
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWater mediated synthesis of dialkylphosphine oxides from white phosphorus and N-(acyloxy)phthalimides†
Guo Tang *ab,
Xuanlin Zhu‡
a,
Jiali He‡a,
Yan Liua and
Yufen Zhaoac
*ab,
Xuanlin Zhu‡
a,
Jiali He‡a,
Yan Liua and
Yufen Zhaoac
aDepartment of Chemistry, College of Chemistry and Chemical Engineering, and the Key Laboratory for Chemical Biology of Fujian Province, Xiamen University, Xiamen, Fujian 361005, China. E-mail: t12g21@xmu.edu.cn
bState Key Laboratory of Chemistry and Utilization of Carbon Based Energy Resources, College of Chemistry, Xinjiang University, Urumqi, 830017 Xinjiang, China
cInstitute of Drug Discovery Technology, Ningbo University, Ningbo, Zhejiang 315221, PR China
First published on 5th February 2025
Abstract
The multicomponent synthesis of dialkylphosphine oxides from P4 without a chlorination step is reported. With the use of N-(acyloxy)phthalimides (NHPI esters) as the alkylation reagents, H2O as the oxygen source, tris(2,2′-bipyridine)ruthenium dichloride as a photocatalyst, 2,6-lutidine as a base, and N,N-dimethylacetamide as a solvent, the reactions are performed under green light irradiation, yielding the desired products in moderate to good yields. Notably, this catalytic system is also capable of synthesizing α-hydroxy phosphine oxides from P4 in one-pot. This two step-economic approach, which directly utilizes P4 as the P-atom source, avoids the traditional chlorination stage and oxidation processes.
Introduction
Organophosphorus compounds (OPCs) containing C–P bonds are ubiquitous in both industrial and academic fields, such as materials science, pharmaceutical chemistry, catalysis, and coordination chemistry.1 In general, the preparation of OPCs predominately relies on white phosphorus (P4) as the source of P atoms.2 However, the conversion of P4 to OPCs involves (oxy)chlorination process and uses corrosive chlorine gas (Cl2) and intermediates such as PCl3/PCl5/POCl3. Another approach toward OPCs is the hydrophosphinylation of alkenes with toxic and explosive PH3.3 To circumvent the use of PCl3 and PH3, significant progress has been made in forming of heteroatom–P bonds, such as S–P bonds4 or O–P bonds5 from P4 (Scheme 1).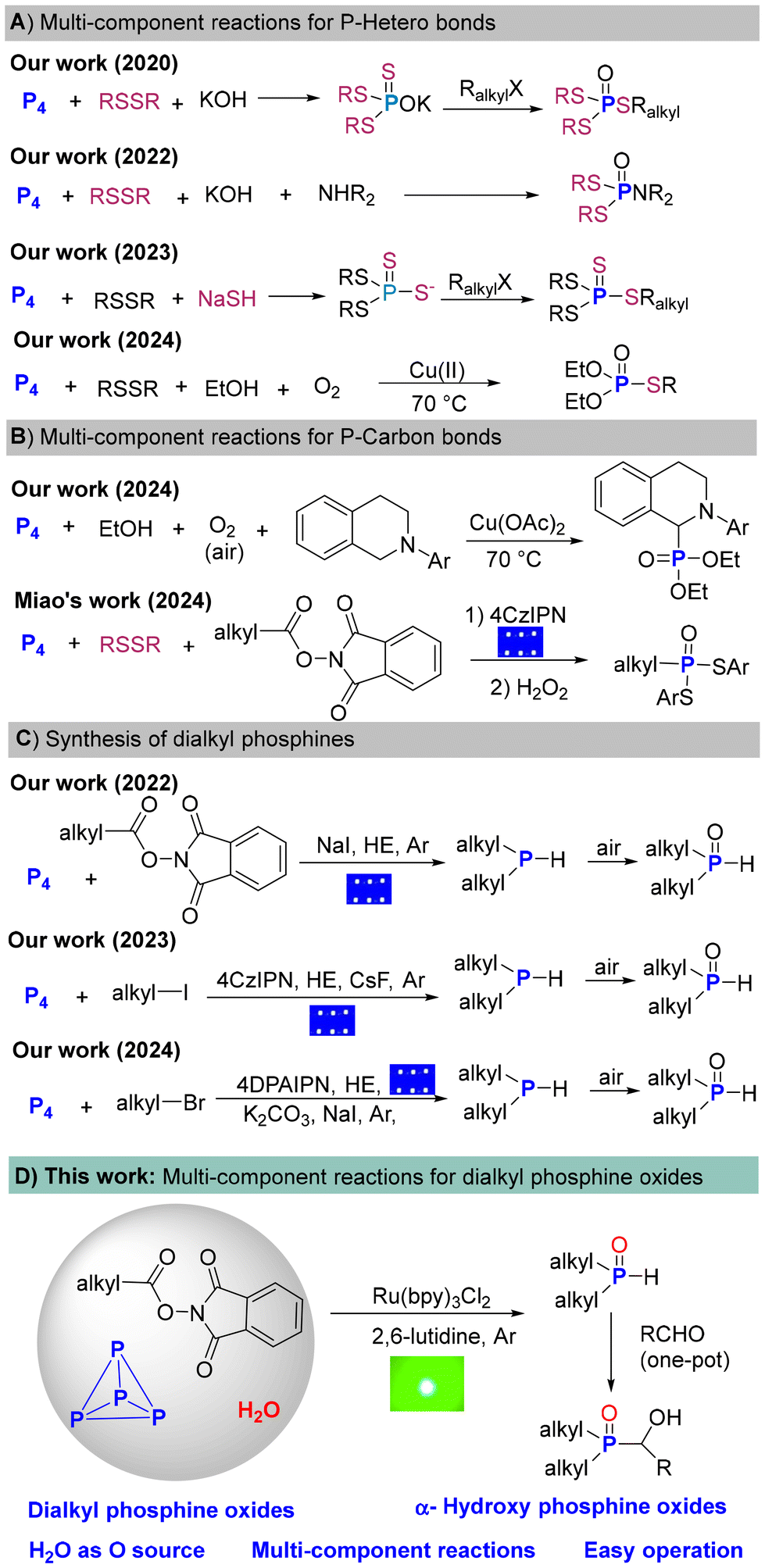 | ||
| Scheme 1 Multi-component reactions with P4. | ||
