
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
AI molecular catalysis: where are we now?
Zhenzhi
Tan
 a,
Qi
Yang
*ab and
Sanzhong
Luo
*a
a,
Qi
Yang
*ab and
Sanzhong
Luo
*a
aCenter of Basic Molecular Science, Department of Chemistry, Tsinghua University, Beijing, 100084, China. E-mail: luosz@tsinghua.edu.cn
bHaihe Laboratory of Sustainable Chemical Transformations, Tianjin, 300192, China. E-mail: yangqi@hlsct.cn
First published on 11th February 2025
Abstract
Artificial intelligence (AI) is transforming molecular catalysis by addressing long-standing challenges in retrosynthetic design, catalyst design, reaction development, and autonomous experimentation. AI-powered tools enable chemists to explore high-dimensional chemical spaces, optimize reaction conditions, and accelerate novel reaction discovery with unparalleled efficiency and precision. These innovations are reshaping traditional workflows, transitioning from expert-driven, labor-intensive methodologies to intelligence-guided, data-driven processes. Despite these transformative achievements, significant challenges persist. Critical issues include the demand for high-quality, reliable datasets, the seamless integration of domain-specific chemical knowledge into AI models, and the discrepancy between model predictions and experimental validation. Addressing these barriers is essential to fully unlock AI's potential in molecular catalysis. This review explores recent advancements, enduring challenges, and emerging opportunities in AI-driven molecular catalysis. By focusing on real-world applications and highlighting representative studies, it aims to provide a clear and forward-looking perspective on how AI is redefining the field and paving the way for the next generation of chemical discovery.
10th anniversary statementI am deeply honored to have published 10 research papers in *organic chemistry frontiers* (OCF) since its inception in 2015. This journey has been immensely rewarding, as the journal's rapid publication process and global reach have significantly enhanced the visibility and impact of my work. Over the past decade, OCF has consistently served as a premier platform for cutting-edge research and innovation in organic chemistry. Its dedication to high-quality, interdisciplinary science has driven groundbreaking discoveries in synthesis, catalysis, and materials chemistry, profoundly shaping the future of the field.I am thrilled to contribute my 11th paper, a perspective titled “AI molecular catalysis: where are we now?”, to this special issue celebrating OCF's 10th anniversary. This milestone reflects the journal's pivotal role in advancing organic chemistry and nurturing a dynamic scientific community. Here's to celebrating a decade of excellence and looking forward to many more years of innovation and discovery! |
Introduction
Artificial intelligence (AI) has emerged as one of the most transformative technologies of the 21st century, revolutionizing a wide range of scientific research.1 In chemistry, where the complexity of molecular interactions often challenges conventional methods, AI provides a powerful framework for addressing problems that have previously relied on chemists’ intuition and trial-and-error approaches. By integrating computational modelling, data-driven insights, and automation, AI is transforming how researchers design, analyze, and optimize chemical systems.2,3As a cornerstone of modern chemistry, molecular catalysis exemplifies this transformation. Historically, progress in catalysis relied on fundamental principles, experimental ingenuity, and serendipity.4,5 Classical models, such as linear free energy relationships (LFERs),6 provided elegant but simplified structure–activity relationships (SAR) based on limited datasets. These models, including the Brønsted catalysis law,7 Hammett equation,8 Taft equation9 and Mayr equation,10,11 guided decades of catalytic research and synthetic design. However, as chemical systems have grown more complex,12,13 these traditional tools have struggled to address the intricate interplay of reaction conditions, multi-scale dynamics, and diverse molecular interaction.
The emergence of AI has paralleled the rise of data-driven approaches in molecular catalysis, where the synergy between machine learning (ML) and chemical data presents unparalleled opportunities for discovery.14 Unlike traditional approaches that depend on experiment-derived heuristics or predefined theoretical frameworks,15 AI excels at identifying patterns and predicting outcomes directly from high-dimensional, complex datasets. This capability enables chemists to explore vast chemical spaces with increased efficiency and precision.16
As illustrated in Fig. 1, AI integration throughout the molecular catalysis workflow fosters innovation at every stage. Retrosynthetic analysis models can quickly propose optimal synthetic routes, helping chemists efficiently prepare catalysts and target molecules. AI-guided catalyst design, informed by chemical knowledge and historical data, facilitates the development of catalysts with enhanced performance. In reaction studies, AI accelerates the optimization of conditions and delineates the scope and limitations of reactions. Furthermore, advanced autonomous experimentation allows chemists to perform experiments with significantly greater efficiency and reproducibility. By seamlessly integrating datasets, models, robots, and experiments, AI is transforming traditional expert-driven, labor-intensive workflows into intelligence-guided, data-driven processes.
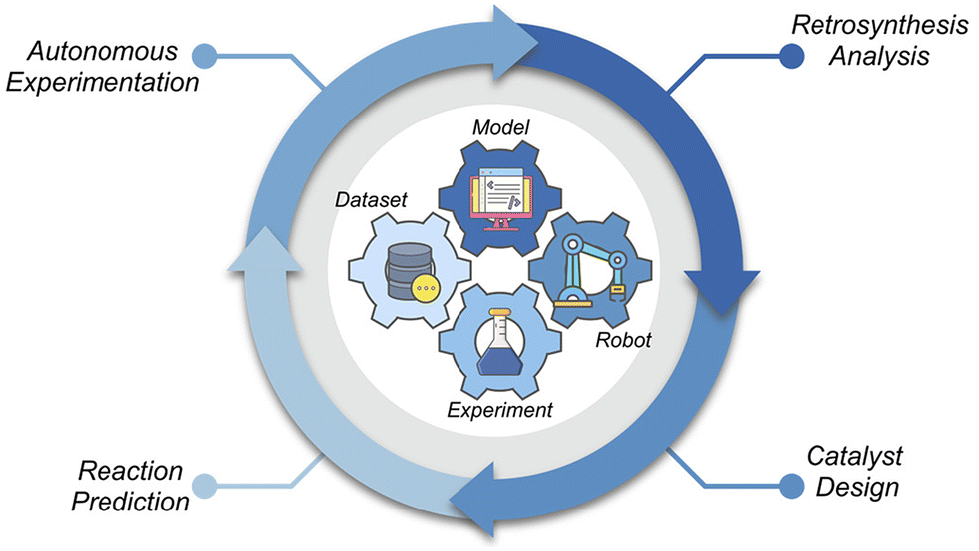 | ||
| Fig. 1 The integration of AI into molecular catalysis workflows. | ||
This review examines the current landscape of AI in molecular catalysis by addressing three key questions: What are the major advancements? What challenges remain? What opportunities lie ahead? We highlight representative studies in retrosynthetic design, catalyst design, reaction development, and autonomous experimentation, providing a focused perspective on how AI is transforming molecular catalysis. Rather than offering a comprehensive overview of AI methodologies, this review emphasizes their practical integration into chemical workflows and their implications for researchers. For readers seeking in-depth discussions on AI applications in chemistry, several notable reviews are recommended.17–20
Retrosynthesis analysis
The essence of organic chemistry lies in synthesis, where efficient and concise synthetic routes often deliver outcomes that far exceed the effort invested. A notable example is Robinson's total synthesis of Tropinone, which transformed a laborious 17-step process into a single Mannich Reaction.21 However, as the complexity of target molecules increases, the design of synthetic routes becomes increasingly challenging. While legendary figures in synthetic chemistry, such as Robert Burns Woodward, have exemplified the extraordinary heights of human ingenuity in the synthesis of highly complex natural products,22 such accomplishments often lack scalability and broad applicability in more generalized contexts.The retrosynthetic analysis framework proposed by E. J. Corey provided chemists with a systematic and rational approach to deconstructing complex molecules into simpler precursors, significantly reducing the difficulty of synthesis.23 Around the same time, the field of computer-aided synthesis planning (CASP) began to emerge, promoting the use of computational tools for retrosynthetic analysis. With the advancement of AI technology, modern retrosynthetic analysis tools have also undergone substantial development, further enhancing the efficiency and feasibility of designing synthetic routes (Fig. 2).24
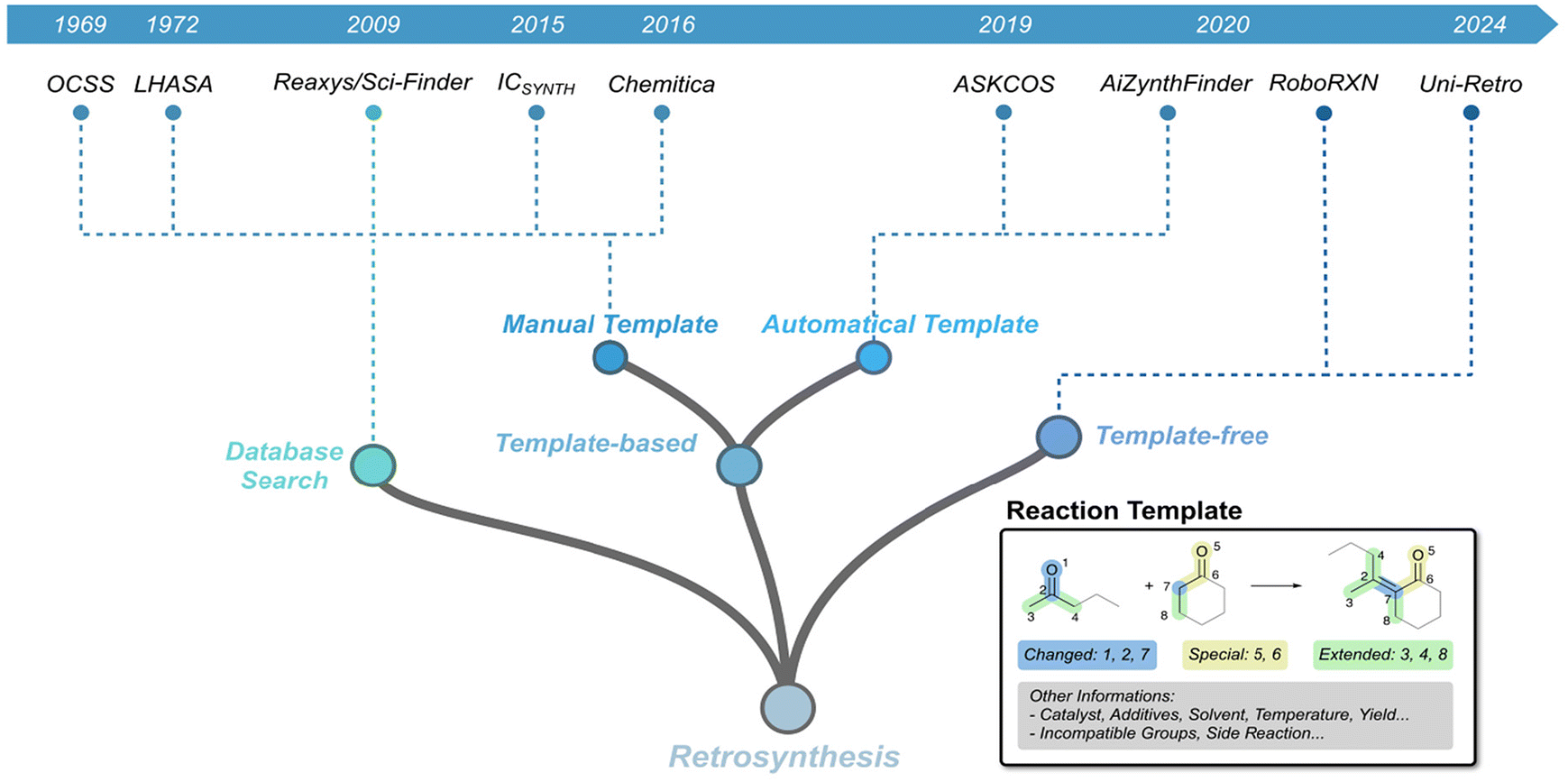 | ||
| Fig. 2 Timeline of key milestones for CASP. Examples of reaction templates are illustrated within the diagram. | ||
Before the advent of sophisticated retrosynthesis tools, chemists primarily relied on database search engines such as Reaxys and SciFinder to retrieve reaction information. These platforms remain widely used for retrosynthetic planning, offering comprehensive access to published reactions and experimental data. However, their utility is limited to recorded reactions, often failing to guide unreported or novel transformations.
AI-guided retrosynthesis planning can be categorized into single-step and multi-step retrosynthesis. Single-step retrosynthesis involves a single disconnection or transformation of a molecule, and repeating this process iteratively until commercial substrates leads to multi-step retrosynthesis. Although significant progress has been made in developing advanced algorithms for single-step disconnections,25–28 practical synthetic applications demand more than just effective disconnection strategies—directional guidance in choosing disconnections is equally or even more crucial.29 Therefore, transitioning from single-step to multi-step retrosynthesis requires models to adopt a broader, long-term perspective, enabling them to approach reaction pathway design with a more holistic, globally optimized strategy.
The introduction of reaction templates marked a significant step forward in retrosynthetic analysis by formalizing chemical reasoning into a structured framework. A reaction template encodes the core structural transformation of a reaction, capturing critical features such as bond changes, functional group compatibility, and mechanistic insights. As shown in Fig. 2, template-based methods rely on curated libraries of reaction templates, enabling retrosynthetic analysis to extend beyond the confines of recorded reactions in databases. By systematically applying these templates, chemists can design synthetic routes for complex molecules, including many natural products and pharmaceuticals.
The first major template-based retrosynthesis software, OCSS,30 was developed by Corey and Wipke and later evolved into tools like LHASA31 and SECS.32 Since then, a series of retrosynthesis programs have been developed, such as SYNLMA,33 SYNCHEM,34 SYNGEN,35 IGOR,36 WODCA37 and CHIRON.38 These pioneering systems laid the foundation for computer-aided retrosynthesis by automating reaction rule application.
