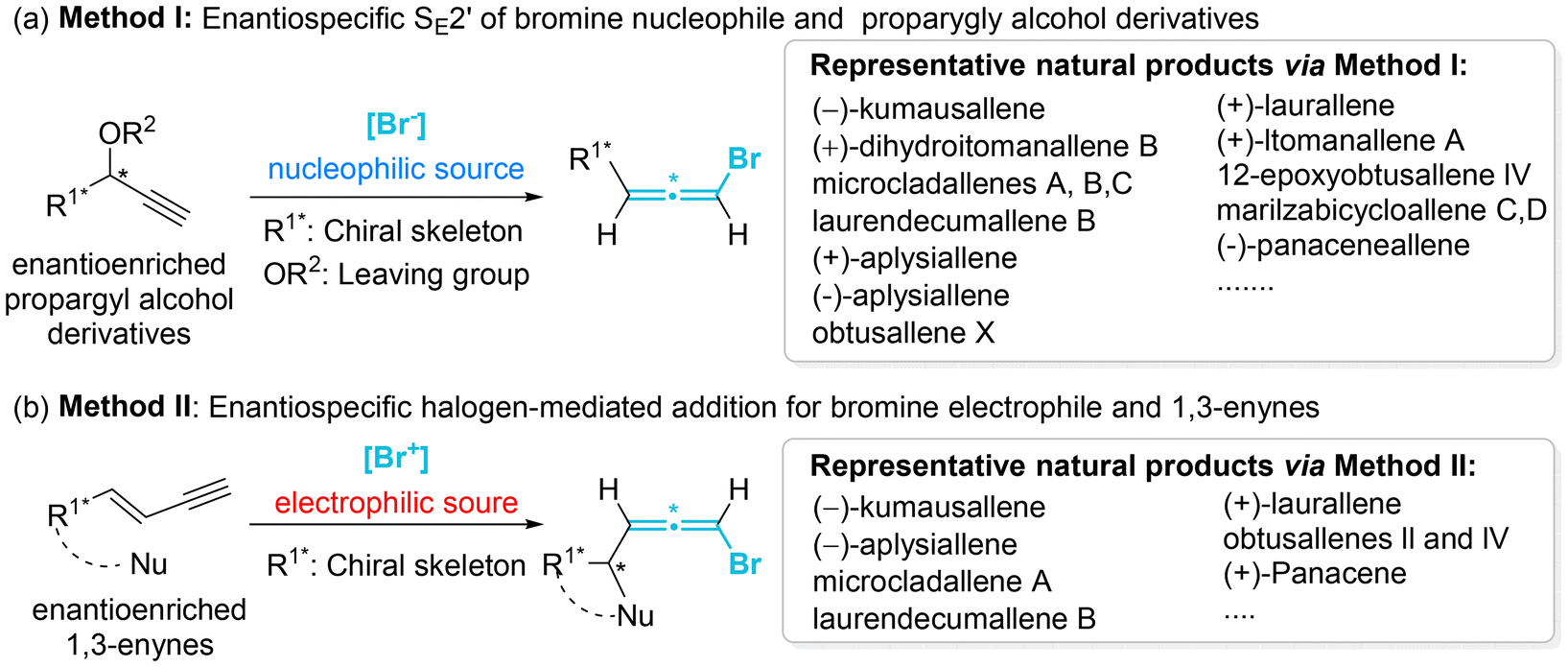
Despite the synthetic and pharmaceutical importance of heteroatom-substituted axially chiral allenes, the development of methods for their asymmetric synthesis remains challenging. In the early stages, the most reliable strategy for accessing these valuable chiral frameworks primarily relied on the use of chiral substrates through chirality transfer, involving stereospecific nucleophilic displacement, rearrangement, or elimination. To date, this traditional strategy is still employed for the assembly of HACA natural products and their synthetic precursors (Scheme 3).
Catalytic asymmetric synthesis is widely recognized as one of the most effective strategies for the preparation of chiral molecules in enantiomeric excess. In recent years, numerous innovative organic chemists have discovered a variety of stereoselective reactions utilizing diverse asymmetric catalysis, ingeniously achieving the asymmetric synthesis of some significant heteroatom-substituted axially chiral allenes. Thus, to raise awareness and facilitate the development of these chiral allenes, herein, we summarize the enantioselective synthetic methods for axially chiral allenes substituted with heteroatoms, including silicon (Si), stannum (Sn), nitrogen (N), phosphorus (P), oxygen (O), sulfur (S), bromine (Br), chlorine (Cl), fluorine (F), and boron (B).9 These catalytic methodologies include enzyme catalysis, chiral organocatalysis, and transition metal catalysis. Additionally, this review covers some elegant transformations involving achiral substrates mediated by stoichiometric chiral reagents. Meanwhile, we present some interesting asymmetric synthetic applications related to heteroatom-substituted axially chiral allenes, highlighting the unique reactivity imparted by the introduced heteroatoms.
2. Asymmetric synthesis of heteroatom-substituted axially chiral allenes
2.1 Asymmetric synthesis of allenylsilanes
Due to the fact that carbon and silicon belong to the same group in the periodic table, the “carbon–silicon switch” has become an important approach for modifying the chemical, physical, and biological characteristics of organic compounds. Meanwhile, the length of the C–Si bond is 1.87 Å, which is longer than that of the C–C bond (1.54 Å). This longer or fixed bond length contributes to the richer reactivity of organosilicon compounds. Thus, the asymmetric synthesis of allenylsilanes has received increasing attention, together with their synthetic applications.10–12
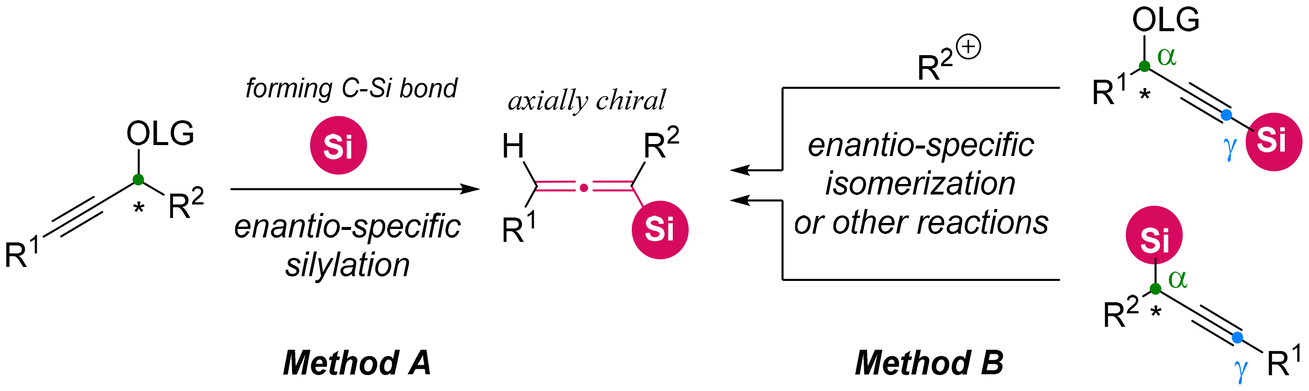
In the early stages, the asymmetric synthesis of axially chiral allenylsilanes predominantly depended on chirality transfer from various enantioenriched propargylic substrates. Two primary categories of substrates have been well-explored.11,12 The first category is the formation of C–Si bonds through the direct silylation of enantioenriched propargylic substrates utilizing silylboranes (Method A: Scheme 6).11 The second category starts from silyl-containing chiral propargylic substrates, utilizing a range of stereospecific transformations, such as nucleophilic displacement, isomerization, and elimination (Method B: Scheme 6).12
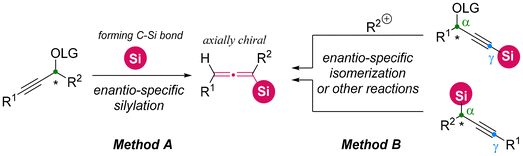 |
| | Scheme 6 Two general stereo-specific methods to access axially chiral allenylsilanes. | |
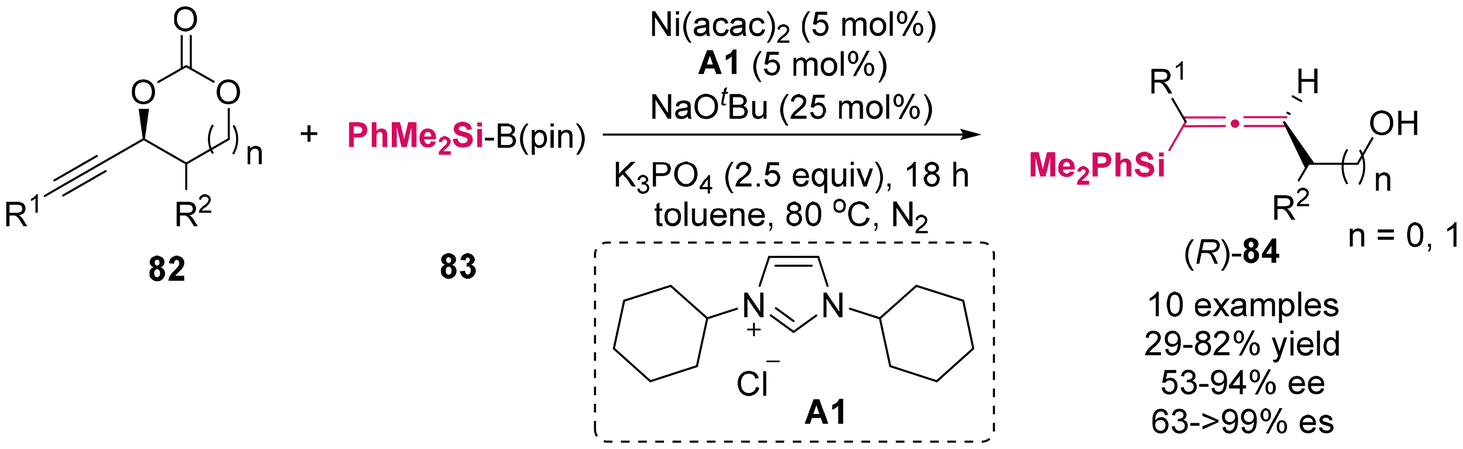
Utilizing stereospecific Method A,11 Kieij and colleagues recently synthesized a novel class of alkynyl cyclic carbonates 82 as enantioenriched substrates to investigate silylation with silylboranes 83, employing the Ni(acac)2/ICy·HCl A1 complex as the catalyst (Scheme 7).11f This protocol demonstrated a moderate to high enantiospecificity, affording axially chiral allenylsilanes 84 bearing a functional hydroxyl group. This methodology encompassed a decarboxylative silylation process via point-to-axial chirality transfer.
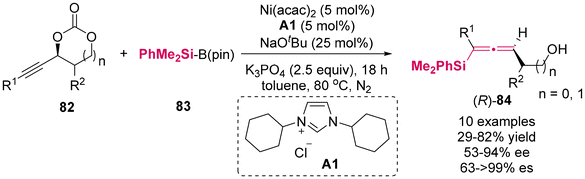 |
| | Scheme 7 Ni-catalyzed decarboxylative silylation of alkynyl carbonates: access to chiral allenes via enantiospecific conversions. | |
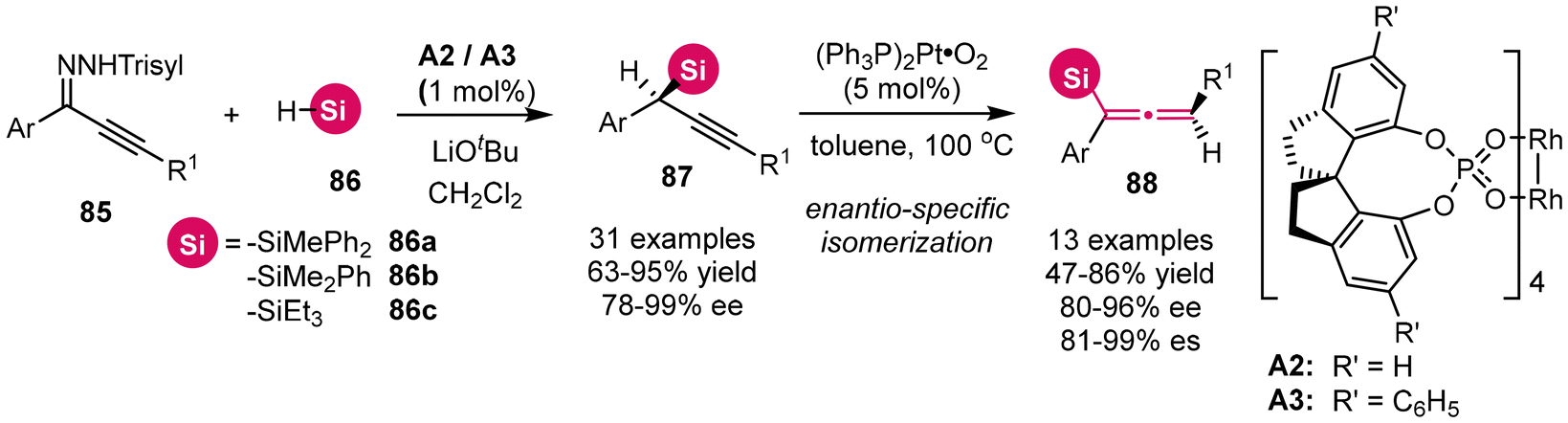
Utilizing stereospecific Method B, numerous studies have focused on γ-silylated propargylic alcohol derivatives, specifically with the silyl substituent located on the terminal alkyne motif.12 In contrast, propargylsilanes, which feature the silyl group at the a-site, have received comparatively less attention. In 2021, Zhou and Zhu's group disclosed the rhodium-catalyzed asymmetric insertion of alkynyl carbenes into the Si–H bonds of silanes, employing alkynyl N-trisylhydrazones as precursors for the alkynyl carbenes (Scheme 8).12g This approach resulted in the synthesis of a collection of challenging chiral propargylsilanes with remarkable stereoselectivity. Notably, the treatment of these chiral propargylsilanes with an in situ generated nanoparticulate catalyst (Ph3P)2Pt·O2 resulted in smooth isomerization to axially chiral allenylsilanes, achieving a commendable to high degree of enantiospecificity.
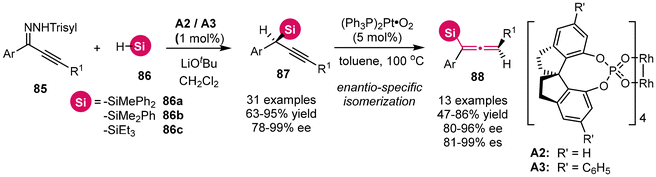 |
| | Scheme 8 Enantioselective insertion of alkynyl carbenes into Si–H bonds: efficient access to chiral propargylsilanes and allenylsilanes. | |
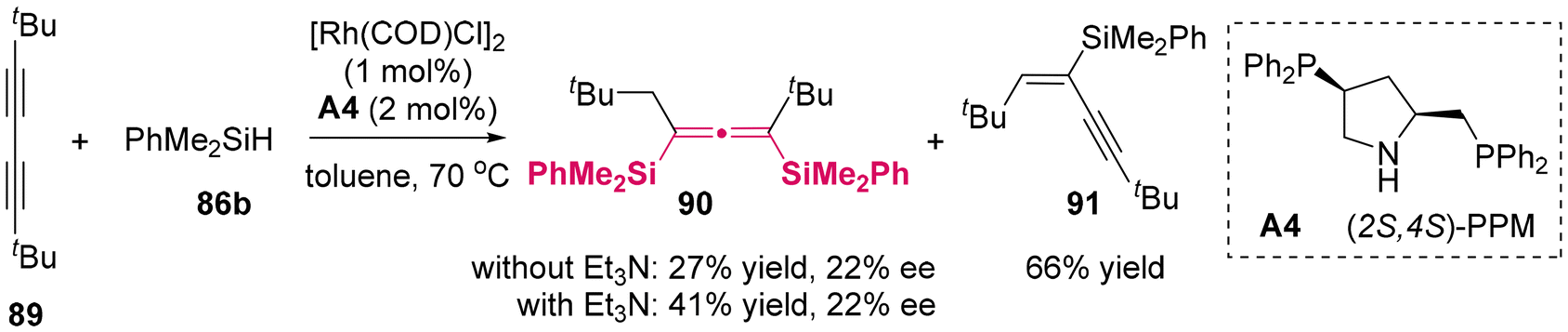
2.1.1 Catalytic enantioselective silylation reaction. In 1999, Tillack was the first to demonstrate the asymmetric synthesis of allenylsilanes through the hydrosilylation of butadiynes 89 using chiral phosphine A4/Rh(I) complexes as the catalyst (Scheme 9).13 However, the enantioselectivity achieved was limited, with an enantiomeric excess of only 22% and the best yield was moderate. Although the addition of triethylamine (Et3N) greatly promoted the second hydrosilylation, the yield of allenesilane 90 only reached 41%.
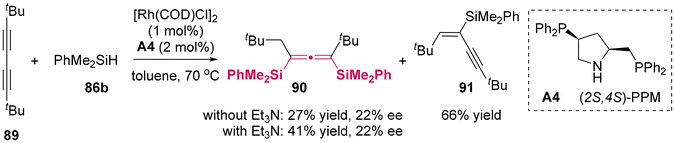 |
| | Scheme 9 Catalytic asymmetric hydrosilylation of butadiynes: new synthesis of optically active allenes. | |
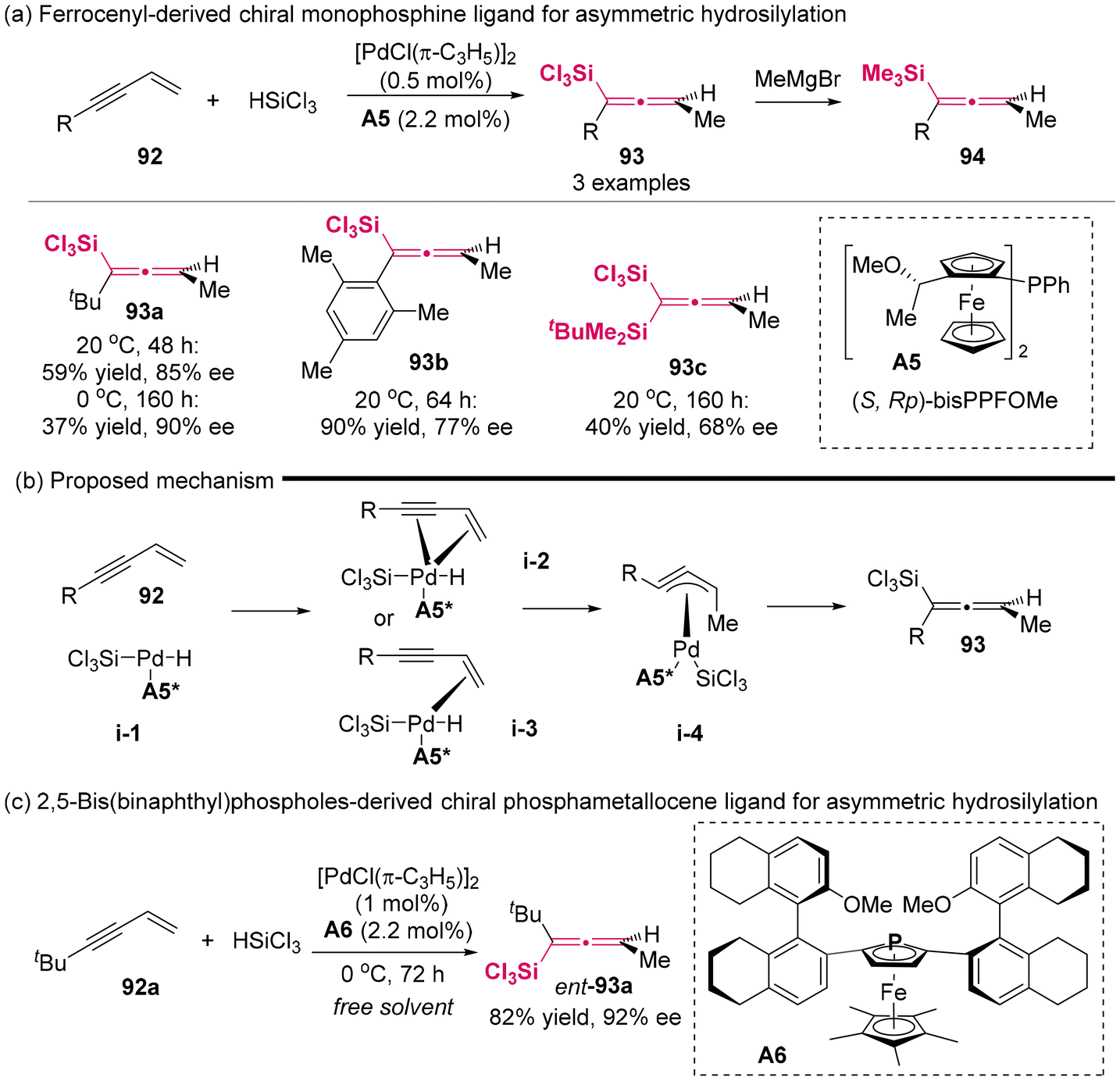
In 2001, the Hayashi group developed novel chiral monophosphine ligand A5 based on a ferrocenyl framework (Scheme 10a).14a Utilizing ligand A5 in coordination with palladium salt as the catalyst, the first successful instance of the preparation of axially chiral allenylsilanes in asymmetric catalysis was demonstrated through the asymmetric hydrosilylation of 1,3-enynes 92 with trichlorosilanes. The enantioselectivity achieved for the allenylsilanes reached up to 90% ee. To streamline the separation procedure and facilitate the analytical determination, the resulting allenyl(trichloro)silanes were treated with MeMgBr, giving relatively stable trimethyl(allenyl)silanes. Furthermore, the authors proposed a plausible mechanism to elucidate the generation of axially chiral allenylsilanes (Scheme 10b),14b which primarily involved the hydropalladation of the terminal alkene, resulting in π-propargyl(silyl)palladium intermediate i-4. The authors observed that the steric bulkiness of substituent R in compound 92 inhibited the hydropalladation of the alkyne moiety, thereby facilitating the synthesis of allenylsilanes. In 2006, Hayashi synthesized a new enantiomerically pure phosphametallocene ligand A6 derived from chiral phospholes that featured axially chiral biaryl substituents at the 2- and 5-positions. Using the novel ligand A6/Pd complex as a catalyst, the asymmetric hydrosilylation of t-butyl-substituted 1,3-enyne 92a gave rise to the corresponding axially chiral allenylsilane 93a with a high enantioselectivity of up to 92% ee and 82% yield (Scheme 10c).
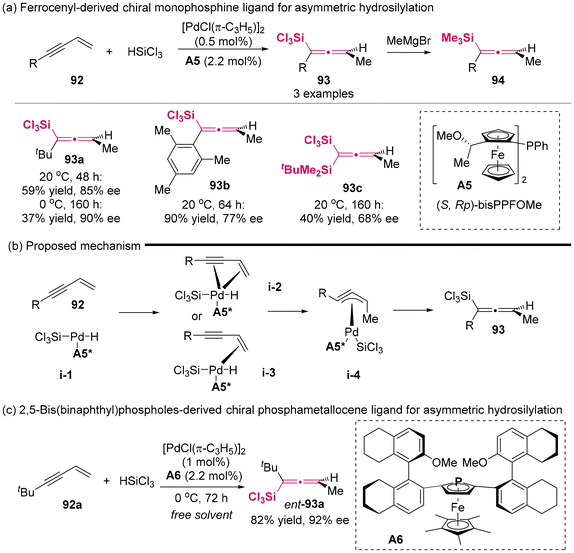 |
| | Scheme 10 Palladium-catalyzed asymmetric hydrosilylation of 4-substituted 1-buten-3-ynes. Catalytic asymmetric synthesis of axially chiral allenylsilanes. | |
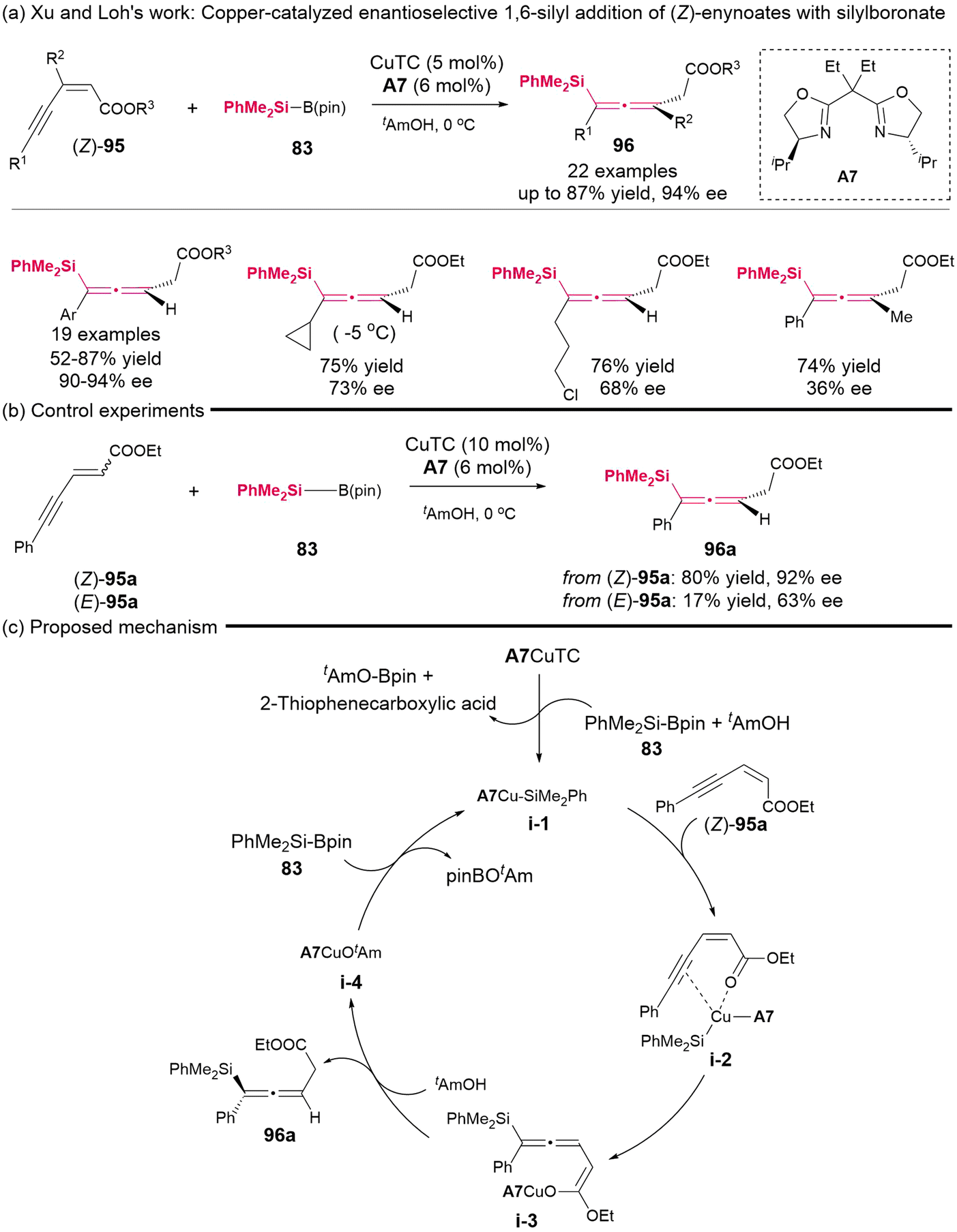
In 2015, Xu and Loh's group demonstrated a copper-catalyzed enantioselective 1,6-silyl addition involving (Z)-enynoates 95 and silylboronate 83, resulting in the formation of highly substituted axially chiral allenylsilanes (Scheme 11).15 The majority of these synthesized products exhibited moderate yields and high enantioselectivities; however, the enantiocontrol for the alkyl-substituted and tetra-substituted allenylsilanes was found to be unsatisfactory. Experiments indicated that (Z)-enynoates have superior reactivity and enantioselectivity compared to (E)-enynoates. Specifically, (Z)-enynoates yielded the desired product in 80% yield with 92% ee, whereas (E)-enynoates resulted in a poor yield of 17% with a moderate ee of 63%. Consequently, the authors confirmed the existence of crucial coordinated intermediate i-2, which is formed from the interaction of (Z)-enynoates with CuTC-A7 complex. The putative mechanism for this reaction is illustrated in Scheme 11c.
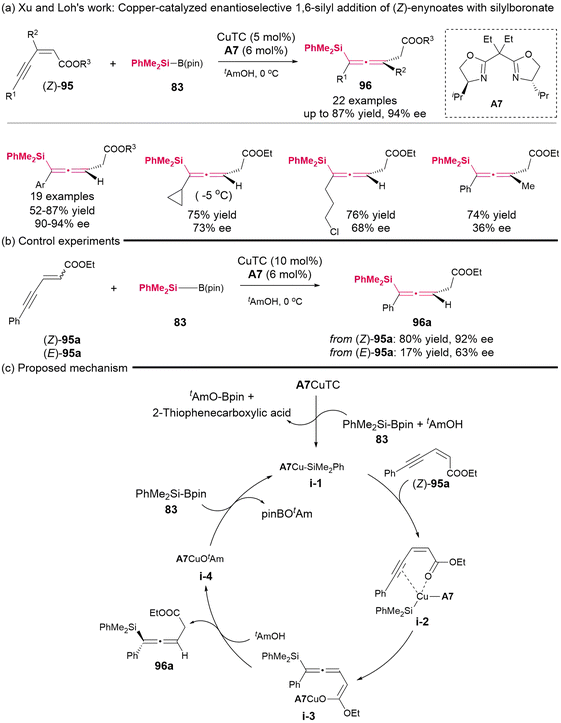 |
| | Scheme 11 Synthesis of highly substituted racemic and enantioenriched allenylsilanes via copper-catalyzed hydrosilylation of (Z)-2-alken-4-ynoates with silylboronate. | |
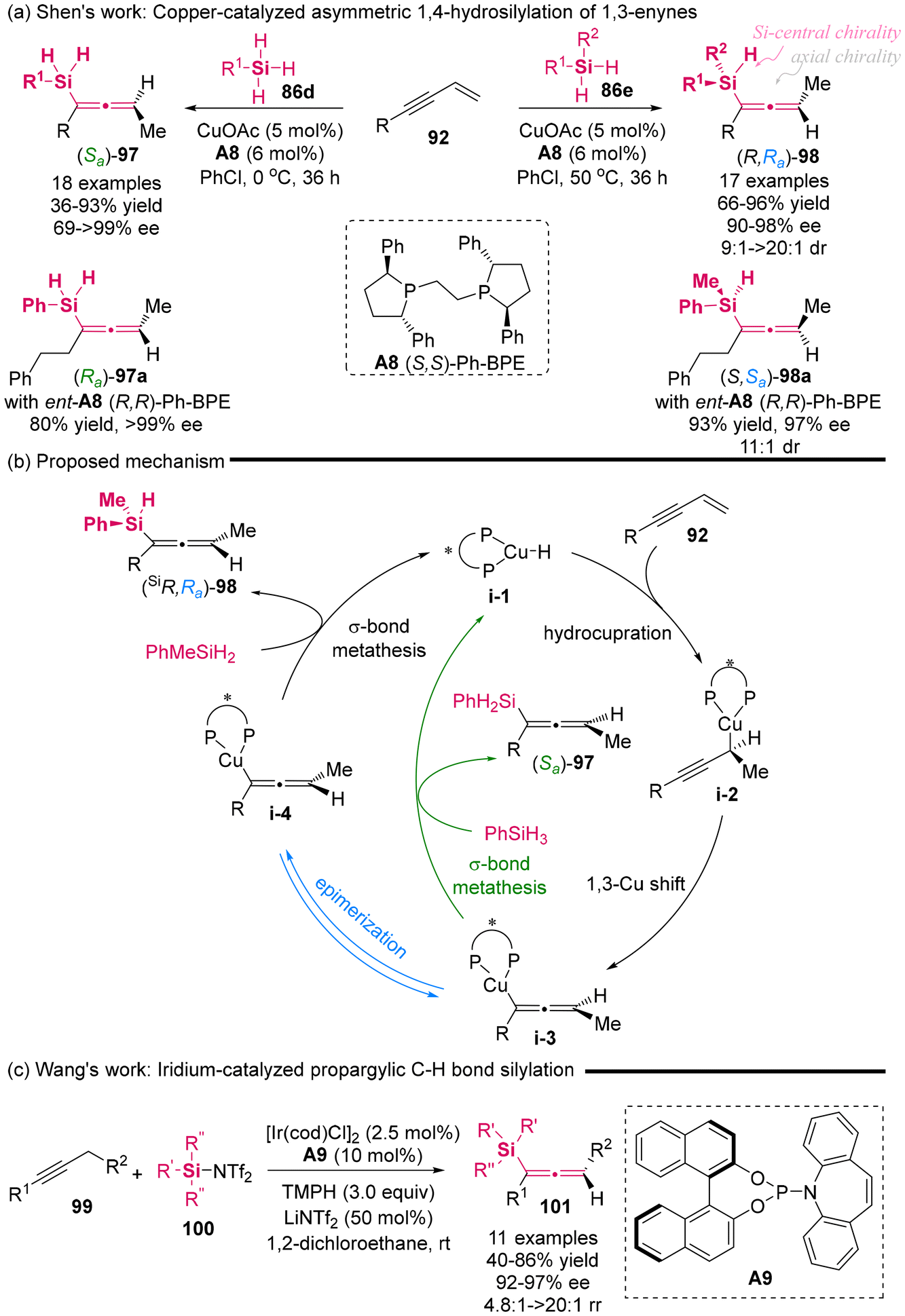
In 2023, Shen's group published the copper-catalyzed asymmetric 1,4-hydrosilylation of 1,3-enynes under the complex catalysis of CuOAc and (S,S)-Ph-BPE A8 (Scheme 12a).16 This protocol provided access to two distinct types of axially chiral silylallenes with excellent yield and high enantioselectivity. Notably, in addition to the chiral silylallenes featuring a single stereogenic axis derived from silanes 86d, this protocol was the first to employ prochiral unsymmetric di-substituted silanes 86e, thereby enabling the generation of chiral silylallenes 98 that possess both a stereogenic silicon center and a stereogenic axis, with excellent enantiocontrol and diastereocontrol. An interesting observation was made regarding the axial chirality of compounds 97 and 98, which were produced from the same chiral Cu/ligand complex, revealing a reversal in axial chirality. Furthermore, the use of the opposite enantiomers of chiral ligand ent-A8 (R,R)-Ph-BPE also resulted in the reversal of axial chirality for the corresponding products. As illustrated in Scheme 12b, the mechanistic studies suggested that the axial chirality reversal for compound 98 arises from the epimerization of chiral allenyl-cooper intermediate i-3 via a DYKAT process involving prochiral silanes 86e. In contrast, silanes 86d preferentially undergo a σ-bond metathesis step, leading to the formation of (Sa)-allene products 97. Recently, Wang's group utilized a phosphor-amidite-ligated iridium catalyst to synthesize axially chiral silylallenes 101 via the enantioselective propargylic silylation of alkynes (Scheme 12c).17 This reaction exhibited excellent enantioselectivity and the use of silyl bistriflimides 100 is crucial for the generation of silylallenes.
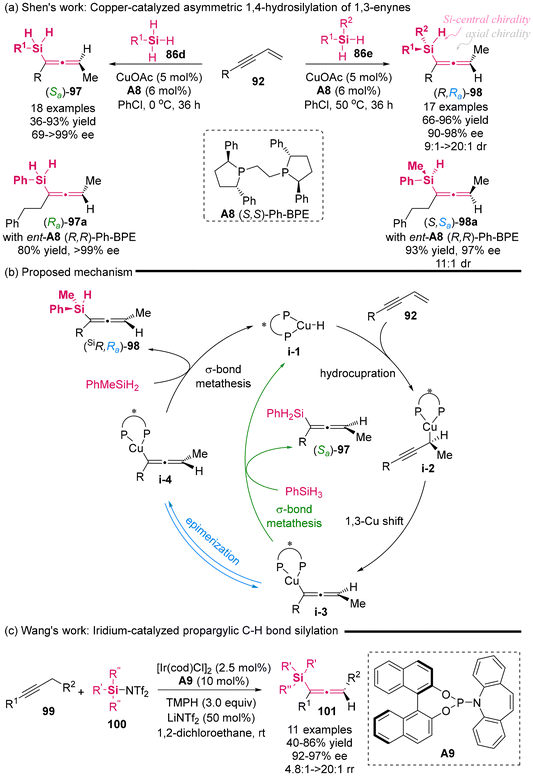 |
| | Scheme 12 Axial chirality reversal and enantioselective access to Si-stereogenic silylallene. | |
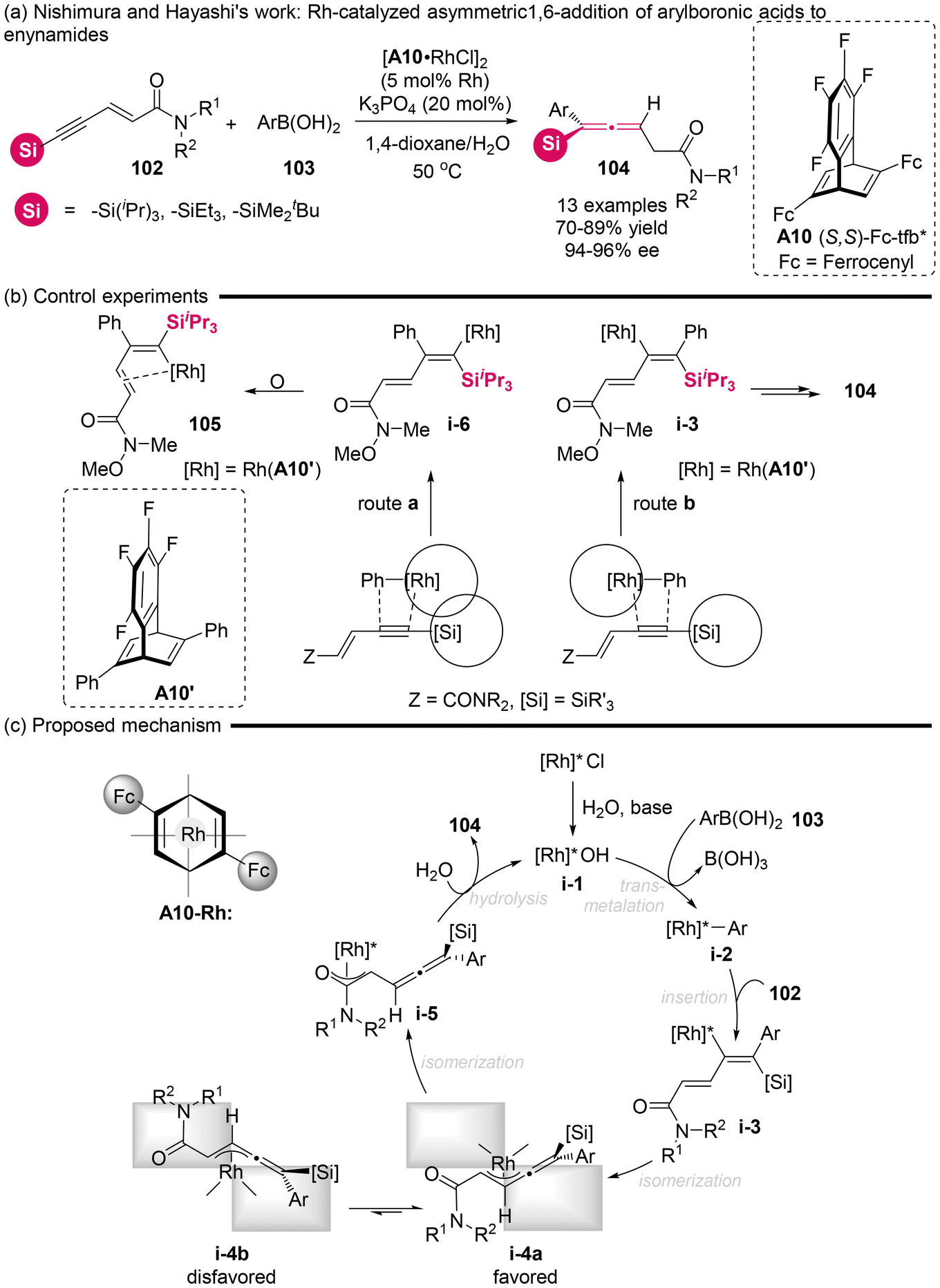
2.1.2 Catalytic asymmetric transformation of silyl-substituted substrates. In 2010, the Nishimura and Hayashi group successfully synthesized axially chiral trisubstituted allenylsilanes with a high degree of enantioselectivity (Scheme 13).18 This synthesis employed silyl-substituted enynamides 102 to achieve highly regioselective 1,6-addition with arylboronic acids 103, enabled by a rhodium/chiral diene complex catalyst. The proposed mechanism for this reaction likely involved several key steps, including transmetalation from species i-1 to i-2, a regioselective migratory insertion from i-2 to i-3, isomerization to form benzylidene-π-allylrhodium (i-3 to i-4a), and subsequent hydrolysis from i-4a/4b to 104 and recycled i-1. Based on the X-ray crystallographic analysis of alkenylrhodium complex 105 generated from a smaller substituent (phenyl) in ligand A10′ and the substrate, the authors believed that the observed excellent regioselectivity via route b involving migratory insertion step is attributed to the steric hindrance arising from the bulky substituent (Fc: ferrocenyl) of ligand A10 interacting with the silyl group of substrate 102.
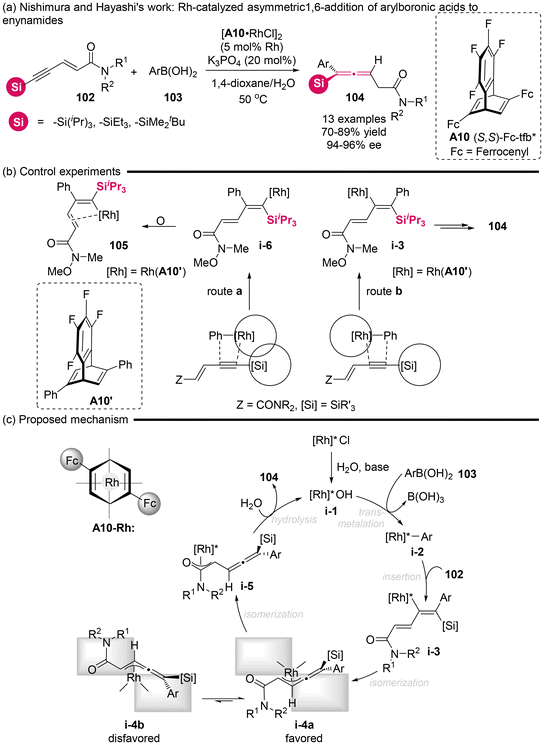 |
| | Scheme 13 Rhodium-catalyzed enantioselective 1,6-addition of arylboronic acids to enynamides: asymmetric synthesis of axially chiral allenylsilanes. | |
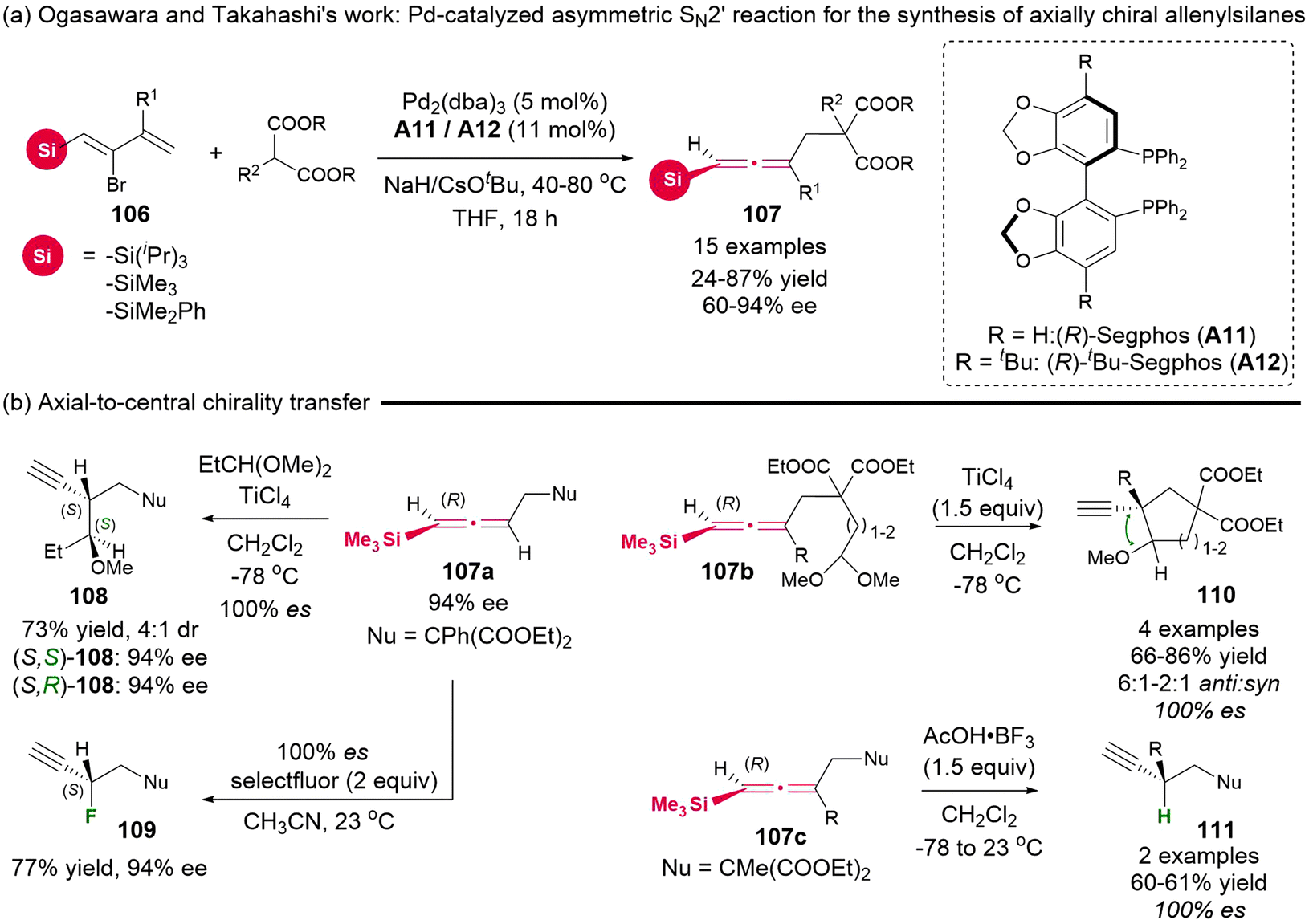
In 2010, Ogasawara and Takahashi's group reported the synthesis of a novel type of substrate, 2-bromo-1-silyl-1,3-dienes 106, which had not been previously documented.19 Utilizing this newly synthesized substrate, they established a palladium-catalyzed asymmetric SN2′ reaction with dimethyl acetal as the nucleophile, which afforded functional axially chiral allenylsilanes 107 in up to 94% ee and 87% yield (Scheme 14). Additionally, several enantio-specific desilylative SE2′ reactions of these chiral allenylsilanes were demonstrated, enabling the construction of tertiary and quaternary stereogenic centers through complete chirality transfer (100% es).
