
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular supramolecular capsules based on α,γ-cyclic peptide dimers for the development of confined catalysis†
Ezequiel Troncoso Mondragón ,
Alberto Fuertes,
Martín Calvelo,
Ángel L. Fuentes de Arriba‡
*,
Manuel Amorín and
Juan R. Granja*
,
Alberto Fuertes,
Martín Calvelo,
Ángel L. Fuentes de Arriba‡
*,
Manuel Amorín and
Juan R. Granja*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CIQUS) and Organic Chemistry Department, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: juanr.granja@usc.es
First published on 24th January 2025
Abstract
A modular strategy is designed for the preparation of supramolecular capsules in the search for new functional containers. The strategy is based on the use of the self-assembly properties of an α,γ-cyclic octapeptide to form toroidal dimers in which two reactive points are incorporated to anchor molecular caps in the last stage of the synthesis. In this way, the used caps, which are decorated with a catalytic metal complex, are easily interchangeable allowing searching for new catalytic properties.
Introduction
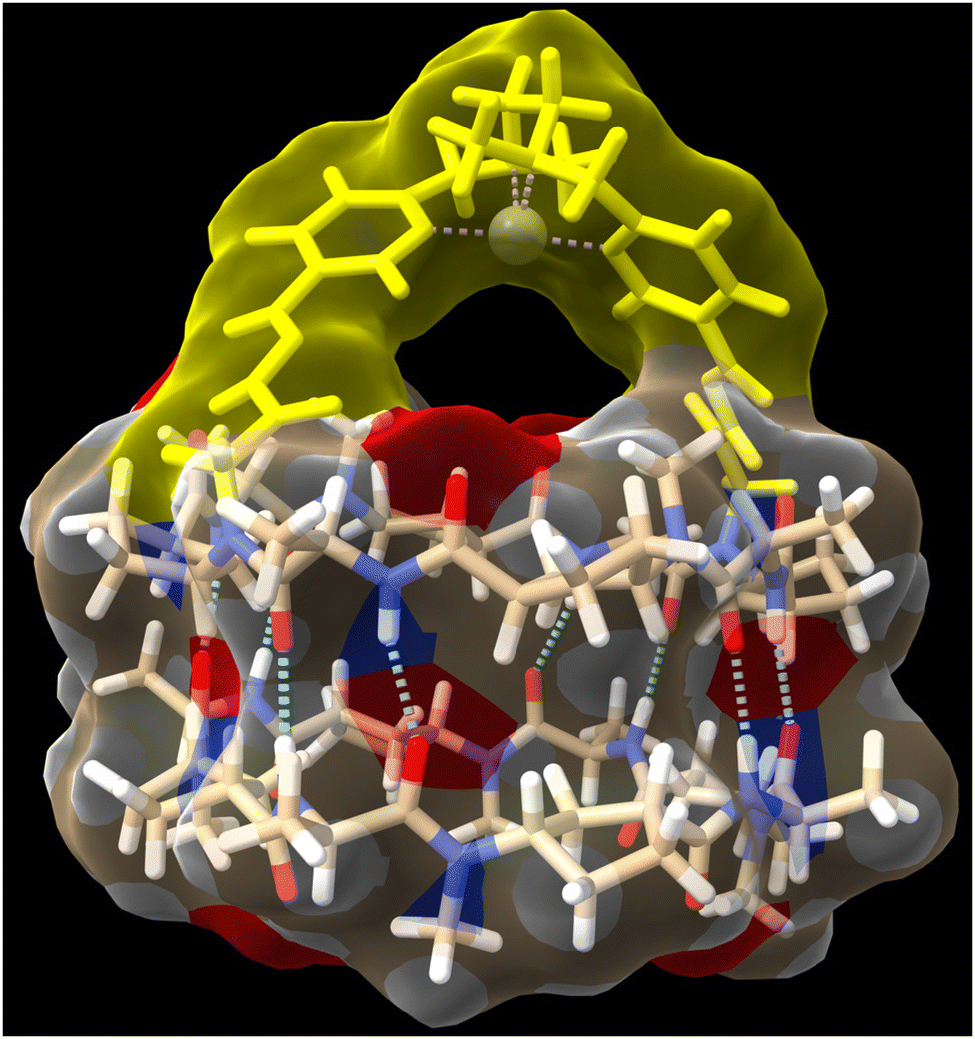
Self-assembled molecular capsules with catalytic properties are emerging as a powerful tool to carry out chemo-, regio-, and stereoselective transformations on challenging substrates.1 They can differentiate almost identical chemical functions in molecules with different sizes, since, in principle, only those that can be confined in the inner space are susceptible to react. Therefore, supramolecular containers can develop a unique selectivity unattainable with conventional catalysts.2 However, molecular capsules are complex structures whose precise and custom-made preparation, generally, requires considerable synthetic effort, therefore, new approaches for their preparation have yet to be developed.3An alternative to facilitate the synthesis of these structures is the use of specific molecular skeletons whose preparation and assembling properties are well stablished and, at the same time, allow ad hoc modifications at the end of the synthesis to accommodate new and specific functional properties. The formation of amide bonds to prepare peptides is an easy, rapid and consistent reaction that allows the creation of an almost infinite number of peptide structures.4 Furthermore, the possibility of using non-natural amino acids expands this repertoire, allowing the preparation of structures with customized functionalities and, consequently, properties not previously seen in nature. In addition, peptides are very versatile molecules that, in addition to forming different 3D structures, such as α-helix, β-sheet or turns, thanks to their ability to fold, can also form other more complex species, i.e., nanotubes, capsids, fibres, etc., through molecular assembly processes.5 All this allows the creation of almost an infinite variety of shapes and structures according to the required functional needs. In particular, planar cyclic peptides are supramolecular building blocks that self-assemble into nanotubes by staking these rings guided by the establishment of hydrogen bond interactions (Fig. 1).6 This method of nanotube formation provides the opportunity to control size and properties of their internal cavity by changing the number and type of amino acids used in the cyclic peptide.7 In addition, control over amino acid sequence allows to tune the external properties according to the characteristics of amino acid side chains.6–10 Methylation of alternating amide functions in the cyclic peptide (BG for blocking groups), those with the same chirality, restricts assembly to discrete dimers or, in some cases, other structures, rather than forming endless nanotubes.8,9 The use of these cyclic peptide dimers allowed to clarify key aspect of nanotube formation, as well as the development of new applications.10 Using these hollow supramolecular aggregates, we have already explored the encapsulation of different molecules, such as xenon atoms,11 C60 fullerene,7a cis-platinum complex,12 carbonate,9b or silver ions,13 etc. Furthermore, the group responsible for preventing nanotube growth, the amide substituent, can also carry additional functional groups to tune the properties at the pore entrance.14 All these features make cyclic peptides a versatile tool for the construction of functional supramolecular components. At this respect, our group has taken a new step in this direction by incorporating molecular caps on cyclic peptide dimers thanks to the use of complementary reactive groups (anchoring sites) in both components (Fig. 1).15,16 The use of click-chemistry-based connectors allowed the incorporation of these molecular caps at the end of the synthesis.17
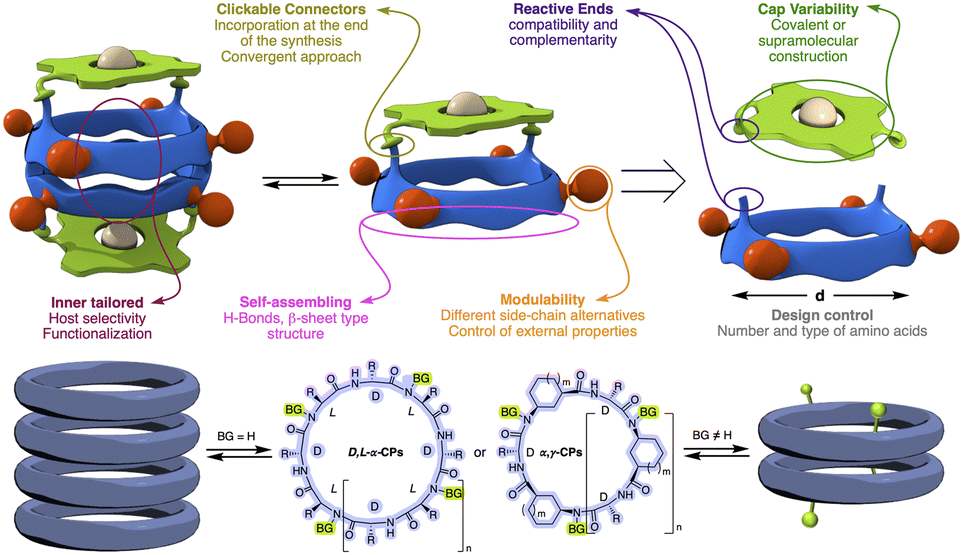 | ||
| Fig. 1 Top, basic properties of self-assembled capsule designs based on dimerization properties of flat cyclic peptides. Bottom, cyclic peptide models and corresponding dimers or nanotubes depending on the substitution (BG, blocking groups) of peptide bonds. | ||
This convergent and modular strategy facilitates the construction of a variety of capsules with different properties from a common adhesive component, the β-sheet type interactions of CPs. Using this approach we have reported the synthesis of supramolecular capsules made up of cyclic peptides dimers decorated with Zn-porphyrins for the encapsulation of bipyridines,15 or tris-triazolylamine components for the recognition of bis-nitriles or water-anion clusters.16 We envisioned that the incorporation of active metal complexes with the molecular cap could represent an attractive alternative for the construction of nanoreactors that catalyse chemical reactions within them (Fig. 2). The modular design proposed here, based on the simple addition of the metallocaps at the end of the process, not only facilitates its synthesis, but also allows the study of different reactions. In this article, we would like to report as a proof of concept our initial findings of our modular approach of nanoreactors.