Dialkyl phosphine oxides [R2P(O)H] containing reactive P(O)–H bonds are versatile building blocks for synthesizing trialkyl phosphine oxides such as α-hydroxy phosphine oxides. The direct transformation of P4 into tertiary phosphines (PR3) and their oxides containing the same three alkyl/ary groups was investigated by the Cummins, Wolf, and Zhang groups and others.6 Due to the complex P–P bond breaking patterns, it is still challenging to construct R2P(O)H containing both C–P and P(O)–H bonds directly from P4 with good selectivity. In 2022, our group made a breakthrough and published the paper entitled “Decarboxylative Selective Phosphorylation of Aliphatic Acids: A Transition-Metal- and Photocatalyst-Free Avenue to Dialkyl and Trialkyl Phosphine Oxides from White Phosphorus”.7a Since then, we have reported the selective construction of R2P(O)H from P4 with alkyl bromides and iodides.7b,c All these experiments required excess alkylation reagents. Furthermore, strict anhydrous operation and subsequent oxidation workup were needed (Scheme 1B).
One-pot multicomponent reactions enable the efficient and atom-economical synthesis of OPCs directly from P4. Significant progress has been made in multicomponent reactions for the synthesis of P–heteroatom bonds (Scheme 1A).8 In 2020, the first general synthesis of mixed phosphorotrithioates involving P4, disulfides, KOH, and alkyl halides was presented.8a In 2022, we described a four-component functionalization of P4 with disulfides, amines and KOH to synthesize phosphoramidodithioates.8b In 2023, we published a one-pot direct synthesis of tetrathiophosphates from P4, NaSH, disulfides and alkyl halides with sodium alkyltetrathiophosphates as the key intermediates.8c In 2024, we presented the first copper-catalyzed three-component reaction for generating phosphorothioates from P4, disulfides, and alcohols in a single reaction step.8d
To construct C–P bonds, many novel phosphorus transfer reagents have been developed in recent years.9 From the perspective of step-economy, it is highly appealing to directly convert P4 into OPCs in one-pot process without the use of phosphorus transfer reagents. Compared to the research of P–heteroatom bonds formation, few multicomponent reactions for the construction of C–P bonds have been explored (Scheme 1C). In 2024, the first multicomponent oxidative α-phosphonylation of amines with P4 and alcohols was developed.10a In the same year, Miao et al. reported a visible light-induced three-component reaction integrating P4, disufides, and N-(acyloxy)phthalimides (NHPI esters) to produce alky-substituted phosphonodithioates.10b
As a continuum of our efforts in P4 chemistry, we envisioned that with the use of H2O as the oxygen source, and N-(acyloxy)phthalimides as the carbon source, R2P(O)H could be directly synthesized from P4 under an argon atmosphere without additional oxidation processes (Scheme 1D).
