
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ generated cobalt(I) catalyst for the efficient synthesis of novel pyridines: revisiting the mechanism of [2 + 2 + 2] cycloadditions†
Susana García-Abellán a,
Asier Urriolabeitiab,
Victor Polo*b and
Manuel Iglesias*a
a,
Asier Urriolabeitiab,
Victor Polo*b and
Manuel Iglesias*a
aInstituto de Síntesis Química y Catálisis Homogénea (ISQCH), CSIC-Universidad de Zaragoza, C/Pedro Cerbuna 12, 50009-Zaragoza, Spain. E-mail: miglesia@unizar.es
bInstituto de Biocomputación y Física de Sistemas Complejos (BIFI), Departamento de Química Física, Universidad de Zaragoza, 50009 Zaragoza, Spain. E-mail: vipolo@unizar.es
First published on 19th February 2025
Abstract
The [2 + 2 + 2] cycloaddition of alkynes and nitriles is an efficient and atom-economic method for the synthesis of pyridines. However, most of the examples so far reported entail the use of diynes, which circumvents selectivity issues but limits the scope of the reaction—with examples of discrete alkynes being scarce. Moreover, the most widely used catalysts are Co(I) complexes featuring Cp or Cp* ligands, which are either too unstable to store or require harsh conditions to promote the cycloaddition reaction. This work describes a mild method for the preparation of a wide range of pyridines employing a Co(I) active species generated in situ from a well-defined, air-stable Co(III) complex—namely, [CoCp*(CH3CN)(P–N)][BF4]2—upon treatment with NaBEt3H. This complex, which contains a hemilabile P–N ligand, has been found to be substantially more active than complexes featuring monodentate or bidentate phosphanes. This behavior has been ascribed to the inadequate stabilization of the resulting Co(I) species for the former, or overstabilization of the Co(III) complex in the case of the latter. A comprehensive DFT study has been conducted to elucidate the experimentally observed chemo- and regioselectivity by examining the competitive pathways following the oxidative coupling of CoCp*(bisalkyne) complex, taking under account the participation of triplet states and intersystem crossing points.
Introduction
Functionalized pyridines are ubiquitous motifs in materials,1 natural products and active pharmaceutical ingredients (APIs).2,3 Therefore, the development of methodologies for the construction of pyridine scaffolds, especially those that allow access to novel molecules, is a continuous endeavor. Among currently available synthetic methods for the preparation of pyridines,4 [2 + 2 + 2] cycloaddition reactions catalyzed by transition metal complexes have received much attention owing to the mild reaction conditions and high atom economy of the process. Rh,5–7 Ru,8–13 Ir,14,15 Ni,16–18 Fe19,20 and Co21–30 complexes have been reported to catalyze this reaction, with the latter arguably being the most successful so far. In particular, complexes based on the CpCo(I) scaffold (Cp = cyclopentadienyl) proficiently catalyze [2 + 2 + 2] cycloadditions, and are widely regarded as the prevailing type of catalysts for these transformations. However, these catalysts have drawbacks related to their synthesis and the balance between stability and activity. [CoCp(CO)2] is one of the most widely employed catalysts for cyclotrimerization reactions, but the generation of the active species requires high temperatures and irradiation.31 [CoCp(Me3SiC2H4)2], on the other hand, is the most active precatalyst for the cycloaddition of alkynes and nitriles, but its handling is difficult due to its low thermal stability, decomposing at temperatures above −30 °C.21,32 The exchange of one of the trimethylvinylsilane ligands by a phosphite has proved to enhance the stability of the catalyst, with complex [CoCp(Me3SiC2H4){P(OPh)3}] showing the best activity–stability compromise.22 Other remarkable examples of active precatalysts for this transformation are [CoCp(PPh3)2], [CoCp(COD)], [CoCp(CO)(dmfu)] (dmfu = dimethyl fumarate) and [CoCp(C2H4)2]. The latter has exhibited the highest activities of the series, but it necessitates meticulous handling under inert conditions and must be stored in an ethylene atmosphere to prevent decomposition. The related bis(olefin) complex [CoCp(COD)] (COD = 1,5-cyclooctadiene) is more stable and can be exposed to air for brief periods, but irradiation or elevated temperatures are needed to prompt good activities.33The overwhelming majority of these transformations involve intramolecular reactions that make use of α,ω-diynes5–32 (Scheme 1A) or cyanoalkynes34 (Scheme 1B) as starting materials; however, reports on the intermolecular cycloaddition of two discrete alkynes and a nitrile are scarce (Scheme 2). The dearth of examples on this topic is likely due to the challenging chemo- and regioselectivity of the reaction, which may render an intricate mixture of products, e.g., alkyne cyclotrimerization products,35 diazines (pyrimidines),36 or different isomeric pyridines.13,37 Wan et al. reported on an FeI2/dppp (1,3-bis(diphenylphosphino)propane) system reduced in situ with Zn that is able to efficiently catalyze the [2 + 2 + 2] cycloaddition of diynes and nitriles, but also proved to be active for the cycloaddition of benzylacetylene and acetonitrile (Scheme 2A).13 However, this is the only example of the intermolecular version of the reaction provided in this work. A NbCl5 catalyst reported by Obora et al. proved active for the intermolecular [2 + 2 + 2] cycloaddition of tert-butylacetylene and arylnitriles in the presence of over stoichiometric amounts of Zn and Ph2Si(OMe)2 as additive (Scheme 2B); however, significant amounts of trimerization product were obtained.35 Liu et al. described a gold catalyst, [AuClPPh3], that is capable of transforming nitriles and two discrete ynamides into diaminopyridines in the presence of catalytic amounts of AgNTf2 (Scheme 2C). Selectivity issues with the formation of pyrimidines as by-products were described for this system.36 The Co(I) catalyst [CoCp(Me3SiC2H4){P(OPh)3}] reported by Hapke et al.—proficient for the cycloaddition of diynes and nitriles—is able to catalyze the intermolecular cycloaddition of 3-hexyne and benzonitrile (Scheme 2D).22 A variety of Co(I) catalysts were explored by Ward et al. employing a combinatorial synthesis approach.24 Photochemical cobalt-catalyzed [2 + 2 + 2] cycloaddition of alkynes and nitriles under mild conditions was demonstrated by Heller and co-workers for a wide variety of nitriles (Scheme 2E).38 Fatland and Eaton achieved the thermal chemoselective synthesis of highly functionalized pyridines using a water-soluble Co(I) catalyst (Scheme 2F).39 Kinetic studies explained the absence of benzene by-products, suggesting that the rate-determining step involves associative coordination of the nitrile. Additionally, a double isotopic crossover experiment revealed that pyridine coordination to cobalt is resistant to displacement by alkyne but can be displaced by nitriles. Recently, Wang and co-workers synthesized pyridines using Fe(II)–Co(II) catalysts (Scheme 2G), where nitrile activation occurs through coordination to the Fe(II) complex.40 The proposed mechanism involves oxidative coupling of alkynes at the Cp*Co complex (Cp* = pentamethylcyclopentadienyl), followed by nitrile migratory insertion and reductive elimination. For terminal alkynes and nitriles, a mixture of two regioisomers was obtained, while high regioselectivity was observed for reactions with pivalonitrile and ethyl propiolate, highlighting the role of steric hindrance in governing regioselectivity. Similarly, Yoshikai and co-workers described a robust cobalt-diphosphine complex capable of catalyzing [2 + 2 + 2] cycloaddition of internal alkynes and unactivated nitriles (Scheme 2H).41 DFT calculations suggest that both (dppp)Co0 and (dppp)Co+ can promote oxidative coupling of alkynes, with the highest activation energy associated with this step.