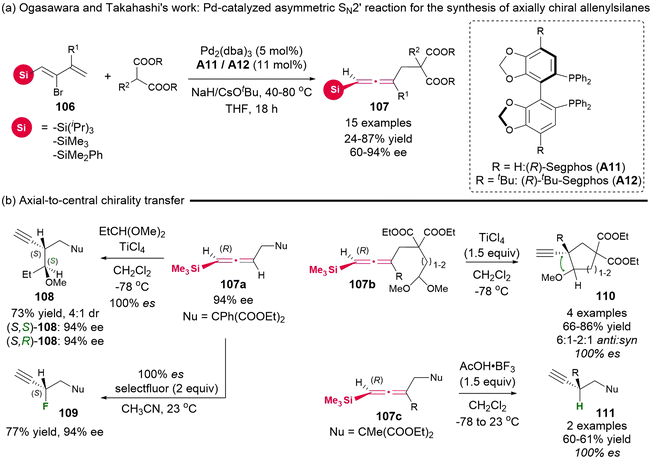 |
| | Scheme 14 Palladium-catalyzed asymmetric synthesis of axially chiral allenylsilanes and their application in SE2′ chirality transfer reactions. | |
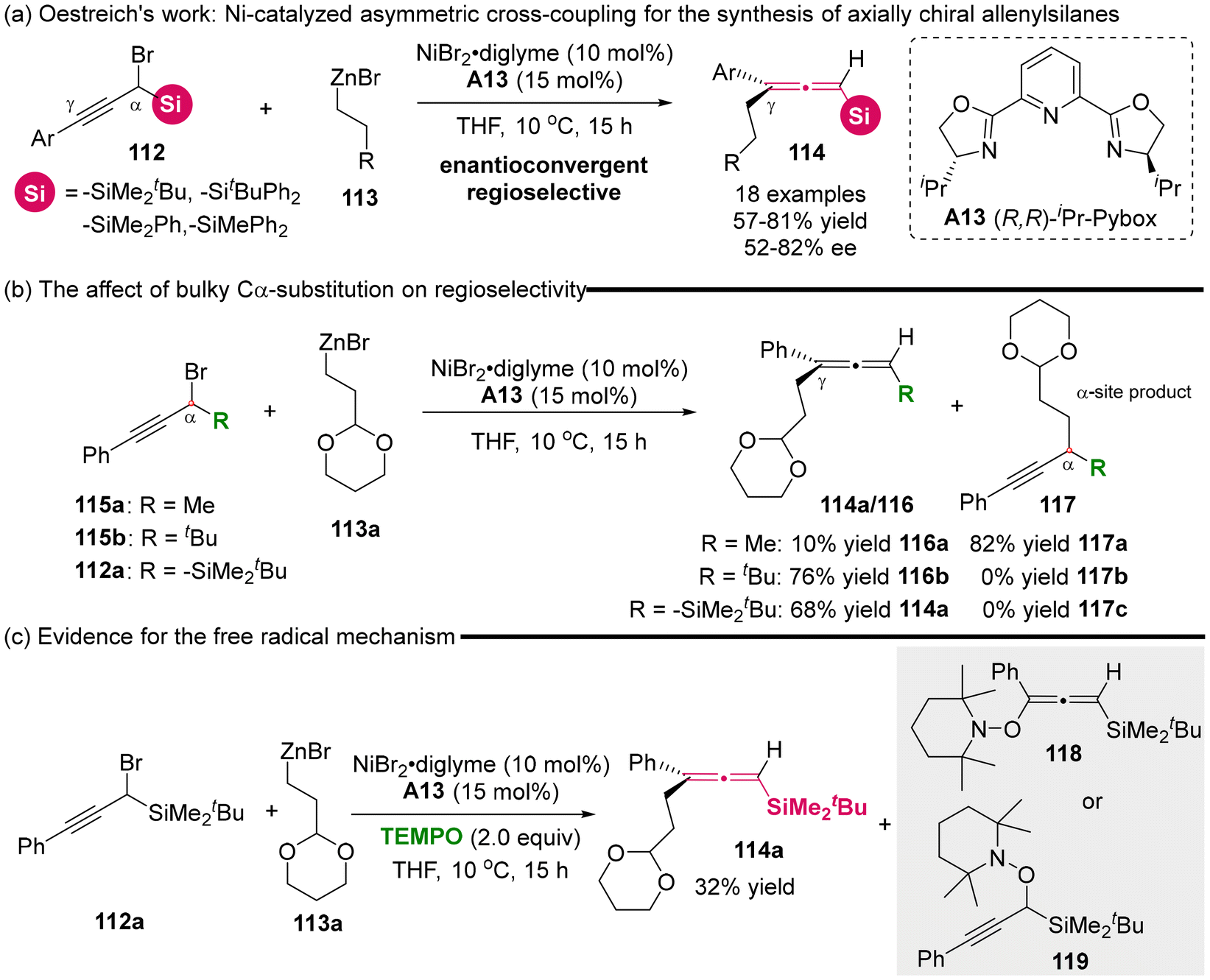
In 2021, the Oestreich group reported the development of a nickel-catalyzed asymmetric cross-coupling involving racemic α-silylated propargylic bromides 112 and primary alkylzinc reagents 113, giving access to axially chiral allenylsilanes 114 (Scheme 15).20 Moderate enantioselectivity was achieved using a chiral Pybox ligand/Ni complex. The control experiments revealed that the presence of the bulky silyl group acted as a steering group, promoting the high γ-selectivity and effectively restricting the formation of the C α-selective propargylic product 117c (Scheme 15b). Besides, the authors observed that the addition of radical trapping reagent (TEMPO) to the reaction mixture allowed the detection of TEMPO adducts 118 or 119 through GC-MS analysis, while the desired cross-coupling reaction was seriously inhibited (Scheme 15c, 114a: 32%). Consequently, they concluded that the nickel-catalyzed cross-coupling process is mediated by a radical-mediated mechanism.
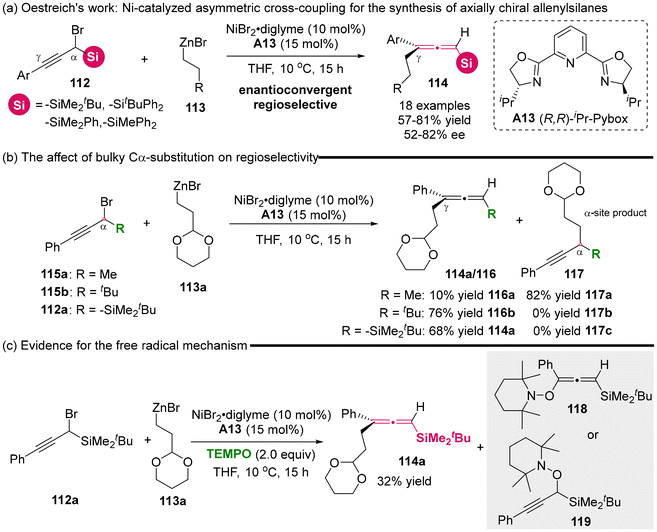 |
| | Scheme 15 Enantioconvergent and regioselective synthesis of allenylsilanes via nickel-catalyzed C(sp2)–C(sp3) cross-coupling starting from racemic α-silylated propargylic bromides. | |
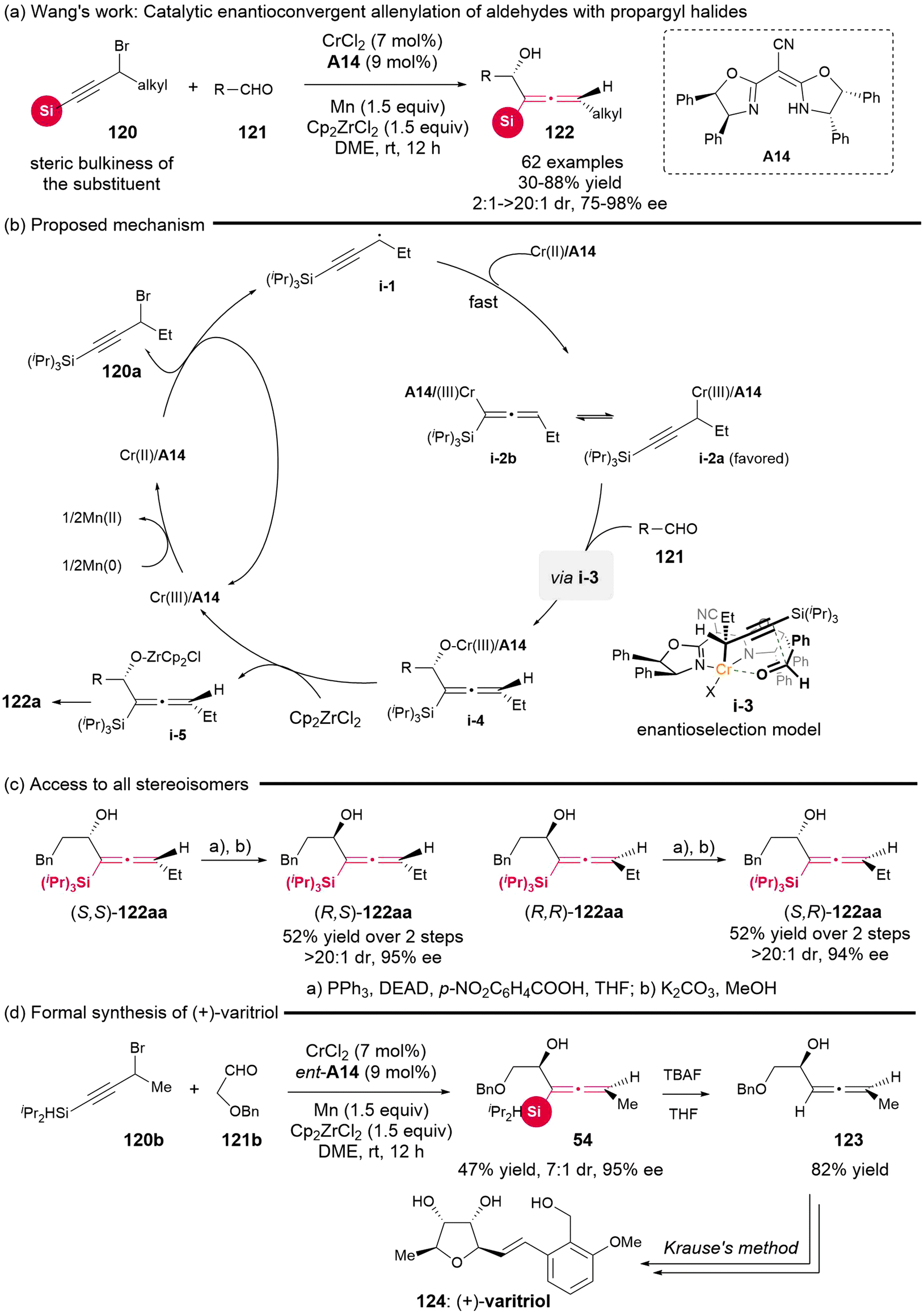
In 2022, the Wang group successfully established an enantioselective allenylation reaction involving propargyl halides 120 and aldehydes 121 (Scheme 16), which involved the asymmetric Nozaki–Hiyama–Kishi (NHK) coupling reaction with the catalyst of CrCl2/cyano-bisoxazo-line A14 complex.21 This protocol showed a broad substrate scope and excellent enantioselectivity, offering a straightforward pathway to a variety of significant chiral silyl-substituted α-allenols 122, which possess both adjacent axial and central chiralities. The authors observed that the presence of bulky substituents (such as tBu and Ph) replacing the silyl groups plays a crucial role in the selective formation of the allenylated products, owing to the existence of steric repulsion between the substrate and catalyst in the transition state, leading to the propargylated product.22a Based on the experimental data and previous related reports, the authors proposed the mechanistic pathway (Scheme 16b). Initially, propargyl radical i-1, generated through the single-electron reduction of propargyl bromide with Cr(II)/A14, is captured by Cr(II)/A14 to give two equilibrated species, propargyl chromium i-2a and allenyl chromium i-2b. To avoid the unfavorable steric repulsion interaction, i-2a as the preferred species undergoes nucleophilic attack at the γ-site from the Re-face of the aldehydes, leading to the formation of six-membered ring transition state i-3. Ultimately, this process yields allenol adduct i-4, which subsequently dissociates in the presence of Cp2ZrCl2 and undergoes hydrolysis to produce the final products. Furthermore, the sequential Mitsunobu reaction and hydrolysis enable the epimerization of central chirality, thereby converting one special stereoisomer of chiral α-allenols into the corresponding diastereomers (Scheme 16c). This strategic approach allows access to all four stereoisomers of the chiral α-allenols. Additionally, when the available chiral silyl-substituted α-allenol 54 was treated with TBAF, a desilylation reaction occurs, yielding α-allenol 123, which is a known important precursor for the total synthesis of (+)-varitriol.22b,c
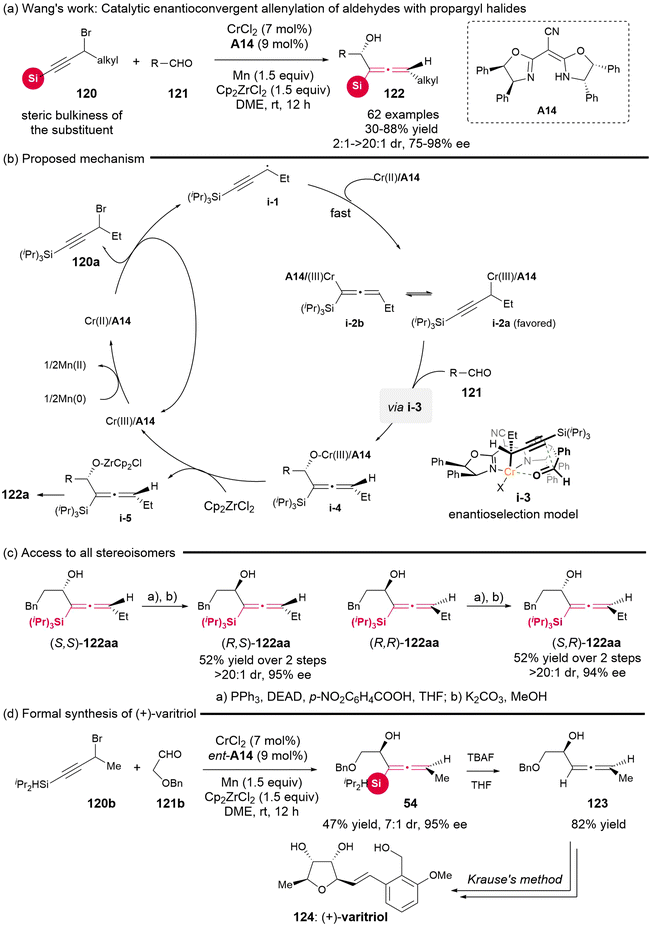 |
| | Scheme 16 Catalytic enantioconvergent allenylation of aldehydes with propargyl halides. | |
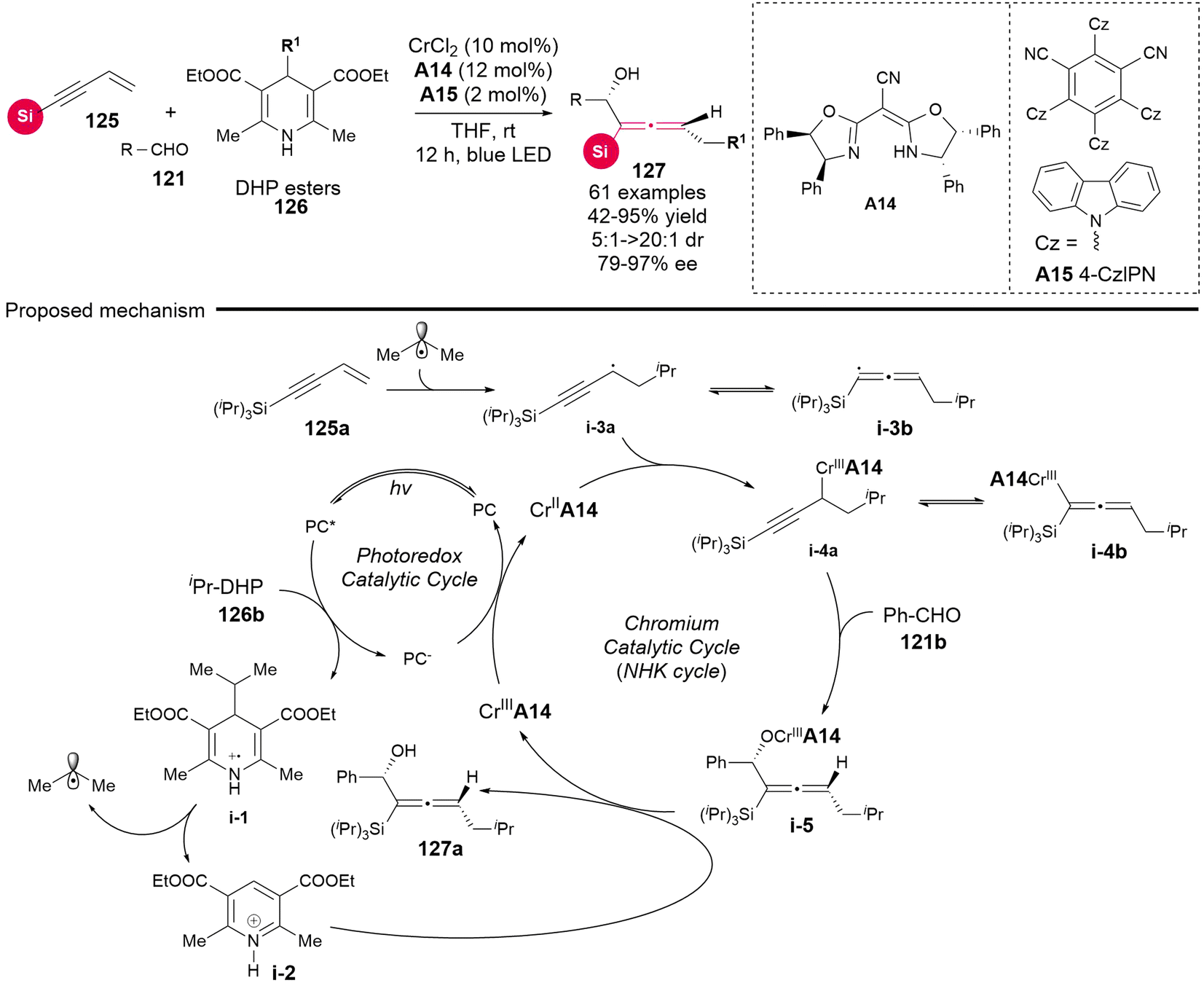
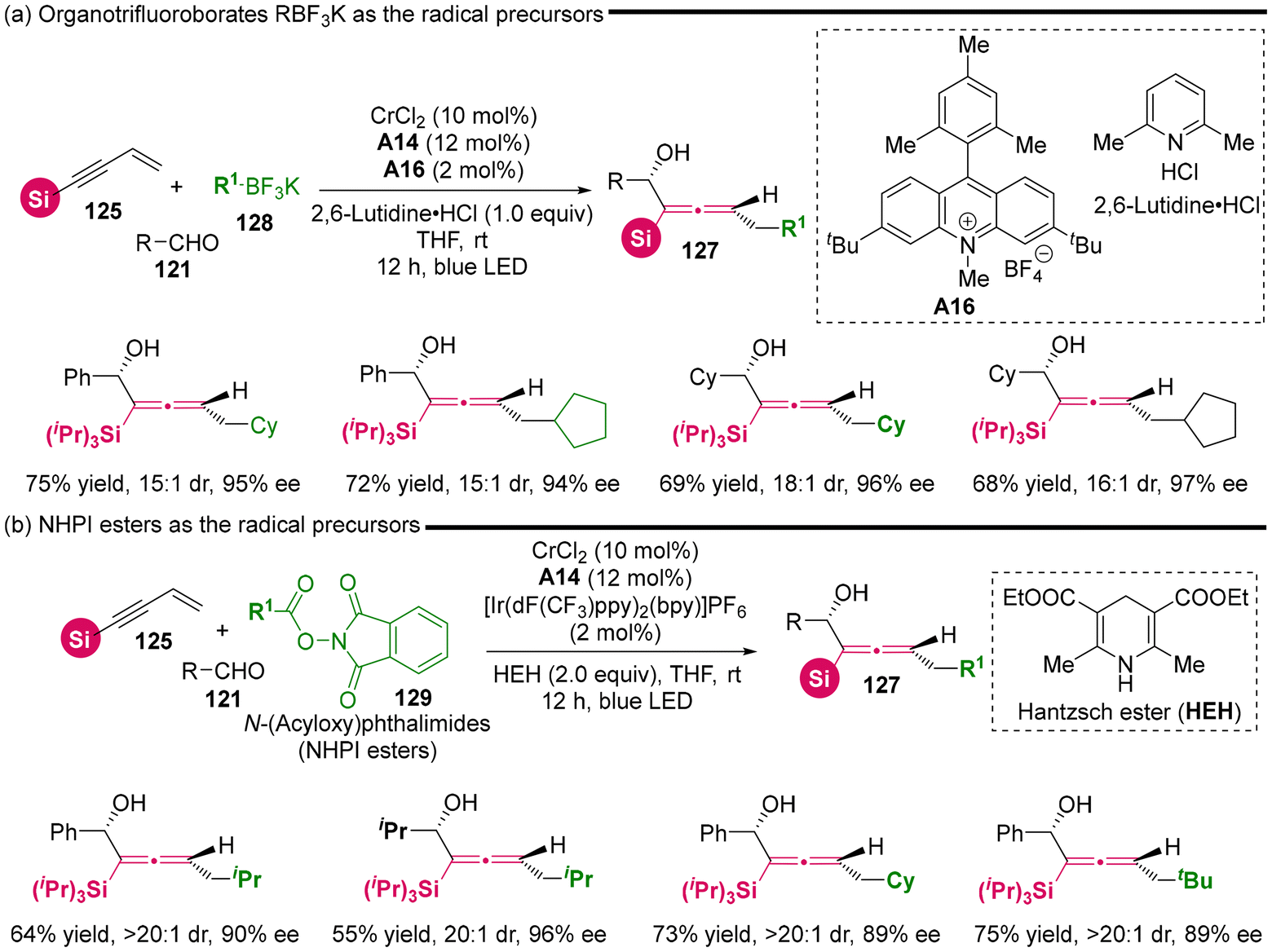
Considering the distinctive capability of photoredox catalysis, Wang's group utilized DHP esters 126 as radical precursors in dual photoredox and chromium catalysis (Scheme 17), establishing the radical-involved asymmetric 1,4-difunctionalization of silyl-substituted 1,3-enynes 125 with aldehydes 121.23 This methodology also resulted in the synthesis of various chiral silyl-substituted α-allenols bearing both a stereogenic center and a stereogenic axis. In comparison to the recently reported NHK cross-coupling method, the three-component photoredox-catalyzed NHK is characterized by avoiding the need for stoichiometric amounts of metal reducing agent and dissociation reagents, thus operating under an economically favorable redox-neutral condition. The mechanism investigations indicate that the reaction proceeds according to the proposed pathway, as follows: (a) upon excitation of the photocatalyst, DHP esters generate radical cation i-1, which subsequently undergoes fragmentation via C–C bond cleavage to yield an isopropyl radical and pyridinium i-2. (b) Subsequently, the isopropyl radical adds to the terminus of 1,3-enyne, resulting in the formation of adduct radical i-3a and i-3b. (c) These adducts enter the Cr-catalyzed NHK cycle, which is similar to the previously reported mechanisms.21 Notably, the in situ generated pyridinium i-2 facilitates the dissociation of intermediate i-5, leading to the final chiral allenols. Besides DHP esters 126 as radical precursors, the authors identified that organotrifluoroborates 128 and NHPI esters 129 can also serve as radical precursors for this reaction under modified conditions, which necessitate both additive dissociation reagents and an appropriate photoredox catalyst (Scheme 18). Additionally, the other bulky acetylenic substituents, including alkyl and aryl nonsilica groups, are also compatible, rendering the chiral allenol product devoid of a silyl group.
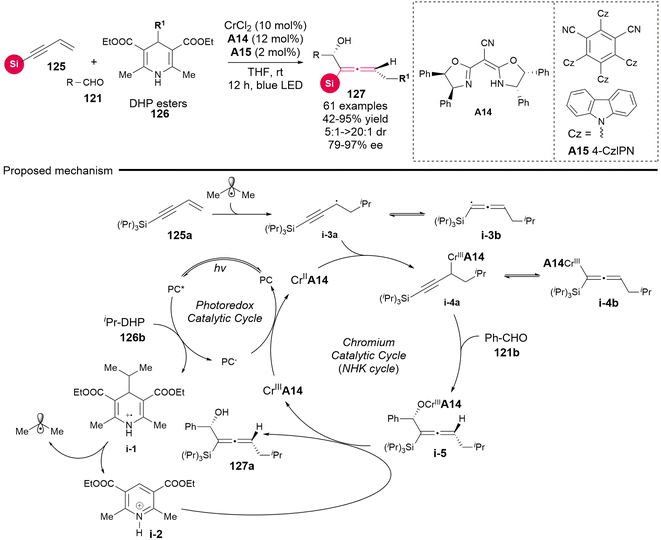 |
| | Scheme 17 Asymmetric 1,4-functionalization of 1,3-enynes via dual photoredox and chromium catalysis. | |
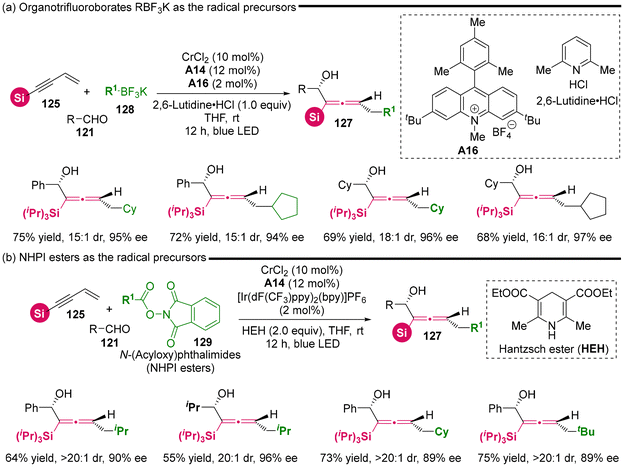 |
| | Scheme 18 Asymmetric 1,4-functionalization of 1,3-enynes via dual photoredox and chromium catalysis. | |
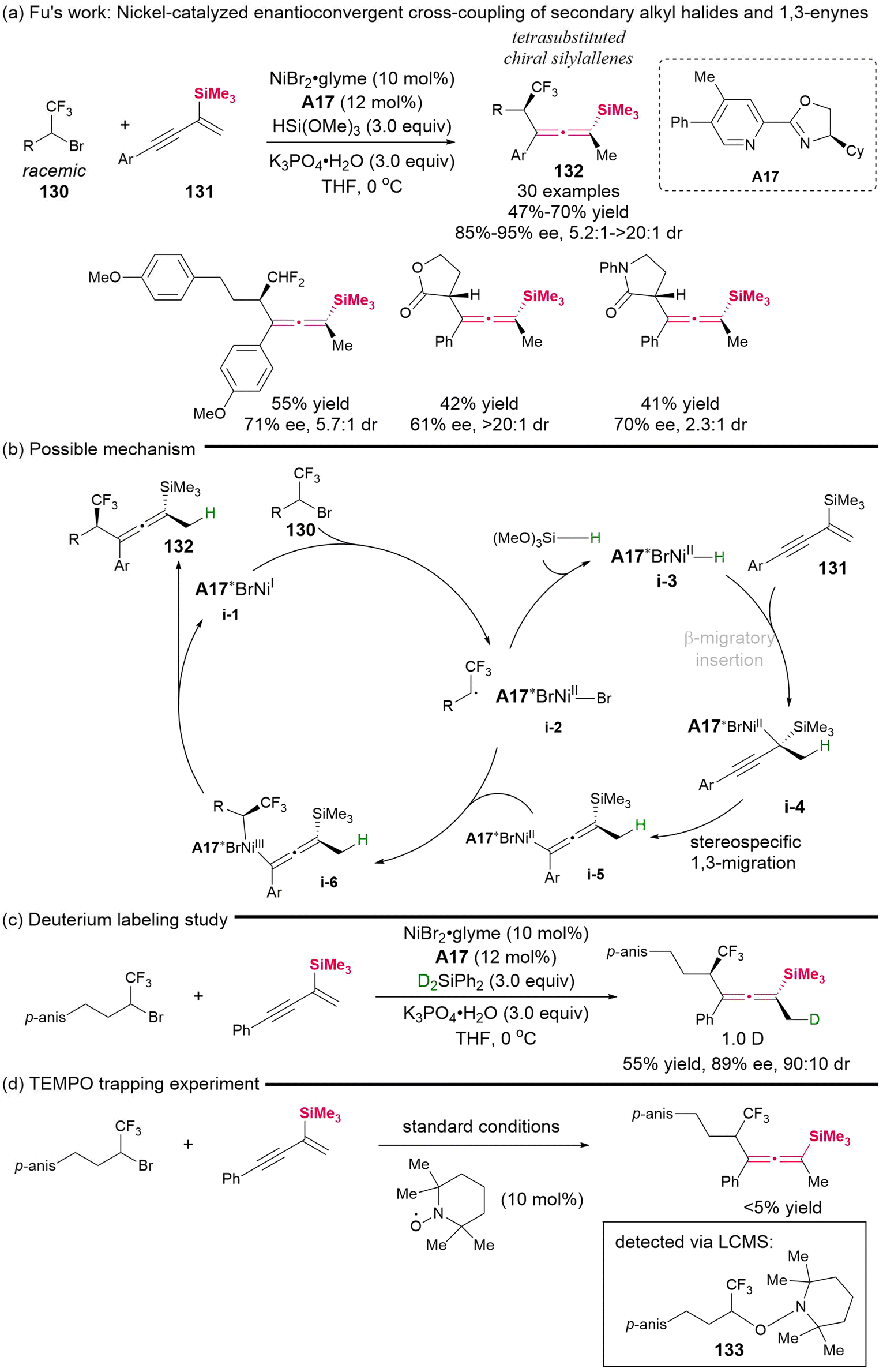
In 2024, Fu's group demonstrated a nickel-catalyzed enantioconvergent cross-coupling reaction involving secondary alkyl halides 130 and silyl-substituted 1,3-enynes 131 (Scheme 19).24 This represents the first successful catalytic asymmetric protocol that allows access to chiral tetrasubstituted silylallenes, which feature both central and axial chiralities and equipped with a versatile trifluoromethyl group. Besides the CF3-substituted secondary alkyl bromides as the competent electrophilic radical precursors, this methodology is also applicable to the analogous CF2H-substitued alkyl bromides, as well as α-bromolactone and α-bromolactam, delivering the desired products in moderate yields, albeit with varying but useful stereoselectivity. The control experiments suggested a radical-involved cross-coupling reaction mechanism, as illustrated in Scheme 19b–d. This mechanism primarily entails the selective β-migratory insertion of chiral Ni-hydride intermediate i-3, followed by stereospecific 1,3-migration, leading to the formation of the allenyl-Ni intermediate (i-4 to i-5), and subsequent coupling with a secondary radical via intermediate i-6 to give the final products.
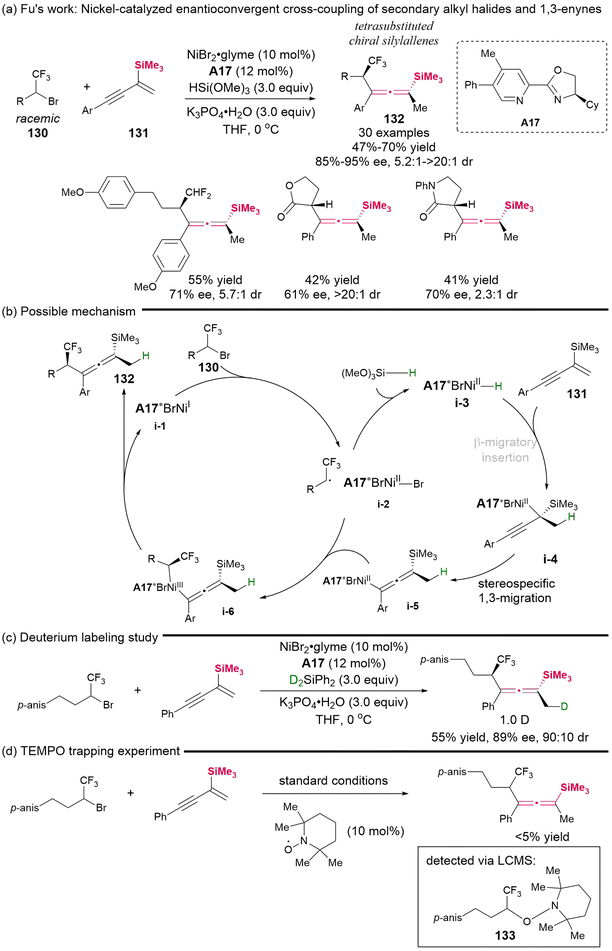 |
| | Scheme 19 Nickel-catalyzed enantioconvergent and diastereoselective allenylation of alkyl electrophiles: simultaneous control of central and axial chirality. | |
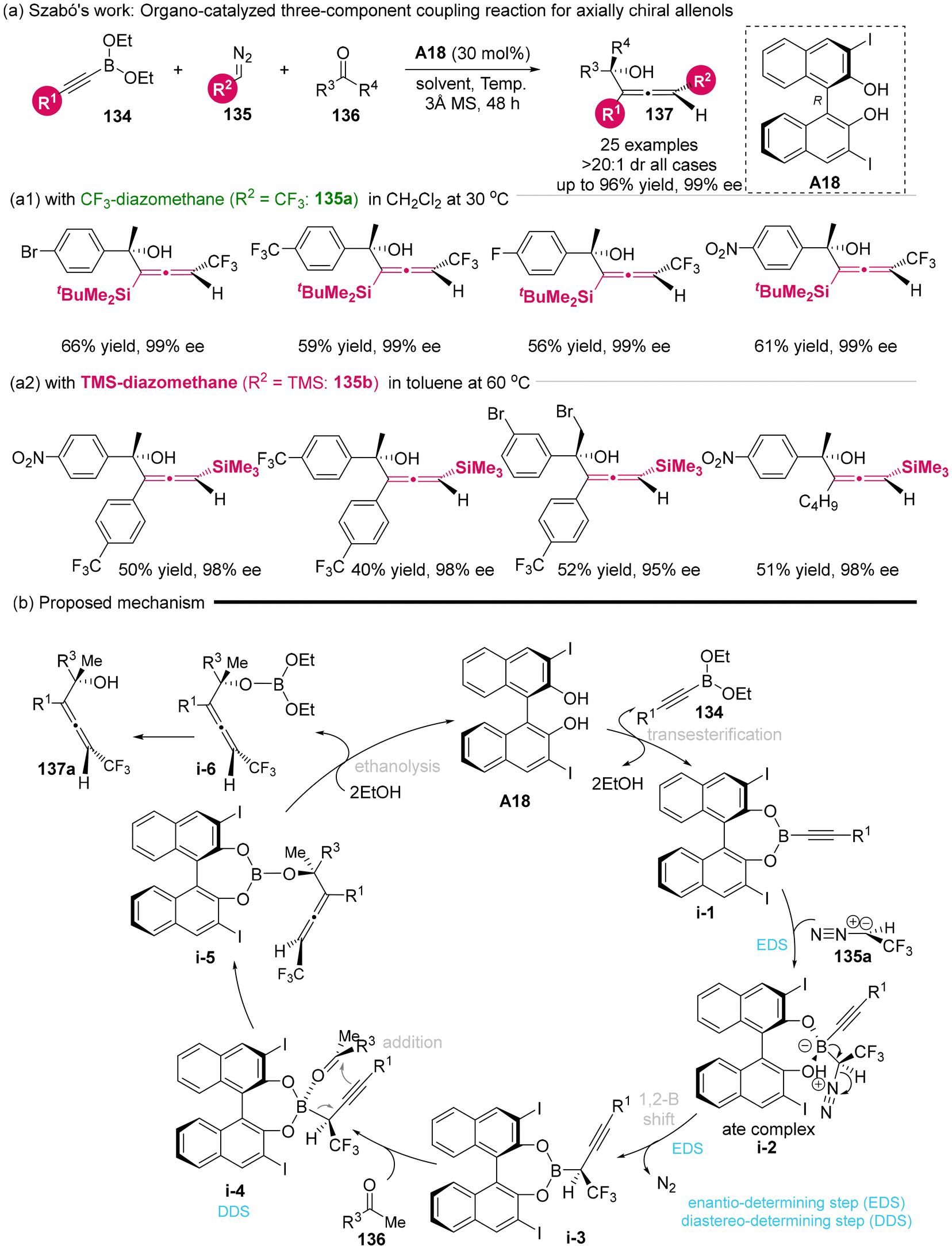
In 2023, Szabó's group developed an organocatalytic three-component coupling reaction involving alkynyl boronates 134, diazomethanes 135 and ketones 136 to construct densely functionalized axially chiral allenols utilizing the catalyst of BINOL derivatives A18.25 When silyl-substituted alkynyl boronates was used to react with CF3-diazomethane 135a and various ketones, the axially chiral CF3-allenylsilanes was produced with moderate yields (56%–66%) and excellent stereoselectivities (99% ee, >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) (Scheme 20a1). When TMS-diazomethane 135b was used to replace CF3-diazomethane, the silylated allenols was obtained with 40–52% yield and 95%–98% ee (Scheme 20a2). Based on experimental data and previous studies, the author proposed a putative mechanism, which involves several key steps: the transesterification of boronates 134 with catalyst A18 to give intermediate i-1, the enantioselective formation of a chiral boronate complex (i-1 to i-2), an enantioselective 1,2-borotropic rearrangement from i-2 to i-3, the diastereoselective addition of a ketone to i-3 via a Zimmerman–Traxler-type transition state i-4 involving point-to-axial chirality transfer, and finally, the ethanolysis of i-5 to yield the final product along with the regeneration of the catalyst.
:1 dr) (Scheme 20a1). When TMS-diazomethane 135b was used to replace CF3-diazomethane, the silylated allenols was obtained with 40–52% yield and 95%–98% ee (Scheme 20a2). Based on experimental data and previous studies, the author proposed a putative mechanism, which involves several key steps: the transesterification of boronates 134 with catalyst A18 to give intermediate i-1, the enantioselective formation of a chiral boronate complex (i-1 to i-2), an enantioselective 1,2-borotropic rearrangement from i-2 to i-3, the diastereoselective addition of a ketone to i-3 via a Zimmerman–Traxler-type transition state i-4 involving point-to-axial chirality transfer, and finally, the ethanolysis of i-5 to yield the final product along with the regeneration of the catalyst.
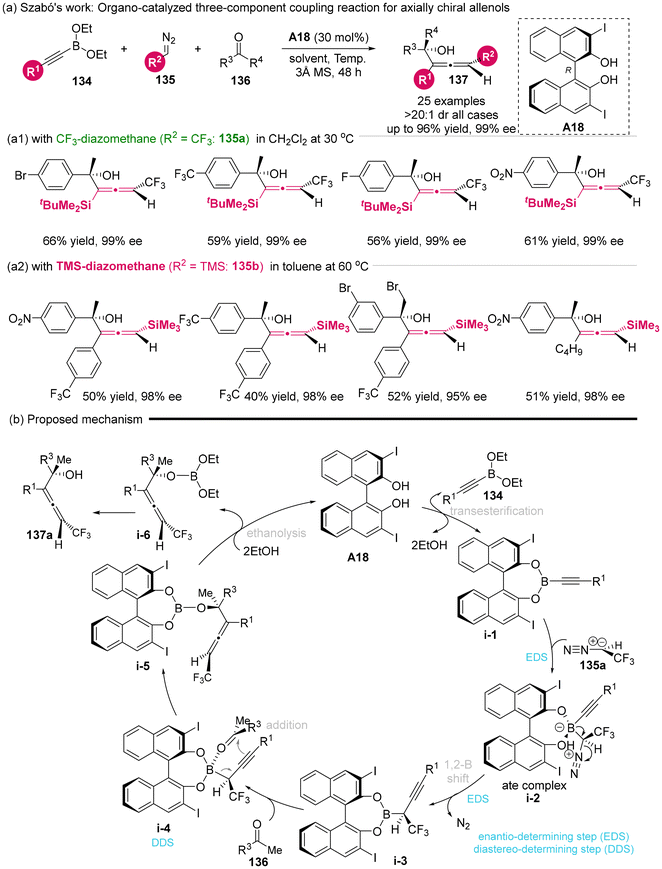 |
| | Scheme 20 Three-component approach to densely functionalized trifluoromethyl allenols by asymmetric organocatalysis. | |
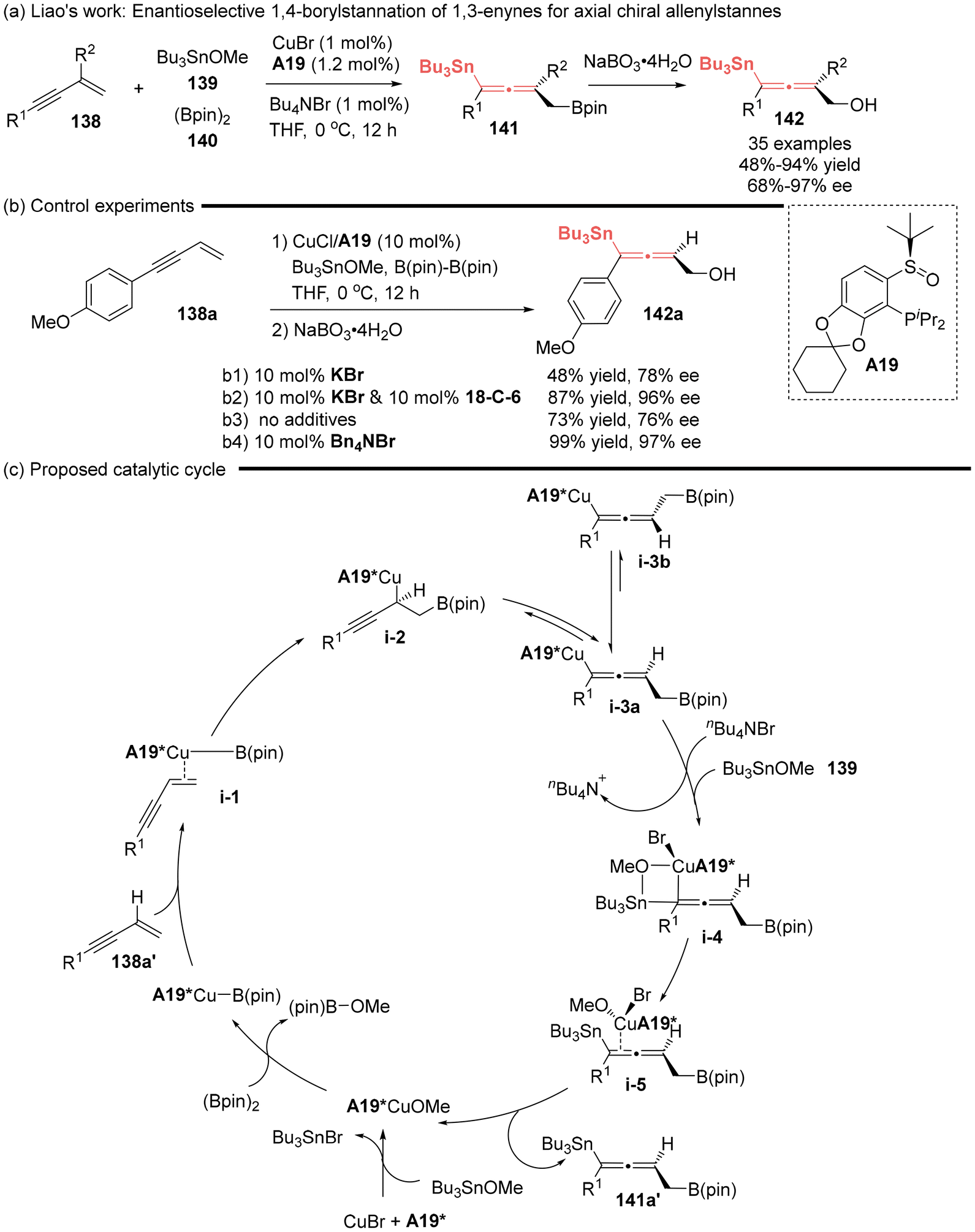
2.2 Asymmetric synthesis of allenylstannes
The significance of the 1,4-functionalization of unactivated 1,3-enynes has been underscored by the Liao group, pioneering the first asymmetric catalytic method for the synthesis of axial chiral allenylstannes 141.26,45,47 This was established by the asymmetric 1,4-borylstannation of 1,3-enynes utilizing a chiral sulfoxide phosphine (SOP A19)/Cu complex as the catalyst (Scheme 21). Notably, the authors observed an interesting phenomenon, wherein the presence of a halogenated salt can significantly improve the stereocontrol (Scheme 21b). The comprehensive mechanistic investigations, involving both experimental and theoretical approaches, revealed that the coordination of a soluble halide anion to copper(I) intermediate i-3a facilitated the formation of four-membered ring transition state i-4. This interaction efficiently avoided or significantly decelerated the racemization process via i-3a to i-3b, thereby greatly improving both the yield and stereoselectivity. Consequently, the catalytic cycle was proposed, as shown in Scheme 21c.
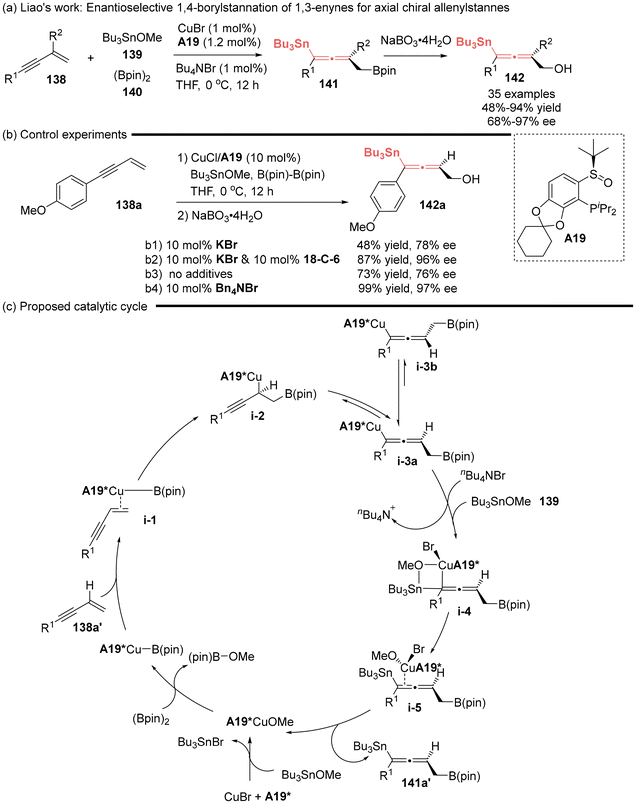 |
| | Scheme 21 Halogenated salt-assisted Cu-catalyzed asymmetric 1,4-borylstannation of 1,3-enynes: enantioselective synthesis of allenylstannes. | |
2.3 Asymmetric synthesis of aza-substituted allenes
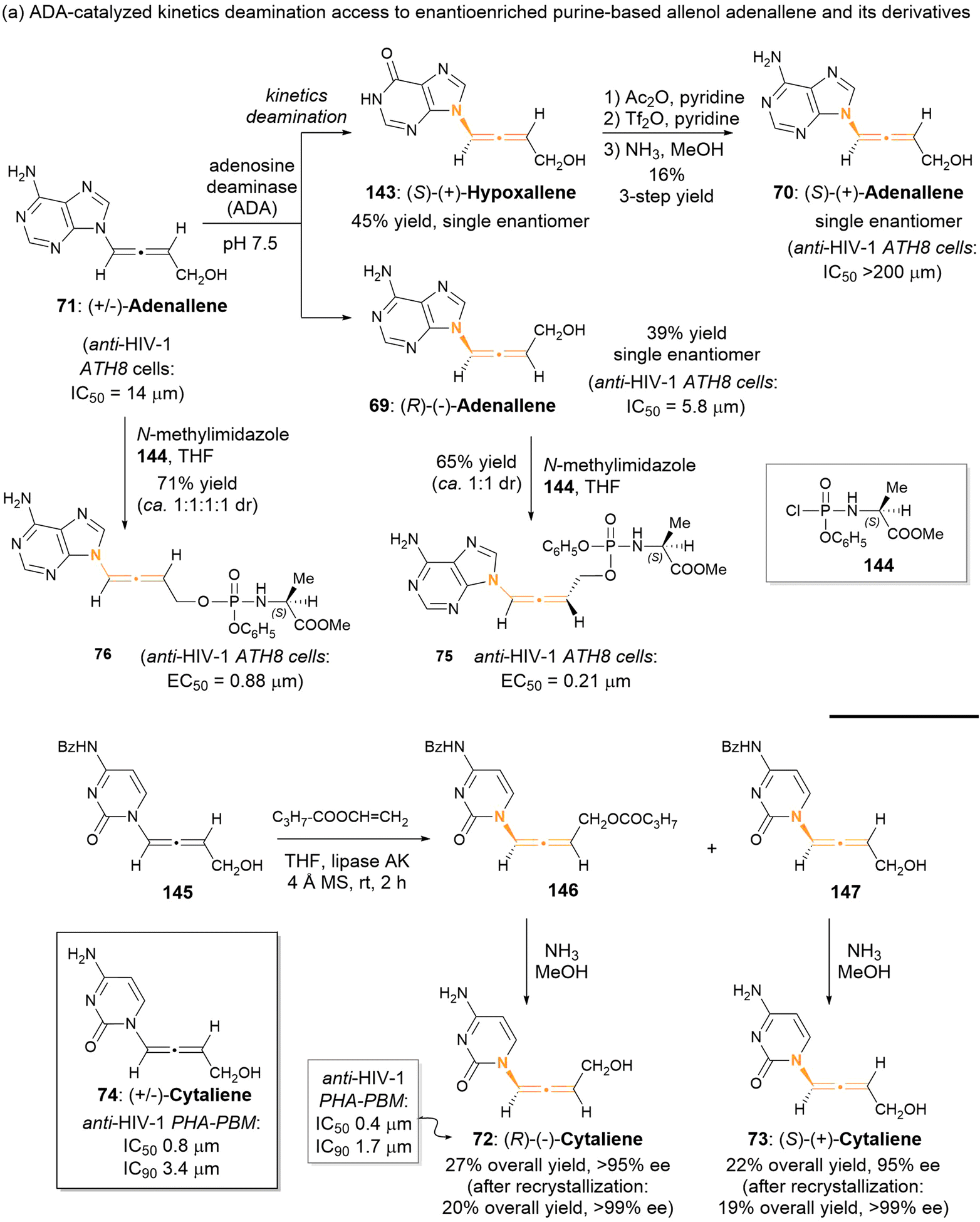
2.3.1 Enzyme-catalyzed kinetics resolution for aza-substituted allenes. Since 1992, Zemlicka has identified two enzyme-catalyzed kinetic resolutions of racemic allenic nucleoside analogues, specifically adenallene 71 and cytaliene 74 (Scheme 22).27 The kinetic deamination for adenallene, facilitated by adenosine deaminase, furnished the axially chiral N-containing allenes (R)-adenallene 69 and (S)-hypoxallene 143 each as a single enantiomer. Subsequently, (S)-hypoxallene 143 underwent a series of reactions including esterification, ammonolysis, and ester hydrolysis, finally leading to the synthesis of (S)-adenallene 70. Notably, enantiomer (R)-adenallene 69 exhibited superior anti-HIV-1 activity (IC50 = 5.8 μm) compared to (S)-adenallene 70.