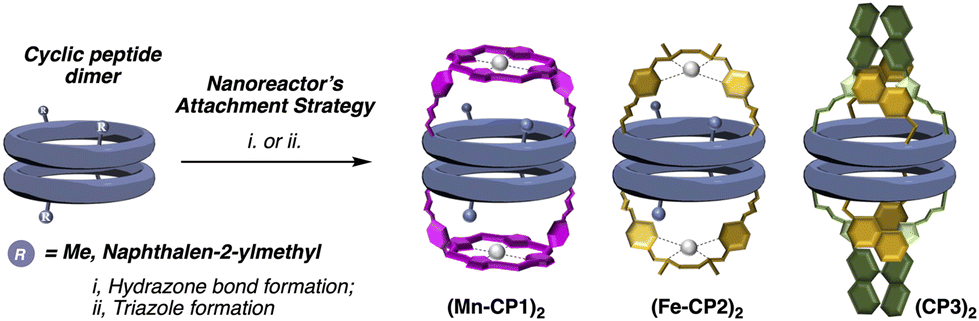 | ||
| Fig. 2 Overview of modular molecular capsules described in this work. | ||
Results and discussion
Inspired by Nolte's toroidal18 and Collman's “picnic basket”19 porphyrins (Fig. 3), we decided to transform the previous described cyclic peptide capsule topped with porphyrin caps into a multifaceted nanoreactor.15 To do this, we envisaged that the simple replacement of inert Zn by Mn in the porphyrin cap should create a catalytic centre capable of performing oxidation reactions within the capsule.20 In this way, size-selective oxidations could be carried out, since only those molecules that enter the reactor cavity would be oxidized.21 Furthermore, taking into account the anisotropic shape of the cavity, a certain regioselectivity could also be expected, since this would preferably occur in the substrate position closest to the catalytic centre. | ||
| Fig. 3 (A) Molecular capsules designed for selective epoxidations. (B) Cyclic peptide-based capsule designed for bipyridine encapsulation. (C) Molecular capsules for selective epoxidations designed in this work. | ||
Cyclic peptide 1 and Mn porphyrin 2 were synthesized following the literature procedure and condensed by mixing both components in dichloromethane (Schemes S1 and S2†), to provide the corresponding hemicapsule Mn-CP1 in almost quantitative yield (Fig. 4A).15 The 1H NMR spectrum of this compound presented most of its signals broad and poorly defined, even worse than that already observed with the previous Zn derivative, as a consequence not only of the conformational and dynamic complexity of this species but also of the paramagnetic properties of Mn(III) complexes.15 Consequently, the catalytic activity of the nanoreactor was addressed next. To ensure that substrate oxidation took place within the capsule, the outer face of the manganese porphyrin needs to be blocked. For this purpose, sterically hindered ligands were used, following the precedent of Nolte with his toroidal reactor.18 Therefore, to carry out these studies we decided to use the m-disubstituted ligands L1 and L2 (Fig. 4B), which, in principle, should not be able to coordinate with manganese via their internal face.
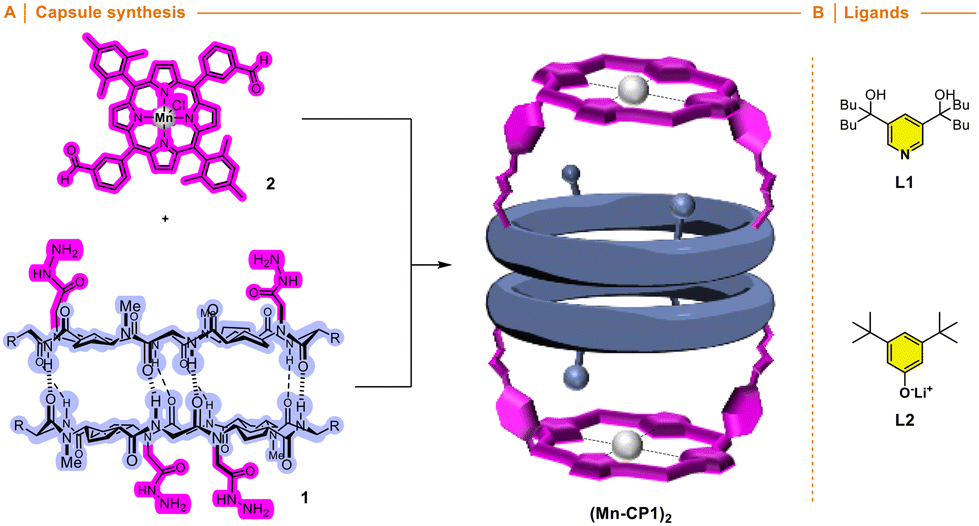 | ||
| Fig. 4 (A) Synthesis of a molecular capsule based on α,γ-cyclic peptide functionalized with Mn-porphyrin. (B) Hindered ligands. | ||
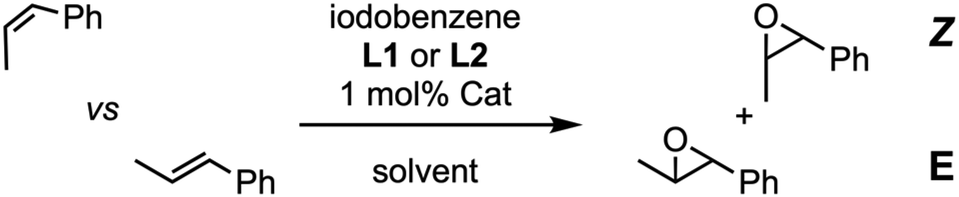
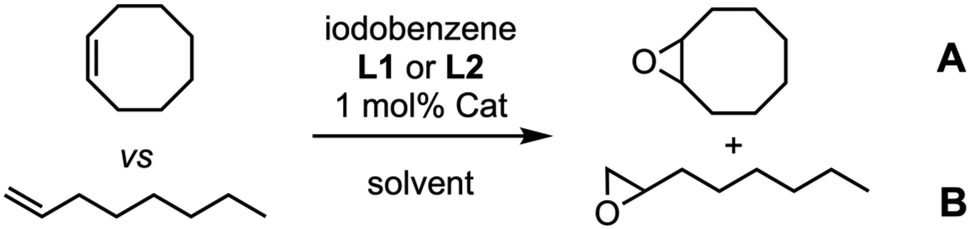
In first place, we tackled the alkene epoxidation using (Mn-CP1)2 as catalyst and iodosobenzene as stoichiometric oxidant. Both isomers of prop-1-en-1-ylbenzene were used as substrate together with Mn-porphyrin 2 that was used as control catalyst. Excess of pyridine ligand L1 was employed due to the low association constant of pyridines by MnIII as contrast by ZnII (Kass = 60 M−1, about ca. 10 times lower than the association constant of the corresponding ZnII porphyrin).18 To our delight, both isomers were oxidized in good yields (55%). Once confirmed the efficient catalytic activity, the competitive epoxidation was tackled. Our first hypothesis was that less hindered substrates such as the Z-isomer would be more easily encapsulated than more hindered ones. Therefore, Z- and E-isomers of prop-1-en-1-ylbenzene were compared using ligand L1 (Table 1, entries 1 and 2), showing a sluggish higher preference for Z-isomer when compared with control model 2 (1.9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 vs. 1.7:1).
:1 vs. 1.7:1).
|
||||
|---|---|---|---|---|
| Entry | Cat | Ligand | Yield (%) | Ratio Z vs. E |
| a Reaction conditions: both alkenes (0.13 mmol each), iodosobenzene (0.13 equiv.), ligand L1 (25–500 equiv.) and catalyst (1 mol%) were dissolved in CDCl3 and stirred overnight and then transferred to an NMR tube. Yields and ratios were determined by 1H NMR.b Reaction conditions: both alkenes (0.1 mmol each), iodosobenzene (0.1 equiv.), ligand L2 (20 equiv.) and catalyst (1 mol%) were dissolved in CD3CN and stirred overnight, and then transferred to an NMR tube. Yields and ratios of epoxides were determined by 1H NMR. | ||||
| 1 | 2 | L1a | 33 | 1.7:1 |
| 2 | (Mn-CP1)2 | L1a | 55 | 1.9.1 |
| 3 | 2 | L2b | 22 | 1:1 |
| 4 | (Mn-CP1)2 | L2b | 37 | 1.2:1 |
Due to the low association constant of Mn-porphyrins with pyridine derivatives, L1 was replaced by the hindered phenoxide L2, whose increased affinity for manganese complex should ensure that oxidation took place on the inner face of the catalyst.19,22 In fact, UV studies showed that the Soret band of the porphyrin did not change appreciably after adding L1, but it did so after the addition of phenoxide L2 (Fig. S1 and S2†).18a,22 Therefore, we decided to study L2 as ligand in competitive epoxidation of both isomers of prop-1-en-1-ylbenzene. Under these conditions, a slight preference for epoxidation of the Z-isomer (1.2:1) was found when (Mn-CP1)2 was employed as catalyst (Table 1, entries 3 and 4). No selectivity and lower yields were obtained when porphyrin 2 was used as catalyst. Therefore, the Mn capsules are more reactive than porphyrin model regardless of the ligand used, but unfortunately the selectivity achieved, although slightly better, is not very significant.
