
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymer-bridged nanofibrils in a high-molar-mass polyester via co-assembly of benzenetricarboxamide end groups and an additive†
Sophia Thiele ,
Michael Giffin,
Matthieu Wendling,
Daniel Görl,
Christopher J. G. Plummer and
Holger Frauenrath*
,
Michael Giffin,
Matthieu Wendling,
Daniel Görl,
Christopher J. G. Plummer and
Holger Frauenrath*
École Polytechnique Fédérale de Lausanne (EPFL), Institute of Materials, 1015 Lausanne, Switzerland. E-mail: holger.frauenrath@epfl.ch
First published on 13th May 2025
Abstract
Benzenetricarboxamide (BTA) derivatives are versatile compounds widely employed as nucleating agents in commercial semicrystalline plastics and as supramolecular ligands in self-assembling telechelic polymer-based organogels, hydrogels, and bulk elastomers. However, their effectiveness as supramolecular modifiers is typically limited to low-molar-mass apolar polymers. Here, we report the supramolecular aggregation of a BTA-end-functionalized semicrystalline aliphatic polyester with a number-average molar mass several times its entanglement molar mass, blended with a matching low-molar-mass BTA additive. In these blends, the BTA end groups and additive co-assemble to form a new phase comprising a network of polymer-bridged nanofibrils. This network gives rise to a high-melt-strength rubbery regime that is absent from the pure telechelic polyester but extends to temperatures well above its melting point in the blends. Moreover, the nanofibrils prove to be highly efficient nucleating agents for crystallization of the polyester, significantly outperforming bulk additive precipitates. Our findings hence demonstrate that the co-assembly of polymer end groups with a low-molar-mass additive may facilitate supramolecular aggregate formation in polymer matrices where end-modification alone is insufficient, leading to materials with increased melt strength, crystallization rates, thermal dimensional stability, and valuable benefits for industrial applications.
Introduction
Supramolecular functional motifs that self-assemble in a controlled and reversible manner into complex hierarchical structures1–3 are used in a wide range of applications, including soft robotics,4,5 targeted drug delivery systems,6 soft pliable adhesives,7 and tissue-mimicking materials.8 An important class of materials based on such motifs are supramolecular polymers, which have evolved from small molecules that aggregate into materials with polymer-like properties,9–13 to telechelic polymers end-modified with supramolecular functional motifs,14,15 improving toughness, enabling self-healing, and facilitating recycling.10,16–21Many of the supramolecular polymers and networks based on telechelic polymers reported are derived from multivalent hydrogen-bonded motifs that we refer to here as “inherently monotopic”, that is, ligands designed to form dimers with one complementary partner. Examples of such ligands include the self-complementary 2-ureido-4-pyrimidone (UPy) motif,9 and hetero-complementary donor/acceptor systems such as uracil/2,6-diacyl-amino-pyridine,22,23 other nucleobases,24 UPy/2,7-diamido-1,8-naphthyridine,25,26 Hamilton receptor/barbituric acid,27–30 and isophthalic acid/pyridine.31 In some cases, these dimers may further undergo microphase separation to form larger clusters with irregularly shaped domains analogous to those observed in thermoplastic elastomers containing hard and soft segments.32
By contrast, “inherently ditopic” hydrogen-bonded motifs bind simultaneously to two other ligands to form one-dimensionally extended aggregates with well-defined lateral dimensions on the nanometer scale.33,34 The vast majority of such ditopic motifs rely on multivalent hydrogen bonding between amide,35–39 urethane,7,40 or urea41–43 functionalities that show cooperative self-assembly due to electronic coupling in the extended N–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bond strands within the aggregates.2,12,44 In polymers end-modified with UPy ligands with additional lateral ditopic urea45 or urethane46 motifs, for instance, it is the combination of the ditopic hydrogen bonds of the urea or urethane motifs and the π-interactions between UPy units that promote the formation of one-dimensionally extended aggregates with well-defined lateral dimensions.47 Telechelic polymers modified in this way are reported to show significantly increased tensile strength.45,46 These examples highlight the importance of synergistic and/or competing interactions for the design of materials with precisely controlled, hierarchical morphologies.48
C hydrogen bond strands within the aggregates.2,12,44 In polymers end-modified with UPy ligands with additional lateral ditopic urea45 or urethane46 motifs, for instance, it is the combination of the ditopic hydrogen bonds of the urea or urethane motifs and the π-interactions between UPy units that promote the formation of one-dimensionally extended aggregates with well-defined lateral dimensions.47 Telechelic polymers modified in this way are reported to show significantly increased tensile strength.45,46 These examples highlight the importance of synergistic and/or competing interactions for the design of materials with precisely controlled, hierarchical morphologies.48
Well-established supramolecular motifs for the reliable formation of nanofibrils include β-sheet-forming oligopeptides,35,49,50 oligourethanes,51 oligoureas,52 and their hybrids,40,53–55 as well as benzene-1,3,5-tricarboxamide (BTA) derivatives.37,38 The last of these have been shown to form nanofibrils by three-fold hydrogen bonding of the amide functions combined with π-interactions between the benzene cores, resulting in extended 1D stacks. The geometric discrepancy between the length of the constituting unit involved in C–N–H⋯OC hydrogen bonding and the π-stacking distance causes a rotational offset of BTA units along the stack, leading to a helical arrangement of the peripheral amide functions.38,56 This disrupts lateral aggregation and crystallization into hexagonal or pseudo-hexagonal columnar phases in dilute solutions of BTA derivatives57 and BTA-functionalized polymers37,58 in favor of nanoscopic BTA aggregates. For instance, apolar low-molar-mass polymers, such as poly(ethylene-ran-butylene), which otherwise behave as viscous fluids, become soft elastomers when end-modified with BTA motifs.37 By contrast, BTA end-group aggregation is not observed in polymers with higher polarity, such as aliphatic polyesters.59
The self-assembly of end groups also generally becomes less favorable with increasing polymer molar mass.42 Previous investigations of supramolecular polymers and networks have therefore largely excluded polymers with molar masses high enough for the formation of a robust entanglement network. This is a critical limitation given that entanglement is generally key to high extensibility and toughness in the solid state,60,61 and the melt properties required for industrially relevant processing techniques, such as thermoforming, film blowing, and extrusion.60,62–64 The few notable exceptions make use of specific polymer architectures to increase the local concentration of aggregating units. For instance, materials with increased toughness are obtained from high molar mass polyisoprene (PI) block copolymers in which the central PI block is well above its entanglement molar mass and the terminal blocks are modified with oligoalanine side groups so that their high local concentration renders aggregation favorable.65,66 By contrast, when the aggregating motifs are incorporated into the corresponding random copolymers, the reduced segment length between physical crosslinks results in materials with reduced tensile strength and extensibility.65
Unlike supramolecular networks based on telechelic polymers end-modified with inherently monotopic ligands, which tend to break down upon the addition of a low-molar-mass ligand,9 the high-aspect-ratio, one-dimensionally extended aggregates formed by intrinsically ditopic end groups may be significantly reinforced by co-assembly with a corresponding low-molar-mass ligand. For instance, co-assembly of a low-molar-mass BTA derivative with BTA-modified polyethylene glycol (PEG) telechelics, which form a micellar fluid in the absence of the additive, has been reported to result in stable viscoelastic hydrogels containing fibrillar domains.67,68
We recently demonstrated that a similar approach was effective for high-molar-mass polymers where the end group concentration might otherwise be insufficient for aggregation to occur. To this end, we used the co-assembly of polymer end groups and a low-molar-mass additive, both based on a β-sheet-forming oligopeptide motif, to create a robust supramolecular network in poly(ε-caprolactone) (PCL), with a molar mass several times the entanglement threshold.50 The resulting high melt elasticity, melt extensibility, and melt strain hardening enable the preparation of highly oriented films by melt drawing, with a correspondingly high room-temperature yield stress.50 However, to the best of our knowledge, the analogous use of a low-molar-mass BTA additive to modify the viscoelastic properties of well-entangled BTA-end-functionalized polymers in the bulk melt state has not yet been reported.
Low-molar-mass BTA derivatives are used as ferroelectric switches,69 emitter materials in organic diodes,70 organogelators,71,72 and hydrogel-promoters with low cytotoxicity for tissue engineering.58,68,73,74 They are of particular interest in the plastics industry as nucleating agents for the crystallization of semicrystalline polymers such as isotactic polypropylene (iPP),75–77 polyvinylidene fluoride (PVDF),78 polybutylene terephthalate (PBT),79 polylactic acid (PLA),80–82 and the semiconductor poly(3-hexylthiophene) (P3HT).83 BTA nucleating agents are hence important processing additives in many packaging materials, and some are considered safe for use in contact with food by the US Food and Drug Administration (FDA) and the European Food Safety Authority (EFSA).84 They typically show high-aspect-ratio morphologies, for instance, needle-like crystals, and their nucleation efficiency increases with their specific active surface area.85 Systematic studies of the concentration dependence of BTA structure formation and its influence on polymer crystallization have been used to construct phase diagrams for binary polymer/BTA nucleating agent blends.86 However, blends of BTA derivatives with telechelic polymers modified with end groups that potentially co-assemble to form nanoscopic structures with significantly higher surface-to-volume ratios than conventional BTA nucleating agents, have not yet been investigated for their nucleation efficiency.
In the present work, we explore the behavior of PCL-BTA, a high-molar-mass PCL end-modified with a BTA-based motif, in the presence of the matching low-molar-mass BTA derivative B (Fig. 1). We establish a phase diagram for this binary system and show that, below a certain threshold B concentration, co-assembly of the PCL-BTA end groups with B results in the precipitation of nanophase-separated fibrillar BTA aggregates upon cooling from the melt. In this low-concentration regime, the nanoscopic fibrillar BTA aggregates act as more efficient nucleating agents for PCL crystallization than the bulk BTA precipitates observed in reference blends of B with unmodified PCL. Moreover, the physical network formed by the nanofibrils and PCL manifests itself as a rubbery plateau in dynamic shear rheometry temperature sweeps that extends to well above the PCL crystallization temperature. This leads to improved thermal dimensional stability and increased melt elasticity, which facilitates thermoforming. We hence overcome previous limitations for the assembly of BTA-based end groups in high-molar-mass, polar polymers.
 | ||
| Fig. 1 (a) Chemical structure of end-modified PCL-BTA, and matching additive B. Upon cooling, the end groups of PCL-BTA/B co-assemble with B by three-fold hydrogen bonding into (b) polymer-tethered nanofibrils comprising one-dimensional stacks of BTA end groups and additive molecules. (c) In the bulk materials, the nanofibrils are bridged by the polymer segments and (d) coexist with crystalline PCL lamella upon cooling to T < Tm. For clarity, the alkyl side chains of B have been omitted in (b–d). | ||
Results and discussion
We have carried out a systematic investigation of blends of the low-molar-mass additive N,N′,N′′-tris(isopentyl)-1,3,5-benzenetricarboxamide (B) with PCL-BTA, a telechelic PCL end-modified with N′,N′′-bis(isopentyl)-1,3,5-benzenetricarboxamide ligands, the overall number-average molar mass of which,![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n = 110
n = 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, is about 37 times the entanglement molar mass of PCL (Me ≈ 3000 g mol−1).87 We focus on the phase behavior, melt properties, thermal dimensional stability, and nucleation efficiency of these materials, using blends of B with unmodified PCL of the same molar mass as reference materials.