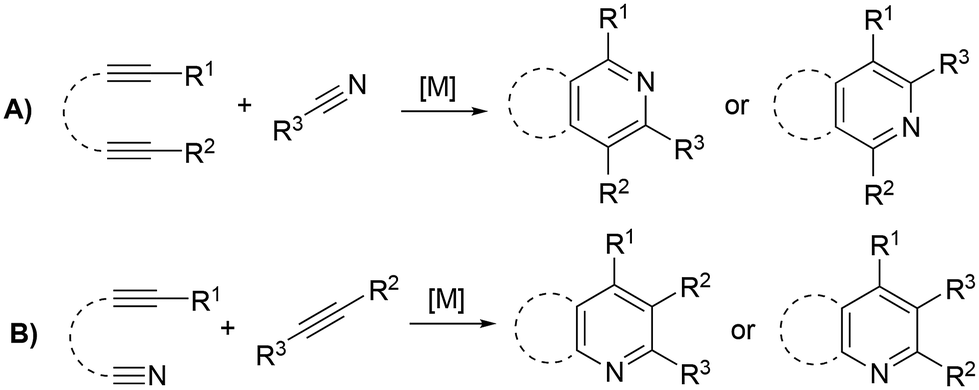 | ||
| Scheme 1 Intramolecular [2 + 2 + 2] cycloaddition reactions of alkynes and nitriles catalyzed by transition metal complexes. | ||
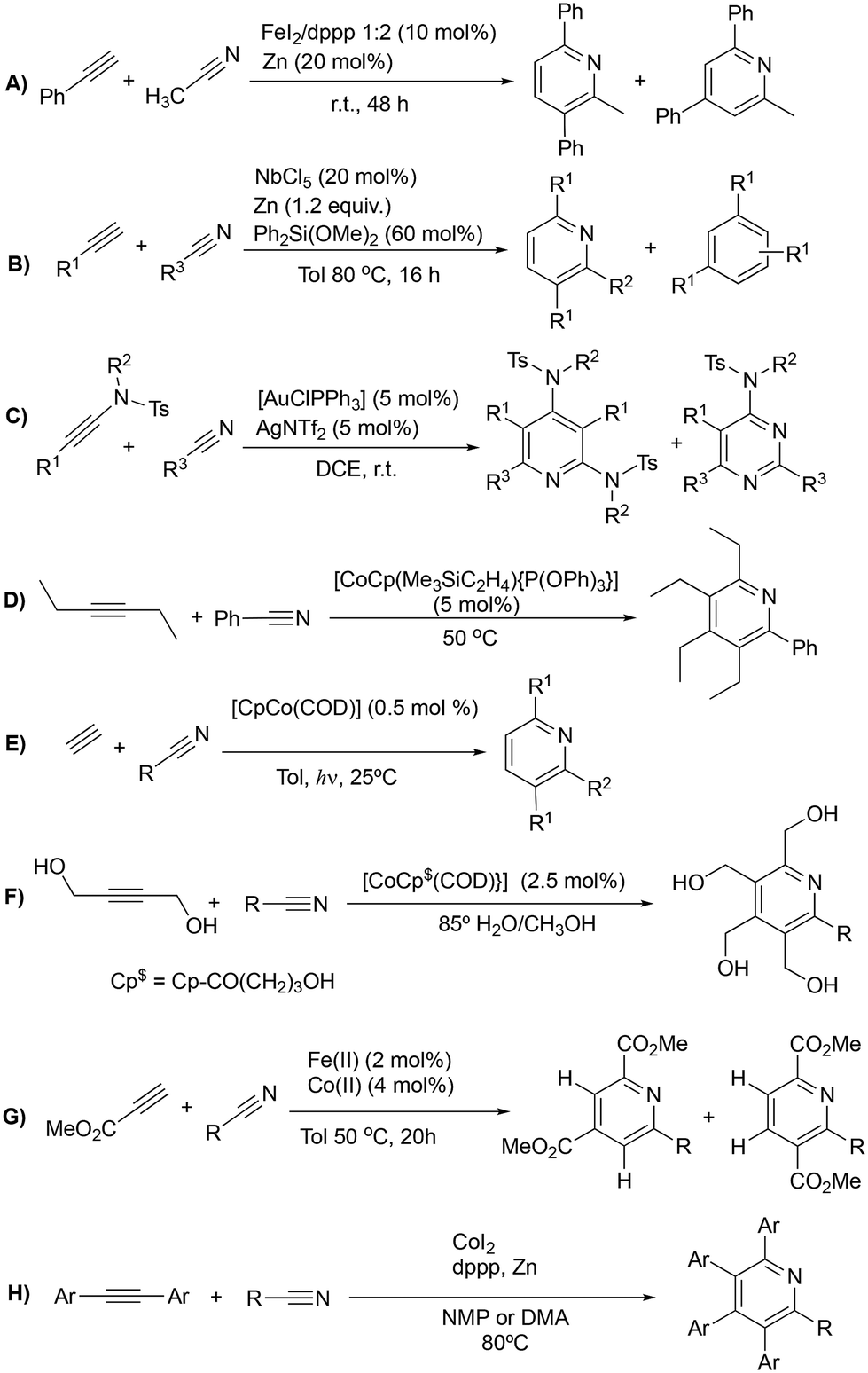 | ||
| Scheme 2 Intermolecular [2 + 2 + 2] cycloaddition reactions of alkynes and nitriles catalyzed by transition metal complexes. | ||
Other works for diynes and nitrile cyclotrimerization provide insights into the relationship between catalyst design and mechanistic insights. Hence, substituted chiral Cp ligands have been utilized in asymmetric cobalt-catalyzed cocyclotrimerization of various diynes and functionalized nitriles under photochemical conditions, achieving high yields and enantiomeric excesses of up to 94%.42 Regioselective 3- or 4-aminopyridines were achieved by Gandon and co-workers due to the substitution pattern at the yne-ynamide for a broad range of nitriles.23 More recently, the importance of cobalt ligands in selectivity has been further emphasized by Li and co-workers.43 Using an NPN*-cobalt complex, they achieved regio- and enantioselective synthesis of pyridines with all-carbon quaternary centers. The regioselectivity towards pyridine formation is primarily influenced by the inherent nature of terminal alkynes and the presence of bulky substituents on the metal ligand. Finally, early work by Diversi et al. describes the catalytic activity of Rh(I) and Co(I) systems, featuring Cp and Cp* ligands, for the synthesis of pyridines from L-alkynes and nitriles.37,44
These systems show that higher ratios of the least sterically hindered pyridines and lower amounts of benzene by-products were obtained with Cp* ligands.
The origin of the chemo- and regioselectivity in [2 + 2 + 2] cycloadditions is difficult to assess, as it is governed by several competitive pathways that are influenced by subtle electronic and steric variations within the system.45–47 In this work, we present a novel procedure for the in situ generation of Cp*Co(I) species capable of efficiently catalyzing the intermolecular [2 + 2 + 2] cycloaddition of a variety of discrete alkynes and nitriles, which has allowed for the isolation and characterization of a range of new functionalized pyridines. The active species was prepared via reduction of the parent Cp*Co(III) complex with 2 equivalents of NaBEt3H in toluene. The Co(III) precatalyst is a well-defined diamagnetic complex that can be straightforwardly characterized by NMR, and, more importantly, it is air-stable and can be stored indefinitely. Moreover, in order to address the observed chemoselectivity and stereoselectivity, stoichiometric experiments and a comprehensive DFT study have been performed to elucidate the reaction mechanism and the role of competitive pathways.
Results and discussion
Study of the catalytic activity of Co(III) precursors
The activity of Co(III) precatalyst 1, [CoCp*(CH3CN)(P–N)][BF4]2 (P–N = 1-((diphenylphosphaneyl)methyl)-1H-benzo-1,2,3-triazole), was explored in the [2 + 2 + 2] cycloaddition reaction of alkynes and nitriles. This complex, previously reported by us,48 presents a phosphane equipped with a triazole moiety that is capable of acting as a hemilabile ligand. The formation of the Co(I) active species was achieved by in situ reduction of 1 with 2 equivalents of NaBEt3H.The initial evaluation of the catalytic activity was conducted using a catalyst loading of 5 mol%, 10 mol% of the reducing agent (NaBEt3H), and benzylacetylene and acetonitrile as substrates in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Scheme 3). The reaction, carried out at 80 °C in C6D6, resulted in quantitative conversion after 72 h. It is noteworthy that the formation of two isomeric species, 2,4-dibenzyl-6-methylpyridine (py-1a) and 3,6-dibenzyl-2-methylpyridine (py-1b), in a 2:1 ratio (Scheme 3), respectively, was observed, which is consistent with the only example found in the literature.24 The exclusive formation of these two products against the array of combinations that could potentially be obtained considering an intermolecular cycloaddition of discrete alkynes and nitriles will be further analyzed later alongside the proposed reaction mechanism.
:1 ratio (Scheme 3). The reaction, carried out at 80 °C in C6D6, resulted in quantitative conversion after 72 h. It is noteworthy that the formation of two isomeric species, 2,4-dibenzyl-6-methylpyridine (py-1a) and 3,6-dibenzyl-2-methylpyridine (py-1b), in a 2:1 ratio (Scheme 3), respectively, was observed, which is consistent with the only example found in the literature.24 The exclusive formation of these two products against the array of combinations that could potentially be obtained considering an intermolecular cycloaddition of discrete alkynes and nitriles will be further analyzed later alongside the proposed reaction mechanism.
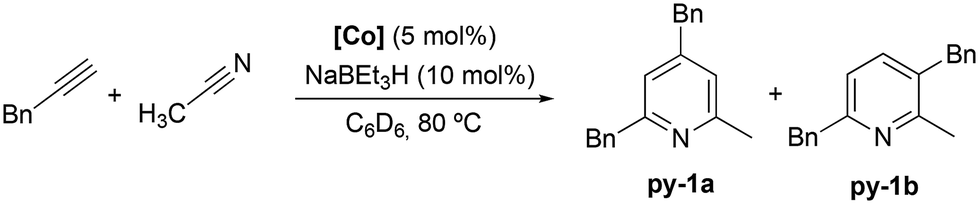 | ||
| Scheme 3 Intermolecular [2 + 2 + 2] cycloaddition reaction catalyzed by 1. | ||
To confirm the need for the cobalt precatalyst and to rule out that the catalytic activity was solely due to the presence of NaBEt3H, a control experiment was performed using conditions analogous to those described earlier, but in the absence of cobalt precatalyst. Under these conditions, no formation of cycloaddition products was observed.
Similarly, to assess the need for the reduction of 1 to the Co(I) active species, the reaction was performed in the presence of the precatalyst but without NaBEt3H. Under such conditions, no reactivity was observed.
With the aim of evaluating the importance of the presence of the triazol group, other CoIIICp* catalytic precursors were studied (Table 1), specifically, [CoCp*(CH3CN)3][BF4]2 (2), [CoCp*(CH3CN)2(PMePh2)][BF4]2 (3), and [CoCp*(CH3CN)(dppe)][BF4]2 (4; dppe = 1,2-bis(diphenylphosphino)ethane). The conversions observed after 24 and 72 h with 2 and 3 (entries 2 and 3, Table 1) were significantly lower than those obtained with 1. These low conversion values can be attributed to the reduced stability of the Co(I) species formed in the absence of a stabilizing bidentate ligand. Conversely, the use of 4 as precatalyst leads to the lowest conversions, showing barely any catalytic activity under these conditions (entry 4, Table 1). This behavior is plausibly a consequence of the strong bidentate coordination of the dppe ligand in the Co(III) precatalyst, which hinders its reduction to Co(I).
| Entry | Catalytic precursor | Conversion (%) | |
|---|---|---|---|
| 24 h | 72 h | ||
| Reaction conditions: 0.15 mmol de benzylacetylene, 0.15 mmol of CH3CN, 10 mol% of NaBEt3H, in C6D6 at 80 °C. Conversions calculated by integration of representative resonances in the 1H NMR spectra. | |||
| 1 | 1 | 70 | 99 |
| 2 | 2 | 17 | 20 |
| 3 | 3 | 11 | 20 |
| 4 | 4 | 0 | 6 |
It is noteworthy that the selectivity of the reaction was not modified by the use of different precatalysts, resulting in all cases in a mixture of py-1a/py-1b in a 2:1 ratio, which suggests an analogous reaction mechanism for all of them.