By leveraging the network of organic chemistry (NOC), which comprises over 10 million compounds and their reaction relationships, Grzybowski et al. introduced synthesis optimization with constraints (SOCS).39 A constrained search algorithm was developed using cost and popularity functions, alongside a manually curated database of more than 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 reaction templates specifying scope and conflict groups. This expert-designed template set enabled the creation of Chemitica, a computer-aided tool for de novo synthetic design. Compared to earlier synthesis software, Chemitica demonstrated efficacy comparable to that of human chemists, supported by extensive chemical reaction data. Complete synthetic routes for complex natural products, including (–)-Dauricine, Tacamonidine, and Lamellodysidine A, were successfully designed and experimentally validated (Fig. 3).40,41 The results of Turing test demonstrated that even experienced chemists are unable to distinguish between synthesis routes generated by Chemitica and those reported in the literature. Subsequent advancements incorporated molecular force field-level parameters to refine synthesis design and introduced the Stereofix module for improved stereochemical handling, establishing Chemitica as one of the most robust retrosynthesis tools. Similarly, the InfoChem group developed ICSYNTH, demonstrating its innovative capabilities in the de novo design of complex spiro and aromatic heterocyclic compounds.42
000 reaction templates specifying scope and conflict groups. This expert-designed template set enabled the creation of Chemitica, a computer-aided tool for de novo synthetic design. Compared to earlier synthesis software, Chemitica demonstrated efficacy comparable to that of human chemists, supported by extensive chemical reaction data. Complete synthetic routes for complex natural products, including (–)-Dauricine, Tacamonidine, and Lamellodysidine A, were successfully designed and experimentally validated (Fig. 3).40,41 The results of Turing test demonstrated that even experienced chemists are unable to distinguish between synthesis routes generated by Chemitica and those reported in the literature. Subsequent advancements incorporated molecular force field-level parameters to refine synthesis design and introduced the Stereofix module for improved stereochemical handling, establishing Chemitica as one of the most robust retrosynthesis tools. Similarly, the InfoChem group developed ICSYNTH, demonstrating its innovative capabilities in the de novo design of complex spiro and aromatic heterocyclic compounds.42
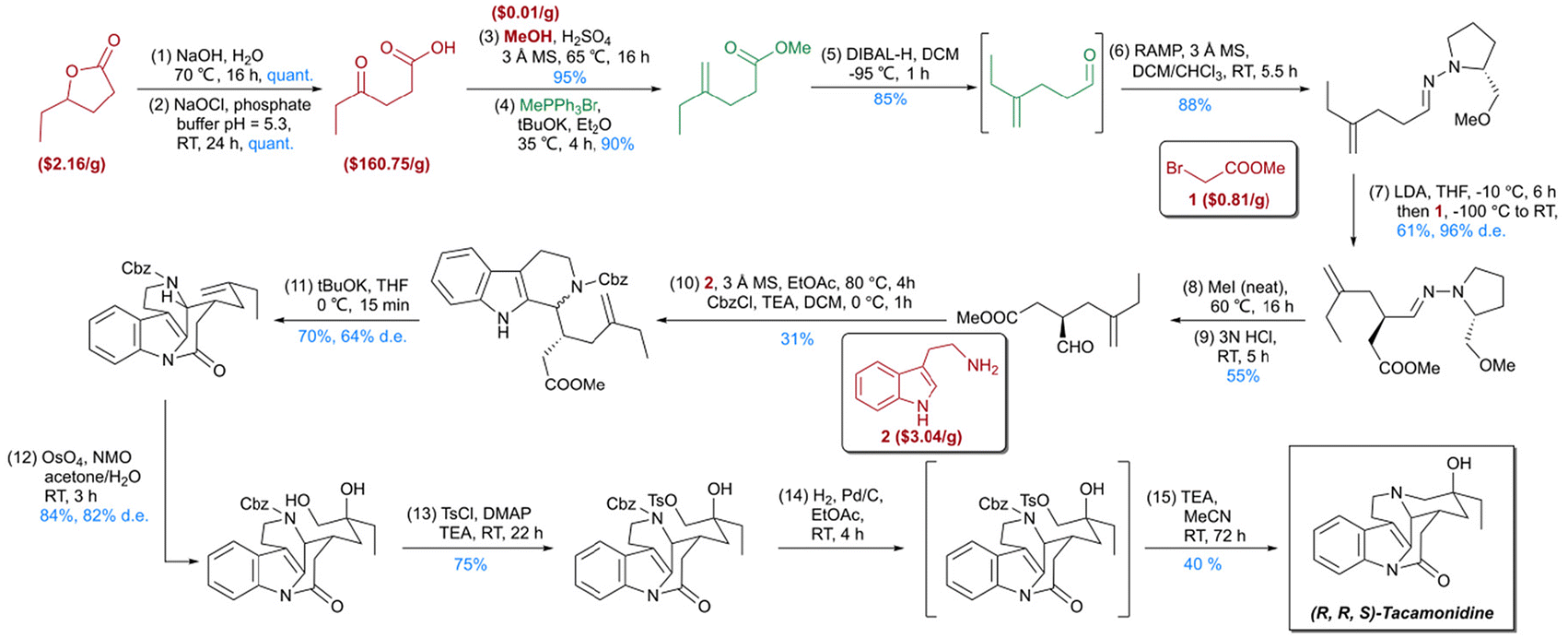 | ||
| Fig. 3 Total synthesis route of (R,R,S)-Tacamonidine designed by Chemitica. Red structures indicate commercially available compounds with their corresponding prices. Green structures represent chemicals with existing synthetic information available in databases. | ||
Despite the success of template-based methods in retrosynthesis, concerns remain about the breadth and generality of manually curated templates. Kayala and Baldi et al. argued that expanding template libraries exponentially increases conflicts among templates, which in turn hinders the generalization of existing templates.43 To address this, Green and Jensen developed RDChiral, an automated template extraction tool based on RDKit.44 Using this toolkit, they derived 163723 reaction rules, including stereochemical details, from 12.5 million single-step reactions in the Reaxys database and USPTO. These templates were incorporated into a neural network model for template matching and integrated with Monte Carlo tree search (MCTS)45 for substrate identification in retrosynthetic planning. The resulting system, ASKCOS, was evaluated on a robotic flow chemistry platform (see section 5.2) using 15 small molecule examples, showcasing its robustness and efficiency in synthetic route design (Fig. 4).46 Similarly, Genheden and Bjerrum developed AiZynthFinder, an open-source retrosynthesis tool that demonstrated superior performance in specific synthetic scenarios compared to ASKCOS.47–49
 | ||
| Fig. 4 Retrosynthetic planning of selected small molecular drugs using ASKCOS. | ||
Template-based retrosynthesis methods, though effective in many cases, are computationally intensive, particularly during route searches. As the molecular complexity of the target increases, the computation time scales up significantly. Furthermore, these methods are inherently constrained by their reliance on predefined reaction templates, making them unable to predict transformations beyond their established scope. This limitation is especially problematic when encountering novel or less-studied reactions, as they fall outside the boundaries of existing templates.
To address these limitations, template-free retrosynthesis approaches have emerged as a promising alternative. These data-driven methods leverage ML models trained on extensive reaction datasets, allowing the prediction of synthetic routes without relying on predefined templates. By directly extracting reaction knowledge from vast collections of literature data, template-free methods significantly enhance the scope and flexibility of retrosynthetic planning, offering the potential to design synthetic pathways for virtually any molecule.50
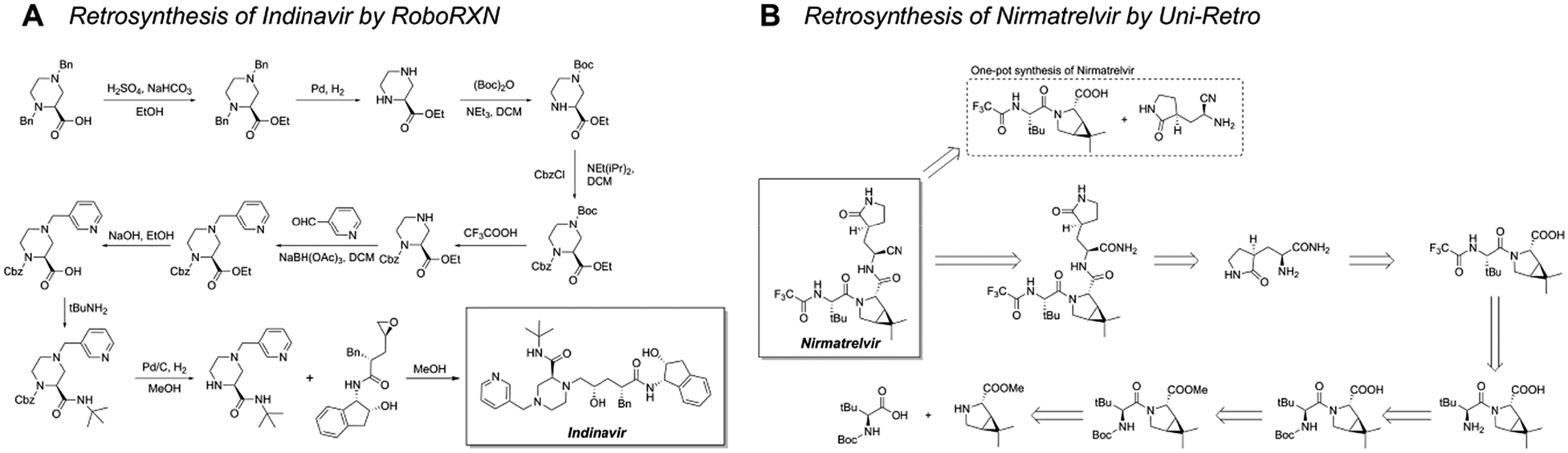
Simplified molecular input line entry system (SMILES)51 encoding is a widely used data representation in template-free retrosynthesis methods, enabling to transfer of retrosynthesis planning into a natural language processing (NLP) problem.52,53 This allows the application of advanced NLP frameworks. Schwaller et al. utilized the reduced USPTO-50k dataset to frame retrosynthesis as a language translation task involving the SMILES strings of reactants, reagents, and products. This work led to the development of the Molecular Transformer model.54 Building on this model, they optimized evaluation metrics and introduced a hypergraph exploration strategy, resulting in the creation of a publicly available retrosynthesis platform, RoboRXN.55 Their study presented retrosynthesis route designs for several complex molecules, demonstrating the power of models (Fig. 5A).
 | ||
| Fig. 5 Retrosynthetic routes of drug molecules designed by template-free models. (A) Retrosynthesis of Indinavir designed by RoboRXN. (B) Retrosynthesis of Nirmatrelvir designed by Uni-Retro. The first step of the retrosynthesis presents two possible pathways. | ||
Beyond sequence-based models, graph neural networks (GNNs) offer another promising direction for template-free reaction prediction. Compared with SMILES-based methods, the graph structure provides richer information for the model.29 Ke et al. developed the node-aligned graph-to-graph (NAG2G) model, which integrates 2D molecular graphs and 3D conformations to capture comprehensive molecular details.56 The model ensures reaction validity by establishing atom mappings between reactants and products. Its capability to design synthetic routes for pharmaceutical molecules highlights its potential for complex synthesis challenges (Fig. 5B).
Template-free methods can leverage the full potential of AI in processing chemical data, offering significant promise for future development. However, compared to template-based methods, their ability to accurately capture and predict complex reactions remains a challenge. Furthermore, most current template-free predictions lack experimental validation, leaving their reliability to be further substantiated.
Retrosynthetic methods have made remarkable progress in recent years, significantly advancing the field of chemical synthesis. However, several challenges remain that hinder their broader application. One major limitation lies in the limitations of available datasets, which often lack critical details on reaction conditions—such as specific additives, solvents, and their precise quantities—thereby necessitating expert intervention to bridge the gap between computational predictions and practical execution. In this regard, template-based methods such as Chemitica have demonstrated notable advantages over template-free methods, as they inherently capture detailed reaction conditions when encoding reaction templates. By contrast, template-free models struggle to accurately infer such information from existing data. Although large-scale reaction databases such as USPTO contain a fraction of entries with recorded catalysts and solvents, these entries typically lack crucial information on reactant quantities and categories. For example, RoboRXN occasionally includes additives and solvents in the output, but they are presented in the same manner as reactants, lacking additional details, which can complicate the interpretation of the procedure as recommended by the model.
Additionally, the datasets used for training retrosynthesis models are significantly smaller and less diverse than the extensive synthetic knowledge available in the literature. Their accuracy is also a significant concern, as Grzybowski et al. reported that 60% of reactions from patents are either incorrect or highly questionable, while 28% of literature-reported reactions are deemed unreliable.57 These data issues limit the robustness and generalizability of current models.
Transforming AI-assisted retrosynthesis into a truly practical tool for chemists will require addressing these challenges, particularly in improving dataset quality, diversity, and the incorporation of actionable reaction details. Despite these limitations, the potential demonstrated by current methods inspires confidence that AI-driven retrosynthesis will ultimately revolutionize chemical synthesis, addressing challenges beyond the capabilities of traditional approaches.
Catalyst design
Catalysts play a central role in determining the performance of organic reactions. Rational design offers a more efficient approach to developing high-performance catalysts compared to traditional trial-and-error screening. Successful design typically relies on a thorough understanding of electronic and spatial effects, as well as the underlying reaction mechanisms. Advances in density functional theory (DFT) have enabled researchers to predict catalyst properties before synthesis.58However, experience-based design is often limited by cognitive biases, which may potentially hinder the exploration of unconventional catalyst structures. Moreover, for complex reactions involving intricate electronic or steric interactions, traditional rational design methods frequently fail to identify optimal catalyst candidates, highlighting the need for more systematic and predictive strategies.