000 g mol−1, is about 37 times the entanglement molar mass of PCL (Me ≈ 3000 g mol−1).87 We focus on the phase behavior, melt properties, thermal dimensional stability, and nucleation efficiency of these materials, using blends of B with unmodified PCL of the same molar mass as reference materials.
PCL is an aliphatic, semi-crystalline polyester with a degree of crystallinity of about 40%, a glass transition temperature, Tg, of −62 °C (measured by DSC at a cooling rate of 10 °C min−1), and a nominal melting temperature, Tm, of about 58 °C, above which it transforms into a low-viscosity liquid for the range of molar masses considered here (ESI Fig. S1†). The low melt strength of PCL is incompatible with many important industrial processes, and its inadequate dimensional stability at temperatures above Tm has largely restricted its use to biomedical applications.88
We begin by discussing the behavior of the PCL/B reference blends in order to establish a benchmark for the interpretation of the results for the modified blends. The additive B used in our study forms extended 1D stacks in its bulk state at room temperature, in common with a wide range of other BTA derivatives. These stacks adopt a pseudo-hexagonal parallel close-packed crystalline structure with a monoclinic unit cell, the lattice parameters of which vary somewhat depending on the crystallization conditions, characterized by strong Bragg peaks with d-spacings, dB, of around 1.3 nm and intercolumnar spacings in the range of 1.25–1.55 nm. Upon heating, this structure first undergoes an endothermal transition to a columnar mesophase at Tmeso = 207 °C, followed by a transition to the isotropic melt at its dissociation temperature, Td0 = 269 °C (ESI Fig. S2†), consistent with previous observations.89
In the range of 0.5–10 wt%, the PCL/B reference blends show an intense exothermic transition in DSC cooling scans at 10 °C min−1, corresponding to crystallization of PCL at Tc = 35 °C, but also an additional weaker exothermic transition at higher temperatures, Tagg, corresponding to the aggregation of B (Fig. 2a and ESI Fig. S3†). Subsequent heating scans reveal the PCL melting transition at Tm = 58 °C and an endothermic transition at higher temperatures, Td, attributed to dissociation of the B precipitates. Dissociation nevertheless occurs at significantly lower temperatures than that for the pure additive B (Td0 = 269 °C) and blends of B with isotactic polypropylene for a given B concentration,86 owing to the comparatively high solubility of B in the polar PCL matrix. Tagg and Td both increase systematically with B concentration, but Td is 6–26 °C higher than Tagg, with the highest supercoolings being observed at the lowest B concentrations. Aggregation of B is therefore inferred to be kinetically controlled under these conditions, so that Td is assumed to be more representative of equilibrium conditions than Tagg.
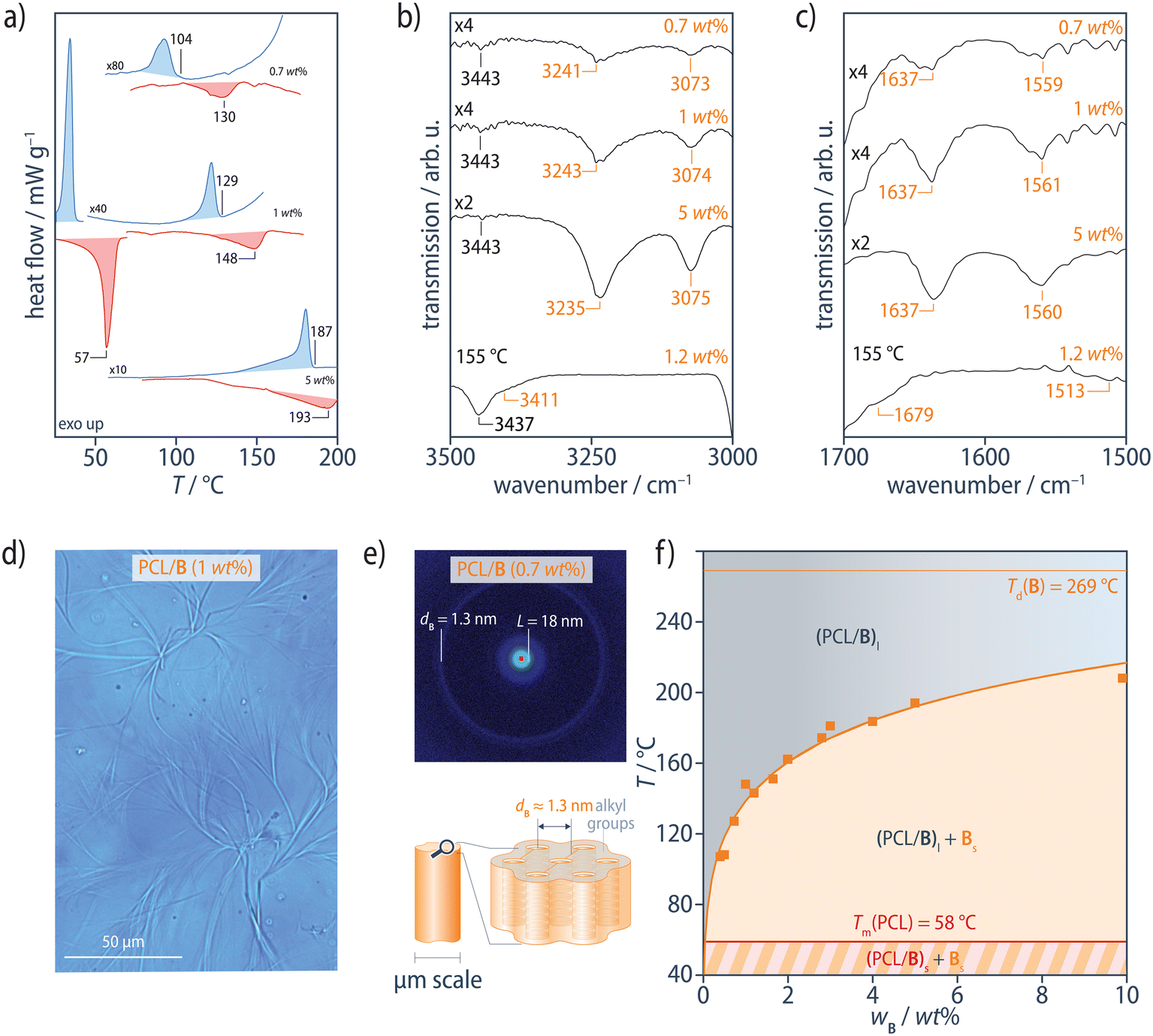 | ||
| Fig. 2 (a) DSC heating (red) and cooling (blue) scans of PCL/B at 10 °C min−1 for different B concentrations. For clarity, the PCL melting and crystallization transitions at Tm = 58 °C and Tc = 35 °C, respectively, are only shown for 1 wt% B. (b) ATR FTIR spectra at room temperature show the N–H stretching band at 3235–3243 cm−1, (c) the CO stretching band at 1637 cm−1, and the combined N–H bending/C–N stretching band at 1559–1561 cm−1, characteristic of strongly hydrogen-bonded amides in extended aggregates of B, all of which shift to positions corresponding to free amides90 in FTIR transmission spectra recorded at 155 °C from a film of PCL/B (1.2 wt%) on KBr. (d) Optical micrographs show micrometer-sized fibrous precipitates of B, formed by lateral aggregation of the individual stacks, giving (e) Bragg reflections in the neighborhood of q ≈ 0.24 Å−1 (dB ≈ 1.3 nm) in XRD patterns obtained at room temperature. (f) Phase diagram for the PCL/B reference blends, constructed from Td determined from DSC heating scans. The PCL/B blends form an optically transparent, homogeneous melt, (PCL/B)l, at sufficiently high temperatures. Solid bulk additive domains (microfibers), Bs, precipitate from the liquid phase upon cooling and persist to below the polymer melting temperature (58 °C), where crystalline lamellae begin to form in the PCL matrix (PCL/B)s. The solid orange curve represents fitting of the Flory–Huggins model to the data. See ESI Figs. S3–S7† for more information. | ||
In Fourier transform infrared (FTIR) spectra recorded in the attenuated total reflection (ATR) configuration at room temperature, the N–H stretching band at 3232–3240 cm−1, the CO stretching band at 1637–1641 cm−1, and the combined N–H bending/C–N stretching band at 1560–1561 cm−1 are characteristic of strongly hydrogen-bonded BTA aggregates in the PCL/B blends, regardless of the B concentration (Fig. 2b, c and ESI Fig. S4†).89,90 FTIR spectra recorded in transmission mode at temperatures above Td show the N–H stretching band at 3411 cm−1, the CO stretching band at 1679 cm−1, and the combined N–H bending/C–N stretching band at 1513 cm−1, all of which indicate the presence of free amide groups, as described previously for BTA derivatives in dilute solution (ESI Fig. S5†).89 B may hence be assumed to be homogeneously dispersed in the PCL matrix above Td.