In situ generation of the active CoICp* species
Experimentally, we observe that the addition of NaBEt3H to a red suspension of 1 in C6D6 results in immediate gas evolution and the formation of a blue solution. Conversely, in the case of 2 and 3, the reaction with NaBEt3H leads to the formation of a brown suspension. The reduction of 4, in contrast, is very slow, and barely any changes or gas evolution are observed. This behavior agrees with the relative activity described above for the different Co(III) catalyst precursors, suggesting that the lower activities observed for 2 and 3 are a result of the partial decomposition of the catalyst, while the overstabilization resulting from the presence of the bidentate phosphane ligand in 4 leads to a sluggish reduction process.To shed light on the catalyst activation process, stoichiometric experiments were carried out with 1, replicating the in situ reduction conditions used in catalysis, monitoring its evolution by 1H, 1H{31P}, and 31P{1H} NMR spectroscopy (Scheme 4).
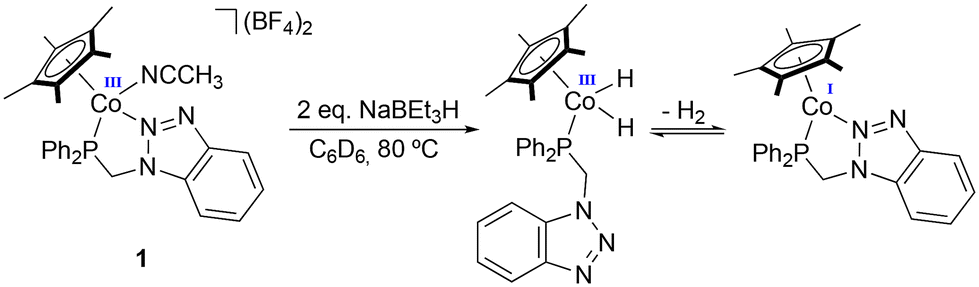 | ||
| Scheme 4 In situ reduction of 1 and proposed active species. | ||
NaBEt3H was added to a suspension of 1 in C6D6 in a Young NMR tube under an inert atmosphere. The 1H NMR spectrum of the reaction mixture shows the appearance of a doublet at δ −16.6 ppm, corresponding to the formation of a dihydride complex, plausibly [CoCp*(H)2(P–N)], with a coupling constant of 83.6 Hz with the phosphorus nucleus (Fig. S69a†). In the 1H{31P} NMR spectrum, this resonance appears as a singlet (Fig. S69b†), confirming the coupling of the hydrides with the phosphorus nucleus of P–N. The shift and coupling constants are consistent with those described by Brookhart et al. for the related complex [CoCp*(H)2(PMe3)], showing a doublet for the hydride ligands at δ −18.09 ppm with a 2JP–H of 89 Hz. In the 1H NMR spectra in C6D6, a singlet at δ 4.47 ppm due to the formation of free H2 is observed, which agrees with the rapid bubbling observed after the addition of NaBEt3H (Fig. S70†). In the 31P{1H} NMR spectrum, the signal initially observed for 1 at δ 62.1 ppm shifts to low field, appearing as a broad signal at δ 81.1 ppm (Fig. S71†).
Upon release of H2 from the NMR tube, both the H2 signal and the doublet corresponding to the hydrides disappear in the 1H NMR spectra. Additionally, a low-intensity broad doublet emerges at δ −6.48 ppm with a coupling constant H–P of 21.6 Hz (Fig. S72†). Both δ and J values are consistent with those of a dihydrogen ligand coordinated to Co(I). This suggests that both species observed in solution are in equilibrium, shifting towards the formation of the Co(I) complex in an open system. In the 1H NMR spectrum, the major methylene signal appears as a doublet at δ 3.39 ppm—which becomes a singlet in the 1H{31P} spectrum. This signal was present before H2 release, but its intensity increases after purging with Ar (Fig. S73†). This resonance presents a 2JH–P of 6.4 Hz, and its integration is consistent with those of the most intense aromatic signals and the one associated with the Cp* methyl groups at δ 1.85 ppm. In this case, the methylene protons do not show diastereotopic nature despite P–N being bidentately coordinated, since this species is a Co(I) pentacoordinated complex with C2v symmetry.
These experiments suggest that, after the addition of NaBEt3H to 1, the dissociation of the triazol group occurs followed by the coordination of two hydrides to the metal center to form the dihydride complex [CoCp*(H)2{κ1-P-(P–N)}]. This complex would be in equilibrium with the Co(I) species, [CoCp*{κ2-P,N-(P–N)}], formed after the release of H2 via reductive elimination. In this pentacoordinated species, the ligand is likely to coordinate in a bidentate manner to occupy the coordination vacancy generated in the process. Albeit to a lesser extent, the formation of intermediate species [CoCp*(η2-H2){κ1-P-(P–N)}] is also detected, where P–N would be coordinated κ1-P, which is consistent with the hemilabile nature of the ligand.
Optimization of the reaction conditions
In view of the results described above, the optimization of reaction conditions was carried out using 1 as the catalytic precursor. Firstly, the amount of nitrile present in the reaction medium was optimized by exploring the impact of using 3 and 6 equivalents of acetonitrile with respect to the alkyne, instead of 1 (Table 2). The results showed a significant improvement with the use of 3 equivalents of acetonitrile, achieving quantitative conversion after 24 hours. Conversely, the use of 6 equivalents showed poorer conversions at initial times. At longer times, the activity improved in both cases compared to that obtained with 1 equivalent, with both reactions finishing after 72 hours. Henceforth, 3 equivalents of nitrile with respect to the alkyne were used.| Entry | CH3CN | Conversion (%) | ||||
|---|---|---|---|---|---|---|
| 1 h | 2 h | 24 h | 48 h | 72 h | ||
| Reaction conditions: 0.15 mmol of benzylacetylene, 10 mol% of NaBEt3H in C6D6 at 80 °C. Conversions calculated by integration of the 1H NMR spectrum. | ||||||
| 1 | 1 equiv. | 43 | 49 | 70 | 88 | 99 |
| 2 | 3 equiv. | 52 | 60 | 99 | — | — |
| 3 | 6 equiv. | 23 | 38 | 86 | 90 | 97 |
Next, we carried out the optimization of the reaction temperature, evaluating the catalyst activity at 80, 100, and 120 °C (Table 3). After 24 hours at 80 and 100 °C, similar conversions were obtained, 97 and 99%, respectively. However, at shorter reaction times, the conversion is significantly higher at 100 °C. At 120 °C, the activity undergoes a significant decrease compared to 80 and 100 °C, conceivably due to catalyst instability at elevated temperatures.
:3 ratio catalyzed by 1 (5 mol%) at different reaction temperatures
| Entry | Temperature (°C) | Conversion (%) | |||
|---|---|---|---|---|---|
| 1 h | 2 h | 5 h | 24 h | ||
| Reaction conditions: 0.15 mmol of benzylacetylene, 0.45 mmol de acetonitrile, 10 mol% of NaBEt3H in C6D6 at 80, 100 and 120 °C. Conversions calculated by integration of the 1H NMR spectrum. | |||||
| 1 | 80 | 52 | 60 | 79 | 97 |
| 2 | 100 | 62 | 71 | 88 | 99 |
| 3 | 120 | 27 | 38 | 54 | 67 |
Under the optimized reaction conditions (0.15 mmol of benzylacetylene, 0.45 mmol of acetonitrile, and 10 mol% of NaBEt3H in C6D6 at 100 °C), the same selectivity described above was observed for the isomers py-1a and py-1b.
Evaluation of the substrate scope
The catalytic activity of 1 was explored using various families of alkynes and nitriles as substrates under optimized reaction conditions.Initially, while maintaining the use of acetonitrile, a variety of terminal alkynes were studied (Scheme 5 and Table 4). Using benzylacetylene, 1-hexyne, and cyclopropylacetylene, quantitative conversions were achieved in less than 24 hours (entries 1, 3, and 4, Table 4). However, the use of methylpropargyl ether (entry 5, Table 4), also an aliphatic alkyne, led to a significantly lower conversion (64%). The use of an aromatic alkyne such as 4-tolylacetylene (entry 2, Table 4) resulted in an even lower conversion (54%). This lower conversion can be ascribed to steric hindrance, which may hamper coordination. It is worth mentioning that the isomer ratio does not vary significantly upon modification of the alkyne.