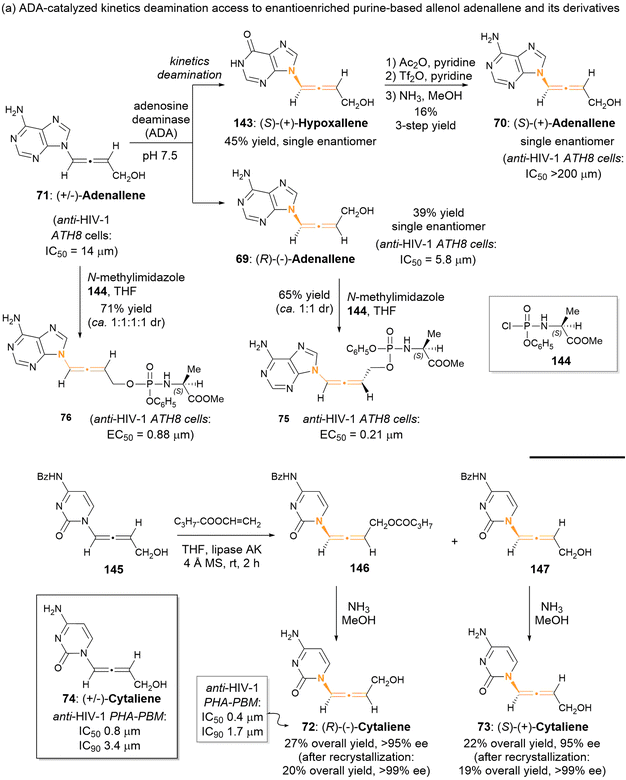 |
| | Scheme 22 Access to axially chiral allenic nucleoside analogues through enzyme-catalyzed kinetic resolution. | |
In a subsequent investigation,28 it was observed that phosphorylated adenallene derivatives 75 and 76 demonstrated anti-HIV-1 activities that were 16-times and 28-times greater than that of their parents 69 and 71, respectively. Cytaliene 74 represents another potent bioactive allenic nucleoside analogue. Due to the ineffective resolution of racemic cytaliene 74 by adenosine deaminase, the authors developed a lipase-catalyzed enantioselective acylation of N-Bz cytaliene 145 as an alternative approach. Following deprotection and recrystallization, optically pure (R)-cytaliene 72 and (S)-cytaliene 73 were successfully obtained. Between these enantiomers, (R)-cytaliene 72 was found to be a more effective inhibitor of HIV-I, while (S)-cytaliene 73 exhibited no activity.29
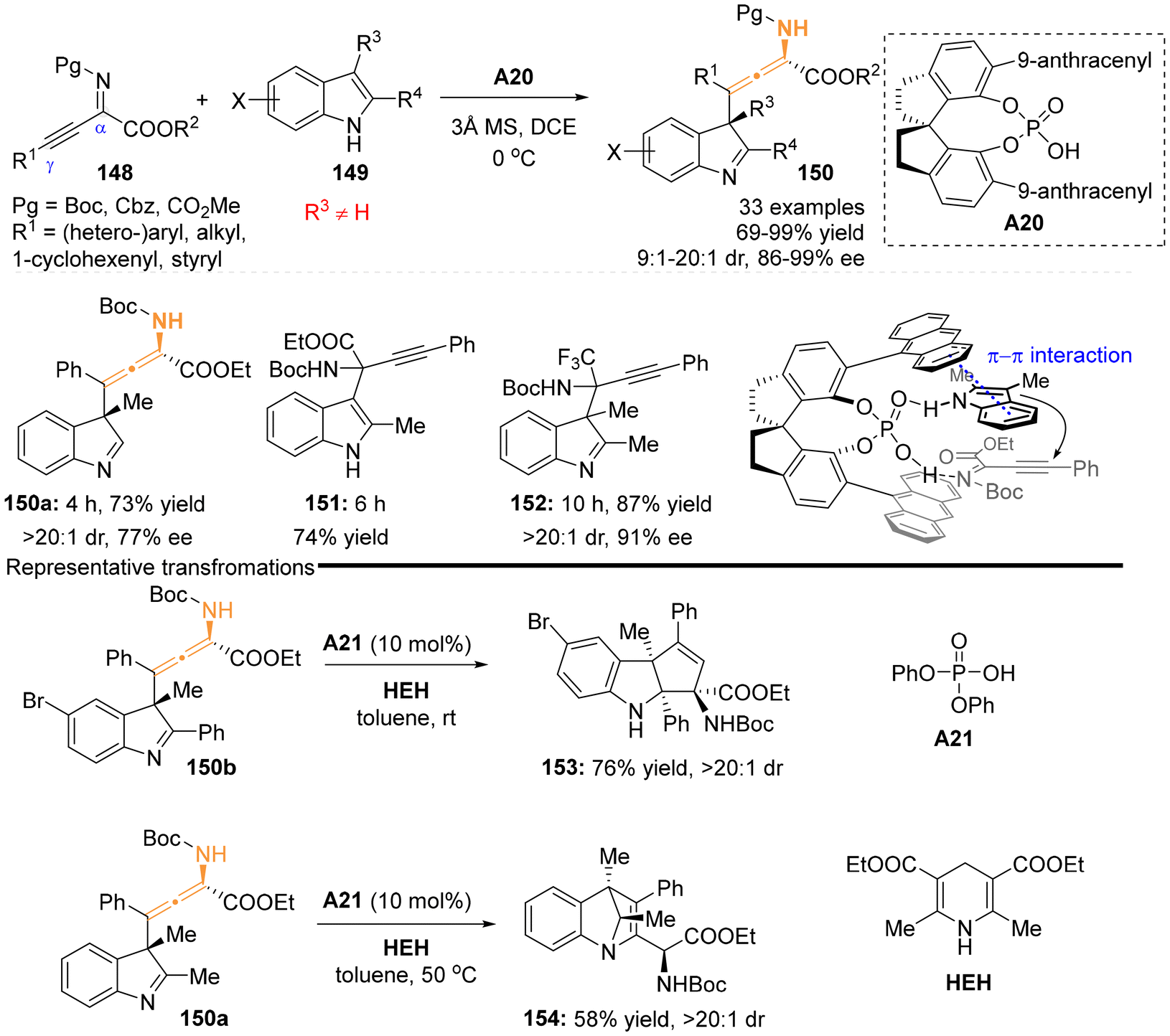
2.3.2 Organocatalysis for aza-substituted allenes. In 2019, the group of Sun and Wang developed a highly enantioselective dearomative γ-addition reaction involving β,γ-alkynyl-α-imino esters 148 and 2,3-disubstituted indoles 149, utilizing the catalyst of chiral spiro-phosphoric acids A20.30 This method proved to be an efficient approach for synthesizing a series of chiral N-containing tetrasubstituted allenes, achieving high yields and excellent stereoselectivities (Scheme 23). The presence of a substituent at the C3 position of the indole significantly influences the γ-regioselectivity of β,γ-alkynyl-α-imino esters due to steric hindrance. Notably, this catalytic method was ineffective for indoles lacking a substituent at the C3 position (2-methyl indoles) or alkynyl trifluoro-methyl ketimines, which only underwent α-addition reactions to yield products 151 and 152. The mechanistic study, supported by density functional theory (DFT) calculations, proposed a kinetically favored transition state, in which the phosphoric acid catalyst facilitates the activation of both substrates through H-bonding interactions. Furthermore, the chiral backbone and steric anthracene substituent of the catalyst were found to control the diastereoselectivity and enantioselectivity through steric effects and π–π stacking interactions. The resulting chiral N-containing tetrasubstituted allenes were identified as versatile building blocks. When subjected to diphenyl phosphate A21 as the catalyst and a Hantzsch ester (HEH) as a reductive reagent, the corresponding allenes 150a and 150b could perform two distinct reductive cyclizations, yielding two important aza-heterocycle scaffolds, 4H-1,4-methanoquinoline 154 and tetrahydrocyclopenta[b]indoline 153, respectively (Scheme 23b).
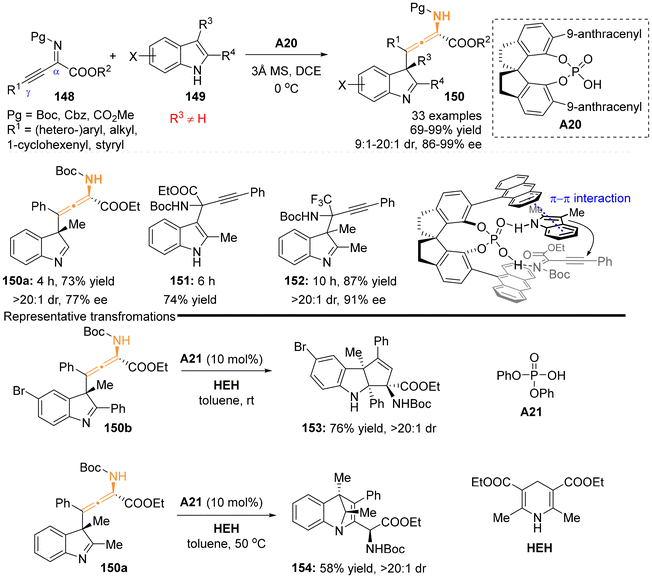 |
| | Scheme 23 Organocatalytic enantioselective synthesis of tetrasubstituted α-amino allenoates via dearomative γ-addition of 2,3-disubstituted indoles to β,γ-alkynyl-α-imino esters. | |
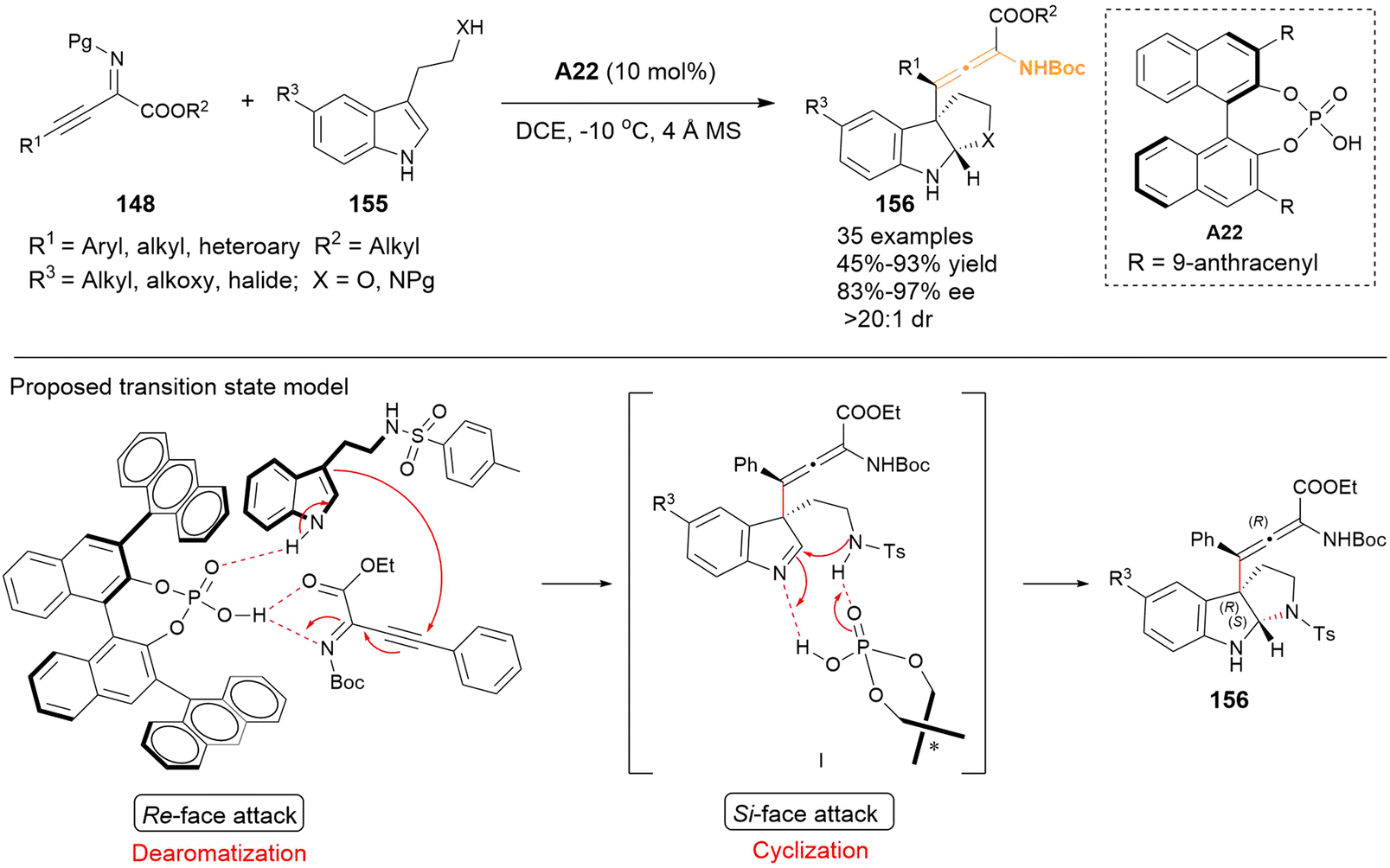
In 2024, Singh's group employed tryptamines and analogous 155 to establish dearomative γ-addition with β,γ-alkynyl-α-imino esters 148, thereby resulting in the synthesis of functional tetrasubstituted α-amino allenoates in high yields and excellent stereoselectivities (Scheme 24).31 The resulting allenes 156 featured a hexahydropyrrolo[2,3-b]indole group and contained one axial and two central stereogenic chirality. Based on the experimental data and previous works, they proposed a possible transition state for the reaction. Catalyst A22 was found to activate both substrates 148 and 155 simultaneously through hydrogen bonding interaction. The C3 position of substrate 155 approached 148 from the Re-face, thereby facilitating the dearomatization of tryptamine 148. Following this, the Si-face of the resulting intermediate I underwent stereoselective intramolecular cyclization, leading to the formation of tetrasubstituted allene 156.
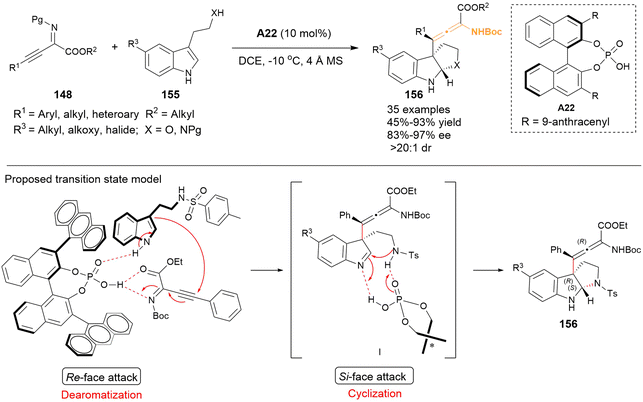 |
| | Scheme 24 Asymmetric cascade dearomatization-cyclization reaction of tryptamines with β,γ alkynyl-α-imino esters to access hexahydropyrrolo[2,3-b]indole-containing tetrasubstituted α-amino allenoates. | |
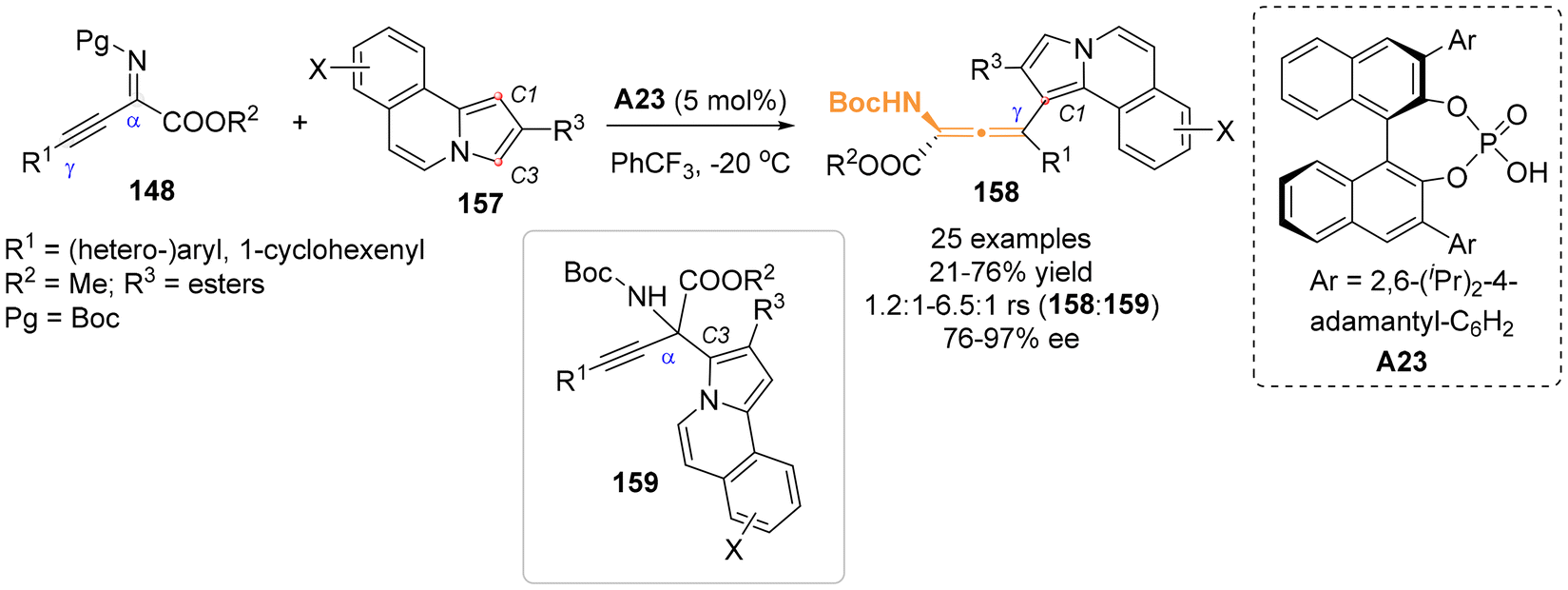
Recently, Song and Ni et al. reported their findings on the asymmetric arylation of β,γ-alkynyl-α-imino esters 148 with C2-substituted pyrrolo[2,1-a]isoquinoline 157 as nucleophiles, enabled by the catalyst of chiral phosphoric acid A23 (Scheme 25).32 Their study revealed that the sterically hindered C1-position of pyrrolo[2,1-a]isoquinoline 157 preferentially attacked the γ-site of β,γ-alkynyl-α-imino esters 148. This reaction predominately yielded N-containing axially chiral allenes 158 with yields reaching up to 76% yield and 97% ee. However, this process was also accompanied by the formation of a competitive C3-regioselective product 159.
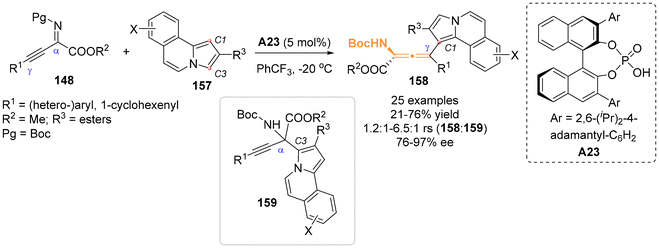 |
| | Scheme 25 Organocatalytic regio- and enantioselective C1-arylation of β,γ-alkynyl-α-imino esters with pyrrolo[2,1-α]isoquinolines. | |
Then, in 2021, Lin's group successfully utilized 1-substituted 2-naphthols 160 to develop an asymmetric dearomative γ-addition reaction involving β,γ-alkynyl-α-imino esters 148 in the presence of chiral phosphoric acids A24 catalyst (Scheme 26).33 This approach resulted in high regio-, diastereo-, and enantioselectivities for the corresponding axially chiral N-containing tetrasubstituted allene-derived naphthalenones.
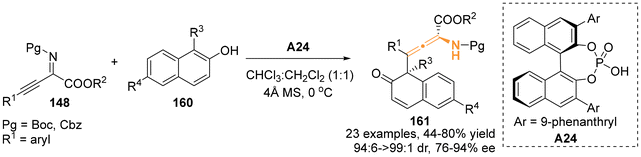 |
| | Scheme 26 Organocatalytic asymmetric dearomatization reaction for the synthesis of axial chiral allene-derived naphthalenones bearing quaternary stereocenters. | |
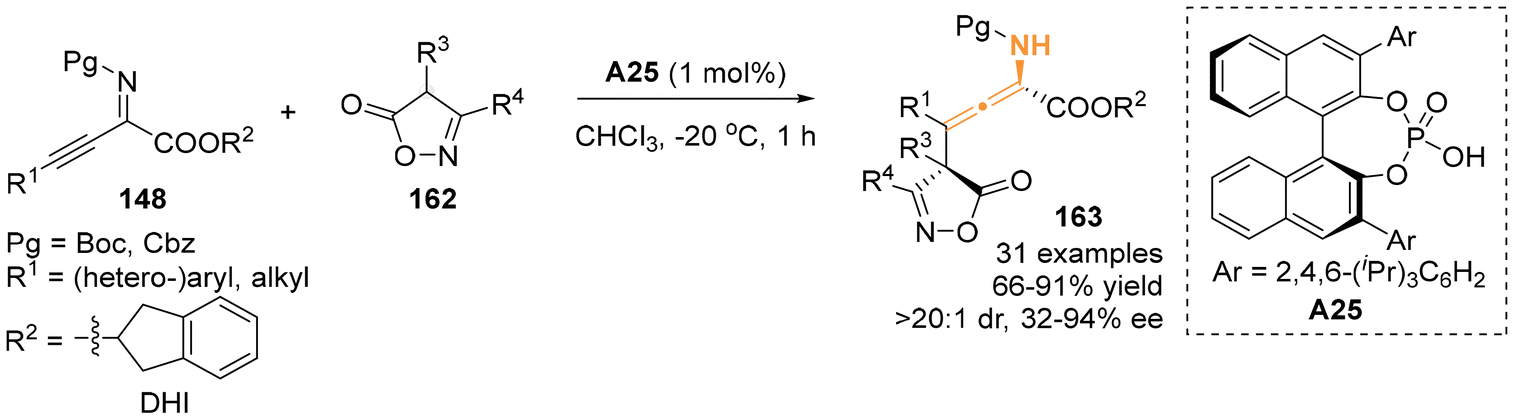
Following the above-mentioned pioneering enantioselective dearomative γ-addition reaction using various arenes as nucleophiles, 4-substituted isoxazol-5(4H)-ones 162 were identified by Li's group to be a compatible nucleophile, smoothly reacting with β,γ-alkynyl-α-imino esters 148 (Scheme 27).34 In the presence of chiral phosphoric acids A25 as a catalyst, the γ-addition reaction exhibited favorable outcomes, resulting in the assembly of chiral N-containing tetrasubstituted allenes bearing an adjacent quaternary carbon stereocenter in high yields.
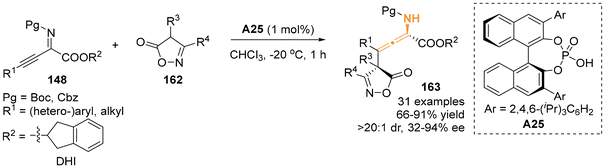 |
| | Scheme 27 Organocatalytic asymmetric γ-addition of isoxazol-5(4H)-ones to β,γ-alkynyl-α-imino esters. | |
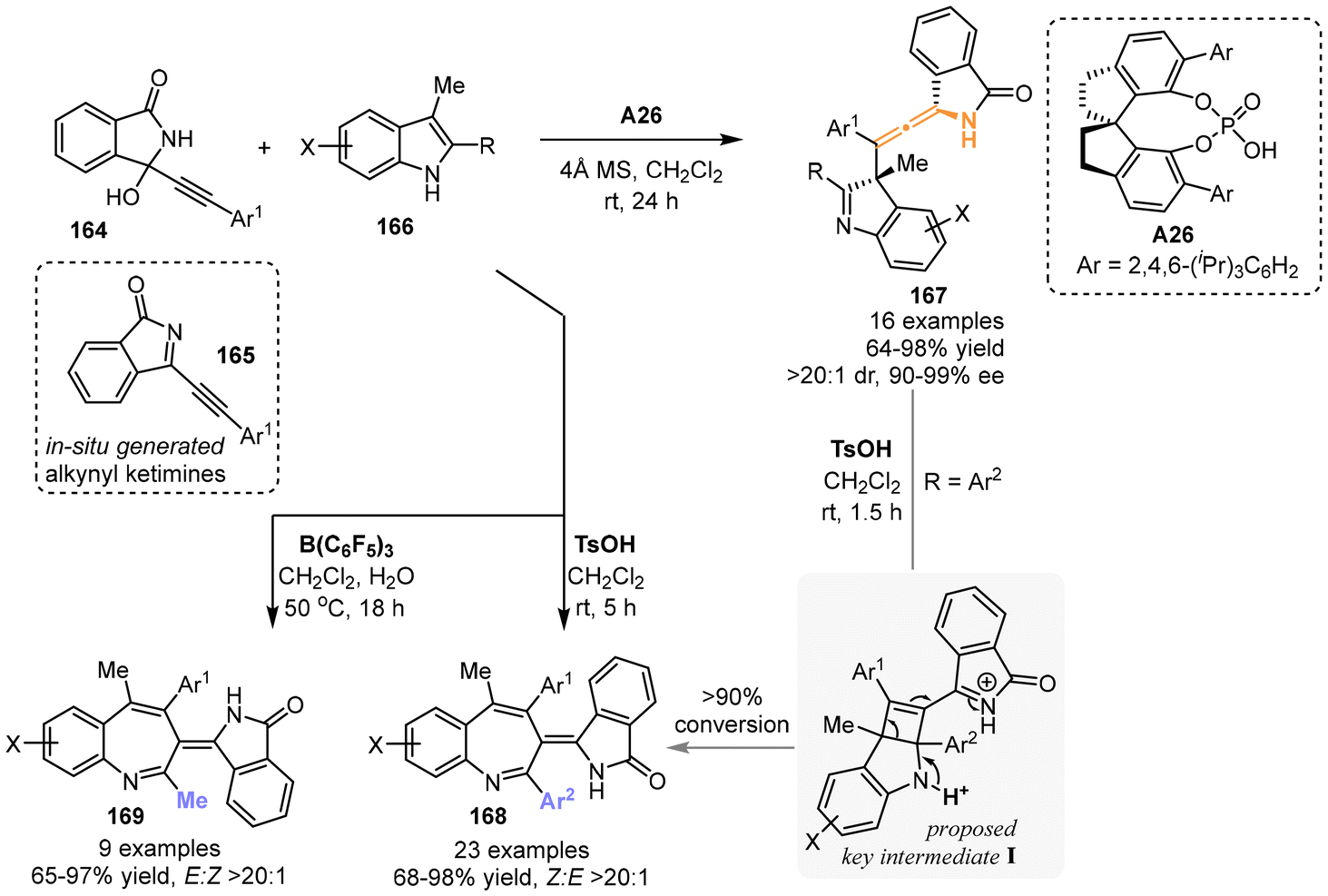
In 2022, the group of Sun and Li utilized in situ-generated alkynyl cyclic-ketimines 165, which were derived from 3-alkynyl-3-hydroxyisoindolinones 164, to react with 2,3-disubstituted indoles 166 (Scheme 28).35 This reaction was facilitated by chiral spiro-phosphoric acid catalyst A26, resulting in the construction of chiral N-containing tetrasubstituted allenes 167 featuring a 5-membered ring. The yields of these allenes ranged from 64% to 98%, accompanied by remarkable diastereoselectivity and enantioselectivity. When stronger acids, such as TsOH and B(C6F5)3, were used, the reaction afforded the 3H-benzo[b]azepines 168 or 169 with excellent geometric selectivity, respectively. The researchers believed that the 3H-benzo[b]azepine products were produced from N-substituted chiral tetrasubstituted allenes 167, probably involving the rearrangement of four-membered intermediate I through C2–C3 bond cleavage in indoles. Moreover, the crossover experiment proved that the dearomative γ-addition step is reversible when catalyzed by TsOH.
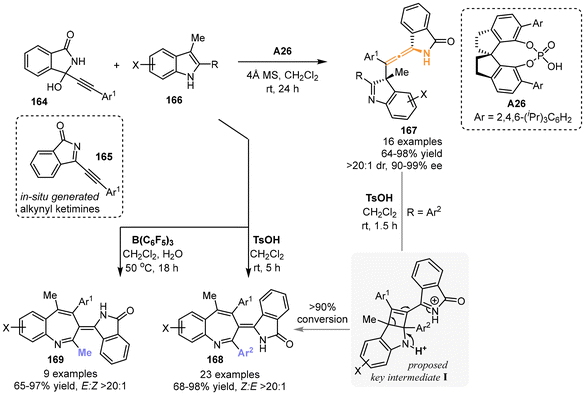 |
| | Scheme 28 Catalyst-controlled divergent reactions of 2,3-disubstituted indoles with propargylic alcohols for the synthesis of 3H-benzo[b]azepines and axially chiral tetrasubstituted allenes. | |
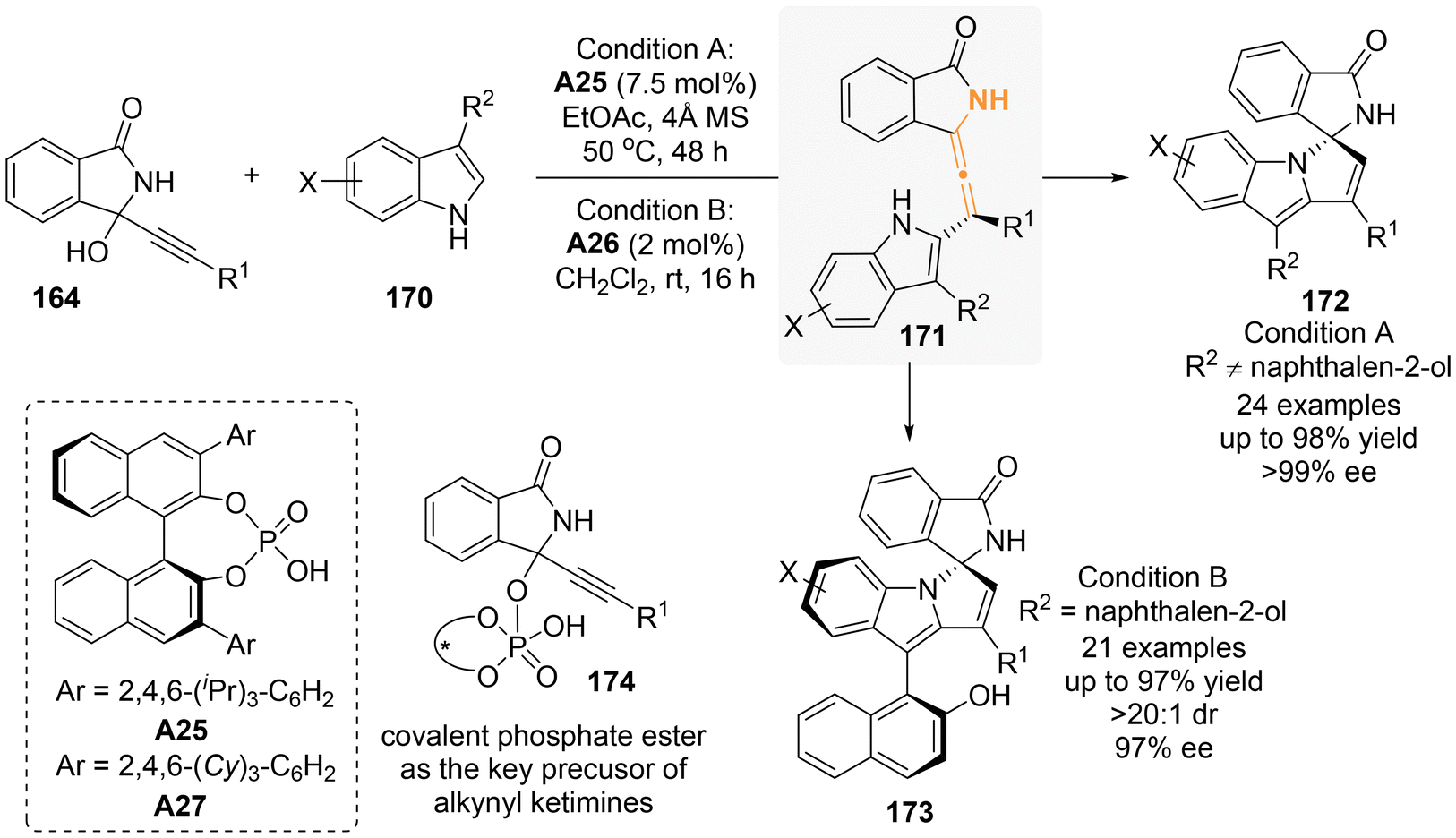
In another study involving 3-substituted indoles that lack substitution at the C2-position, it was demonstrated that these compounds can act as nucleophiles in a chiral phosphoric acid (CPA)-catalyzed [3 + 2]-annulation with 3-alkynyl-3-hydroxyisoindolinones (Scheme 29: condition A).36 The reaction yielded enantioenriched spiro isoindolinone-indolines with excellent yields and stereoselectivities. The authors believed that axially chiral tetrasubstituted allene intermediates 171 were likely involved in the reaction mechanism. Additionally, they observed the existence of 174 through NMR analysis, which served as the key precursor for alkynyl ketimines 165. Following the finding of Sun and Li, Dong and Li's group later incorporated a 2-naphthalenol moiety at the C3-position of indole to perform asymmetric [3 + 2]-annulation, resulting in spiro products 173 characterized by atropisomeric chirality and good diastereoselectivity (condition B).37
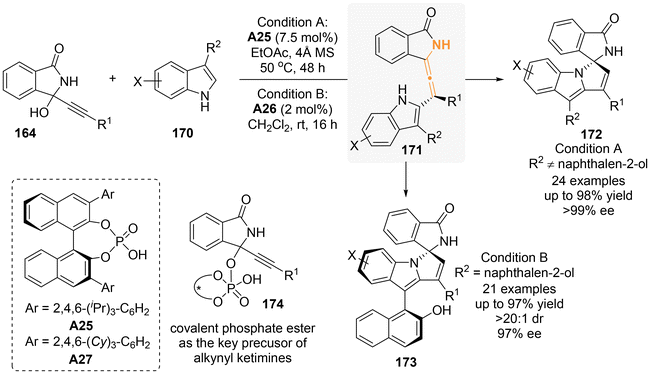 |
| | Scheme 29 Asymmetric organocatalytic [3 + 2] annulation of propargylic alcohols with indolylnaphthalenols: synergistic construction of axial and central chirality. | |
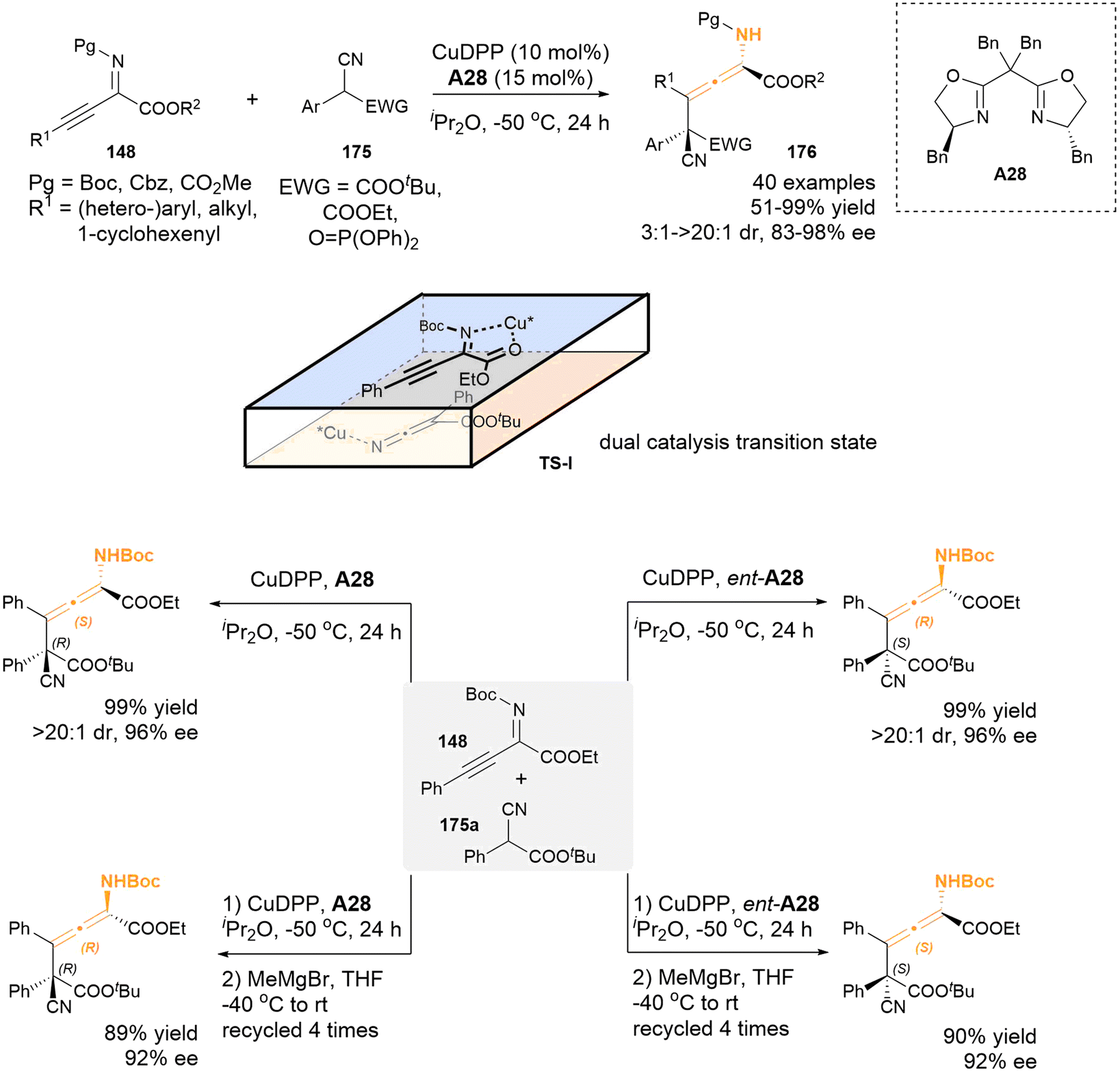
2.3.3 Transitional-metal catalysis for aza-substituted allenes. In 2023, a dual copper-catalysis approach was successfully employed by Zhang's group to establish the asymmetric γ-addition of β,γ-alkynyl-α-imino esters 148 with the readily accessible α-substituted cyanoacetates 175 as nucleophiles (Scheme 30).38 This methodology presents a mild and general strategy to construct chiral tetrasubstituted N-substituted allenes, which feature a vicinal acyclic all-carbon quaternary stereocenter, achieving high yields alongside excellent diastereoselectivities and enantioselectivities (51–99% yield, 3:1–>20:1 dr, 83–99% ee). The proposed transition state TS-I was supported by a series of control experiments and density functional theory (DFT) calculations, in which the chiral copper catalysts played a double role to activate both substrates for the asymmetric γ-addition reactions. Furthermore, an innovative strategy was developed to afford all four stereoisomers of the chiral N-substituted allene products through the iterative addition of Grignard reagent. Specifically, the Grignard reagent MeMgBr acts as a base, facilitating the deprotonation of the carbamate NH group of compound 176 and promoting the epimerization process of the axial chirality motif.
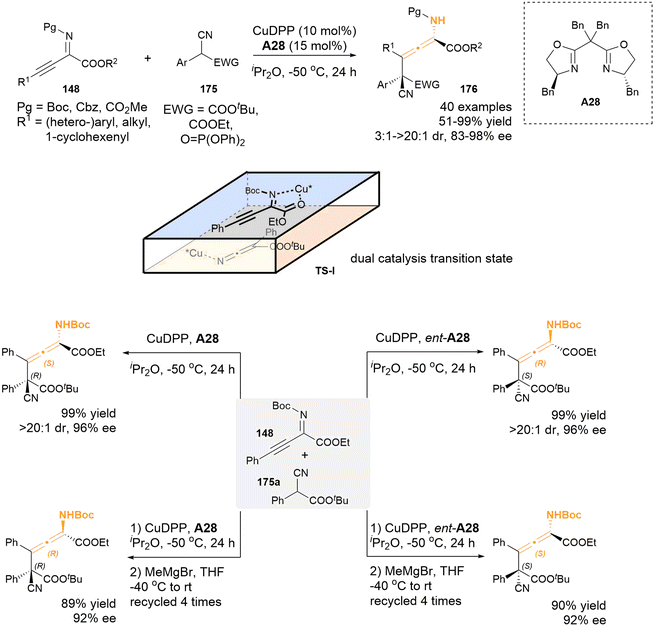 |
| | Scheme 30 Enantio- and diastereoselective synthesis of chiral tetrasubstituted α-amino allenoates bearing a vicinal all-carbon quaternary stereocenter with dual-copper-catalysis. | |
2.4 Asymmetric synthesis of allenylphosphines
Chiral organophosphorus compounds exhibit a broad application across various fields, including catalysis,39a chemical synthesis, materials science, medicinal chemistry, and agricultural science. Among them, axially chiral allenylphosphines represent a charming subclass that has been recognized for their utility as ligands and catalysts in asymmetric synthesis. The traditional synthetic approaches for the preparation of allenylphosphines is predominantly the stereospecific [2,3]-sigmatropic rearrangements of enantioenriched propargyl phosphonates.39b–e Only recently the enantioselective synthesis of axially chiral allenyl phosphorus compounds via catalytic asymmetric methods has been successfully accomplished by Ma and Zhang,40 Guo,41 Zhang42 and Wang.43
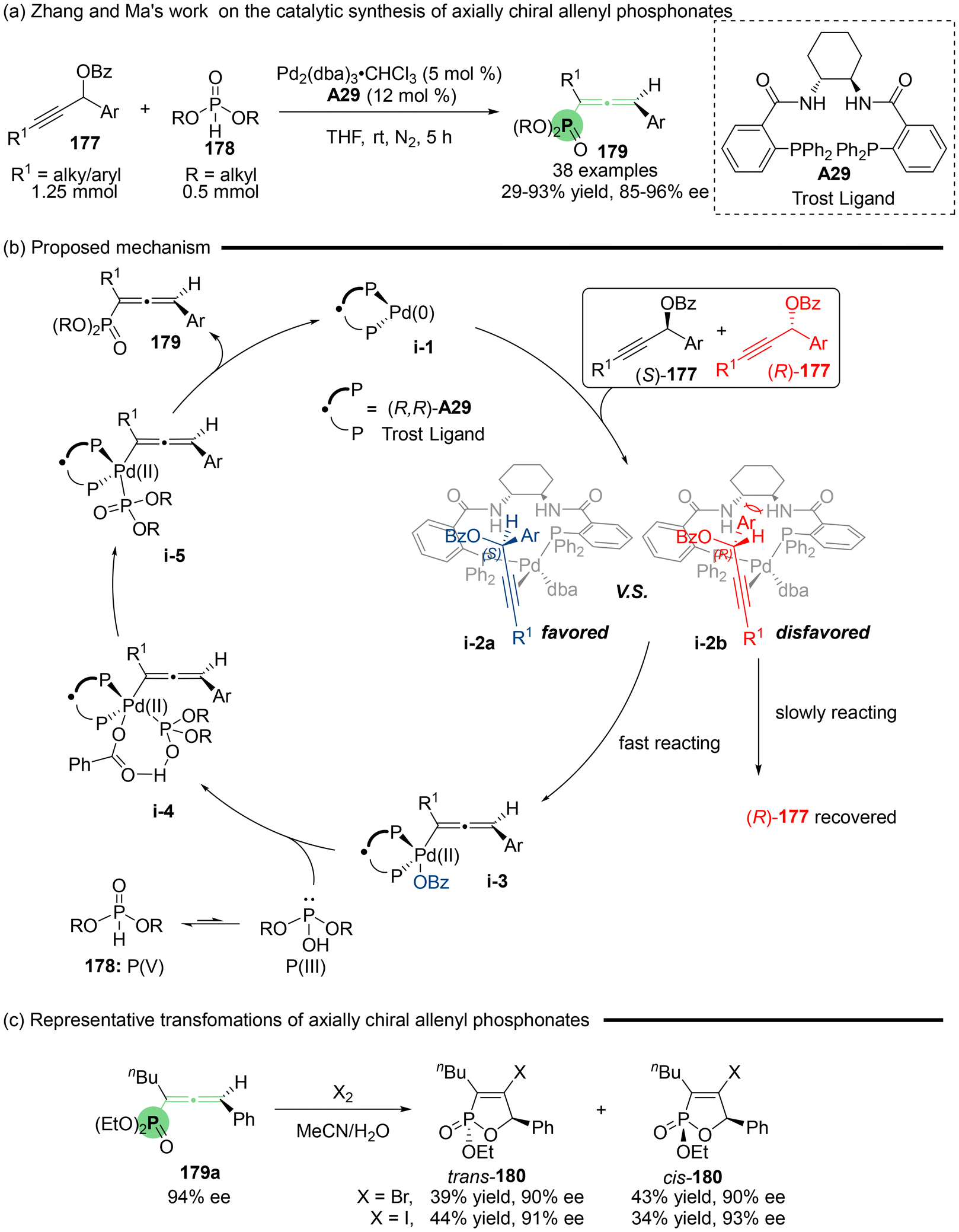
Specifically, the team of Zhang and Ma developed highly enantioselective palladium-catalyzed allenylation utilizing the readily accessible propargylic benzoates 177 and dialkyl phosphites (H-phosphonates: 178) as phosphorus-centered nucleophiles, which involved allenylic C–P bond formation (Scheme 31). This protocol enjoys high efficiency and regioselectivity in the synthesis of axially chiral allenyl phosphonates 179, effectively inhibiting the undesired propargylic substitution reactions. The rational reaction mechanism was proposed (Scheme 31b), which begins with the coordination of Pd2(dba)3·CHCl3 with the Trost ligand A29 to give active chiral palladium(0) species i-1. Subsequently, this species adds to 177 in an anti-oxidative manner. This step entails a kinetic resolution process. Due to the unfavorable steric hindrance present in transition state i-2b involving (R)-177 and the catalyst, the reaction is preferentially directed toward oxidative addition via i-2a involving (S)-177, ultimately resulting in the formation of (Ra)-allenes.40 Additionally, when allenyl phosphonate 179a was subjected to Br2 or I2, halocylization furnished the chiral Br- or I-containing oxaphosphole 180, respectively.
