
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Beyond HAT: harnessing TBADT for photocatalyzed Giese-type C(sp3)–C(sp3) bond formation through reductive decarboxylation†
Matteo Leone ab,
Dalila Arnaldia and
Maurizio Fagnoni*a
ab,
Dalila Arnaldia and
Maurizio Fagnoni*a
aPhotoGreen Lab, Department of Chemistry, University of Pavia, Viale Taramelli 12, 27100 Pavia, Italy. E-mail: maurizio.fagnoni@unipv.it
bDepartment of Industrial Chemistry “Toso Montanari”, University of Bologna, 40129 Bologna, Italy
First published on 3rd May 2025
Abstract
The decatungstate anion as a tetrabutylammonium salt (TBADT) facilitates a variety of chemical transformations under mild conditions, primarily through hydrogen atom transfer (HAT) and marginally through single electron transfer (SET) mechanisms. This study explores the dual ability of TBADT to cleave C–H bonds and initiate SET processes, leading to efficient C(sp3)–C(sp3) coupling reactions. We address the main limitations of direct HAT by leveraging the doubly-reduced form of TBADT [W10O32]6− to activate redox-active esters (RAEs), enabling the formation of alkyl radicals for Giese-type additions. An extensive screening of various hydrogen donors showed their pivotal role in the selective generation of the reduced form of TBADT and in suppressing any undesired C–H activation. Our optimized conditions, using γ-terpinene as the hydrogen donor, gave high yields in alkylations of various olefins, demonstrating the versatility and robustness of the proposed strategy. This methodology extends the application of TBADT in sustainable organic synthesis and in late-stage functionalization of complex molecules for the synthesis of pharmaceutical building blocks.
Introduction
In recent years, photocatalysis has emerged as a powerful and sustainable approach for developing novel and efficient synthetic routes.1 Among various photocatalysts, the decatungstate anion [W10O32]4− (mainly as a tetrabutylammonium salt, TBADT) has garnered significant attention due to its exceptional photochemical activity, particularly its ability to facilitate a wide range of chemical reactions under mild conditions.2 Since the earliest reports,3,4 TBADT has entered its golden age in photocatalytic organic transformations thanks to its peculiar properties. Mechanistic studies have shown that in its excited state, TBADT undergoes a ligand-to-metal charge transfer (LMCT) mechanism, imparting partial radical character to the monocoordinated oxygen centers. This enables it to homolytically cleave C(sp3)–H and formylic C(sp2)–H bonds in various aliphatic derivatives through a hydrogen atom transfer (HAT) mechanism (Scheme 1A).5 For many years, this feature has been exploited for the direct activation of C–H bonds under mild conditions, facilitating effective C–C couplings,6 C–N couplings,7 fluorinations,8 oxygenations,9 and dehydrogenations.10 Additional studies evidenced that in the presence of TBADT, synergistic control by polar and steric effects allows, to some extent, selective C–H functionalization at competitive sites.11 The ability of TBADT to homolytically cleave unactivated C–H bonds with high bond dissociation energies (BDEs) has (in part) restricted its use in the functionalization of complex molecules, where the presence of multiple active sites can lead to competing reactions.12 Nevertheless, the application of TBADT is thus mostly confined to HAT processes hiding its propensity toward photoredox catalyzed processes. In fact, TBADT has a remarkably high redox potential in the excited state (E([W10O32]4−*/[W10O32]5−) ≈ +2.5 V vs. SCE),13 comparable to the most commonly used strong photo-oxidants,14 making it ideal for activating a vast array of substrates via oxidative single-electron transfer (SETox, Scheme 1B).15 To this end, TBADT has been applied to photoredox promoted desilylation of alkyl16a and acyl16b silanes, decarboxylative benzylations17a and ring opening of cyclopropanols.17b However, one of the major limitations of this approach is the potential competition between HAT and SET processes deriving from the same excited state, which could lead to unpredictable reactivity.18 On the other hand, the final catalytic step of TBADT reveals intriguing characteristics for SET processes that circumvent this limitation, a feature that was largely overlooked until recent years. In fact, the reduced form of the decatungstate anion [W10O32]5− can disproportionate, regenerating the ground state photocatalyst [W10O32]4− and forming doubly-reduced decatungstate [W10O32]6−, a good reductant (Ered1/2([W10O32]5−/[W10O32]6−) = −1.48 V vs. SCE)19 (Scheme 1C). The reducing potential of TBADT was useful when merging a photocatalyst with a transition-metal (TM) catalyst in dual catalyzed cross-couplings.20 The authors proposed that [W10O32]6− undergoes SET with TMn+1 to restore the active TMn. Inspired by this, numerous novel transformations have been