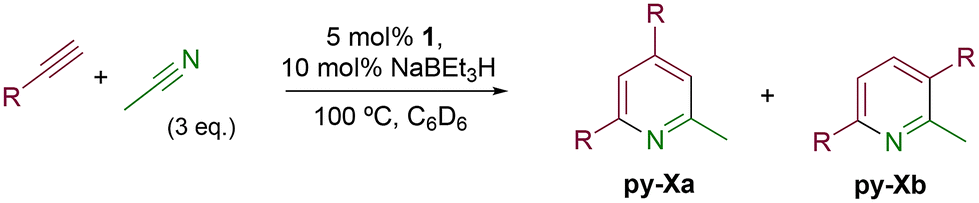 | ||
| Scheme 5 Intermolecular [2 + 2 + 2] cycloaddition reaction with different alkynes (X = 1–5). | ||
| Entry | Alkyne | Conv % (h) | py-Xa: py-Xb ratio |
Yield py-Xa + py-Xb (%) | Products |
|---|---|---|---|---|---|
| Reaction conditions: 0.15 mmol of alkyne, 0.45 mmol of acetonitrile, 10 mol% of NaBEt3H, and 5 mol% of 1 in C6D6 at 100 °C. Conversions calculated by integration of the 1H NMR spectrum using 1,3,5-triazine as an internal standard. | |||||
| 1 | 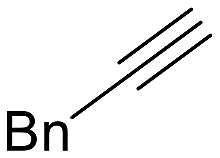 |
>99 (24) | 2:1 |
93 | py-1a: py-1b |
| 2 | 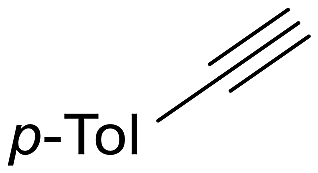 |
54 (24) | 2:1 |
44 | py-2a: py-2b |
| 3 | 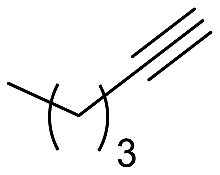 |
>99 (24) | 2:1 |
90 | py-3a: py-3b |
| 4 |  |
>99 (5) | 1.5:1 |
90 | py-4a: py-4b |
| 5 | 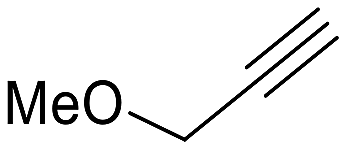 |
64 (24) | 2:1 |
58 | py-5a: py-5b |
Next, the use of different aromatic and aliphatic nitriles was studied while maintaining benzylacetylene as the substrate (Scheme 6 and Table 5).
 | ||
| Scheme 6 Intermolecular [2 + 2 + 2] cycloaddition reaction with different nitriles (X = 6–9). | ||
| Entry | Nitrile | Conv % (h) | py-Xa:py-Xb ratio |
Yield py-Xa + py-Xb (%) | Products |
|---|---|---|---|---|---|
| Reaction conditions: 0.15 mmol of alkyne, 0.45 mmol of acetonitrile, 10 mol% of NaBEt3H, and 5 mol% of 1 in C6D6 at 100 °C. Conversions calculated by integration of the 1H NMR spectrum using 1,3,5-triazine as an internal standard. | |||||
| 1 | 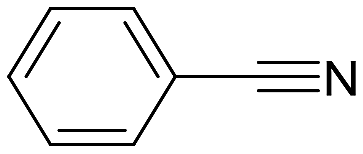 |
>99 (2) | 2:1 |
95 | py-6a: py-6b |
| 2 | 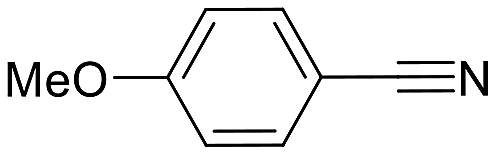 |
95 (24) | 2:1 |
87 | py-7a: py-7b |
| 3 | 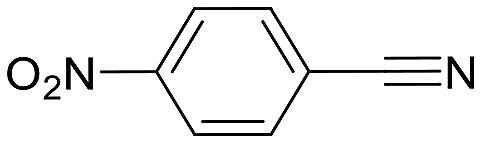 |
0 (24) | — | — | — |
| 4 |  |
0 (24) | — | — | — |
| 5 | 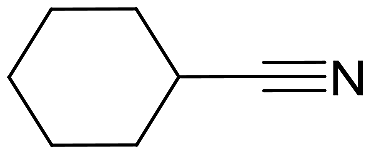 |
96 (5) | 2:1 |
90 | py-8a: py-8b |
| 6 | 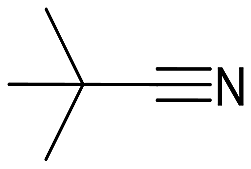 |
44 (24) | 1:0 |
35 | py-9a: py-9b |
The reaction with benzonitrile results in the formation of the corresponding pyridines quantitatively in 2 hours (entry 1, Table 5). The use of an electron-donating group, as in the case of 4-methoxybenzonitrile (entry 2, Table 5), leads to a noticeable change in activity, requiring 24 h to achieve a 95% conversion. On the other hand, the presence of an electron-withdrawing group (4-nitrobenzonitrile) completely inhibits the activity (entry 3, Table 5), likely due to a weaker interaction with the metal center.
The use of 2-(pyridin-2-yl)acetonitrile also resulted in no detectable product formation (entry 4, Table 5), plausibly as a result of a lower access to vacant coordination sites due to substrate coordination.
Cyclohexanecarbonitrile is converted into the corresponding pyridines in almost quantitative conversions in less than 5 h (entry 5, Table 5). Trimethylacetonitrile (entry 6, Table 5), in contrast to cyclohexanecarbonitrile (also an aliphatic nitrile), led to the formation of a single isomer, 2,4-dibenzyl-6-(tert-butyl)pyridine (py-9a), although in lower yield. This selectivity may be due to the steric hindrance between the tert-butyl and benzyl groups, which would prevent the formation of 3,6-dibenzyl-2-(tert-butyl)pyridine (py-9b). 1 also exhibited catalytic activity with internal alkynes; namely, 3-hexyne, yielding a single reaction product (Table 6). The yields were moderate, around 50% after 24 h, both with acetonitrile and benzonitrile.
:3 ratio, catalyzed by 1 (5 mol%)
| Entry | Alkyne | Nitrile | Conversion % (h) | Yield (%) |
|---|---|---|---|---|
| Reaction conditions: 0.15 mmol of 3-hexyne, 0.45 mmol of nitrile, 10 mol% of NaBEt3H in C6D6 at 100 °C. Conversions calculated by integration of the 1H NMR spectrum using 1,3,5-triazine as internal standard. | ||||
| 1 | 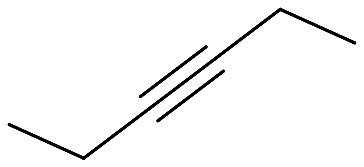 |
 |
53 (24) | 52 |
| 2 |  |
 |
44 (24) | 42 |
Mechanistic considerations
The chemoselectivity of the reaction is remarkable, as no products resulting from alkyne cyclotrimerization or diazine formation are observed in the reaction crude. Furthermore, regarding the regioselectivity of the reaction, only two isomeric pyridines are obtained as products (py-Xa and py-Xb), despite the fact that the [2 + 2 + 2] cycloaddition reaction of two discrete terminal alkynes and a nitrile could potentially lead to the formation of 4 structural isomers (see Scheme 7).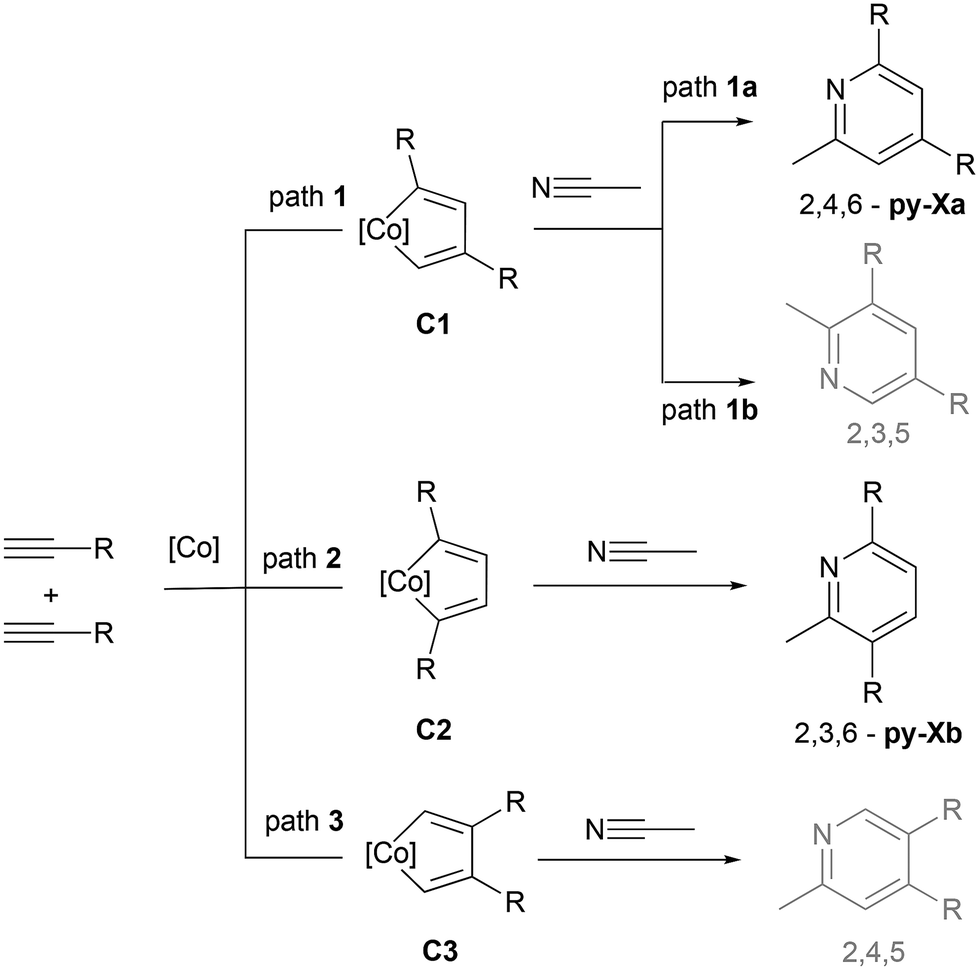 | ||
| Scheme 7 Possible isomeric pyridines obtained from the [2 + 2 + 2] cycloaddition reaction of two discrete terminal alkynes and acetonitrile. Unobserved 2,3,5- and 2,4,5-pyridines are marked in grey. | ||
For precatalyst 1, the selectivity is maintained even after variations in temperature or changes to the nitrile and alkyne substituents, typically yielding a mixture of isomers py-Xa and py-Xb in a 2:1 ratio. Additionally, experiments were carried out to shed light on key points of the reaction mechanism, such as the participation of triplet species or the role of the P–N ligand.