Fortunately, the availability of extensive literature data has enabled data-driven approaches in rational catalyst design. Among these, insights from physical organic chemistry have proven particularly impactful in guiding molecular catalysis. Electronic effects such as pKa,59 hydrogen bond lengths,60,61 infrared vibration frequencies62 and intensities and non-covalent interaction parameters,63 along with steric parameters such as Taft parameters, Sterimol parameters, and %Vbur,64 have played a crucial role in guiding catalyst design. For example, Sigman et al. studied asymmetric propargylation reactions catalyzed by chiral chromium complexes and used a binary substitution matrix to analyze the steric and electronic effects of ligands.65
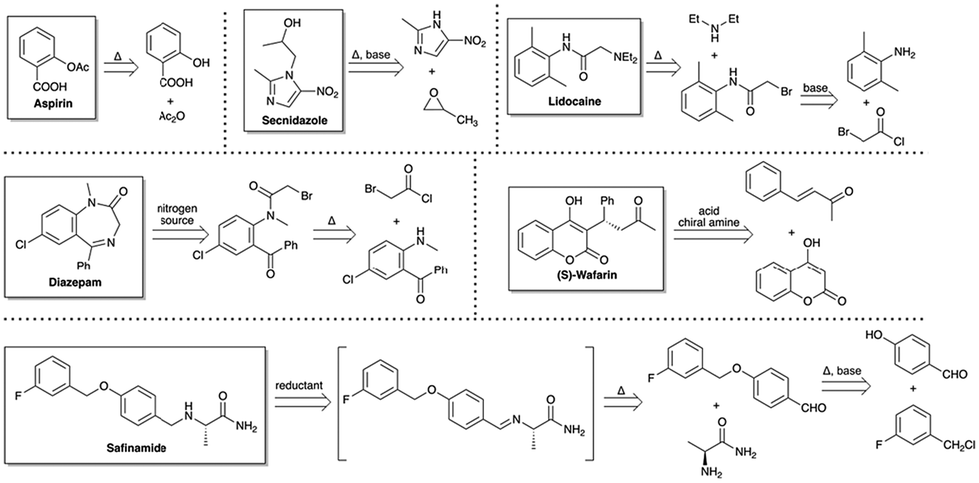
While physical organic chemistry has provided critical insights into catalyst design, its application in practical reaction development remains fraught with significant challenges. Chemists often require several weeks or even months to synthesize numerous catalysts in order to derive physical organic parameters of a certain reaction. However, these principles and parameters are often applicable to a limited range of catalysts, thereby reducing the overall efficiency of the discovery process and restricting the breadth of exploration in catalyst design. By integrating chemists’ expertise with AI's capacity for large-scale data exploration, more expansive catalyst candidates can be investigated. Coupled with optimization algorithms, AI facilitates the efficient identification of optimal catalyst structures, presenting significant potential to accelerate catalyst discovery. The general workflow for AI-assisted catalyst design and optimization is illustrated in Fig. 6. The process begins with applying clustering algorithms to the catalyst library, grouping catalysts into distinct categories based on their properties. From these clusters, representative candidates are selected for experimental testing. Optimization algorithms are then employed to refine both the catalyst structures and reaction conditions, followed by experimental validation to discover new catalysts.
 | ||
| Fig. 6 The general workflow for virtual ligand screening using AI. | ||
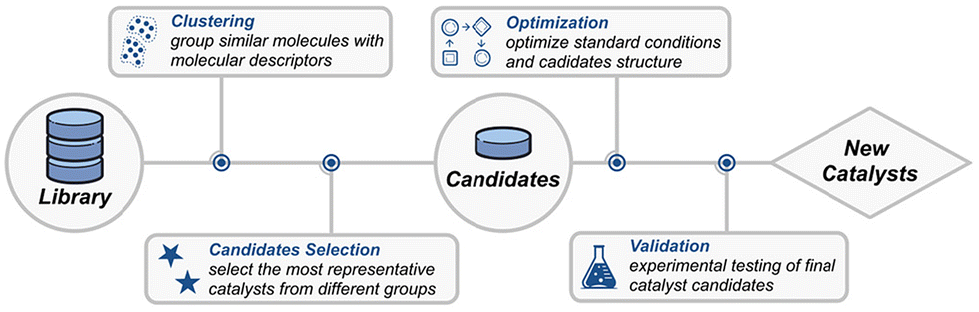
Schoenebeck et al. applied a clustering algorithm to identify potential Pd(I) dimer catalysts from 348 phosphine ligand pairs in the LKB-P ligand library (Fig. 7A).66 Using K-Means clustering algorithms, the ligands were initially reduced to 25%, followed by further selection based on 42 problem-specific descriptors calculated via DFT. Experimental validation of the cluster containing previously reported Pd(I) dimers led to the identification of 8 Pd(I) dimer catalysts with minimal experimental effort. Similarly, Denmark et al. utilized clustering to optimize peptide catalysts for the reaction of aldehydes with nitroethylene.67 They utilized an algorithm to select 161 tripeptides from a library constructed from 174 commercially available amino acids, which were then synthesized and experimentally tested. The optimal catalyst identified through this process improved the enantioselectivity from 86% to 91% ee.
 | ||
| Fig. 7 Examples of AI-assisted catalyst design. (A) Discovery of dinuclear Pd(I) complexes through data collection and clustering, with the figure depicting results from the second round of clustering.Reprinted (adapted) with permission from ref. 66, copyright 2021, American Association for the Advancement of Science. (B) Optimization of trisubstituted alkene selectivity using the Kraken virtual screening library. Reprinted (adapted) with permission from ref. 72, copyright 2024, American Chemical Society. | ||
Clustering enables the rapid identification of structurally or functionally similar catalysts from literature data, facilitating the extraction of highly reactive candidates from databases. Catalyst libraries for clustering can be generated using templates, with molecular properties characterized through fingerprinting methods or DFT calculations. However, unsupervised clustering approaches often require experimental validation to refine results, and the random selection of representative molecules for synthesis from clusters can significantly influence the overall outcomes.
Moreover, employing structure generation strategy to construct a comprehensive library of organocatalysts with their molecular properties, represents a pivotal advancement in accelerating catalyst discovery. Traditionally, manual screening relies on the design of potential catalyst candidates based on empirical knowledge or physical organic principles, resulting in catalyst libraries that are inherently prone to oversight and cognitive bias. In contrast, divergent structure generation methods start from a limited set of molecular scaffolds and systematically combine them with common functional groups to generate a large and structurally diverse set of molecules. This approach produces libraries with broad coverage, enabling a more extensive and unbiased exploration of the potential catalyst space.
The Kraken platform developed by Aspuru-Guzik et al. is a possible catalyst library paradigm.68 They collected 1556 commercially available ligands for PR3 phosphine catalysts and constructed 331776 virtual catalysts containing at least two identical substituents and more than 1.9 million different substituents from a unique 576 substituent R. The prediction of properties of these molecules can be predicted by their proposed BoS and other molecular fingerprints, to predict new organic reactions. Some studies have demonstrated that Kraken can be used to optimize reaction performance.69 or validate reaction mechanisms70,71 through virtual ligand screening. For example, Doyle and Sigman et al. utilized Kraken as a virtual screening library in their study on the nickel-catalyzed reduction of enol tosylates to selectively form trisubstituted alkene products (Fig. 7B).72 Kraken provides a large pool of monophosphine ligands as candidates for the optimal catalyst. By employing an optimization algorithm, they successfully identified phosphine ligands suitable for the selective formation of E- and Z- trisubstituted alkenes, significantly accelerating the screening process.
Despite the availability of comprehensive libraries for phosphine ligands, many other ligand and catalyst frameworks still lack corresponding screening libraries. Expanding virtual catalyst databases is therefore essential to explore a broader range of scaffolds and their derivatives. However, current catalyst design remains limited by manually defined scaffold structures rather than fully mechanism-driven approaches. AI-powered generative molecular design provides a promising solution for rational catalyst design.73 Significant progress in AI-assisted small molecule drug design, where AI analyses target sites to generate protein-binding molecules, has already revolutionized drug discovery.74,75 Given the structural parallels, organocatalysis, which similarly relies on “pocket” architectures, stands to benefit greatly from AI-driven strategies.
Reaction development
While catalyst design is critical for the performance of catalytic reactions, successful reaction development can only be achieved when paired with the appropriate reagent combinations and substrate types. Traditionally, reaction optimization has relied heavily on chemists’ intuition and the sequential adjustment of single variables to infer trends in reactivity. Empirical rules for organic reactions, for their simplicity, interpretability, and effectiveness within specific reaction types, have long provided intuitive guidance for reaction discovery and optimization. However, these traditional approaches face substantial limitations when applied to complex or novel reaction systems. As reaction complexity increases, predictions based purely on intuition or simple heuristic rules often become unreliable. Additionally, a wealth of chemical knowledge is dispersed across the literature, awaiting systematic curation and analysis. Given the human inability to process and synthesize such vast datasets efficiently, many critical insights risk being overlooked.With the fast growth of reaction data, AI-driven approaches are showcasing remarkable capabilities in reaction prediction. By leveraging AI models and extensive literature data, these methods enable the accurate prediction of reaction conditions,76–78 products,79,80 regioselectivity,81,82 yields83–85 and stereoselectivity.86,87 However, in the discovery of novel catalytic reactions, the lack of related reaction data often limits the models’ generality. To address the challenge of making predictions in zero or low-data scenarios, active learning or active sampling strategies have been developed.88,89 This section highlights two representative challenges in molecular catalysis research – optimizing reaction conditions and exploring substrate scopes – and highlights AI-assisted solutions that address these challenges.
Condition optimization
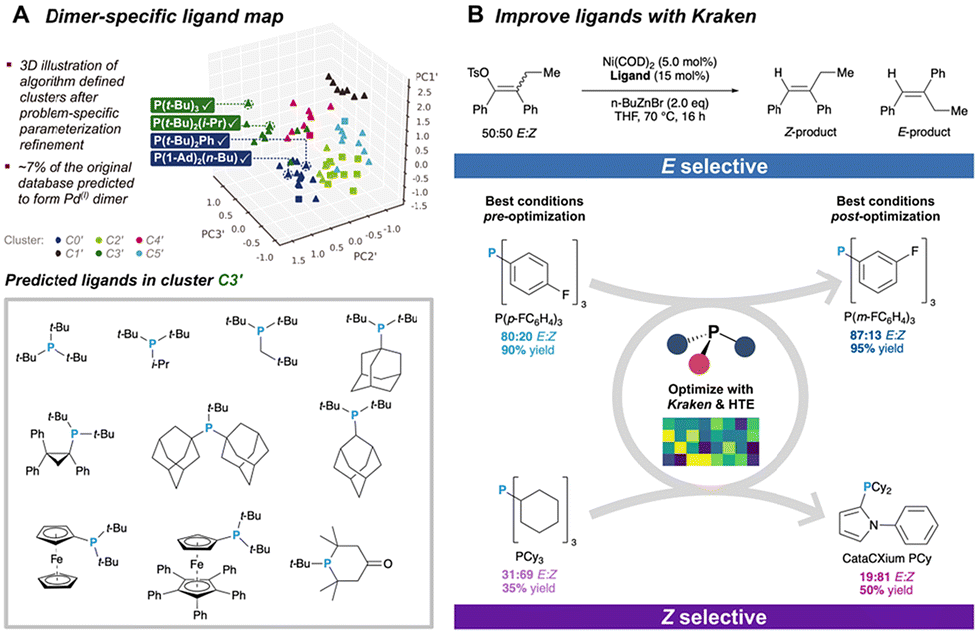
Optimizing reaction conditions is a critical step in molecular catalysis, as selecting appropriate additives, solvents, and catalysts can significantly impact reaction performance. Traditionally, this process relies on manual experimentation, which is time-consuming, inefficient, and prone to human bias. ML algorithms address these challenges by efficiently exploring vast screening spaces and minimizing experimental effort. Common approaches include Bayesian optimization (BO), reinforcement learning, genetic algorithms, particle swarm optimization, simulated annealing, and differential evolution. These methods enable the identification of optimal reaction conditions through data-driven strategies, providing significant advantages over manual approaches.90Among those algorithms, BO is widely used for its high sample efficiency, integration of prior knowledge, and strong high-dimensional space searchability.91 As illustrated in Fig. 8, the general workflow for employing BO in organic reaction condition optimization begins with defining the screening space of reaction conditions and establishing the optimization target. Initial conditions are then selected, either algorithmically or based on chemists’ expertise. Experimental results are iteratively input into the BO model to refine predictions, allowing the process to progressively converge toward optimal conditions that meet the defined target.
 | ||
| Fig. 8 Workflow for reaction condition optimization using BO algorithm. | ||
In 2021, Aspuru-Guzik et al. developed Gryffin, a BO framework capable of handling both discrete and continuous variables.92 To demonstrate its effectiveness, they applied it to optimize Suzuki–Miyaura reaction conditions using 88 reactions and their turnover numbers (TON) as inputs. Bayesian neural networks were used to predict TON across the reaction space, demonstrating Gryffin's superiority over traditional BO methods in achieving higher yields and faster optimization.