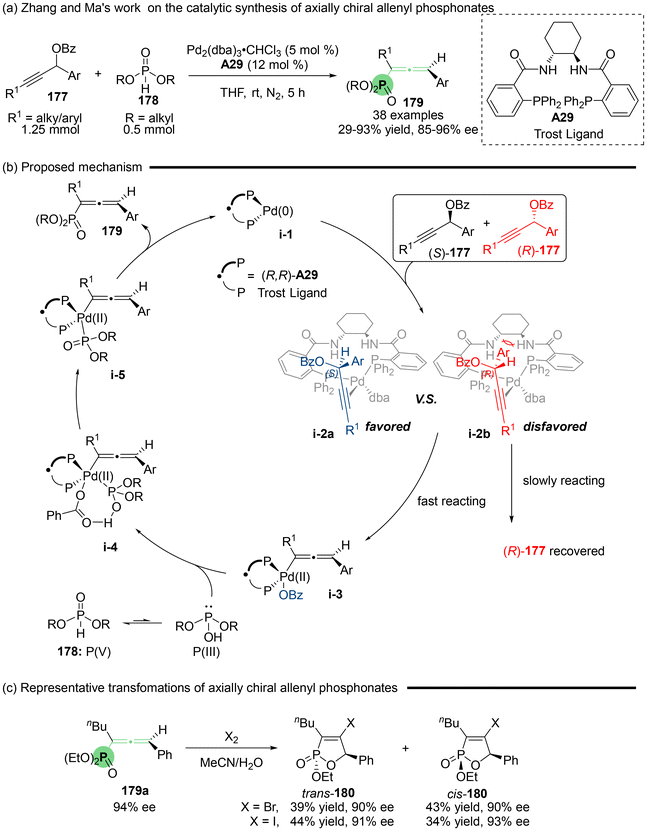 |
| | Scheme 31 Palladium-catalyzed enantioselective synthesis of axially chiral allenyl phosphonates. | |
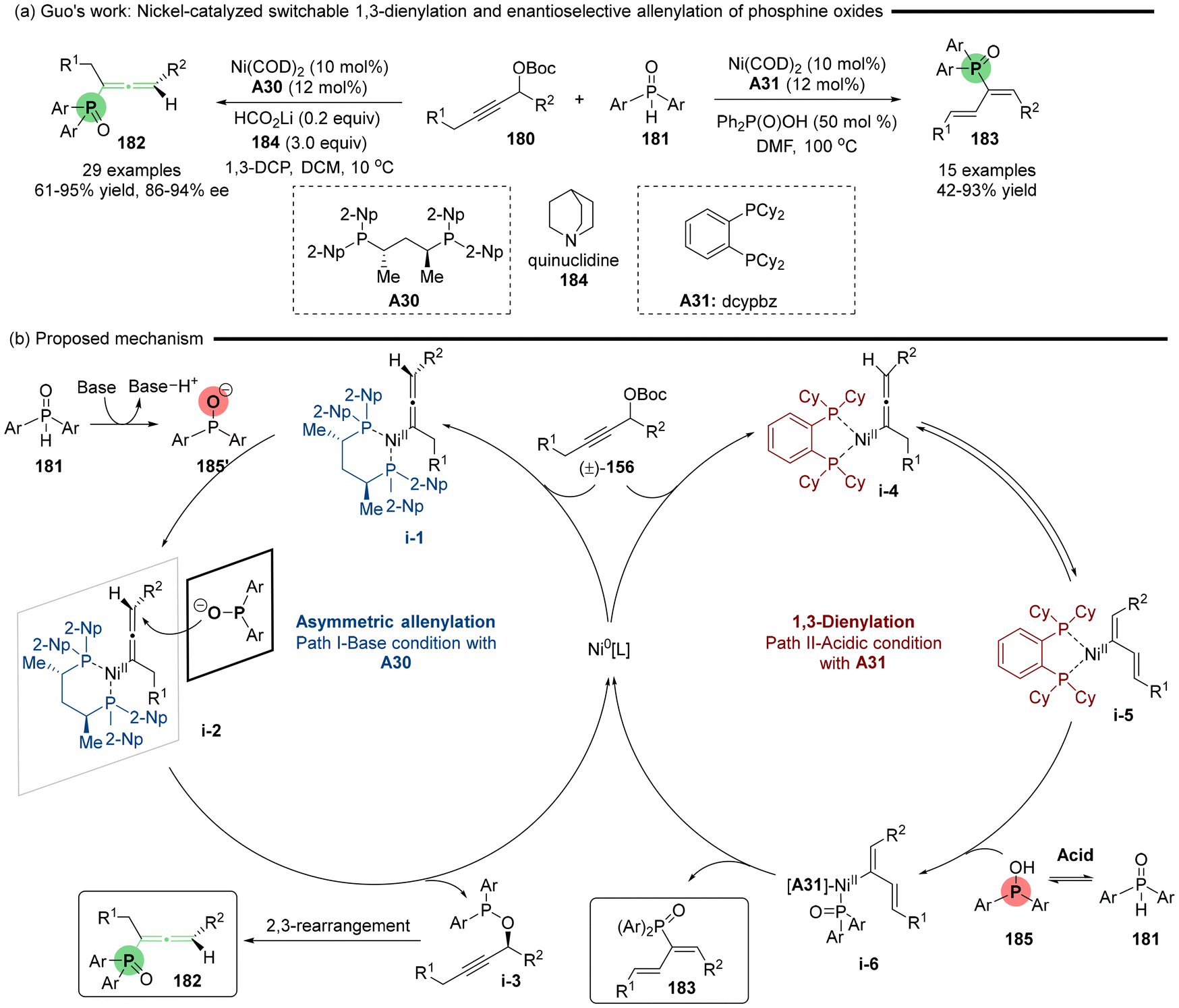
Subsequent, Guo reported a Ni-catalyzed asymmetric allenylation reaction that yields axially chiral allenylphosphine oxides.41 In this reaction, phosphine oxides 181 serve as P-nucleophiles, engaging with propargylic carbonates 180 in the presence of a newly developed BDPP-type chiral ligand A30 under basic conditions (Scheme 32). By modifying the conditions to utilize an achiral phosphine ligand (A31: dcypbz) along with diphenylphosphinic acid 184 as an additive, the same set of coupling substrates underwent 1,3-dienylation. This adjustment offers a complementary approach for synthesizing functionalized phosphinoyl 1,3-butadienes. The mechanistic studies indicate that the structures of the ligands and additives significantly influence the selectivity between the allenylation and 1,3-dienylation pathways (Scheme 32b). Basic additives in cooperation with ligand A30 facilitate the deprotonation of pentavalent R2P(O)H, resulting in the generation of trivalent phosphinite anion 185′, in which the oxygen anion exhibits greater nucleophilicity than the phosphine atom. Consequently, enantioselective O-addition leads to the formation of chiral propargyl phosphinate i-3, which subsequently undergoes a [2,3]-sigmatropic rearrangement to deliver allenylphosphine oxide 182. Conversely, the acidic additives and ligand A31 induce tautomeric equilibrium between the pentavalent R2P(O)H 181 and trivalent R2POH 185, wherein the phosphine atom (P) more readily coordinates with the Ni-centers in i-5 than the oxygen atom, delivering transition state i-6. Subsequently, reductive elimination of i-6 directly produces the 1,3-diene products 183.
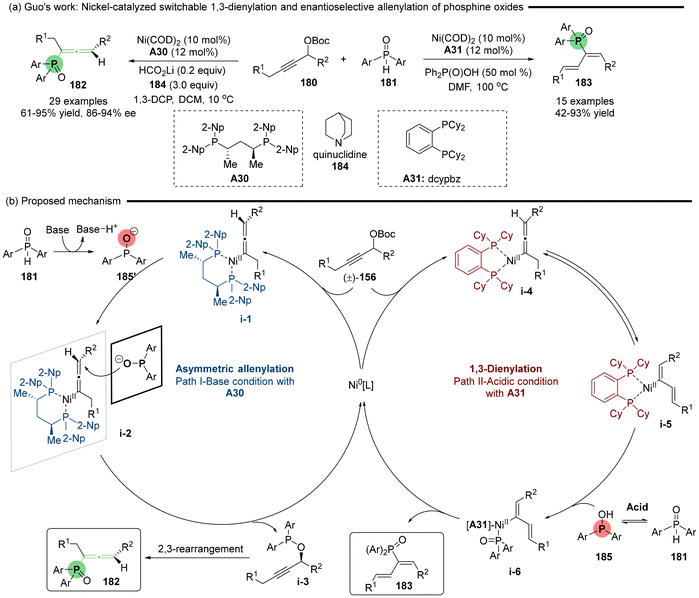 |
| | Scheme 32 Nickel-catalyzed switchable 1,3-dienylation and enantioselective allenylation of phosphine oxides. | |
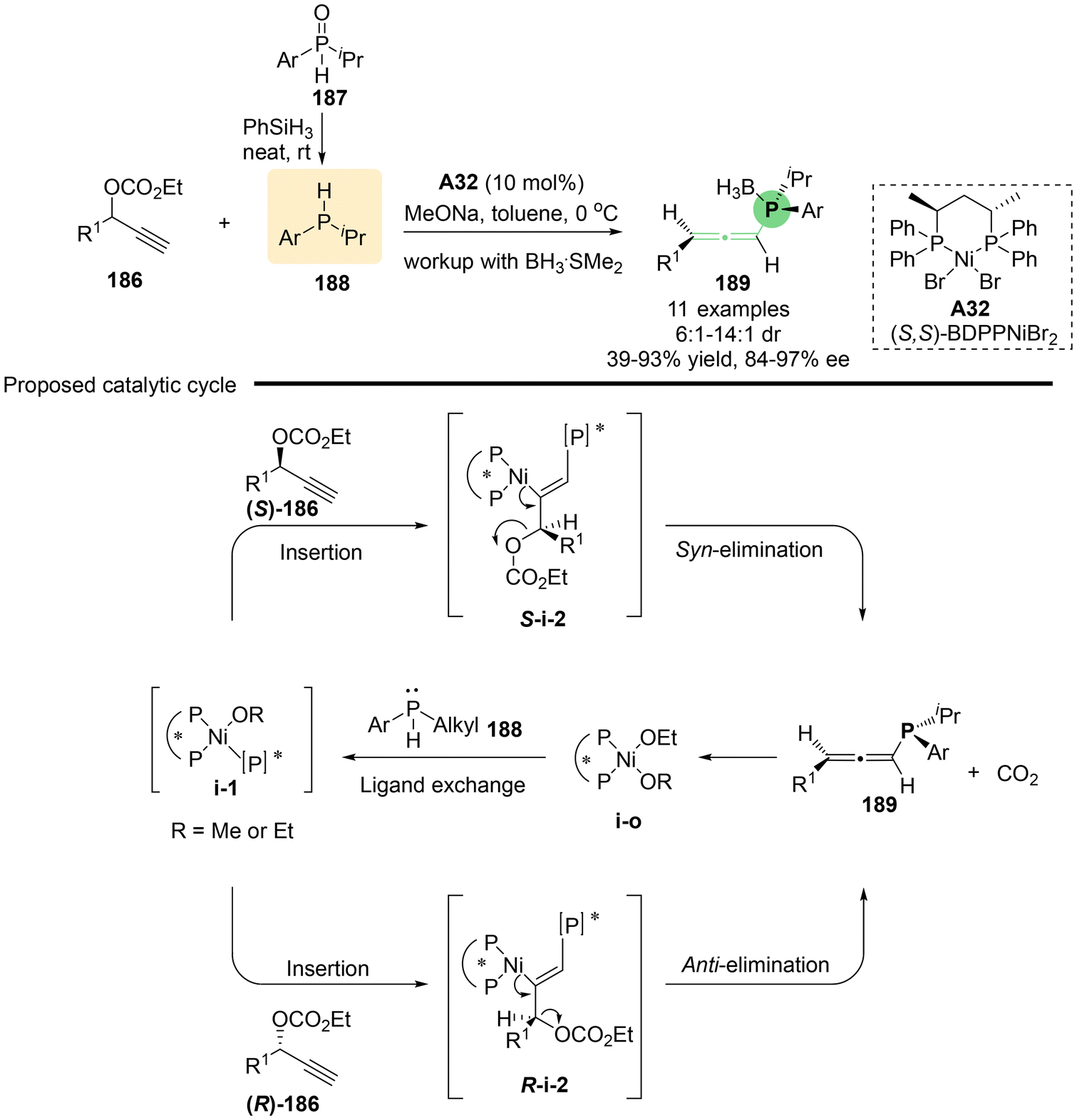
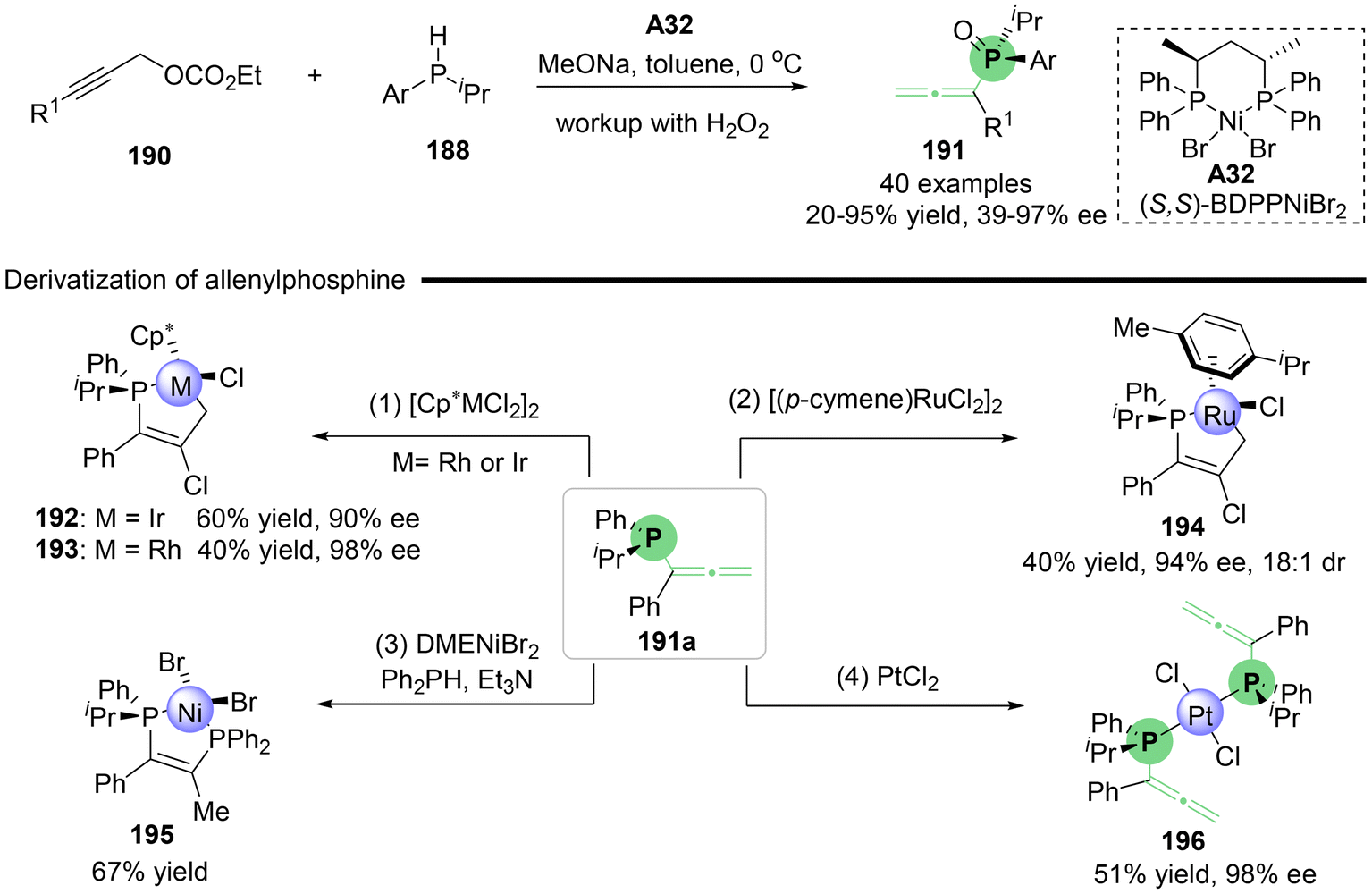
Utilizing the rapid racemization of secondary phosphines in transition metals, Zhang's group reported the nickel-catalyzed asymmetric propargylic substitution reaction of branched propargylic carbonates 186 with in situ generated secondary phosphines as the nucleophile (Scheme 33).42 This method provides an efficient approach for the synthesis of P, axial-stereogenic allenylphosphine products 189 that feature both P-central and axial chirality. Through a series of comprehensive control experiments, the authors proposed a possible reaction mechanism (Scheme 33b). Initially, chiral complex catalyst A32, (S,S)-BDPPNiBr2, and secondary phosphine 188 undergo ligand exchange to give nickel phosphido complex i-1, which then inserts into compound 186 to yield alkenyl-nickel complex i-2. Both S-i-2 and R-i-2 lead to the common (Ra)-product, with S-i-2 proceeding via syn-elimination and R-i-2 via anti-elimination, both processes accompanied by the release of CO2 and the regeneration of the nickel ethoxide catalyst. This transformation is also applicable to linear propargylic carbonates 190, which produce P-stereogenic allenylphosphines 191 without axial chirality (Scheme 34). Additionally, several interesting derivatizations of P-stereogenic allenylphosphines 191a have been demonstrated, enabling access to a series of versatile five-membered transition metal phosphine complexes 192–195, including metals such as Ir, Rh, Ru and Ni. When allenylphosphine 191a was subjected to PtCl2, complex compound 196 containing two allene units was obtained.
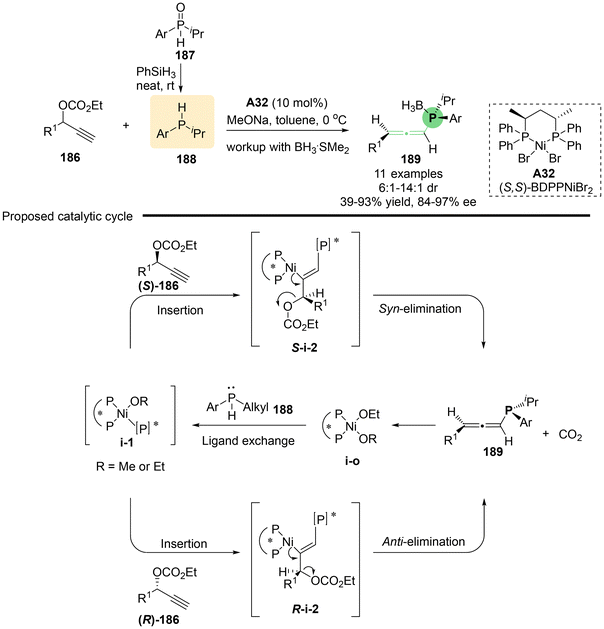 |
| | Scheme 33 Enantioselective synthesis of P, axial-stereogenic allenylphosphines through Ni-catalyzed propargylic substitution. | |
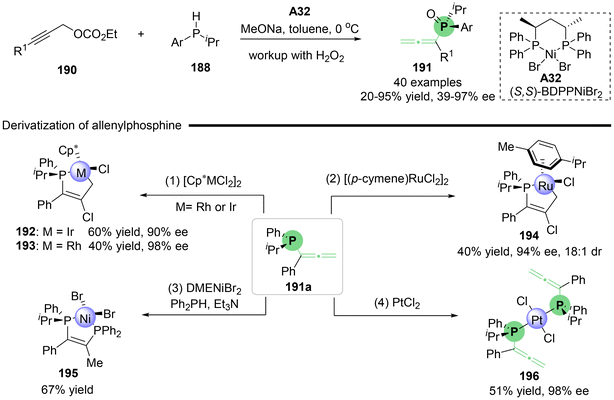 |
| | Scheme 34 Enantioselective synthesis of P-stereogenic allenylphosphines through Ni-catalyzed propargylic substitution. | |
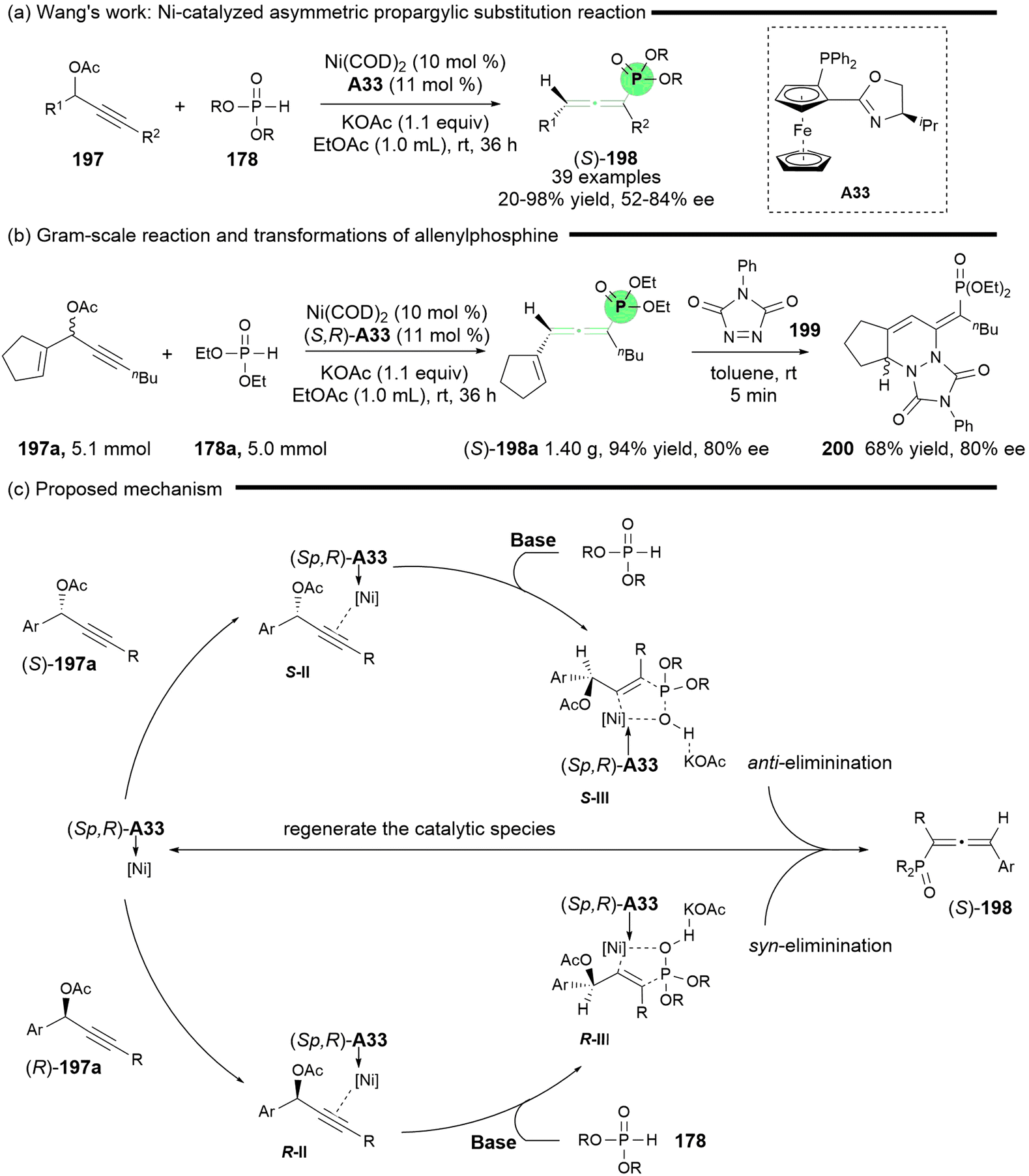
In light of the abundant availability of various phosphorus nucleophiles, the Wang group conducted an extensive investigation into allenylphosphines, as reported in 2024 (Scheme 35).43 Their study detailed the reaction between propargylic carbonate and a range of nucleophilic phosphorus compounds, including phosphine oxides, phosphinates, and phosphonates, leading to the formation of racemic multi-substituted phosphorus-containing allenes, particularly achieving tetrasubstituted phosphines that were not previously reported. In this study, the asymmetric synthesis of axially chiral allenylphosphines was achieved through the reaction of phosphonates 178 and propargyl acetates 197 in the presence of ligand A33 and Ni(COD)2 (Scheme 35a). To demonstrate the practicality of this reaction, a gram-scale reaction for the synthesis of chiral (S)-198a (80% ee) was successfully performed, followed by a Diels–Alder reaction with triazolinone 199, yielding cycloadducts 200 with an enantiomeric excess of 80% (Scheme 35b). The authors proposed a plausible mechanism, which is similar to that of Zhang's methodology (Scheme 35c).42
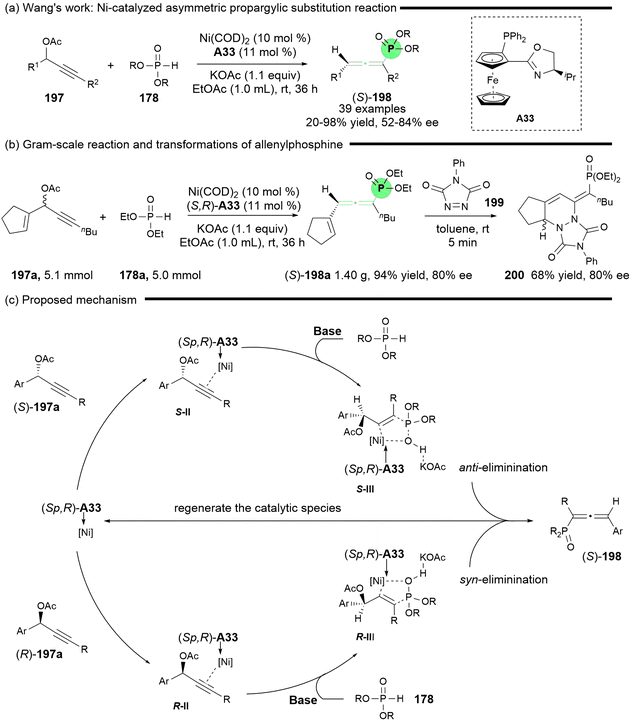 |
| | Scheme 35 Ni-catalyzed propargylic substitution reaction: a general and versatile tool to assemble axially chiral phosphorus-containing allenes. | |
2.5 Asymmetric synthesis of oxygen-substituted allenes
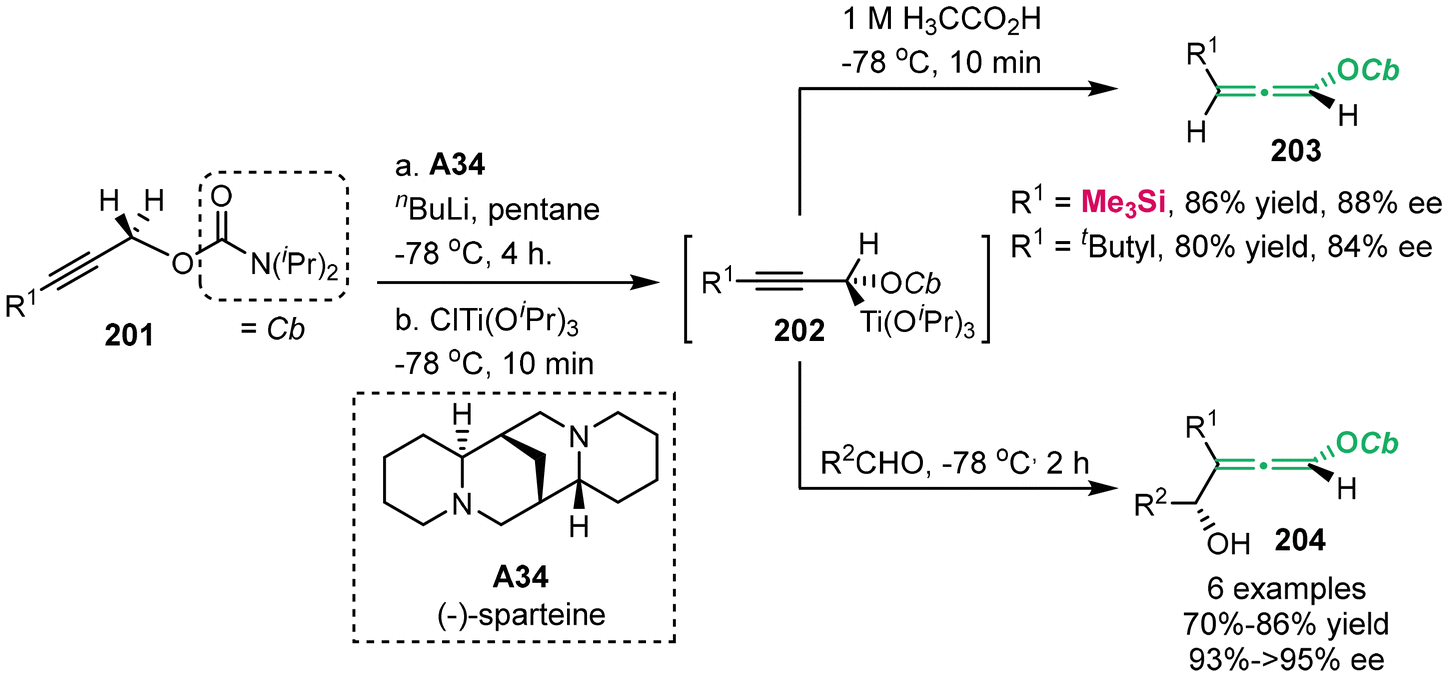
In 2001, Hoppe's group successfully synthesized enantioenriched 4-hydroxyallenyl carbamates 203 and 204 (Scheme 36).44–46 This process involved the deprotonation of alkynyl carbamates 201 using chiral base (−)-sparteine A34 and n-butyllithium. This step was followed by transmetallization with ClTi(OiPr)3. Ultimately, this reaction performed a substitution process with aldehydes, producing allene compounds 204. Meanwhile, treatment with acetic acid furnished the formation of allenes 203.
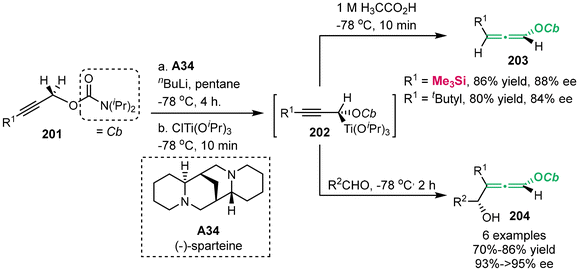 |
| | Scheme 36 Synthesis of enantiomerically enriched allenes by (−)-sparteine-mediated lithiation of alkynyl carbamates. | |
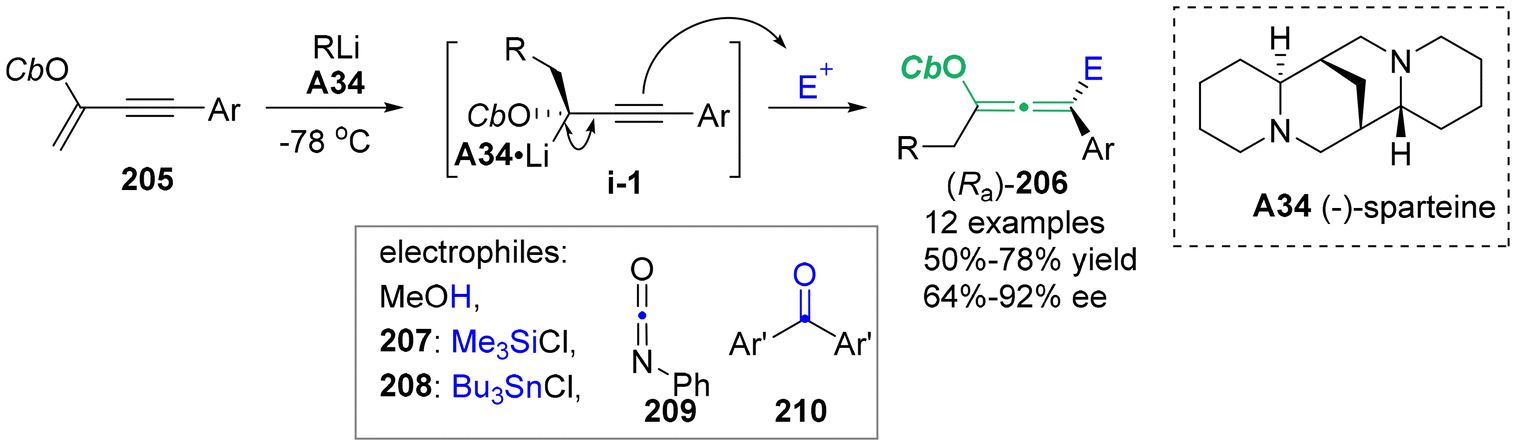
In 2011, Yoshida's group employed a flow microreactor system to produce a series of different enantioenriched chiral allenes 206 (Scheme 37).47 Conjugated enyne 205 with a directing group of carbamoyloxy group generated chiral organolithium intermediates i-1 in the presence of nBuLi/chiral ligand complex A34. This process was characterized by precise control of the residence time, allowing the effective trapping of intermediates i-1 with several compatible electrophiles, including MeOH, chlorotrimethylsilane 207, tributylchlorostannane 208, phenyl isocyanate 209, and benzophenone 210. The final stereospecific anti-SE2′ substitution procedure yielded oxygen-containing chiral allenes 206 with enantiomeric excesses of up to 92%.
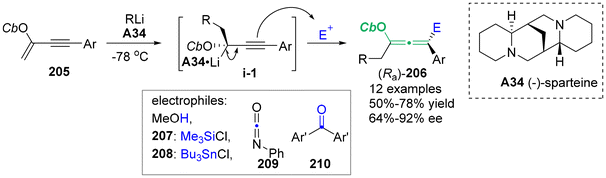 |
| | Scheme 37 Asymmetric carbolithiation of conjugated enynes: a flow microreactor enables the use of configurationally unstable intermediates before they epimerize. | |
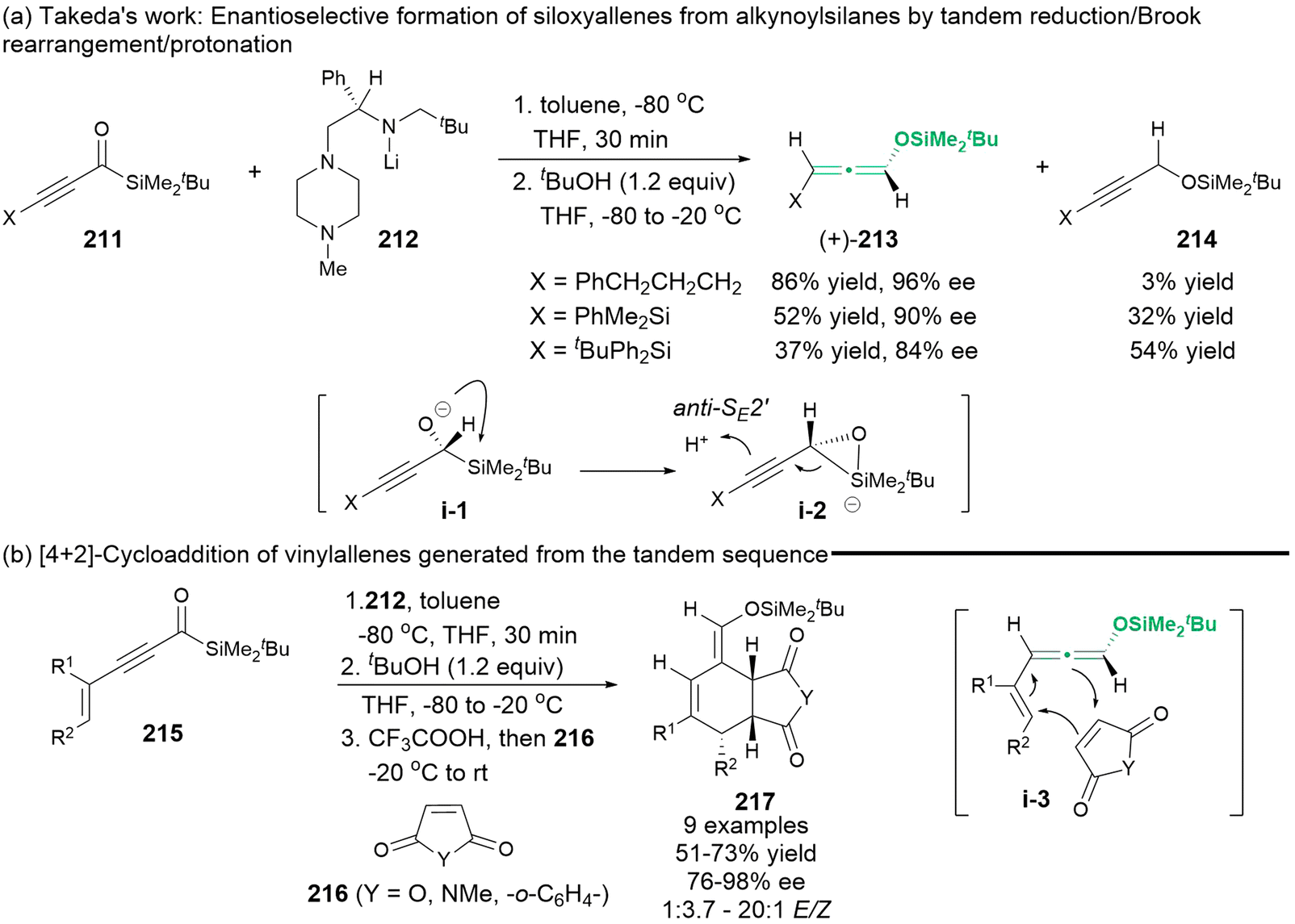
In 2011, Takeda's group employed chiral lithium amide 212 to reduce alkynoylsilanes 211, yielding chiral α-silyl alkoxide intermediate i-1. Subsequently, this intermediate underwent a Brook rearrangement, which was mediated by anti-SE2′ electrophilic substitution in the presence of an additive alcohol, resulting in the formation of enantioenriched 1,3-substituted siloxyallenes (+)-213 (Scheme 38).48 The large bulky substituent X led to the direct formation of Brook rearrangement product 214. The in situ-generated chiral siloxyallenes were capable of undergoing a tandem [4 + 2]-type cycloaddition with different dienophiles 216, finally affording cyclized products 217 with a high level of stereospecificity.
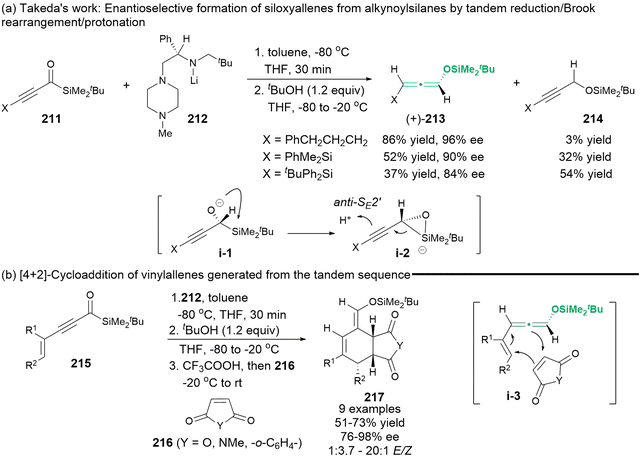 |
| | Scheme 38 Enantioselective synthesis of siloxyallenes from alkynoylsilanes by reduction and a Brook rearrangement and their subsequent trapping in a [4 + 2] cycloaddition. | |
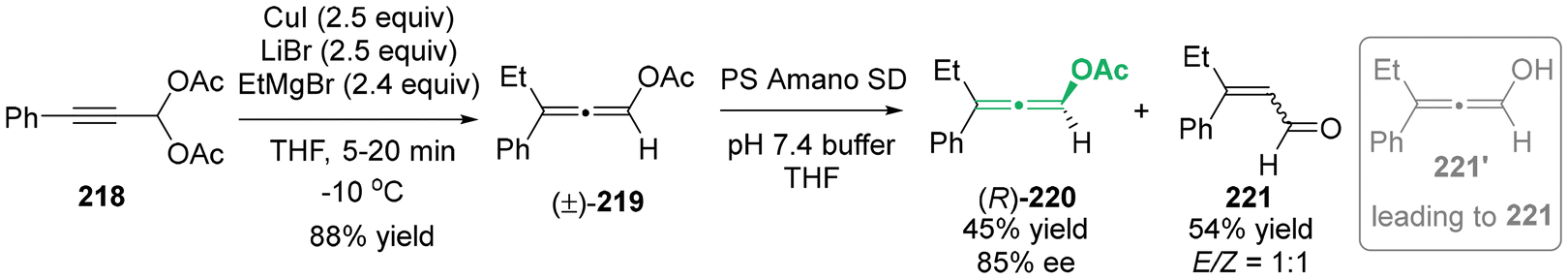
In 2007, Woodward developed an elegant SN2′ addition of propargylic diacetates 218 for the synthesis of racemic allenyl acetates 219 (Scheme 39).49 Furthermore, the lipase-catalyzed kinetic resolution of readily available racemic allenyl acetates 219 was reported, resulting in the production of enantioenriched allenyl acetates 220 in a yield of 45% and enantiomeric excess of 85%.
 |
| | Scheme 39 Enantioselective synthesis of siloxyallenes from alkynoylsilanes by reduction and a brook rearrangement and their subsequent trapping in a [4 + 2] cycloaddition. | |
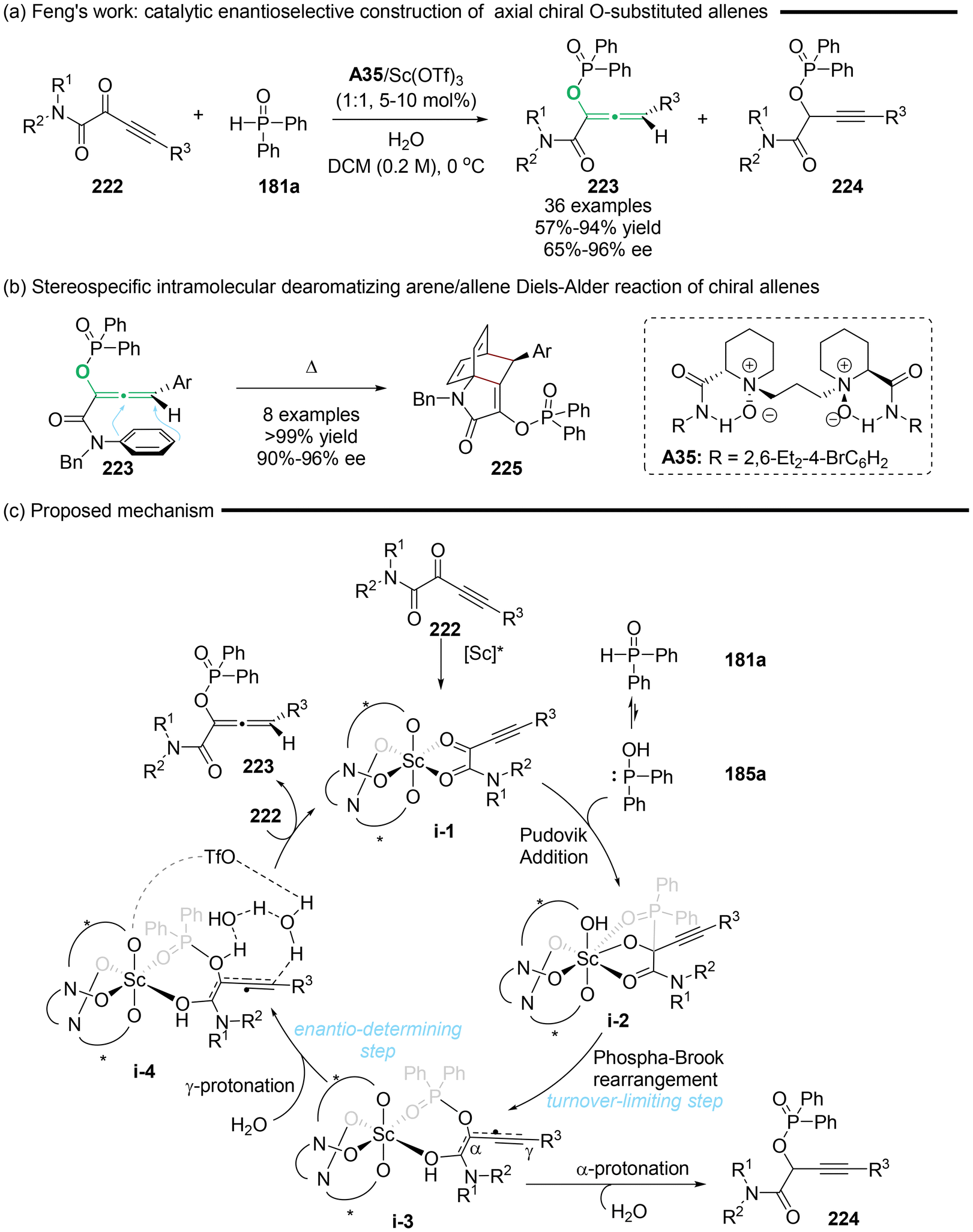
In 2022, Feng's group pioneered the catalytic enantioselective synthesis of axial chiral O-substituted allenes 223 (Scheme 40).50 This method involved asymmetric Pudovik addition, followed by [1,2]-phospha-Brook rearrangement of α-alkynylketoamides 222 and diarylphosphine oxides 181a utilizing an N,N′-dioxide/Sc(III) complex as the catalyst. According to the mechanistic studies including control experiments and DFT calculations, the authors concluded that the reaction rate is determined by the [1,2]-phospha-Brook rearrangement, while the enantioselectivity is primarily influenced by the final protonation step. Furthermore, they identified that the counterion of the Lewis acid and the presence of water as an additive exert a synergistic and significant impact on both the regioselectivity and enantioselectivity in the synthesis of enantioenriched allenes. With these functional chiral allenes linked with a phenylamine moiety in hand, they displayed a notable stereospecific cyclization, Himbert's intramolecular dearomatizing arene/allene Diels–Alder reaction (Scheme 40b). Ultimately, the cyclization gives rise to complex bridged polycyclic compounds in a stereospecific manner.
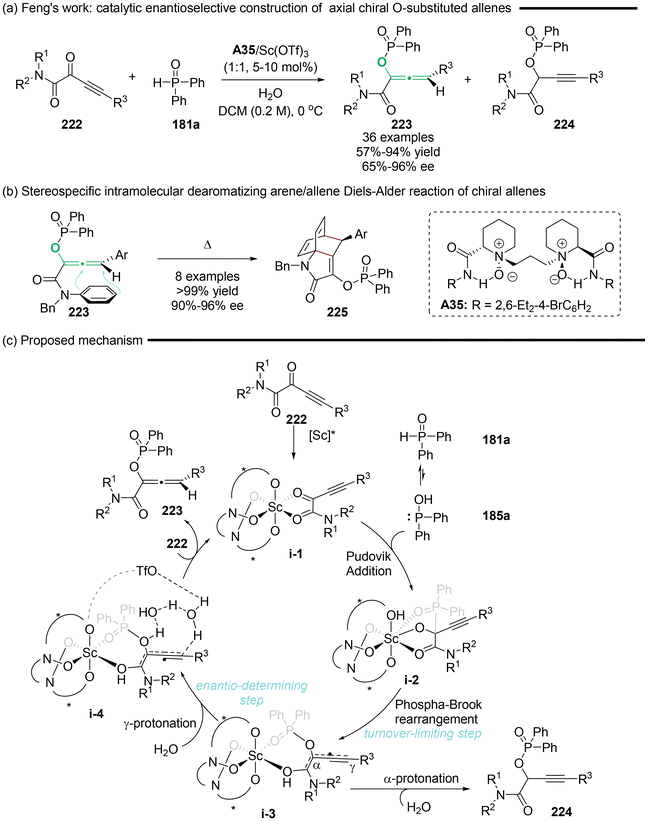 |
| | Scheme 40 Catalytic regio- and enantioselective protonation for the synthesis of chiral allenes: synergistic effect of the counterion and water. | |
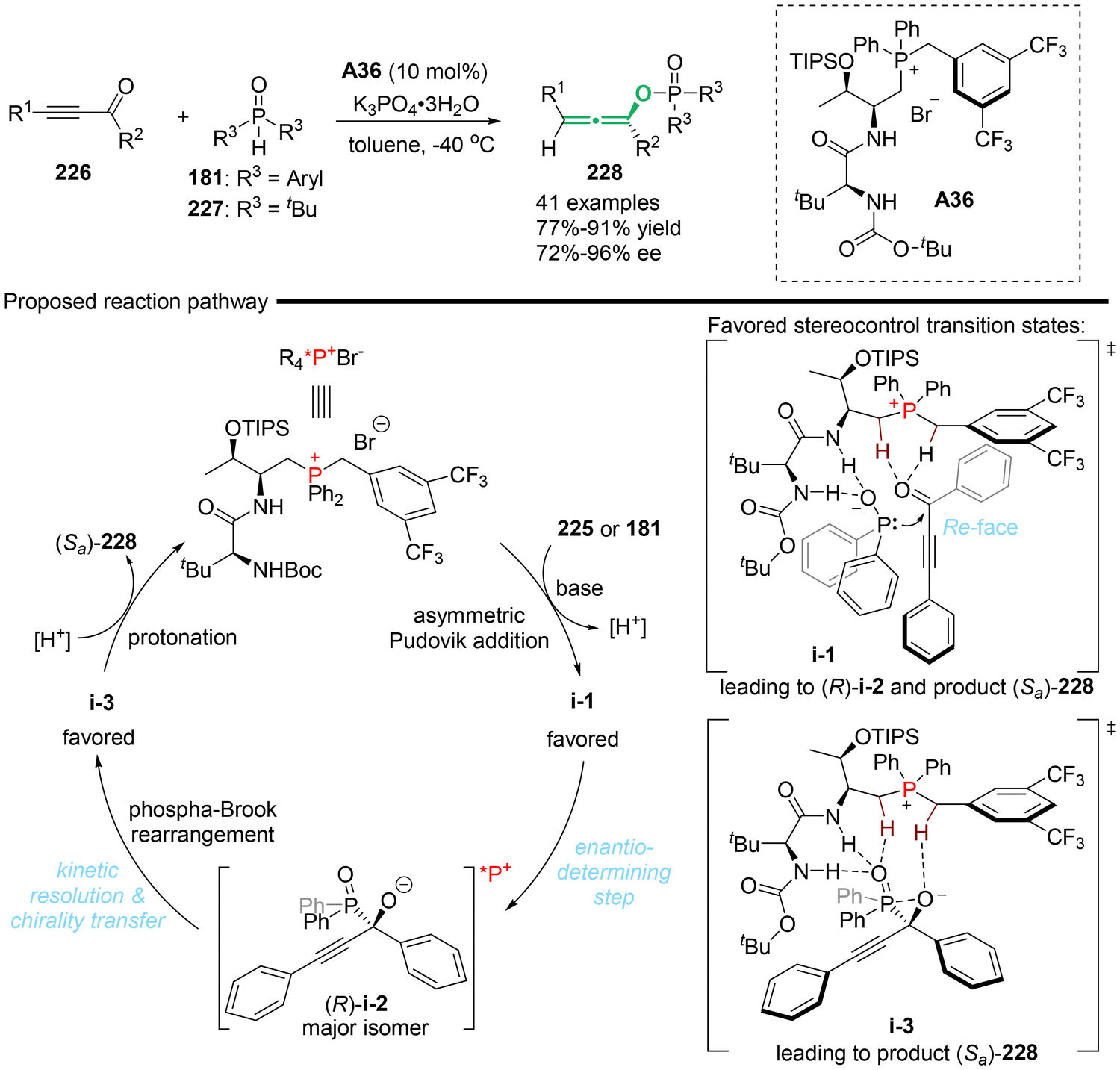
Recently, Wang's group utilized the peptide-mimic phosphonium salt (PPS) as a privilege organocatalyst to synthesize axially chiral allenyl phosphorus compounds 228, achieving high yields and excellent stereoselectivities (Scheme 41).51 A series of mechanistic investigations was conducted to support the proposed mechanism, which involved an enantio-determining step involving an asymmetric Pudovik addition reaction between ethynyl ketones 226 and phosphine oxides in PPS catalysis. This was followed by a tandem phospha-Brook rearrangement via central-to-axial chirality transfer. According to the experimental data, the authors concluded that the rearrangement process is a kinetic resolution process enabled by the PPS catalyst, wherein (R)-i-2 was identified as the favorable and matched intermediate species.