Results and discussion
To verify our conjecture, we commenced our project by employing H218O, and 4-phenylbutanic acid NHPI ester (2a) as model substrates to direct functionalization of P4 (1). When a solution of P4 (6.20 mg), H218O (5 μL), 2a (0.5 mmol, 2.5 equivalents per P-atom), 2,6-lutidine in N,N-dimethylacetamide (DMA) was irradiated by green LEDs at room temperature for 6 h in the presence of Ru(bpy)3Cl2 under argon atmosphere, 3a-18O was obtained as the main product (Scheme 2).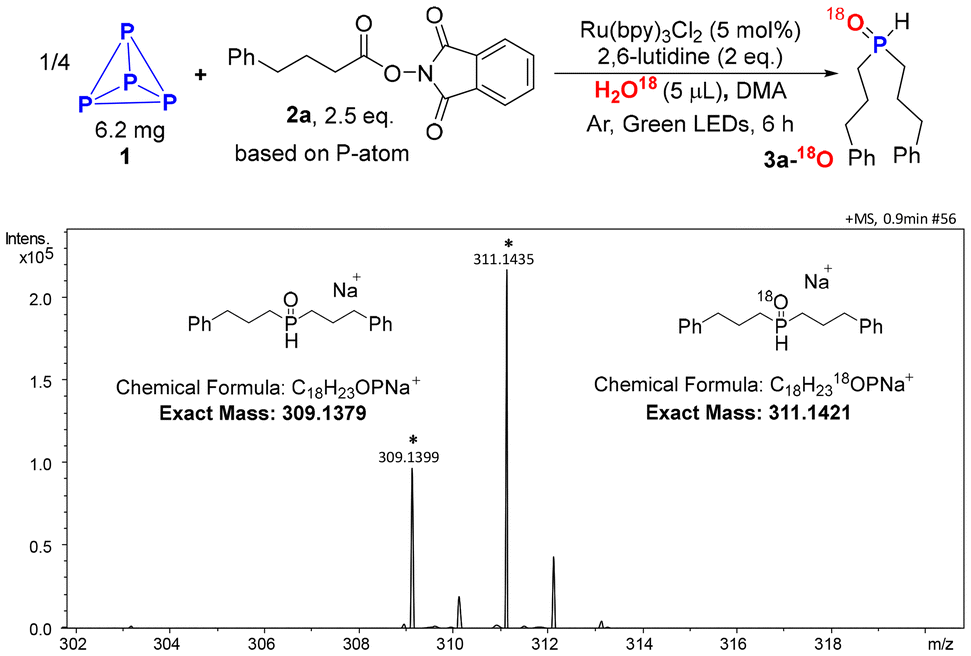 | ||
| Scheme 2 Water-18O as the O-resource. | ||
Inspired by this result, we began optimizing the conditions for the synthesis of R2P(O)H 3a using H2O as the oxygen source. After extensive screening of bases, solvents, and catalysts, the desired product 3a was obtained in 90% yield (entry 1 in Table 1). No R2P(O)H and R2PH were obtained in the absence of H2O (entry 2). Increasing the amount of H2O to 0.5 equivalents gave 3a but in only 13% yield (entry 3). Using H218O and H2O as the O-resouces verifed that water was the key starting material in this transformation. Other bases, such as DIPEA and DABCO, resulted in lower yield of 3a (entry 4). DMA and NMP were found to be good solvents for this reaction (entry 5), while other polar solvents, such as DMF, DMSO, and MeCN, led to much lower yields (entry 6). When transition-metal-free photocatalysts were tested, most performed poorly (entry 7), although Na2-eosin Y gave the product in 70% yield. No C–P bond formation was observed when the reaction was conducted under air conditions (entry 8). The reaction was inhibited in the absence of either photocatalyst or light (entry 9).
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield (%) |
| a Reaction conditions: P4 (6.20 mg, 0.20 mmol of P atom, a 0.74 M solution of P4 in toluene, 0.27 mL), 2a (2.5 equivalents per P-atom), Ru(bpy)3Cl2 (0.01 mmol, 5 mol%), H2O (1.1 eq., 4 μL), 2,6-lutidine (0.40 mmol, 2 eq.) in DMA (1 mL) irradiated by green LEDs (2 × 3 W, 565–575 nm) at room temperature for 6 h under argon atmosphere. Yield of 3a was determined by the 31P{1H} NMR analysis of the crude reaction mixture using (C6H5O)3P(O) as an internal standard.b Isolated yield in parenthesis. PC = photocatalyst. | ||
| 1b | Standard conditions | 94 (90) |
| 2 | Without H2O | Trace |
| 3 | H2O (0.5 eq.) | 13 |
| 4 | DIPEA, DABCO instead of 2,6-lutidine | 60–70 |
| 5 | NMP instead of DMA | 89 |
| 6 | DMF, DMSO, MeCN instead of DMA | 32–49 |
| 7 | Other organo-PC | 0–70 |
| 8 | Air instead of Ar | 0 |
| 9 | No light or PC | 0 |
After optimizing the reaction conditions for the synthesis of 3a, we explored the scope of carboxylic acids for the synthesis of structurally diverse R2P(O)H 3 (Scheme 3). Under photoirradiation conditions, various alkyl acid NHPI esters can participate in this process, providing the desired products in good yields. 2-Cyclopentylacetic acid and 3-cyclohexylpropanoic acid NHPI esters gave products 3b and 3c in 73% and 70% yields, respectively. Linear acid NHPI esters with heterocyclic rings such as O-heterocycle (3d), S-heterocycle (3e) underwent phosphonylation without difficulty. Aryl-substituted linear acid NHPI esters with electron donating groups (methyl, methoxy, naphthalene) or electron withdrawing group (bromo) on the benzene ring gave 3f–3i in 80–88% yields. Simple primary saturated fatty acid ester gave 3j in 85% yield. Furthermore, a range of synthetically useful functional groups, such as C(sp3)–Cl, C(sp3)–Br, and ester (3m and 3n), were also compatible with the mild conditions, providing respective dialkyl phosphine oxides in 71–85% yields. Due to steric hindrance, cyclohexanecarboxylic acid ester produced dicyclohexylphosphine oxide in a low yield (3o, 40%). When linear secondary or tertiary carbon radical precursors were employed, no C–P bonds were formed. In these failed experiments, white phosphorus remained completely (δ = −529 ppm). It is speculated that the sharp decrease in the yield of secondary and tertiary carbon free radical reactions may be due to the stability and steric hindrance effect of their free radicals.