To further extend the application of BO to real experimental scenarios, Doyle et al. developed EDBO (experimental design via Bayesian optimization) to guide the direct arylation of imidazoles.93 By comparing EDBO with manual optimization, the study demonstrated its superior efficiency in identifying optimal conditions. The authors noted that while machines may initially lack the “smartness” of human intuition due to limited prior knowledge, EDBO was able to converge on optimal conditions more rapidly. Subsequently, they introduced EDBO+, which enabled simultaneous optimization of ee value and reaction yield, achieving a 10% yield improvement for the cross-coupling of styrene oxide with aryl iodides (Fig. 9A ).94 More recently, they explored reaction condition optimization as a multi-arm bandit problem, applying the Bayesian UCB algorithm to efficiently identify optimal conditions for palladium-catalyzed C–H arylation, anilamide coupling, and phenol alkylation.95
).94 More recently, they explored reaction condition optimization as a multi-arm bandit problem, applying the Bayesian UCB algorithm to efficiently identify optimal conditions for palladium-catalyzed C–H arylation, anilamide coupling, and phenol alkylation.95
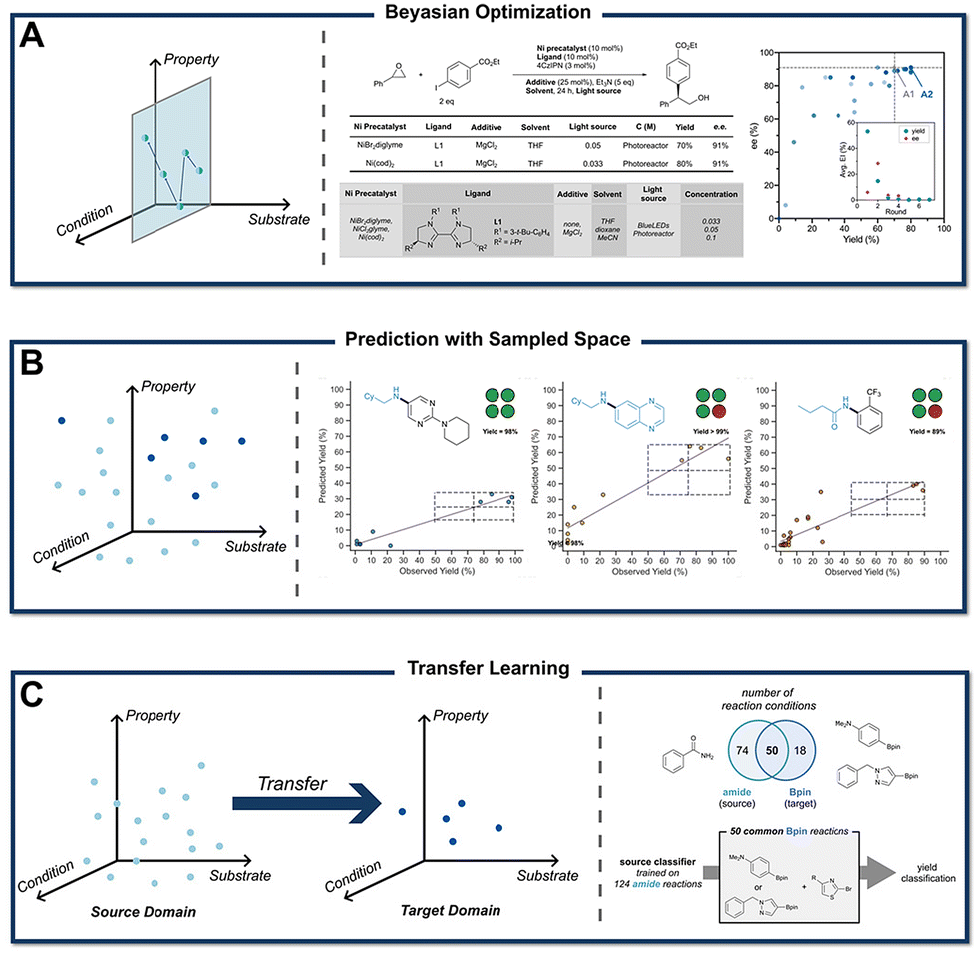 | ||
| Fig. 9 Schematic representation of three approaches for reaction condition optimization. (A) BO only optimizes conditions for a single substrate. Condition optimization with BO can achieve rapid improvements in yield and ee within 7 iterations. Reprinted with permission from ref. 94, copyright 2022, American Chemical Society. (B) Uniform sampling of the reaction space, combined with experimental results for modeling, allows prediction across different substrates and conditions. Plots demonstrate that model-predicted optimal conditions often perform well in actual synthesis (blue structures indicate this fragment is not included in the training set. Reprinted (adapted) with permission from ref. 96, copyright 2023, American Association for the Advancement of Science. (C) TL improves efficiency by leveraging prior knowledge, but its performance is limited by domain discrepancy. A general working framework for transfer learning in condition optimization is shown in the figure. Reprinted (adapted) with permission from ref. 97, copyright 2022, Royal Society of Chemistry. | ||
Although BO is widely used for optimizing organic reaction conditions, its current applications often focus narrowly on specific substrate templates, which can result in overfitting and limit the transferability of optimized conditions to broader substrate scopes. To address this, Bigler and Denmark et al. developed an ML-guided tool for predicting optimal conditions across a user-defined reaction space (Fig. 9B).96 They evaluated 121 substrates and 24 reaction conditions through 3300 carefully selected experiments. Using these data, a neural network model was trained to predict yields for different substrate groups. In out-of-sample validation, the model demonstrated a reasonable ability to predict optimal conditions, even though the exact predicted yields were not always accurate. However, this approach involves over 3000 experiments to evaluate the reaction scope. For more complex chemical reaction optimizations, it would be necessary to further reduce the experimental data-to-reaction space ratio.
Leveraging literature data can significantly reduce the experimental burden in reaction condition screening. Transfer learning (TL), an algorithm that applies patterns learned from existing experimental data to similar reaction systems, utilizes prior knowledge to minimize trial-and-error efforts. For example, Zimmerman et al. demonstrated the transferability of TL between Suzuki (C–C bond formation) and Buchwald–Hartwig (C–N bond formation) reactions under comparable conditions (Fig. 9C).97 However, their analysis revealed that condition predictions remain largely confined to relatively similar reaction types. To address this limitation, adaptive tree-based learning (ATL) models have been introduced to improve the predictive capabilities of TL, offering a promising strategy for extending its applicability to more diverse reaction spaces.
Although reaction optimization models have demonstrated their effectiveness in global optimization within complex chemical spaces, challenges remain in conditional screening. Selecting the appropriate number of optimization cycles is particularly challenging, especially in large reaction space where too few cycles may obscure meaningful trends, while excessive cycles increase the experimental burden. The inefficiency of manual screening further underscores the need for autonomous experimentation to enhance optimization efficiency (see section 5). For reactions with large chemical spaces and unclear mechanisms, combining AL with high-throughput experimentation (HTE) offers a promising solution, which is likely to shape the future of organic laboratory workflows.
Reaction scope
Organic reactions, even within the same class of substrates, can exhibit significant variations in efficiency. As a result, reaction conditions optimized for the template reaction may fail to perform consistently with other similar substrates. To address this, exploring the scope of catalysts and reaction conditions across a range of substrates is essential. Such investigations not only define the capability boundaries of catalysts but also offer valuable insights into underlying reaction mechanisms.Establishing reaction scope requires selecting a representative set of substrates. Over the past decades, the number of reaction examples required to establish reaction scope has nearly doubled, significantly increasing the workload for organic chemists.98 However, substrate selection has traditionally relied on subjective choices made by researchers, introducing potential biases in both the selection and reporting of results. To present reactions in a more favorable light, researchers may exclude incompatible substrates and selectively highlight optimal outcomes, resulting in skewed evaluations of reaction performance. The integration of intelligence-guided, data-driven approaches into reaction scope studies offers a powerful solution to mitigate human-induced biases. By leveraging ML and data analytics, these approaches can provide a more comprehensive and objective assessment of a reaction's true capabilities, ensuring a more accurate representation of its versatility and limitations.
One of the most commonly used substrate selection algorithms is clustering, which selects a few representative substrate molecules from a large pool of candidates based on their features.99 In the study of Ni/photoredox cross-coupling, Doyle et al.100 utilized a data-driven approach to explore the scope of aryl bromide substrates (Fig. 10A). They searched Reaxys for all aryl bromide substrates and, after a simple screening process, obtained a dataset of 2600 members. Using auto-qchem, they calculated the DFT descriptors of the molecules. These descriptors were then used for dimensionality reduction via uniform manifold approximation and projection (UMAP) and hierarchical clustering, resulting in 15 representative aryl bromide molecules selected for evaluation. The results demonstrated that, compared to traditional literature-based methods, the data science-driven approach could cover a broader substrate space with fewer experiments, providing a comprehensive overview of the reaction scope.
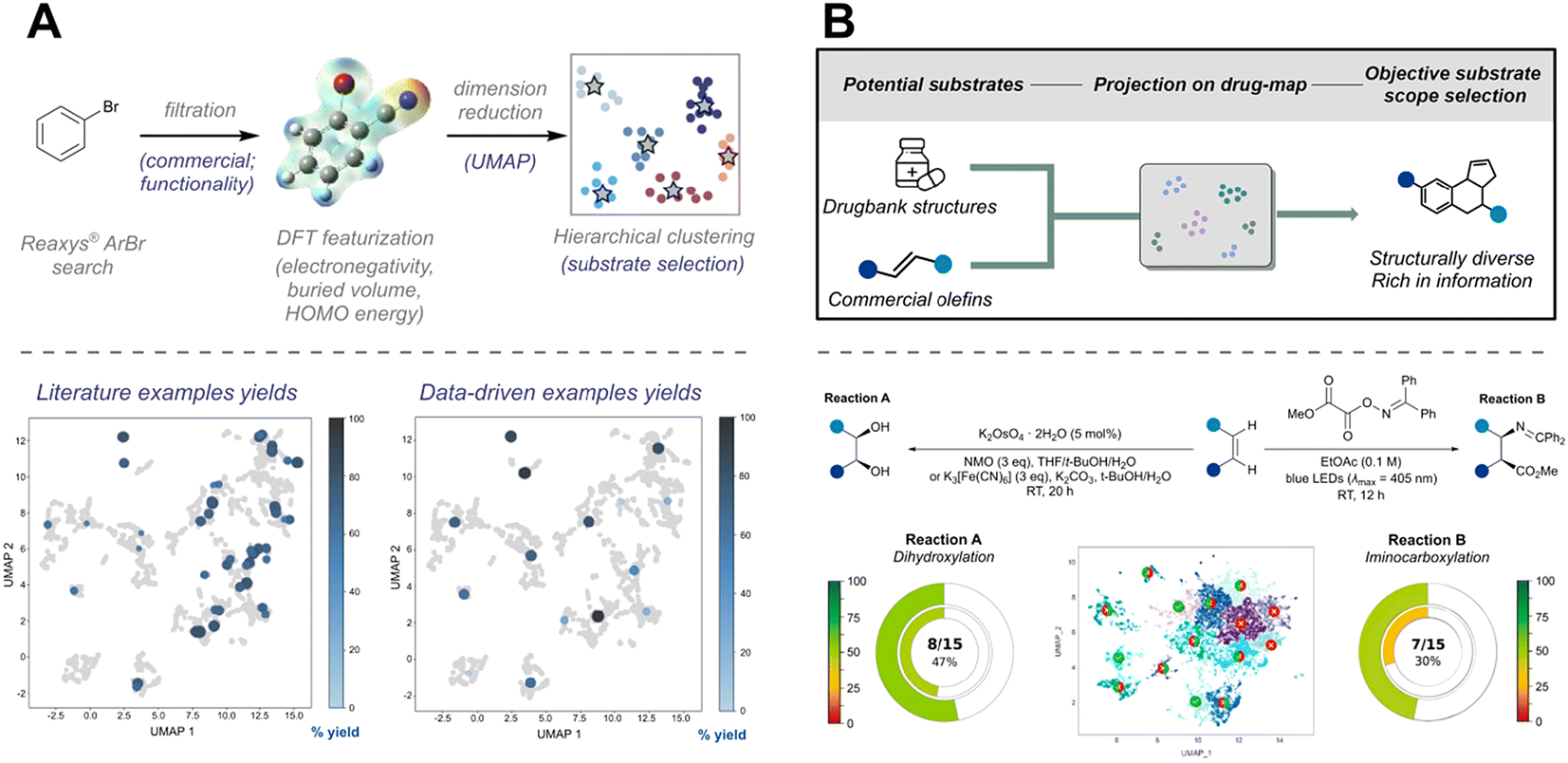 | ||
| Fig. 10 (A) Clustering of aryl bromides based on DFT descriptors, enabling a reduction in the number of experimental points while achieving results comparable to literature scope exploration. Reprinted with request permission from ref. 100, copyright 2022, American Chemical Society. (B) Exploration of diverse alkene categories using the DrugBank dataset as the background. The reactivity of the two reaction types is systematically compared. Reprinted (adapted) with request permission from ref. 101, copyright 2024, American Chemical Society. | ||
While AI-guided approaches effectively reduce selection and reporting biases, they often struggle to capture the complex interactions among functional groups in real chemical systems. To address these challenges, Glorius et al. developed a universal substrate screening strategy to minimize such biases (Fig. 10B).101 Using the DrugBank database as a representative dataset of diverse drug-like molecules, they described molecular structures with extended-connectivity fingerprints (ECFP) and applied K-means clustering to group the substrates into 15 spatially distinct classes, visualized in two dimensions via UMAP. To evaluate reaction scope, the substrate space of a given reaction was mapped onto this same descriptor framework, and 15 olefins closest to each cluster center were experimentally tested. This method was successfully demonstrated for the photochemical imino-carboxylation of olefins and osmium-catalyzed dihydroxylation, revealing the extent to which reported conditions are applicable in diverse chemical spaces.
In practice, selecting the optimal number of candidate substrates presents a significant challenge. A small set of substrates may fail to represent the diversity of chemical space adequately, whereas an excessive number can render experimental validation impractical. Although methods like elbow or silhouette plots provide some guidance, their effectiveness in practical applications remains limited.102 Another challenge in reaction evaluation lies in reporting and selection bias. Researchers often prioritize favorable results for publication, while unbiased, algorithmically designed experiments may produce outcomes that appear less impressive. As a result, negative or suboptimal data are often underreported, leading to a skewed perception of reaction performance. To address this issue, it is crucial to emphasize the systematic inclusion and transparent reporting of unbiased experiments and negative data. This approach will enable a more accurate and comprehensive understanding of reaction robustness and generalizability.103–105
AI has demonstrated significant potential in accelerating reaction condition optimization and unbiased reaction scope analysis. However, for complex chemical systems, even active learning often fails to substantially reduce experimental workloads. Additionally, the limited interpretability of current models hinders their ability to generate new chemical insights. Despite these limitations, continued advancements in AI algorithms and the growing adoption of AI technologies by researchers are expected to accelerate progress in reaction optimization and scope, and efficient and insightful exploration of reaction space.
Autonomous experimentation
Despite a century of rapid development in chemical theory and significant advancements in compound analysis techniques, the fundamental methods for synthesizing organic molecules in laboratories have seen little change. Manual processes—such as weighing, dosing, separation, and analysis—remain central to experimental workflows. This reliance on manual experimentation not only increases researchers’ workload but also slows the pace of reaction exploration. Human error compromises the reliability of experimental data, further limiting the accuracy and reproducibility of manual experimentation.The automation of chemical synthesis offers a promising solution to these challenges. In 1965, Merrifield introduced automated solid-phase peptide synthesis, laying the foundation for automatic synthesis.106 This milestone was followed by the development of automated analytical systems107 and algorithm-driven chemical synthesis platforms.108 However, the complexity of chemical reactions and the limitations of automatic technologies have hindered the widespread adoption of these innovations.