The lack of a clear selectivity that could be attributed to the preferential confinement of a substrate in the nanoreactor (Mn-CP1)2, led us to carry out new competitive experiments, in this case comparing two different substrates. For this experiment, mono- and disubstituted olefines were compared using 1-octene and Z-cyclooctene. A preferential epoxidation of the cyclic derivative was already found with the model porphyrin 2 and the pyridine ligand L1, which was slightly enhanced when the capsule (Mn-CP1)2 (8:1) was used (Table 2, entries 1 and 2). Epoxidation in the presence of bulky phenolic ligand L2 resulted in a significant reduction in yield, although selectivity was improved when the model porphyrin was used (Table 2, entry 3). Once again, the epoxidation catalysed with (Mn-CP1)2 gave better yields than 2, but the selectivity remained similar. This could suggest that the use of stronger coordinating ligands, although it reduces their activity, increases their selectivity. However, the incorporation of the size selectivity was not observed with a preferent epoxidation of the more substituted olefine.
|
||||
|---|---|---|---|---|
| Entry | Cat | Ligand | Yieldc (%) | Ratio A vs. B |
| a Reaction conditions: both alkenes (0.13 mmol each), iodosobenzene (0.13 equiv.), ligand L1 (25–500 equiv.) and catalyst (1 mol%) were dissolved in CDCl3 and stirred overnight. Then it was transferred to an NMR tube.b Reaction conditions: both alkenes (0.1 mmol each), iodosobenzene (0.1 equiv.), ligand L2 (20 equiv.) and catalyst (1 mol%) were dissolved in CD3CN and stirred overnight and then transferred to an NMR tube.c Yields and ratios were determined by 1H NMR. | ||||
| 1 | 2 | L1a | 53 | 7.5:1 |
| 2 | (Mn-CP1)2 | L1a | 56 | 8:1 |
| 3 | 2 | L2b | 12 | 10.5:1 |
| 4 | (Mn-CP1)2 | L2b | 16 | 10.6:1 |
To try to shed some light on these results, we modelled the capsule using DFT methods to understand the catalytic properties of the molecular cap in relation to the capsule cavity (see the Computational methods in ESI†). As can be seen in Fig. 5, using a Zn porphyrin model to simplify the calculations, there is a large distance between the cyclic peptide backbone and the porphyrin cap, which creates large channels through which substrates can enter the reaction chamber without requiring the disassembly of the dimer but can also react without being completely encapsulated. Therefore, the size or shape of the substrates cannot be determinant of the reaction selectivity.
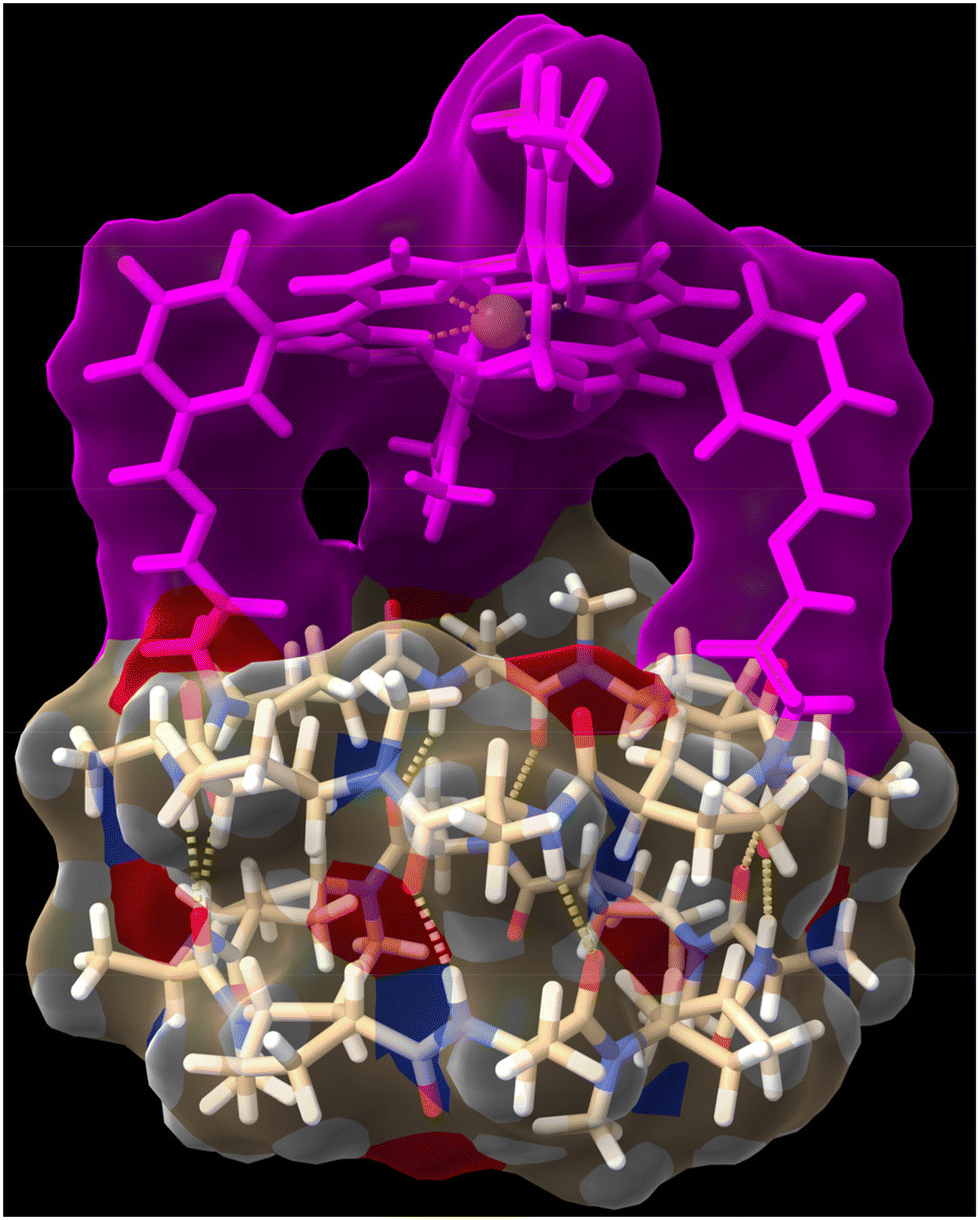 | ||
| Fig. 5 Side view of computer-minimized [B3LYP/6-31G(d) (LANL2DZ for Zn)] model of hemicapsule Zn-CP1 structure using a heterodimeric model by combining it with cyclic octapeptide c-[(D-Ala-(1R,3S)-MeN-Acp-)4]. | ||
As alternative to Mn-porphyrin, several groups have developed nonheme iron oxygenases-like catalysts, which are based on tetradentate aminopyridine ligands for FeII.23 These catalysts have found successful applications in the enantioselective epoxidation of alkenes and C–H oxidation of hydrocarbon chains.24
We envisioned that the functionalization of this kind of ligands with two carbonyl moieties pointing in the same direction, such as iron complex 3a, would allow to condense with hydrazide moieties of cyclic peptide 1 (Fig. 6) forming a new nanoreactor. Therefore, we expected that the catalytic centre would be oriented towards the internal cavity of the reactor if the ligand had formyl groups in the meta-position of the pyridine nucleus. The synthesis of ligand 4 was carried out by nucleophilic substitution of methyl 6-(chloromethyl)-nicotinate and N,N′-dimethylethane-1,2-diamine and subsequent transformation of the ester group into the corresponding aldehyde (Scheme S4†). Compound 3b resulting from the condensation of 4 with phenylhydrazine was used as an additional model catalyst together with commercial White–Chen catalyst 5 (Fig. 6B).