Consistent with the DSC results, solid bulk precipitates, Bs, in the form of microfibers are observed by bright-field optical microscopy (OM) between Tagg and Tc during cooling (Fig. 2d and ESI Fig. S6†), but are obscured by the PCL crystallinity at temperatures below Tc, (PCL/B)s, and disappear upon heating to above Td, where OM indicates a single-phase, amorphous melt, (PCL/B)l, for the range of B concentrations investigated. Room-temperature X-ray diffraction (XRD) patterns of all the blends show an intense Bragg reflection at q < 0.2 Å−1, which corresponds to the lamellar long period of PCL, L, of 17.5–18.3 nm.91 The additional, markedly less intense Bragg reflections in the neighborhood of q ≈ 0.24 Å−1 (dB ≈ 1.3 nm) are characteristic of the bulk B crystal structure (Fig. 2e and ESI Fig. S7†).89
The Td values determined from DSC heating scans were fitted using the Flory–Huggins model, as described in our previous study of oligoalanine aggregates in PCL,50 assuming a Flory–Huggins interaction parameter in the form of
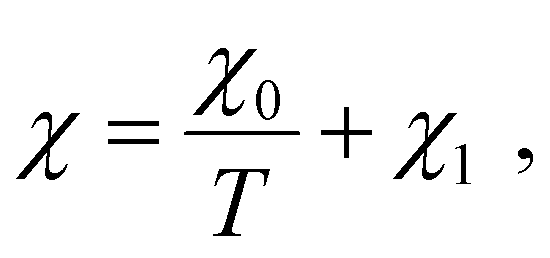 | (1) |
The end-modified PCL-BTA used in the investigation of the corresponding supramolecular PCL-BTA/B blends is obtained from commercial telechelic PCL with a degree of (difunctional) hydroxyl end-group functionalization of at least 92%, determined by comparing n obtained from gel permeation chromatography (GPC) with the end-group content according to the peak integrals in 1H nuclear magnetic resonance (NMR) spectroscopy (ESI Fig. S8†). The hydroxyl end groups were then quantitatively modified by Steglich esterification with Fmoc-protected glycine, as confirmed by the absence of the peak at δ = 3.63 ppm assigned to the CH2–OH end groups and the presence of peaks characteristic of the Fmoc protection group in the range of δ = 7–8 ppm in the 1H NMR spectrum of PCL-GlyFmoc (ESI Fig. S9†). Subsequent deprotection yields NH2-terminated PCL, which is functionalized via a standard peptide coupling reaction with 3,5-bis(isopentylcarbamoyl)benzoic acid (ESI Scheme S1†). By comparing n obtained from GPC with the end-group content determined from peak integrals in 1H NMR spectroscopy, we estimate a degree of BTA end-group functionalization of at least 95% (ESI Fig. S10†). Pure PCL-BTA, which has an end-group concentration of 0.63 wt%, shows no evidence of end-group aggregation and its melt behavior is therefore very similar to that of unmodified PCL of the same molar mass (ESI Fig. S1c and d†), as generally observed for high-molar-mass polymers42,50 and low-molar-mass polymers with high backbone polarity, including polyesters.59
In what follows, unless mentioned otherwise, we compare PCL/B and PCL-BTA/B blends with equal wB, given in wt% (for the corresponding effective total concentration of BTA-based moieties, wB+BTA, in wt%, [B] and [B + BTA] in mol L−1, and the additive-to-end-group ratio, [B]/[BTA], in mol mol−1, see Table 1 in the Materials and methods). Upon addition of B at concentrations in the range of 0.6–5 wt% investigated, the resulting PCL-BTA/B blends again form a single-phase, amorphous melt (PCL-BTA/B)l at sufficiently high temperatures, based on their homogeneous appearance in OM. However, FTIR spectra recorded of PCL-BTA/B (1 wt%) in the melt state at temperatures up to 155 °C (Fig. 3) now show the main N–H stretching band at 3396 cm−1, the CO stretching band at 1672–1677 cm−1, and the combined N–H bending/C–N stretching band at 1520–1524 cm−1, indicating weakly hydrogen-bonded BTA amides, as reported previously for optically homogeneous melts of pure BTA-derivatives.89 The CO stretching band assigned to the polymer backbone ester groups at 1733 cm−1 remains unchanged over the whole temperature range, showing that these ester groups do not interact significantly with B (Fig. 3d). Hence, in marked contrast with the PCL/B reference blends, the IR results for the modified PCL-BTA/B blends suggest that at temperatures immediately above the Td implied by DSC scans, B is present in the form of colloidal aggregates, characterized by a weak, discontinuous hydrogen bond network, and stabilized by the end-modified PCL-BTA, which acts as a surfactant. Hence, under these conditions, Td does not mark a transition to a fully dispersed state but rather a melting transition to a disordered, but still phase-separated state.
 | ||
| Fig. 3 FTIR transmission spectra of a PCL-BTA/B (1 wt%) film on KBr, recorded at 25, 79, 101, 119, 126, 132, and 155 °C, showing (a) the N–H stretching band, with examples of the deconvolution of the spectra at 25 and 155 °C, and (b) the CO stretching band and the combined N–H bending/C–N stretching band. The ensemble of deconvoluted spectra is shown in ESI Fig. S11.† The sharp absorption signal visible in all the spectra at 3435–3445 cm−1 is assigned to water absorbed by PCL.92 (c) The area of the N–H stretching peak obtained from the deconvoluted spectra of PCL-BTA/B (1 wt%) (blue) and PCL/B (1.2 wt%) (orange), normalized with respect to the area of the PCL C–H2 stretching peak (not shown). The dashed curves serve as guides to the eye. (d) The CO stretching band of the PCL backbone does not change with temperature. | ||
| wB wt% | PCL/B | PCL-BTA/B | ||
|---|---|---|---|---|
| [B] (mol L−1) | wB+BTA (wt%) | [B + BTA] (mol L−1) | [B]/[BTA] (mol mol−1) | |
| 0 | 0 | 0.63 | 0.02 | 0 |
| 0.6 | 0.02 | 1.23 | 0.04 | 0.79 |
| 1 | 0.03 | 1.63 | 0.05 | 1.32 |
| 2 | 0.05 | 2.63 | 0.08 | 2.63 |
| 4 | 0.11 | 4.63 | 0.13 | 5.27 |
DSC scans of the PCL-BTA/B blends reveal two distinct types of behavior depending on the B concentration. In a high-concentration regime corresponding to wB ≥ 1.5 wt% ([B]/[BTA] ≥ 1.98 mol mol−1, see Materials and methods), we observe results similar to those of the PCL/B reference blends, that is, an exothermic transition at Tagg > Tc in cooling scans and an endothermic transition at Td > Tm in subsequent heating scans, corresponding to association and dissociation of B aggregates, respectively (Fig. 4a). OM shows that this leads to solid bulk additive precipitates, Bs, in the form of microfibers similar to those observed in the PCL/B reference blends according to XRD and FTIR (Fig. 4b–f, ESI Fig. S12†). The absolute values of Td of the PCL-BTA/B blends are also similar to those of the reference blends, indicating the end groups to have little effect on the global solubility of B in PCL-BTA. Tagg values determined from the DSC cooling scans at a cooling rate of 10 °C min−1 are some 13–21 °C lower than the corresponding Td in this concentration regime; the highest supercoolings are observed at B concentrations immediately above 1.5 wt%.
 | ||
| Fig. 4 (a) DSC heating and cooling scans (10 °C min−1, exothermal transitions up) from the PCL-BTA/B blends for B concentrations ≥ 1.5 wt%. For clarity, the PCL melting and crystallization transitions at Tm = 58 °C and Tc = 35 °C, respectively, are only shown for one composition. (b and c) Cross-polarized (top) and bright-field (bottom) optical micrographs of PCL-BTA/B recorded at T < Tagg during cooling at 10 °C min−1 from the melt state. PCL-BTA/B blends containing ≥1.5 wt% B show microfibrous precipitates (see also ESI Fig. S12†). (d) The N–H stretching band, and (e) the CO stretching band and the combined N–H bending/C–N stretching band from ATR FTIR spectra of PCL-BTA/B recorded at 25 °C. The sharp absorption signal visible in all the spectra at 3435–3445 cm−1 is assigned to water absorbed by PCL.92 (f) The corresponding structures show Bragg reflections in the neighborhood of q ≈ 0.24 Å−1 (dB ≈ 1.3 nm) in XRD patterns obtained at room temperature. | ||
Despite these similarities in the high-concentration regime, the modified PCL-BTA/B blends behave very differently from the unmodified PCL/B reference blends in the low-concentration regime corresponding to wB ≤ 1.1 wt% ([B]/[BTA] ≥ 1.45 mol mol−1, see Materials and methods), where two endothermic transitions are observed in the DSC heating scans at temperatures above Tm. The first of these transitions occurs at temperatures 18–35 °C below Td in PCL/B for a given B concentration (Fig. 5a), which we refer to as Td,NF, and is identified by the co-aggregation of B with the chain ends to form a distinct nanofibrillar phase upon cooling. Although OM suggests the melt remains optically homogeneous at temperatures down to Tc for PCL upon cooling and immediately above Tm upon heating (Fig. 5b, ESI Fig. S12†), FTIR spectra exclusively show sharp absorption signals at about 3238 cm−1 (N–H stretching) and 1642 cm−1 (CO stretching); this proves that B and the PCL-BTA end groups nevertheless quantitatively assemble into strongly hydrogen-bonded BTA aggregates, with no indication of weakly hydrogen-bonded, disordered aggregates (Fig. 5c and d).90 Moreover, the ratio of the intensity of the 3235 cm−1 peak in the temperature-dependent IR spectra (Fig. 3c) of PCL-BTA/B (1 wt%) below 90 °C to that in PCL/B (1.2 wt%) is close to the ratio of the combined concentrations of the PCL-BTA end groups and additive B in the modified blend to the B concentration in the reference blend. This indicates that practically all the end groups in PCL-BTA/B take part in aggregation.