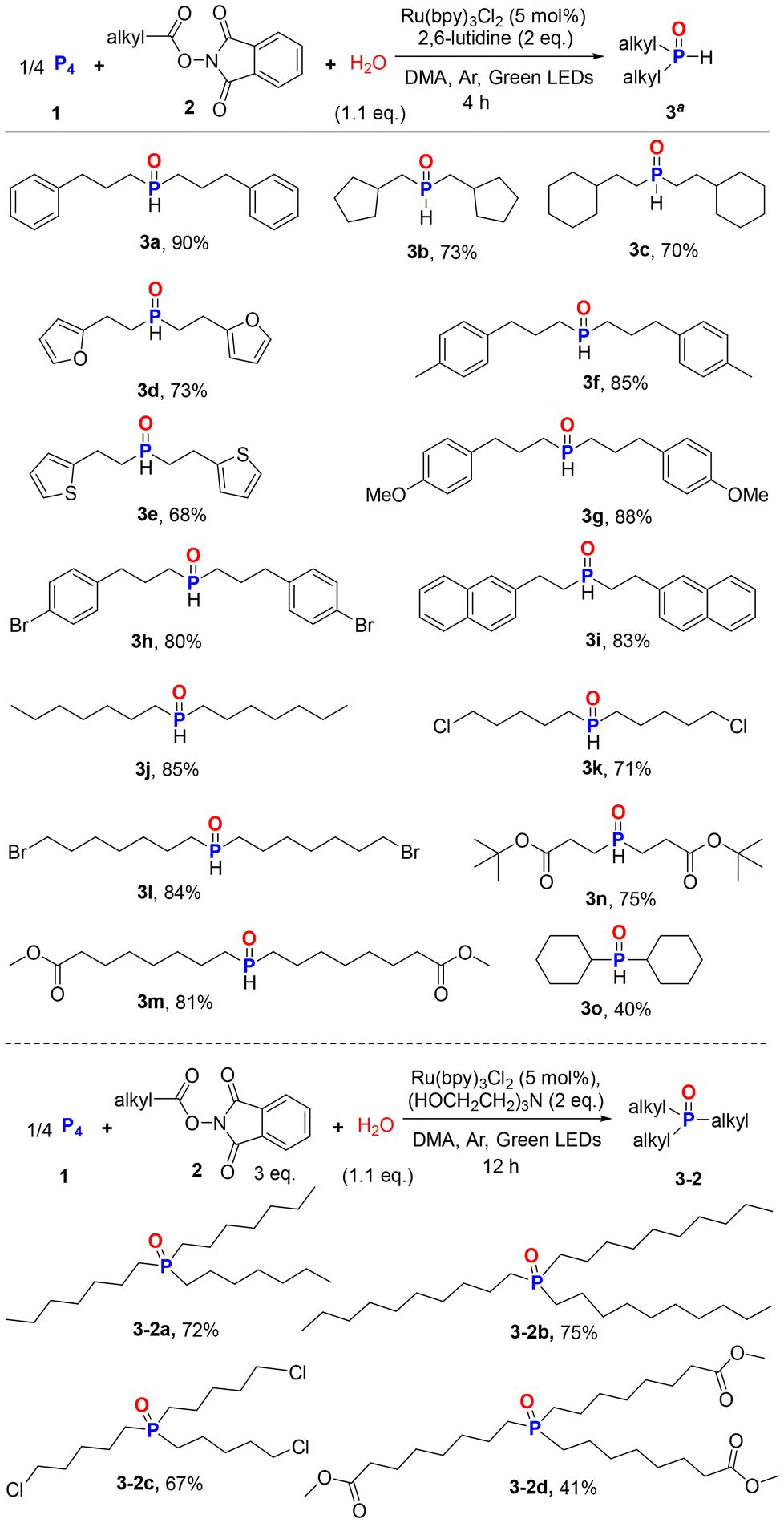 | ||
| Scheme 3 Scope of alkyl acid N-hydroxyphthalimide esters for the synthesis of dialkyl/trialkyl phosphine oxides. | ||
We then speculated about whether this water mediated multicomponent strategy could be used to produce valuable trialkyl phosphine oxides (Scheme 3). With Ru(bpy)3Cl2 as the best catalyst and trolamine as the best base, the reaction was performed for 12 h to afford the desired product 3-2a in 72% yield. A series of chain primary fatty acid ester selected as substrates provided the corresponding trialkyl phosphine oxides 3-2b–3-2d in satisfactory yields.