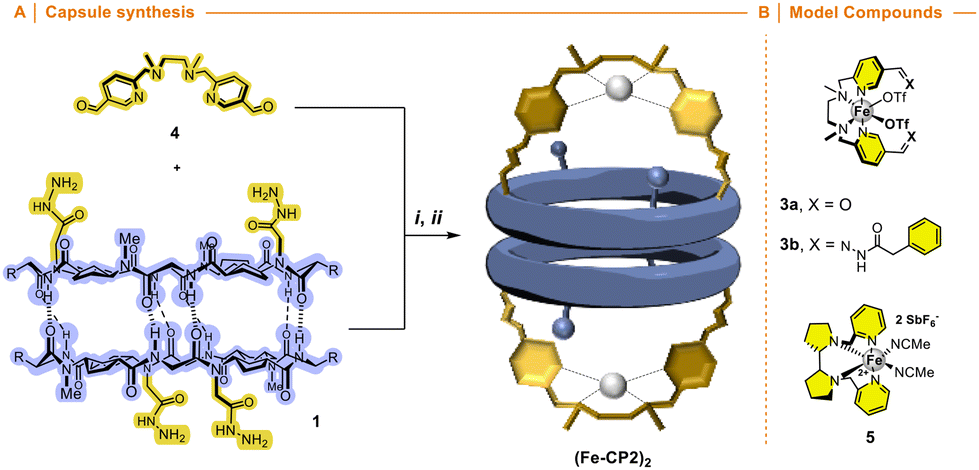 | ||
| Fig. 6 (A) Synthesis of the molecular capsule (Fe-CP2)2 based on α,γ-cyclic peptide functionalized with tetradentate aminopyridine Fe complex. Reagents i. 4, 1, DCM, rt; ii. Fe(OTf)2·2MeCN, THF; (B) structure of model catalysts 3a, 3b and 5. | ||
Condensation of 4 with 1 and subsequent metalation with Fe(OTf)2·2MeCN provided the second catalytic capsule (Fe-CP2)2 in almost quantitative yield (Fig. 6). As in precedent works, the catalytic activity of (Fe-CP2)2 was evaluated and then competitive epoxidation studies between oct-1-ene and E-oct-4-ene were carried out. In this case, H2O2 was employed as stoichiometric oxidant. Low discrimination was observed when capsule (Fe-CP2)2 was used as a catalyst (Table 3, entry 3). It is notorious that the incorporation of electron withdrawing substituents at the pyridine moieties induces a severe reduction in the catalytic activity of these complexes compared with White–Chen catalyst (Table 3, entry 1). In any case, slightly better ratio in favour of the internal alkene was obtained when model catalyst 3b and commercial catalyst 5 were employed, suggesting a minor preference for the more nucleophilic olefin. In any case, the comparation between olefines with different reactivities seems to hinder the selectivity derived from size restrictions due to confinement.
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield (%) | Ratio C vs. D |
| a Reaction conditions: a stock solution of 1-octene and (E)-oct-4-ene was prepared in MeCN-d3. Then, it was added over a mass vial containing the catalyst (3 mol%). H2O2 (30%, 1.2 equiv.) and AcOH (0.5 equiv.) were added, and the sample was analysed by 1H NMR after 30 minutes. | |||
| 1 | 5 | 51 | 1:1.4 |
| 2 | 3b | 36 | 1:1.4 |
| 3 | (Fe-CP2)2 | 10 | 1:1.1 |
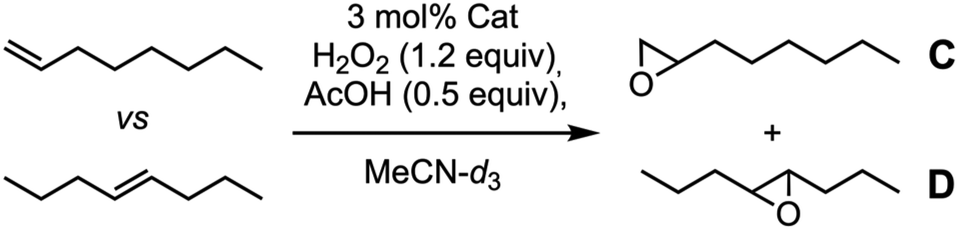
We reasoned that these results could be due to the use of two substrates of similar sizes, so we decided to evaluate other alternatives. Thus, we challenged the competitive epoxidation comparing reactivity of a cyclic derivative (cyclohexene) with a linear one [(E)-oct-4-ene]. Our hypothesis was to promote the preferential epoxidation of the small cyclohexene molecule which should fit better within the capsule than the linear E-alkene. Again, control tests were made with commercial White–Chen catalyst 5 and model dihydrazone 3b. According to the results showed in Table 4, White–Chen catalyst already showed a 3-fold preference for cyclic alkene (Table 3, entry 1). This preference increases to 4.2 when model hydrazone 3b was used. Probably, the hydrazone 3b creates some steric hindrance around the iron ion that favours the epoxidation of small cyclic alkene (Table 3, entry 2). However, once again, epoxidation yields decrease with respect to White–Chen catalyst from 58% to 30%. Epoxidation with (Fe-CP2)2 showed higher selectivity for cyclohexene, up to 7.5:1, which, as expected, could mean that cyclohexene encapsulation is favoured. However, the oxidation yield was only 15% (Table 4, entry 3).
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield (%) | Ratio F vs. G |
| a Reaction conditions: a stock solution of cyclohexene and (E)-oct-4-ene was prepared in MeCN-d3. Then, it was added over a mass vial containing the catalyst (3 mol%). H2O2 (30%, 1.2 equiv.) and AcOH (0.5 equiv.) were added, and the sample was analysed by 1H NMR after 30 minutes. | |||
| 1 | 5 | 58 | 3.4:1 |
| 2 | 4 | 30 | 4.2:1 |
| 3 | (Fe-CP2)2 | 15 | 7.5:1 |
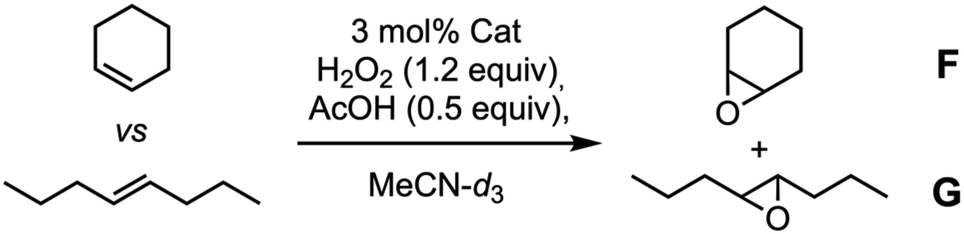
DFT calculations were also carried out to assess the capsule structure (Fig. 7, see the Computational methods in ESI†). In this case, although the ligand is closer to the cyclic peptide backbone, there are two side channels through which substrates can approach the catalytic centre from outside the capsule. Unfortunately, although better selectivity was achieved with the supramolecular capsule, the use of electron-withdrawing linkers reduces its catalytic efficiency, limiting its use. Therefore, we decided to develop a new capsule model to also show the versatility of the catalytically active supramolecular capsules approach.
 | ||
| Fig. 7 Side view of computer-minimized [B3LYP/6-31G(d) (LANL2DZ for Fe)] model of hemicapsule Fe-CP2 structure using a heterodimeric model by combining it with cyclic octapeptide c-[(D-Ala-(1R,3S)-MeN-Acp-)4]. The trifluoromethanesulfonate counterions were removed for better visualization the metal complex accessibility to carry out catalysis. | ||
For the new design, we consider that the observed low selectivity may derive form the high mobility of the cap because of the linkers used to connect both components. In this way, the catalytic centre is quite far from the internal cavity and, in addition, can oscillate over the channel entrance (Fig. 8), leaving the metal centre exposed to the medium and, therefore, being able to catalyse reactions without the full inclusion of the substrate. Additionally, N-acyl hydrazone isomers might provide different environments to the catalyst reducing its selectivity.
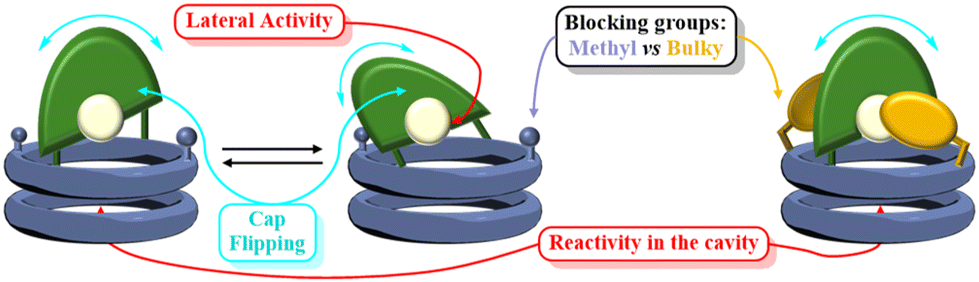 | ||
| Fig. 8 Cap flipping which explains the low selectivity observed. | ||
To overcome these drawbacks, we decided to introduce some modifications in the capsule structure. The modular character of the nanoreactor allows to make these changes without interfering in the self-assembly properties. To restrict the oscillation of the catalyst at the entrance of the capsule, we anticipated that the incorporation of a bulkier group in place of the methyl moieties would reduce this mobility, fixing it in front of the entrance of the cavity. Therefore, two naphthalen-2-ylmethyl moieties were incorporated onto a cyclic octapeptide to block the free space between the catalytic centre and the capsule to avoid the mentioned lateral reactivity. Furthermore, the conformationally and configurationally flexible hydrazone linker was replaced by a triazole moiety to provide increased rigidity. Therefore, we decided to use a copper catalysed alkyne-azide [3 + 2] cycloaddition (CuAAC) to connect both components (cap and CP) in the last step of the synthesis. Finally, a BINOL was used as a new cap. The two phenolic groups of BINOL should be pointing towards the cavity to ensure that the reaction takes place inside the capsule. For the preparation of the substituted Acp (3-aminocyclohexanecarboxylic acid) derivatives we used the alkylation of the nosyl-protected derivative 6 (Fig. 9).14c,15,25 After deprotection of the amino group with thiophenol in basic conditions, the resulting compounds were coupled with Boc-protected D-Leu. The resulting dipeptides (dp1 and dp2) were coupled to form the tetrapeptide tp1. This compound was divided into two fractions, and each was separately deprotected at its N- or C-terminus and the resulting tetrapeptides were coupled under standard conditions. Finally, the octapeptide was cyclized, after the corresponding removal of methyl ester and t-butyl carbamate, under diluted conditions to provide 7 in 45% yield.