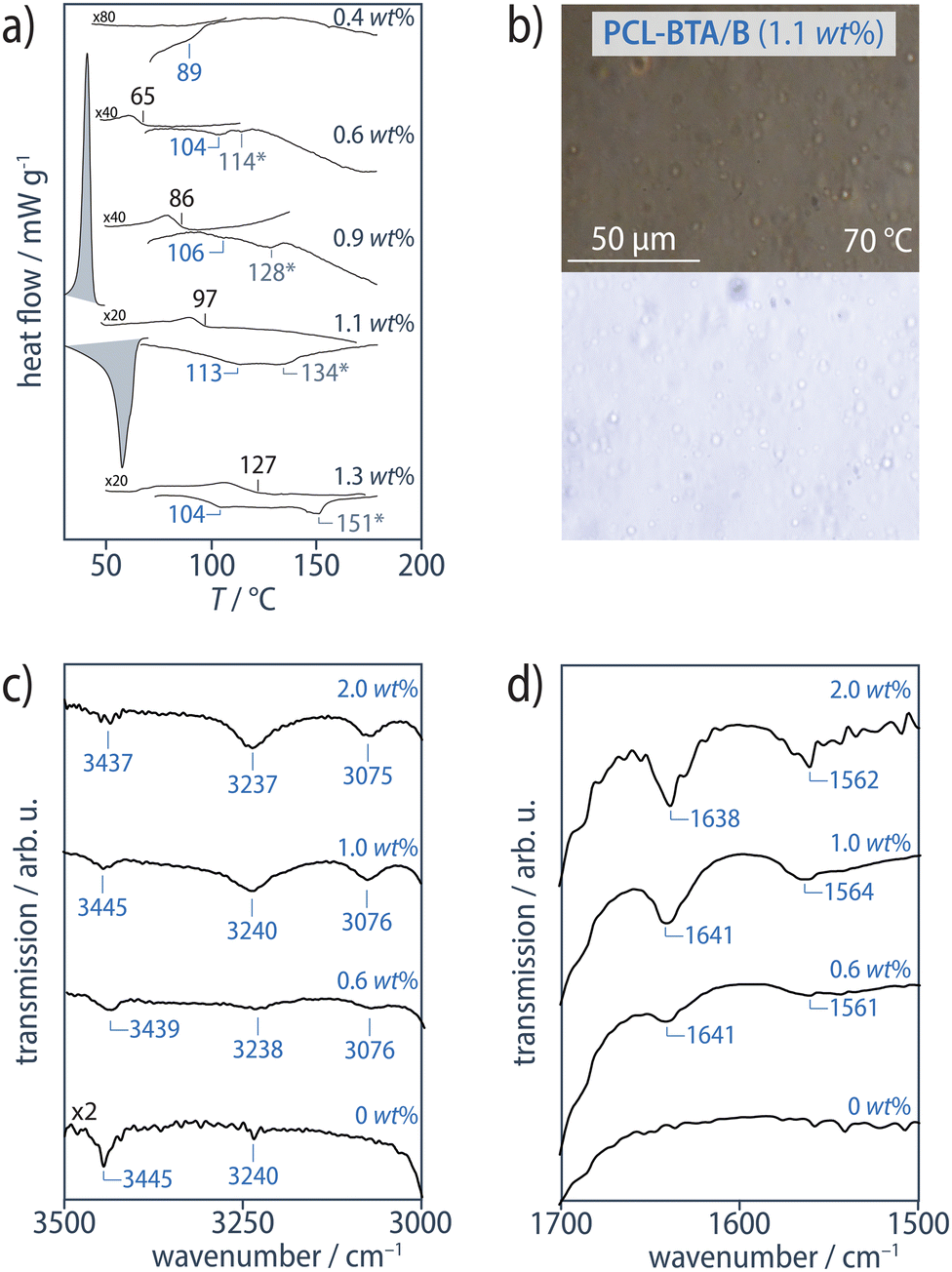 | ||
| Fig. 5 (a) DSC heating and cooling scans (10 °C min−1, exothermal transitions up) from PCL-BTA/B blends. For clarity, the PCL melting and crystallization transitions at Tm = 58 °C and Tc = 35 °C, respectively, are only shown for one composition. (b) Cross-polarized (top) and bright-field (bottom) optical micrographs of PCL-BTA/B recorded at T < Tagg during cooling at 10 °C min−1 from the melt state. PCL-BTA/B blends at wB ≤ 1.1 wt% remain optically homogeneous (see also ESI Fig. S12†), but ATR FTIR spectra of PCL-BTA/B recorded at 25 °C contain (c) the N–H stretching band and (d) the CO stretching band and the combined N–H bending/C–N stretching band indicative of BTA aggregates at all B concentrations investigated. The sharp absorption signal visible in all the spectra at 3435–3445 cm−1 is assigned to water absorbed by PCL.92 | ||
The nanofibrillar nature of the resulting structures is confirmed by AFM images recorded for PCL-BTA/B (0.6 wt%) heated to between Tm and Td,NF, which are dominated by high-aspect-ratio nanofibrils with diameters in the nanometer size range, bearing in mind that the radius of the curvature of the AFM tip may be up to 7 nm, locally aligned with interfibrillar spacings in the range of 10–20 nm (Fig. 6 and ESI Fig. S13†). The nanofibril spacing is hence considerably greater than the intercolumnar spacings characteristic of the crystalline bulk B precipitates, but comparable with a value of 13 nm estimated from volumetric considerations for an ideal dispersion of single stacks of co-assembled B and PCL-BTA end groups (see Materials and methods for details). However, it generally remains smaller than the root-mean-square end-to-end distance of the present PCL of about 25 nm,93 consistent with substantial incorporation of the chain ends in the nanofibrils and implying significant bridging of these by the PCL chains or via trapped entanglements.
 | ||
| Fig. 6 Intermittent-contact AFM phase images of PCL-BTA/B (0.6 wt%) recorded at 70 °C at different magnifications. See ESI Fig. S13.† | ||
In XRD scans of PCL-BTA/B for B concentrations in the range of 0.6–1.1 wt%, Bragg peaks corresponding to bulk B precipitates are still present but overlap to form a single broad peak, in marked contrast to the PCL/B reference materials where the three reflections remain well-resolved even at the lowest B concentrations (Fig. 7a–d). The resulting relatively large effective XRD peak widths at half-maximum, β, may reflect reduced crystal sizes, D, according to the Scherrer equation
 | (2) |
 | ||
| Fig. 7 (a) 1D SAXS diffractograms of PCL-BTA/B blends, measured on flat specimens obtained by cooling from the melt at −10 °C min−1, normalized with respect to the intense low q Bragg peaks corresponding to the lamellar long period, L, of the PCL matrix at about 17–18 nm. (b) Enlargement of the Bragg reflections in the neighborhood of q ≈ 0.24 Å−1 (dB ≈ 1.3 nm). (c and d) Corresponding 1D SAXS diffractograms of PCL/B blends. (e) Total area of the Bragg peaks in the neighborhood of q ≈ 0.24 Å−1 divided by the B concentration and plotted against the B concentration, and (f) the crystallite size, D, estimated from β for the Bragg reflections at dB ≈ 1.3 nm using the Scherrer equation, eqn (2), in both the PCL/B (orange symbols) and PCL-BTA/B blends (blue symbols – filled symbols for the NF regime, open symbols for the regime in which bulk precipitates become visible in OM); the dashed line corresponds to D estimated from β for bulk B, for which extrinsic factors such as instrumental broadening are assumed be significant. | ||
A new phase region, denoted NF and bounded by Td,NF therefore appears in the pseudo-phase diagram for PCL-BTA/B in the B concentration range of 0.6–1.1 wt%, which gradually gives way to bulk B precipitation (bounded by Td) as the B concentration increases beyond this range. In this region, we propose that the polymer-stabilized, weakly hydrogen-bonded colloidal aggregates revealed by FTIR are converted into a phase characterized by a network of strongly hydrogen-bonded polymer-bridged nanofibrils that persist upon PCL crystallization (Fig. 8).
 | ||
| Fig. 8 Phase diagram of PCL-BTA/B blends constructed from Td* (open circles), Td,NF (filled squares) and Td (open squares) obtained from DSC heating scans of PCL-BTA/B blends. (PCL-BTA/B)l – homogeneous melt; NF – nanofibrils; Bs – bulk B precipitates; (PCL-BTA/B)s – solid solution. The grey dashed line indicates fitting of the Flory–Huggins model to Td for PCL/B (see Fig. 2). | ||
In addition to the dissociation transition of the nanofibrils at Td,NF, we observe an endothermic transition at higher temperatures, Td*, which is very close to Td of the corresponding PCL/B blends for a given B concentration. However, no corresponding exothermic transition is observed in the cooling scans. In view of the supercooling associated with bulk B precipitation in the high-concentration regime, we therefore assume it to be kinetically suppressed at low B concentrations in favor of a direct transition from the PCL-BTA-stabilized colloidal aggregates to co-assembled nanofibrils. However, B molecules released from the nanofibrils upon heating through Td,NF are inferred to reorganize into bulk precipitates that subsequently melt at Td*. It is noteworthy that the Tagg values determined from DSC cooling scans at 10 °C min−1 are also some 16–40 °C lower than Td,NF in this regime (Fig. 5a). At lower cooling rates, however, Tagg approaches Td,NF, confirming that this is a kinetic effect, and the cooling rate has little effect on the values of Td,NF and Td* observed in subsequent heating scans at 10 °C min−1 (ESI Fig. S14†).
The melt properties of the PCL-BTA/B blends are compared with those of the PCL/B reference blends based on oscillatory shear rheometry temperature sweeps at a frequency of 1 rad s−1 and a scanning rate of 10 °C min−1, in which the specimens were cooled from the homogeneous melt state (180–200 °C, depending on the B content) to 30 °C and heated back to the melt state. In what follows, we focus on the results for PCL-BTA/B obtained from the heating scans (Fig. 9, ESI Fig. S15†), which show one or more of the following features as the temperature is raised: (i) a steep drop in the storage modulus, G′, upon melting of the PCL matrix at its Tm of about 58 °C; (ii) a rubbery plateau at which G′ decreases slowly from a value in the range of 0.1–2.7 MPa (depending on the B content of the material, Fig. 9c); (iii) a crossover temperature referred to as the softening temperature, Ts (Fig. 9d), above which G′ < G′′, marking the upper limit of the rubbery plateau; (iv) a steep decrease in both G′ and G′′ towards a “rheological” dissociation temperature, Td,rheo (Fig. 9e and f), above which G′ < 3.5 kPa and the behavior approaches that of a low-viscosity fluid (so that G′ falls below the sensitivity limit of the present setup).
 | ||
| Fig. 9 (a and b) Representative plots of the storage (G′, filled squares) and loss (G′′, open squares) moduli obtained from oscillatory shear rheometry heating scans for the PCL-BTA/B (blue) and PCL/B blends (orange) at a frequency of 1 rad s−1 and a scanning rate of 10 °C min−1. Data for other B contents are shown in ESI Fig. S15.† (c) G′ at 70 °C increases systematically with wB as do (d) the softening temperature, Ts, above which G′ > G′′ (not observed for PCL/B) and (e) Td,rheo. (f) In PCL-BTA/B, Td,rheo corresponds to either Td,NF, for wB ≤ 1.1 wt%, or Td for wB ≥ 1.5 wt% determined from DSC heating scans, but is slightly lower in PCL/B particularly at low B concentrations. Similar trends are also observed for G′ at 70 °C, Ts, and Td,rheo when plotted against the total concentration of BTA moieties, including the end groups, wB+BTA (see ESI Fig. S16†). The dashed curves in all panels serve as guides to the eye. | ||
In the PCL-BTA/B blends, Td,rheo is generally close to either Td,NF, for wB ≤ 1.1 wt%, or Td for wB ≥ 1.5 wt% (Fig. 9f), implying that the aggregates act as a reinforcement at temperatures T < Td,rheo. The absence of significant reinforcement between Td,rheo and Td* suggests that the nanofibrils make a dominant contribution, where they are present, and that the structural reorganization that gives rise to Td* has no significant effect. In the PCL/B reference blends, Td,rheo is generally somewhat lower than Td, but the two temperatures remain strongly correlated over the whole composition range. It follows that Td,rheo decreases strongly with decreasing B concentration in both PCL-BTA/B and PCL/B (Fig. 9e).