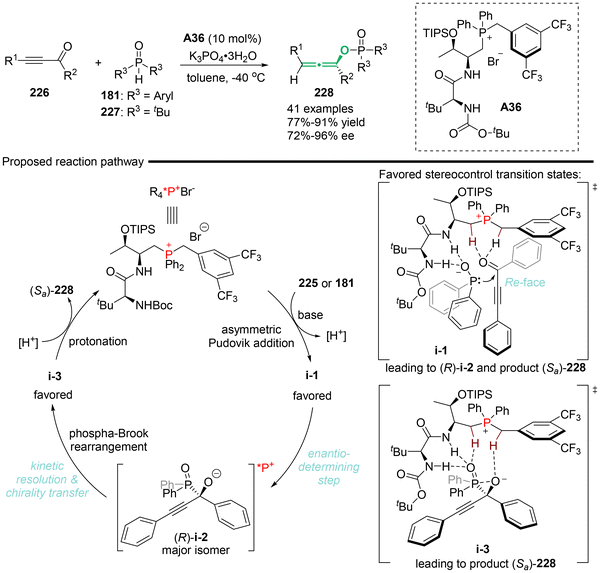 |
| | Scheme 41 Peptide-mimic phosphonium salt (PPS)-catalyzed Pudovik addition/phospha-Brook rearrangement towards chiral trisubstituted allenes. | |
2.6 Asymmetric synthesis of sulfur-substituted allenes
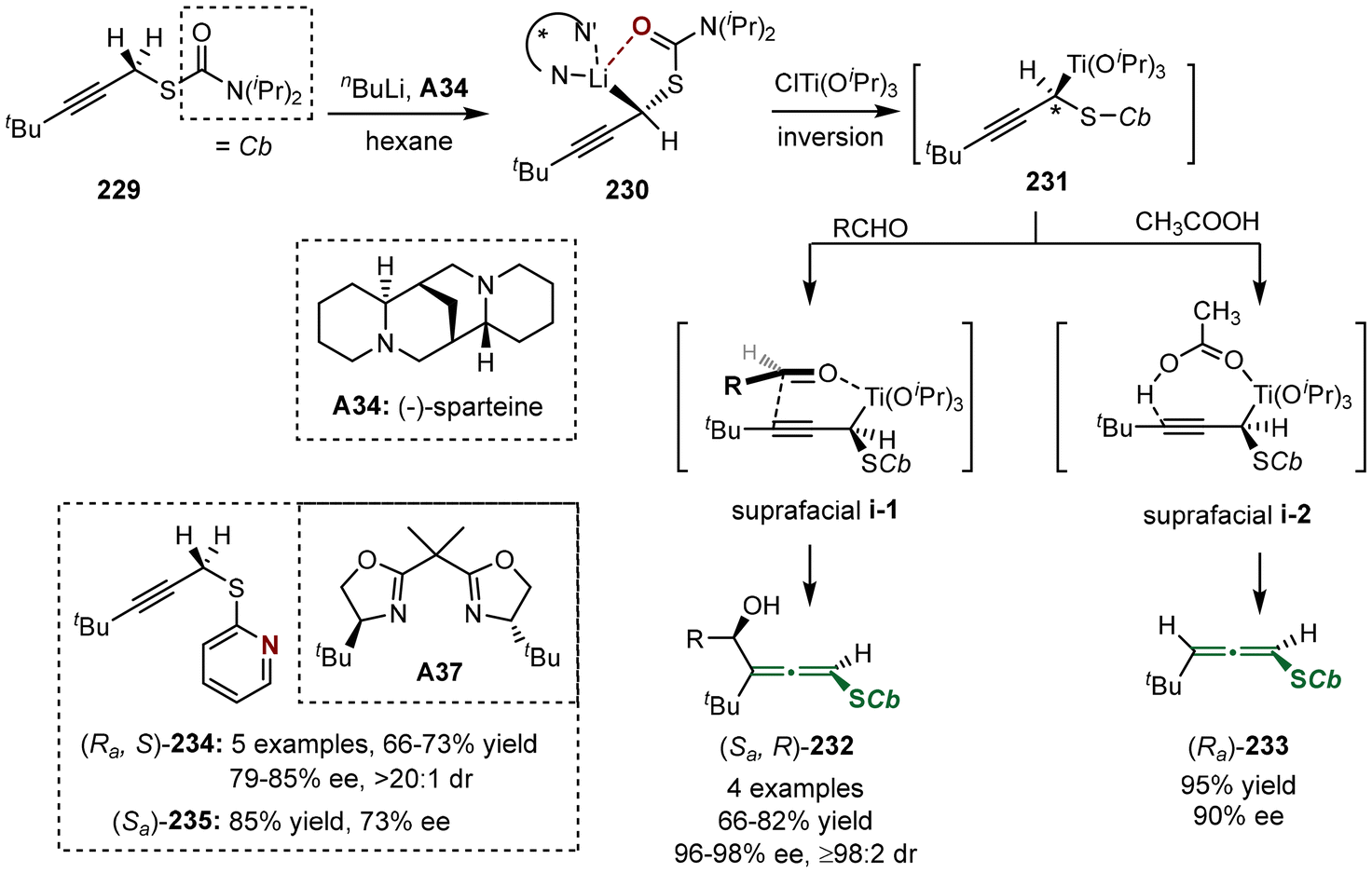
In 2005, the group of Hoppe reported the asymmetric synthesis of axially chiral sulfur-containing allenes 232 and 233, utilizing prochiral propargyl sulfides 229 as substrates through an asymmetric lithiation process (Scheme 42).52 The reaction was initiated with the generation of propargyllithium complex 230, which was subsequently subjected to transmetallation with ClTi(OiPr)3, followed by suprafacial addition with aldehydes or acetic acid. When aldehydes were employed in the reaction, the method yielded compound 232 via preferred transition state i-1. Alternatively, when acetic acid was used instead of aldehydes, the reaction also proceeds through suprafacial addition via i-2, resulting in the formation of sulfur-containing disubstituted allene 233. Additionally, the authors later identified that bis(oxazoline) ligand A37 is also effective in facilitating the synthesis of these two types of axially chiral sulfur-containing allenes (Ra,S)-234 and (Sa)-235 with moderate enantioselectivities.
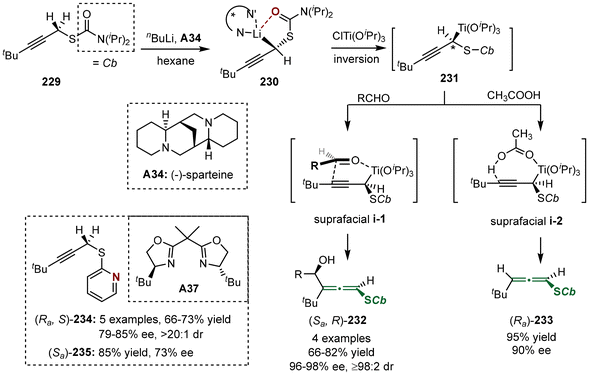 |
| | Scheme 42 Asymmetric lithiation of 2-alkynyl aryl sulfides-enantio-and diastereoselective formation of allenyl aryl sulfides. | |
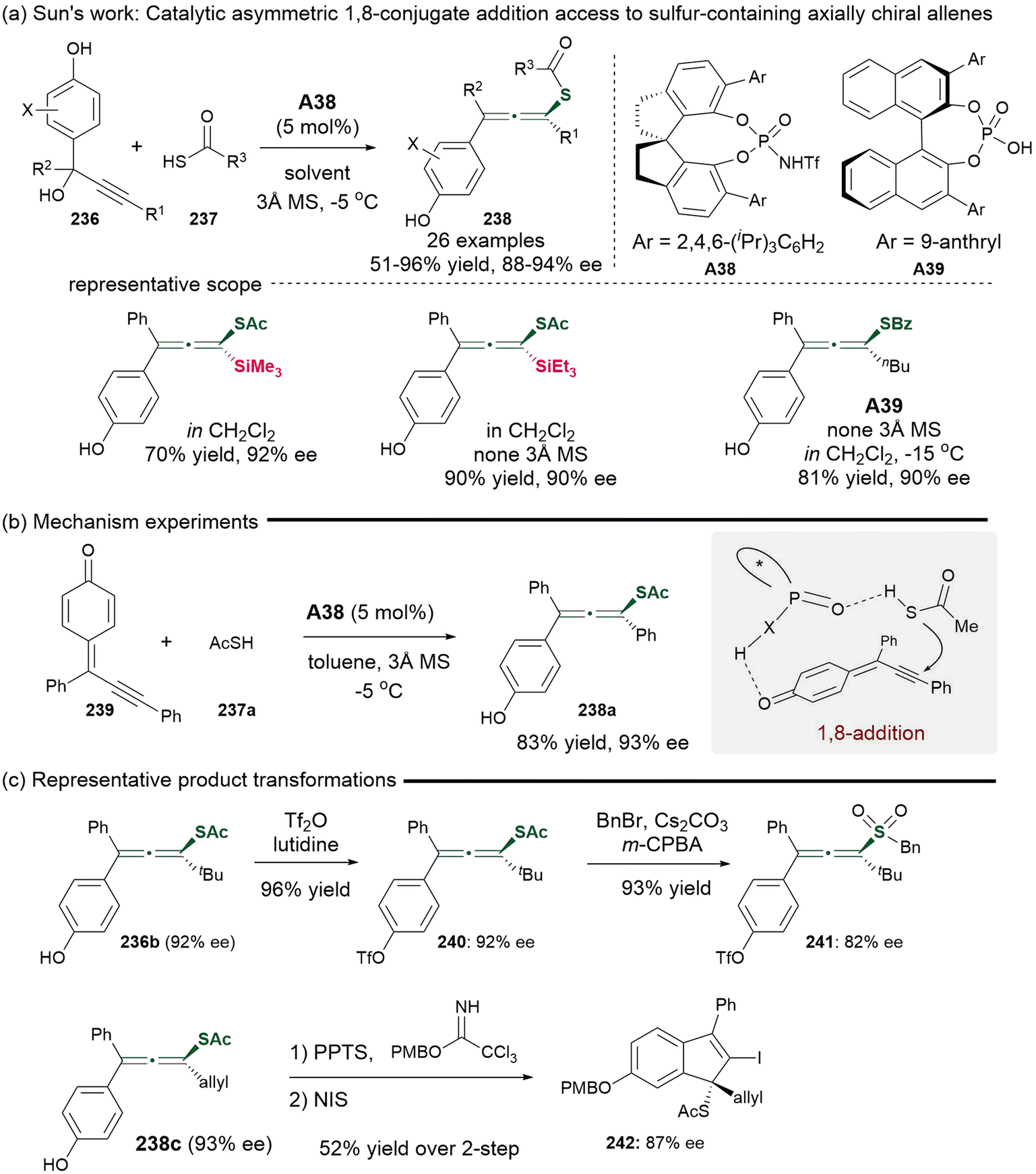
In 2017, Sun's group demonstrated a groundbreaking approach for the asymmetric synthesis of sulfur-containing chiral allenes 238 utilizing catalytic methods (Scheme 43).53 This process involved a catalytic asymmetric 1,8-addition enabled by the catalyst of chiral N-triflylphosphoramides A38 or A39, which employed racemic propargylic alcohols 236 as precursors for alkynyl-substituted para-quinone methides 239 to react with thioacid nucleophiles 237a. This procedure could tolerate various substituents at positions R1, R2 and X. Additionally, thiobenzoic acid was identified as an effective partner in the presence of chiral phosphoric acid A39 catalyst. To validate the mechanism, alkynyl-substituted para-quinone methides 239 were prepared and subjected to the identified standard conditions, giving consistent results that supported the 1,8-addition mechanism. The resulting sulfur-containing tetrasubstituted allenes could subsequently be converted into allenyl sulfones with minimal loss of enantioselectivity. Furthermore, the allene products underwent iodocyclization, resulting in the formation of chiral indenes featuring sulfur-substituted quaternary carbon centers through axial-to-central chirality transfer. Besides the synthesis of S-containing allenes, using 1,3-dicarbonyl compounds as nucleophiles led to the generation of axial chiral tetrasubstituted allenes, which could arise from either propargylic cation intermediates via chiral ion-pairing interactions or through 1,8-conjugate addition of para-quinone methides. Notably, this study represents the first methodology for establishing axial chirality using quinone methides, as opposed to central chirality.
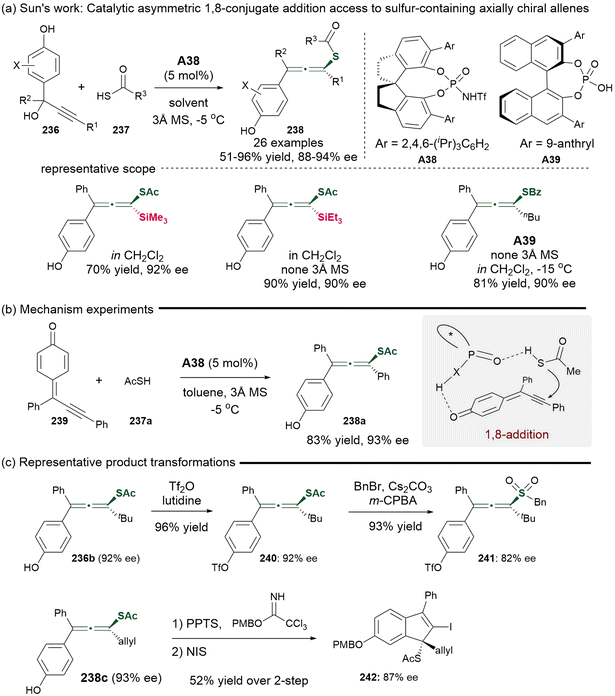 |
| | Scheme 43 Organocatalyzed enantioselective 1,8-addition of alkynyl-substituted para-quinone methides and thioacid nucleophiles. | |
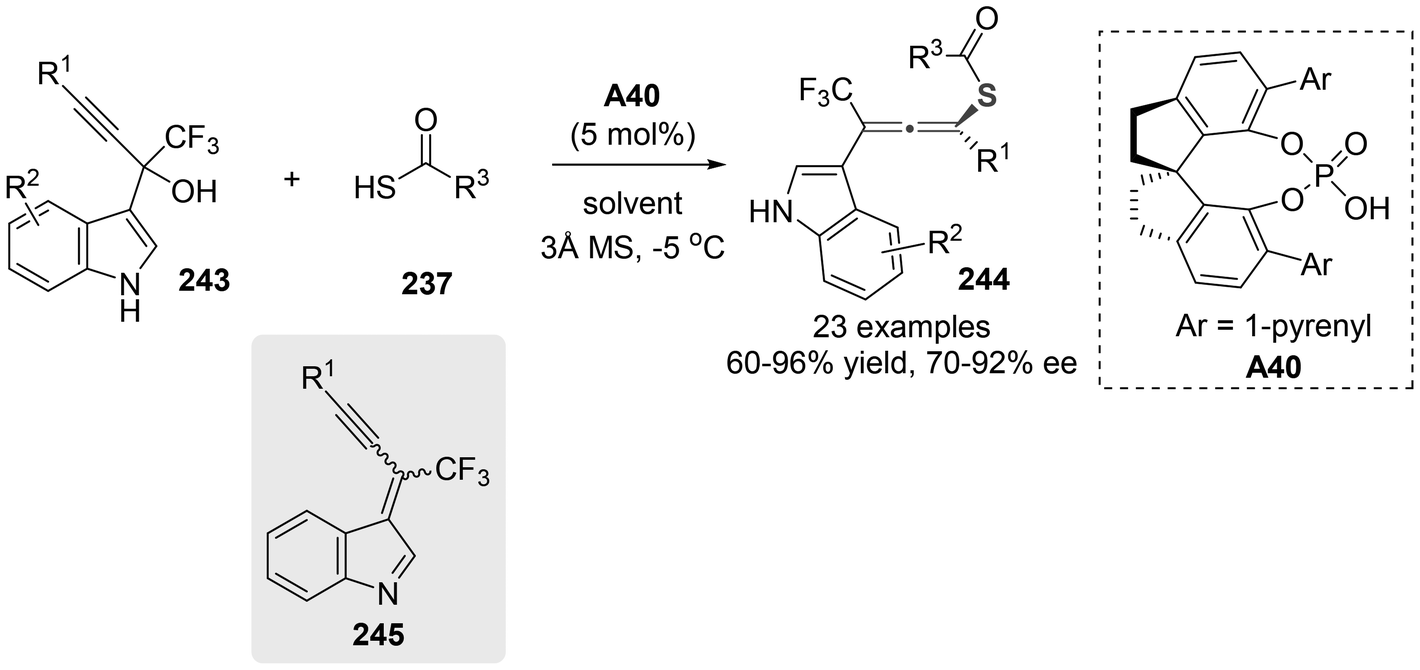
In 2021, Li and colleagues documented the synthesis of axially chiral sulfur-containing tetrasubstituted CF3-allenes 244 through a chiral phosphoric acid-catalyzed symmetric 1,6-addition involving thiolacetic acid 237 and α-(3-indolyl) propargylic alcohols 243 (Scheme 44).54 This method exhibited good yield and excellent stereoselectivity. The authors believed that this method involved the 1,6-conjugate addition of alkynyl indole-imine-methides 245 in situ generated from propargylic alcohols 243.
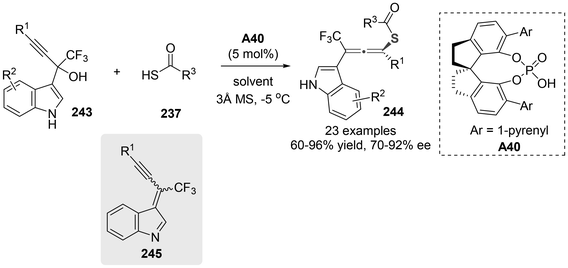 |
| | Scheme 44 Organocatalytic enantioselective 1,6-addition of thiolacetic acids to alkynyl indole imine methides. | |
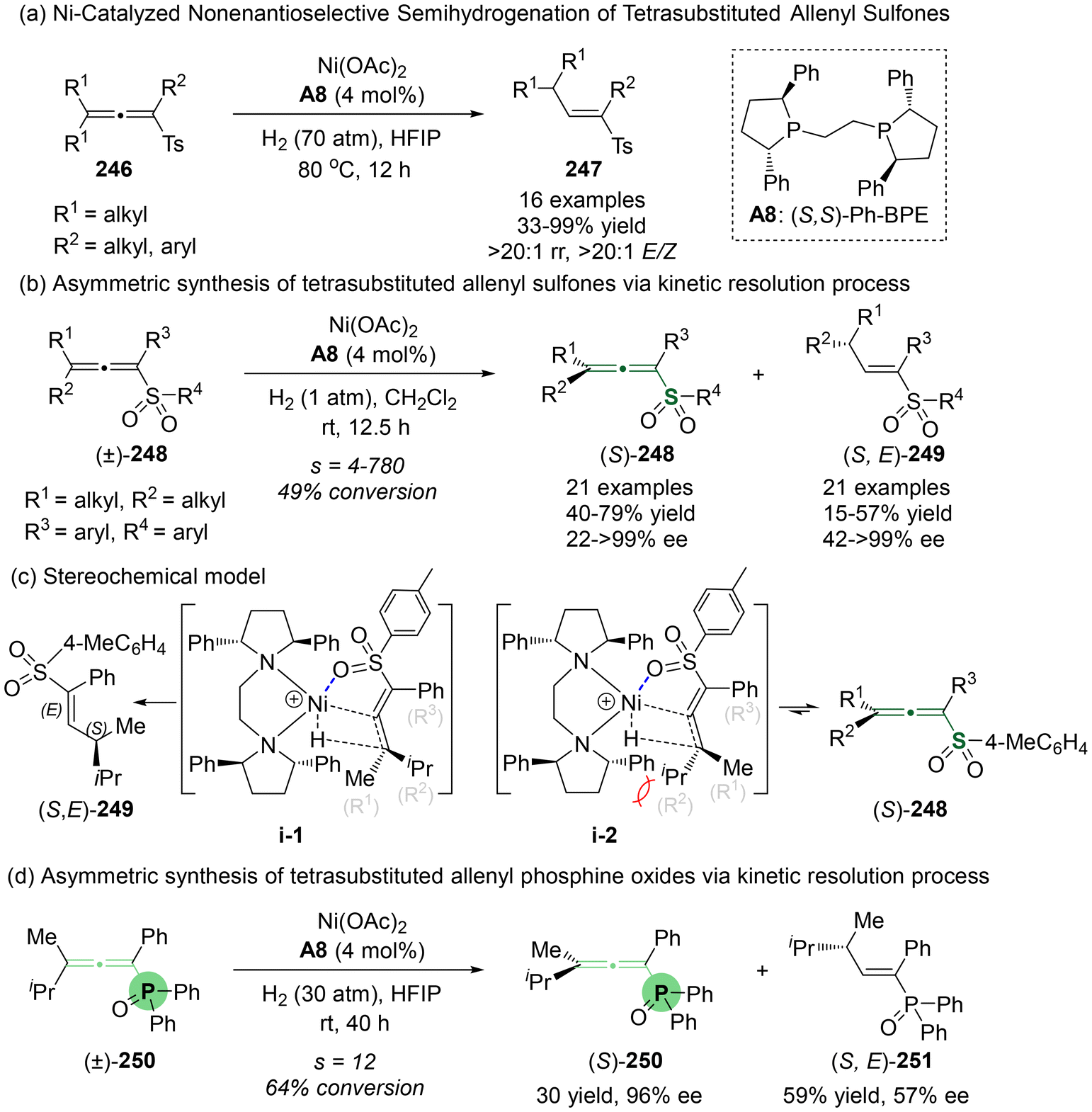
Recently, Zhang and Dong's group reported the first Ni-catalyzed semi-hydrogenation of racemic sulfur-containing tetrasubstituted allenes (Scheme 45).55 Although this reaction yielded racemic products with remarkable efficacy under conditions of 70 atm H2 at 80 °C (Scheme 45a), the condition of 1 atm H2 at room temperature allowed a kinetic resolution process (Scheme 45b). This resulted in the recovery of enantioenriched sulfur-containing tetrasubstituted allenes (Sa)-248, together with the formation of semi-hydrogenation (S,E)-products 249. To gain further insight into the reaction mechanism, the authors performed density functional theory (DFT) calculations and deuterium labeling experiments focused on the kinetic separation process. These investigations revealed that the two hydrogen atoms involved in the semi-hydrogenation originated from hydrogen gas. The sulfone group of the substrates served as a directing group, coordinating with a chiral Ni–H complex to give transition states i-1 and i-2. Due to steric hindrance between the chiral ligand and substrate (S,E)-249 in i-2, enantioenriched (S,E)-249 exhibited a reduced thermodynamical efficiency for hydrogenation, finally leading to the enrichment of (Sa)-248 via a kinetic resolution process. Furthermore, the authors observed that this kinetic protocol is also effectively applicable to the asymmetric synthesis of chiral tetrasubstituted allenyl phosphine oxides 250 (Scheme 45d).
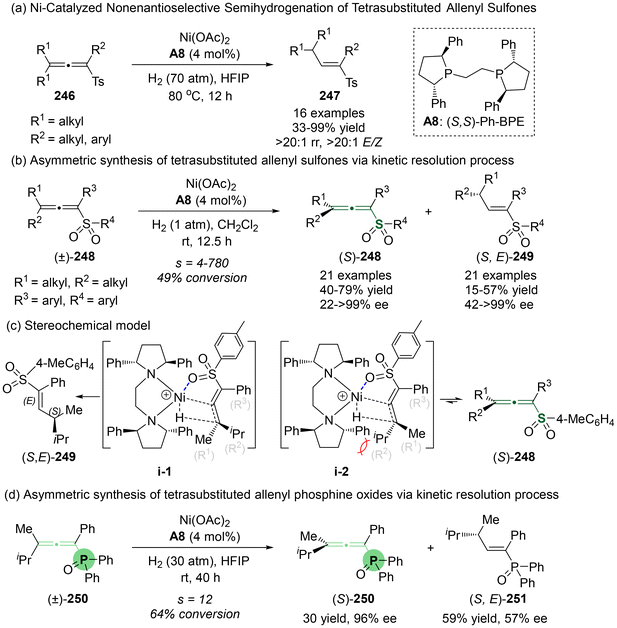 |
| | Scheme 45 Challenging task of Ni-catalyzed highly regio-/enantioselective semi-hydrogenation of racemic tetrasubstituted allenes via a kinetic resolution process. | |
2.7 Asymmetric synthesis of haloallenes
Organohalides represent a significant category of readily accessible and highly versatile building blocks in organic chemistry. Due to the rich and tunable reactivity associated with carbon–halogen (C–X) bonds, numerous novel reagents and transformations involving organohalides have been developed.56 For instance, Grignard reagent (RMgX) is prepared from organohalides (RX) and magnesium (Mg). The well-known phosphorous ylides, which are commonly employed for the preparation of alkenes through the Wittig reaction, are produced from alkyl halides with PPh3. The claim that organohalides serve as the backbone of transition metal chemistry is not an exaggeration. A variety of prominent cross-coupling reactions, including Suzuki–Miyaura, Sonogashira, Still, Negishi, Hiyama and Heck cross-coupling reactions, have been developed from organohalides, which have emerged as the most powerful strategy for constructing new carbon–carbon and carbon–heteroatom bonds. As a result, the synthesis of structurally diverse organohalides is still of significant interest in organic synthesis.
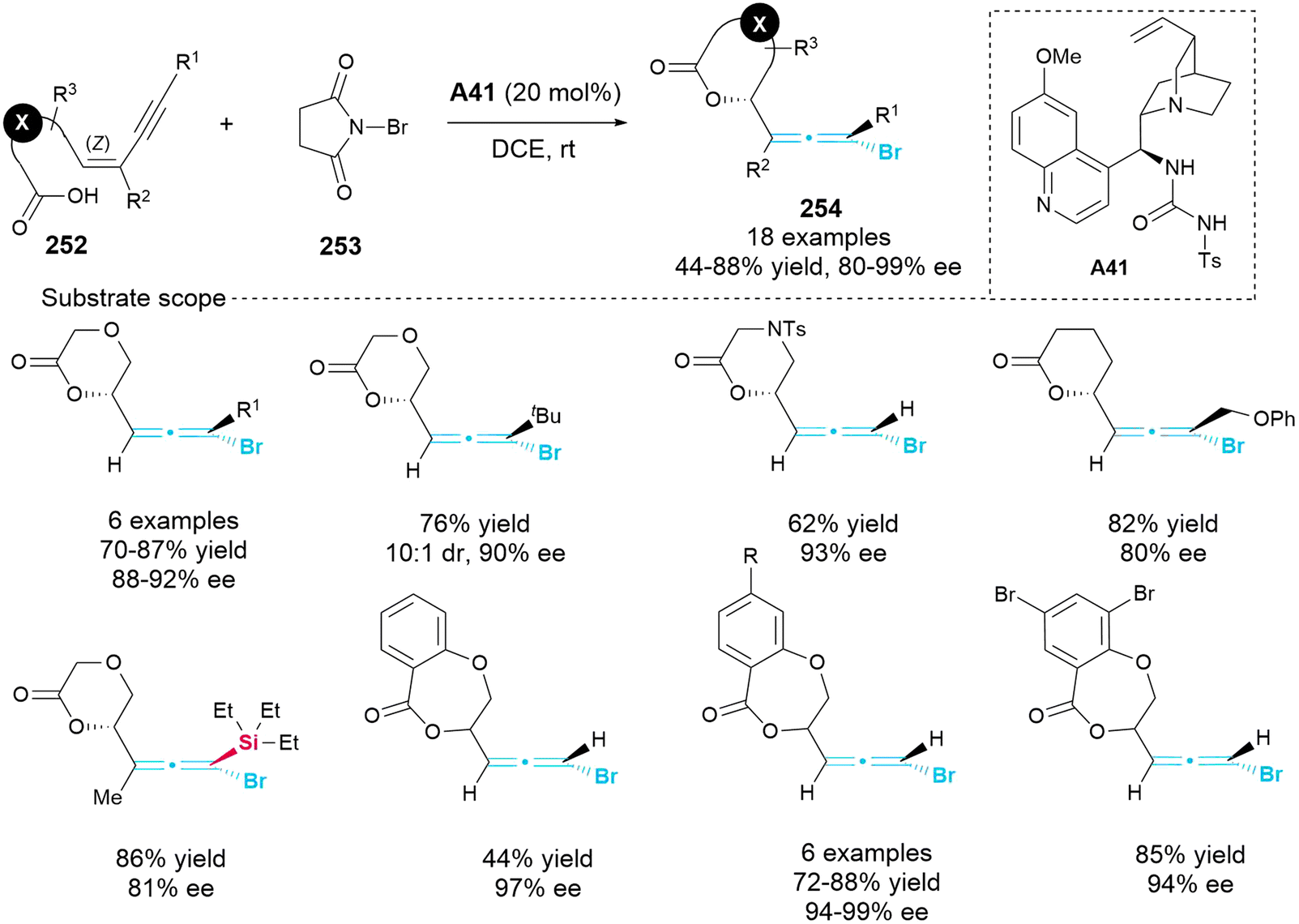
2.7.1 Asymmetric synthesis of bromo-substituted allenes. In 2011, Tang's group published findings on the enantioselective bromolactonization of conjugated (Z)-enynes 252 in the presence of bifunctional catalyst A41, aiming to prepare of axially chiral bromoallenes 254 bearing lactone heterocycles (Scheme 46).57 This methodology had a wide tolerance for various functional groups, allowing the incorporation of oxygen, nitrogen and carbon linkers. With the exception of one instance that exhibited a diastereomeric ratio (dr) of 10:1, all other synthesized products achieved a dr greater than 20:1. Furthermore, this methodology was successfully adapted to produce heptalactone-containing bromoallenes with high enantioselectivity and excellent conversion.
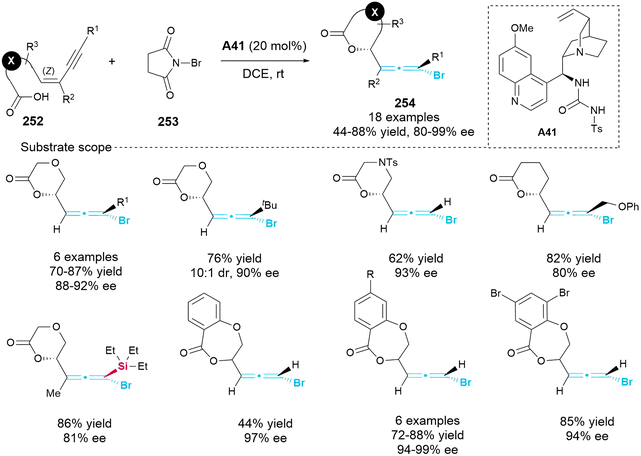 |
| | Scheme 46 Enantioselective bromolactonization of conjugated (Z)-enynes. | |
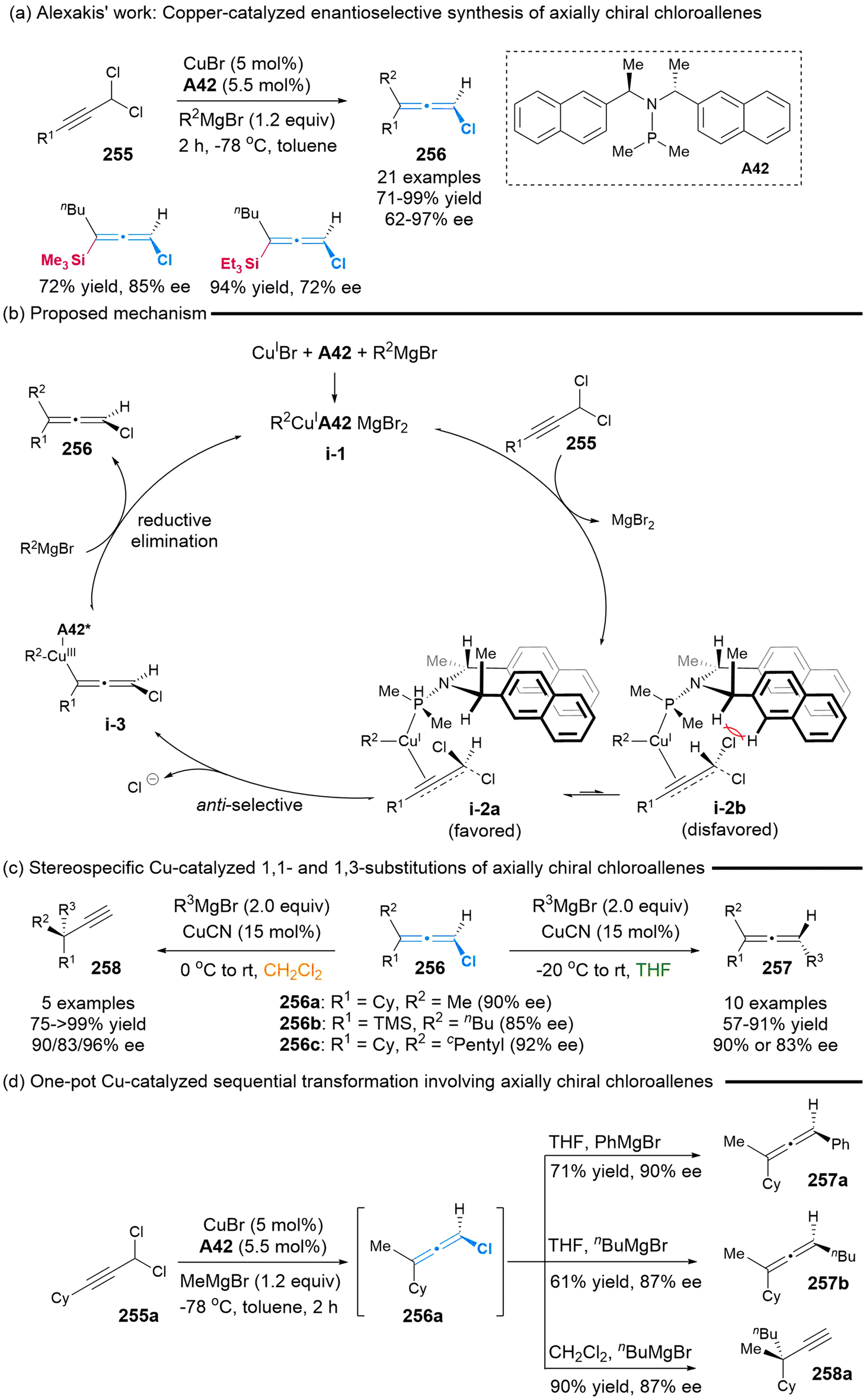
2.7.2 Asymmetric synthesis of chloro-substituted allenes. In 2012, Alexakis’ group employed the chiral SimplePhos A42/CuBr complex as a catalyst to facilitate the synthesis of the axially chiral chloroallenes 256, achieving yields in the range of 71% to 99% and enantiomeric excess (ee) values in the range of 62% to 97% (Scheme 47).58a This represents the first report on the use of asymmetric catalysis to realize the synthesis of axially chiral chloroallenes. Subsequently, the authors proposed the plausible reaction mechanism, involving an anti-SN2′/reductive elimination sequence associated with key preferred transition state i-2a.58b Furthermore, the stereospecific copper-catalyzed 1,1- and 1,3-substitutions of synthesized chloroallenes 256 were demonstrated by altering the solvent from tetrahydrofuran (THF) to dichloromethane (DCM). In THF, regioselective 1,1-substitution was carried out, allowing access to axially chiral trisubstituted allenes 257. When DCM was employed as the solvent, propargylic chiral quaternary carbon compounds 258 were obtained through regioselective 1,3-substiution, while maintaining the ee value. To simplify this sequential synthetic procedure, they also provided the one-pot process utilizing a single copper catalysis, wherein axially chiral chloroallenes 256a served as the key intermediates (Scheme 47d).
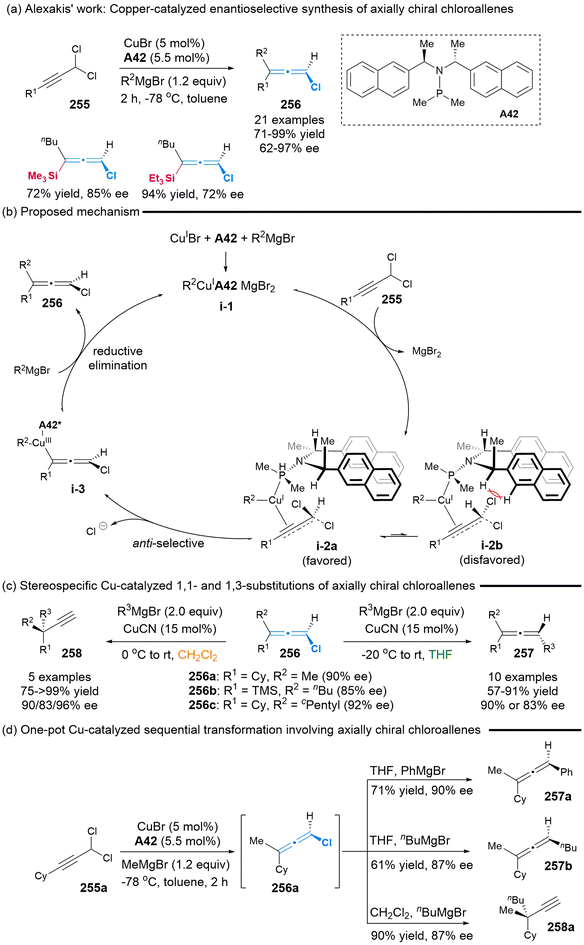 |
| | Scheme 47 Alexakis’ work on the enantioselective synthesis of axially chiral chloroallenes enabled by copper-catalyzed propargylic substitution of prochiral dichloro substrates. | |
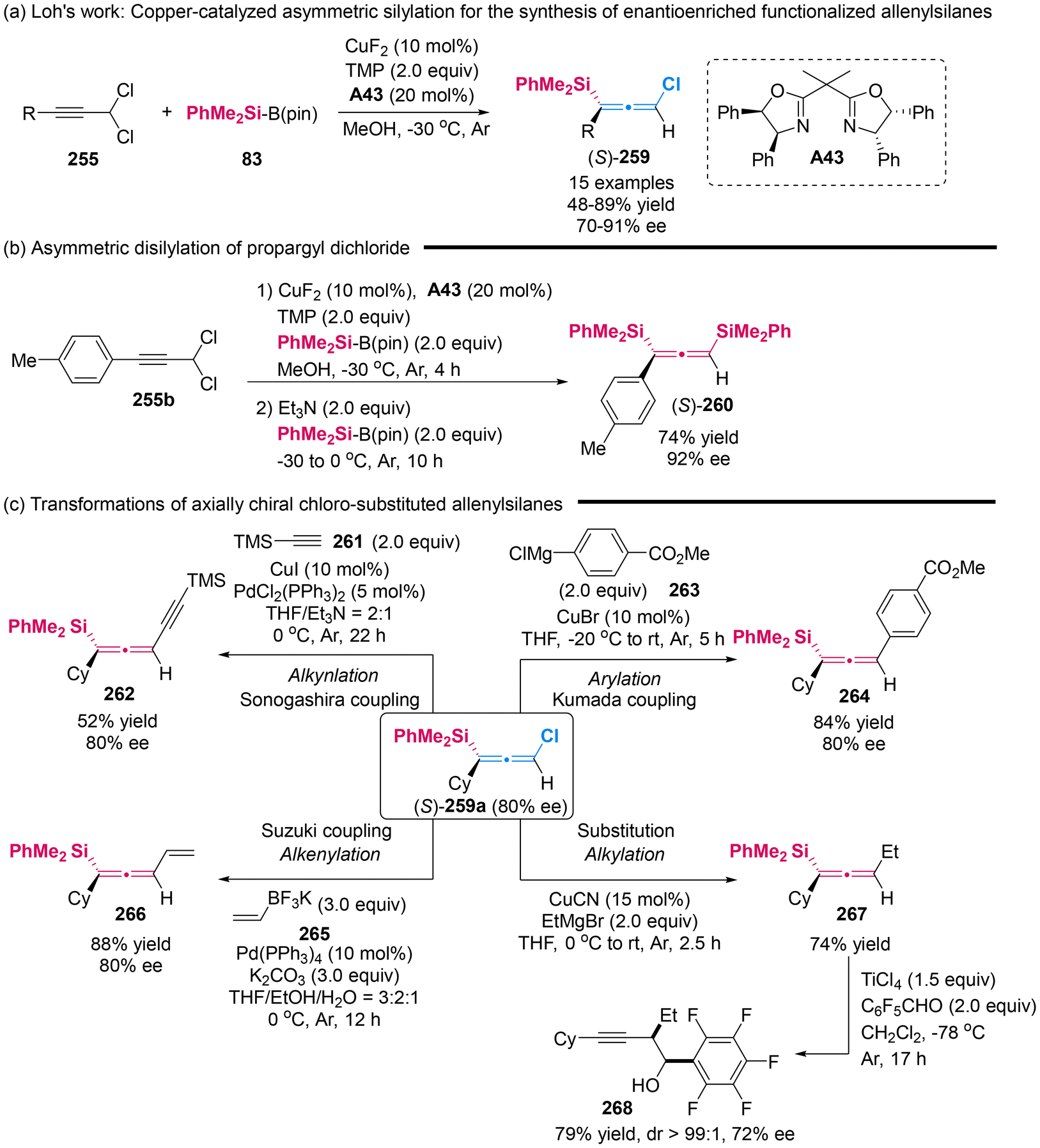
In 2019, Xu's group introduced a copper-catalyzed asymmetric silylation for prochiral propargylic dichlorides 255, efficiently providing access to chloro-substituted allenylsilanes 259 with high yields and good enantioselectivities (Scheme 48).59 They also demonstrated the synthetic potential of these chiral allenes. Initially, they displayed the synthesis of axially chiral disilyl-substituted allenes 260 (74% yield, 92% ee) through a one-pot asymmetric disilylation of propargyl dichloride (Scheme 48b). Then, they conducted the direct stereospecific functionalization of the C–Cl bond in chloro-substituted allenylsilanes 259a, employing methodologies such as Sonogashira, Suzuki and Kumada cross-couplings as well as Cu-catalyzed substitutions to yield the corresponding C–C bond formation products 262, 264, 266 and 267, respectively. Additionally, upon treatment of pentafluorobenzaldehyde with TiCl4, allenylsilanes 267 were further transformed to homopropargylic alcohol 268 with a slight reduction in enantiopurity but excellent diastereoselectivity (72% ee, >99:1 dr).
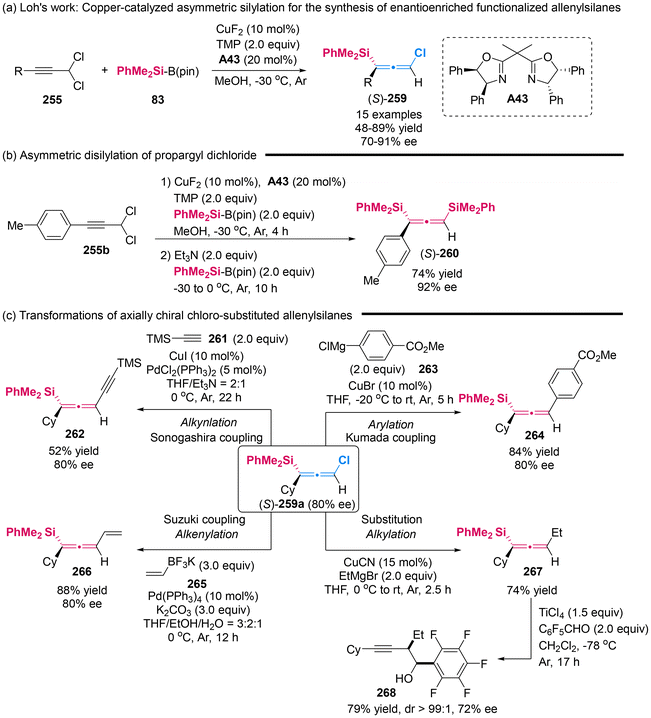 |
| | Scheme 48 Copper-catalyzed asymmetric silylation of propargyl dichlorides to attain obtain enantioenriched functionalized allenylsilanes. | |
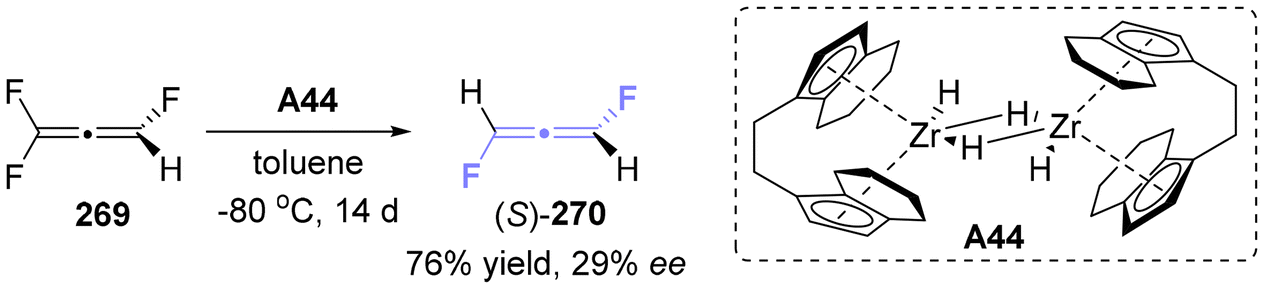
2.7.3 Asymmetric synthesis of fluoro-substituted allenes. In 2012, Lentz successfully synthesized axially chiral 1,2-difluoroallene 270, which is recognized as the smallest axially chiral allenes besides isotopologues (Scheme 49).60 This synthesis exhibited a modest but noteworthy enantiomeric excess of 29%. This methodology utilized a chiral zirconocene hydride (A44) as the reducing agent to reduce trifluoroallene 269.
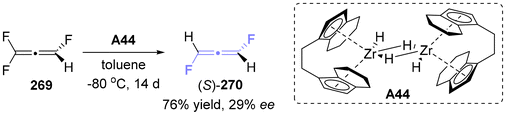 |
| | Scheme 49 Synthesis of the smallest axially chiral molecule by asymmetric carbon–fluorine bond activation. | |
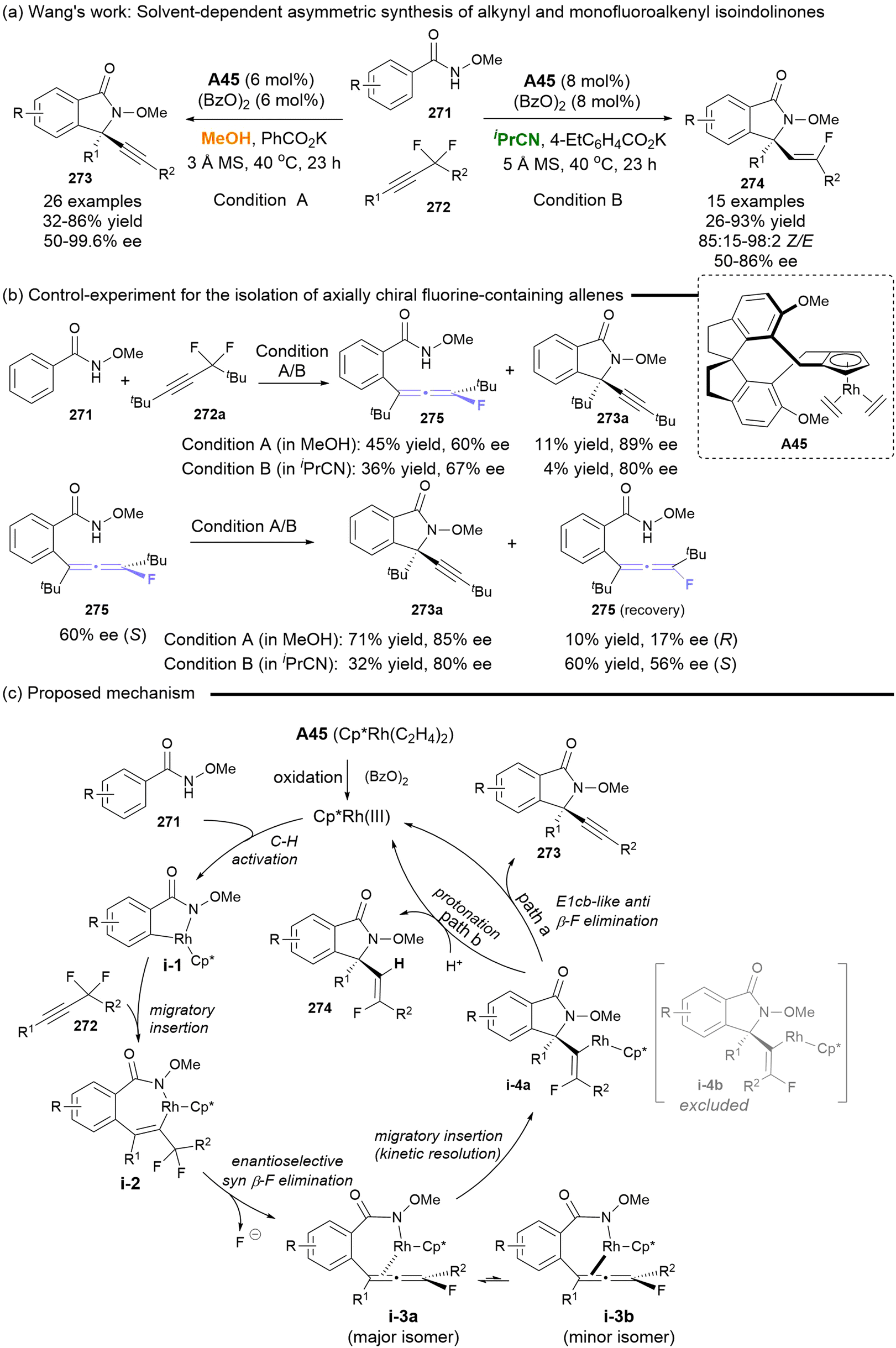
In 2018, Wang disclosed the solvent-directed asymmetric synthesis of alkynyl and monofluoroalkenyl isoindolinones, starting from N-methoxy benzamides and difluoromethylene alkynes with the catalyst of Cp*Rh(III) (Scheme 50).61 The introduction of the bulky tBu group in the difluoromethylene alkynes resulted in the isolation of axially chiral fluoro allenes 275, which served as key common chiral intermediates for 273 and 274, albeit with a moderate ee (Scheme 50b). The comprehensive mechanism studies demonstrated that the reaction commenced with Rh(III)-catalyzed C–H activation. This was followed by migratory insertion and enantioselective syn β-F elimination, leading to the synthesis of axially chiral fluoro allenes i-3a as the major isomer. A subsequent migratory insertion, occurring through a kinetic resolution process, produced intermediate i-4a. In MeOH, i-4a underwent E1cb-like anti-elimination of fluorine, resulting in the formation of alkynes 273. In isopropyl cyanide (iPrCN), the solvent coordinated with the rhodium centers, facilitating direct protonation to afford monofluoroalkenyl isoindolinones 274. Despite the moderate enantiocontrol for axially chiral fluoro allenes, this work represents the first catalytic asymmetric synthetic strategy. Furthermore, the solvent-directed asymmetric synthesis of two structural divergent isoindolinones based on the axially chiral fluoro-substituted allenes constitutes a significant contribution.