developed, merging TBADT with transition-metal catalysis (Scheme 1D, left part).21 We were, however, surprised by the limited applications of TBADT as a versatile direct reductant of organic compounds22 even though this approach could overcome the inherent limitations of direct HAT.23 As a matter of fact, this strategy could (i) eliminate dependence on bond dissociation energy (BDE), (ii) reduce the need for excess precious hydrogen donors, (iii) give access to the more elusive 1° radicals and (iv) enable late-stage functionalization (LSF) without site competition in complex molecules (Scheme 1D, right part). Additionally, the redox potential of the doubly-reduced form of TBADT is suitable for most SOMOphiles, since these are mostly difficult to reduce compared to the most commonly used redox active functional groups, preventing unwanted side-reduction processes.24 On this basis, we envisioned the forging of new C(sp3)–C(sp3) bonds starting from ubiquitous alkyl carboxylic acids, ideal starting materials present in natural products and pharmaceuticals.25 | ||
| Scheme 1 A) Application of TBADT in HAT transformations. (B) TBADT mediated photo-oxidations. (C) General mechanism for the photocatalytic pathways of TBADT. (D) TBADT mediated photo-reductions. (E) Development of a novel approach for TBADT photocatalyzed Giese type additions. | ||
We hypothesized that in the presence of a suitable hydrogen donor (D-H), [W10O32]6− could accumulate in solution and may reduce various redox-active esters (RAEs) via SETred (Scheme 1E).26 Hydroxyphthalimide (NHPI) esters are well-suited for the reaction thanks to their reduction potential (Ered1/2 ≈ −1.3 V vs. SCE).27 The resulting alkyl radicals, derived from the mesolytic cleavage of the N–O bond followed by CO2 loss, could then undergo Giese-type additions with a wide range of SOMOphiles, potentially yielding the target molecules of pharmaceutical interest. Photocatalyzed reductive decarboxylation is a well-established and extensively studied method for generating carbon-centered radicals, widely applied in Giese-type additions.28 Therefore, we envisioned this transformation as an excellent platform to investigate the reactivity of TBADT as a photoreductant.
Results and discussion
Building on previous studies and our expertise29 it is apparent that the choice of the sacrificial D-H would be critical in the reaction mechanism for effectively generating the desired product.30 An efficient initial hydrogen atom abstraction is the key step to accumulate the blue colored species ([W10O32]6−) in the reaction medium, thus promoting the entire process. Therefore, we aimed to select an ideal D-H that should have very labile C–H bonds, facilitating the initial HAT step.30 The donor (a commercially available and cost-effective compound) should be easily separable from the reaction medium and the resulting radical (D˙) should not interfere with the reaction course. Saying so, we began our study with a model reaction using NHPI ester 1a as the radical precursor, 2a as the radical acceptor, and TBADT as the photocatalyst (Table 1). In our initial attempts, we screened various alcohols (e.g. 3a-H, BDE(C–H) ca. 94 kcal mol−1)31a as H-donors32 (Table S1 ESI†), but these mostly resulted in competitive radical addition onto 2a, leading to low yields of the desired product 4a. Consequently, we shifted our focus to more stabilized allylic radical precursors,33 hypothesizing that the stability of the resulting radical may hamper the interaction with the SOMOphiles. With tetramethylethylene 3f-H (BDE(C–H) ca. 85 kcal mol−1)31b and cyclohexadiene 3g-H (BDE(C–H) = 75 kcal mol−1),31a we observed a clean reaction, free from competitive addition to 2a, yielding 4a in promising amounts (entries 2 and 3). However, due to the high cost and volatility of these alkenes, which could result in partial evaporation, we turned our attention to low volatility γ-terpinene 3h-H. γ-Terpinene, a natural product found in medicinal and aromatic plants,34 is commercially available (0.53€ per g)35 and possesses all the required features for our target reaction (calculated BDE(C–H) is ca. 70 kcal mol−1).33b Accordingly, the reaction carried out in an acetonitrile/dichloromethane (ACN/DCM) 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture gave 4a in 92% yield (entry 4). When shifting to a greener solvent, such as acetone,36 the yield slightly increased (95%, entry 5). Conducting the reaction under 390 nm Kessil lamp irradiation for 24 h resulted in incomplete conversion of 1, although no significant side products were observed, indicating that the reaction proceeds at a slower rate (entry 6). Further variation by adjusting the catalyst loading or the equivalents of 2a or 3h-H led to lower yields (see Table S2, ESI†). Control experiments confirmed that the reaction was completely inhibited in the absence of light, the photocatalyst, or the D-H (entries 7–9).