Dialkyl phosphine oxides are often used as starting materials for the preparation of various organophosphorus compounds. Encouraged by the findings described above, we continued to explore the one-pot synthesis of α-hydroxy phosphine oxides from P4, avoiding the purification process of R2P(O)H and the use of dangerous Cl2 and PCl3. Pleasingly, we added aldehydes and Na2CO3 to the crude product 3a solution to produce α-hydroxy phosphine oxides in good yields (Scheme 4). Benzaldehyde with electron withdrawing group (–Cl, –CN, and –NO2) and electron donating groups (–OCOCH3, –OCH3, and –SCH3) provided the corresponding products in yields of 51–76% (4a–4f). When aromatic heterocyclic formaldehydes such as furan-2-carbaldehyde, thiophene-2-carbaldehyde and pyridine-2-carbaldehyde were employed as substrates, products 4g–4i were obtained in the range of 52–64%. Cinnamaldehyde, with a double bond, afforded the product 4j in 52% yield. This method was found to be compatible with primary saturated fatty acid esters. Decarboxylative phosphorylation of propionic acid NHPI ester followed by nucleophilic addition reaction of aldehyde gave product 4k in 41% yield. Long-chain n-octoic acid NHPI ester successfully participated in this one-pot synthesis of α-hydroxy phosphine oxide 4l.
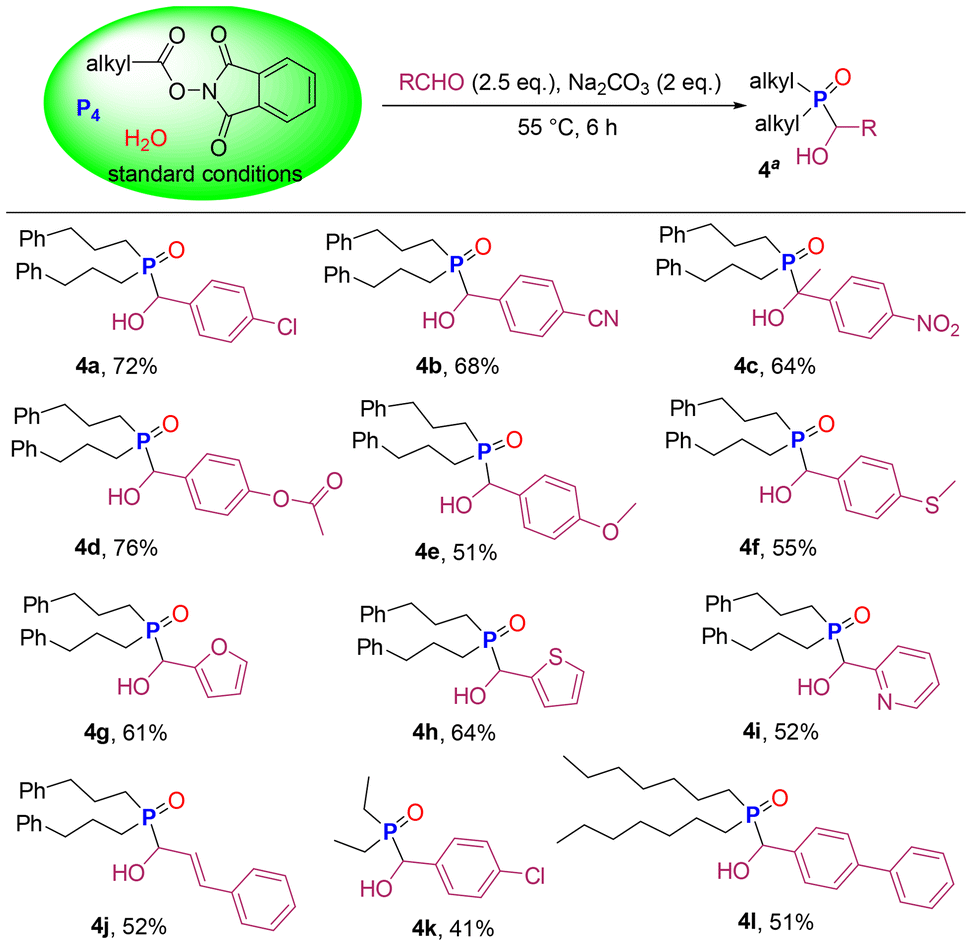 | ||
| Scheme 4 One-pot synthesis of α-hydroxy phosphine oxides from P4. | ||
Furthermore, in situ NMR studies on the reaction of NHPI ester 2a and P4 were conducted and the corresponding 31P{1H} NMR spectra were shown in Fig. 1–3 (see ESI†). When the reaction mixture was irradiated by white light-emitting diodes (LEDs) for 30 min, the signal of the product 3a and byproduct (RP(O)(OH)H) appeared. No RPH2 or R2PH was detected. Based on these results and previous studies,7 a tentative mechanism for the photoinduced functionalization of P4 was proposed (Scheme 5). Initially, water breaks the P–P bond of P4 in a manner like super-basic systems (OH− in DMSO) to yield intermediate A.11 Subsequently, the alkyl radical breaks the P–P bond of intermediate A to give an unstable intermediate B, which then reacts with excited PC* and base to form the phosphinoyl radical D. Radical D can directly combine with another R˙ yields R2P(O)H as the main product of the reaction.