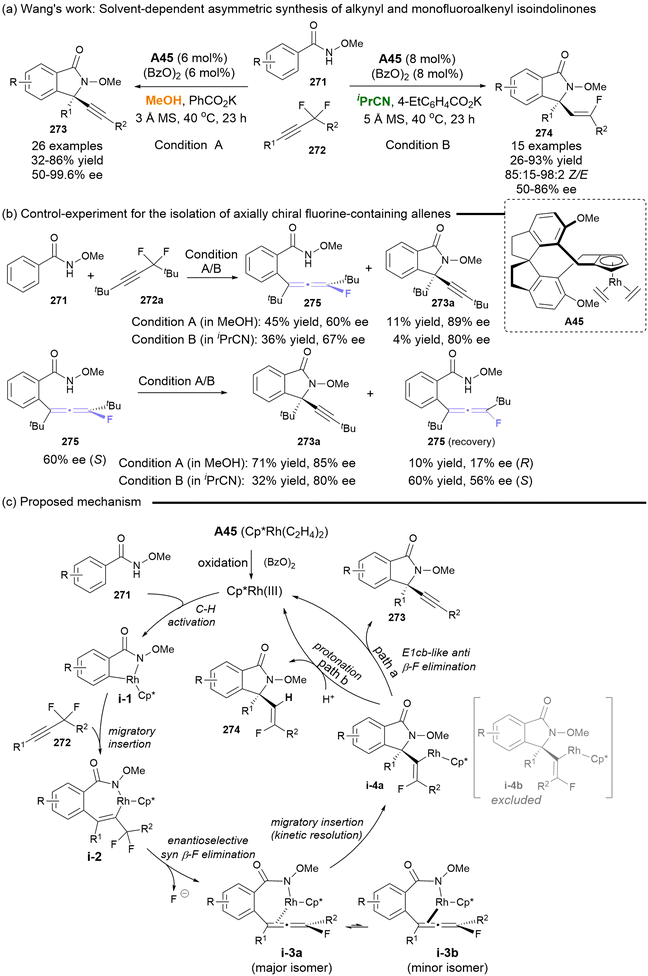 |
| | Scheme 50 Solvent-dependent asymmetric synthesis of alkynyl and monofluoroalkenyl isoindolinones. | |
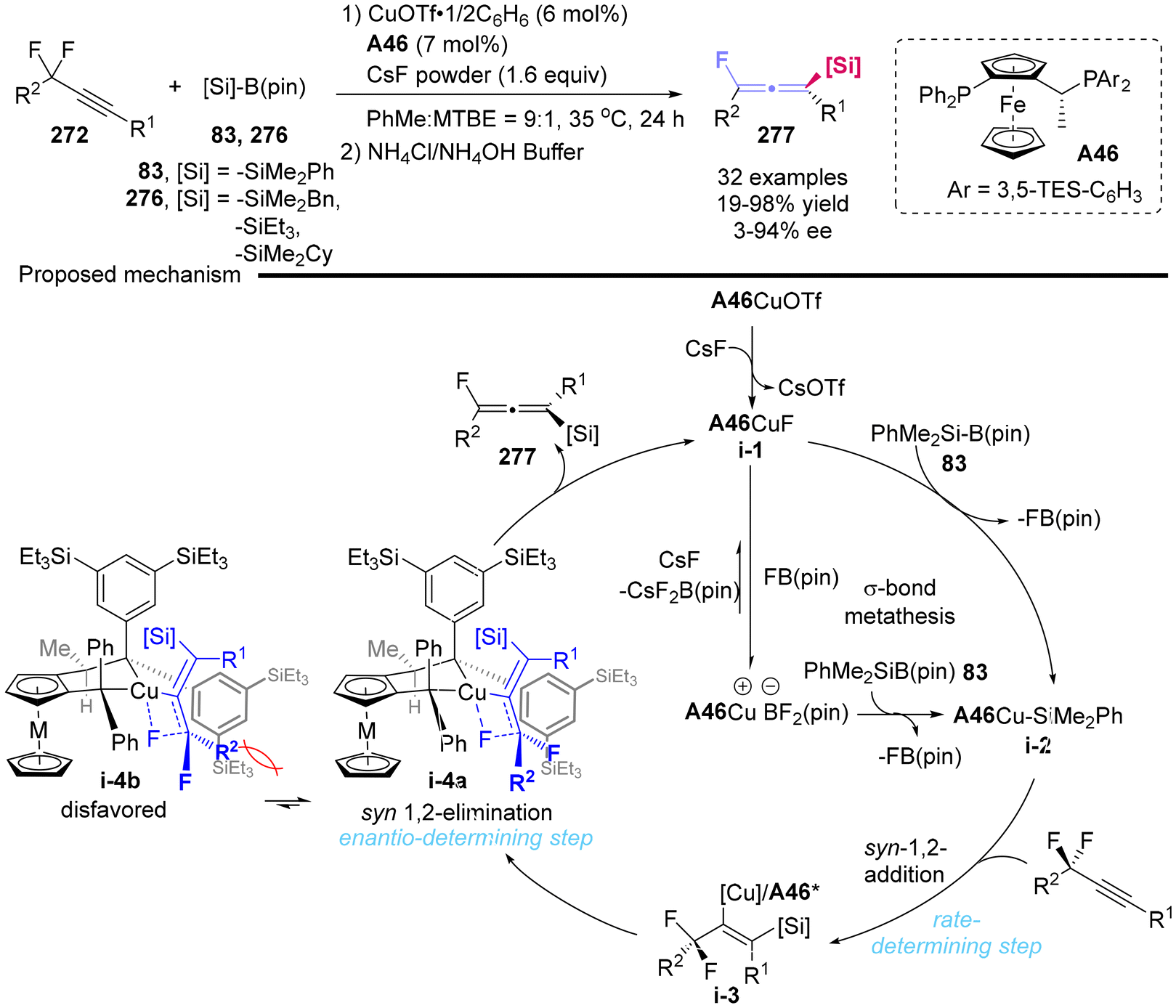
In 2021, Toste's group reported the development of a copper-catalyzed asymmetric silylation process for prochiral propargylic difluorides 272, which yielded the fluorine-containing, chiral tetrasubstituted allenylsilanes in up to 98% yield and 94% ee (Scheme 51).62 The mechanism studies demonstrated that the silylation of the alkyne constituted the rate-determining step, while the syn β-fluoride elimination, rather than an anti-elimination pathway, was identified as the enantio-determining step. Furthermore, the reduced Pauli repulsion (steric repulsion) observed between ligand A46 and substrate in i-4a compared to that in i-4b facilitated the syn-elimination pathway, ultimately leading to the formation of (S)-allenes 277.
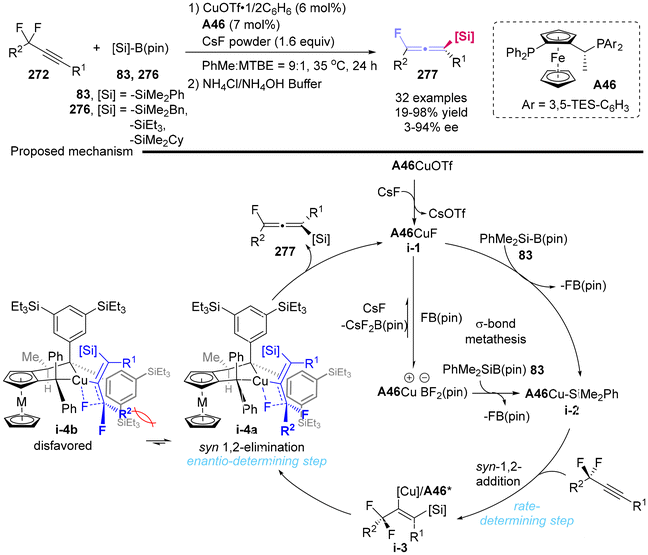 |
| | Scheme 51 Generation of axially chiral fluoroallenes through a copper-catalyzed enantioselective β-fluoride elimination. | |
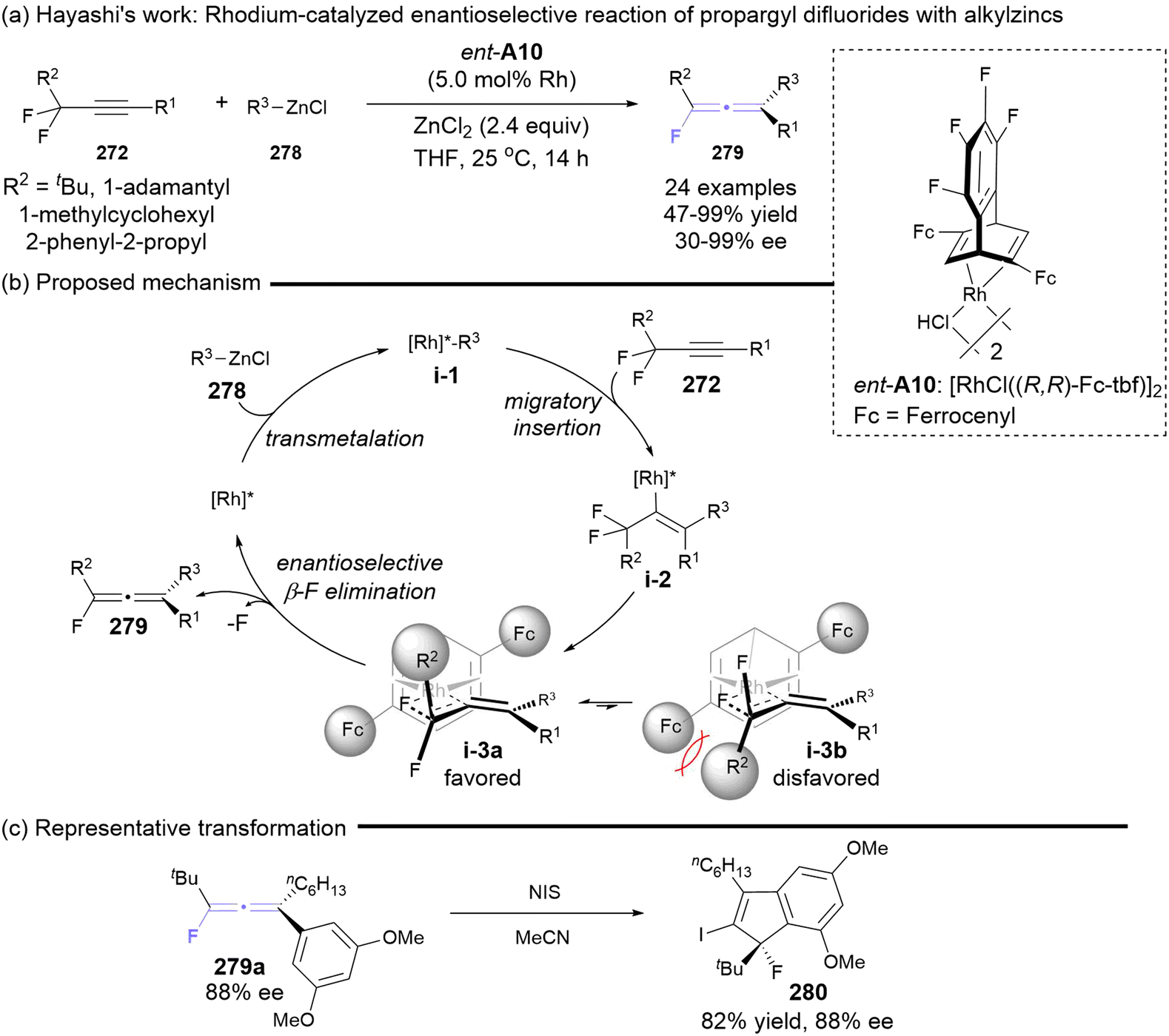
Almost simultaneously, the Hayashi group reported a rhodium-catalyzed asymmetric reaction involving prochiral propargyl difluorides 272 and alkylzinc reagents 278, enabling access to axially chiral monofluorinated allenes 279 in up to 99% yield and 99% ee (Scheme 52).63 The introduction of sterically demanding substituents R1 on propargyl difluorides 272, such as tBu, 1-adamantlyl, 1-methylcyclohexyl and 2-phenyl-2-propyl group, was essential for achieving favorable enantioselectivity. In contrast, the use of smaller substituents (cyclohexyl and isopropyl) resulted in diminished or negligible enantioselectivity. The authors inferred that the more severe steric repulsion between ligand A10 and the R1 substituent in intermediate i-3b would drive the enantioselective β-fluoride elimination from alkenyl-Rh intermediate i-3a. Furthermore, the iodonium-induced cyclization of chiral fluorinated allenes 279a was performed to construct chiral indene 280, which features a fluorinated quaternary carbon center, achieving a maintained ee value of 88% in 82% yield.
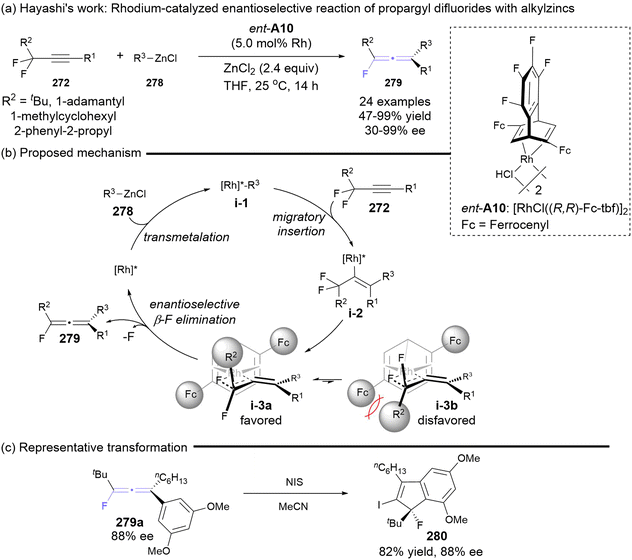 |
| | Scheme 52 Asymmetric synthesis of fluorinated allenes through rhodium-catalyzed enantioselective alkylation/defluorination of propargyl difluorides with alkylzincs. | |
2.8 Asymmetric synthesis of allenylborons
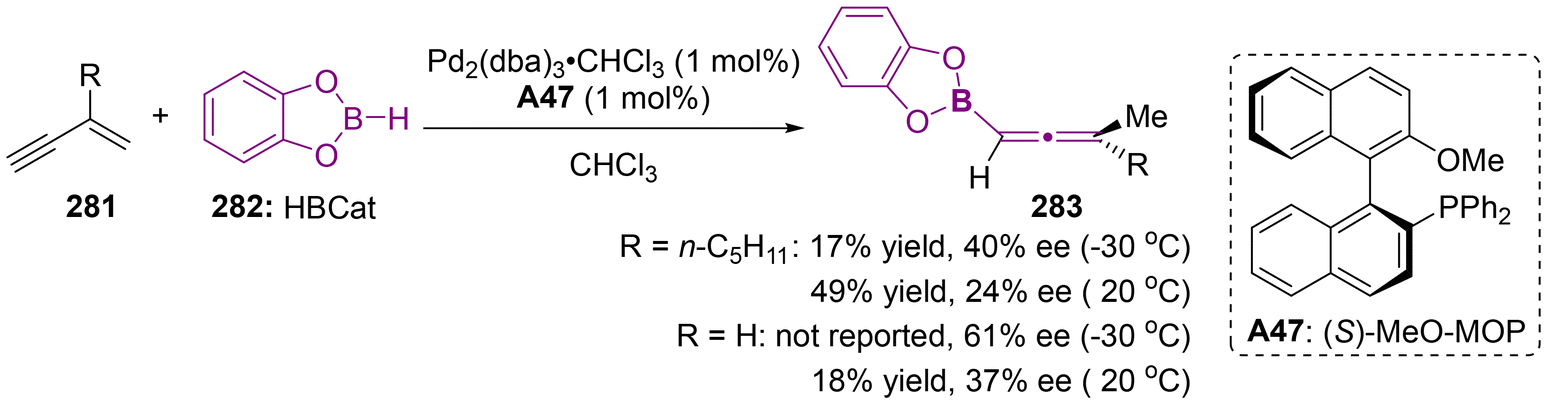
Organoboron compounds represent a fundamental class of platform molecules in synthetic chemistry due to their environmentally benign nature, high stability, broad compatibility with functional groups and easy accessibility. The significance of organoboron compounds has also promoted the advancement of axially chiral allenylboron compounds.9,11d,64 Notably, in 1993, the Hayashi group reported the synthesis of enantioenriched axially chiral allenylboranes through the palladium-catalyzed asymmetric hydroboration of but-1-en-3-ynes 281 (Scheme 53).65 Although the enantioselectivity observed was moderate and the yield was suboptimal, this work marked a pioneering effort in the realm of asymmetric catalysis. Subsequently, some highly stereospecific SN2′-type reactions have emerged as a reliable strategy for the construction of axially chiral allenylboronates, utilizing various enantiomerically enriched propargylic substrates through the process of chirality transfer.
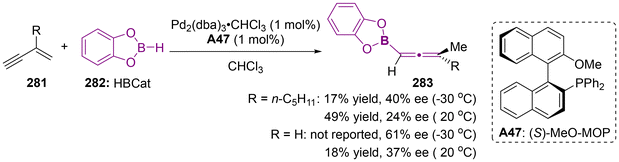 |
| | Scheme 53 Palladium-catalyzed hydroboration of but-I-en-3-ynes access to afford axially chiral allenylboranes. | |
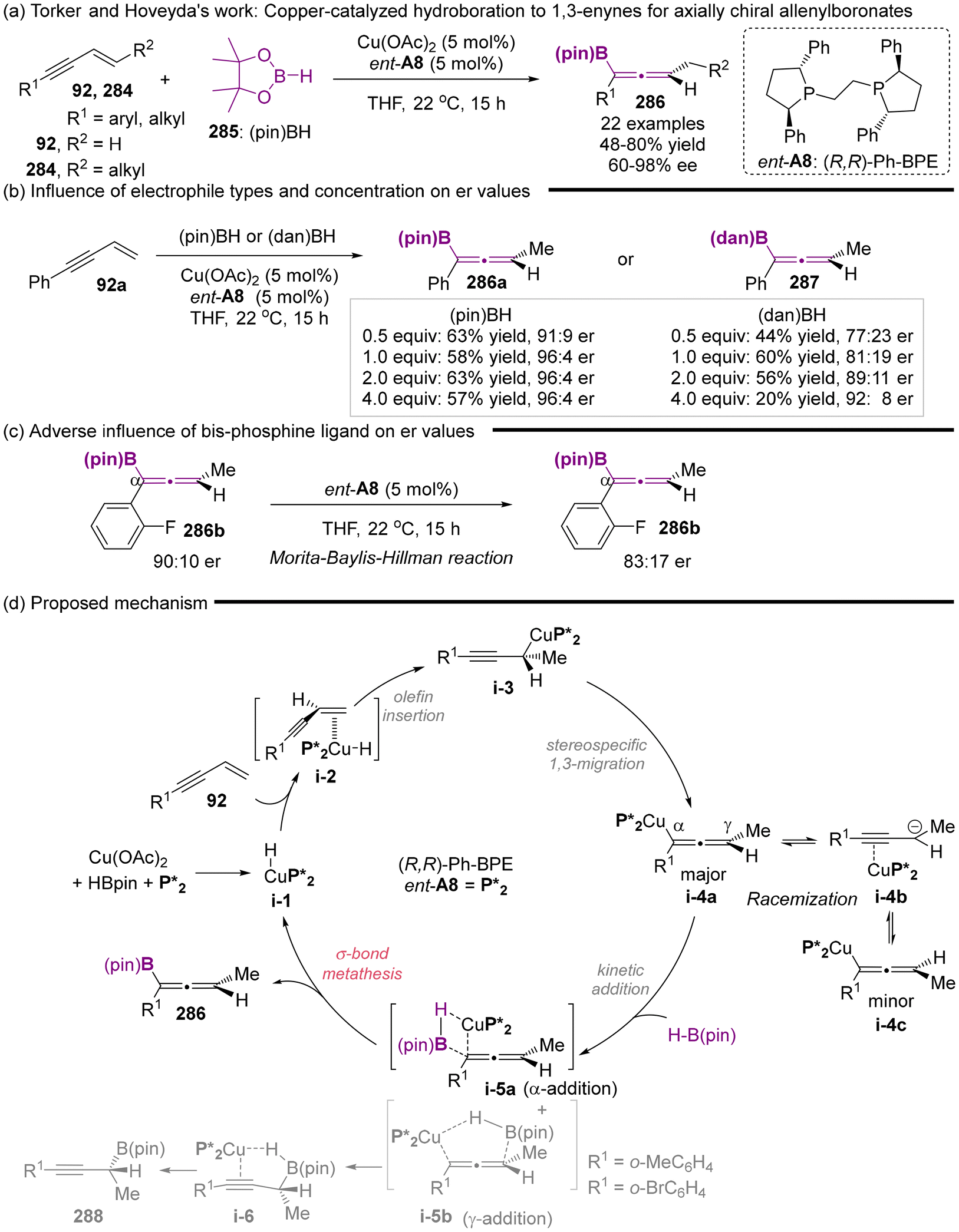
In 2018, Torker and Hoveyda's group reported the copper-catalyzed enantioselective hydroboration of 1,3-enynes with the catalyst of Cu(OAc)2/chiral ligand ent-A8 complex (Scheme 54).66 This study represents the first successful application of a catalytic asymmetric strategy for the synthesis of chiral allenylboronates, achieving excellent enantioselectivity and reactivity. The described protocol enjoys atom-economy, and a wide substrate scope, which includes both alkyl and aryl-substituted 1,3-enynes. Moreover, a series of comprehensive experiments was conducted (Scheme 54b and c), elucidating the clear mechanistic pathway (Scheme 54d). The findings indicated that ortho-substituted aryl enynes would result in the formation of propargyl-B(pin) 288 in varying proportions. Particularly, when substituents R1 are o-BrC6H4 or o-MeC6H4, propargyl-B(pin) 288 emerges as the predominate products. Using the strongly electron-deficient aryl-substituted enynes, less reactive electrophiles such as H–B(dan), lower concentrations of electrophiles or an excess of bis-phosphines ligand resulted in a reduced er values. The observed diminution in enantioselectivity primarily arises from the competitive isomerization process of allenyl-Cu species i-4a to i-4c. Therefore, the authors concluded that appropriately adjusting the reaction parameters such as employing highly reactive H–B(pin), increasing the concentration of H–B(pin) or utilizing a relative lower equivalent of enynes-one can enhance the rate of kinetic Cα-addition, thereby achieving a higher degree of enantioselectivity.
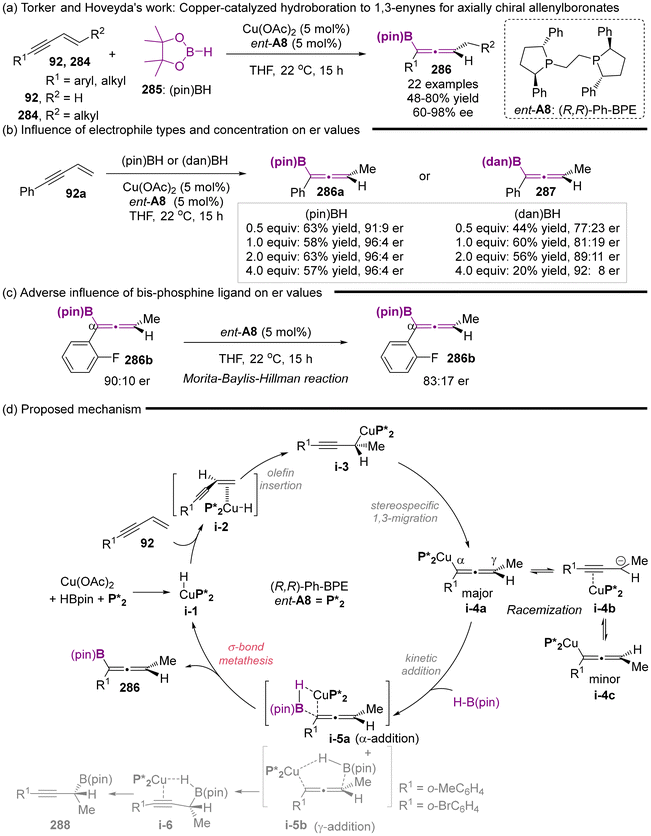 |
| | Scheme 54 Enantioselective synthesis of trisubstituted allenyl-B(pin) compounds via phosphine-Cu-catalyzed 1,3-enyne hydroboration and insights regarding stereochemical integrity of Cu-allenyl intermediates. | |
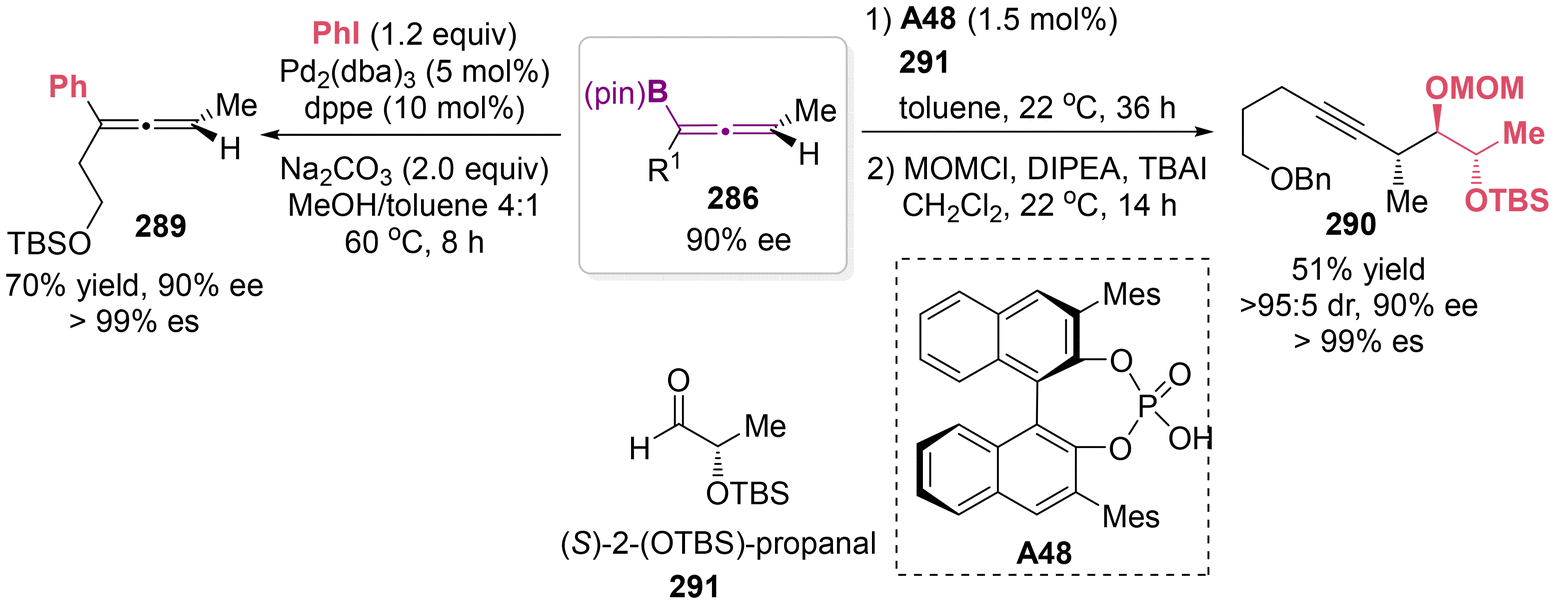
To illustrate the synthetic utility of the available allenylboronate products, a diastereoselective addition to aldehydes 291 catalyzed by chiral phosphoric acid A48 was conducted to access homopropargyl alcohols 290, which serve as a versatile anti–anti stereotriad unit (Scheme 55).67 Besides, allenylboronate 286 can be transformed to arylated allenes 289, while maintaining enantiomeric purity via a palladium-catalyzed cross-coupling reaction with phenyl iodide.
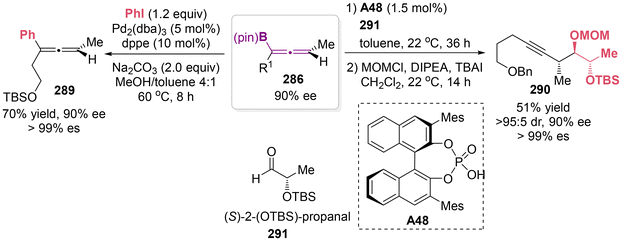 |
| | Scheme 55 Representative transformations of axially chiral allenylboronates. | |
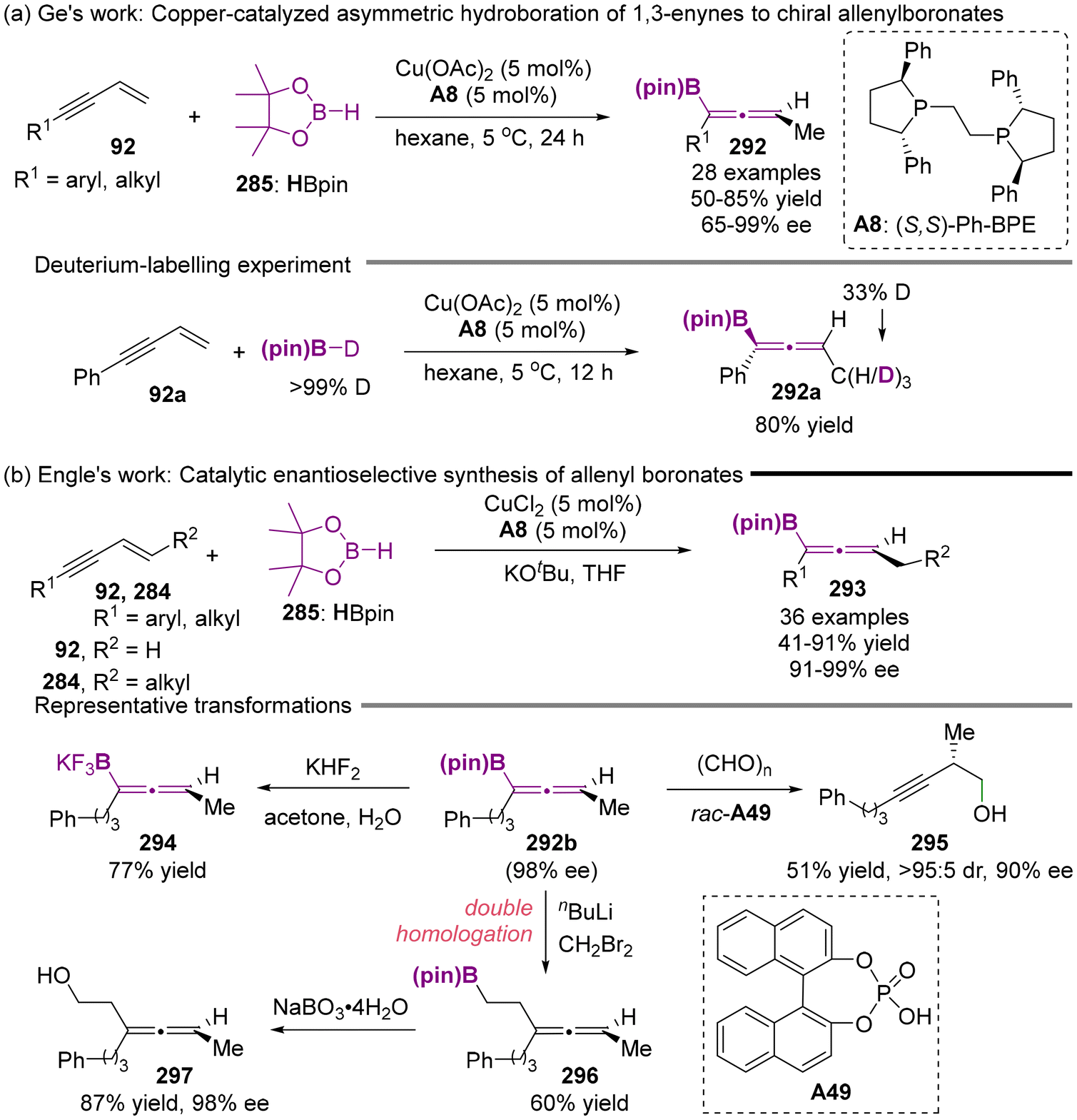
Almost simultaneously, the Ge group68 and Engle group69 reported the catalytic asymmetric synthesis of axially chiral allenyl boronates with the Cu(II)/(S,S)-Ph-BPE A8 complex as catalyst. This process involves the enantioselective hydroboration of 1,3-enynes, as illustrated in Scheme 56. A deuterium-labeling experiment revealed that the reaction proceeds through the generation of the Cu–H intermediate derived from HBpin, followed by 1,2-migratory insertion into the olefin motif of 1,3-enynes. This mechanism is consistent with the reaction pathway previously established by Torker and Hoveyda.66
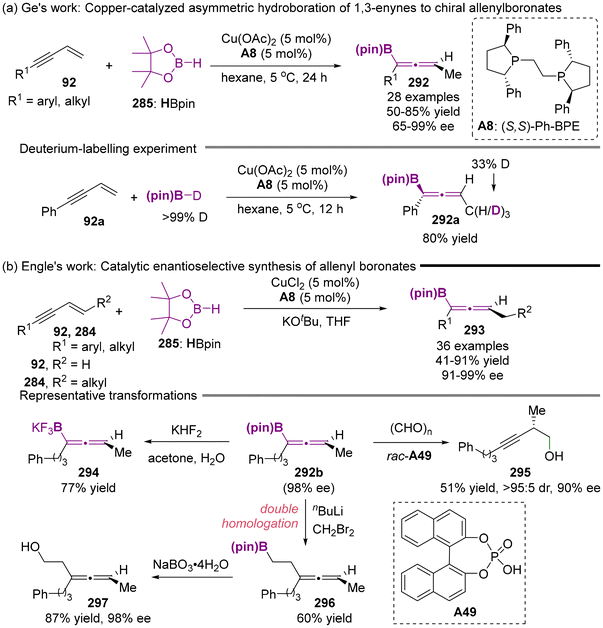 |
| | Scheme 56 Copper-catalyzed asymmetric hydroboration of 1,3-enynes with pinacolborane to access chiral allenylboronates. | |
As depicted in Scheme 57b, in addition to the synthesis of valuable chiral homopropargylic alcohols 295 by reacting with paraformaldehyde with catalyst A49, axially chiral allenyl boronates 296b can also react with KHF2 to deliver the corresponding trifluoroborate salt 294. When subjected to nBuLi and CH2Br2, the one-pot introduction of the two carbon atoms into the allenyl boronates via double homologation occurred, following oxidative hydrolysis to produce compound 297.
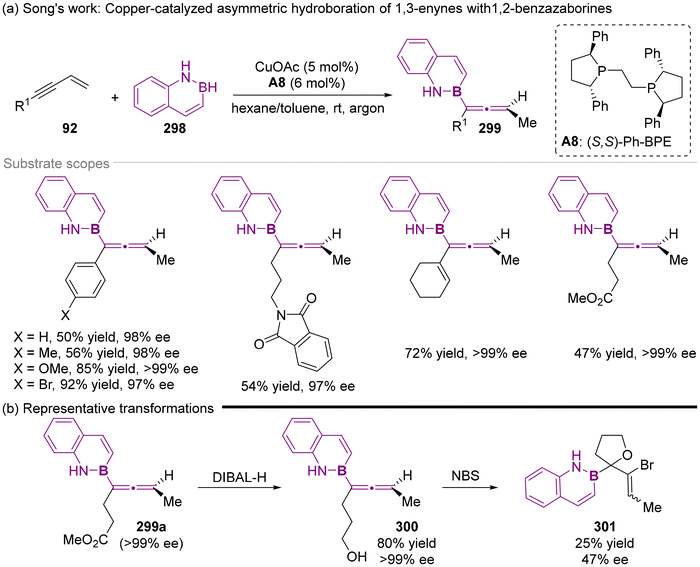 |
| | Scheme 57 Copper-catalyzed asymmetric hydroboration of alkenes with 1,2-benzazaborines to access chiral naphthalene isosteres. | |
Inspired by the principle of bioisosteric replacement, 1,2-benzazaborines have emerged as a promising isostere of naphthalene, attracting increasing attention in organic synthesis. To investigate the potential of 1,2-benzazaborines in asymmetric synthesis, the Song group recently demonstrated the copper-catalyzed enantioselective hydroboration of alkenes with the catalyst of Cu/chiral bis-phosphines complex, providing a mild and efficient approach access to diverse chiral 1,2-benzazaborine compounds with excellent enantioselectivity.70 When the substrate scope was extended from general alkenes to 1,3-enynes 92 under modified reaction conditions, the catalytic asymmetric hydroboration proceeded effectively, furnishing the axially chiral allenyl 1,2-benzazaborines with excellent enantiocontrol (Scheme 57). This finding represents an advancement, corroborating the earlier discovery by Torker and Hoveyda that the relatively less reactive electrophiles 1,2-benzazaborines could reach nearly perfect levels of enantioselection. Additionally, the ester functionality of axially chiral allenyl 1,2-benzazaborine 299a can be selectively reduced using DIBAL-H, followed by subsequent bromoetherification to produce 1,2-benzazaborine-substituted tetrahydrofuran 301.
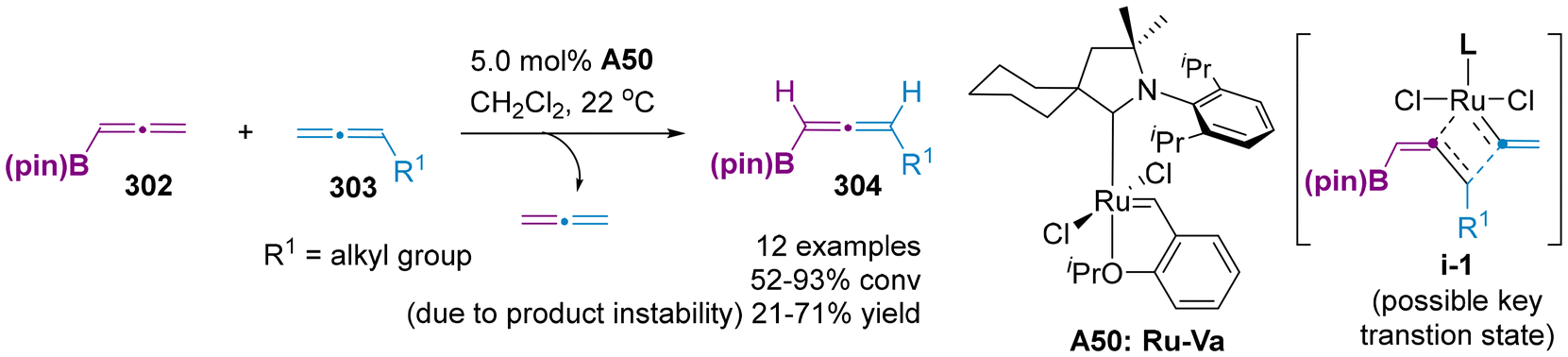
Despite these intellectual advancements in different axially chiral allenylboron compounds, these methods predominately involved the catalytic enantioselective hydroboration of 1,3-enynes and utilized the common linear chiral bis-phosphines ligands. Thus, to address the demand for synthetic applications involving a broader range of structurally diverse organoboron compounds, it is imperative to explore alternative catalytic systems, as well as the development of novel ligands and new reactions. Recently, Hoveyda et al. disclosed the first cross-metathesis reaction between two distinct symmetric allenes 302 and 303, thereby providing a novel strategy access to unsymmetrical allenylboronates 304 (Scheme 58).71 This methodology involved the formation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds through the proposed transition state i-1, representing a direct strategy for allene skeletal editing, rather than peripheral editing. Nevertheless, the asymmetric variant of this methodology remains unexplored.
C bonds through the proposed transition state i-1, representing a direct strategy for allene skeletal editing, rather than peripheral editing. Nevertheless, the asymmetric variant of this methodology remains unexplored.
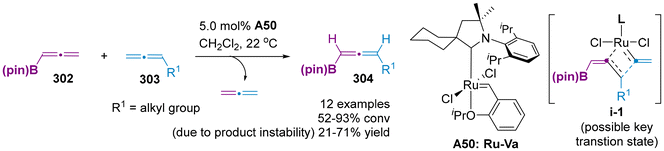 |
| | Scheme 58 Cross-metathesis of allenes for the synthesis of unsymmetrical allenylboronates. | |
3. Asymmetric synthetic applications: from simple to complex
The incorporation of heteroatom in allenes endows them with some unique reactivities. Various heteroatom-substituted axially chiral allenes have demonstrated their utility as versatile propargylated synthons.9,10,11b,19,66,67,72 Haloallenes are capable of undergoing reaction with transition-metal catalysts through an oxidation addition step, allowing cross-coupling reactions. The silicon hyperconjugation has been shown to direct an unprecedented [3 + 2]-annulation of allenylsilanes. These noteworthy reactions have enabled the development of high-efficient and concise protocols for the construction of significant chiral molecules or frameworks. Notably, all the heteroatom-substituted axially chiral allenes utilized in these synthetic processes are derived from chiral materials through chirality transfer.
3.1 Nucleophilic addition with organometal reagents
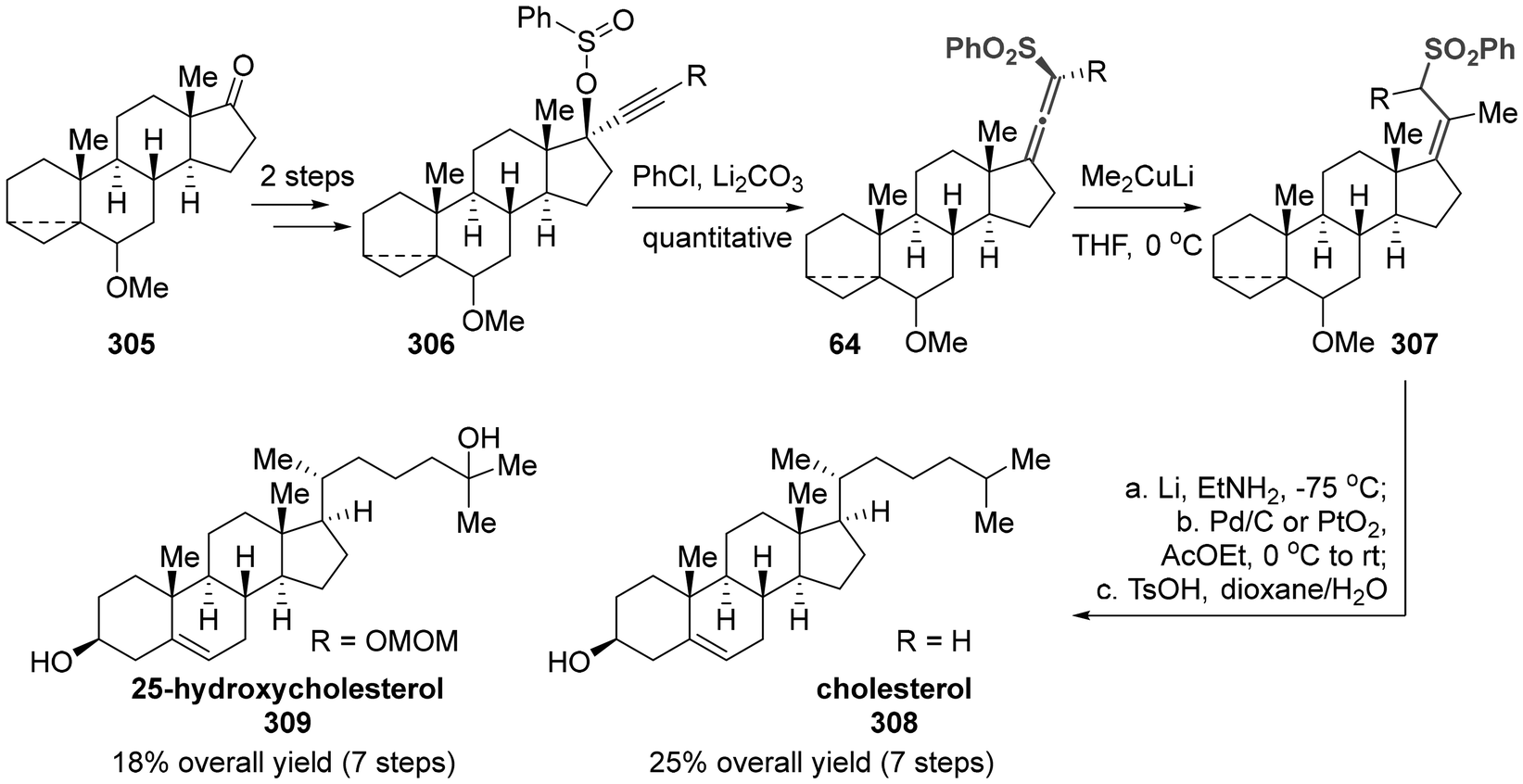
The earliest report we identified involved the use of axially chiral sulfonyl allenes as electrophilic acceptors to introduce the requisite functional group. Finally, this protocol accomplished the synthesis of cholesterol (308) and 25-hydroxycholesterol (309),73 which are important fundamental life-supporting materials in mammals (Scheme 59). The synthesis of axially chiral sulfonyl allenes commenced with the readily available 17-oxosteroid 305, which underwent alkynylation of the ketone group, sulfination of the hydroxyl group, and thermal [2,3]-sigmatropic rearrangement of compound 306. Subsequently, the key step involved the nucleophilic addition of Me2CuLi to sulfonyl allenes for the introduction of a methyl group, followed by a sequential process of reductive desulfonylation, hydrogenation and fragmentation, ultimately yielding the desired products.