To investigate the involvement of triplet species in the reaction mechanism, radical quenching experiments using (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) were conducted. The standard catalytic test, involving benzylacetylene and acetonitrile in a 1:3 ratio (Table 4, entry 1) in the presence of 1 equivalent of TEMPO, resulted in a significant reduction in catalytic activity, with the conversion decreasing from quantitative to 19%. This suggests that TEMPO reacts with the triplet species of the catalytic cycle, inhibiting the catalytic activity. The impact of P–N dissociation during the catalytic cycle was evaluated, using the cycloaddition of benzylacetylene and acetonitrile (Table 4, entry 1) as a test reaction, adding an excess of P–N (25 mol%). This results in a significant decrease in conversion, from quantitative to 37%, which can be ascribed to the increased difficulty in generating the necessary coordination vacancy.
DFT theoretical studies based on experimental evidences of [2 + 2 + 2] cobalt-catalyzed cycloadditions have been carried out to clarify the reaction mechanism. Early computational studies on trimerization of acetylenes49,50 and nitriles42 were carried out, but they only considered closed-shell species. The role of two-state reactivity (TSR) processes was pointed out by Koga et al.52 for acetylene trimerization to yield benzene. The importance of considering parallel reaction pathways arising at the CoCp metallacyclodiene intermediate was highlighted by Aubert et al.,53 concluding that benzene is formed from triplet cobaltacyclodiene intermediate after a spin-state change and subsequent alkyne ligation, although it can be trapped by σ-donor coordinating ligands of the appropriate size. In the case of cycloaddition of alkynes and nitriles to yield pyridines, the number of possible competitive reaction pathways increase after formation of the CoCp cyclometalladiene intermediate. Hence, it was found for 2-methylpyridine that internal [4 + 2] cycloaddition is the preferred pathway over migratory insertion into the metallacycle and external [4 + 2] cycloaddition for acetylene and acetonitrile.54 However, depending on the substituents, insertion of the nitrile on the cobaltacyclodiene may occur following the migratory insertion pathway.23 Based on previous studies on model systems, the chemoselectivity of the reaction cannot be explained invoking this mechanism, as the insertion of an alkyne into the metallacycle is more favorable than that of the nitrile according to the theoretical calculations hitherto reported52 and a thorough study of alkyne and nitrile competing pathways is required.
Early computational studies using semiempirical methods on stereoselectivity in cyclocobaltadiene formation, via the oxidative addition of substituted alkynes, revealed a preference for α-carbon positions within the metallacycle, driven by the steric effects of bulky substituents.55 Electronic effects appear to be less significant, with electron-donating substituents favoring the β-position of the metallacycle due to the larger orbital lobe at the substituted carbon atom.56 Based on these studies, steric hindrance favors the formation of intermediate C2, leading to 2,3,6-pyridine (py-Xb), while electronic effects should promote path 3, leading to the unobserved 2,4,5-pyridine. A systematic DFT study of the regioselectivity of CoCp(bisacetylene) showed the preference for 1,4-coupling for methyl substituents44,57 (path 2 in Scheme 7).
In order to understand the observed chemoselectivity and regioselectivity, a thorough DFT study considering all possible reaction pathways for the full cycle of CoCp*(P–N) catalyzed [2 + 2 + 2] cycloaddition of benzylacetylene and acetonitrile, depicted in Scheme 8, has been conducted.
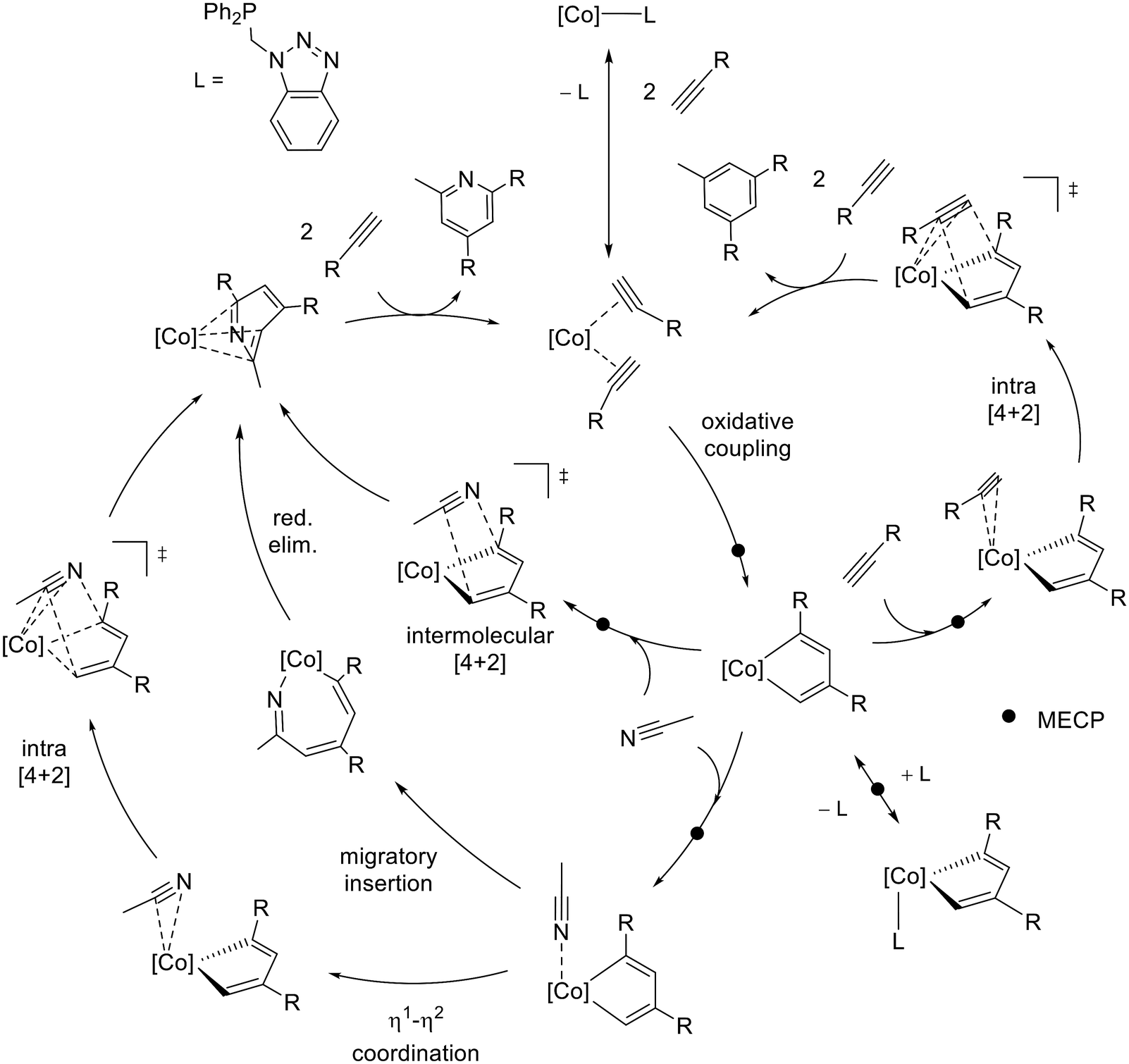 | ||
| Scheme 8 Catalytic cycle proposed for the [2 + 2 + 2] cycloaddition of benzylacetylene (R = –CH2–Ph) and acetonitrile catalyzed by CoCp*(P–N) leading to 1,3,5-trisubstituted benzene and 2,4,6-trisubstituted pyridine. | ||
DFT Study on the catalytic cycle
According to previous studies,49 the initial step in the [2 + 2 + 2] cycloaddition process catalyzed by the CoCp(PH3)2 complex involves the ligand exchange of phosphanes by alkynes, followed by an oxidative coupling step that produces a cobaltacyclediene intermediate.51In order to discard other possible pathways, the oxidative coupling between two benzylacetylene molecules enabled by the [CoCp*{κ1-P-(P–N)}] intermediate was considered (see Fig. S74†), concluding that the activation energies for this process were higher and full dissociation of the P–N ligand is required.
The DFT-calculated free energy profile for the full catalytic cycle shown in Scheme 8 is presented in Fig. 1 and 2. Fig. 1 considers precatalyst activation and the oxidative coupling step leading to the formation of the CoCp*-cyclopentadiene intermediate, while Fig. 2 outlines the possible competing pathways that yield trisubstituted pyridine and benzyne products. The catalytic cycle starts with the formation of CoCp*(bisalkyne) via stepwise dissociation of P–N from the metal to afford intermediate B, an exergonic process by 13.2 kcal mol−1 (see Fig. 1). According to Scheme 7, regioselective oxidative coupling of two mono-substituted alkynes can lead to regioisomers C1, C2 and C3 due to head-to-tail (1,3-), head-to-head (1,4-) and tail-to-tail (2,3-) coupling. The corresponding transition states are TSBC1, TSBC2, and TSBC3, presenting energetic barriers of 28.3 kcal mol−1, 29.4 kcal mol−1, and 34.6 kcal mol−1, respectively. In order to understand the lower activation energy for the 1,3-coupling, non-covalent interactions have been analyzed at the transition states using NCI methodology (see Fig. S75†).
 | ||
| Fig. 1 DFT calculated Gibbs free energy profile (in kcal mol−1) for the ligand exchange and oxidative coupling of bis(benzylacetylene) catalyzed by complex CoCp*(P–N) (A). Regioselective pathways are color-coded as follows: black for 1,3-coupling, blue for 1,4-coupling, and red for 2,4-coupling. | ||
 | ||
| Fig. 2 DFT calculated Gibbs energy profile (in kcal mol−1, energies relative to A and isolated species) for reaction of CoCp*cyclodienes (C1 and 3C1) and benzylacetylene (-A path, orange line), acetonitrile (-N path, black line) and P–N (-P path, violet line). | ||
NCI plots reveal that steric repulsion between groups attached to the carbon atoms forming the C–C bond is significant for 2,3-coupling, but almost negligible for 1,4- and 1,3-coupling. Interestingly, the 1,3-coupling provides greater conformational flexibility to the –CH2–Ph groups of both benzylacetylenes, allowing favorable π-methyl interactions between the terminal phenyl groups and the methyl groups of the Cp* ligand. In contrast, the 1,4-coupling does not permit such interactions.