Both PCL-BTA/B and PCL/B also show an increase in G′ with decreasing temperature towards a limiting value immediately above Tm, which again decreases with decreasing B concentration, as reflected by 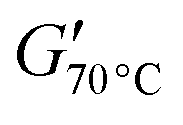 , the value of G′ measured at 70 °C (Fig. 9c). However,
, the value of G′ measured at 70 °C (Fig. 9c). However,  is about six times higher in PCL-BTA/B than in PCL/B for a given wB, levelling off at about 2 MPa in PCL-BTA/B (4 wt%). Moreover, only PCL-BTA/B shows a true rubbery plateau delimited by Tm and Ts; the latter is undefined for PCL/B, which remains a viscoelastic fluid (G′ < G′′) at all temperatures T > Tm, regardless of the B concentration in the range investigated.
is about six times higher in PCL-BTA/B than in PCL/B for a given wB, levelling off at about 2 MPa in PCL-BTA/B (4 wt%). Moreover, only PCL-BTA/B shows a true rubbery plateau delimited by Tm and Ts; the latter is undefined for PCL/B, which remains a viscoelastic fluid (G′ < G′′) at all temperatures T > Tm, regardless of the B concentration in the range investigated.
The additional rheological transition at Ts in the PCL-BTA/B modified blends, their rubbery elastic response in the temperature range Tm < T < Ts, and their significantly increased G′ compared with the PCL/B reference blends are consistent with the formation of a network of polymer-bridged nanofibrils94 due to the incorporation of the PCL-BTA end groups in the B aggregates formed during cooling. This is strongly reminiscent of the behavior of the oligoalanine-based system described previously,50 but the transition temperatures Td, Td,rheo, and Ts are significantly higher in the PCL-BTA/B blends than in the oligoalanine-based materials, not least because the Td value of the pure B additive is higher than that of the oligoalanine additive. The PCL-BTA/B materials are therefore expected to be processable by techniques that require a high melt strength and elasticity over a correspondingly wider temperature range.60 Moreover, thermal degradation of oligopeptides (decomposition temperature in air, Tdec ≈ 250 °C) limits their potential for high-temperature polymer processing, whereas the BTA-based materials show much greater stability (Tdec ≈ 350 °C).
To demonstrate the improved processability, we successfully processed PCL-BTA/B (1 wt%) films into cup-shaped specimens by thermoforming at 70 °C (Fig. 10); this is not possible for free-standing unmodified PCL films. When filled with boiling water, the PCL-BTA/B cups become transparent due to the loss of crystallinity in the PCL matrix at temperatures above Tm = 58 °C but retain their shape and support the weight of water, thanks to the elastic response of the nanofibrillar network at this temperature.
 | ||
| Fig. 10 (a) PCL-BTA/B (1 wt%) has been processed into cup-shaped objects by thermoforming, whereas pristine PCL can only be thermoformed with the aid of a paper support. (b) The PCL-BTA/B cups were filled with boiling water. Defects associated with the connection of the mold to the vacuum pump during thermoforming resulted in water leakage after 12 s but the cups remained globally intact. Scale bars represent 1 cm. | ||
BTA derivatives are well known to be efficient nucleating agents for polymer crystallization. In iPP, they typically form microfibrous or needle-like crystalline bulk precipitates with high surface-to-volume ratios,75 similar to the structures observed in the PCL/B reference blends. The much higher surface-to-volume ratio areas of the nanofibrils formed in the PCL-BTA/B blends may further enhance their nucleation effect. To investigate this, we determined the crystallization half time (τ1/2) in isothermal DSC experiments at 48 °C after cooling from the melt. While τ1/2 is reduced by a factor of up to 5.6 in PCL/B reference blends with respect to pristine PCL, it is reduced by a factor of 11 in PCL-BTA/B, for all wB ≥ 1 wt% (Fig. 11a and b). The resulting crystallinity, determined by integrating the PCL melting peak in heating scans at 10 °C min−1 immediately after the isothermal step and assuming the melting enthalpy of an ideal PCL crystal to be 139.5 J g−1,95 is around 40% at wB ≤ 2 wt% but decreases somewhat at higher B concentrations (Fig. 11c). Hence, the nanofibrils in the PCL-BTA/B materials are confirmed to nucleate PCL crystallization more effectively than the bulk additive. This may be due to their high degree of dispersion and/or the covalent attachment of the polymer chains.96
 | ||
| Fig. 11 (a) Examples of isothermal DSC traces for PCL-BTA/B (blue), PCL/B (orange) and pristine PCL (grey) at 48 °C after cooling from 200 °C at 10 °C min−1 to 70 °C, followed by rapid cooling to 48 °C, from which (b) the crystallization half times, τ1/2, have been determined. τ1/2 is decreased by a factor of up to 5.6 in PCL/B (orange), and up to 11 in blends of PCL-BTA/B (blue) compared with pristine PCL (grey). The dashed curves serve as guides to the eye. (c) The degree of crystallinity of the PCL matrix, as determined by integrating the PCL melting peak in heating scans at 10 °C min−1 recorded immediately after the isothermal step, assuming the melting enthalpy of an ideal PCL crystal to be 139.5 J g−1.95 | ||
To emphasize the importance of using high molar mass polymers, we also studied BTA-end-modified PCL of n = 24900 g mol−1 (PCL25-BTA), with an end-group concentration of 3.2 wt%. There is no evidence from DSC, FTIR, or shear rheology experiments for end-group aggregation in PCL25-BTA in the absence of B, consistent with previous reports on BTA-modified PCL.59 However, PCL25-BTA/B blends contain co-assembled nanoscopic aggregates similar to those in PCL-BTA/B. Moreover, due to its higher end-group concentration, the threshold for the onset of bulk B precipitation increases to wB ≥ 3 wt% in PCL25-BTA, indicating that more B participates in the formation of the nanoscopic aggregates (ESI Fig. S17†). In agreement with our previously reported findings for low-n PCL modified with oligo-L-alanine end groups,50 PCL25-BTA/B remains brittle (ESI Fig. S17f†) despite the presence of the reinforcing network, because the molar mass of the base polymer is too low for the establishment of well-developed, stable entanglements.
Conclusions
We have shown that high-molar-mass PCL-BTA, modified with BTA-based self-assembling end groups, undergoes nanophase separation when blended with the low-molar-mass BTA derivative B in the low-concentration regime, wB ≤ 1.1 wt%. In this regime, we observe the quantitative formation of strongly hydrogen-bonded nanoscopic aggregates by co-assembly of the end groups and additive. This is particularly remarkable considering not only the high molar mass of the PCL but also its polarity, which has been reported to render supramolecular network formation difficult, even for substantially lower molar masses.59 Accordingly, pure PCL-BTA shows no evidence of end-group assembly. Moreover, the bulk B precipitates seen in unmodified PCL/B reference blends in this concentration range are absent from PCL-BTA/B. Indeed, weakly hydrogen-bonded colloidal B domains persist in PCL-BTA/B at temperatures well above the dissociation temperature of the aggregates, presumably stabilized by the PCL-BTA end groups. This may impede the bulk precipitation of B upon cooling, instead providing precursors for the formation of well-defined, polymer-bridged, high-aspect-ratio BTA nanofibrils.In the bulk melt state, the resulting physically crosslinked, reinforced polymer network manifests itself as a rubbery plateau in rheometry temperature sweeps. Depending on the B content, this plateau extends up to about 90 °C above the PCL melting point and is associated with a storage modulus more than two orders of magnitude higher than that of pristine PCL or PCL-BTA in this temperature range, and some six times higher than that of PCL/B containing the same amount of B. The resulting materials show significantly enhanced thermal stability and facilitate processing by thermoforming, for instance. In addition, the polymer-tethered BTA nanofibrils are far more efficient nucleating agents for polymer crystallization than the corresponding bulk B precipitates, with the potential to reduce industrial process cycle times and hence manufacturing costs. Moreover, a comparison with blends using modified PCL of lower molar mass demonstrates that it is precisely the combination of a well-entangled base polymer and the polymer-bridged nanofibril network that renders PCL-BTA/B technologically interesting. Finally, the concept of end-group and additive co-assembly may in principle be extended to the many other aliphatic polyesters currently considered to be more sustainable alternatives to conventional plastics, but which often lack sufficient melt strength for certain key processing techniques, thus broadening manufacturing options.
Materials and methods
Materials
The following chemicals were obtained from commercial sources and used without further purification: PCL,n = 80000 g mol−1 (Sigma), triethylamine ≥ 99.5% (Sigma), N,N′-dicyclohexylcarbodiimide (DCC) 99% (Sigma), 3-methylbutylamine ≥ 98% (Sigma), 1,3,5-benzenetricarbonyl trichloride 98% (Sigma), N,N-diisopropylethylamine (DIPEA) 99% (abcr), trimethyl benzene-1,3,5-tricarboxylate 98% (abcr), 4-dimethyl aminopyridine (DMAP, Fluorochem), 1H-benzotriazol-1-yloxytripyrrolidinophosphoniumhexafluorophosphate (PyBOP, Fluorochem), N-(9-fluorenylmethoxycarbonyl)-glycine (GlyFmoc) ≥98% (novabiochem), piperidine 99% (Acros Organics), DMSO-d6 99.8% D (ZEOtope), CDCl3 99.8% D, stabilized with Ag (ZEOtope), diethyl ether (Reactolab), ethanol (EtOH, Reactolab), hexane (Reactolab), ethyl acetate (EtOAc, Reactolab), argon 99.999% (Carbagas), HCl 25% (Sigma), KOH (Reactolab), NaOH (Reactolab), LiOH ≥98% (Fisher Chemicals). Tetrahydrofuran (THF), methanol (MeOH), and CHCl3 (Reactolab) were distilled prior to use. Dichloromethane (DCM) ≥99.9% stabilized with amylene (Sigma) was distilled prior to use.