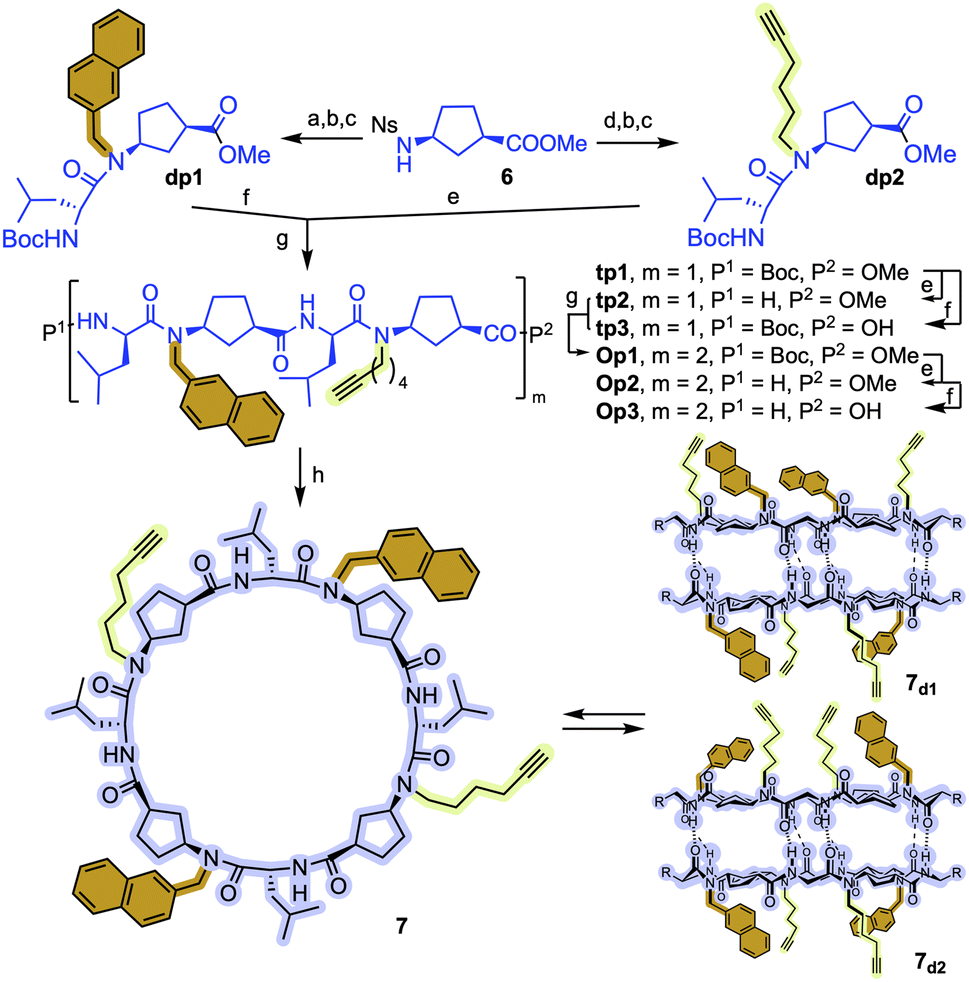 | ||
Fig. 9 Synthetic scheme used for the preparation of CP3. Reaction conditions: (a) NaphCH2Br, K2CO3, DMF, 75%; (b) PhSH, K2CO3, DMF, 55–89%; (c) Boc-D-Leu-OH, N-HATU, DIEA, CH2Cl2, 79–92%; (d) HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C(CH2)4-OTs, K2CO3, DMF, 97%; (e) TFA, CH2Cl2; (f) LiOH, MeOH; (g) N-HBTU, DIEA, CH2Cl2, 83–86%; (h) N-TBTU, DIEA, CH2Cl2, 45%. C(CH2)4-OTs, K2CO3, DMF, 97%; (e) TFA, CH2Cl2; (f) LiOH, MeOH; (g) N-HBTU, DIEA, CH2Cl2, 83–86%; (h) N-TBTU, DIEA, CH2Cl2, 45%. | ||
As expected, in chloroform, 7 formed the two corresponding dimers, the alternated and the eclipsed one (7d1 and 7d2), depending on the matching or mismatching pairing of N-substituted Acp residues (Fig. S3†). This was clearly denoted by the splitting of the amide proton signals into two pairs of doublets. Both isomeric dimers are formed in almost 1:2 ratio, although we were unable to assign the structure of each dimer.
The CuAAC between diazidobinol 8 (Scheme S4†) and 7 provided the corresponding hemicapsule CP3 in 62% yield (Fig. 10). Although this compound can also form the corresponding alternated or eclipsed dimers (CP3)2, it seems that mostly only one of them is formed (Fig. S4†). Unfortunately, as already mentioned for the previous capsule models, the signals are broad and not very well defined, making difficult to differentiate between both species. Finally, MOM groups were removed by treatment with hydrochloric acid in a mixture of methanol and DCM to give rise CP4. Once again, NMR studies did not allow full characterization of the dimeric equilibrium and structure (Fig. S5†), although it seems that one of the dimers is predominantly formed. Semiempirical calculations (PM7)26 of both dimeric models suggest the preferential formation of the alternating dimer (Fig. S6†). In both structures naphthalen moieties are oriented towards the interior of the assembling filling their cavities.
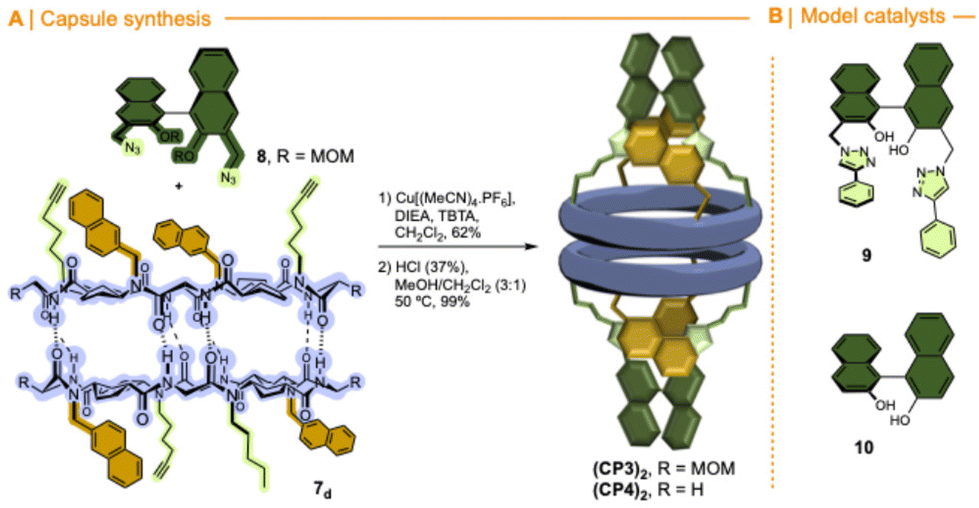 | ||
| Fig. 10 (A) Synthesis of a molecular capsule based on α,γ-cyclic peptide functionalized with (S)-BINOL ligand. (B) Control compounds. | ||
In any case, the presence of these two species should not affect its reactivity, so the study of (CP4)2 was carried out. To do that, we were inspired on Mancheño metallic complexes of Ti(O-iPr)4 and BINOL decorated with triazoles to catalyse the enantioselective alkylation of aldehydes with diethylzinc.27 The best results were obtained when using non-polar solvents as toluene, observing a decrease in yields and enantiomeric excess as the polarity of the solvent increases. For example, when using dichloromethane the yields drop to 35% and almost a racemic mixture was obtained. We envisioned that capsule (CP4)2 could be used to catalyse the same reaction while preferring small aldehydes over bulky ones. Unfortunately, the capsule was not soluble in toluene, forcing us to carry out the studies in dichloromethane although these were not the optimal conditions. DFT calculations of a heterodimeric model combining Ti-CP4 with c-[(D-Leu-(1R,3S)-MeN-Acp-)4] were performed to understand the structure of this complex (Fig. 11). The presence of titanium complex expels the naphthyl moieties from the internal cavity, liberating it for the encapsulation of reagents and facilitating the catalytic process. Furthermore, these groups are arranged perpendicularly to the disk-shaped CPs, generating “molecular walls” around the Ti complex that must restrict the access of the substrates to the active site. In any case, the length of the linkers between the CP and the cap still seems quite long to fully protect the Ti-BINOL complex and prevent it from reacting with substrates outside the capsule. To confirm the formation of the titanium complex within the capsule, titanium tetraisopropoxide was added to a solution of (CP4)2 in dichloromethane and studied by UV. The characteristic band shift was observed after the addition of one equivalent of Ti, confirming the formation of (Ti-CP4)2 (Fig. 12, left).