Unsaturated Co(III)Cp*-cyclopentadiene intermediates C1, C2, and C3 present unstable singlet electronic wavefunctions with respect to their corresponding triplet state species 3C1, 3C2 and 3C3 at 2.9, 2.6 and 0.7 kcal mol−1, respectively. The geometries of singlet and triplet species differ: singlet cobaltacyclodienes exhibit a puckered conformation of the five-membered ring, whereas the triplet state displays a planar arrangement. Singlet–triplet minimum energy crossing points (MECP), CP(C1), CP(C2), and CP(C3), were identified within an accessible energy range, allowing for efficient surface hopping between electronic states. According to the DFT energies presented in Fig. 1, the catalyst induces kinetic control of the regioselectivity in the bis-alkyne coupling step, favoring the formation of C1 as the major intermediate.
Since the predominant product observed experimentally requires head-to-tail coupling of two alkyne molecules, only the pathway involving the C1 regioisomer will be discussed in the following section. Starting at C1 and 3C1 intermediates, the reaction proceeds through coordination of one of three different molecules to the metal (see Fig. 2): acetonitrile (-N path), benzylacetylene (-A path), or the P–N ligand previously dissociated from A (-L path). Acetonitrile and benzylacetylene are present in high concentrations, whereas P–N is found at a significantly lower concentration. Transition states were found for the coordination of benzylacetylene, acetonitrile, and phosphane to the metal vacancy at singlet C1. These early transition states correspond to the geometric rearrangement of the metallic complex from a puckered to a planar conformation, while the coordinated molecule interacts weakly with the metal center. These transition states lead to the formation of coordinatively saturated Co(III) complexes which are exergonic, D1-A −7.5 kcal mol−1, D1-N −9.8 kcal mol−1, and D1-L −27.1 kcal mol−1. As the corresponding triplet states for the D1 intermediates are significantly higher in energy than the singlet states, a surface hopping process between the 3C1 and D1 intermediates may occur.
Intersystem crossing points were found for the three competitive pathways, presenting relative Gibbs free energies of 11.1 kcal mol−1 for CP(D1-A), 6.9 kcal mol−1 for CP(D1-N), and 0.9 kcal mol−1 for CP(D1-P), respectively. The energies of MECPs are lower than the transition states found for singlet states, thus we can assume that the reaction proceeds through 3C1 → CP(D1) → D1. The lower value of CP(D1-P) indicates that the most favorable pathway goes through the phosphane σ-coordination to the metal. Previous studies49,53,58 showed that bulky phosphanes actively catalyze alkyne cyclotrimerization, while compact phosphanes inhibit catalysis. Our results are in this line and support the fact that the experimental excess of P–N (25 mol%) leads to a significant decrease in conversion (vide supra).
Considering the competition between acetonitrile and benzylacetylene towards metal coordination at intermediate 3C1, the nitrile presents a lower energetic barrier (6.9 kcal mol−1 CP(D1-N)) than the alkyne (11.1 kcal mol−1 CP(D1-A)). Inspection of the geometrical structures reveals that the coordination of the alkyne is hindered by steric clashes between the terminal hydrogen and the methyl groups of the Cp* ligand, while the nitrogen atom of the nitrile can easily access the metal center. 1,3,5-trisubstituted benzene can be formed from D1-A following an intramolecular [4 + 2] mechanism facilitated by the metal, characterized by TSDE1-A. This transition state has an energetic barrier of 1.1 kcal mol−1 relative to D1-A and results in η4-type arene coordination to the metal, E1-A. A stepwise exchange of the arene with two benzylacetylene molecules generates intermediate B, thereby completing the catalytic cycle. The formation of pyridine from the D1-N intermediate requires consideration of three competing reaction mechanisms proposed in the literature (see Fig. S76†). The intermolecular [4 + 2] mechanism between the cyclometalladiene and acetonitrile, can be excluded due to its very high relative energy of 27.3 kcal mol−1. Migratory insertion of the η1-coordinated nitrile into one of the Co–C bond shows an energetic barrier of 17.9 kcal mol−1 (from D1-N), and leads to the seven-member puckered metallacycle G1-N (−5.3 kcal mol−1). Then, the pyridine can be formed through reductive elimination with a low energetic barrier of 3.2 kcal mol−1 from G1-N. The last possible pathway, the intramolecular [4 + 2] mechanism, involves a change in the coordination mode of the nitrile, from η1 (D1-N) to η2 (E1-N), characterized by TSDE1-N. The concerted addition of the metal-coordinated nitrile to the cyclometalladiene may occur in two different orientations (paths 1a and 1b described in Scheme 7) leading to 2,4,6- and 2,3,5- regioisomers. Calculation of transition states shows that path 1a is energetically favored, the relative energy being 1.8 kcal mol−1 for TSEF1a-N, while path 1b is energetically unaffordable. The final 2,4,6-trisubstituted pyridine can be released through stepwise ligand exchange with two benzylacetylene molecules forming intermediate B and completing the catalytic cycle. EF1b-N may also undergo a coordination mode change from η4 to η6, which is energetically favorable due to the triplet instability in the CoCp*(η6-pyridine) wavefunction and the corresponding crossing point that allows surface hopping (Fig. S77†).
According to the DFT Gibbs free energetic profile, chemoselectivity is governed by the lower energy of CP(D1-N) compared to CP(D1-A), allowing a faster coordination of nitrile to CoCp*cyclopentadiene intermediate in the triplet state. The regioselectivity is controlled in two steps. First, the oxidative coupling of two alkynes favors 1,3-alkyne coupling, although the transition state leading to 1,4-coupling is only 1.1 kcal mol−1 higher. This energetic difference leads to 81.5:18.5 proportion of products, which is in good agreement with the value of 2:1 obtained experimentally. Second, the 2,3,5-pyridiyne is not observed due to the energetic difference at the metal-mediated [4 + 2] addition of the nitrile to the diene (TSEF1b-N and TSEF1a-N). However, it should be noted that DFT free energy profiles do not consider concentration effects.59 The concentration of phosphane is smaller than nitrile and alkyne in the competitive step of ligand coordination to the metal in 3C1 intermediate and also in the oxidative coupling of the two alkynes.
Conclusions
We have developed a catalytic system for the straightforward preparation of novel functionalized pyridines via the [2 + 2 + 2] cycloaddition of nitriles and alkynes. The precatalyst is an air-stable CoCp*(III) complex stabilized by a hemilabile P–N ligand. In situ reduction of this complex with NaBEt3H generates the active Co(I) species. Remarkably, related systems with strongly coordinating bisphosphane ligands or monodentate phosphanes (lacking the stabilizing effect of the hemilabile moiety) fail to efficiently generate the active species, leading to a sharp reduction in catalytic activity. This behavior suggests that the hemilabile P–N ligand resides in the “Goldilocks zone” for precatalyst stabilization. Moreover, an in-depth computational study at the DFT level of the reaction mechanism has clarified the key steps driving the chemo- and stereoselectivity of the reaction, providing essential insights into the factors governing the competitive pathways arising from alkyne, nitrile, or phosphane coordination in a two-state reactivity scenario. Non-covalent interactions between alkyne substituents and the methyl groups of the Cp* ligand favor 1,3-regioisomer as the major intermediate in the oxidative coupling step, while selective metal-assisted [4 + 2] cycloaddition of acetonitrile to the cyclometalladiene yields the final products. Notably, the coordination of acetonitrile to the metal is favored over that of benzylacetylene, due to a more encumbered transition state for the latter, thus explaining the selective formation of pyridines over arenes.This catalytic method holds significant potential as a tool for constructing complex pyridines, which are commonly found in bioactive molecules and functional materials. Furthermore, the mechanistic insights presented here lay the foundation for the development of more efficient catalysts for [2 + 2 + 2] cycloaddition reactions. On these grounds, further improvements in selectivity and substrate scope require fine-tuning of the electron density at the metal center and, especially, the steric hindrance of the ligand system. A decrease in the steric hindrance around the metal center is likely to improve the catalyst's activity, leading to higher conversions and a broader substrate scope, but the selectivity is expected to be affected, likely increasing alkyne trimerization (as suggested by the proposed reaction mechanism).