Synthetic procedures
:3) as the eluent. 1H NMR (400 MHz, DMSO-d6): δ/ppm = 8.76 (t, 2H), 8.55 (d, 1H), 8.53 (d, 2H), 3.93 (s, 3H), 3.32 (m, 4H), 1.63 (m, 2H), 1.45 (q, 4H), 0.92 (d, 12H). 13C NMR (100 MHz, DMSO- d6): δ/ppm = 161.03, 160.95, 130.77, 126.22, 125.62, 124.98, 47.87, 33.92, 33.64, 22.16, 17.71. HRMS (nanochip-ESI/LTQ-Orbitrap) m/z: [M + H]+ calcd for C20H30N2O4+ 363.2206; found: 363.2274. For HRMS, 1H NMR and 13C NMR spectra, see ESI Figs. S22–S24.†:2) had been completely removed. Pure 3 was obtained as a white powder (8.0 g, 80%). 1H NMR (400 MHz, toluene-d8, 375 K): δ/ppm = 12.72 (br, 1H), 8.82 (s, 2H), 8.75 (s, 1H), 8.09 (br, 2H), 3.43 (q, J = 6.9 Hz, 4H), 1.63 (m, 2H), 1.50 (q, J = 6.9 Hz, 4H), 0.88 (d, J = 6.5 Hz, 12H). 13C NMR (100 MHz, DMSO-d6): δ/ppm = 166.52, 164.86, 135.34, 131.10, 130.35, 130.08, 37.99, 37.60, 25.24, 22.41. HRMS (nanochip-ESI/LTQ-Orbitrap) m/z: [M + H]+ calcd for C19H29N2O4+ 349.2122; found 349.2118. EA: calcd for C19H28N2O4: C, 65.49; H, 8.10; N, 8.04. Found: C, 65.31; H, 8.10; N, 8.01. For HRMS, 1H NMR, and 13C NMR spectra, see ESI Figs. S25–S27.†n = 112000 g mol−1, was determined by GPC (polystyrene calibration). The number-average molar mass, n = 103000 g mol−1, was also calculated from the ratio of CH2–OH end group signals to all PCL backbone peak integrals, implying an end group content of ≥92% OH end groups. 1H NMR (400 MHz, CDCl3, 512 scans): δ/ppm = 4.04 (1817 H, t, CH2OCOPCL), 3.63 (4 H, t, CH2–OH), 2.29 (1813 H, t, CH2COOPCL), 1.68 (3714 H, m, CH2,PCL), 1.37 (1813 H, m, CH2,PCL), GPC: n = 112000, w = 164000, Đ = 1.47. GPC traces are shown in ESI Fig. S28.†n = 107000, w = 166000, Đ = 1.54. GPC traces are shown in ESI Fig. S28.†n = 24900, w = 33100, Đ = 1.33. GPC traces are shown in ESI Fig. S28.† For the 1H NMR spectrum, see ESI Fig. S31.†Methods
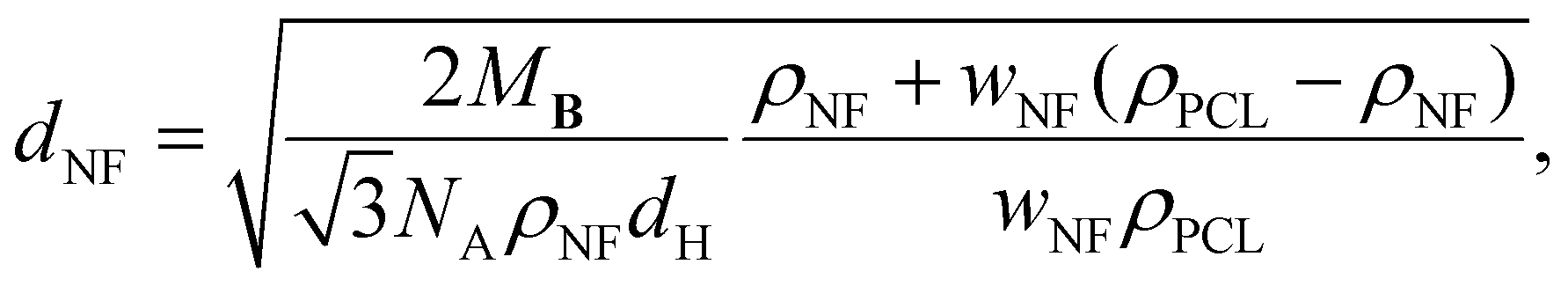 | (3) |
n, the weight-average molar mass, w, and the dispersity, Đ, were determined by dissolving a 3–5 mg sample in 1 mL THF and filtering the solution through a 0.220 μm PTFE filter before injection. Elution was performed in THF at 40 °C at a flow rate of 1 mL min−1 using an Agilent 1260 Infinity instrument incorporating the 390-MDS detector train equipped with a refractive index detector, one PSS SDV precolumn and either two PLgel 5 μm MIXED-C Analytical columns or two PSS SDV Analytical Linear XL columns. Calibration was performed with polystyrene standards with n in the range of 682–2520000 g mol−1.
 | (4) |
Author contributions
H.F. and D.G. developed the research concept and designed the experiments. S.T. carried out the DSC, FTIR, OM, XRD, and rheology experiments and analyzed the data. M.G. developed the synthesis procedure for the BTA-based reagent for polymer end modification and additive B. M.W. performed the temperature-dependent FTIR measurements and the Flory–Huggins analysis. C.J.G.P. carried out the AFM experiments. S.T., D.G., C.J.G.P., and H.F. wrote the manuscript.Data availability
The authors declare that the data supporting the findings of this study are available within the paper and its ESI.†Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
This project was funded by the SNSF BRIDGE program under grant agreement no. 40B2-0_211501. We would like to thank Dr S. Koppenhoefer for photography and Dr Y. Hryshunin for carrying out the thermoforming experiments.References
- S.-L. Li, T. Xiao, C. Lin and L. Wang, Advanced supramolecular polymers constructed by orthogonal self-assembly, Chem. Soc. Rev., 2012, 41, 5950–5968 RSC.
- C. Rest, R. Kandanelli and G. Fernández, Strategies to create hierarchical self-assembled structures via cooperative non-covalent interactions, Chem. Soc. Rev., 2015, 44, 2543–2572 RSC.
- H. Frauenrath and E. Jahnke, A General Concept for the Preparation of Hierarchically Structured π-Conjugated Polymers, Chem. – Eur. J., 2008, 14, 2942–2955 CrossRef CAS PubMed.
- C. Li, A. Iscen, H. Sai, K. Sato, N. A. Sather, S. M. Chin, Z. Álvarez, L. C. Palmer, G. C. Schatz and S. I. Stupp, Supramolecular–covalent hybrid polymers for light-activated mechanical actuation, Nat. Mater., 2020, 19, 900–909 CrossRef CAS PubMed.
- S. M. Chin, C. V. Synatschke, S. Liu, R. J. Nap, N. A. Sather, Q. Wang, Z. Álvarez, A. N. Edelbrock, T. Fyrner, L. C. Palmer, I. Szleifer, M. Olvera de la Cruz and S. I. Stupp, Covalent-supramolecular hybrid polymers as muscle-inspired anisotropic actuators, Nat. Commun., 2018, 9, 2395 CrossRef.
- M. J. Webber and R. Langer, Drug delivery by supramolecular design, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- D. Gao, G. Thangavel, J. Lee, J. Lv, Y. Li, J.-H. Ciou, J. Xiong, T. Park and P. S. Lee, A supramolecular gel-elastomer system for soft iontronic adhesives, Nat. Commun., 2023, 14, 1990 CrossRef CAS.
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, From supramolecular polymers to multi-component biomaterials, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. K. Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Reversible Polymers Formed from Self-Complementary Monomers Using Quadruple Hydrogen Bonding, Science, 1997, 278, 1601–1604 CrossRef CAS PubMed.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Self-healing and thermoreversible rubber from supramolecular assembly, Nature, 2008, 451, 977–980 CrossRef CAS.
- L. Voorhaar and R. Hoogenboom, Supramolecular polymer networks: hydrogels and bulk materials, Chem. Soc. Rev., 2016, 45, 4013–4031 RSC.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Supramolecular Polymerization, Chem. Rev., 2009, 109, 5687–5754 CrossRef CAS.
- A. Lavrenova, D. W. R. Balkenende, Y. Sagara, S. Schrettl, Y. C. Simon and C. Weder, Mechano- and Thermoresponsive Photoluminescent Supramolecular Polymer, J. Am. Chem. Soc., 2017, 139, 4302–4305 CrossRef CAS.
- J. H. K. K. Hirschberg, F. H. Beijer, H. A. van Aert, P. C. M. M. Magusin, R. P. Sijbesma and E. W. Meijer, Supramolecular Polymers from Linear Telechelic Siloxanes with Quadruple-Hydrogen-Bonded Units, Macromolecules, 1999, 32, 2696–2705 CrossRef.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Supramolecular Polymer Materials: Chain Extension of Telechelic Polymers Using a Reactive Hydrogen-Bonding Synthon, Adv. Mater., 2000, 12, 874–878 CrossRef CAS.
- N. Roy, V. Schädler and J.-M. Lehn, Supramolecular Polymers: Inherently Dynamic Materials, Acc. Chem. Res., 2024, 57, 349–361 CrossRef CAS PubMed.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Mechanically robust, readily repairable polymers via tailored noncovalent cross-linking, Science, 2018, 359, 72–76 CrossRef CAS.
- J.-C. Lai, X.-Y. Jia, D.-P. Wang, Y.-B. Deng, P. Zheng, C.-H. Li, J.-L. Zuo and Z. Bao, Thermodynamically stable whilst kinetically labile coordination bonds lead to strong and tough self-healing polymers, Nat. Commun., 2019, 10, 1164 CrossRef PubMed.
- Z. Huang, X. Chen, S. J. K. O'Neill, G. Wu, D. J. Whitaker, J. Li, J. A. McCune and O. A. Scherman, Highly compressible glass-like supramolecular polymer networks, Nat. Mater., 2022, 21, 103–109 CrossRef CAS.
- T. Aida, E. W. Meijer and S. I. Stupp, Functional Supramolecular Polymers, Science, 2012, 335, 813–817 CrossRef CAS.
- J. Sautaux, F. Marx, I. Gunkel, C. Weder and S. Schrettl, Mechanically robust supramolecular polymer co-assemblies, Nat. Commun., 2022, 13, 356 CrossRef CAS PubMed.
- C. Fouquey, J. Lehn and A. Levelut, Molecular recognition directed self–assembly of supramolecular liquid crystalline polymers from complementary chiral components, Adv. Mater., 1990, 2, 254–257 CrossRef CAS.
- J.-M. Lehn, Supramolecular Chemistry, Science, 1993, 260, 1762–1763 CrossRef CAS.
- S. Sivakova and S. J. Rowan, Nucleobases as supramolecular motifs, Chem. Soc. Rev., 2005, 34, 9–21 RSC.
- K. E. Feldman, M. J. Kade, E. W. Meijer, C. J. Hawker and E. J. Kramer, Phase Behavior of Complementary Multiply Hydrogen Bonded End-Functional Polymer Blends, Macromolecules, 2010, 43, 5121–5127 CrossRef CAS.
- G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, Complementary Quadruple Hydrogen Bonding in Supramolecular Copolymers, J. Am. Chem. Soc., 2005, 127, 810–811 CrossRef CAS.
- S. K. Chang and A. D. Hamilton, Molecular recognition of biologically interesting substrates: synthesis of an artificial receptor for barbiturates employing six hydrogen bonds, J. Am. Chem. Soc., 1988, 110, 1318–1319 CrossRef CAS.
- W. H. Binder, L. Petraru, T. Roth, P. W. Groh, V. Pálfi, S. Keki and B. Ivan, Magnetic and Temperature-Sensitive Release Gels from Supramolecular Polymers, Adv. Funct. Mater., 2007, 17, 1317–1326 CrossRef CAS.
- F. Herbst, S. Seiffert and W. H. Binder, Dynamic supramolecular poly(isobutylene)s for self-healing materials, Polym. Chem., 2012, 3, 3084–3092 RSC.