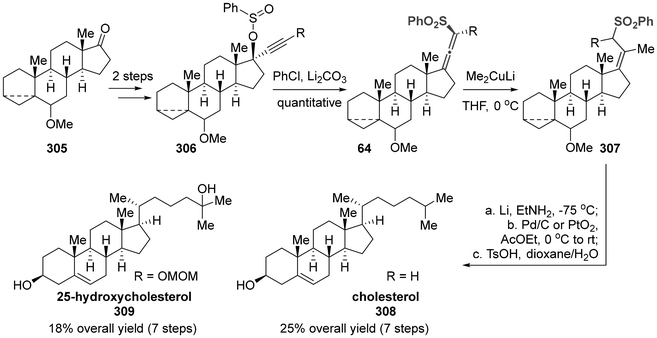 |
| | Scheme 59 Stereo-controlled synthesis of steroid side chain: stereoselective synthesis of cholesterol and 25-hydroxycholesterol. | |
3.2 Propargylation with imines
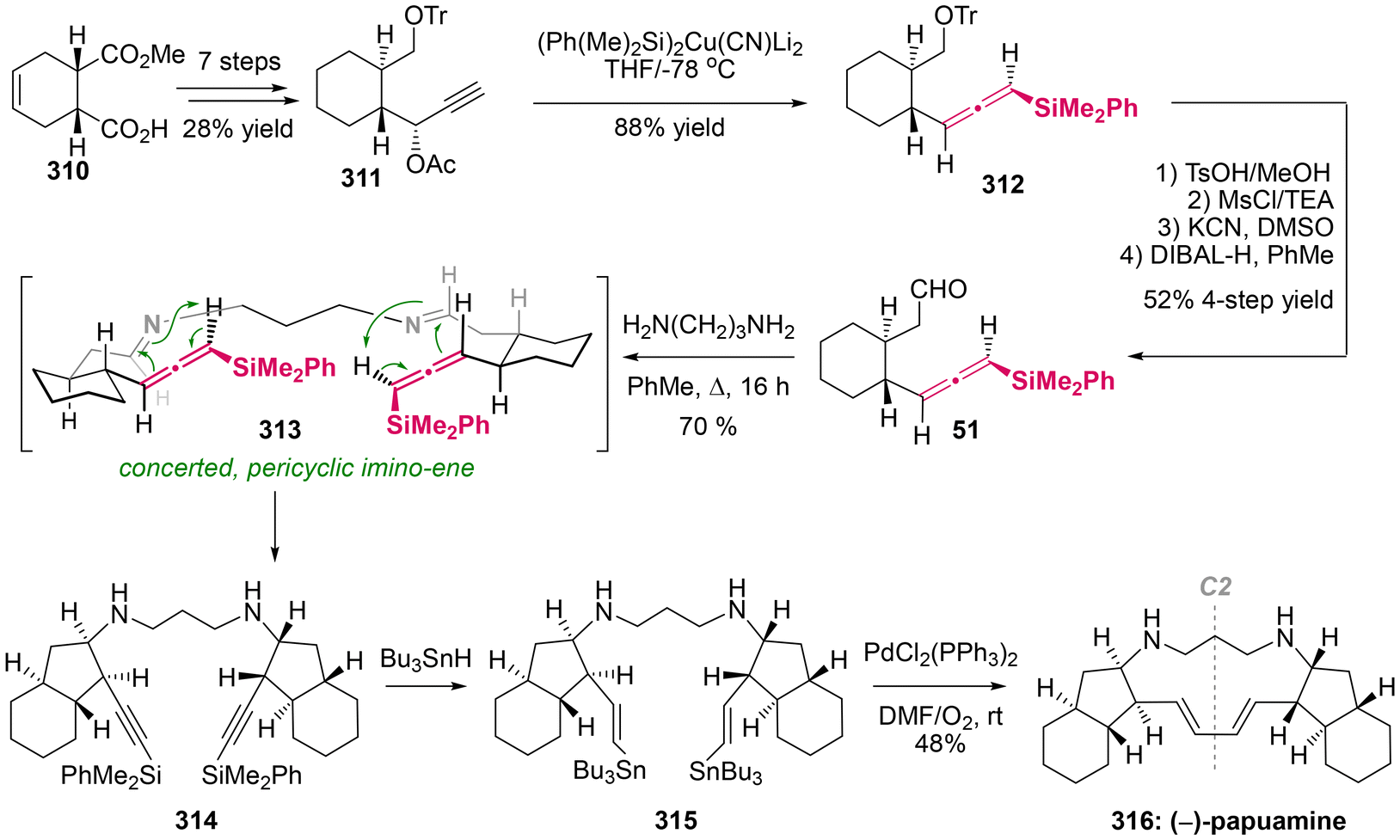
The imino-ene reaction, as an extension of the carbonyl–ene reaction and closely related to the Prins reaction, involves the coupling of readily accessible imines with allenes, thereby serving as a valuable method for the assembly of homopropargylic amines through C–C bond formation. This reaction has been utilized as a pivotal step in the total synthesis of various natural products.74
Papuamine, a C2-symmetric pentacyclic alkaloid with antibacterial and antifungal activities, was originally isolated from the marine sponges of the genus Haliclona. In 1994, Weinreb demonstrated the first asymmetric total synthesis of the natural enantiomer of (−)-papuamine 316,75 which prominently featured a double intramolecular imino-ene reaction involving axially chiral allenylsilane as the key step (Scheme 60). This synthesis was accomplished in 16 steps, commencing with the readily available enantiopure acid ester 310. This study revealed that the imino-ene reaction proceeded effectively in refluxing toluene without the need for a Lewis acid, stereospecifically yielding cyclopentylamine 313 as the sole stereoisomer in 70% yield. The findings supported the notion that the thermal imino-ene reaction is a concerted pericyclic process, rather than a well-known stepwise ionic mechanism.76 Besides the thermal activation, the imino-ene reaction can also be promoted by SnCl4 at relatively lower temperatures. Moreover, the presence of a silyl group on the allenes is essential for their reactivity, and this method demonstrates broad compatibility with various substituents on the tether. In the final step of this synthesis, Pd(II)-catalyzed coupling was employed to finish the macrocyclization through the formation of a C(sp2)–C(sp2) bond. Although a moderate yield of 48% was obtained, preparative thin-layer chromatography (TLC) and an ion exchange column were necessary for the purification of the free alkaloid.
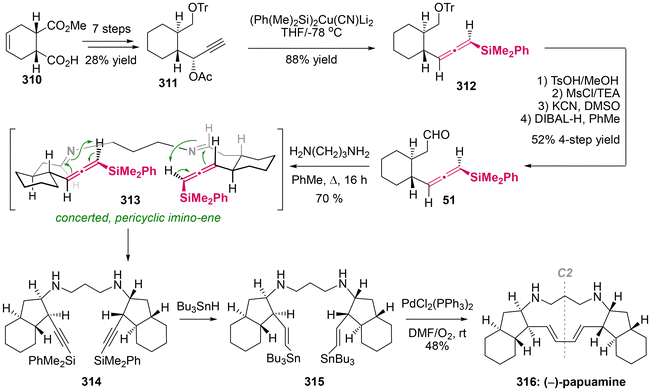 |
| | Scheme 60 Asymmetric total synthesis of (−)-papuamine via the stereospecific intramolecular imino-ene reaction of an allenylsilane. | |
5,11-Methanomorphanthridine alkaloids represent a distinct subclass of Amaryllidaceae alkaloids, distinguished by their unique bridged pentacyclic structure. In 1997, Weinreb successfully accomplished the enantioselective total synthesis of several 5,11-methanomorphanthridine alkaloids, specifically (−)-coccinine 330 and (−)-montanine 331, as well as the formal synthesis of (−)-brunsvigine 327 and (−)-pancracine 329.77,78 As illustrated in Scheme 61, this was achieved through the utilization of concerted thermal imino-ene cyclization as the crucial synthetic step. This divergent synthetic strategy commenced with the readily accessible optically pure hydroxy epoxide 317. The highly stereospecific anti-SN2′-type silacupration of 318, followed by the reduction of the nitrile group mediated by DIBAL-H directly yielded the key compound, (Ra)-allenylsilane aldehyde 52. Following that, key intermediate 320a, imino-allenesilane, was synthesized through the aza-Wittig reaction. Intermediate 320a underwent thermal imino-ene cyclization via transition states i-1 or i-2, followed by desilylation, leading to the formation of highly substituted cyclohexylamine 320c, which features an adjacent propargylic group in a syn configuration. Furthermore, the synthetic steps, including an intramolecular Heck coupling, an intramolecular Mitsunobu reaction and Saegusa-oxidation, were critical in constructing the core pentacyclic framework and introducing the necessary enone functionality (320c to 324). This synthetic protocol effectively yielded the known compounds 32679 and 328,80,81 which are important precursors for the total synthesis of (−)-brunsvigine 327 and (−)-pancracine 329, respectively. Moreover, by adjusting the sequence of the reduction and desilylation processes, access to (−)-coccinine 330 and (−)-montanine 331 was achieved in moderate to good yields.
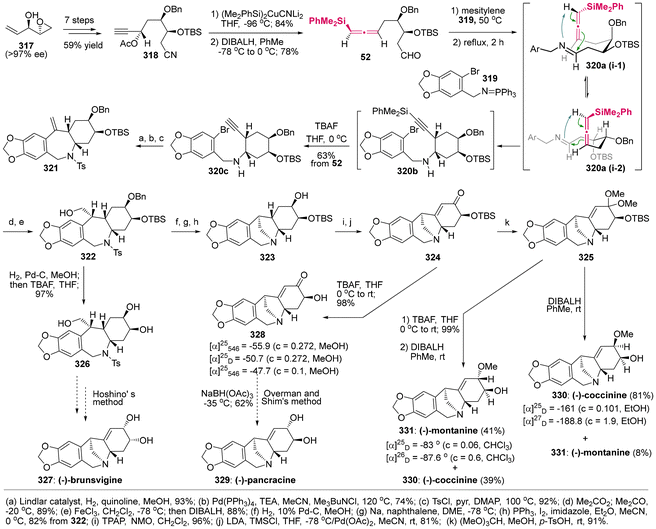 |
| | Scheme 61 Application of a stereospecific intramolecular allenylsilane imino-ene reaction for the enantioselective total synthesis of the 5,11-methanomorphanthridine alkaloids. | |
In 2009, Chemla et al. demonstrated a highly diastereoselective addition involving optically pure tert-butylsulfinylimine 332 and racemic allenylzinc reagent 56 (Scheme 62). Allenylzinc 56 serves as a nucleophilic partner and can be readily prepared from the 3-trimethylsilylpropargyl methyl ether.82 The stereospecific addition process entails kinetic resolution of racemic allenylzinc reagent 56, thereby providing access to the chiral propargylated products with an anti-selective 1,2-amino alcohol framework. By employing this addition and a following ring-closing metathesis as the key steps, these researchers successfully achieved the divergent asymmetric synthesis of four sphingoid-type alkaloids,83 specifically spisulosines ES271 (337) and ES285 (336)), sphinganine (338), N-deacetyl clavaminol H (339), as well as a significant advanced intermediate 342,84 which is the common key precursor for several non-peptide human NK-1 receptor antagonists, including (+)-CP-99994 (343), (+)-CP-122721 (344), (+)-LP-733060 (345) and the leading drug azaspirodecane (346). Additionally, utilizing a similar synthetic approach, (+)-6-epi-castanospermine (351) was prepared via the diastereoselective addition of chiral tert-butylsulfinylimine (S,S)-347 and allenylzinc 56 in a total of 8 steps with a total yield of 14% (Scheme 63).85
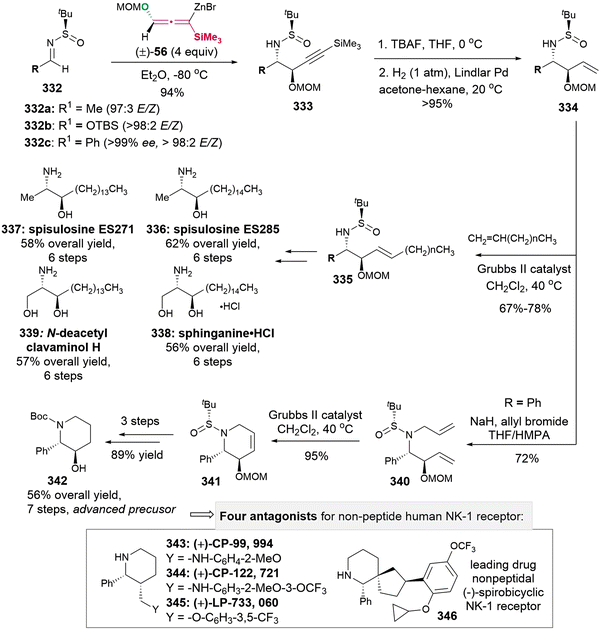 |
| | Scheme 62 Asymmetric synthesis of sphingoid-type alkaloids and the common advanced intermediate of several human NK-1 receptor antagonists. | |
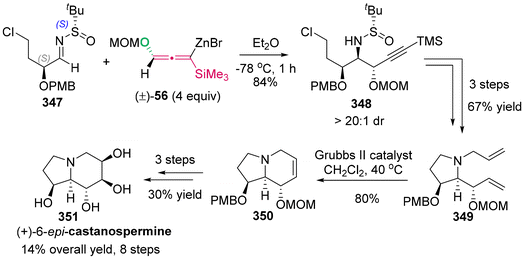 |
| | Scheme 63 Asymmetric total synthesis of (+)-6-epi-castanospermine. | |
3.3 Propargylation with carbonyl compounds
In 2004, Fleming successfully completed the total synthesis of 2-epi-ebelactone A (356) in a convergent synthetic strategy (Scheme 64).86 One of the key fragments 355 was prepared from the TiCl4-catalyzed diastereoselective addition of chiral allenylsilanes 5512a,87 to chiral aldehydes 352, followed by the hydrosilylation of alkyne and iodo-desilylation. In 2007, Mulzer and colleagues identified that this chiral fragment 355 exhibits potential for the synthesis of the antibiotic branimycin (357).88
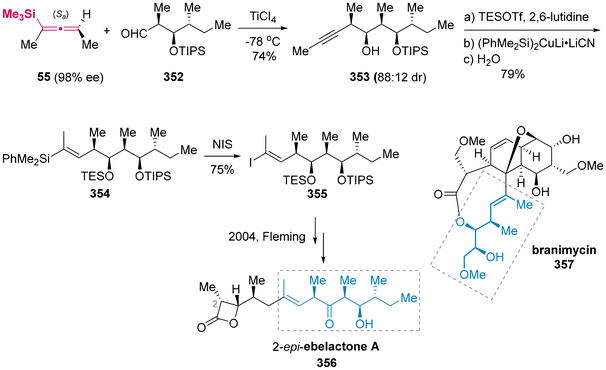 |
| | Scheme 64 Synthesis of the key fragments ebelactone A and branimycin. | |
NFAT-68 (364) is a highly effective immunosuppressant, exhibiting an IC50 value of less than 1 μg mL−1, with no apparent cytotoxic effects at this concentration. In 2016, the Panek group published a total five-step sequential methodology for the synthesis of both enantiomers of NFAT-68 (363 and 364).89 As demonstrated in Scheme 65, this process utilized the enantioenriched axially chiral allenylsilanes as starting materials, employing the stereospecific Prins reaction and titanium-mediated reductive coupling reaction as pivotal steps. The authors provided a detailed discussion on the asymmetric synthesis of ent-NFAT-68 (363) from axially chiral allenylsilanes (Sa)-55 (>98% ee). Chiral allenylsilanes (Sa)-55 were synthesized from the corresponding enantiopure (S)-but-3-yn-2-ol 358. Subsequently, the synthesis of syn-homopropargylic ethers 360 was achieved with a high degree of enantiospecificity through a three-component propargylation involving (Sa)-55, aldehydes 359 and (benzyloxy)trimethylsilane (TMSOBn). The nitro group on the aromatic ring was converted to N-acylated compound 361 using in situ-generated sulfurated borohydride (NaBH2S3) under acylation condition. Following this transformation, compound 361 underwent a titanium alkoxide-mediated alkyne–alkyne reductive coupling reaction through the proposed transition state, followed by a deprotection step to yield the final product ent-NFAT-68 (363). Utilizing the same protocol, chiral allenylsilanes (Ra)-55 were subjected to the five-step sequential procedure, delivering NFAT-68 (364) in an overall yield of 14%.
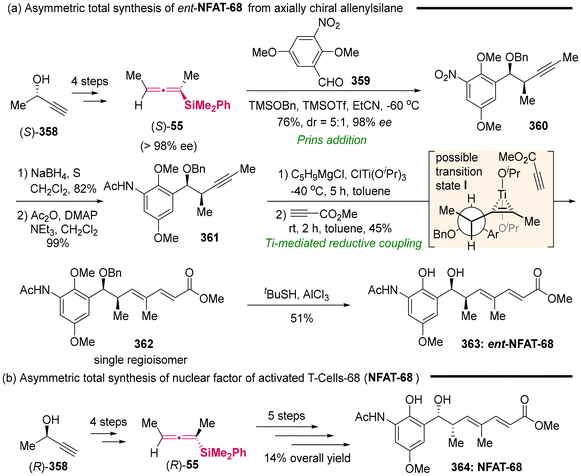 |
| | Scheme 65 Asymmetric synthesis of NFAT-68. | |
In 2004, Hegedus described a seven-step synthetic protocol for the synthesis of chiral 1-deoxy-D-galactohomonojirimycin (368).90 As illustrated in Scheme 66, a pivotal aspect of this protocol involves a Lewis acid-catalyzed reaction between optically pure allenylstannane 6191 and L-lactate-derived aldehyde 365, which produced the syn-amino alcohol with a high degree of diastereoselectivity.
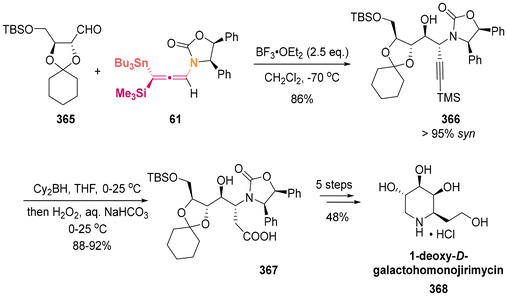 |
| | Scheme 66 Asymmetric synthesis of 1-deoxy-D-galactohomonojirimycin. | |
In 1999, Marshall et al. discovered reactive axially chiral allenylzinc reagent 59, which was generated in situ from enantioenriched propargylic mesylates and diethyl zinc in the presence of a Pd(0)-phosphine catalyst (Scheme 67: Solution A).92 Chiral allenylzinc reagent 59 can perform the SE2′ additions with various aldehydes, effectively providing access to the corresponding anti-homopropargylic alcohols. Given the efficiency in synthesizing anti-homopropargylic alcohols, the authors subsequently employed a sequential addition of allenylstannanes and allenylzinc to the corresponding chiral aldehydes to construct fragment 372.93 Ultimately, the sp2–sp3 Suzuki coupling of fragment 373, as the key step, enabled the total synthesis of (−)-callystatin A (374).
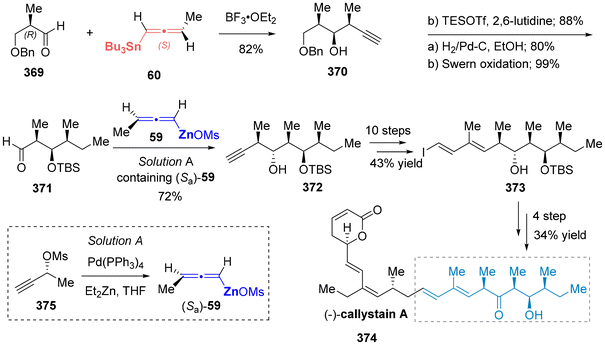 |
| | Scheme 67 Asymmetric synthesis of (−)-callystatin A. | |
In a subsequent study, Marshall et al. discovered that using indium iodide in place of diethyl zinc results in the formation of enantioenriched allenylindium reagents 58, which also are derived from chiral propargylic mesylates 381 with the Pd(0)-phosphine complex catalyst (Scheme 68b).94 Allenylindium 58 exhibits reactivity comparable to that of allenylzinc, readily performing the highly syn-selective addition with chiral aldehydes. Based on this reactivity, as illustrated in Scheme 68a, the researchers developed a sequential approach involving two propargylation reactions of (Sa)-allenylstannanes (60) and (Ra)-allenylindium (58) to produce the desired enantiomeric precursor 379 for (−)-membrenone C (380).95 Conversely, the reverse sequence of propargylation, which involved the use of (Sa)-allenylindium (58) followed by compound (Ra)-allenylstannanes (57: enantiomer of 60), yielded the opposite enantiomer 379 for (+)-membrenone C (380) (Scheme 68c). Both asymmetric syntheses commenced with the same (S)-propanal 376. Utilizing the suitable sequence of propargylations, the total synthesis of both enantiomers of membrenone C was efficiently completed through subsequent bis-intramolecular hydrosilylation–oxidation and a bis-intramolecular aldol reaction as two additional key steps.
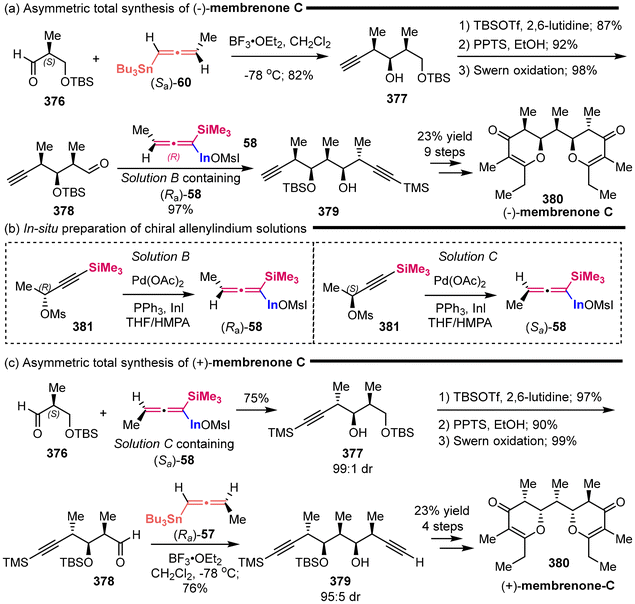 |
| | Scheme 68 Asymmetric synthesis of (−)-membrenone C and (+)-membrenone C. | |
Besides the aforementioned important aldehydes, ketones have also been proven to be effective partners to react with allenylmetal reagent. In 2007, Caddick documented the synthesis of epoxydiynes 384 through the addition of allenylzinc 382 to propargylic ketone 381, which was followed by KF-promoted epoxidation. As illustrated in Scheme 69, epoxydiyne unit 384 is a key fragment of the natural neocarzinostatin chromophore.96
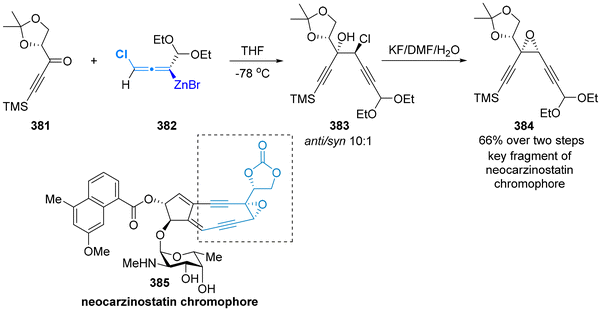 |
| | Scheme 69 Asymmetric synthesis of the epoxydiyne fragment for neocarzinostatin chromophore. | |
3.4 Cyclization with alkenes
In 1988, the Okamura group established a concise and efficient method for the synthesis of (+)-sterpurene (392).97 As illustrated in Scheme 70, this synthesis begins with the coupling of chiral compounds (−)-387 and 386, resulting in the formation of diene propargyl alcohol 388. Subsequently, the alcohol is treated with benzenesulfenyl chloride to yield sulfoxides 389. These sulfoxides readily undergo [2,3]-sigmatropic rearrangement under identical conditions at elevated reaction temperatures, leading to the preparation of vinylallene sulfoxides 65. Following this, an intramolecular Diels–Alder reaction was conducted, leading to the formation of tricyclic compound 390 with complete stereoselectivity. Finally, compound 391 can be readily converted into (+)-sterpurene (392).
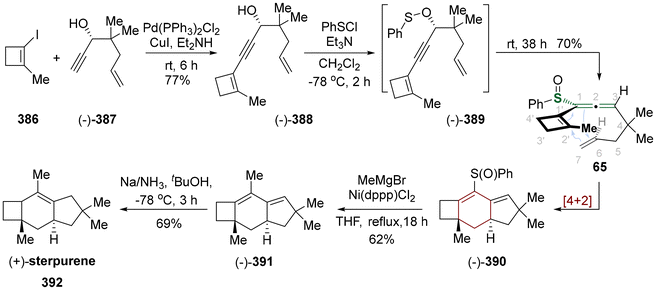 |
| | Scheme 70 Enantioselective central-axial-central chiral element transfer process leading to a concise synthesis of (+)-sterpurene: intramolecular Diels–Alder reactions of vinylallene sulfoxides. | |
In 2024, Li et al. introduced an elegant intramolecular [3 + 2]-annulation involving an unsymmetric allenylsilane-ene (type II),98 which efficiently constructed the synthetically important and challenging bicyclo[3.2.1] framework (Scheme 71a). This protocol displayed a broad substrate scope, excellent diastereoselectivity, and high stereospecificity together with complete axial-to-point chirality transfer from the enantioenriched axially chiral allenylsilanes. The proposed mechanism is illustrated in Scheme 71b. The [3 + 2]-annulation process mainly involved the Lewis acid-promoted conjugate addition of allenylsilane to enones (i-1 to i-2), the β-effect of silicon on the carbocation (i-2), and intramolecular SN2-type nucleophilic attack (i-3 to i-4). Notably, a cascade DMP-mediated oxidation/[3 + 2] procedure for compound 397 could yield bicyclo[3.2.1] compound 398, even in the absence of a Lewis acid.
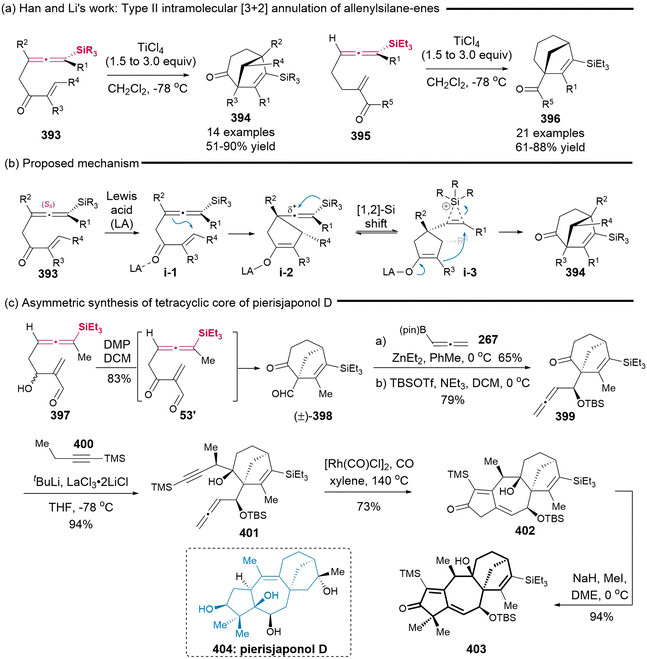 |
| | Scheme 71 Diastereoselective type II intramolecular [3 + 2] annulation of unsymmetric allenylsilane-ene. | |
The bicyclo[3.2.1] scaffold is ubiquitous in numerous natural products and biologically active synthetic molecules, such as pierisjaponol D (404) and strepsesquitriol (411 and 413). Employing intramolecular [3 + 2] annulation as a pivotal step, the authors successfully synthesized the tetracyclic core of pierisjaponol D (404) in six steps, which included several critical processes, such as sequential 1,2-addition, the Pauson–Khand reaction and dimethylation (Scheme 71c). However, the total synthesis of pierisjaponol D (404) was not elaborated.
Instead, the authors accomplished the first asymmetric total synthesis of both (+)-strepsesquitriol (411) and (−)-strepsesquitriol (413) through the utilization of chiral bicyclo[3.2.1] compound 405, which is a single enantiomer of 398 (Scheme 72).98 This chiral compound can be generated from the corresponding enantiomers of either axially chiral allenylsilane 397 or 53. This protocol featured several essential steps, including the sequential chemo- and diastereo-selective 1,2-addition of Grignard reagents and olefin cross-metathesis reaction to complete the three-ring core of strepsesquitriol. Subsequently, the key sequential processes involved SeO2-mediated allylic oxidative rearrangement, O3-mediated oxidative cleavage of the exocyclic olefin motif, and SmI2-mediated C–O bond cleavage to introduce the ketone functionality group onto the bicyclic scaffold, followed by Cα-dimethylation, ketone addition and deprotection to afford the corresponding enantiomer of strepsesquitriol.
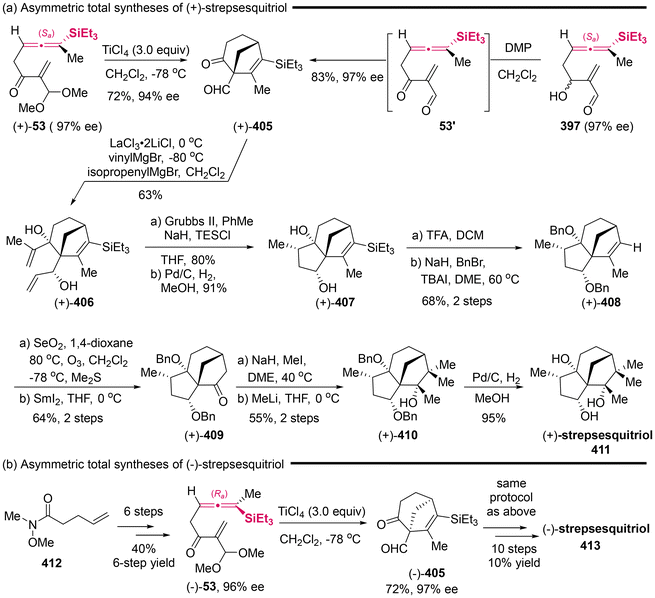 |
| | Scheme 72 Asymmetric total synthesis of (+)- and (−)-strepsesquitriol. | |
3.5 Cross-coupling reaction
Compared to the highly predictable reactivity observed in palladium-catalyzed cross-coupling reactions involving aryl, alkenyl, or alkyl halides, the reactivity and stereoselectivity of axially chiral haloallenes in the catalytic variant remain intricate. The early studies conducted by Vermeer indicated that the stereo-retention and stereo-inversion of palladium-catalyzed cross-coupling reactions involving the enantiopure halogenated allenes and organozinc reagents were determined by the type of halogen atom present.99 Specifically, brominated allenes resulted in stereochemical inversion, while iodo-substituted allenes exhibited stereochemical retention. In 2005, de Lera's group observed analogous stereochemical outcomes in the Still cross-coupling reaction of chiral enantiopure brominated allenes 67 with alkenylstannanes 418.100 They employed the stereo-inversion coupling reaction as the cornerstone, followed by Julia condensation and two additional Still-coupling procedures to complete the synthesis of 6′-epi-peridinin (426) (Scheme 73). This compound was isolated from the iodine-assisted photoisomerization of natural peridinin, which is originally isolated from the planktonic algae Dinoflagellates and serves as a significant carotenoid involved in photosynthesis.101 Furthermore, it has demonstrated notable antitumor and antilipoperoxidant activities. Structurally, 6′-epi-peridinin features a γ-alkylidene-butenolide ring and a conjugated polyene chain, along with a sensitive and synthetically challenging chiral allene axis.
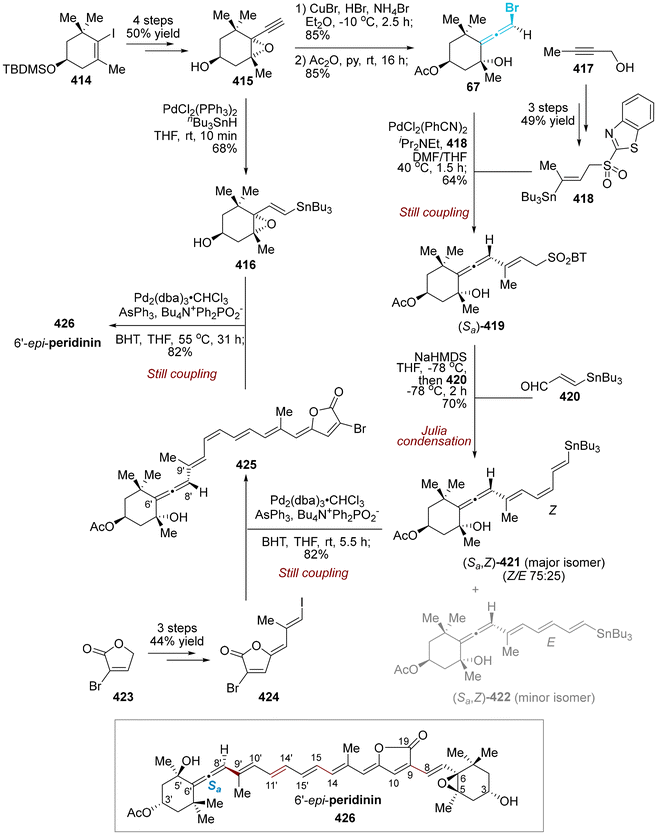 |
| | Scheme 73 Asymmetric total synthesis of 6′-epi-peridinin. | |
To elucidate the intriguing stereo-inversion observed in the coupling reaction of halogenated allenes, a series of mechanistic studies was performed.102 The experimental findings revealed that various factors, such as the type of halogen, the presence of a free hydroxyl group in the allene substrate, type of palladium catalyst, the coordinating ligand and the solvent, significantly influenced the reaction stereoselectivity. As illustrated in Scheme 74, two competing reaction pathways were identified. The direct oxidative addition of a carbon–halogen bond (C–X) to the palladium complex leads to the retention of stereochemistry, whereas stereo-chemical inversion occurs through a tandem process involving an indirect anti-SN2′-type oxidative addition, followed by a suprafacial [1,3]-shift of propargyl to the allenyl palladium species.
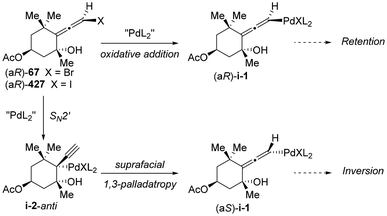 |
| | Scheme 74 Proposed mechanism for stereochemistry. | |
The Suzuki–Miyaura cross-coupling reaction has experienced significant advancements over the past few decades, primarily due to its high synthetic efficiency and extensive compatibility with various substrates, including organic halides and readily accessible organoboronic acid derivatives. A notable development in this field was introduced by Burke, who proposed an iterative Suzuki cross-coupling strategy. The essence of this strategy generally involves the use of two distinct boron groups, i.e., one that is masked and temporarily inactive, and another that is unmasked and capable of preferentially participating in the desired coupling reaction. In 2010, Burke successfully employed this iterative Suzuki cross-coupling strategy in a convergent approach to achieve the first fully stereo-controlled synthesis of (−)-peridinin 440 (Scheme 75).103 The precise stereocontrol of the allenic axial chirality in this synthesis is attributed to the stereoretentive Suzuki–Miyaura coupling of chiral iodo-substituted allenes 68, which, despite demonstrating good stereo-retention, proved to be ineffective and complex when applied in the Still coupling with stannanes 418.100
 |
| | Scheme 75 Stereoretentive Suzuki–Miyaura coupling of haloallenes enables fully stereo-controlled access to (−)-peridinin. | |
Pachastrissamine (445), a marine natural product, was synthesized by the group of Fujii and Ohno in a total of 11 steps with an overall yield of 11% (Scheme 76).104 The pivotal step of this synthesis involved the development of a novel ring-construction and stereoselective functionalization cascade, which was facilitated by the palladium(0)-catalyzed bis-cyclization of optically pure bromoallenes 66 containing two nucleophilic hydroxy and benzamide groups. This bis-cyclization occurs in a highly selective sequence, efficiently constructing the functionalized tetrahydrofuran ring equipped with a vicinal syn-amino alcohol motif.
 |
| | Scheme 76 Ring-construction/stereoselective functionalization cascade: total synthesis of pachastrissamine. | |
3.6 Epoxidation-mediated transformation
In 2009, Williams developed a highly effective stereospecific double epoxidation method involving enantiopure chiral allenylsilanes (Scheme 77), yielding spirodiepoxides 446 with good diastereoselectivity. Based on this methodology, a sequential regioselective eliminative opening was employed to produce chiral diols 447 in the presence of organolithium reagents. Utilizing chiral allenylsilanes 55, the one-pot sequential process of epoxidation, eliminative opening, and desilylation resulted in chiral diols 448 with a yield of 69%. Following this, an additional sequence involving olefin cross-metathesis and a Wittig reaction was developed, culminating in the first total synthesis of the fungal metabolite epi-citreodiol (450).105
 |
| | Scheme 77 Synthesis of epi-citreodiol. | |
The Nazarov cyclization of allene ethers represents a powerful electrocyclization strategy for constructing valuable functionalized cyclopentanones.106 In 2009, Frontier described the novel oxidation-initiated Nazarov cyclization of the stannyl alkoxyallene 62, which enabled access to the synthetically important cis-benzofuran-fused cyclopentanones 453 (Scheme 78).107 This transformation served as a crucial step in the total synthesis of the natural products aglafolin (455) and rocaglamide (457), accomplished in 11 and 13 synthetic steps, respectively, starting from the readily accessible benzofuranone 451. The initial objective of this study was to achieve a seemingly more efficient Nazarov cyclization using Lewis acids as catalysts (Scheme 78b). However, all attempts were unsuccessful and no desired cyclized product was obtained due to the polarity mismatch. This matter was adeptly addressed by the oxidation-initiated Nazarov cyclization of allene esters. It is regrettable that the enantiospecific cyclization variant involving chiral stannyl alkoxyallene 62 has not yet been discussed, which may provide an opportunity access the chiral aglafolin (455) and rocaglamide (457).
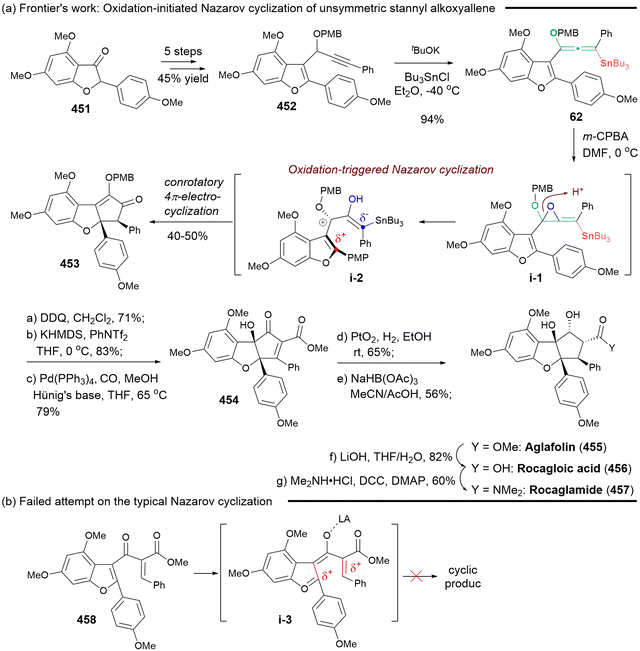 |
| | Scheme 78 Total synthesis of (±)-rocaglamide via oxidation-initiated Nazarov cyclization. | |
3.7 Isomerization-mediated transformation
Salinomycin (464), isolated from Streptomyces albus, is widely recognized as a commercial anti-coccidial drug and has demonstrated a promising inhibitory effect against various types of cancer stem cells. In 1998, Kocieński presented the elegant asymmetric total synthesis of salinomycin using a convergent strategy that incorporated three chiral fragments (Scheme 79).108 The synthesis of one of these fragments, 463, commenced with the preparation of allenol ether 63, which served as a crucial intermediate. Allenol ether 63 was synthesized through the base-promoted isomerization of chiral alkyne 461. Following its formation, allenol ether 63 was subjected to n-BuLi, leading to its coupling with lactones 462, followed by an acid-promoted isomerization and spirocyclization, ultimately resulting in the formation of dispiroacetal 463. Dispiroacetal 463 underwent an additional 13-step process, culminating in the synthesis of salinomycin (464). Due to the susceptibility of salinomycin to decomposition during chromatographic purification, the authors finally isolated salinomycin methyl ester 465 through treatment with diazomethane.
 |
| | Scheme 79 Synthesis of salinomycin. | |
4. Summary and outlook
In recent years, significant breakthroughs and progress have been made in the asymmetric synthesis of heteroatom-substituted axially chiral allenes, especially through catalytic enantioselective approaches. A wide variety of significant axially chiral allenes, which are substituted with diverse heteroatomic groups incorporating boron (B), nitrogen (N), oxygen (O), fluorine (F), bromine (Br), chlorine (Cl), sulfur (S), phosphorus (P), silicon (Si), and stannum (Sn), has been synthesized through catalytic asymmetric synthetic methods. The majority of these asymmetric methods are catalyzed by either transition-metal catalysts or organocatalysts alone. Although a limited number of enzyme-catalyzed methods have been developed, which involve the kinetic resolution of racemic heteroatom-substituted allene substrates, their inherent compatibility limitation and partially moderate stereoselectivity cannot satisfy the demands of synthetic applications. Synergistic dual catalysis is recognized as a highly promising approach for enhancing and expanding chemical reactions, but it has only been documented in a single report. It involved dual photocatalysis and transition metal catalysis for the synthesis of silyl-substituted allenols. The exploration of dual catalysis in the asymmetric synthesis of heteroatom-substituted axially chiral allenes is both highly desirable and remains an area for further investigation.
Compared to the development of general chiral allenes, the catalytic asymmetric synthesis of heteroatom-substituted axially chiral allenes remains in its infancy, with numerous unexplored areas and persistent limitations. For instance, although iodo-substituted allenes have been proven to be important building blocks, there is no report detailing their synthesis through a catalytic asymmetric protocol. Also, although over 50 brominated allenes natural products have been isolated, only one report exists on organocatalyzed bromolactonization that facilitates the synthesis of axially chiral brominated allenes, which is difficult for application in the synthesis of complex natural products. Furthermore, the exploration of axially chiral allenes substituted with other more significant heteroatoms, such as selenium (Se), germanium (Ge), arsenic (As), and tellurium (Te), remains largely unaddressed. Many synthetic methodologies tend to encounter competitive regioselective pathways, wherein the desired allenylation process contends with the side propargylation process. The existing literature on heteroatom-substituted allenes predominantly emphasizes peripheral modifications, while the de novo skeletal editing of allenes in an enantioselective manner, such as through cross-metathesis and Witting reaction, presents a formidable challenge and has been infrequently investigated. Besides the constant catalysis systems available for the analogous transformations, the type of accessible transformations and novel substrates for individual heteroatom-substituted allenes are notably limited. These limitations further restrict the structural diversity of the resulting heteroatom-substituted allene products.
Alternatively, we delineated the proposed mechanisms of several representative methods, with the intention of providing guidance for the design of more efficient or innovative strategies for the asymmetric synthesis of heteroatom-substituted allenes. The introduction of heteroatoms endowed these allenes with richer reactivity and broader synthetic applications. Numerous significant and structurally complex molecules, as well as natural products, have been synthesized by using enantioenriched heteroatom-substituted allenes as key intermediate materials. In these illustrated synthetic applications, axially chiral heteroatom-substituted allenes exhibit unique reactivity and stereospecificity, which are distinct from the conventional chiral heteroatom compounds and allene compounds. Moreover, we anticipate that the design and synthesis of synthetically important and structurally diverse axially chiral heteroatom-substituted allenes will be achieved through simple or innovative asymmetric reactions, thereby facilitating the exploration of their unprecedented reactivity and enhancing their synthetic utility. Given the promising bioactivities associated with certain reported axially chiral heteroatom-substituted allenes, future biological applications are also highly anticipated.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22171240, 22371248, 22261053), Yunnan Fundamental Research Projects (202301AS070021, 202101AT070156), Project of Innovative Research Team of Yunnan Province (202405AS350010), and Yunnan Provincial Science and Technology Project at Southwest United Graduate School (202302AP370004). We thank Advanced Analysis and Measurement Center of Yunnan University for the sample testing service.
References
- Selected references:
(a) H. F. Schuster and G. M. Coppola, Allenes in Organic Synthesis, Wiley, New York, 1984 Search PubMed;
(b) B. S. Burton and H. v. Pechmann, Ueber die Einwirkung von Chlorphosphor auf Acetondicarbonsäureäther, Ber. Dtsch. Chem. Ges., 1887, 20, 145–149 CrossRef;
(c) E. R. H. Jones, G. H. Mansfield and M. L. H. Whiting, Researches on Acetylenic Compounds. Part XLVII, The Prototropic Rearrangements of Some Acetylenic Dicarboxylic Acids, J. Chem. Soc., 1954, 3208–3212 RSC.
-
(a) A. Hoffmann-Röder and N. Krause, Synthesis and Properties of Allenic Natural Products and Pharmaceuticals, Angew. Chem., Int. Ed., 2004, 43, 1196–1216 CrossRef;
(b) S. Yu and S. Ma, Allenes in Catalytic Asymmetric Synthesis and Natural Product Syntheses, Angew. Chem., Int. Ed., 2012, 51, 3074–3112 CrossRef CAS;
(c) H. Wu, Z. Zheng, K. Zhang, J. Kajanus, M. J. Johansson, A. Córdova and J.-E. Bäckvall, Heterogeneous Copper-Catalyzed Cross-Coupling for Sustainable Synthesis of Chiral Allenes: Application to the Synthesis of Allenic Natural Products, Angew. Chem., Int. Ed., 2023, 62, e202314512 CrossRef CAS.
-
(a) V. M. Dembitsky and T. Maoka, Allenic and Cumulenic Lipids, Prog. Lipid Res., 2007, 46, 328–375 CrossRef CAS;
(b) P. Rivera-Fuentes and F. Diederich, Allenes in Molecular Materials, Angew. Chem., Int. Ed., 2012, 51, 2818–2828 (Angew. Chem., 2012, 124, 2872–2882) CrossRef CAS PubMed.
-
(a) S. Ma, Some Typical Advances in the Synthetic Applications of Allenes, Chem. Rev., 2005, 105, 2829–2871 CrossRef PubMed;
(b) R. K. Neff and D. E. Frantz, Recent Applications of Chiral Allenes in Axial-to-Central Chirality Transfer Reactions, Tetrahedron, 2005, 71, 7–18 CrossRef;
(c) L. Liu, R. M. Ward and J. M. Schomaker, Mechanistic Aspects and Synthetic Applications of Radical Additions to Allenes, Chem. Rev., 2019, 119, 12422–12490 CrossRef CAS;
(d) R. Guo and M. K. Brown, Lewis Acid-Promoted [2 + 2] Cycloadditions of Allenes and Ketenes: Versatile Methods for Natural Product Synthesis, Acc. Chem. Res., 2023, 56, 2253–2264 CrossRef CAS.
-
(a) P. Maitland and W. H. Mills, Experimental Demonstration of the Allene Asymmetry, Nature, 1935, 135, 994 CrossRef CAS;
(b) M. Ogasawara, Catalytic Enantioselective Synthesis of Axially Chiral Allenes, Tetrahedron: Asymmetry, 2009, 20, 259–271 CrossRef CAS;
(c) R. K. Neff and D. E. Frantz, Recent Advances in the Catalytic Syntheses of Allenes: A Critical Assessment, ACS Catal., 2014, 4, 519–528 CrossRef CAS;
(d) S. Ye and S. Ma, Conquering Three-Carbon Axial Chirality of Allenes, Org. Chem. Front., 2014, 1, 1210–1224 RSC;
(e) W. Xiao and J. Wu, Recent Advances in the Metal-Catalyzed Asymmetric Synthesis of Chiral Allenes, Org. Chem. Front., 2022, 9, 5053–5073 RSC;
(f) T. T. Nguyen, Organocatalytic synthesis of axially chiral tetrasubstituted allenes, Org. Biomol. Chem., 2023, 21, 252–272 RSC.
-
(a) X. Pu, X. Qi and J. M. Ready, Allenes in Asymmetric Catalysis: Asymmetric Ring Opening of meso-Epoxides Catalyzed by Allene-Containing Phosphine Oxides, J. Am. Chem. Soc., 2009, 131, 10364–10365 CrossRef CAS;
(b) F. Cai, X. Pu, X. Qi, V. Lynch, A. Radha and J. M. Ready, Chiral Allene-Containing Phosphines in Asymmetric Catalysis, J. Am. Chem. Soc., 2011, 133, 18066–18069 CrossRef CAS PubMed.
-
(a) K. Kokkotou, E. Ioannou, M. Nomikou, F. Pitterl, A. Vonaparti, E. Siapi, M. Zervou and V. Roussis, An Integrated Approach using UHPLC-PDA-HRMS and 2D HSQC NMR for the Metabolic Profiling of the Eed Alga Laurencia: Dereplication and Tracing of Natural Products, Phytochemistry, 2014, 108, 208–219 CrossRef CAS PubMed;
(b) S. Sutour, B. Therrien, S. H. von Reuss and F. Tomi, Halogenated C(15) Acetogenin Analogues of Obtusallene III from a Laurenciella sp. Collected in Corsica, J. Nat. Prod., 2018, 81, 279–285 CrossRef CAS;
(c) A. Koutsaviti, M. G. Daskalaki, S. Agusti, S. C. Kampranis, C. Tsatsanis, C. M. Duarte, V. Roussis and E. Ioannou, Thuwalallenes A-E and Thuwalenynes A-C: New C(15) Acetogenins with Anti-Inflammatory Activity from a Saudi Arabian Red Sea Laurencia sp, Mar. Drugs, 2019, 17, 644 CrossRef CAS PubMed;
(d) S. Perdikaris, A. Mangoni, L. Grauso, P. Papazafiri, V. Roussis and E. Ioannou, Vagiallene, a Rearranged C(15) Acetogenin from Laurencia Obtusa, Org. Lett., 2019, 21, 3183–3186 CrossRef CAS PubMed;
(e) M. Harizani, D. I. Diakaki, S. Perdikaris, V. Roussis and E. Ioannou, New C(15) Acetogenins from Two Species of Laurencia from the Aegean Sea, Molecules, 2022, 27, 1866 CrossRef CAS PubMed.