With the advent of the AI era, the development of versatile mechanical systems and advanced algorithms has empowered chemists to design automated setups for complex organic reactions at significantly reduced costs. This progress has paved the way for autonomous experimentation, a process in which AI and automation systems independently plan, execute, analyze, and optimize experiments with minimal human intervention. By enhancing experimental efficiency, reducing human error, and enabling a systematic exploration of chemical space, autonomous experimentation is poised to revolutionize the field of chemistry, transforming how discoveries are made and accelerating the pace of innovation.
Automatic experimental technique
Autonomous experimental techniques involve the use of automated systems and technologies to perform experimental procedures with minimal human intervention. In chemical instrumentation, limited automation, such as auto-samplers in analytical devices, has already been widely adopted. In chemical synthesis, automation primarily takes the form of HTE and flow chemistry, which replace manual operations in experimental workflows.The use of multiparallel batch reactors is an effective approach to automate reactions and improve efficiency in HTE. Typically conducted in 96-, 384-, or 1536-well plates, HTE employs automated dosing systems to accelerate reaction screening through parallel processing and batch analysis. This approach is widely used in synthesizing large screening libraries for drug discovery and has also been applied in molecular catalysis for condition screening109,110 and reaction dataset construction (Fig. 11A).111 Despite its advantages, such as low material consumption and high parallelism, HTE faces challenges with compatibility for complex conditions and limitations in screening diverse substrates due to analytical constraints. Consequently, its adoption in organic synthesis laboratories remains limited.
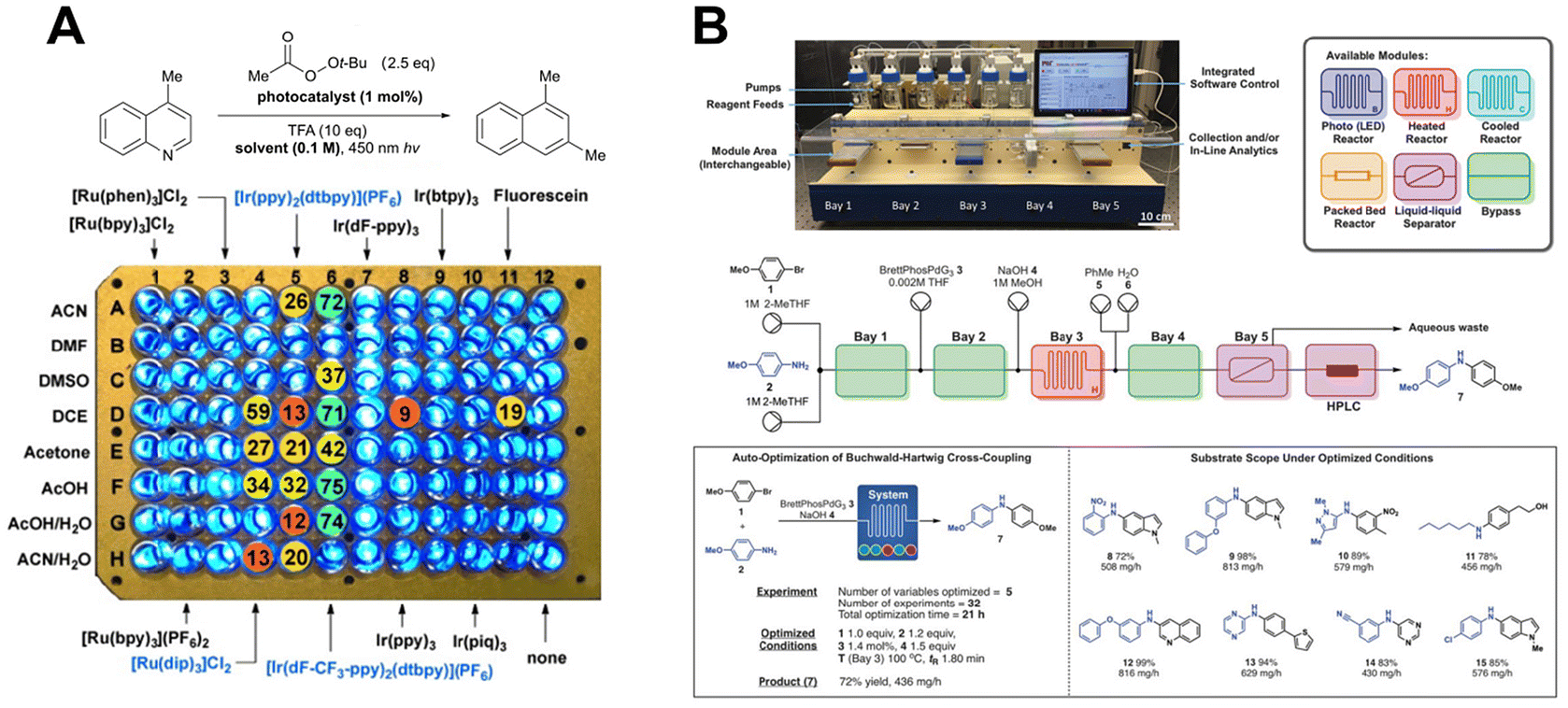 | ||
| Fig. 11 Examples of autonomous experimental techniques. (A) HTE applied to photoreaction condition screening. Reprinted with permission request from ref. 111, copyright 2023, Royal Society of Chemistry (B) Flow chemistry systems employed for Buchwald–Hartwig coupling reactions. Reprinted with permission request from ref. 116, copyright 2018, American Association for the Advancement of Science. | ||
Another approach to automating experiments is flow chemistry. Flow chemistry uses syringe pumps for sample injection, allowing reactions to occur in flow loops or reactors. One major advantage of flow chemistry is its compatibility with analytical techniques such as HPLC, enabling efficient compound synthesis and analysis. Additionally, by combining multiple flow loops in series or parallel, multistep synthesis can be achieved.
Although a single flow setup is typically optimized for one reaction type, innovations like the “radial synthesizer” have enabled multistep syntheses and optimizations for specific target molecules without requiring complex reconfigurations. For example, Jensen et al. designed a plug-and-play continuous-flow chemical synthesis system, where users can control reagent selection, unit operations (e.g., reactors and separators), and reaction analytics through a graphical interface. Catalytic reactions such as Buchwald–Hartwig cross-coupling, Horner–Wadsworth–Emmons (HWE) olefination, reductive amination, aromatic nucleophilic substitution, photoredox reactions, and [2 + 2] cycloaddition have been successfully demonstrated. Additionally, methods for conducting photoelectrochemical reactions using flow chemistry have also been developed. However, flow chemistry still has limitations. Due to reaction and analysis times, its efficiency is not significantly superior to manual experimentation. Furthermore, metal-based flow loops may influence reaction outcomes, potentially reducing reproducibility.
Flow chemistry represents another approach to automating experiments.112–114 Using syringe pumps for sample injection, reactions occur in flow loops or reactors. A key advantage is its compatibility with analytical techniques like HPLC, enabling efficient compound synthesis and analysis. By integrating multiple flow loops in series or parallel, multistep syntheses can also be achieved.
Innovations such as the “radial synthesizer” have further expanded the potential of flow chemistry, enabling multistep syntheses and optimization for target molecules without complex reconfigurations.115 For example, Jensen et al. developed a plug-and-play continuous-flow system, allowing users to control reagents, unit operations, and analytics through a graphical interface.116 This system has been successfully applied to catalytic reactions such as Buchwald–Hartwig cross-coupling, HWE olefination, reductive amination, photoredox reactions, and [2 + 2] cycloadditions (Fig. 11B). Additionally, photoelectrochemical reactions have been demonstrated using flow chemistry.117 However, flow chemistry has limitations. Its efficiency is not markedly superior to manual experimentation due to reaction and analysis times. Meanwhile, metal-based flow loops can influence reaction outcomes, leading to results that may not be easily reproducible in traditional synthetic laboratories.118,119
To better align with existing synthesis techniques in the laboratory, Cronin et al. reported a novel chemical synthesis framework called Chemputer.120–122 They reimagined chemical synthesis as like computer programming, using sensors and control components to manage hardware operations and flow pumps and valves for system interconnections. The χDL framework acts as a “code” to translate chemical reactions into executable steps. This approach successfully enabled the automation of various organic reactions and was extended to perform parallel reactions.123 While the ChemPU requires complex equipment, it partially overcomes the compatibility challenges often faced by flow chemistry systems.
Automatic experimental techniques for laboratory synthesis have made significant strides, but challenges remain due to the complexity of organic reactions. Reactions demanding strict anhydrous or anaerobic conditions, or those involving insoluble or viscous substances, often surpass the capabilities of standard systems. Additionally, the limited expertise in adapting reactions for automation presents an additional barrier to widespread adoption. Compact and affordable devices with lower automation levels but greater compatibility could help overcome these barriers, enabling traditional synthesis laboratories to transition toward modern workflows and expanding automation's accessibility in research.
Closed-loop experimentation
Closed-loop experimentation integrates experimental design, execution, and analysis into a unified process for reaction discovery and optimization. By leveraging decision-making AI and automated experimental systems, this approach enables the execution of experiments, collection of reaction data, and data-driven recommendations for subsequent experiments in a seamless loop. Some automated devices, such as automated column chromatography systems, have already demonstrated elements of closed-loop functionality. Extending this methodology to molecular catalysis holds great promise for significantly accelerating the efficiency of reaction discovery and optimization. In practical applications, closed-loop experiments have been conducted using flow chemistry or robotic chemists, demonstrating significant advancements in both reaction condition optimization and molecular synthesis (Fig. 12). | ||
| Fig. 12 Various experimental setups for closed-loop experimentation. The four systems on the left illustrate the use of flow chemistry to achieve automated experimentation. Closed-loop workflow (Reprinted with permission request from ref. 124, copyright 2022, American Association for the Advancement of Science) and RoboChem (Reprinted with permission request from ref. 125, copyright 2024, American Association for the Advancement of Science) are designed for closed-loop condition optimization, while the robotic platform for flow synthesis (Reprinted with permission request from ref. 46, copyright 2019, American Association for the Advancement of Science) and Chemputer (Reprinted with permission request from ref. 126, copyright 2019, American Association for the Advancement of Science) enable closed-loop molecular synthesis. The figure on the right illustrates mobile robotic chemist (Reprinted with permission request from ref. 128 copyright 2020, Springer Nature) capable of performing human-like tasks, offering high adaptability for both reaction condition optimization and molecular synthesis. | ||
Integrating automation with optimization algorithms can significantly enhance the efficiency of reaction optimization. Grzybowski and Burke et al. successfully utilized a closed-loop system to optimize conditions for the Suzuki–Miyaura cross-coupling reaction, achieving a significantly broader substrate scope compared to previously published methods.124 Similarly, Noël et al. demonstrated the use of a robotic platform, RoboChem, for the self-optimization and scale-up of photochemical reactions.125 This system, integrating components such as continuous flow photoreactors and benchtop NMR spectrometers, optimized diverse photochemical reactions. The approach not only accelerated the procedure to identify improved reaction conditions but also highlighted the potential of NMR as an alternative to traditional liquid chromatography for yield determination.
Beyond reaction condition optimization, closed-loop systems have also been applied to molecular synthesis. A notable example is the robotic platform developed by Green and Jensen.46 This platform, enabled by ASKCOS software, facilitates the automated flow synthesis of small-molecule pharmaceuticals. Another notable example is the Chemputer, developed by Cronin et al., which incorporates extensive sensor integration to replicate the human-like execution of routine organic operations, such as extraction and evaporation.126 Demonstrated through the synthesis of 3 pharmaceutical molecules, the Chemputer showcases the potential of automated synthesis for general applications. While the system currently relies on chemists to encode reaction steps for synthesis planning, it holds the promise of integrating automated retrosynthetic planning software, paving the way for a fully autonomous closed-loop reaction planning framework.
Robotic chemists, designed to handle samples similar to human chemists, are well-suited for traditional organic laboratories and experimental setups, offering greater scalability for addressing complex synthesis and optimization challenges.127 For example, Cooper et al. developed a mobile robotic chemist guided by Bayesian search algorithms, capable of autonomously conducting large-scale experiments and identifying optimal reaction conditions.128 By integrating mobile robots, automated synthesis platforms, and analytical tools such as LC-MS and NMR, they established an end-to-end workflow for autonomous reaction optimization and substrate identification.129 Despite these advances, robotic chemists are generally limited to performing one reaction at a time, with throughput comparable to human chemists, and they still rely on fixed programming methods. These limitations highlight the gap in efficiency compared to flow chemistry and HTE systems, as well as the need for greater adaptability to varying reaction conditions in future developments.
Closed-loop optimization can be applied not only to the selection of reaction conditions but also to molecular discovery. Li and Cooper et al. demonstrated a two-step data-driven approach to screen and synthesize organic conjugated photocatalysts (OCPs) from a virtual molecular library.130 However, the vastness of chemical space highlights a limitation of this approach: the inefficiency of human-driven synthesis hinders large-scale exploration, even with modern ML algorithms. To tackle these challenges, Burke and Aspuru-Guzik et al. proposed asynchronous cloud-based delocalized closed-loop (ACDC) optimization, applying it to discover organic solid-state laser gain materials.131 By screening 92800 molecules in silico and employing BO to recommend candidates, they synthesized and characterized 12 superior compounds using automatic platforms.132 While human involvement remains crucial in molecular catalysis, such automated platforms are essential for advancing data-driven closed-loop discovery.
While closed-loop reaction optimization strategies is promising, significant challenges remain in fully integrating these approaches into organic reaction research. The ultimate goal of closed-loop systems is to achieve seamless integration of AI and automation for reaction discovery and optimization. Burke and Aspuru-Guzik et al. offer a glimpse into this goal.133 As shown in Fig. 13, phase I utilized Bayesian optimization (BO) to guide the synthesis of photo-stable molecules, with interpretable machine learning (ML) models based on DFT descriptors highlighting the critical role of solvents. In phase II, additional experiments confirmed the impact of solvents, and phase III focused on solvent optimization, further enhancing photostability. This workflow enables researchers to uncover new insights and iteratively apply them to molecular design, overcoming the limitations of traditional hypothesis-driven trial-and-error approaches.