- S. Chen and W. H. Binder, Dynamic Ordering and Phase Segregation in Hydrogen-Bonded Polymers, Acc. Chem. Res., 2016, 49, 1409–1420 CrossRef CAS PubMed.
- L. Montero de Espinosa, S. Balog and C. Weder, Isophthalic Acid–Pyridine H-Bonding: Simplicity in the Design of Mechanically Robust Phase-Segregated Supramolecular Polymers, ACS Macro Lett., 2014, 3, 540–543 CrossRef CAS PubMed.
- A. Jangizehi, M. Ahmadi and S. Seiffert, Emergence, evidence, and effect of junction clustering in supramolecular polymer materials, Mater. Adv., 2021, 2, 1425–1453 RSC.
- C. B. Cooper and Z. Bao, Using Periodic Dynamic Polymers to Form Supramolecular Nanostructures, Acc. Mater. Res., 2022, 3, 948–959 CrossRef CAS.
- W. P. J. Appel, G. Portale, E. Wisse, P. Y. W. Dankers and E. W. Meijer, Aggregation of Ureido-Pyrimidinone Supramolecular Thermoplastic Elastomers into Nanofibers: A Kinetic Analysis, Macromolecules, 2011, 44, 6776–6784 CrossRef CAS.
- E. Croisier, S. Liang, T. Schweizer, S. Balog, M. Mionić, R. Snellings, J. Cugnoni, V. Michaud and H. Frauenrath, A toolbox of oligopeptide-modified polymers for tailored elastomers, Nat. Commun., 2014, 5, 4728 CrossRef CAS PubMed.
- J. Scavuzzo, S. Tomita, S. Cheng, H. Liu, M. Gao, J. P. Kennedy, S. Sakurai, S. Z. D. Cheng and L. Jia, Supramolecular Elastomers: Self-Assembling Star–Blocks of Soft Polyisobutylene and Hard Oligo(β-alanine) Segments, Macromolecules, 2015, 48, 1077–1086 CrossRef CAS.
- J. Roosma, T. Mes, P. Leclère, A. R. A. Palmans and E. W. Meijer, Supramolecular Materials from Benzene-1,3,5-tricarboxamide-Based Nanorods, J. Am. Chem. Soc., 2008, 130, 1120–1121 CrossRef CAS PubMed.
- S. Cantekin, T. F. A. de Greef and A. R. A. Palmans, Benzene-1,3,5-tricarboxamide : a versatile ordering moiety for supramolecular chemistry, Chem. Soc. Rev., 2012, 41, 6125–6137 RSC.
- L. Albertazzi, F. J. Martinez-Veracoechea, C. M. A. Leenders, I. K. Voets, D. Frenkel and E. W. Meijer, Spatiotemporal control and superselectivity in supramolecular polymers using multivalency, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 12203–12208 CrossRef CAS.
- Y. Song, Y. Liu, T. Qi and G. L. Li, Towards Dynamic but Supertough Healable Polymers through Biomimetic Hierarchical Hydrogen-Bonding Interactions, Angew. Chem., Int. Ed., 2018, 57, 13838–13842 CrossRef CAS.
- S. Pensec, N. Nouvel, A. Guilleman, C. Creton, F. Boué and L. Bouteiller, Self-Assembly in Solution of a Reversible Comb-Shaped Supramolecular Polymer, Macromolecules, 2010, 43, 2529–2534 CrossRef CAS.
- C. Véchambre, X. Callies, C. Fonteneau, G. Ducouret, S. Pensec, L. Bouteiller, C. Creton, J.-M. Chenal and L. Chazeau, Microstructure and Self-Assembly of Supramolecular Polymers Center-Functionalized with Strong Stickers, Macromolecules, 2015, 48, 8232–8239 CrossRef.
- J. Courtois, I. Baroudi, N. Nouvel, E. Degrandi, S. Pensec, G. Ducouret, C. Chanéac, L. Bouteiller and C. Creton, Supramolecular Soft Adhesive Materials, Adv. Funct. Mater., 2010, 20, 1803–1811 CrossRef CAS.
- M. M. J. Smulders, M. M. L. Nieuwenhuizen, M. Grossman, I. A. W. Filot, C. C. Lee, T. F. A. De Greef, A. P. H. J. Schenning, A. R. A. Palmans and E. W. Meijer, Cooperative Two-Component Self-Assembly of Mono- and Ditopic Monomers, Macromolecules, 2011, 44, 6581–6587 CrossRef CAS.
- H. Kautz, D. J. M. van Beek, R. P. Sijbesma and E. W. Meijer, Cooperative End-to-End and Lateral Hydrogen-Bonding Motifs in Supramolecular Thermoplastic Elastomers, Macromolecules, 2006, 39, 4265–4267 CrossRef CAS.
- D. J. M. van Beek, A. J. H. Spiering, G. W. M. Peters, K. te Nijenhuis and R. P. Sijbesma, Unidirectional Dimerization and Stacking of Ureidopyrimidinone End Groups in Polycaprolactone Supramolecular Polymers, Macromolecules, 2007, 40, 8464–8475 CrossRef CAS.
- M. M. L. Nieuwenhuizen, T. F. A. de Greef, R. L. J. van der Bruggen, J. M. J. Paulusse, W. P. J. Appel, M. M. J. Smulders, R. P. Sijbesma and E. W. Meijer, Self-Assembly of Ureido-Pyrimidinone Dimers into One-Dimensional Stacks by Lateral Hydrogen Bonding, Chem. – Eur. J., 2010, 16, 1601–1612 CrossRef CAS PubMed.
- M. Muthukumar, C. K. Ober and E. L. Thomas, Competing Interactions and Levels of Ordering in Self-Organizing Polymeric Materials, Science, 1997, 277, 1225–1232 CrossRef CAS.
- A. Levin, T. A. Hakala, L. Schnaider, G. J. L. Bernardes, E. Gazit and T. P. J. Knowles, Biomimetic peptide self-assembly for functional materials, Nat. Rev. Chem., 2020, 4, 615–634 CrossRef CAS.
- D. Görl, S. Haraguchi, Y. Hryshunin, S. Thiele, G. Scetta, A. Simula, M. Wendling, O. Oguz, N. Candau, T. Tänzer, M. Liebi, C. J. G. Plummer and H. Frauenrath, Supramolecular modification of sustainable high-molar-mass polymers for improved processing and performance, Nat. Commun., 2025, 16, 217 CrossRef.
- R. Guo, Q. Zhang, Y. Wu, H. Chen, Y. Liu, J. Wang, X. Duan, Q. Chen, Z. Ge and Y. Zhang, Extremely Strong and Tough Biodegradable Poly(urethane) Elastomers with Unprecedented Crack Tolerance via Hierarchical Hydrogen-Bonding Interactions, Adv. Mater., 2023, 35, 2212130 CrossRef CAS.
- S. Das, I. Yilgor, E. Yilgor, B. Inci, O. Tezgel, F. L. Beyer and G. L. Wilkes, Structure–property relationships and melt rheology of segmented, non-chain extended polyureas: Effect of soft segment molecular weight, Polymer, 2007, 48, 290–301 CrossRef CAS.
- J. C. Johnson, N. D. Wanasekara and L. T. J. Korley, Influence of secondary structure and hydrogen-bonding arrangement on the mechanical properties of peptidic-polyurea hybrids, J. Mater. Chem. B, 2014, 2, 2554–2561 RSC.
- L. E. Matolyak, J. K. Keum, K. M. V. de Voorde and L. T. J. Korley, Synthetic approach to tailored physical associations in peptide-polyurea/polyurethane hybrids, Org. Biomol. Chem., 2017, 15, 7607–7617 RSC.
- Z. Li, Y.-L. Zhu, W. Niu, X. Yang, Z. Jiang, Z.-Y. Lu, X. Liu and J. Sun, Healable and Recyclable Elastomers with Record-High Mechanical Robustness, Unprecedented Crack Tolerance, and Superhigh Elastic Restorability, Adv. Mater., 2021, 33, 2101498 CrossRef CAS.
- M. P. Lightfoot, F. S. Mair, R. G. Pritchard and J. E. Warren, New supramolecular packing motifs: π-stacked rods encased in triply-helical hydrogen bonded amide strands, Chem. Commun., 1999, 1945–1946 RSC.
- R. P. M. Lafleur, S. Herziger, S. M. C. Schoenmakers, A. D. A. Keizer, J. Jahzerah, B. N. S. Thota, L. Su, P. H. H. Bomans, N. A. J. M. Sommerdijk, A. R. A. Palmans, R. Haag, H. Friedrich, C. Böttcher and E. W. Meijer, Supramolecular Double Helices from Small C3-Symmetrical Molecules Aggregated in Water, J. Am. Chem. Soc., 2020, 142, 17644–17652 CrossRef CAS PubMed.
- S. Hafeez, M. C. Decarli, A. Aldana, M. Ebrahimi, F. A. A. Ruiter, H. Duimel, C. van Blitterswijk, L. M. Pitet, L. Moroni and M. B. Baker, In Situ Covalent Reinforcement of a Benzene-1,3,5-Tricarboxamide Supramolecular Polymer Enables Biomimetic, Tough, and Fibrous Hydrogels and Bioinks, Adv. Mater., 2023, 35, 2301242 CrossRef CAS.
- T. Mes, M. M. J. Smulders, A. R. A. Palmans and E. W. Meijer, Hydrogen-Bond Engineering in Supramolecular Polymers: Polarity Influence on the Self-Assembly of Benzene-1,3,5-tricarboxamides, Macromolecules, 2010, 43, 1981–1991 CrossRef CAS.
- X. Colin and J. Verdu, Polymer degradation during processing, C. R. Chim., 2006, 9, 1380–1395 CrossRef CAS.
- R. J. Young and P. A. Lovell, Introduction to Polymers, CRC Press, 2011 Search PubMed.
- R. S. Porter and J. F. Johnson, The Entanglement Concept in Polymer Systems, Chem. Rev., 1966, 66, 1–27 CrossRef.
- L. Chen, X. Sun, Y. Ren, R. Wang, M. Sun and W. Liang, Enhancing melt strength of polyglycolic acid by reactive extrusion with chain extenders, J. Appl. Polym. Sci., 2022, 139, 51796 CrossRef CAS.
- A. Y. Malkin, A. Arinstein and V. G. Kulichikhin, Polymer extension flows and instabilities, Prog. Polym. Sci., 2014, 39, 959–978 CrossRef CAS.
- M. Tang, R. Zhang, S. Li, J. Zeng, M. Luo, Y.-X. Xu and G. Huang, Towards a Supertough Thermoplastic Polyisoprene Elastomer Based on a Biomimic Strategy, Angew. Chem., Int. Ed., 2018, 57, 15836–15840 CrossRef CAS PubMed.