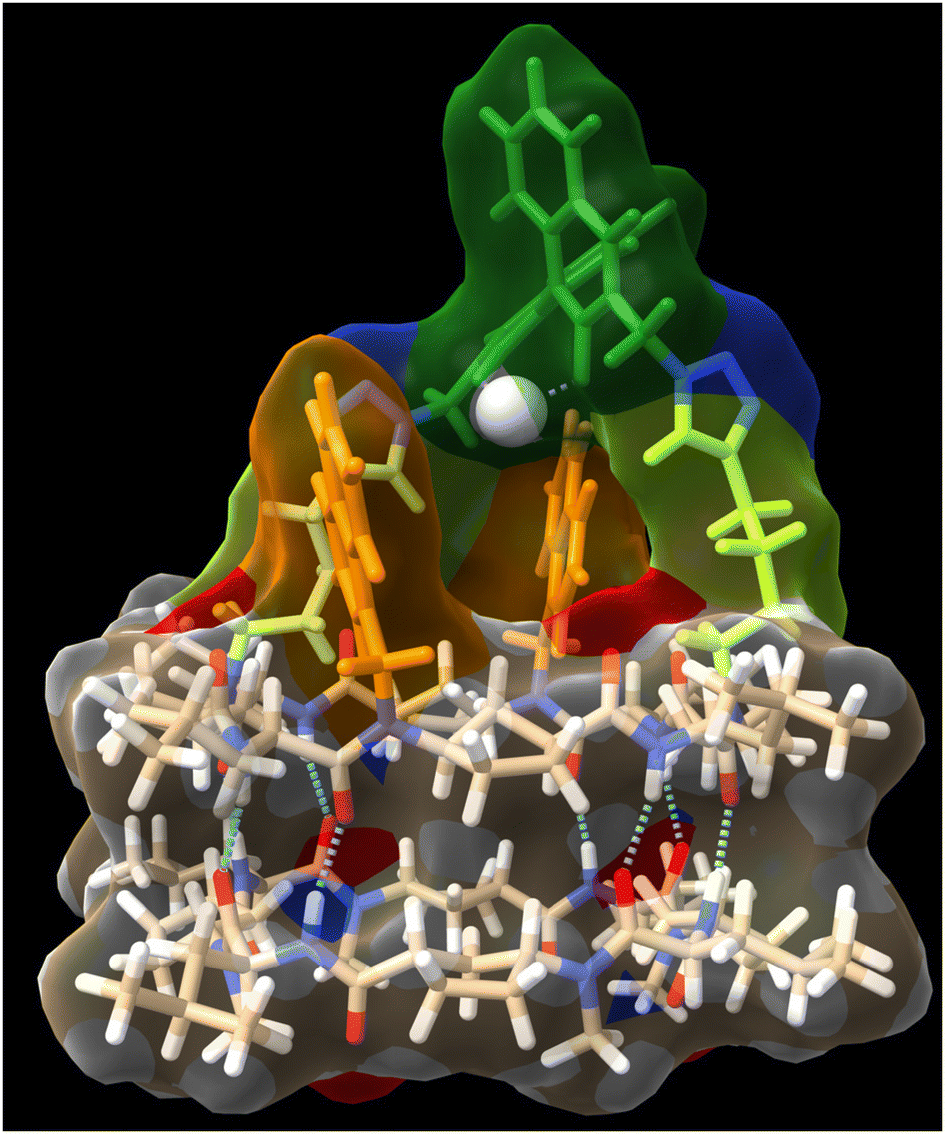 | ||
| Fig. 11 Side view of computer-minimized [B3LYP/6-31G(d) (LANL2DZ for Ti)] model of a heterodimeric complex of titanium coordinated cyclic peptide bearing a BINOL cap (Ti-CP4) with c-[(D-Leu-(1R,3S)-MeN-Acp-)4], confirming the role of naphthalen-2-ylmethyl moieties forming lateral molecular walls around the BINOL cap. Isopropoxide were removed for better visualization of the metal complex responsible for catalysis. | ||
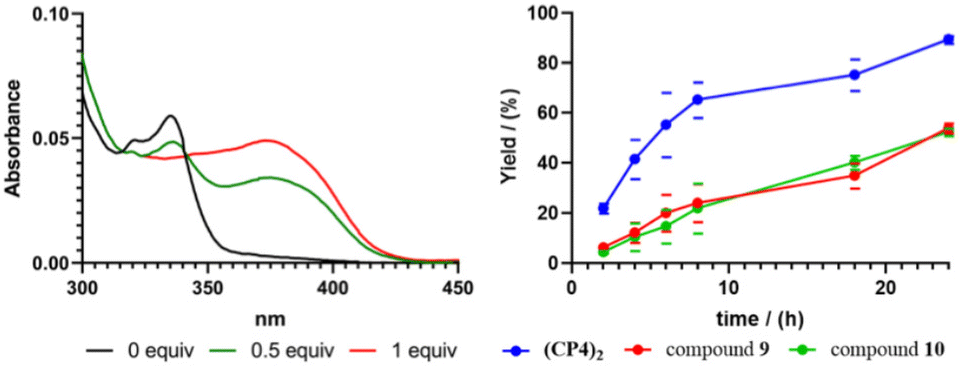 | ||
| Fig. 12 Left, UV studies to evaluate the Ti coordination of (CP4)2 in which the blue shift of the absorption upon addition of Ti(O-iPr)4 is observed. Right, reaction kinetics for benzaldehyde alkylation (reaction catalysed by (CP4)2 in blue, 9 in red and 10 in green). | ||
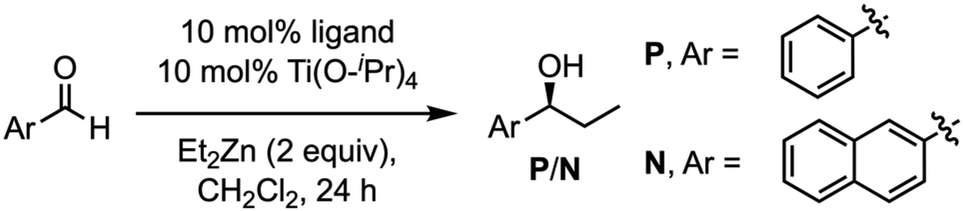
One confirmed the ability of CP4 to coordinate Ti, the reactivity studies were addressed. As it is shown in Table 5, using CH2Cl2 as a solvent, alkylation reaction provided better yields when hemicapsule CP4 was used as the ligand in comparison with (S)-BINOL (10) or Mancheño's ligand 9. Furthermore, competitive experiments between benzaldehyde and 2-naphthaldehyde were also carried out. To our delight, a clear increase in selectivity from 1.5 to 5 was obtained for the alkylation of the small aldehyde when nanoreactor (CP4)2 was used as catalyst. Clearly, this selectivity can be attributed to the expected size selection in which the aldehydes that fit better inside the cavity are more reactive. Enantioselectivity also showed a sluggish increase when the capsule was used as a ligand, although, in any case, this was too low to be considered relevant, further studies should be carried out.
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Yield (%) | Ratio P vs. N | ee (P) |
| a Reaction conditions: ligand (10 mol%) and Ti(O-iPr)4 (10 mol%) were stirred in CH2Cl2 for 30 min. After this time, the aldehyde (0.25 mmol, 0.3 M) was added and, 30 min later, Et2Zn (0.50 mmol, 2 equiv., 1 M in hexanes) was also added. The reaction was then stirred at r.t. for 24 h. | ||||
| 1 | 9 | 56 | 1.5:1 |
6% |
| 2 | 10 | 57 | 2:1 |
— |
| 3 | (CP4)2 | 84 | 5:1 |
15% |
Kinetics experiments to evaluate the influence of the capsule model on the reaction rates shown a large increase in product formation when using (CP4)2, compared with simple models (Fig. 11, right). It appears that after 8 h a yield of 70% is already achieved when using the nanocontainer, while with the other two simpler catalysts less than 20% is formed. These differences in reaction rates could explain the low selectivity found between benzaldehyde and 2-naphthaldehyde. Most likely, after 8 h, most of the benzaldehyde has already been consumed while the catalyst is still able to catalyse the transformation of the larger derivative during the rest of the reaction time. The mismatch between rate enhancement and enantiomeric excess might suggest, as already reported by Mancheño, that the position of triazoles and its substituent with respect to BINOL is important to generate a better chiral environment. In any case, this represents the first step to the development of more efficient catalysts.