:1 mixture gave 4a in 92% yield (entry 4). When shifting to a greener solvent, such as acetone,36 the yield slightly increased (95%, entry 5). Conducting the reaction under 390 nm Kessil lamp irradiation for 24 h resulted in incomplete conversion of 1, although no significant side products were observed, indicating that the reaction proceeds at a slower rate (entry 6). Further variation by adjusting the catalyst loading or the equivalents of 2a or 3h-H led to lower yields (see Table S2, ESI†). Control experiments confirmed that the reaction was completely inhibited in the absence of light, the photocatalyst, or the D-H (entries 7–9).
|
||||
|---|---|---|---|---|
| Entry | H-donor | Solvent (0.1 M) | Yielda (%) | Deviations |
| Reaction conditions: 1a (0.1 mmol), 2a (0.12 mmol), 3-H (1.0 mmol) and TBADT (2 mol%) in 1.0 mL of solvent under N2. Reactions were irradiated with a 40 W Kessil lamp (370 nm) for 24 h.a 1H NMR yield of 4a was determined by using CH2Br2 as an internal standard. | ||||
| 1 | 3a-H | ACN | 20% | |
| 2 | 3f-H | ACN | 41% | |
| 3 | 3g-H | ACN/DCM 9/1 | 84% | |
| 4 | 3h-H | ACN/DCM 9/1 | 92% | |
| 5 | 3h-H | Acetone | 95% | |
| 6 | 3h-H | ACN/DCM 9/1 | 53% | 390 nm |
| 7 | 3h-H | ACN/DCM 9/1 | — | No photocatalyst |
| 8 | 3h-H | ACN/DCM 9/1 | — | No light |
| 9 | — | ACN/DCM 9/1 | — | No H-donor |
 |
||||
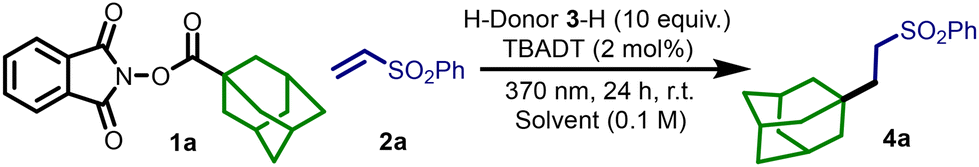
Encouraged by these results, we proceeded to explore the addition of radicals derived from NHPI esters onto phenyl vinylsulfone 2a or benzylidenemalononitrile 2b (Scheme 2). We began by screening the reactivity of primary (1°) radicals, which are challenging to generate and capture through direct HAT.37
 | ||
| Scheme 2 Reaction conditions: 1 (0.2 mmol), 2a (0.24 mmol), 3h-H (2.0 mmol) and TBADT (2 mol%) in 2.0 mL of acetone under N2. Reactions were irradiated with a 40 W Kessil lamp (370 nm) for 24 h; isolated yields. aReaction performed in ACN/DCM 9/1 (0.1 M). b2-Benzylmalononitrile was isolated in ca. 10% yield. | ||
We successfully isolated products 4b and 4c in moderate to good yields. In the latter case, the formation of 4c benefits from substantial stabilization of the radical from the adjacent nitrogen group. Notably, switching to the more reactive radical trap 2b38 enabled the incorporation of additional primary radicals, affording products 4j and 4k in good yields. Interestingly, the use of 2b, a more easily reducible SOMOphile [E1/2 = −1.35 V vs. SCE],39 led to the formation of benzylmalononitrile as a byproduct in approximately 10% yield. This observation suggests a possible SET event between the doubly reduced form of TBADT and the radical trap.