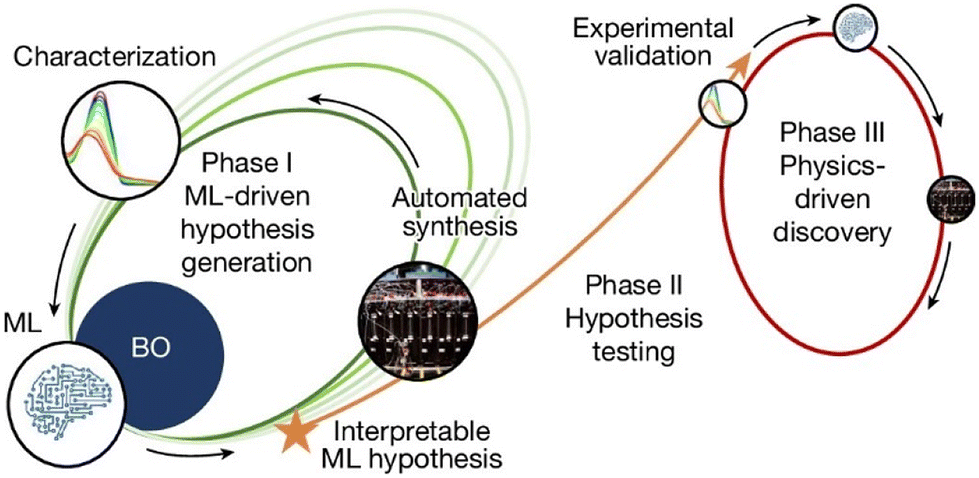 | ||
| Fig. 13 The closed-loop transfer (CLT) diagram. Phase I involves ML-driven reaction discovery and optimization. In phase II, ML-generated hypotheses are experimentally validated, leading to phase III, where physics-driven discovery generates new insights into molecular catalysis. Reprint with permission request from ref. 133, copyright 2024, Springer Nature. | ||
In summary, autonomous experimentation is transforming molecular catalysis by integrating closed-loop optimization with AI, driving advances in reaction discovery and optimization. However, as previously discussed, limitations in AI modeling and automation technology constrain the broader application of closed-loop systems in molecular catalysis. Overcoming these barriers requires not only the adoption of AI by synthetic chemists but also the development of more user-friendly and cost-effective hardware and software solutions with interdisciplinary collaboration.
Conclusion and outlook
Returning to the core question, “AI molecular catalysis: where are we now?”—For retrosynthesis, AI-driven approaches have already demonstrated substantial efficacy in retrosynthetic route design for simple molecules. Even for more complex natural products, AI has begun to offer valuable guidance for human chemists. For catalyst design, virtual library screening has emerged as a promising tool, though its current application remains confined to few catalyst scaffolds. For reaction development, AI has shown impressive performance in optimizing well-established reactions, with the prospect of extending these methods to the discovery of entirely new reactions appearing increasingly attainable. Furthermore, autonomous experimentation enabled by flow chemistry and robotic chemists has been validated across a few selected cases. With the continued development and commercialization of these technologies, they are likely to become standard tools in laboratories in the near future.However, for AI-driven methods to be deeply integrated into molecular catalysis, several challenges must still be addressed. As highlighted in this review, current models have yet to match or surpass human chemists in terms of domain-specific knowledge within chemical systems. While data distribution and quality are critical factors, a deeper challenge lies in the unmatched ability of human chemists to systematically acquire and apply chemical domain knowledge. While improving molecular representations and model performance are straightforward technical solutions, a more fundamental approach lies in embedding chemical domain knowledge into the development of AI models.18 Additionally, the high cost and limited applicability of automation devices further constrain their utility for a broad range of organic reactions, limiting widespread adoption in laboratories.
Despite the challenges ahead, the collaborative progress between AI and molecular catalysis is poised to usher in a new chapter in the development of organic chemistry. High-quality databases will serve as the foundation for robust and reliable AI-driven predictions. More intuitive and accessible AI tools will empower chemists from diverse backgrounds to integrate ML into their research, breaking down technical barriers. In parallel, cultivating a new generation of talent with expertise in both chemistry and AI will be crucial for bridging the gap between these fields and fostering transformative innovation. Finally, interdisciplinary collaborations, combining the strengths of chemistry, computer science, physics, and engineering, will drive the creation of novel AI-powered methodologies and paradigms, accelerating progress in molecular catalysis.
At the threshold of a new era, the integration of AI and chemistry holds the promise of transformative advancements in molecular catalysis. This paradigm shift is poised to redefine the landscape of chemical research, accelerating discoveries and innovations once deemed beyond reach. By delivering unprecedented efficiency, precision, and exploratory power, it will pave the way for a future where molecular catalysis achieves unparalleled heights of possibility and impact.
Author contributions
Conceptualization – S.L. and Q.Y.; writing – original draft, Z.T.; writing – review and editing, Q.Y., Z.T., and S.L.; funding acquisition, Q.Y. and S.L. All authors have given approval to the final version of the manuscript.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Natural Science Foundation of China (22031006, 22373056 and 22393891), the National Key R&D Program of China (2023YFA1506401 and 2023YFA1506402), Tsinghua University Initiative Scientific Research Program and Haihe Laboratory of Sustainable Chemical Transformations for financial support.References
- Y. Xu, X. Liu, X. Cao, C. Huang, E. Liu, S. Qian, X. Liu, Y. Wu, F. Dong, C.-W. Qiu, J. Qiu, K. Hua, W. Su, J. Wu, H. Xu, Y. Han, C. Fu, Z. Yin, M. Liu, R. Roepman, S. Dietmann, M. Virta, F. Kengara, Z. Zhang, L. Zhang, T. Zhao, J. Dai, J. Yang, L. Lan, M. Luo, Z. Liu, T. An, B. Zhang, X. He, S. Cong, X. Liu, W. Zhang, J. P. Lewis, J. M. Tiedje, Q. Wang, Z. An, F. Wang, L. Zhang, T. Huang, C. Lu, Z. Cai, F. Wang and J. Zhang, Artificial intelligence: A powerful paradigm for scientific research, The Innovation, 2021, 2, 100179 CrossRef PubMed.
- Z. J. Baum, X. Yu, P. Y. Ayala, Y. Zhao, S. P. Watkins and Q. Zhou, Artificial Intelligence in Chemistry: Current Trends and Future Directions, J. Chem. Inf. Model., 2021, 61, 3197–3212 CrossRef CAS PubMed.
- V. P. Ananikov, Top 20 influential AI-based technologies in chemistry, Artif. Intell. Chem., 2024, 2, 100075 CrossRef.
- L. Gomez, Decision Making in Medicinal Chemistry: The Power of Our Intuition, ACS Med. Chem. Lett., 2018, 9, 956–958 CrossRef CAS PubMed.
- S. Copeland, On serendipity in science: discovery at the intersection of chance and wisdom, Synthese, 2017, 196, 2385–2406 CrossRef.
- P. R. Wells, Linear Free Energy Relationships, Chem. Rev., 1963, 63, 171–219 CrossRef CAS.
- J. N. Brønsted and K. Pedersen, Die katalytische Zersetzung des Nitramids und ihre physikalisch-chemische Bedeutung, Z. Phys. Chem., 1924, 108U, 185–235 CrossRef.
- L. P. Hammett, The effect of structure upon the reactions of organic compounds. Benzene derivatives, J. Am. Chem. Soc., 1937, 59, 96–103 CrossRef CAS.
- R. W. Taft Jr, Polar and steric substituent constants for aliphatic and o-Benzoate groups from rates of esterification and hydrolysis of esters1, J. Am. Chem. Soc., 1952, 74, 3120–3128 CrossRef.
- H. Mayr, CC Bond Formation by Addition of Carbenium Ions to Alkenes: Kinetics and Mechanism, Angew. Chem., Int. Ed., 2003, 29, 1371–1384 CrossRef.
- H. Mayr and M. Patz, Scales of Nucleophilicity and Electrophilicity: A System for Ordering Polar Organic and Organometallic Reactions, Angew. Chem., Int. Ed., 2003, 33, 938–957 CrossRef.
- J. Li and M. D. Eastgate, Current complexity: a tool for assessing the complexity of organic molecules, Org. Biomol. Chem., 2015, 13, 7164–7176 RSC.
- F. Provost and T. Fawcett, Data Science and its Relationship to Big Data and Data-Driven Decision Making, Big Data, 2013, 1, 51–59 CrossRef PubMed.
- W. L. Williams, L. Zeng, T. Gensch, M. S. Sigman, A. G. Doyle and E. V. Anslyn, The Evolution of Data-Driven Modeling in Organic Chemistry, ACS Cent. Sci., 2021, 7, 1622–1637 CrossRef CAS PubMed.
- N. Graulich, H. Hopf and P. R. Schreiner, Heuristic thinking makes a chemist smart, Chem. Soc. Rev., 2010, 39, 1503–1512 RSC.
- T. F. G. G. Cova and A. A. C. C. Pais, Deep Learning for Deep Chemistry: Optimizing the Prediction of Chemical Patterns, Front. Chem., 2019, 7, 809 CrossRef CAS.
- R. S. Aal E Ali, J. Meng, M. E. I. Khan and X. Jiang, Machine learning advancements in organic synthesis: A focused exploration of artificial intelligence applications in chemistry, Artif. Intell. Chem., 2024, 2, 100049 CrossRef.
- S. Q. Zhang, L. C. Xu, S. W. Li, J. C. A. Oliveira, X. Li, L. Ackermann and X. Hong, Bridging Chemical Knowledge and Machine Learning for Performance Prediction of Organic Synthesis, Chemistry, 2023, 29, e202202834 CrossRef CAS.
- X. Hong, Q. Yang, K. Liao, J. Pei, M. Chen, F. Mo, H. Lu, W.-B. Zhang, H. Zhou, J. Chen, L. Su, S.-Q. Zhang, S. Liu, X. Huang, Y.-Z. Sun, Y. Wang, Z. Zhang, Z. Yu, S. Luo, X.-F. Fu and S.-L. You, AI for organic and polymer synthesis, Sci. China: Chem., 2024, 67, 2461–2496 CrossRef CAS.
- A. F. de Almeida, R. Moreira and T. Rodrigues, Synthetic organic chemistry driven by artificial intelligence, Nat. Rev. Chem., 2019, 3, 589–604 CrossRef.
- R. Robinson, LXIII.—A synthesis of tropinone, J. Chem. Soc., Trans., 1917, 111, 762–768 RSC.
- K. C. Nicolaou and E. J. Sorensen, Classics in total synthesis: targets, strategies, methods, John Wiley & Sons, 1996 Search PubMed.
- E. J. Corey, General methods for the construction of complex molecules, Pure Appl. Chem., 1967, 14, 19–38 CrossRef CAS.
- Y. Jiang, Y. Yu, M. Kong, Y. Mei, L. Yuan, Z. Huang, K. Kuang, Z. Wang, H. Yao and J. Zou, Artificial intelligence for retrosynthesis prediction, Engineering, 2023, 25, 32–50 CrossRef.
- S. Chen and Y. Jung, Deep Retrosynthetic Reaction Prediction using Local Reactivity and Global Attention, JACS Au, 2021, 1, 1612–1620 CrossRef CAS PubMed.
- X. Wang, Y. Li, J. Qiu, G. Chen, H. Liu, B. Liao, C.-Y. Hsieh and X. Yao, RetroPrime: A Diverse, plausible and Transformer-based method for Single-Step retrosynthesis predictions, Chem. Eng. J., 2021, 420, 129845 CrossRef CAS.
- S.-W. Seo, Y. Y. Song, J. Y. Yang, S. Bae, H. Lee, J. Shin, S. J. Hwang and E. YangGTA: Graph Truncated Attention for RetrosynthesisProceedings of the AAAI Conference on Artificial Intelligence 2021, vol. 35, pp. 531–539 Search PubMed.
- M. Sacha, M. Błaz, P. Byrski, P. Dabrowski-Tumanski, M. Chrominski, R. Loska, P. Włodarczyk-Pruszynski and S. Jastrzebski, Molecule edit graph attention network: modeling chemical reactions as sequences of graph edits, J. Chem. Inf. Model., 2021, 61, 3273–3284 CrossRef CAS PubMed.
- Z. Zhong, J. Song, Z. Feng, T. Liu, L. Jia, S. Yao, T. Hou and M. Song, Recent advances in deep learning for retrosynthesis, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2024, 14, e1694 Search PubMed.
- E. J. Corey and W. T. Wipke, Computer-assisted design of complex organic syntheses, Science, 1969, 166, 178–192 CrossRef CAS PubMed.
- E. J. Corey, W. T. Wipke, R. D. Cramer and W. J. Howe, Computer-assisted synthetic analysis. Facile man-machine communication of chemical structure by interactive computer graphics, J. Am. Chem. Soc., 2002, 94, 421–430 CrossRef.
- W. T. Wipke, G. I. Ouchi and S. Krishnan, Simulation and evaluation of chemical synthesis—SECS: An application of artificial intelligence techniques, Artif. Intell., 1978, 11, 173–193 CrossRef.
- P. Y. Johnson, I. Burnstein, J. Crary, M. Evans and T. Wang, in Expert System Applications in Chemistry, American Chemical Society, 1989, vol. 408, ch. 9, pp. 102–123 Search PubMed.
- H. L. Gelernter, A. F. Sanders, D. L. Larsen, K. K. Agarwal, R. H. Boivie, G. A. Spritzer and J. E. Searleman, Empirical Explorations of SYNCHEM, Science, 1977, 197, 1041–1049 CrossRef CAS.
- J. B. Hendrickson and A. G. Toczko, SYNGEN program for synthesis design: basic computing techniques, J. Chem. Inf. Comput. Sci., 2002, 29, 137–145 CrossRef.