- Method I:
(a) T. Saitoh, T. Suzuki, M. Sugimoto, H. Hagiwara and T. Hoshi, Total Synthesis of (+)-Laurallene, Tetrahedron Lett., 2003, 44, 3175–3178 CrossRef CAS;
(b) J. Boukouvalas, M. Pouliot, J. Robichaud, S. MacNeil and V. Snieckus, Asymmetric Total Synthesis of (-)-Panacene and Correction of Its Relative Configuration, Org. Lett., 2006, 8, 3597–3599 CrossRef CAS PubMed;
(c) J. Wang and B. L. Pagenkopf, First Total Synthesis and Structural Reassignment of (-)-Aplysiallene, Org. Lett., 2007, 9, 3703–3706 CrossRef CAS;
(d) W. Jeong, M. J. Kim, H. Kim, S. Kim, D. Kim and K. J. Shin, Substrate-Controlled Asymmetric Total Synthesis and Structure Revision of (+)-Itomanallene A, Angew. Chem., Int. Ed., 2010, 49, 752–756 CrossRef CAS PubMed;
(e) M. J. Kim, T. I. Sohn, D. Kim and R. S. Paton, Concise Substrate-Controlled Asymmetric Total Syntheses of Dioxabicyclic Marine Natural Products with 2,10-Dioxabicyclo-[7.3.0]dodecene and 2,9-Dioxabicyclo[6.3.0]undecene Skeletons, J. Am. Chem. Soc., 2012, 134, 20178–20188 CrossRef CAS PubMed;
(f) Y. Yoshimitsu, S. Inuki, S. Oishi, N. Fujii and H. Ohno, Palladium-Catalyzed Medium-Ring Formation for Construction of the Core Structure of Laurencia Oxacycles: Synthetic Study of Laurendecumallene B, Org. Lett., 2013, 15, 3046–3049 CrossRef CAS;
(g) T. Umezawa, Y. Oguri, H. Matsuura, S. Yamazaki, M. Suzuki, E. Yoshimura, T. Furuta, Y. Nogata, Y. Serisawa, K. Matsuyama-Serisawa, T. Abe, F. Matsuda, M. Suzuki and T. Okino, Omaezallene from Red Alga Laurencia sp: Structure Elucidation, Total Synthesis, and Antifouling Activity, Angew. Chem., Int. Ed., 2014, 53, 3909–3912 CrossRef CAS;
(h) T. I. Sohn, D. Kim and R. S. Paton, Substrate-Controlled Asymmetric Total Syntheses of Microcladallenes A, B, and C Based on the Proposed Structures, Chem. – Eur. J., 2015, 21, 15988–15997 CrossRef CAS PubMed;
(i) J. Clarke, K. J. Bonney, M. Yaqoob, S. Solanki, H. S. Rzepa, A. J. White, D. S. Millan and D. C. Braddock, Epimeric Face-Selective Oxidations and Diastereodivergent Transannular Oxonium Ion Formation Fragmentations: Computational Modeling and Total Syntheses of 12-Epoxyobtusallene IV, 12-Epoxyobtusallene II, Obtusallene X, Marilzabicycloallene C, and Marilzabicycloallene D, J. Org. Chem., 2016, 81, 9539–9552 CrossRef CAS PubMed;
(j) F. Yoshimura, T. Okada and K. Tanino, Asymmetric Total Synthesis of Laurallene, Org. Lett., 2019, 21, 559–562 CrossRef CAS;
(k) I. Shin, H. Jang, S. Y. Kwak, Y. Park, D. Lee, H. Kim and D. Kim, Highly Stereodivergent Construction of a C(2)-Symmetric cis, cis- and trans, trans-2,6-Dioxabicyclo[3.3.0]octane Framework by Double Intramolecular Amide Enolate Alkylation: Total Synthesis of (+)-Laurenidificin and (+)-Aplysiallene, Org. Lett., 2022, 24, 8780–8785 CrossRef CAS PubMed;
(l) R. A. Fernandes, A. Kumar and R. S. Pathare, Asymmetric Total Synthesis
of (+)-Dihydroitomanallene B and Formal Synthesis of (-)-Kumausallene, Chem. Commun., 2022, 58, 11921–11924 RSC Method II:
(m) J. Lshihara, Y. Shimada, N. Kanoh, Y. Takasugi, A. Fukuzawa and A. Murai, Conversion of Prelaureatin into Laurallene, a Bromo-Allene Compound, by Enzymatic and Chemical Bromo-Etherification Reactions, Tetrahedron, 1997, 53, 8371–8382 CrossRef;
(n) P. Evans, V. S. Murthy, J. D. Roseman and A. L. Rheingold, Enantioselective Total Synthesis of the Nonisoprenoid Sesquiterpene (-)-Kumausallene, Angew. Chem., Int. Ed., 1999, 38, 3175–3177 CrossRef CAS;
(o) D. C. Braddock, R. Bhuva, D. S. Millan, Y. Perez-Fuertes, C. A. Roberts, R. N. Sheppard, S. Solanki, E. S. E. Stokes and A. J. P. White, A Biosynthetically-Inspired Synthesis of the Tetrahydrofuran Core of Obtusallenes II and IV, Org. Lett., 2007, 9, 445–448 CrossRef CAS;
(p) J. B. Werness and W. Tang, Stereoselective Total Synthesis of (-)-Kumausallene, Org. Lett., 2011, 13, 3664–3666 CrossRef CAS;
(q) M. Yamakawa, T. Kurachi, Y. Yoshikawa, M. Arisawa, Y. Okino, K. Suzuki and H. Fujioka, Stereoselective Construction of 2,7-Disubstituted Fused-Bis Tetrahydrofuran Skeletons: Biomimetic-Type Synthesis and Biological Evaluation of (±)- and (-)-Aplysiallene and Their Derivatives, J. Org. Chem., 2015, 80, 10261–10277 CrossRef CAS;
(r) N. Alnafta, J. P. Schmidt, C. L. Nesbitt and C. S. P. McErlean, Total Synthesis of (+)-Panacene, Org. Lett., 2016, 18, 6520–6522 CrossRef CAS;
(s) Y.-A. Zhang, N. Yaw and S. A. Snyder, General Synthetic Approach for the Laurencia Family of Natural Products Empowered by a Potentially Biomimetic Ring Expansion, J. Am. Chem. Soc., 2019, 141, 7776–7788 CrossRef CAS;
(t) C. A. Taylor, Y.-A. Zhang and S. A. Snyder, The Enantioselective Total Synthesis of Laurendecumallene B, Chem. Sci., 2020, 11, 3036–3041 RSC.
- Review on the allenylboron compounds: S. Manna, K. K. Das, D. Aich and S. Panda, Synthesis and Reactivity of Allenylboron Compounds, Adv. Synth. Catal., 2021, 363, 2444–2463 CrossRef CAS.
- Review on the allenylsilanes: M. J. Curtis-Long and Y. Aye, Vinyl-, Propargyl-, and Allenylsilicon Reagents in Asymmetric Synthesis: A Relatively Untapped Resource of Environmentally Benign Reagents, Chem. – Eur. J., 2009, 15, 5402–5416 CrossRef CAS PubMed.
- Selected examples on the asymmetric synthesis of alleneylsilanes via method A, please see:
(a) I. Fleming, K. Takaki and A. P. Thomas, Synthesis of Allenylsilanes frorn Propargyl Carbarnates, J. Chem. Soc., Perkin Trans. 1, 1987, 2269–2273 RSC;
(b) J. A. Marshall and K. Maxson, Stereoselective Synthesis of Stereotriad Subunits of Polyketides through Additions of Nonracemic Allenylsilanes to (R)- and (S)-2-Methyl-3-oxygenated Propanals, J. Org. Chem., 2000, 65, 630–633 CrossRef CAS PubMed;
(c) M. Shimizu, T. Kurahashi, H. Kitagawa and T. Hiyama, gem-Silylborylation of an sp Carbon: Novel Synthesis of 1-Boryl-1-silylallenes, Org. Lett., 2003, 5, 225–227 CrossRef CAS PubMed;
(d) H. Ohmiya, H. Ito and M. Sawamura, General and Functional Group-Tolerable Approach to Allenylsilanes by Rhodium-Catalyzed Coupling between Propargylic Carbonates and a Silylboronate, Org. Lett., 2009, 11, 5618–5620 CrossRef CAS;
(e) D. J. Vyas, C. K. Hazra and M. Oestreich, Copper(I)-Catalyzed Regioselective Propargylic Substitution Involving Si-B Bond Activation, Org. Lett., 2011, 13, 4462–4465 CrossRef CAS;
(f) K. Guo, Q. Zeng, A. Villar-Yanez, C. Bo and A. W. Kleij, Ni-Catalyzed Decarboxylative Silylation of Alkynyl Carbonates: Access to Chiral Allenes via Enantiospecific Conversions, Org. Lett., 2022, 24, 637–641 CrossRef CAS PubMed.
- Selected examples on the asymmetric synthesis of alleneylsilanes via method B, please see:
(a) M. J. C. Buckle and I. Fleming, Accurate Determination of the Extent to which the SE2′ Reactions of an Allefiylsilaxik are Stereospecifically anti, Tetrahedron Lett., 1993, 34, 2383–2386 CAS;
(b) M. Suginome, A. Matsumoto and Y. Ito, Palladium-Catalyzed Intramolecular Bis-Silylation of Propargylic Alcohols: A New Stereospecific Access to Chiral Allenylsilanes, J. Org. Chem., 1996, 61, 4884–4885 CrossRef CAS;
(c) I. Fleming and K. L. C. Pang, Determination of the Extent to which an SE2′ Reaction of a Propargylsilane is anti, Tetrahedron Lett., 2002, 43, 5985–5988 CrossRef CAS;
(d) R. A. Brawn and J. S. Panek, Preparation and Use of Enantioenriched Allenylsilanes for the Stereoselective Synthesis of Homopropargylic Ethers, Org. Lett., 2007, 9, 2689–2692 CrossRef CAS PubMed;
(e) M. R. Uehling, S. T. Marionni and G. Lalic, Asymmetric Synthesis of Trisubstituted Allenes: Copper-Catalyzed Alkylation and Arylation of Propargylic Phosphates, Org. Lett., 2012, 14, 362–365 CrossRef CAS PubMed;
(f) U. Yokobori, H. Ohmiya and M. Sawamur, Synthesis of Allenylsilanes through Copper-Catalyzed γ-Selective Coupling between γ-Silylated Propargylic Phosphates and Alkylboranes, Organometallics, 2012, 31, 7909–7913 CrossRef CAS;
(g) L.-L. Yang, J. Ouyang, H.-N. Zou, S.-F. Zhu and Q.-L. Zhou, Enantioselective Insertion of Alkynyl Carbenes into Si-H Bonds: An Efficient Access to Chiral Propargylsilanes and Allenylsilanes, J. Am. Chem. Soc., 2021, 143, 6401–6406 CrossRef CAS PubMed.
- A. Tillack, D. Michalik, C. Koy and M. Michalik, Catalytic Asymmetric Hydrosilylation of Butadiynes: A New Synthesis of Optically Active Allenes, Tetrahedron Lett., 1999, 40, 6567–6568 CrossRef CAS.
-
(a) J. W. Han, N. Tokunaga and T. Hayashi, Palladium-Catalyzed Asymmetric Hydrosilylation of 4-Substituted 1-Buten-3-Ynes. Catalytic AsymmetricSynthesis of Axially Chiral Allenylsilanes, J. Am. Chem. Soc., 2001, 123, 12915–12916 CrossRef CAS PubMed;
(b) M. Ogasawara, A. Ito, K. Yoshida and T. Hayashi, Synthesis of 2,5-Bis(binaphthyl)phospholes and Phosphametallocene Derivatives and Their Application in Palladium-Catalyzed Asymmetric Hydrosilylation, Organometallics, 2006, 25, 2715–2718 CrossRef CAS.
- M. Wang, Z.-Li Liu, X. Zhang, P.-P. Tian, Y.-H. Xu and T.-P. Loh, Synthesis of Highly Substituted Racemic and Enantioenriched Allenylsilanes via Copper-Catalyzed Hydrosilylation of (Z)-2-Alken-4-Ynoates with Silylboronate, J. Am. Chem. Soc., 2015, 137, 14830–14833 CrossRef CAS.
- C. Jin, X. He, S. Chen, Z. Guo, Y. Lan and X. Shen, Axial Chirality Reversal and Enantioselective Access to Si-Stereogenic Silylallene, Chem, 2023, 9, 2956–2970 CAS.
- J. Zhu, H. Xiang, H. Chang, J. C. Corcoran, R. Ding, Y. Xia, P. Liu and Y.-M. Wang, Enantioselective and Regiodivergent Synthesis of Propargyl- and Allenylsilanes through Catalytic Propargylic C-H Deprotonation, Angew. Chem., Int. Ed., 2024, 63, e202318040 CrossRef CAS PubMed.
- T. Nishimura, H. Makino, M. Nagaosa and T. Hayashi, Rhodium-Catalyzed Enantioselective 1,6-Addition of Arylboronic Acids to Enynamides: Asymmetric Synthesis of Axially Chiral Allenylsilanes, J. Am. Chem. Soc., 2010, 132, 12865–12867 CrossRef CAS PubMed.
- M. Ogasawara, A. Okada, V. Subbarayan, S. Sörgel and T. Takahashi, Palladium-Catalyzed Asymmetric Synthesis of Axially Chiral Allenylsilanes and Their Application to SE2′ Chirality Transfer Reactions, Org. Lett., 2010, 12, 5736–5739 CrossRef CAS.
- Y. Xu, H. Yi and M. Oestreich, Enantioconvergent and Regioselective Synthesis of Allenylsilanes by Nickel-Catalyzed C(sp2)–C(sp3) Cross-Coupling Starting from Racemic α-Silylated Propargylic Bromides, Organometallics, 2021, 40, 2194–2197 CrossRef CAS.
-
(a) F.-H. Zhang, X. Guo, X. Zeng and Z. Wang, Catalytic Enantioconvergent Allenylation of Aldehydes with Propargyl Halides, Angew. Chem., Int. Ed., 2022, 61, e202117114 CrossRef CAS PubMed;
(b) X. Zeng, F.-H. Zhang and Z. Wang, Cr-Catalyzed Chiral Allenone Synthesis via Sequential Radical-Polar Crossover and Oppenauer Oxidation, Org. Chem. Front., 2023, 10, 310–316 RSC.
-
(a) X. Guo, Z. Shi, F.-H. Zhang and Z. Wang, Cr-Catalyzed
Regio-, Diastereo-, and Enantioselective Reductive Couplings of Ketones and Propargyl Halides, ACS Catal., 2023, 13, 3170–3178 CrossRef CAS;
(b) J. M. Alonso and P. Almendros, Deciphering the Chameleonic Chemistry of Allenols: Breaking the Taboo of a Onetime Esoteric Functionality, Chem. Rev., 2021, 121, 4193–4252 CrossRef CAS;
(c) T. Sun, C. Deutsch and N. Krause, Combined Coinage Metal Catalysis in Natural Product Synthesis: Total Synthesis of (+)-Varitriol and Seven Analogs, Org. Biomol. Chem., 2012, 10, 5965–5970 RSC.
- F.-H. Zhang, X. Guo, X. Zeng and Z. Wang, Asymmetric 1,4-Functionalization of 1,3-Enynes via Dual Photoredox and Chromium Catalysis, Nat. Commun., 2022, 13, 5036 CrossRef CAS.
- A. Hossain, R. L. Anderson, C. S. Zhang, P.-J. Chen and G. C. Fu, Nickel-Catalyzed Enantioconvergent and Diastereoselective Allenylation of Alkyl Electrophiles: Simultaneous Control of Central and Axial Chirality, J. Am. Chem. Soc., 2024, 146, 7173–7177 CrossRef CAS PubMed.
- M. Deliaval, R. Jayarajan, L. Eriksson and K. J. Szabó, Three-Component Approach to Densely Functionalized Trifluoromethyl Allenols by Asymmetric Organocatalysis, J. Am. Chem. Soc., 2023, 145, 10001–10006 CrossRef CAS PubMed.
- J. Ye, Y. Liao, H. Huang, Y. Liu, D. Fang, M. Wang, L. Hu and J. Liao, Halogenated Salt Assisted Cu-Catalyzed Asymmetric 1,4-Borylstannation of 1,3-Enynes: Enantioselective Synthesis of Allenylstannes, Chem. Sci., 2020, 12, 3032–3038 RSC.
- S. Megati, Z. Goren, J. V. Silverton, J. Orlina, H. Niahimura, T. Shiraaaki, H. Mitsuya and J. Zemlicka, (R)-(-)- and (S)-(+)-Adenallene: Synthesis, Absolute Configuration, Enantioselectivity of Antiretroviral Effect, and Enzymic Deamination, J. Med. Chem., 1992, 35, 4098–4104 CrossRef CAS.
- H. Winter, Y. Maeda, H. Mitsuya and J. Zemlicka, Phosphodiester Amidates of Allenic Nucleoside Analogues: Anti-HIV Activity and Possible Mechanism of Action, J. Med. Chem., 1996, 39, 3300–3306 CrossRef CAS PubMed.
- B. C. N. M. Jones, J. V. Silverton, C. Simons, S. Megati, H. Nishimura, Y. Maeda, H. Mitsuya and J. Zemlicka, Synthesis, Absolute Configuration, and Enantioselectivity of Antiretroviral Effect of (R)-(-)- and (S)-(+)-Cytallene. Lipase-Catalyzed Enantioselective Acylations of (±)-N4-Acylcytallenes, J. Med. Chem., 1995, 38, 1397–1405 CrossRef CAS.
- J. Yang, Z. Wang, Z. He, G. Li, L. Hong, W. Sun and R. Wang, Organocatalytic Enantioselective Synthesis of Tetrasubstituted α-Amino Allenoates by Dearomative γ-Addition of 2,3-Disubstituted Indoles to β, γ-Alkynyl-α-Imino Esters, Angew. Chem., Int. Ed., 2020, 59, 642–647 CAS.
- C. Khajuria, N. Saini, P. Subba and V. K. Singh, Asymmetric Cascade Dearomatization-Cyclization Reaction of Tryptamines with β,γ-Alkynyl-α-Imino Esters: Access to Hexahydropyrrolo[2,3-b]indole-Containing Tetrasubstituted α-Amino Allenoates, J. Org. Chem., 2024, 89, 10148–10162 CrossRef CAS.
- X. Zhang, X. Song and Q. Ni, Organocatalytic Regio- and Enantioselective C1-Arylation of β,γ-Alkynyl-α-Imino Esters with Pyrrolo[2,1-a]isoquinolines, Chem. Commun., 2024, 60, 831–834 RSC.
- A. G. Woldegiorgis, Z. Han and X. Lin, Organocatalytic Asymmetric Dearomatization Reaction for the Synthesis of Axial Chiral Allene-Derived Naphthalenones Bearing Quaternary Stereocenters, Org. Lett., 2021, 23, 6606–6611 CrossRef CAS PubMed.
- F. Li, S. Liang, Y. Luan, X. Chen, H. Zhao, A. Huang, P. Li and W. Li, Organocatalytic Regio-, Diastereo- and Enantioselective γ-Additions of Isoxazol-5(4H)-ones to β,γ-alkynyl-α-imino Esters for the Synthesis of Axially Chiral Tetrasubstituted α-Amino Allenoates, Org. Chem. Front., 2021, 8, 1243–1248 RSC.
- C. Qian, T. Huang, J. Sun and P. Li, Catalyst-Controlled Divergent Reactions of 2,3-Disubstituted Indoles with Propargylic Alcohols: Synthesis of 3H-Benzo[b]azepines and Axially Chiral Tetrasubstituted Allenes, Org. Lett., 2022, 24, 6472–6476 CrossRef CAS PubMed.
- C. Qian, M. Liu, J. Sun and P. Li, Chiral Phosphoric Acid-Catalyzed Regio- and Enantioselective Reactions of Functionalized Propargylic Alcohols, Org. Chem. Front., 2022, 9, 1234–1240 RSC.
- Y. Xia, M. Liu, C. Qian, P. Li, M. Dong and W. Li, Asymmetric Organocatalytic (3 + 2) Annulation of Propargylic Alcohols with Indolylnaphthalenols: Synergistic Construction of Axial and Central Chirality, Org. Chem. Front., 2023, 10, 30–34 RSC.
- C. Sheng, Z. Ling, J. Xiao, K. Yang, F. Xie, S. Ma and W. Zhang, Enantio– and Diastereoselective Synthesis of Chiral Tetrasubstituted α–Amino Allenoates Bearing a Vicinal All–Carbon Quaternary Stereocenter with Dual–Copper–Catalysis, Angew. Chem., Int. Ed., 2023, 62, e202305680 CrossRef PubMed.
-
(a) N. Brodta and J. Niemeyer, Chiral organophosphates as ligands in asymmetric metal catalysis, Org. Chem. Front., 2023, 10, 3080–3109 RSC;
(b) A. P. Boisselle and N. A. Meinhardt, Acetylene-Allene Rearrangements. Reactions of Trivalent Phosphorus Chlorides with α-Acetylenic Alcohols and Glycols, J. Org. Chem., 1962, 27, 1828–1833 Search PubMed;
(c) V. Mark, A Facile SNi′ Rearrangement: the Formation of 1,2 Alkadienylphosphonates from 2-Alkynyl Phosphites, Tetrahedron Lett., 1962, 3, 281–285 Search PubMed;
(d) W.-S. Li, W. S. Lam, K.-C. Liu, C.-H. Wang, H.-C. Chang, Y. C. Jen, Y. T. Hsu, S. S. Shivatare and S. C. Jao, Overcoming the Drug Resistance in Breast Cancer Cells by Rational Design of Efficient Glutathione S-Transferase Inhibitors, Org. Lett., 2010, 12, 20–23 CrossRef CAS;
(e) M. Kaleka and J. Stawinski, Novel, Stereoselective and Stereospecific Synthesis of Allenyl-phosphonates and Related Compounds via Palladium-Catalyzed Propargylic Substitution, Adv. Synth. Catal., 2011, 353, 1741–1755 CrossRef.
- H. Wang, H. Qian, J. Zhang and S. Ma, Catalytic Asymmetric Axially Chiral Allenyl C-P Bond Formation, J. Am. Chem. Soc., 2022, 144, 12619–12626 CrossRef CAS PubMed.
- J. Zhang, X. Chang, X. Xu, H. Wang, L. Peng and C. Guo, Nickel-Catalyzed Switchable 1,3-Dienylation and Enantioselective Allenylation of Phosphine Oxides, Nat. Commun., 2022, 13, 7049 CrossRef CAS PubMed.
- W.-H. Wang, Y. Wu, H.-T. Wang, P.-J. Qi, W.-N. Lan and Q.-W. Zhang, Enantioselective Synthesis of P-Stereogenic Allenylphosphines through Ni-catalysed Propargylic Substitution, Nat. Synth., 2022, 1, 738–747 CrossRef CAS.
- X.-D. Gu, K. Y. Ngai, W. Wang, B. Li and J. Wang, Ni-Catalyzed Propargylic Substitution Reaction: A General and Versatile Tool to Assemble Axially Chiral Phosphorus-Containing Allenes, Chem. Catal., 2024, 4, 100903 CrossRef CAS.
- C. Schultz-Fademrecht, B. Wibbeling, R. Frö1hlich and D. Hoppe, Synthesis of Enantiomerically Enriched Allenes by (-)-Sparteine-Mediated Lithiation of Alkynyl Carbamates, Org. Lett., 2001, 3, 1221–1224 CrossRef CAS PubMed.
- R. B. Chedid, M. Brümmer, B. Wibbeling, R. Fröhlich and D. Hoppe, Stereo- and Regiochemical Divergence in the Substitution of a Lithiated Alk-1-en-3-yn-2-yl Carbamate: Synthesis of Highly Enantioenriched Vinylallenes or Alk-3-en-5-yn-1-ols, Angew. Chem., Int. Ed., 2007, 46, 3131–3134 CrossRef CAS PubMed.
- M. Zimmermann, B. Wibbeling and D. Hoppe, Synthesis of Enantiomerically and Diastereomerically Pure 4-Hydroxy-1,2-alkadienyl Carbamates and Their Application in a Modified Nazarov Cyclization Towards Chiral Cyclopentenones, Synthesis, 2004, 765–774 CAS.
- Y. Tomida, A. Nagaki and J. Yoshida, Asymmetric Carbolithiation of Conjugated Enynes: a Flow Microreactor Enables the Use of Configurationally Unstable Intermediates before They Epimerize, J. Am. Chem. Soc., 2011, 133, 3744–3747 CrossRef CAS.
- M. Sasaki, Y. Kondo, M. Kawahata, K. Yamaguchi and K. Takeda, Enantioselective Synthesis of Siloxyallenes from Alkynoylsilanes by Reduction and a Brook Rearrangement and Their Subsequent Trapping in a [4 + 2] Cycloaddition, Angew. Chem., Int. Ed., 2011, 50, 6375–6378 CrossRef CAS.
- M. Asikainen, W. Lewis, A. J. Blake and S. Woodward, An SN2′ Displacement Approach to Allenyl Acetates, Tetrahedron Lett., 2010, 51, 6454–6456 CrossRef CAS.
- Q. Lin, S. Zheng, L. Chen, J. Wu, J. Li, P. Liu, S. Dong, X. Liu, Q. Peng and X. Feng, Catalytic Regio– and Enantioselective Protonation for the Synthesis of Chiral Allenes: Synergistic Effect of the Counterion and Water, Angew. Chem., Int. Ed., 2022, 61, e202203650 CrossRef CAS PubMed.
- J.-Y. Zheng, F. Wang, Y. Zhang, Z. Zheng, J.-H. Wu, X. Ren, Z. Su, W. Chen and T. Wang, Novel Stereo–Induction Pattern in Pudovik Addition/Phospha–Brook Rearrangement Towards Chiral Trisubstituted Allenes, Angew. Chem., Int. Ed., 2024, 63, e202403707 CrossRef CAS PubMed.
-
(a) R. Otte, R. Fröhlich, B. Wibbeling and D. Hoppe, Solid-State Structure and Enantioselective Reactions of a Complex of a 1-Thio-Substituted Propargyllithium and (-)-Sparteine, Angew. Chem., Int. Ed., 2005, 44, 5492–5496 CrossRef CAS;
(b) R. Otte, B. Wibbeling, R. Fröhlich, S. Nakamura, N. Shibata, T. Torub and D. Hoppe, Asymmetric Lithiation of 2-Alkynyl Aryl Sulfides-Enantio- and Diastereoselective Formation of Allenyl Aryl Sulfides and Their Application in Nickel-Catalyzed Cross-Coupling Reactions, Tetrahedron Lett., 2007, 48, 8636–8642 CrossRef CAS.
- D. Qian, L. Wu, Z. Lin and J. Sun, Organocatalytic Synthesis of Chiral Tetrasubstituted Allenes from Racemic Propargylic Alcohols, Nat. Commun., 2017, 8, 567 CrossRef PubMed.
- Z. Wang, X. Lin, X. Chen, P. Li and W. Li, Organocatalytic Stereoselective 1,6-Addition of Thiolacetic Acids to Alkynyl Indole Imine Methides: Access to Axially Chiral Sulfur-Containing Tetrasubstituted Allenes, Org. Chem. Front., 2021, 8, 3469–3474 RSC.
- G. Liu, X. Yang, P. Gu, M. Wang, X. Zhang and X.-Q. Dong, Challenging Task of Ni-Catalyzed Highly Regio-/Enantioselective Semihydrogenation of Racemic Tetrasubstituted Allenes via a Kinetic Resolution Process, J. Am. Chem. Soc., 2024, 146, 7419–7430 CrossRef CAS.
-
(a) A. R. Pinder, The Hydrogenolysis of Organic Halides, Synthesis, 1980, 425–452 CrossRef CAS;
(b) D. C. Billington, π-Allylnickel Halides as Selective Reagents in Organic Synthesis, Chem. Soc. Rev., 1985, 14, 93–120 RSC;
(c) F. Alonso, I. P. Beletskaya and M. Yus, Metal-Mediated Reductive Hydrodehalogenation of Organic Halides, Chem. Rev., 2002, 102, 4009–4091 CrossRef CAS;
(d) M. K. Guptaa and T. P. O'Sullivan, Recent Applications of Gallium and Gallium Halides as Reagents in Organic Synthesis, RSC Adv., 2013, 3, 25498–25522 RSC;
(e) R. Lekkala, R. Lekkala, B. Moku, K. P. Rakesh and H.-L. Qin, Recent Developments in Radical-Mediated Transformations of Organohalides, Eur. J. Org. Chem., 2019, 2769–2806 CrossRef CAS.
- W. Zhang, S. Zheng, N. Liu, J. B. Werness, I. A. Guzei and W. Tang, Enantioselective Bromolactonization of Conjugated (Z)-Enynes, J. Am. Chem. Soc., 2010, 132, 3664–3665 CrossRef CAS.
-
(a) H. Li, D. Müller, L. Guénée and A. Alexakis, Copper-Catalyzed Enantioselective Synthesis of Axially Chiral Allenes, Org. Lett., 2012, 14, 5880–5883 CrossRef CAS;
(b) H. Li, D. Grassi, L. Guénée, T. Bürgi and A. Alexakis, Copper-Catalyzed Propargylic Substitution of Dichloro Substrates: Enantioselective Synthesis of Trisubstituted Allenes and Formation of Propargylic Quaternary Stereogenic Centers, Chem. – Eur. J., 2014, 20, 16694–16706 CrossRef CAS.
- Z.-L. Liu, C. Yang, Q.-Y. Xue, M. Zhao, C.-C. Shan, Y.-H. Xu and T.-P. Loh, Copper-Catalyzed Asymmetric Silylation of Propargyl Dichlorides: Access to Enantioenriched Functionalized Allenylsilanes, Angew. Chem., Int. Ed., 2019, 58, 16538–16542 CrossRef CAS.
- M. F. Kuehnel, T. Schloder, S. Riedel, B. Nieto-Ortega, F. J. Ramirez, J. T. Navarrete, J. Casado and D. Lentz, Synthesis of the Smallest Axially Chiral Molecule by Asymmetric Carbon-Fluorine Bond Activation, Angew. Chem., Int. Ed., 2012, 51, 2218–2220 CrossRef CAS PubMed.
- T. Li, C. Zhou, X. Yan and J. Wang, Solvent-Dependent Asymmetric Synthesis of Alkynyl and Monofluoroalkenyl Isoindolinones by CpRhIII-Catalyzed C-H Activation, Angew. Chem., Int. Ed., 2018, 57, 4048–4052 CrossRef CAS.
- T. J. O'Connor, B. K. Mai, J. Nafie, P. Liu and F. D. Toste, Generation of Axially Chiral Fluoroallenes through a Copper-Catalyzed Enantioselective β-Fluoride Elimination, J. Am. Chem. Soc., 2021, 143, 13759–13768 CrossRef PubMed.
- J. S. Ng and T. Hayashi, Asymmetric Synthesis of Fluorinated Allenes by Rhodium-Catalyzed Enantioselective Alkylation/Defluorination of Propargyl Difluorides with Alkylzincs, Angew. Chem., Int. Ed., 2021, 60, 20771–20775 CrossRef CAS PubMed.
-
(a) H. Ito, Y. Sasaki and M. Sawamura, Copper(I)-Catalyzed Substitution of Propargylic Carbonates with Diboron: Selective Synthesis of Multisubstituted Allenylboronates, J. Am. Chem. Soc., 2008, 130, 15774–15775 CrossRef CAS PubMed;
(b) T. S. N. Zhao, Y. Yang, T. Lessing and K. J. Szabo, Borylation of Propargylic Substrates by Bimetallic Catalysis. Synthesis of Allenyl, Propargylic, and Butadienyl Bpin Derivatives, J. Am. Chem. Soc., 2014, 136, 7563–7566 CrossRef CAS;
(c) A. Bermejo-López, W.-J. Kong, P. J. Tortajada, D. Posevins, B. Martín-Matute and J.-E. Bäckvall, Iron-Catalyzed Borylation of Propargylic Acetates for the Synthesis of Multisubstituted Allenylboronates, Chem. – Eur. J., 2023, 29, e202203130 CrossRef PubMed;
(d) J. Qian, Z.-H. Chen, Y. Liu, Y. Li, Q. Li, S.-L. Huang and H. Wang, Synthesis of Allenyl-B(MIDA) via Hydrazination/Fragmentation Reaction of B(MIDA)-Propargylic Alcohol, Chin. Chem. Lett., 2023, 34, 107479 CrossRef CAS.
- Y. Matsumoto, M. Naito, Y. Uozumi and T. Hayashi, Axially Chiral Allenylboranes: Catalytic Asymmetric Synthesis by Palladium-catalysed Hydroboration of But-I-en-3-ynes and their Reaction with an Aldehyde, J. Chem. Soc., Chem. Commun., 1993, 1468–1469 RSC.
- Y. Huang, J. del Pozo, S. Torker and A. H. Hoveyda, Enantioselective Synthesis of Trisubstituted Allenyl-B(pin) Compounds by Phosphine-Cu-Catalyzed 1,3-Enyne Hydroboration. Insights Regarding Stereochemical Integrity of Cu-Allenyl Intermediates, J. Am. Chem. Soc., 2018, 140, 2643–2655 CrossRef CAS PubMed.
-
(a) M. Chen and W. R. Roush, Enantioselective Synthesis of anti- and syn-Homopropargyl Alcohols via Chiral Brønsted Acid Catalyzed Asymmetric Allenylboration Reactions, J. Am. Chem. Soc., 2012, 134, 10947–10952 CrossRef CAS;
(b) J.-L. Han, M. Chen and W. R. Roush, Diastereo- and Enantioselective Synthesis of (E)-2-Methyl-1,2-syn-and (E)-2-Methyl-1,2-anti-3-pentenediols via Allenylboronate Kinetic Resolution with (dIpc)2BH and Aldehyde Allylboration, Org. Lett., 2012, 14, 3028–3031 CrossRef CAS.
- H. L. Sang, S. Yu and S. Ge, Copper-catalyzed Asymmetric Hydroboration of 1,3-Enynes with Pinacolborane to Access Chiral Allenylboronates, Org. Chem. Front., 2018, 5, 1284–1287 RSC.
- D. W. Gao, Y. Xiao, M. Liu, Z. Liu, M. K. Karunananda, J. S. Chen and K. M. Engle, Catalytic, Enantioselective Synthesis of Allenyl Boronates, ACS Catal., 2018, 8, 3650–3654 CrossRef CAS PubMed.
- W. Su, J. Zhu, Y. Chen, X. Zhang, W. Qiu, K. Yang, P. Yu and Q. Song, Copper-Catalysed Asymmetric Hydroboration of Alkenes with 1,2-Benzazaborines to Access Chiral Naphthalene Isosteres, Nat. Chem., 2024, 16, 1312–1319 CrossRef CAS.
- S. A. Gonsales, Z. C. Mueller, F. Zhao, P. H. S. Paioti, L. Karmazin, J. Wan, F. Liu, K. N. Houk and A. H. Hoveyda, Cross-metathesis of Allenes. Mechanistic Analysis and Identification of a Ru-CAAC as the Most Effective Catalyst, J. Am. Chem. Soc., 2021, 143, 20640–20644 CAS.
- J. A. Marshall, Chiral Allylic and Allenic Metal Reagents for Organic Synthesis, J. Org. Chem., 2007, 72, 8153–8166 CrossRef CAS PubMed.
- M. Ohmori, S. Yamada, H. Takayama and K. Ochi, Stereocontrolled Synthesis of Steroid Side Chain; Stereoselective Syntheses of Cholesterol and 25-Hydroxycholesterol, Tetrahedron Lett., 1962, 23, 4709–4712 Search PubMed.
- S. M. Weinreb, Synthetic Applications of a Novel Pericyclic Imino Ene Reaction of Allenyl Silanes, J. Heterocycl. Chem., 1996, 33, 1429–1436 CAS.
- R. M. Borzilleri and S. M. Weinreb, Total Synthesis of Papuamine via a Stereospecific Intramolecular Imino Ene Reaction of an Allenylsilane, J. Am. Chem. Soc., 1994, 116, 9789–9790 CrossRef CAS.
- R. M. Borzilleri, S. M. Weinreb and M. Parvez, Total Synthesis of the Unusual Marine Alkaloid (-)-Papuamine Utilizing a Novel Imino Ene Reaction, J. Am. Chem. Soc., 1995, 117, 10905–10913 CAS.
- J. Jin and S. M. Weinreb, Application of a Stereospecific Intramolecular Allenylsilane Imino Ene Reaction to Enantioselective Total Synthesis of the 5,11-Methanomorphanthridine Class of Amaryllidaceae Alkaloids, J. Am. Chem. Soc., 1997, 119, 2050–2051 CrossRef CAS.
- J. Jin and S. M. Weinreb, Enantioselective Total Syntheses of the 5,11-Methanomorphanthridine Amaryllidaceae Alkaloids (-)-Pancracine and (-)-Coccinine, J. Am. Chem. Soc., 1997, 119, 5773–5784 CAS.
-
(a) M. Ishizaki, O. Hoshino and Y. Iitaka, Total Synthesis of Montanine-type Amaryllidaceae Alkaloids, which Possess a 5,11-Methanomorphanthridine Ring System, through Cyclization with Sodium bis(2-methoxyethoxy)aluminum Hydride (SMEAH): the First Stereoselective Total Syntheses of (±)-Montanine, (±)-Coccinine, (±)-O-acetylmontanine, (±)-Pancracine, and (±)-Brunsvigine, J. Org. Chem., 1992, 57, 7285–7295 CAS;
(b) M. Ishizaki, K. I. Kurihara, E. Tanazawa and O. Hoshino, Radical-mediated Synthesis of the 5,11-Methanomorphanthridine Ring System: Formal Total synthesis of Montanine-type Amaryllidaceae Alkaloids, (±)-Montanine, (±)-Coccinine and (±)-Pancracine, J. Chem. Soc., Perkin Trans. 1, 1993, 101–110 RSC.
- L. E. Overman and J. Shim, Synthesis Applications of Cationic Aza-Cope Rearrangements. 23. First Total Synthesis of Amaryllidaceae Alkaloids of the 5,11-Methano Morphanthridine Type. An Efficient Total Synthesis of (±)-Pancracine, J. Org. Chem., 1991, 56, 5005–5007 CrossRef CAS.
- L. E. Overman and J. Shim, Synthesis Applications of Cationic Aza-Cope Rearrangements. Part 25. Total Synthesis of Amaryllidaceae Alkaloids of the 5,11-Methanomorphanthridine Type. Efficient Total Syntheses of (-)-Pancracine and (±)-Pancracine, J. Org. Chem., 1993, 58, 4662–4672 CrossRef CAS.
- A. Voituriez, A. Pérez-Luna, F. Ferreira, C. Botuha and F. Chemla, Stereo- and Enantioselective Synthesisof Acetylenic 2-Amino-1,3-diol Stereotriads, Org. Lett., 2009, 11, 931–934 CrossRef CAS PubMed.
- C. Séguin, F. Ferreira, C. Botuha, F. Chemla and A. Péerez-Luna, High-Yielding Synthesis of Sphingoid-Type Bases, J. Org. Chem., 2009, 74, 6986–6992 CrossRef.
- B. Hélal, F. Ferreira, C. Botuha, F. Chemla and A. Pérez-Luna, Concise Synthesis of (2S,3R)-3-Hydroxy-2-phenylpiperidine: An Advanced Key Intermediate of Human Non-Peptide NK-1 Receptor Antagonists, Synlett, 2009, 3115–3118 Search PubMed.
- J. Louvel, C. Botuha, F. Chemla, E. Demont, F. Ferreira and A. Pérez-Luna, Asymmetric Total Synthesis of (+)-6-epi-Castanospermine by the Stereoselective Formation of a syn, anti Acetylenic 2-Amino-1,3-diol Stereotriad, Eur. J. Org. Chem., 2010, 2921–2926 CrossRef CAS.
- S. C. Archibald, D. J. Barden, J. F. Y. Bazin, I. Fleming, C. F. Foster, A. K. Mandal, A. K. Mandal, D. Parker, K. Takaki, A. C. Ware, A. R. B. Williams and A. B. Zwicky, Stereocontrol in Organic Synthesis using Silicon-Containing Compounds. Studies Directed Towards the Synthesis of Ebelactone A, Org. Biomol. Chem., 2004, 2, 1051–1064 RSC.
- A. B. Bahadoor, A. Flyer and G. C. Micalizio, A Pentenyl Dianion-Based Strategy for Convergent Synthesis of Ene-1,5-diols, J. Am. Chem. Soc., 2005, 127, 3694–3695 CrossRef CAS PubMed.
- W. Felzmann, D. Castagnolo, D. Rosenbeiger and J. Mulzer, Crotylation versus Propargylation: Two Routes for the Synthesis of the C13-C18 Fragment of the Antibiotic Branimycin, J. Org. Chem., 2007, 72, 2182–2186 CrossRef CAS.
- B. Cai, R. W. Evans, J. Wu and J. S. Panek, Total Synthesis of Nuclear Factor of Activated T-Cells-68 (NFAT-68): Sequential Use of Chiral Allenylsilane and Titanium Alkoxide-Mediated Reductive Coupling Bond Construction, Org. Lett., 2016, 18, 4304–4307 CrossRef CAS PubMed.
- M. Achmatowicz and L. S. Hegedus, Synthesis of 1-Deoxy-D-galactohomonojirimycin via Enantiomerically Pure Allenylstannanes, J. Org. Chem., 2004, 69, 2229–2234 CrossRef CAS PubMed.
-
(a) C. J. T. Hyland and L. S. Hegedus, Boron-Mediated Stereoselective Syntheses of γ,γ-Disubstituted Allenamides, J. Org. Chem., 2005, 70, 8628–8630 CAS;
(b) C. de los Rios and L. S. Hegedus, Reaction of Optically Active α-Aminoallenylstannanes with Aldehydes Formed in Situ from the Lewis-Acid-Catalyzed Rearrangement of Epoxides, J. Org. Chem., 2005, 70, 6541–6543 Search PubMed;
(c) C. J. T. Hyland and L. S. Hegedus, Gold-Catalyzed and N-Iodosuccinimide-Mediated Cyclization of γ-Substituted Allenamides, J. Org. Chem., 2006, 71, 8658–8660 CAS.
- J. A. Marshall and N. D. Adams, Addition of Allenylzinc Reagents, Prepared in Situ from Nonracemic Propargylic Mesylates, to Aldehydes. A New Synthesisof Highly Enantioenriched Homopropargylic Alcohols, J. Org. Chem., 1999, 64, 5201–5204 Search PubMed.
- J. A. Marshall and M. P. Bourbeau, Total Synthesis of (-)-Callystatin A, J. Org. Chem., 2002, 67, 2751–2754 Search PubMed.
- J. A. Marshall, H. R. Chobanian and M. M. Yanik, Lipase-Mediated Resolution of 4-TMS-3-butyn-2-ol and Use of the Mesylate Derivatives as a Precursor to a Highly Stereoselective Chiral Allenylindium Reagent, Org. Lett., 2001, 3, 3369–3372 CAS.
- J. A. Marshall and K. C. Ellis, Total Synthesis of (-)- and (+)-Membrenone C, Org. Lett., 2003, 5, 1729–1732 CAS.
- O. Thominet, J. R. Baker, H. Britton, Z. C. Etheridge, M. G. Sosciab and S. Caddick, Synthetic Strategies to Epoxydiynes and a Key Synthon of the Neocarzinostatin Chromophore, Org. Biomol. Chem., 2007, 5, 3703–3712 RSC.
- R. A. Gibbs, K. Bartels, R. W. K. Lee and W. H. Okamura, An Enantioselective Central-Axial-Central Chiral Element Transfer Process Leading to a Concise Synthesis of (+)-Sterpurene: Intramolecular Diels-Alder Reactions of Vinylallene Sulfoxides, J. Am. Chem. Soc., 1989, 111, 3717–3725 CrossRef CAS.
-
(a) L.-Z. Li, Y.-R. Huang, Z.-X. Xu, H.-S. He, H.-W. Ran, K.-Y. Zhu, J.-C. Han and C.-C. Li, Synthesis of Bridged Five-Membered Ring Systems by Type II [3 + 2] Annulation of Allenylsilane-ene, J. Am. Chem. Soc., 2024, 146, 24782–24787 CrossRef CAS;
(b) N. Lv, J.-C. Han, P. Zhang, Y.-R. Huang, Z.-X. Xu, K. Xu, X.-F. Wang, X. Li, L. W. Chung and C.-C. Li, Intramolecular [3 + 2] Annulation of Allenylsilane-Enes: Direct Synthesis of Highly Strained trans-Fused 5/5 Ring Systems, Chem, 2024, 10, 190–198 CrossRef CAS.
-
(a) K. Ruitenberg, H. Kleijn, C. J. Elsevier, J. Meijer and P. Vermeer, Palladium(0)-Promoted Synthesis of Functionally Substituted Allenes by Means of Organozinc Compounds, Tetrahedron Lett., 1981, 22, 1451–1452 CrossRef CAS;
(b) C. J. Elsevier, H. H. Mooiweer, H. Kleijn and P. Vermeer, Stereochemistry of the Pd(PPh3)4-Catalyzed Conversion of 1-Bromoallenes into Phenyl Substituted Allenes, Tetrahedron Lett., 1984, 25, 5571–5572 CrossRef CAS;
(c) C. J. Elsevier and P. Vermeer, Stereochemistry of the Palladium(0)-Catalyzed Phenylation of 1-Halooallenes, J. Org. Chem., 1985, 50, 3042–3045 CrossRef CAS.
- B. Vaz, M. Domínguez, R. Álvarez and A. R. de Lera, Total Synthesis of Peridinin and Related C37-Norcarotenoid Butenolides, Chem. – Eur. J., 2007, 13, 1273–1290 CrossRef CAS PubMed.
-
(a) Y. Yamano, S. Sumiya, K. Suzuki, Y. Kurimoto, Y. Koyama, T. Shimamura and M. Ito, A Novel Photoproduct of Peridinin, A 6S Allenic Isomer, Tetrahedron Lett., 1992, 33, 2991–2994 CrossRef CAS;
(b) J. A. Havgan, G. Englert, T. Aakermann, E. Glinz and S. Liaaen-Jensen, Algal Carotenoids 58.* Isomerization Studies on Peridinin, Acta Chem. Scand., 1994, 48, 769–779 CrossRef.
- B. Vaz, R. Pereira, M. Pérez, R. Álvarez and A. R. de Lera, Stereoselective Stille Coupling of Enantiopure Haloallenes and Alkenylstannanes for the Synthesis of Allenyl Carotenoids. Experimental and Computational Studies, J. Org. Chem., 2008, 73, 6534–6541 CrossRef CAS.
-
(a) E. P. Gillis and M. D. Burke, A Simple and Modular Strategy for Small Molecule Synthesis: Iterative Suzuki-Miyaura Coupling of B-Protected Haloboronic Acid Building Blocks, J. Am. Chem. Soc., 2007, 129, 6716–6717 CrossRef CAS PubMed;
(b) S. J. Lee, K. C. Gray, J. S. Paek and M. D. Burke, Simple, Efficient, and Modular Syntheses of Polyene Natural Products via Iterative Cross-Coupling, J. Am. Chem. Soc., 2008, 130, 466–468 CrossRef CAS PubMed;
(c) E. M. Woerly, A. H. Cherney, E. K. Davis and M. D. Burke, Stereoretentive Suzuki-Miyaura Coupling of Haloallenes Enables Fully Stereocontrolled Access to (-)-Peridinin, J. Am. Chem. Soc., 2010, 132, 6941–6943 CrossRef CAS.
- S. Inuki, Y. Yoshimitsu, S. Oishi, N. Fujii and H. Ohno, Ring-Construction/Stereoselective Functionalization Cascade: Total Synthesis of Pachastrissamine (Jaspine B) through Palladium-Catalyzed Bis-cyclization of Bromoallenes, Org. Lett., 2009, 11, 4478–4481 CrossRef CAS.
- P. Ghosh, J. R. Cusick, J. Inghrim and L. J. Williams, Silyl-Substituted Spirodiepoxides: Stereoselective Formation and Regioselective Opening, Org. Lett., 2009, 11, 4672–4675 CrossRef CAS PubMed.
-
(a) M. A. Tius, Allene Ether Nazarov Cyclization, Chem. Soc. Rev., 2014, 43, 2979–3002 RSC;
(b) A. J. Frontier and J. J. Hernandez, New Twists in Nazarov Cyclization Chemistry, Acc. Chem. Res., 2020, 53, 1822–1832 CrossRef CAS.
-
(a) J. A. Malona, K. Cariou and A. J. Frontier, Nazarov Cyclization Initiated by Peracid Oxidation: The Total Synthesis of (±)-Rocaglamide, J. Am. Chem. Soc., 2009, 131, 7560–7561 CrossRef CAS;
(b) J. A. Malona, K. Cariou, W. T. Spencer III and A. J. Frontier, Total Synthesis of (±)-Rocaglamide via Oxidation-Initiated Nazarov Cyclization, J. Org. Chem., 2012, 77, 1891–1908 CrossRef CAS PubMed.
- P. J. Kocieński, R. C. D. Brown, A. Pommier, M. Procter and B. Schmidt, Synthesis of Salinomycin, J. Chem. Soc., Perkin Trans. 1, 1998, 9–39 RSC.
|
| This journal is © the Partner Organisations 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *a and
Zhihui Shao
*a and
Zhihui Shao