Next, we were particularly interested in applying our strategy to compounds where TBADT might activate multiple C–H sites, potentially leading to several side products. Accordingly, we screened a diverse range of secondary (2°) radicals, achieving good to excellent yields of products 4d–4g. Satisfactorily, we observed no competitive formation of undesired α-amido radicals via direct HAT, due to the efficient role of γ-terpinene as a H-donor which completely quenched the catalytic activity of TBADT as a hydrogen atom abstractor. We were then able to synthesize the desired products 4f and 4g, which are difficult to obtain through direct HAT when omitting the NHPI moiety. Building on these promising outcomes with various carboxylic acid derived radical precursors, we further investigated the functionalization of potential bioactive compounds, such as clofibric acid40 and a dipeptide (Boc-Ala-Ala-OH).41 In both cases, we successfully isolated the desired products 4h and 4i in more than 80% yield. We then investigated the feasibility of our strategy for the alkylation of various α,β-unsaturated ketones, esters, nitriles etc. (Scheme 3). Notably, the Giese adducts (4l–4q) were formed in a shorter reaction time (16 h) mostly in >70% yield, with product 4n being obtained in nearly quantitative yield. Lower yields were observed for 4r, attributed to the lower electrophilicity of the olefin, which led to partial reduction of 1g to N-Boc piperidine. Remarkably, the reaction could also be applied to more complex molecules having stereogenic centers, such as menthol 4s, cholesterol 4t, and cedrol 4u derivatives. These desired products were isolated with exceptional yields, without unwanted C–H activations (or racemization) catalyzed by TBADT of starting 1g and 2. These findings underscore the potential of this strategy for late-stage functionalization of complex molecules by maintaining their structural features. We then extended our methodology to the synthesis of promising commercially relevant molecules. Considering the growing global demand for unnatural α-amino acids, and their potential as valuable building blocks in bioactive compounds,42 we explored the possibility of applying our approach for their preparation in one step and under mild conditions by adopting suitable radical acceptors (Scheme 4A). To this end, we screened various NHPI esters in the presence of 2l and 2m, as α-amino acid precursors (Scheme 4B). In positive case, this methodology may lead to the formation of racemic products but may represent the starting point for future synthesis of enantiopure α-amino acids by adopting an enantioselective variant.43 We evaluated the behaviour of primary (1°), secondary (2°), and tertiary (3°) radicals. To our delight, we successfully incorporated 1° radicals (compounds 4v and 4w). Subsequently, screening different 2° radicals yielded excellent results for 4x–4ad. In particular, carboxylic acids with multiple functional groups exhibited high reactivity, producing good yields for 4x–4z and showing excellent compatibility with our methodology. Finally, 3° radicals (4ae–4ag) gave the desired products in up to 70% yields. As for compounds 4aa–4ad it was possible to obtain derivatives showing orthogonality of the protecting groups of the nitrogen atoms.44 To further demonstrate the practicality of this protocol, we scaled-up the synthesis of 4a up to a 1 mmol scale using different setups (see Fig. S2 and S3 ESI†). The reaction performed well under both batch and flow conditions (>80% yield), thereby confirming the robustness of the methodology (Scheme 5).