- I. Ugi, J. Bauer, K. Bley, A. Dengler, A. Dietz, E. Fontain, B. Gruber, R. Herges, M. Knauer, K. Reitsam and N. Stein, Computer–Assisted Solution of Chemical Problems—The Historical Development and the Present State of the Art of a New Discipline of Chemistry, Angew. Chem., Int. Ed., 2003, 32, 201–227 CrossRef.
- M. Pförtner and M. Sitzmann, in Handbook of Chemoinformatics, 2003, pp. 1457–1507, DOI:10.1002/9783527618279.ch44a.
- S. Hanessian, J. Franco and B. Larouche, The psychobiological basis of heuristic synthesis planning - man, machine and the chiron approach, Pure Appl. Chem., 1990, 62, 1887–1910 CrossRef CAS.
- S. Szymkuc, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Computer-Assisted Synthetic Planning: The End of the Beginning, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef CAS.
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. J. Trice and B. A. Grzybowski, Efficient Syntheses of Diverse, Medicinally Relevant Targets Planned by Computer and Executed in the Laboratory, Chem, 2018, 4, 522–532 CAS.
- B. Mikulak-Klucznik, P. Golebiowska, A. A. Bayly, O. Popik, T. Klucznik, S. Szymkuc, E. P. Gajewska, P. Dittwald, O. Staszewska-Krajewska, W. Beker, T. Badowski, K. A. Scheidt, K. Molga, J. Mlynarski, M. Mrksich and B. A. Grzybowski, Computational planning of the synthesis of complex natural products, Nature, 2020, 588, 83–88 CrossRef CAS PubMed.
- A. Bøgevig, H.-J. Federsel, F. Huerta, M. G. Hutchings, H. Kraut, T. Langer, P. Löw, C. Oppawsky, T. Rein and H. Saller, Route Design in the 21st Century: The ICSYNTH Software Tool as an Idea Generator for Synthesis Prediction, Org. Process Res. Dev., 2015, 19, 357–368 CrossRef.
- M. A. Kayala and P. Baldi, ReactionPredictor: prediction of complex chemical reactions at the mechanistic level using machine learning, J. Chem. Inf. Model., 2012, 52, 2526–2540 CrossRef CAS.
- C. W. Coley, W. H. Green and K. F. Jensen, RDChiral: An RDKit Wrapper for Handling Stereochemistry in Retrosynthetic Template Extraction and Application, J. Chem. Inf. Model., 2019, 59, 2529–2537 CrossRef CAS.
- M. H. S. Segler, M. Preuss and M. P. Waller, Planning chemical syntheses with deep neural networks and symbolic AI, Nature, 2018, 555, 604–610 CrossRef CAS.
- C. W. Coley, D. A. Thomas 3rd, J. A. M. Lummiss, J. N. Jaworski, C. P. Breen, V. Schultz, T. Hart, J. S. Fishman, L. Rogers, H. Gao, R. W. Hicklin, P. P. Plehiers, J. Byington, J. S. Piotti, W. H. Green, A. J. Hart, T. F. Jamison and K. F. Jensen, A robotic platform for flow synthesis of organic compounds informed by AI planning, Science, 2019, 365, eaax1566 CrossRef CAS PubMed.
- L. Saigiridharan, A. K. Hassen, H. Lai, P. Torren-Peraire, O. Engkvist and S. Genheden, AiZynthFinder 4.0: developments based on learnings from 3 years of industrial application, J. Cheminf., 2024, 16, 57 Search PubMed.
- S. Genheden, A. Thakkar, V. Chadimova, J. L. Reymond, O. Engkvist and E. Bjerrum, AiZynthFinder: a fast, robust and flexible open-source software for retrosynthetic planning, J. Cheminf., 2020, 12, 70 Search PubMed.
- A. Thakkar, T. Kogej, J. L. Reymond, O. Engkvist and E. J. Bjerrum, Datasets and their influence on the development of computer assisted synthesis planning tools in the pharmaceutical domain, Chem. Sci., 2020, 11, 154–168 RSC.
- Y. Sun and N. V. Sahinidis, Computer-aided retrosynthetic design: fundamentals, tools, and outlook, Curr. Opin. Chem. Eng., 2022, 35, 100721 CrossRef.
- D. Weininger, SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules, J. Chem. Inf. Comput. Sci., 1988, 28, 31–36 CrossRef CAS.
- A. Cadeddu, E. K. Wylie, J. Jurczak, M. Wampler-Doty and B. A. Grzybowski, Organic chemistry as a language and the implications of chemical linguistics for structural and retrosynthetic analyses, Angew. Chem., Int. Ed., 2014, 53, 8108–8112 CrossRef CAS.
- B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender and V. Pande, Retrosynthetic reaction prediction using neural sequence-to-sequence models, ACS Cent. Sci., 2017, 3, 1103–1113 CrossRef CAS PubMed.
- P. Schwaller, T. Laino, T. Gaudin, P. Bolgar, C. A. Hunter, C. Bekas and A. A. Lee, Molecular Transformer: A Model for Uncertainty-Calibrated Chemical Reaction Prediction, ACS Cent. Sci., 2019, 5, 1572–1583 CrossRef CAS.
- P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano and T. Laino, Predicting retrosynthetic pathways using transformer-based models and a hyper-graph exploration strategy, Chem. Sci., 2020, 11, 3316–3325 RSC.
- L. Yao, W. Guo, Z. Wang, S. Xiang, W. Liu and G. Ke, Node-Aligned Graph-to-Graph: Elevating Template-free Deep Learning Approaches in Single-Step Retrosynthesis, JACS Au, 2024, 4, 992–1003 CrossRef CAS PubMed.
- S. Szymkuc, T. Badowski and B. A. Grzybowski, Is Organic Chemistry Really Growing Exponentially?, Angew. Chem., Int. Ed., 2021, 60, 26226–26232 CrossRef CAS PubMed.
- P. H.-Y. Cheong, C. Y. Legault, J. M. Um, N. Çelebi-Ölçüm and K. N. Houk, Quantum Mechanical Investigations of Organocatalysis: Mechanisms, Reactivities, and Selectivities, Chem. Rev., 2011, 111, 5042–5137 CrossRef CAS.
- X. Li, H. Deng, B. Zhang, J. Li, L. Zhang, S. Luo and J. P. Cheng, Physical organic study of structure–activity–enantioselectivity relationships in asymmetric bifunctional thiourea catalysis: Hints for the design of new organocatalysts, Chem. – Eur. J., 2009, 2, 450–455 Search PubMed.
- R. R. Knowles and E. N. Jacobsen, Attractive noncovalent interactions in asymmetric catalysis: links between enzymes and small molecule catalysts, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 CrossRef CAS PubMed.
- S. J. Zuend and E. N. Jacobsen, Mechanism of amido-thiourea catalyzed enantioselective imine hydrocyanation: transition state stabilization via multiple non-covalent interactions, J. Am. Chem. Soc., 2009, 131, 15358–15374 CrossRef CAS PubMed.
- A. Milo, E. N. Bess and M. S. Sigman, Interrogating selectivity in catalysis using molecular vibrations, Nature, 2014, 507, 210–214 CrossRef CAS.
- S. E. Wheeler and K. N. Houk, Substituent effects in the benzene dimer are due to direct interactions of the substituents with the unsubstituted benzene, J. Am. Chem. Soc., 2008, 130, 10854–10855 CrossRef CAS PubMed.
- H. W. Liu, P. He, W. T. Li, W. Sun, K. Shi, Y. Q. Wang, Q. K. Mo, X. Y. Zhang and S. F. Zhu, Catalyst-Oriented Design Based on Elementary Reactions (CODER) for Triarylamine Synthesis, Angew. Chem., Int. Ed., 2023, 62, e202309111 CrossRef CAS.
- K. C. Harper and M. S. Sigman, Three-dimensional correlation of steric and electronic free energy relationships guides asymmetric propargylation, Science, 2011, 333, 1875–1878 CrossRef CAS.
- J. A. Hueffel, T. Sperger, I. Funes-Ardoiz, J. S. Ward, K. Rissanen and F. Schoenebeck, Accelerated dinuclear palladium catalyst identification through unsupervised machine learning, Science, 2021, 374, 1134–1140 CrossRef CAS PubMed.
- T. Schnitzer, M. Schnurr, A. F. Zahrt, N. Sakhaee, S. E. Denmark and H. Wennemers, Machine Learning to Develop Peptide Catalysts-Successes, Limitations, and Opportunities, ACS Cent. Sci., 2024, 10, 367–373 CrossRef CAS PubMed.
- T. Gensch, G. Dos Passos Gomes, P. Friederich, E. Peters, T. Gaudin, R. Pollice, K. Jorner, A. Nigam, M. Lindner-D'Addario, M. S. Sigman and A. Aspuru-Guzik, A Comprehensive Discovery Platform for Organophosphorus Ligands for Catalysis, J. Am. Chem. Soc., 2022, 144, 1205–1217 CrossRef CAS.
- W. Matsuoka, Y. Harabuchi, Y. Nagata and S. Maeda, Highly chemoselective ligands for Suzuki-Miyaura cross-coupling reaction based on virtual ligand-assisted screening, Org. Biomol. Chem., 2023, 21, 3132–3142 RSC.
- J. F. Goebel, J. Loffler, Z. Zeng, J. Handelmann, A. Hermann, I. Rodstein, T. Gensch, V. H. Gessner and L. J. Goossen, Computer-Driven Development of Ylide Functionalized Phosphines for Palladium-Catalyzed Hiyama Couplings, Angew. Chem., Int. Ed., 2023, 62, e202216160 CrossRef CAS PubMed.
- K. Feng, E. R. Raguram, J. R. Howard, E. Peters, C. Liu, M. S. Sigman and S. L. Buchwald, Development of a Deactivation-Resistant Dialkylbiarylphosphine Ligand for Pd-Catalyzed Arylation of Secondary Amines, J. Am. Chem. Soc., 2024, 146, 26609–26615 CrossRef CAS PubMed.
- N. P. Romer, D. S. Min, J. Y. Wang, R. C. Walroth, K. A. Mack, L. E. Sirois, F. Gosselin, D. Zell, A. G. Doyle and M. S. Sigman, Data Science Guided Multiobjective Optimization of a Stereoconvergent Nickel-Catalyzed Reduction of Enol Tosylates to Access Trisubstituted Alkenes, ACS Catal., 2024, 14, 4699–4708 CrossRef CAS.
- Y. Du, A. R. Jamasb, J. Guo, T. Fu, C. Harris, Y. Wang, C. Duan, P. Liò, P. Schwaller and T. L. Blundell, Machine learning-aided generative molecular design, Nat. Mach. Intell., 2024, 6, 589–604 CrossRef.
- J. Guo, J. P. Janet, M. R. Bauer, E. Nittinger, K. A. Giblin, K. Papadopoulos, A. Voronov, A. Patronov, O. Engkvist and C. Margreitter, DockStream: a docking wrapper to enhance de novo molecular design, J. Cheminf., 2021, 13, 89 Search PubMed.
- A. A. Sadybekov, A. V. Sadybekov, Y. Liu, C. Iliopoulos-Tsoutsouvas, X.-P. Huang, J. Pickett, B. Houser, N. Patel, N. K. Tran, F. Tong, N. Zvonok, M. K. Jain, O. Savych, D. S. Radchenko, S. P. Nikas, N. A. Petasis, Y. S. Moroz, B. L. Roth, A. Makriyannis and V. Katritch, Synthon-based ligand discovery in virtual libraries of over 11 billion compounds, Nature, 2022, 601, 452–459 CrossRef CAS.
- H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green and K. F. Jensen, Using Machine Learning To Predict Suitable Conditions for Organic Reactions, ACS Cent. Sci., 2018, 4, 1465–1476 CrossRef CAS PubMed.
- Z. Wang, K. Lin, J. Pei and L. Lai, Reacon: a template- and cluster-based framework for reaction condition prediction, Chem. Sci., 2025, 16, 854–866 RSC.
- G. Marcou, J. Aires de Sousa, D. A. Latino, A. de Luca, D. Horvath, V. Rietsch and A. Varnek, Expert system for predicting reaction conditions: the Michael reaction case, J. Chem. Inf. Model., 2015, 55, 239–250 CrossRef CAS.
- C. W. Coley, R. Barzilay, T. S. Jaakkola, W. H. Green and K. F. Jensen, Prediction of Organic Reaction Outcomes Using Machine Learning, ACS Cent. Sci., 2017, 3, 434–443 CrossRef CAS.
- P. Schwaller, T. Gaudin, D. Lanyi, C. Bekas and T. Laino, “Found in Translation”: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models, Chem. Sci., 2018, 9, 6091–6098 RSC.
- Y. Guan, C. W. Coley, H. Wu, D. Ranasinghe, E. Heid, T. J. Struble, L. Pattanaik, W. H. Green and K. F. Jensen, Regio-selectivity prediction with a machine-learned reaction representation and on-the-fly quantum mechanical descriptors, Chem. Sci., 2020, 12, 2198–2208 RSC.
- X. Li, S. Q. Zhang, L. C. Xu and X. Hong, Predicting Regioselectivity in Radical C-H Functionalization of Heterocycles through Machine Learning, Angew. Chem., Int. Ed., 2020, 59, 13253–13259 CrossRef CAS PubMed.
- D. T. Ahneman, J. G. Estrada, S. Lin, S. D. Dreher and A. G. Doyle, Predicting reaction performance in C-N cross-coupling using machine learning, Science, 2018, 360, 186–190 CrossRef CAS PubMed.
- P. Raghavan, A. J. Rago, P. Verma, M. M. Hassan, G. M. Goshu, A. W. Dombrowski, A. Pandey, C. W. Coley and Y. Wang, Incorporating Synthetic Accessibility in Drug Design: Predicting Reaction Yields of Suzuki Cross-Couplings by Leveraging AbbVie's 15-Year Parallel Library Data Set, J. Am. Chem. Soc., 2024, 146, 15070–15084 CrossRef CAS.
- J. Schleinitz, M. Langevin, Y. Smail, B. Wehnert, L. Grimaud and R. Vuilleumier, Machine Learning Yield Prediction from NiCOlit, a Small-Size Literature Data Set of Nickel Catalyzed C-O Couplings, J. Am. Chem. Soc., 2022, 144, 14722–14730 CrossRef CAS PubMed.