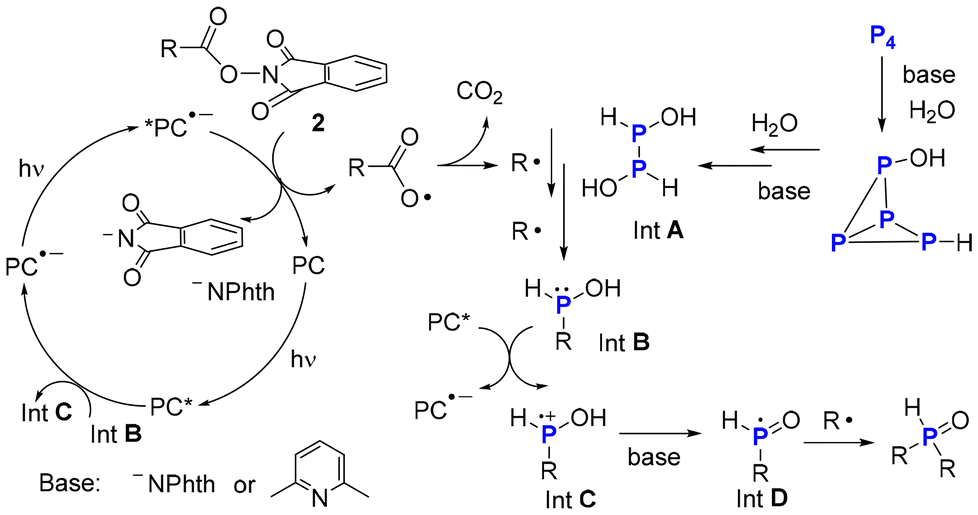 | ||
| Scheme 5 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we developed the first water mediated multicomponent synthesis of dialkylphosphine oxides and α-hydroxy phosphine oxides from P4 without a chlorination step. With the use of N-(acyloxy)phthalimides as the alkylation reagents, H2O as the oxygen source, the reactions are performed under green light irradiation. The desired dialkylphosphine oxides [R2P(O)H] can be directly synthesized from P4 under argon atmosphere without additional oxidation processes in moderate to good yields. Advantageously, the catalytic system can also synthesize α-hydroxy phosphine oxides from P4. These step-economic approaches use P4 as the P-atom source, avoiding the traditional chlorination stage and oxidation processes.Experimental
Synthesis of dialkyl phosphine oxides from white phosphorus
A Schlenk tube containing Ru(bpy)3Cl2 (6.4 mg, 0.01 mmol, 5 mol%) and N-(acyloxy)phthalimides (0.50 mmol, 2.5 eq.) was evacuated and purged with argon three times. P4 (6.20 mg, 0.20 mmol of P atom, a 0.74 M solution of P4 in toluene, 0.27 mL), H2O (1.1 eq., 4 μL), 2,6-lutidine (0.40 mmol, 2 eq.) and DMA (1 mL) were sequentially added to the system at room temperature. Then the reaction mixture was stirred at room temperature under the irradiation of 6 W green LEDs (565–575 nm) for 6 h. Afterwards, the reaction mixture was quenched with the addition of saturated brine (0.5 mL), and extracted with ethyl acetate (3 × 10 mL). The combined organic layer was dried over anhydrous Na2SO4 and then removed in a vacuum. The residue was purified by flash chromatography using DCM/MeOH (from 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 20:1, v/v) as the eluent to afford the desired dialkyl phosphine oxides 3a–3o.