 | ||
| Scheme 3 Reaction conditions: 1g (0.2 mmol), 2 (0.24 mmol), 3h-H (2.0 mmol) and TBADT (2 mol%) in 2.0 mL of acetone under N2. Reactions were irradiated with a 40 W Kessil lamp (370 nm) for 16 h; isolated yields. aEndo configuration bN-Boc piperidine as a byproduct. | ||
 | ||
| Scheme 4 Reaction conditions: 1 (0.2 mmol), 2 (0.24 mmol), 3h-H (2.0 mmol) and TBADT (2 mol%) in 2.0 mL of acetone under N2. Reactions were irradiated with a 40 W Kessil lamp (370 nm) for 16 h; isolated yields. | ||
 | ||
| Scheme 5 Synthesis of 4a on a 1 mmol scale under (a) batch, (b) flow, and (c) solar simulated conditions. Isolated yields of 4a. | ||
We also tested the reaction under solar simulated light and achieved a good yield of 4a (88% yield) (see Fig. S4 ESI†). This suggests that diverse building blocks can be efficiently accessed using sunlight as a green energy source.2a,45
Additionally, a reaction performed with TEMPO (5 equiv.) as a radical scavenger was completely inhibited, indicating that a radical step is involved in the reaction pathway. Finally, a light ON–OFF experiment provided no evidence of a radical chain propagation mechanism (see Fig. S5–S8 ESI†).
On the basis of the experiments performed and literature precedents, we propose the mechanistic scenario detailed in Scheme 6.46 Previous reports point to a smooth hydrogen abstraction of allylic hydrogens in tetramethylethylene and cyclohexene by excited TBADT.47 In addition, cyclohexadiene derivatives are known to be excellent H-donors in photocatalyzed HAT reactions. In fact, the rate constant for abstraction of the C–H bond in 1,4-cyclohexadiene (CHD) by excited TBADT is very high (3 × 108 M−1 s−1).48 Moreover, the rate constant for H-atom abstraction by OH˙ radicals is 1.7 times higher for γ-terpinene than for CHD.49 This opens the way for the use of γ-terpinene as an elective H-donor in photocatalyzed reactions.50 Thus, the TBADT excited state cleaves exclusively the weak allylic C–H bond in γ-terpinene, producing [W10O32]5− and radical 3h˙. Selective HAT from the allylic C–H bonds of 3h-H is due to its high H-donor capability along with its high concentration in solution causing no competitive cleavage of other C–H bonds present in other reaction partners. The so formed 3h˙ is a pro-aromatic radical51 that easily undergoes aromatization to p-cymene 5 (a well-documented process)48,49,52 which was confirmed by GC-MS analysis (see Fig. S9 ESI†). At this stage, direct reduction of NHPI esters seems unlikely due to the insufficient redox potential of the reduced TBADT [W10O32]4−/[W10O32]5− (E = −0.97 V vs. SCE).19a Accordingly, the disproportionation of [W10O32]5− to regenerate [W10O32]4− along with 2H+[W10O32]6− can be envisaged. In its doubly-reduced form, [W10O32]5−/[W10O32]6− (E = −1.48 V vs. SCE),19 TBADT can undergo an exergonic SET with NHPI 1 [Ered ≈ −1.3 V vs. SCE]27 (Scheme 6), while direct reduction of the SOMOphiles was safely excluded due to their higher redox potentials [Ered ≈ −2.5 V vs. SCE]24 except for the case of 2b.39 The resulting alkyl radical 1˙ undergoes a Giese-type addition to the electron-poor olefin 2, forming the radical adduct 6. The presence of an excess of 3h-H did not cause, however, the conversion of 1 to R–H (the rate constant of H-abstraction of alkyl radicals on CHD is ca. 4–5 × 105 M−1 s−1).53 Two pathways may be hypothesized at this stage for the release of the final product 4. First, the interaction of 6 with H+[W10O32]5− by back-HAT or reduction followed by protonation seems more plausible. The occurrence of the latter pathway is witnessed because the redox potential [Ered = −0.97 V vs. SCE] of H+[W10O32]5− is enough to reduce 6 (Ered [≈−0.6 vs. SCE])16b to the corresponding anion. An alternative pathway leveraging hydrogen abstraction from 3h-H by 6, however, may not be safely excluded.54