Conclusions
In summary, a strategy to create modular molecular capsules using dimer-forming α,γ-cyclic peptide was described. For this purpose, the cyclic peptides are provided with pendants placed at the amide nitrogen of two of the amino acids of the same chirality. Those blocking groups are designed to react with different kind of molecular caps through click-type connections. The cyclopeptide skeleton allows the properties of the cavity to be adjusted in terms of its size, polarity and functionalization by choosing the appropriate number and type of amino acids, as well as its external surface according to the side chains of the amino acids used.6 Molecular pendants endowed with reactive groups involved in orthogonal click transformations allow the molecular cap to be rapidly attached to the cyclic peptide. The cap is placed at the entrance of the dimer cavity, forming the supramolecular capsule. This allows to use the same peptide skeleton with different molecular caps, favouring the versatility of the capsule design. By the introduction of a catalytic component into the stopper, molecular capsules have been transformed into nanoreactors. Although the selectivity and efficiency of the studied processes still need to be improved, we believe in the potential of the proposed modular strategy, in which different chemical transformations can be developed using a common capsule model whose catalytic element is incorporated ad hoc at the end of the synthesis according to the synthetic needs of each moment. Therefore, we envisage that the strategy can be used in the functionalization of challenging molecules achieving selectivity in substrates with different functional groups.Author contributions
ETM, AF and ALFA carried out the experiments and analysed the data. MC performed the molecular calculations. MA and JRG designed and supervised the project. ALFA and JRG wrote the manuscript.Data availability
All files needed to reproduce the computational part (3D coordinates and Gaussian inputs and outputs, etc.) are uploaded to the Zenodo repository. These files will be accessible to reviewers at this time through the following link: https://zenodo.org/records/14075004?token=eyJhbGciOiJIUzUxMiJ9.eyJpZCI6ImNkZDE0NDU0LTBmOGQtNGUyYS04MTJlLTIxZjVmZjQ1MDVjYiIsImRhdGEiOnt9LCJyYW5kb20iOiJiYTA3MDQ1NGYzZWM1YThhYWQyNzJmYzFmZjUxMTlmYiJ9.CossHigRLZ5HvCoaYD2y-eip5GvEdcrMnCLb-7kW2_vkuSHckeOkuV1DJlYMd_N4KWUr1hazTi64vRTZB_kaIQConflicts of interest
There are no conflicts to declare.Acknowledgements
The Spanish Agencia Estatal de Investigación (AEI) (PID2019-111126RB-100 and PID2022-142440NB-I00), the Xunta de Galicia (ED431C 2021/21 and Centro de investigación do Sistema Universitario de Galicia accreditation 2023-2027, ED431G 2023/03), and the European Union (European Regional Development Fund -ERDF) are gratefully acknowledge. We also thank the ORFEO-CINCA network and Mineco (RED2022- 134287-T). A. L. F. A. thanks the Spanish Agencia Estatal de Investigación (AEI) for a Juan de la Cierva fellowship (FJCI-2015-26847). E. T. M. thanks the Spanish Ministry of Universities for his FPI contract. Prof. Miquel Costas (University of Girona) is acknowledged for fruitful discussions about the reactivity of iron(II) complexes.References
- R. Saha, B. Mondal and P. S. Mukherjee, Molecular Cavity for Catalysis and Formation of Metal Nanoparticles for Use in Catalysis, Chem. Rev., 2022, 122, 12244–12307 CrossRef CAS PubMed.
- (a) T. Lorenzetto, F. Bordignon, L. Munarin, F. Mancin, F. Fabris and A. Scarso, Substrate Selectivity Imparted by Self-Assembled Molecular Containers and Catalysts, Chem. – Eur. J., 2024, 30, e202301811 CrossRef CAS PubMed; (b) R. G. DiNardi, S. Rasheed, S. S. Capomolla, M. H. Chak, I. A. Middleton, L. K. Macreadie, J. P. Violi, W. A. Donald, P. J. Lusby and J. E. Beves, Photoswitchable Catalysis by a Self-Assembled Molecular Cage, J. Am. Chem. Soc., 2024, 146, 21196–21202 Search PubMed; (c) Y. Xue, X. Hang, J. Ding, B. Li, R. Zhu, H. Pang and Q. Xu, Catalysis within Coordination Cages, Coord. Chem. Rev., 2021, 430, 213656 CrossRef CAS; (d) C. Gaeta, P. La Manna, M. De Rosa, A. Soriente, C. Talotta and P. Neri, Supramolecular Catalysis with Self–Assembled Capsules and Cages: What Happens in Confined Spaces, ChemCatChem, 2021, 13, 1638–1658 CrossRef CAS; (e) M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Advances in Supramolecular Host-Mediated Reactivity, Nat. Catal., 2020, 3, 969–984 CrossRef CAS; (f) L. Catti, Q. Zhang and K. Tiefenbacher, Advantages of Catalysis in Self–Assembled Molecular Capsules, Chem. – Eur. J., 2016, 22, 9060–9066 Search PubMed; (g) S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Transition Metal Catalysis in Confined Spaces, Chem. Soc. Rev., 2015, 44, 433–448 RSC; (h) M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Supramolecular Catalysis. Part 1: Non-Covalent Interactions as a Tool for Building and Modifying Homogeneous Catalysts, Chem. Soc. Rev., 2014, 43, 1660–1733 RSC; (i) R. S. Forgan and G. O. Lloyd, Reactivity in Confined Spaces, Monographs in Supramolecular Chemistry, Royal Society of Chemistry, Croydon, UK, 2021 Search PubMed.
- (a) A. Llamosí, M. P. Szymański and A. Szumna, Molecular Vessels from Preorganised Natural Building Blocks, Chem. Soc. Rev., 2024, 53, 4434–4462 RSC; (b) C. T. McTernan, J. A. Davies and J. R. Nitschke, Beyond Platonic: How to Build Metal–Organic Polyhedra Capable of Binding Low-Symmetry, Information-Rich Molecular Cargoes, Chem. Rev., 2022, 122, 10393–10437 CrossRef CAS PubMed.
- (a) T. Tatsumi, K. Sasamoto, T. Matsumoto, R. Hirano, K. Oikawa, M. Nakano, M. Yoshida, K. Oisaki and M. Kanai, Practical N-to-C Peptide Synthesis with Minimal Protecting Groups, Commun. Chem., 2023, 6, 231 CrossRef CAS PubMed; (b) M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, A Green Chemistry Perspective on Catalytic Amide Bond Formation, Nat. Catal., 2019, 2, 10–17 CrossRef CAS; (c) E. Valeur and M. Bradley, Amide Bond Formation: Beyond the Myth of Coupling Reagents, Chem. Soc. Rev., 2009, 38, 606–631 RSC; (d) A. El-Faham and F. Albericio, Peptide Coupling Reagents, More than a Letter Soup, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- (a) L. Shao, J. Ma, J. L. Prelesnik, Y. Zhou, M. Nguyen, M. Zhao, S. A. Jenekhe, S. V. Kalinin, A. L. Ferguson, J. Pfaendtner, C. J. Mundy, J. J. De Yoreo, F. Baneyx and C.-L. Chen, Hierarchical Materials from High Information Content Macromolecular Building Blocks: Construction, Dynamic Interventions, and Prediction, Chem. Rev., 2022, 122, 17397–17478 CrossRef CAS PubMed; (b) Y. Chen, K. Tao, W. Ji, V. B. Kumar, S. Rencus-Lazar and E. Gazit, Histidine as a Key Modulator of Molecular Self-Assembly: Peptide-Based Supramolecular Materials Inspired by Biological Systems, Mater. Today, 2022, 60, 106–127 CrossRef CAS; (c) T. Li, X.-M. Lu, M.-R. Zhang, K. Hu and Z. Li, Peptide-Based Nanomaterials: Self-Assembly, Properties and Applications, Bioact. Mater., 2021, 11, 268–282 Search PubMed; (d) A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Biomimetic Peptide Self-Assembly for Functional Materials, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS PubMed.
- (a) Q. Song, Z. Cheng, M. Kariuki, S. C. L. Hall, S. K. Hill, J. Y. Rho and S. Perrier, Molecular Self-Assembly and Supramolecular Chemistry of Cyclic Peptides, Chem. Rev., 2021, 121, 13936–13995 CrossRef CAS PubMed; (b) N. Rodríguez-Vázquez, M. Amorín and J. R. Granja, Recent Advances in Controlling the Internal and External Properties of Self-Assembling Cyclic Peptide Nanotubes and Dimers, Org. Biomol. Chem., 2017, 15, 4490–4505 RSC; (c) A. L. Fuentes de Arriba and J. R. Granja, Self-assembling Cyclic Peptide Nanotubes: Methods and Characterization, in Peptide Self–Assembly and Engineering: Fundamentals, Structures, and Applications, ed. X. Yan, Blackwell Verlag GmbH, Weinheim, Germany, 2024, vol. 1, pp. 109–141 Search PubMed.