:1 to 20:1, v/v) as the eluent to afford the desired dialkyl phosphine oxides 3a–3o.
One-pot synthesis of α-hydroxy phosphine oxides from white phosphorus
After the above-mentioned reaction mixture was stirred at room temperature under the irradiation of 6 W green LEDs (565–575 nm) for 6 h without any work-up, aldehyde (0.50 mmol, 2.5 eq.) and Na2CO3 (42.40 mg, 0.4 mmol, 2.0 eq.) were added in air. The reaction mixture was stirred for 22 hours at 55 °C for 6 hours. After completion, saturated brine (0.5 mL) was added; the mixture was diluted by EtOAc (10.0 mL). The mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic layer was dried over anhydrous Na2SO4 and then removed in a vacuum. The residue was purified by flash chromatography using DCM/MeOH (from 40:1 to 10:1, v/v) as the eluent to afford the desired α-hydroxy phosphine oxides 4a–4l.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the National Key Research and Development Program of China (grant no. 2020YFA0608300), the Space Application System of China Manned Space Program (grant no. KJZ-YY-WSM01), and National Natural Science Foundation of China (grant no. 21772163, 21778042, and 41876072).References
- H. Guo, Y. C. Fan, Z. Sun, Y. Wu and O. Kwon, Phosphine organocatalysis, Chem. Rev., 2018, 118, 10049–10293 CrossRef CAS PubMed.
- (a) D. J. Scott, Recent breakthroughs in P4 chemistry: towards practical, direct transformations into P1 compounds, Angew. Chem., 2022, 134, e202205019 CrossRef; (b) X. Huangfu, Z. Wang, Y. Chen, J. Wei, W. Liu and W. Zhang, Recent progress on thefunctionalization of white phosphorus in China, Natl. Sci. Rev., 2024, 11, nwae162 CrossRef PubMed.
- R. Rothfelder, V. Streitferdt, U. Lennert, J. Cammarata, D. J. Scott, K. Zeitler, R. M. Gschwind and R. Wolf, Photocatalytic arylation of P4 and PH3: reaction development through mechanistic insight, Angew. Chem., Int. Ed., 2021, 60, 24650–24658 CrossRef CAS PubMed.
- (a) G. Lu, J. Chen, X. Huangfu, X. Li, M. Fang, G. Tang and Y. Zhao, Visible-light-mediated direct synthesis of phosphorotrithioates as potent anti-inflammatory agents from white phosphorus, Org. Chem. Front., 2019, 6, 190–194 RSC; (b) X. Huangfu, Y. Wang, G. Lu, Y. Cao, G. Tang and Y. Zhao, Direct synthesis of phosphorotrithioites and phosphorotrithioates from white phosphorus and thiols, Green Chem., 2020, 22, 5303–5306 RSC.
- (a) Z. Cai, X. Zeng, Y. Zhang, Y. Liu, G. Tang and Y. Zhao, Direct synthesis of dialkylphosphites from white phosphorus, Adv. Synth. Catal., 2022, 364, 2916–2921 CrossRef CAS; (b) Z. Cai, Y. Zhang, Y. Cao, Y. Liu, G. Tang and Y. Zhao, Ternary photoredox/nickel/halide catalysis for the phosphorylation of alcohols with white phosphorus, ACS Catal., 2023, 13, 8330–8335 CrossRef CAS; (c) Y. Zhang, Z. Cai, Y. Chi, X. Zeng, S. Chen, Y. Liu, G. Tang and Y. Zhao, Diphenyl diselenide-catalyzed synthesis of triaryl phosphites and triaryl phosphates from white phosphorus, Org. Lett., 2021, 23, 5158–5163 CrossRef CAS PubMed.
- (a) B. M. Cossairt and C. C. Cummins, Radical synthesis of trialkyl, triaryl, trisiyl and tristannyl phosphines from P4, New J. Chem., 2010, 34, 1533–1536 RSC; (b) U. Lennert, P. B. Arockiam, V. Streitferdt, D. J. Scott, C. Rödl, R. M. Gschwind and R. Wolf, Direct catalytic transformation of white phosphorus into arylphosphines and phosphonium salts, Nat. Catal., 2019, 2, 1101–1106 CrossRef CAS PubMed; (c) M. Till, V. Streitferdt, D. J. Scott, M. Mende, R. M. Gschwind and R. Wolf, Direct catalytic transformation of white phosphorus into arylphosphines and phosphonium salts, Chem. Commun., 2022, 58, 1100–1103 RSC; (d) X. Huangfu, W. Liu, H. Xu, Z. Wang, J. Wei and W. Zhang, Photochemical benzylation of white phosphorus, Inorg. Chem., 2023, 62, 12009–12017 CrossRef CAS PubMed; (e) Y. Chen, W. Liu, X. Huangfu, J. Wei, J. Yu and W. Zhang, Direct synthesis of phosphoryltriacetates from white phosphorus via visible light catalysis, Chem. – Eur. J., 2023, 29, e202302289 Search PubMed.