 | ||
| Scheme 6 Proposed mechanism. All the redox potentials are referred to the SCE. | ||
Conclusions
In summary, we have developed a practical and sustainable alkylation method based on SET processes with TBADT serving as an efficient reductant. γ-Terpinene is the elective hydrogen donor for the full exploitation of our methodology to synthesize key building blocks (e.g. unnatural α-amino acids) under robust and mild conditions. The reaction could be carried out in a 1 mmol scale (under batch or flow conditions or under solar simulated sunlight). We extended the application of TBADT beyond its conventional role in C–H activation, demonstrating that this photocatalyst has yet to reveal its full potential.Author contributions
This work was conceptualized by Prof. M. F. and experimentation was performed by M. L. and D. A. The first draft of the manuscript was prepared by M. L. and the final version was edited and revised by M. L. and Prof. M. F.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the support from UniPV and MUR through the program ‘Departments of Excellence’ (2023-2027). M. L. is thankful for the financial support provided by the Project PRIN PNRR ‘LIGHT CAT’ (no. P2022RHMCM) supported by the European Commission – NextGeneration EU programme.References
- (a) M. D. Kärkäs, B. S. Matsuura and C. R. J. Stephenson, Enchained by visible light–mediated photoredox catalysis, Science, 2015, 349, 1285–1286 CrossRef PubMed; (b) R. A. Angnes, Z. Li, C. R. D. Correia and G. B. Hammond, Recent synthetic additions to the visible light photoredox catalysis toolbox, Org. Biomol. Chem., 2015, 13, 9152–9167 RSC; (c) M. H. Shaw, J. Twilton and D. W. C. MacMillan, Photoredox Catalysis in Organic Chemistry, J. Org. Chem., 2016, 81, 6898–6926 CrossRef CAS PubMed; (d) D. Ravelli, S. Protti and M. Fagnoni, Carbon–Carbon Bond Forming Reactions via Photogenerated Intermediates, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (e) K. L. Skubi, T. R. Blum and T. P. Yoon, Dual Catalysis Strategies in Photochemical Synthesis, Chem. Rev., 2016, 116, 10035–10074 CrossRef CAS PubMed; (f) S. Crespi and M. Fagnoni, Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy, Chem. Rev., 2020, 120, 9790–9833 CrossRef CAS PubMed.
- (a) D. Ravelli, S. Protti and M. Fagnoni, Decatungstate Anion for Photocatalyzed “Window Ledge” Reactions, Acc. Chem. Res., 2016, 49, 2232–2242 CrossRef CAS PubMed; (b) B.-C. Hong and R. R. Indurmuddam, Tetrabutylammonium Decatungstate (TBADT), a Compelling and Trailblazing Catalyst for Visible-Light-Induced Organic Photocatalysis, Org. Biomol. Chem., 2024, 22, 3799–3842 RSC.
- K. Nomiya, Y. Sugie, T. Miyazaki and M. Miwa, Catalysis by heteropolyacids—ix. Photocatalytic oxidation of isopropyl alcohol to acetone under oxygen using tetrabutylammonium decatungstate, Polyhedron, 1986, 5, 1267–1271 CrossRef CAS.
- D. Dondi, M. Fagnoni and A. Albini, Tetrabutylammonium Decatungstate-Photosensitized Alkylation of Electrophilic Alkenes: Convenient Functionalization of Aliphatic C-H Bonds, Chem. – Eur. J., 2006, 12, 4153–4163 CrossRef CAS PubMed.
- (a) C. Tanielian, Decatungstate Photocatalysis, Coord. Chem. Rev., 1998, 178–180, 1165–1181 CrossRef CAS; (b) D. Ravelli, D. Dondi, M. Fagnoni, A. Albini and A. Bagno, Electronic and EPR Spectra of the Species Involved in [W10O32]4− Photocatalysis. A Relativistic DFT Investigation, Phys. Chem. Chem. Phys., 2013, 15, 2890–2896 RSC.