- (a) A. Lamas, A. Guerra, M. Amorín and J. R. Granja, New Self-Assembling Peptide Nanotubes of Large Diameter Using δ-Amino Acids, Chem. Sci., 2018, 9, 8228–8233 RSC; (b) S. Leclair, P. Baillargeon, R. Skouta, D. Gauthier, Y. Zhao and Y. L. Dory, Micrometer–Sized Hexagonal Tubes Self–Assembled by a Cyclic Peptide in a Liquid Crystal, Angew. Chem., Int. Ed., 2004, 43, 349–353 CrossRef CAS PubMed; (c) N. Khazanovich, J. R. Granja, D. E. McRee, R. A. Milligan and M. R. Ghadiri, Nanoscale Tubular Ensembles with Specified Internal Diameters. Design of a Self-Assembled Nanotube with a 13 Å Pore, J. Am. Chem. Soc., 1994, 116, 6011–6012 CrossRef CAS.
- (a) M. R. Ghadiri, K. Kobayashi, J. R. Granja, R. K. Chadha and D. E. McRee, The Structural and Thermodinamic Basis of Self-Assembled Peptide Nanotubes, Angew. Chem., Int. Ed. Engl., 1995, 34, 93–95 CrossRef CAS; (b) M. Amorín, L. Castedo and J. R. Granja, New Cyclic Peptide Assemblies with Hydrophobic Cavities: The Structural and Thermodynamic Basis of a New Class of Peptide Nanotubes, J. Am. Chem. Soc., 2003, 125, 2844–2845 CrossRef PubMed.
- (a) M. Amorín, L. Castedo and J. R. Granja, Folding Control in Cyclic Peptides through N–Methylation Pattern Selection: Formation of Antiparallel β–Sheet Dimers, Double Reverse Turns and Supramolecular Helices by 3α,γ-Cyclic Peptides, Chem. – Eur. J., 2008, 14, 2100 CrossRef PubMed; (b) M. Amorín, A. Pérez, J. Barberá, H. L. Ozores, J. L. Serrano, J. R. Granja and T. Sierra, Liquid Crystal Organization of Self-Assembling Cyclic Peptides, Chem. Commun., 2014, 50, 688–690 RSC; (c) N. Rodríguez-Vázquez, M. Amorín, I. Alfonso and J. R. Granja, Anion Recognition and Induced Self–Assembly of an α,γ–Cyclic Peptide to Form Spherical Clusters, Angew. Chem., Int. Ed., 2016, 55, 4504–4508 CrossRef PubMed.
- (a) M. Panciera, M. Amorín, L. Castedo and J. R. Granja, Design of Stable β–Sheet–Based Cyclic Peptide Assemblies Assisted by Metal Coordination: Selective Homo– and Heterodimer Formation, Chem. – Eur. J., 2013, 19, 4826–4834 CrossRef CAS PubMed; (b) R. J. Brea, L. Castedo, J. R. Granja, M. A. Herranz, L. Sánchez, N. Martín, W. Seitz and D. M. Guldi, Electron Transfer in Me-Blocked Heterodimeric α,γ-Peptide Nanotubular Donor–Acceptor Hybrids, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5291–5294 Search PubMed; (c) M. J. Pérez-Alvite, M. Mosquera, L. Castedo and J. R. Granja, Toward the Rational Design of Molecular Rotors Ion Sensors Based on α,γ-Cyclic Peptide Dimers, Amino Acids, 2011, 41, 621–628 CrossRef PubMed.
- A. Pizzi, H. L. Ozores, M. Calvelo, R. García-Fandiño, M. Amorín, N. Demitri, G. Terraneo, S. Bracco, A. Comotti, P. Sozzani, C. X. Bezuidenhout, P. Metrangolo and J. R. Granja, Tight Xenon Confinement in a Crystalline Sandwich–like Hydrogen–Bonded Dimeric Capsule of a Cyclic Peptide, Angew. Chem., Int. Ed., 2019, 58, 14472–14476 CrossRef CAS PubMed.
- N. Rodríguez-Vázquez, R. García-Fandiño, M. J. Aldegunde, J. Brea, M. I. Loza, M. Amorín and J. R. Granja, cis-Platinum Complex Encapsulated in Self-Assembling Cyclic Peptide Dimers, Org. Lett., 2017, 19, 2560–2563 CrossRef PubMed.
- N. Rodríguez-Vázquez, R. García-Fandiño, M. Amorín and J. R. Granja, Self-Assembling α,γ-Cyclic Peptides that Generate Cavities with Tunable Properties, Chem. Sci., 2016, 7, 183–187 RSC.
- (a) J. Sánchez-Quesada, M. P. Isler and M. R. Ghadiri, Modulating Ion Channel Properties of Transmembrane Peptide Nanotubes through Heteromeric Supramolecular Assemblies, J. Am. Chem. Soc., 2002, 124, 10004–10005 CrossRef PubMed; (b) A. Fuertes, M. Amorín and J. R. Granja, Membrane Spanners Based on a Dimeric α,-Cyclic Peptide Core, Supramol. Chem., 2020, 32, 239–246 CrossRef CAS; (c) A. Fuertes, H. L. Ozores, M. Amorín and J. R. Granja, Self-Assembling Venturi-Like Peptide Nanotubes, Nanoscale, 2017, 9, 748–753 Search PubMed; (d) A. Fuertes, M. Amorín and J. R. Granja, Versatile Symport Transporters Based on Cyclic Peptide Dimers, Chem. Commun., 2020, 56, 46–49 Search PubMed.
- H. L. Ozores, M. Amorín and J. R. Granja, Self-Assembling Molecular Capsules Based on α,γ-Cyclic Peptides, J. Am. Chem. Soc., 2017, 139, 776–784 Search PubMed.
- V. López-Corbalán, A. Fuertes, A. L. Llamas-Saiz, M. Amorín and J. R. Granja, Recognition of Anion-Water Clusters by Peptide-Based Supramolecular Capsules, Nat. Commun., 2024, 15, 6055 CrossRef PubMed.
- N. K. Devaraj and M. G. Finn, Introduction: Click Chemistry, Chem. Rev., 2021, 121, 6697–6698 Search PubMed.
- (a) P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Epoxidation of Polybutadiene by a Topologically Linked Catalyst, Nature, 2003, 424, 915–918 CrossRef CAS PubMed; (b) X. Chen, Q. Duez, G. L. Tripodi, P. J. Gilissen, D. Piperoudis, P. Tinnemans, J. A. A. W. Elemans, J. Roithová and R. J. M. Nolte, Mechanistic Studies on the Epoxidation of Alkenes by Macrocyclic Manganese Porphyrin Catalysts, Eur. J. Org. Chem., 2022, e202200280 CrossRef CAS PubMed.
- J. P. Collman, X. Zhang, V. J. Lee, E. S. Uffelman and J. Brauman, Regioselective and Enantioselective Epoxidation Catalyzed by Metalloporphyrins, Science, 1993, 261, 1404–1411 Search PubMed.
- (a) M. Costas, Selective C–H Oxidation Catalyzed by Metalloporphyrins, Coord. Chem. Rev., 2011, 255, 2912–2932 CrossRef CAS; (b) M. C. Feiters, A. E. Rowan and R. J. M. Nolte, From Simple to Supramolecular Cytochrome P450 Mimics, Chem. Soc. Rev., 2000, 29, 375–384 Search PubMed.
- S. Mecozzi and J. Rebek Jr., The 55% Solution: A Formula for Molecular Recognition in the Liquid State, Chem. – Eur. J., 1998, 4, 1016–1022 Search PubMed.
- L.-C. Yuan and T. C. Bruice, Influence of Nitrogen Base Ligation and Hydrogen Bonding on the Rate Constant for Oxygen transfer from Percaboxylic Acid and Alkyl Hydroperoxides to meso-Tetraphenylporphyrinato)manganese(III) Chloride, J. Am. Chem. Soc., 1986, 108, 1643–1650 CrossRef CAS.
- S. Kal, S. Xu and L. Que, Bio-inspired Nonheme Iron Oxidation Catalysis: Involvement of Oxoiron(V) Oxidants in Cleaving Strong C−H Bonds, Angew. Chem., Int. Ed., 2020, 59, 7332–7349 CrossRef CAS PubMed.
- G. Olivo, O. Cussó and M. Costas, Biologically Inspired C−H and C=C Oxidations with Hydrogen Peroxide Catalyzed by Iron Coordination Complexes, Chem. – Asian J., 2016, 11, 3148–3158 CrossRef CAS PubMed and references therein.
- T. Fukuyama, C. K. Jow and M. Cheung, 2- and 4-Nitrobenzenesulfonamides: Exceptionally Versatile Means for Preparation of Secondary Amines and Protection of Amines, Tetrahedron Lett., 1995, 36, 6373–6374 Search PubMed.
- J. J. P. Stewart, Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-optimization of Parameters, J. Mol. Model, 2013, 19, 1–32 CrossRef CAS PubMed.
- S. Beckendorf and O. García Mancheño, ‘Click’-BINOLs: A New Class of Tunable Ligands for Asymmetric Catalysis, Synthesis, 2012, 2162–2172 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo02270j |
| ‡ Current address: Organic Chemistry Department, University of Salamanca, 37008 Salamanca, Spain. E-mail: angelfuentes@usal.es |
| This journal is © the Partner Organisations 2025 |