- (a) F. Chen, M. Bai, Y. Zhang, W. Liu, X. Huangfu, Y. Liu, G. Tang and Y. Zhao, Decarboxylative selective phosphorylation of aliphatic acids: a transition-metal- and photocatalyst-free avenue to dialkyl and trialkyl phosphine oxides from white phosphorus, Angew. Chem., Int. Ed., 2022, 61, e202210334 CrossRef CAS PubMed; (b) F. Chen, J. Peng, Y. Ying, Y. Cao, P. Xu, G. Tang and Y. Zhao, Metal-free visible-light-induced phosphorylation of unactivated alkyl iodides with white phosphorus as the P-atom source, Green Chem., 2023, 25, 6629–6634 RSC; (c) J. Peng, A. Wang, Y. Liu, F. Chen, G. Tang and Y. Zhao, Selective functionalization of white phosphorus with alkyl bromides under photocatalytic conditions: a chlorine-free protocol to dialkyl and trialkyl phosphine oxides, Org. Lett., 2024, 26, 9316–9321 CrossRef CAS PubMed.
- (a) X. Huangfu, Y. Zhang, P. Chen, G. Lu, Y. Cao, G. Tang and Y. Zhao, Synthesis of mixed phosphorotrithioates from white phosphorus, Green Chem., 2020, 22, 8353–8359 RSC; (b) Y. Zhang, Y. Cao, Y. Chi, S. Chen, X. Zeng, Y. Liu, G. Tang and Y. Zhao, Formation of N–P(O)–S bonds from white phosphorus via a four-component reaction, Adv. Synth. Catal., 2022, 364, 2221–2226 CrossRef CAS; (c) J. He, S. Shi, Y. Zhang, Y. Zhang, P. Xu, G. Tang and Y. Zhao, Synthesis of tetrathiophosphates from white phosphorus, Chin. J. Chem., 2023, 41, 2311–2316 CrossRef CAS; (d) Y. Cao, M. Bai, J. Huang, F. Chen, Y. Liu, G. Tang and Y. Zhao, Three-component coupling reaction of white phosphorus, alcohols and diaryl disulfides: A chlorine-free avenue for accessing phosphorothioates, Green Chem., 2024, 26, 477–482 RSC.
- (a) D. J. Scott, J. Cammarata, M. Schimpf and R. Wolf, Synthesis of monophosphines directly from white phosphorus, Nat. Chem., 2021, 13, 458–464 CrossRef CAS PubMed; (b) M. Till, J. Cammarata, R. Wolf and D. J. Scott, Photocatalytic stannylation of white phosphorus, Chem. Commun., 2022, 58, 8986–8989 RSC; (c) M. Donath, K. Schwedtmann, T. Schneider, F. Hennersdorf, A. Bauza, A. Frontera and J. J. Weigand, Direct conversion of white phosphorus to versatile phosphorus transfer reagents via oxidative onioation, Nat. Chem., 2022, 14, 384–391 CrossRef CAS PubMed; (d) Y. Mei, Z. Yan and L. L. Liu, Facile synthesis of the dicyanophosphide anion via electrochemical activation of white phosphorus: An avenue to organophosphorus compounds, J. Am. Chem. Soc., 2022, 144, 1517–1522 CrossRef CAS PubMed.
- (a) M. Bai, Y. Cao, J. Huang, Y. Liu, G. Tang and Y. Zhao, Direct synthesis of α-aminophosphonates from amines, alcohols and white phosphorus, CCS Chem., 2024, 6, 91–99 CrossRef CAS; (b) F. Wang, X. Zhang, J. Xu, Q. Shen, B. Jiang and Z. Miao, Metal-free visible-light-induced direct synthesis of alkyl-substituted phosphonodithioates from white phosphorus (P4), Adv. Synth. Catal., 2024 DOI:10.1002/adsc.202401355.
- B. A. Trofimov and N. K. Gusarova, Elemental phosphorus in strongly basic media as phosphorylating reagent: a dawn of halogen-free ‘green’ organophosphorus chemistry, Mendeleev Commmun., 2009, 19, 295–305 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures for the synthesis, spectral data and NMR spectra of compounds 3a–3o and 4a–4l. See DOI: https://doi.org/10.1039/d4qo02447h |
| ‡ These two authors contribute equally. |
| This journal is © the Partner Organisations 2025 |