Experimental
Computational details
All DFT theoretical calculations were carried out using the Gaussian16 program package.60 Geometry optimization and frequency analysis was performed using the M06-L functional,61 which has proven to yield good results for singlet–triplet gap on similar complexes,62 in combination to the def2-SVP basis set63 which include effective core potentials for cobalt atom. Energies were refined by MN15/def2-TZVP single point calculations64 using the SMD approach65 for benzene as implemented in G16. The “ultrafine” grid was employed in all calculations. All reported energies are Gibbs free energies referred to a 1 M standard state at 373.15° K including basis set, solvent, and entropic quasi-harmonic corrections66 as implemented in Goodvibes program.67 Intersystem crossing points where calculated using the MECP program as implemented in EASYMECP68,69 employing the same computational level employed for geometry optimizations and free energy corrections. NCI plots were made using the NCIPLOT software.70Data availability
The data supporting this article have been included as part of the ESI† in two separate files. The first document includes the experimental procedures, DFT calculations and NMR spectra (PDF file), while the second includes the x,y,z coordinates of the DFT intermediates (xyz file).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Grant PID2021-126212OB-I00 funded by MCIN/AEI/10.13039/501100011033 and by “ERDF A way of making Europe”, as well as the “Departamento de Ciencia, Universidad y Sociedad del Conocimiento del Gobierno de Aragón” (group E42_23R) are gratefully acknowledged. Authors would like to acknowledge the use of Servicio General de Apoyo a la Investigación-SAI at the Universidad de Zaragoza and at the ISQCH/CEQMA (CSIC).References
- Comprehensive heterocyclic chemistry II. 5: Six-membered rings with one heteroatom and fused carbocyclic derivatives/vol. ed.: Alexander McKillop, ed. A. MacKillop, Pergamon, Oxford, 1st edn, 1996 Search PubMed.
- A. Nefzi, J. M. Ostresh and R. A. Houghten, The Current Status of Heterocyclic Combinatorial Libraries, Chem. Rev., 1997, 97, 449–472 CrossRef CAS PubMed.
- Recent developments in the synthesis and applications of pyridines, ed. P. Singh, Elsevier, Amsterdam, Netherlands, 2023 Search PubMed.
- M. D. Hill, Recent Strategies for the Synthesis of Pyridine Derivatives, Chem. – Eur. J., 2010, 16, 12052–12062 CrossRef CAS PubMed.
- K. Tanaka, N. Suzuki and G. Nishida, Cationic Rhodium(I)/Modified–BINAP Catalyzed [2 + 2 + 2] Cycloaddition of Alkynes with Nitriles, Eur. J. Org. Chem., 2006, 3917–3922 CrossRef CAS.
- K. Tanaka, H. Hara, G. Nishida and M. Hirano, Synthesis of Perfluoroalkylated Benzenes and Pyridines through Cationic Rh(I)/Modified BINAP-Catalyzed Chemo- and Regioselective [2 + 2 + 2] Cycloaddition, Org. Lett., 2007, 9, 1907–1910 CrossRef CAS PubMed.
- K. Kashima, M. Ishii and K. Tanaka, Synthesis of Pyridylphosphonates by Rhodium–Catalyzed [2 + 2 + 2] Cycloaddition of 1,6− and 1,7−Diynes with Diethyl Phosphorocyanidate, Eur. J. Org. Chem., 2015, 1092–1099 CrossRef CAS.
- Y. Yamamoto, S. Okuda and K. Itoh, Ruthenium(ii)-catalyzed [2 + 2 + 2] cycloaddition of 1,6-diynes with electron-deficient nitriles, Chem. Commun., 2001, 1102–1103 RSC.
- Y. Yamamoto, R. Ogawa and K. Itoh, Significant Chemo- and Regioselectivies in the Ru(II)-Catalyzed [2 + 2 + 2] Cycloaddition of 1,6-Diynes with Dicyanides, J. Am. Chem. Soc., 2001, 123, 6189–6190 CrossRef CAS PubMed.
- Y. Yamamoto, K. Kinpara, H. Nishiyama and K. Itoh, Synthesis of 2−Haloalkylpyridines via Cp*RuCl–Catalyzed Cycloaddition of 1,6−Diynes with α–Halonitriles. Unusual Halide Effect in Catalytic Cyclocotrimerization, Adv. Synth. Catal., 2005, 347, 1913–1916 CrossRef CAS.
- Y. Yamamoto, K. Kinpara, R. Ogawa, H. Nishiyama and K. Itoh, Ruthenium–Catalyzed Cycloaddition of 1,6−Diynes and Nitriles under Mild Conditions: Role of the Coordinating Group of Nitriles, Chem. – Eur. J., 2006, 12, 5618–5631 CrossRef CAS PubMed.
- F. Xu, C. Wang, X. Li and B. Wan, Ruthenium–catalyzed [2 + 2 + 2] Cycloaddition of Diynes with Nitriles in Pure Water, ChemSusChem, 2012, 5, 854–857 CrossRef CAS PubMed.
- C. Wang, X. Li, F. Wu and B. Wan, A Simple and Highly Efficient Iron Catalyst for a [2 + 2 + 2] Cycloaddition to Form Pyridines, Angew. Chem., Int. Ed., 2011, 50, 7162–7166 CrossRef CAS PubMed.
- T. Hashimoto, S. Ishii, R. Yano, H. Miura, K. Sakata and R. Takeuchi, Iridium–Catalyzed [2 + 2 + 2] Cycloaddition of α,ω–Diynes with Cyanamides, Adv. Synth. Catal., 2015, 357, 3901–3916 CrossRef CAS.
- T. Hashimoto, K. Kato, R. Yano, T. Natori, H. Miura and R. Takeuchi, Iridium-Catalyzed Synthesis of Acylpyridines by [2 + 2 + 2] Cycloaddition of Diynes with Acyl Cyanides, J. Org. Chem., 2016, 81, 5393–5400 CrossRef CAS PubMed.
- M. M. McCormick, H. A. Duong, G. Zuo and J. Louie, A Nickel-Catalyzed Route to Pyridines, J. Am. Chem. Soc., 2005, 127, 5030–5031 CrossRef CAS PubMed.
- T. N. Tekavec, G. Zuo, K. Simon and J. Louie, An in Situ Approach for Nickel-Catalyzed Cycloaddition, J. Org. Chem., 2006, 71, 5834–5836 CrossRef CAS PubMed.
- R. M. Stolley, M. T. Maczka and J. Louie, Nickel–Catalyzed [2 + 2 + 2] Cycloaddition of Diynes and Cyanamides, Eur. J. Org. Chem., 2011, 3815–3824 CrossRef CAS PubMed.
- C. Wang, D. Wang, F. Xu, B. Pan and B. Wan, Iron-Catalyzed Cycloaddition Reaction of Diynes and Cyanamides at Room Temperature, J. Org. Chem., 2013, 78, 3065–3072 CrossRef CAS PubMed.
- T. K. Lane, B. R. D'Souza and J. Louie, Iron-Catalyzed Formation of 2-Aminopyridines from Diynes and Cyanamides, J. Org. Chem., 2012, 77, 7555–7563 CrossRef CAS PubMed.
- M. Hapke, N. Weding and A. Spannenberg, Highly Reactive Cyclopentadienylcobalt(I) Olefin Complexes, Organometallics, 2010, 29, 4298–4304 CrossRef CAS.
- I. Thiel, H. Jiao, A. Spannenberg and M. Hapke, Fine–Tuning the Reactivity and Stability by Systematic Ligand Variations in CpCo I Complexes as Catalysts for [2 + 2 + 2] Cycloaddition Reactions, Chem. – Eur. J., 2013, 19, 2548–2554 CrossRef CAS PubMed.
- P. Garcia, Y. Evanno, P. George, M. Sevrin, G. Ricci, M. Malacria, C. Aubert and V. Gandon, Synthesis of Aminopyridines and Aminopyridones by Cobalt–Catalyzed [2 + 2 + 2] Cycloadditions Involving Yne–Ynamides: Scope, Limitations, and Mechanistic Insights, Chem. – Eur. J., 2012, 18, 4337–4344 CrossRef CAS PubMed.
- C. Brändli and T. R. Ward, A Versatile Approach to the Solution-Phase Combinatorial Synthesis of Substituted Pyridines: The Cobalt-Catalyzed Cyclotrimerization of Alkynes with a Nitrile, J. Comb. Chem., 2000, 2, 42–47 CrossRef PubMed.
- K. Kase, A. Goswami, K. Ohtaki, E. Tanabe, N. Saino and S. Okamoto, On-Demand Generation of an Efficient Catalyst for Pyridine Formation from Unactivated Nitriles and α,ω-Diynes Using CoCl2 -6H2 O, dppe, and Zn, Org. Lett., 2007, 9, 931–934 CrossRef CAS PubMed.
- A. Geny, N. Agenet, L. Iannazzo, M. Malacria, C. Aubert and V. Gandon, Air–Stable {(C5 H5)Co} Catalysts for [2 + 2 + 2] Cycloadditions, Angew. Chem., Int. Ed., 2009, 48, 1810–1813 CrossRef CAS PubMed.
- F. Fischer, A. F. Siegle, M. Checinski, C. Fischer, K. Kral, R. Thede, O. Trapp and M. Hapke, Synthesis of Naphthylpyridines from Unsymmetrical Naphthylheptadiynes and the Configurational Stability of the Biaryl Axis, J. Org. Chem., 2016, 81, 3087–3102 CrossRef CAS PubMed.
- M. Hapke, K. Kral, C. Fischer, A. Spannenberg, A. Gutnov, D. Redkin and B. Heller, Asymmetric Synthesis of Axially Chiral 1-Aryl-5,6,7,8-tetrahydroquinolines by Cobalt-Catalyzed [2 + 2 + 2] Cycloaddition Reaction of 1-Aryl-1,7-octadiynes and Nitriles, J. Org. Chem., 2010, 75, 3993–4003 CrossRef CAS PubMed.
- D. D. Young and A. Deiters, A General Approach to Chemo– and Regioselective Cyclotrimerization Reactions, Angew. Chem., Int. Ed., 2007, 46, 5187–5190 CrossRef CAS PubMed.
- K. Cen, M. Usman, W. Shen, M. Liu, R. Yang and J. Cai, A review on the assembly of multi-substituted pyridines via Co-catalyzed [2 + 2 + 2] cycloaddition with nitriles, Org. Biomol. Chem., 2022, 20, 7391–7404 RSC.
- Cobalt catalysis in organic synthesis: methods and reactions, ed. M. Hapke and G. Hilt, Wiley-VCH Verlag GmbH, Weinheim, 2020 Search PubMed.