- M. Tang, R. Xu, R. Zhang, S.-J. Bai, G. Huang and Y.-X. Xu, Bioinspired strategy to tune viscoelastic response of thermoplastic polyisoprene by retarding the dissociation of hydrogen bonding, Polymer, 2021, 212, 123272 CrossRef CAS.
- E. Vereroudakis, M. Bantawa, R. P. M. Lafleur, D. Parisi, N. M. Matsumoto, J. W. Peeters, E. Del Gado, E. W. Meijer and D. Vlassopoulos, Competitive Supramolecular Associations Mediate the Viscoelasticity of Binary Hydrogels, ACS Cent. Sci., 2020, 6, 1401–1411 CrossRef CAS.
- S. Hafeez, F. R. Passanha, A. J. Feliciano, F. A. A. Ruiter, A. Malheiro, R. P. M. Lafleur, N. M. Matsumoto, C. Van Blitterswijk, L. Moroni, P. Wieringa, V. L. S. LaPointe and M. B. Baker, Modular mixing of benzene-1,3,5-tricarboxamide supramolecular hydrogelators allows tunable biomimetic hydrogels for control of cell aggregation in 3D, Biomater. Sci., 2022, 10, 4740–4755 RSC.
- C. F. C. Fitié, W. S. C. Roelofs, M. Kemerink and R. P. Sijbesma, Remnant Polarization in Thin Films from a Columnar Liquid Crystal, J. Am. Chem. Soc., 2010, 132, 6892–6893 CrossRef PubMed.
- I. Bala, H. Kaur, M. Maity, R. A. K. Yadav, J. De, S. P. Gupta, J.-H. Jou, U. K. Pandey and S. K. Pal, Electroluminescent Aggregation-Induced Emission-Active Discotic Liquid Crystals Based on Alkoxy Cyanostilbene-Functionalized Benzenetricarboxamide with Ambipolar Charge Transport, ACS Appl. Electron. Mater., 2022, 4, 1163–1174 CrossRef CAS.
- D. Ogata, T. Shikata and K. Hanabusa, Chiral Amplification of the Structure and Viscoelasticity of a Supramolecular Polymeric System Consisting of N,N‘,N”-Tris(3,7-Dimethyloctyl)Benzene-1,3,5-Tricarboxamide and n-Decane, J. Phys. Chem. B, 2004, 108, 15503–15510 CrossRef CAS.
- T. Shikata, D. Ogata and K. Hanabusa, Viscoelastic Behavior of Supramolecular Polymeric Systems Consisting of N,N‘,N”-Tris(3,7-dimethyloctyl)benzene-1,3,5-tricarboxamide and n-Alkanes, J. Phys. Chem. B, 2004, 108, 508–514 CrossRef CAS.
- S. Hafeez, A. Aldana, H. Duimel, F. A. A. Ruiter, M. C. Decarli, V. Lapointe, C. van Blitterswijk, L. Moroni and M. B. Baker, Molecular tuning of a benzene-1,3,5-tricarboxamide supramolecular fibrous hydrogel enables control over viscoelasticity and creates tunable ECM-mimetic hydrogels and bioinks, Adv. Mater., 2023, 35, 2207053 CrossRef CAS PubMed.
- S. Hafeez, H. W. Ooi, D. Suylen, H. Duimel, T. M. Hackeng, C. van Blitterswijk and M. B. Baker, Desymmetrization via Activated Esters Enables Rapid Synthesis of Multifunctional Benzene-1,3,5-tricarboxamides and Creation of Supramolecular Hydrogelators, J. Am. Chem. Soc., 2022, 144, 4057–4070 CrossRef CAS PubMed.
- M. Blomenhofer, S. Ganzleben, D. Hanft, H.-W. Schmidt, M. Kristiansen, P. Smith, K. Stoll, D. Mäder and K. Hoffmann, “Designer” Nucleating Agents for Polypropylene, Macromolecules, 2005, 38, 3688–3695 CrossRef CAS.
- F. Abraham, S. Ganzleben, D. Hanft, P. Smith and H.-W. Schmidt, Synthesis and Structure–Efficiency Relations of 1,3,5-Benzenetrisamides as Nucleating Agents and Clarifiers for Isotactic Poly(propylene), Macromol. Chem. Phys., 2010, 211, 171–181 CrossRef CAS.
- M. Kristiansen, P. Smith, H. Chanzy, C. Baerlocher, V. Gramlich, L. McCusker, T. Weber, P. Pattison, M. Blomenhofer and H.-W. Schmidt, Structural Aspects of 1,3,5-Benzenetrisamides−A New Family of Nucleating Agents, Cryst. Growth Des., 2009, 9, 2556–2558 CrossRef CAS.
- F. Abraham and H.-W. Schmidt, 1,3,5-Benzenetrisamide based nucleating agents for poly(vinylidene fluoride), Polymer, 2010, 51, 913–921 CrossRef CAS.
- F. Richter and H.-W. Schmidt, Supramolecular Nucleating Agents for Poly(butylene terephthalate) Based on 1,3,5-Benzenetrisamides, Macromol. Mater. Eng., 2013, 298, 190–200 CrossRef CAS.
- H. Bai, W. Zhang, H. Deng, Q. Zhang and Q. Fu, Control of Crystal Morphology in Poly(l-lactide) by Adding Nucleating Agent, Macromolecules, 2011, 44, 1233–1237 CrossRef CAS.
- H. Nakajima, M. Takahashi and Y. Kimura, Induced Crystallization of PLLA in the Presence of 1,3,5-Benzenetricarboxylamide Derivatives as Nucleators: Preparation of Haze-Free Crystalline PLLA Materials, Macromol. Mater. Eng., 2010, 295, 460–468 CrossRef CAS.
- H. Bai, C. Huang, H. Xiu, Q. Zhang, H. Deng, K. Wang, F. Chen and Q. Fu, Significantly Improving Oxygen Barrier Properties of Polylactide via Constructing Parallel-Aligned Shish-Kebab-Like Crystals with Well-Interlocked Boundaries, Biomacromolecules, 2014, 15, 1507–1514 CrossRef CAS PubMed.
- N. D. Treat, J. A. Nekuda Malik, O. Reid, L. Yu, C. G. Shuttle, G. Rumbles, C. J. Hawker, M. L. Chabinyc, P. Smith and N. Stingelin, Microstructure formation in molecular and polymer semiconductors assisted by nucleation agents, Nat. Mater., 2013, 12, 628–633 CrossRef CAS PubMed.
- EFSA Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids (CEF), Scientific Opinion on the safety evaluation of the substance, 1,3,5−tris(2,2−dimethylpropanamido)benzene, CAS No. 745070–61–5, for use in food contact materials, EFSA J., 2013, 11, 3306 Search PubMed.
- W. Wang, A. Saperdi, A. Dodero, M. Castellano, A. J. Müller, X. Dong, D. Wang and D. Cavallo, Crystallization of a Self-Assembling Nucleator in Poly(l -lactide) Melt, Cryst. Growth Des., 2021, 21, 5880–5888 CrossRef CAS PubMed.
- P. M. Kristiansen, A. Gress, P. Smith, D. Hanft and H.-W. Schmidt, Phase behavior, nucleation and optical properties of the binary system isotactic polypropylene/N,N′,N′′-tris-isopentyl-1,3,5-benzene-tricarboxamide, Polymer, 2006, 47, 249–253 CrossRef CAS.
- J. Gimenez, P. Cassagnau and A. Michel, Bulk polymerization of ε-caprolactone: Rheological predictive laws, J. Rheol., 2000, 44, 527–547 CrossRef CAS.
- M. A. Woodruff and D. W. Hutmacher, The return of a forgotten polymer—Polycaprolactone in the 21st century, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS.
- A. Timme, R. Kress, R. Q. Albuquerque and H.-W. Schmidt, Phase Behavior and Mesophase Structures of 1,3,5-Benzene- and 1,3,5-Cyclohexanetricarboxamides: Towards an Understanding of the Losing Order at the Transition into the Isotropic Phase, Chem. – Eur. J., 2012, 18, 8329–8339 CrossRef CAS PubMed.
- P. J. M. Stals, M. M. J. Smulders, R. Martín-Rapún, A. R. A. Palmans and E. W. Meijer, Asymmetrically Substituted Benzene-1,3,5-tricarboxamides: Self-Assembly and Odd–Even Effects in the Solid State and in Dilute Solution, Chem. – Eur. J., 2009, 15, 2071–2080 CrossRef CAS PubMed.
- T. S. Plivelic, S. N. Cassu, M. do Carmo Gonçalves and I. L. Torriani, Structure and Morphology of Poly(ε-caprolactone)/Chlorinated Polyethylene (PCL/PECl) Blends Investigated by DSC, Simultaneous SAXS/WAXD, and Elemental Mapping by ESI-TEM, Macromolecules, 2007, 40, 253–264 CrossRef CAS.
- M. Kawagoe, Y. Doi, N. Fuwa, T. Yasuda and K. Takata, Effects of absorbed water on the interfacial fracture between two layers of unsaturated polyester and glass, J. Mater. Sci., 2001, 36, 5161–5167 CrossRef CAS.
- L. Yuan, N. Hamidi, S. Smith, F. Clemons, A. Hamidi and C. Tang, Molecular characterization of biodegradable natural resin acid-substituted polycaprolactone, Eur. Polym. J., 2015, 62, 43–50 CrossRef CAS.
- M. Ahmadi, A. Jangizehi, K. Saalwächter and S. Seiffert, Effect of Junction Aggregation on the Dynamics of Supramolecular Polymers and Networks, Macromol. Chem. Phys., 2023, 224, 2200389 CrossRef CAS.
- V. Crescenzi, G. Manzini, G. Calzolari and C. Borri, Thermodynamics of fusion of poly-β-propiolactone and poly-ε-caprolactone. comparative analysis of the melting of aliphatic polylactone and polyester chains, Eur. Polym. J., 1972, 8, 449–463 CrossRef CAS.
- Y. Ming, Z. Zhou, T. Hao and Y. Nie, Molecular simulation of polymer crystallization under chain and space confinement, Phys. Chem. Chem. Phys., 2021, 23, 17382–17391 RSC.
- J. Li, A. T.-L. Lam, J. P. W. Toh, S. Reuveny, S. K.-W. Oh and W. R. Birch, Tunable Volumetric Density and Porous Structure of Spherical Poly-ε-caprolactone Microcarriers, as Applied in Human Mesenchymal Stem Cell Expansion, Langmuir, 2017, 33, 3068–3079 CrossRef CAS PubMed.
- M. A. Giffin, PhD thesis, EPFL, 2023.
- W. Ryu, L. Xiang, T. Jeon and M. Ree, Melt density, equilibrium melting temperature, and crystallization characteristics of highly pure cyclic poly(ε-Caprolactone)s, Polymer, 2020, 207, 122899 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5qo00087d |
| This journal is © the Partner Organisations 2025 |