- A. F. Zahrt, J. J. Henle, B. T. Rose, Y. Wang, W. T. Darrow and S. E. Denmark, Prediction of higher-selectivity catalysts by computer-driven workflow and machine learning, Science, 2019, 363, eaau5631 CrossRef CAS PubMed.
- S. W. Li, L. C. Xu, C. Zhang, S. Q. Zhang and X. Hong, Reaction performance prediction with an extrapolative and interpretable graph model based on chemical knowledge, Nat. Commun., 2023, 14, 3569 CrossRef CAS PubMed.
- B. Settles, Active learning literature survey, 2009 Search PubMed.
- Y. Ureel, M. R. Dobbelaere, Y. Ouyang, K. De Ras, M. K. Sabbe, G. B. Marin and K. M. Van Geem, Active Machine Learning for Chemical Engineers: A Bright Future Lies Ahead!, Engineering, 2023, 27, 23–30 CrossRef CAS.
- S. Sun, Z. Cao, H. Zhu and J. Zhao, A Survey of Optimization Methods From a Machine Learning Perspective, IEEE Trans. Cybern., 2020, 50, 3668–3681 Search PubMed.
- K. Wang and A. W. Dowling, Bayesian optimization for chemical products and functional materials, Curr. Opin. Chem. Eng., 2022, 36, 100728 CrossRef.
- F. Häse, M. Aldeghi, R. J. Hickman, L. M. Roch and A. Aspuru-Guzik, Gryffin: An algorithm for Bayesian optimization of categorical variables informed by expert knowledge, Appl. Phys. Rev., 2021, 8, 031406 Search PubMed.
- B. J. Shields, J. Stevens, J. Li, M. Parasram, F. Damani, J. I. M. Alvarado, J. M. Janey, R. P. Adams and A. G. Doyle, Bayesian reaction optimization as a tool for chemical synthesis, Nature, 2021, 590, 89–96 CrossRef CAS.
- J. A. G. Torres, S. H. Lau, P. Anchuri, J. M. Stevens, J. E. Tabora, J. Li, A. Borovika, R. P. Adams and A. G. Doyle, A Multi-Objective Active Learning Platform and Web App for Reaction Optimization, J. Am. Chem. Soc., 2022, 144, 19999–20007 CrossRef CAS.
- J. Y. Wang, J. M. Stevens, S. K. Kariofillis, M. J. Tom, D. L. Golden, J. Li, J. E. Tabora, M. Parasram, B. J. Shields, D. N. Primer, B. Hao, D. Del Valle, S. DiSomma, A. Furman, G. G. Zipp, S. Melnikov, J. Paulson and A. G. Doyle, Identifying general reaction conditions by bandit optimization, Nature, 2024, 626, 1025–1033 CrossRef CAS PubMed.
- N. I. Rinehart, R. K. Saunthwal, J. Wellauer, A. F. Zahrt, L. Schlemper, A. S. Shved, R. Bigler, S. Fantasia and S. E. Denmark, A machine-learning tool to predict substrate-adaptive conditions for Pd-catalyzed C-N couplings, Science, 2023, 381, 965–972 CrossRef CAS.
- E. Shim, J. A. Kammeraad, Z. Xu, A. Tewari, T. Cernak and P. M. Zimmerman, Predicting reaction conditions from limited data through active transfer learning, Chem. Sci., 2022, 13, 6655–6668 RSC.
- M. C. Kozlowski, On the topic of substrate scope, Org. Lett., 2022, 24, 7247–7249 CrossRef CAS.
- L. Rokach and O. Maimon, Clustering methods, Data mining and knowledge discovery handbook, 2005, pp. 321–352 Search PubMed.
- S. K. Kariofillis, S. Jiang, A. M. Zuranski, S. S. Gandhi, J. I. Martinez Alvarado and A. G. Doyle, Using Data Science To Guide Aryl Bromide Substrate Scope Analysis in a Ni/Photoredox-Catalyzed Cross-Coupling with Acetals as Alcohol-Derived Radical Sources, J. Am. Chem. Soc., 2022, 144, 1045–1055 CrossRef CAS PubMed.
- D. Rana, P. M. Pfluger, N. P. Holter, G. Tan and F. Glorius, Standardizing Substrate Selection: A Strategy toward Unbiased Evaluation of Reaction Generality, ACS Cent. Sci., 2024, 10, 899–906 CAS.
- E. Schubert, Stop using the elbow criterion for k-means and how to choose the number of clusters instead, ACM SIGKDD Explorations Newsletter, 2023, 25, 36–42 CrossRef.
- M. P. Maloney, C. W. Coley, S. Genheden, N. Carson, P. Helquist, P.-O. Norrby and O. Wiest, Negative Data in Data Sets for Machine Learning Training, Org. Lett., 2023, 25, 2945–2947 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, F. Sandfort, M. Kühnemund, F. R. Schäfer, H. Kuchen and F. Glorius, Machine Learning for Chemical Reactivity: The Importance of Failed Experiments, Angew. Chem., Int. Ed., 2022, 61, e202204647 CrossRef CAS PubMed.
- T. Gensch and F. Glorius, The straight dope on the scope of chemical reactions, Science, 2016, 352, 294–295 CrossRef CAS PubMed.
- R. B. Merrifield, Automated synthesis of peptides, Science, 1965, 150, 178–185 CrossRef CAS PubMed.
- G. P. Hicks, A. A. Eggert and E. C. Toren, Application of an on-line computer to the automation of analytical experiments, Anal. Chem., 2002, 42, 729–737 CrossRef.
- H. Winicov, J. Schainbaum, J. Buckley, G. Longino, J. Hill and C. E. Berkoff, Chemical process optimization by computer—a self-directed chemical synthesis system, Anal. Chim. Acta, 1978, 103, 469–476 CrossRef CAS.
- Y. Xu, Y. Gao, L. Su, H. Wu, H. Tian, M. Zeng, C. Xu, X. Zhu and K. Liao, High-Throughput Experimentation and Machine Learning-Assisted Optimization of Iridium-Catalyzed Cross-Dimerization of Sulfoxonium Ylides, Angew. Chem., Int. Ed., 2023, 62, e202313638 CrossRef CAS.
- D. A. Dirocco, K. Dykstra, S. Krska, P. Vachal, D. V. Conway and M. Tudge, Late-stage functionalization of biologically active heterocycles through photoredox catalysis, Angew. Chem., Int. Ed., 2014, 53, 4802–4806 CrossRef CAS PubMed.
- Z. Yu, Y. Kong, B. Li, S. Su, J. Rao, Y. Gao, T. Tu, H. Chen and K. Liao, HTE- and AI-assisted development of DHP-catalyzed decarboxylative selenation, Chem. Commun., 2023, 59, 2935–2938 RSC.
- C. P. Breen, A. M. K. Nambiar, T. F. Jamison and K. F. Jensen, Ready, Set, Flow! Automated Continuous Synthesis and Optimization, Trends Chem., 2021, 3, 373–386 CrossRef.
- J. Wegner, S. Ceylan and A. Kirschning, Flow Chemistry – A Key Enabling Technology for (Multistep) Organic Synthesis, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS.
- D. Perera, J. W. Tucker, S. Brahmbhatt, C. J. Helal, A. Chong, W. Farrell, P. Richardson and N. W. Sach, A platform for automated nanomole-scale reaction screening and micromole-scale synthesis in flow, Science, 2018, 359, 429–434 CrossRef CAS.
- S. Chatterjee, M. Guidi, P. H. Seeberger and K. Gilmore, Automated radial synthesis of organic molecules, Nature, 2020, 579, 379–384 CrossRef CAS PubMed.
- A. C. Bedard, A. Adamo, K. C. Aroh, M. G. Russell, A. A. Bedermann, J. Torosian, B. Yue, K. F. Jensen and T. F. Jamison, Reconfigurable system for automated optimization of diverse chemical reactions, Science, 2018, 361, 1220–1225 CrossRef CAS.
- Y. Chen, Y. He, Y. Gao, J. Xue, W. Qu, J. Xuan and Y. Mo, Scalable decarboxylative trifluoromethylation by ion-shielding heterogeneous photoelectrocatalysis, Science, 2024, 384, 670–676 CrossRef CAS.
- M. Z. C. Hatit, L. F. Reichenbach, J. M. Tobin, F. Vilela, G. A. Burley and A. J. B. Watson, A flow platform for degradation-free CuAAC bioconjugation, Nat. Commun., 2018, 9, 4021 CrossRef PubMed.
- A. J. S. Hammer, A. I. Leonov, N. L. Bell and L. Cronin, Chemputation and the Standardization of Chemical Informatics, JACS Au, 2021, 1, 1572–1587 CrossRef CAS PubMed.
- S. Rohrbach, M. Siauciulis, G. Chisholm, P. A. Pirvan, M. Saleeb, S. H. M. Mehr, E. Trushina, A. I. Leonov, G. Keenan, A. Khan, A. Hammer and L. Cronin, Digitization and validation of a chemical synthesis literature database in the ChemPU, Science, 2022, 377, 172–180 CrossRef CAS.
- S. H. M. Mehr, M. Craven, A. I. Leonov, G. Keenan and L. Cronin, A universal system for digitization and automatic execution of the chemical synthesis literature, Science, 2020, 370, 101–108 CrossRef CAS.
- P. S. Gromski, J. M. Granda and L. Cronin, Universal Chemical Synthesis and Discovery with ‘The Chemputer’, Trends Chem., 2020, 2, 4–12 CrossRef CAS.
- M. Siauciulis, C. Knittl-Frank, M. M. Sh, E. Clarke and L. Cronin, Reaction blueprints and logical control flow for parallelized chiral synthesis in the Chemputer, Nat. Commun., 2024, 15, 10261 CrossRef CAS PubMed.
- N. H. Angello, V. Rathore, W. Beker, A. Wolos, E. R. Jira, R. Roszak, T. C. Wu, C. M. Schroeder, A. Aspuru-Guzik, B. A. Grzybowski and M. D. Burke, Closed-loop optimization of general reaction conditions for heteroaryl Suzuki-Miyaura coupling, Science, 2022, 378, 399–405 CrossRef CAS PubMed.
- A. Slattery, Z. Wen, P. Tenblad, J. Sanjose-Orduna, D. Pintossi, T. den Hartog and T. Noël, Automated self-optimization, intensification, and scale-up of photocatalysis in flow, Science, 2024, 383, eadj1817 CrossRef CAS.
- S. Steiner, J. Wolf, S. Glatzel, A. Andreou, J. M. Granda, G. Keenan, T. Hinkley, G. Aragon-Camarasa, P. J. Kitson, D. Angelone and L. Cronin, Organic synthesis in a modular robotic system driven by a chemical programming language, Science, 2019, 363, eaav2211 CrossRef CAS PubMed.
- Q. Zhu, Y. Huang, D. Zhou, L. Zhao, L. Guo, R. Yang, Z. Sun, M. Luo, F. Zhang, H. Xiao, X. Tang, X. Zhang, T. Song, X. Li, B. Chong, J. Zhou, Y. Zhang, B. Zhang, J. Cao, G. Zhang, S. Wang, G. Ye, W. Zhang, H. Zhao, S. Cong, H. Li, L.-L. Ling, Z. Zhang, W. Shang, J. Jiang and Y. Luo, Automated synthesis of oxygen-producing catalysts from Martian meteorites by a robotic AI chemist, Nat. Synth., 2023, 3, 319–328 CrossRef.
- B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick and A. I. Cooper, A mobile robotic chemist, Nature, 2020, 583, 237–241 CrossRef CAS.
- T. Dai, S. Vijayakrishnan, F. T. Szczypinski, J. F. Ayme, E. Simaei, T. Fellowes, R. Clowes, L. Kotopanov, C. E. Shields, Z. Zhou, J. W. Ward and A. I. Cooper, Autonomous mobile robots for exploratory synthetic chemistry, Nature, 2024, 635, 890–897 CrossRef.
- X. Li, Y. Che, L. Chen, T. Liu, K. Wang, L. Liu, H. Yang, E. O. Pyzer-Knapp and A. I. Cooper, Sequential closed-loop Bayesian optimization as a guide for organic molecular metallophotocatalyst formulation discovery, Nat. Chem., 2024, 16, 1286–1294 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, H. Hao, V. Rathore, J. Derasp, T. Gaudin, N. H. Angello, M. Seifrid, E. Trushina, M. Guy, J. Liu, X. Tang, M. Mamada, W. Wang, T. Tsagaantsooj, C. Lavigne, R. Pollice, T. C. Wu, K. Hotta, L. Bodo, S. Li, M. Haddadnia, A. Wolos, R. Roszak, C. T. Ser, C. Bozal-Ginesta, R. J. Hickman, J. Vestfrid, A. Aguilar-Granda, E. L. Klimareva, R. C. Sigerson, W. Hou, D. Gahler, S. Lach, A. Warzybok, O. Borodin, S. Rohrbach, B. Sanchez-Lengeling, C. Adachi, B. A. Grzybowski, L. Cronin, J. E. Hein, M. D. Burke and A. Aspuru-Guzik, Delocalized, asynchronous, closed-loop discovery of organic laser emitters, Science, 2024, 384, eadk9227 CrossRef CAS.
- T. C. Wu, A. Aguilar-Granda, K. Hotta, S. A. Yazdani, R. Pollice, J. Vestfrid, H. Hao, C. Lavigne, M. Seifrid, N. Angello, F. Bencheikh, J. E. Hein, M. Burke, C. Adachi and A. Aspuru-Guzik, A Materials Acceleration Platform for Organic Laser Discovery, Adv. Mater., 2023, 35, e2207070 CrossRef PubMed.
- N. H. Angello, D. M. Friday, C. Hwang, S. Yi, A. H. Cheng, T. C. Torres-Flores, E. R. Jira, W. Wang, A. Aspuru-Guzik, M. D. Burke, C. M. Schroeder, Y. Diao and N. E. Jackson, Closed-loop transfer enables artificial intelligence to yield chemical knowledge, Nature, 2024, 633, 351–358 CrossRef CAS PubMed.
| This journal is © the Partner Organisations 2025 |