- N. Weding, A. Spannenberg, R. Jackstell and M. Hapke, Formation and Reactivity of a Co4 -μ-Alkyne Cluster from a Co(I)-Alkene Complex, Organometallics, 2012, 31, 5660–5663 CrossRef CAS.
- T. Gläsel, B. N. Baumann and M. Hapke, Cobalt Catalysts for [2 + 2 + 2] Cycloaddition Reactions: Isolated Precatalysts and in situ Generated Catalysts, Chem. Rec., 2021, 21, 3727–3745 CrossRef PubMed.
- C. Yuan, C.-T. Chang, A. Axelrod and D. Siegel, Synthesis of (+)-Complanadine A, an Inducer of Neurotrophic Factor Excretion, J. Am. Chem. Soc., 2010, 132, 5924–5925 CrossRef CAS PubMed.
- Y. Satoh and Y. Obora, Low-Valent Niobium-Catalyzed Intermolecular [2 + 2 + 2] Cycloaddition of tert -Butylacetylene and Arylnitriles to Form 2,3,6-Trisubstituted Pyridine Derivatives, J. Org. Chem., 2013, 78, 7771–7776 CrossRef CAS PubMed.
- Y.-L. Chen, P. Sharma and R.-S. Liu, Sulfonamide-directed gold-catalyzed [2 + 2 + 2]-cycloadditions of nitriles with two discrete ynamides to construct 2,4-diaminopyridine cores, Chem. Commun., 2016, 52, 3187–3190 RSC.
- P. Diversi, L. Ermini, G. Ingrosso and A. Lucherini, Electronic and steric effects in the rhodium-complex catalysed co-cyclization of alkynes and nitriles to pyridine derivatives, J. Organomet. Chem., 1993, 447, 291–298 CrossRef CAS.
- B. Heller, B. Sundermann, H. Buschmann, H.-J. Drexler, J. You, U. Holzgrabe, E. Heller and G. Oehme, Photocatalyzed [2 + 2 + 2]-Cycloaddition of Nitriles with Acetylene: An Effective Method for the Synthesis of 2-Pyridines under Mild Conditions, J. Org. Chem., 2002, 67, 4414–4422 CrossRef CAS PubMed.
- A. W. Fatland and B. E. Eaton, Cobalt-Catalyzed Alkyne−Nitrile Cyclotrimerization To Form Pyridines in Aqueous Solution, Org. Lett., 2000, 2, 3131–3133 CrossRef CAS PubMed.
- Y. Xie, C. Wu, C. Jia, C.-H. Tung and W. Wang, Iron–cobalt-catalyzed heterotrimerization of alkynes and nitriles to polyfunctionalized pyridines, Org. Chem. Front., 2020, 7, 2196–2201 RSC.
- C. Wang, Q. Sun, F. García, C. Wang and N. Yoshikai, Robust Cobalt Catalyst for Nitrile/Alkyne [2 + 2 + 2] Cycloaddition: Synthesis of Polyarylpyridines and Their Mechanochemical Cyclodehydrogenation to Nitrogen–Containing Polyaromatics, Angew. Chem., Int. Ed., 2021, 60, 9627–9634 CrossRef CAS PubMed.
- M. Hapke, K. Kral, C. Fischer, A. Spannenberg, A. Gutnov, D. Redkin and B. Heller, Asymmetric Synthesis of Axially Chiral 1-Aryl-5,6,7,8-tetrahydroquinolines by Cobalt-Catalyzed [2 + 2 + 2] Cycloaddition Reaction of 1-Aryl-1,7-octadiynes and Nitriles, J. Org. Chem., 2010, 75, 3993–4003 CrossRef CAS PubMed.
- K. Li, L. Wei, M. Sun, B. Li, M. Liu and C. Li, Enantioselective Synthesis of Pyridines with All–Carbon Quaternary Carbon Centers via Cobalt–Catalyzed Desymmetric [2 + 2 + 2] Cycloaddition, Angew. Chem., Int. Ed., 2021, 60, 20204–20209 CrossRef CAS PubMed.
- P. Diversi, G. Ingrosso, A. Lucherini and D. Vanacore, Cobalt-catalyzed synthesis of pyridines from 1-alkynes and nitriles: substrate structure and regioselectivity, J. Mol. Catal., 1987, 41, 261–270 CrossRef CAS.
- T. Gläsel and M. Hapke, in Cobalt Catalysis in Organic Synthesis, ed. M. Hapke and G. Hilt, Wiley, 1st edn, 2020, pp. 287–335 Search PubMed.
- A. Roglans, A. Pla-Quintana and M. Solà, Mechanistic Studies of Transition-Metal-Catalyzed [2 + 2 + 2] Cycloaddition Reactions, Chem. Rev., 2021, 121, 1894–1979 CrossRef CAS PubMed.
- W. I. Dzik, W. Böhmer and B. De Bruin, in Spin States in Biochemistry and Inorganic Chemistry, ed. M. Swart and M. Costas, Wiley, 1st edn, 2015, pp. 103–129 Search PubMed.
- S. García-Abellán, D. Barrena-Espés, J. Munarriz, V. Passarelli and M. Iglesias, Cobalt-catalysed nucleophilic fluorination in organic carbonates, Dalton Trans., 2023, 52, 4585–4594 RSC.
- J. H. Hardesty, J. B. Koerner, T. A. Albright and G.-Y. Lee, Theoretical Study of the Acetylene Trimerization with CpCo, J. Am. Chem. Soc., 1999, 121, 6055–6067 CrossRef CAS.
- L. F. Veiros, G. Dazinger, K. Kirchner, M. J. Calhorda and R. Schmid, By What Mechanisms Are Metal Cyclobutadiene Complexes Formed from Alkynes?, Chem. – Eur. J., 2004, 10, 5860–5870 CrossRef CAS PubMed.
- G. Dazinger, M. Torres-Rodrigues, K. Kirchner, M. J. Calhorda and P. J. Costa, Formation of pyridine from acetylenes and nitriles catalyzed by RuCpCl, CoCp, and RhCp derivatives – A computational mechanistic study, J. Organomet. Chem., 2006, 691, 4434–4445 CrossRef CAS.
- A. A. Dahy, C. H. Suresh and N. Koga, Theoretical Study of the Formation of a Benzene Cobalt Complex from Cobaltacyclopentadiene and Acetylene, Bull. Chem. Soc. Jpn., 2005, 78, 792–803 CrossRef CAS.
- N. Agenet, V. Gandon, K. P. C. Vollhardt, M. Malacria and C. Aubert, Cobalt-Catalyzed Cyclotrimerization of Alkynes: The Answer to the Puzzle of Parallel Reaction Pathways, J. Am. Chem. Soc., 2007, 129, 8860–8871 CrossRef CAS PubMed.
- A. A. Dahy, K. Yamada and N. Koga, Theoretical Study on the Reaction Mechanism for the Formation of 2-Methylpyridine Cobalt(I) Complex from Cobaltacyclopentadiene and Acetonitrile, Organometallics, 2009, 28, 3636–3649 CrossRef CAS.
- Y. Wakatsuki, O. Nomura, K. Kitaura, K. Morokuma and H. Yamazaki, Cobalt metallacycles. 11. On the transformation of bis(acetylene)cobalt to cobaltacyclopentadiene, J. Am. Chem. Soc., 1983, 105, 1907–1912 CrossRef CAS.
- A. Stockis and R. Hoffmann, Metallacyclopentanes and bisolefin complexes, J. Am. Chem. Soc., 1980, 102, 2952–2962 CrossRef CAS.
- A. A. Dahy and N. Koga, Theoretical Study on the Transformation of Bis(acetylene)cobalt to Cobaltacyclopentadiene and the Regioselectivity in this Transformation, Bull. Chem. Soc. Jpn., 2005, 78, 781–791 CrossRef CAS.
- D. R. McAlister, J. E. Bercaw and R. G. Bergman, Parallel reaction pathways in the cobalt-catalyzed cyclotrimerization of acetylenes, J. Am. Chem. Soc., 1977, 99(5), 1666–1668 CrossRef CAS.
- J. N. Harvey, F. Himo, F. Maseras and L. Perrin, Scope and Challenge of Computational Methods for Studying Mechanism and Reactivity in Homogeneous Catalysis, ACS Catal., 2019, 9, 6803–6813 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- S. García-Abellán, D. Barrena-Espés, J. Munarriz, V. Passarelli and M. Iglesias, Cobalt-catalysed nucleophilic fluorination in organic carbonates, Dalton Trans., 2023, 52, 4585–4594 RSC.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, MN15: A Kohn–Sham global-hybrid exchange–correlation density functional with broad accuracy for multi-reference and single-reference systems and noncovalent interactions, Chem. Sci., 2016, 7, 5032–5051 RSC.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS.
- S. Grimme, Supramolecular Binding Thermodynamics by Dispersion–Corrected Density Functional Theory, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS.
- G. Luchini, J. V. Alegre-Requena, I. Funes-Ardoiz and R. S. Paton, GoodVibes: automated thermochemistry for heterogeneous computational chemistry data, F1000Research, 2020, 9, 291 Search PubMed.
- J. Rodríguez-Guerra, I. Funes-Ardoiz and F. Maseras, EasyMECP, 2018 DOI:10.5281/zenodo.4293421.
- J. N. Harvey, Understanding the kinetics of spin-forbidden chemical reactions, Phys. Chem. Chem. Phys., 2007, 9, 331–343 RSC.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, NCIPLOT: A Program for Plotting Noncovalent Interaction Regions, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, DFT calculations, NMR spectra (PDF), x,y,z coordinates. See DOI: https://doi.org/10.1039/d5qo00222b |
| This journal is © the Partner Organisations 2025 |