- (a) D. Ravelli, A. Albini and M. Fagnoni, Smooth Photocatalytic Preparation of 2−Substituted 1,3−Benzodioxoles, Chem. – Eur. J., 2011, 17, 572–579 CrossRef CAS PubMed; (b) J. J. Murphy, D. Bastida, S. Paria, M. Fagnoni and P. Melchiorre, Asymmetric Catalytic Formation of Quaternary Carbons by Iminium Ion Trapping of Radicals, Nature, 2016, 532, 218–222 CrossRef CAS PubMed; (c) D. Ravelli, M. Zoccolillo, M. Mella and M. Fagnoni, Photocatalytic Synthesis of Oxetane Derivatives by Selective C-H Activation, Adv. Synth. Catal., 2014, 356, 2781–2786 CrossRef CAS; (d) M. Leone, J. P. Milton, D. Gryko, L. Neuville and G. Masson, TBADT–Mediated Photocatalytic Stereoselective Radical Alkylation of Chiral N–Sulfinyl Imines: Towards Efficient Synthesis of Diverse Chiral Amines, Chem. – Eur. J., 2024, 30, e202400363 CrossRef CAS PubMed; (e) Z.-Y. Dai, Z.-S. Nong and P.-S. Wang, Light-Mediated Asymmetric Aliphatic C–H Alkylation with Hydrogen Atom Transfer Catalyst and Chiral Phosphoric Acid, ACS Catal., 2020, 10, 4786–4790 CrossRef CAS; (f) G. Laudadio, Y. Deng, K. Van Der Wal, D. Ravelli, M. Nuño, M. Fagnoni, D. Guthrie, Y. Sun and T. Noël, C(sp3)–H Functionalizations of Light Hydrocarbons Using Decatungstate Photocatalysis in Flow, Science, 2020, 369, 92–96 CrossRef CAS PubMed; (g) M. C. Quattrini, S. Fujii, K. Yamada, T. Fukuyama, D. Ravelli, M. Fagnoni and I. Ryu, Dehydrogenative Coupling of Heteroaromatics and Hydrogen Donors via Decatungstate Photocatalysis, Chem. Commun., 2017, 53, 2335–2338 RSC; (h) W. Ma, M. Leone, E. Derat, P. Retailleau, C. R. Reddy, L. Neuville and G. Masson, Photocatalytic Asymmetric Acyl Radical Truce–Smiles Rearrangement for the Synthesis of Enantioenriched α–Aryl Amides, Angew. Chem., Int. Ed., 2024, 63, e202408154 CrossRef CAS PubMed.
- (a) I. Ryu, A. Tani, T. Fukuyama, D. Ravelli, S. Montanaro and M. Fagnoni, Efficient C–H/C–N and C–H/C–CO–N Conversion via Decatungstate-Photoinduced Alkylation of Diisopropyl Azodicarboxylate, Org. Lett., 2013, 15, 2554–2557 CrossRef CAS PubMed; (b) C. R. Zwick and H. Renata, Remote C–H Hydroxylation by an α-Ketoglutarate-Dependent Dioxygenase Enables Efficient Chemoenzymatic Synthesis of Manzacidin C and Proline Analogs, J. Am. Chem. Soc., 2018, 140, 1165–1169 CrossRef CAS PubMed; (c) Y.-C. Lu, S.-C. Kao and J. G. West, Decatungstate-Photocatalysed C(sp3)–H Azidation, Chem. Commun., 2022, 58, 4869–4872 RSC; (d) P. Xie, S. Shi, X. Hu, C. Xue and D. Du, Sunlight Photocatalytic Synthesis of Aryl Hydrazides by Decatungstate–Promoted Acylation under Room Temperature, ChemistrySelect, 2021, 6, 3922–3925 CrossRef CAS; (e) T. Wan, L. Capaldo, G. Laudadio, A. V. Nyuchev, J. A. Rincón, P. García-Losada, C. Mateos, M. O. Frederick, M. Nuño and T. Noël, Decatungstate–Mediated C(sp3)–H Heteroarylation via Radical–Polar Crossover in Batch and Flow, Angew. Chem., Int. Ed., 2021, 60, 17893–17897 CrossRef CAS PubMed.
- (a) S. D. Halperin, D. Kwon, M. Holmes, E. L. Regalado, L.-C. Campeau, D. A. DiRocco and R. Britton, Development of a Direct Photocatalytic C–H Fluorination for the Preparative Synthesis of Odanacatib, Org. Lett., 2015, 17, 5200–5203 CrossRef CAS PubMed; (b) M. B. Nodwell, A. Bagai, S. D. Halperin, R. E. Martin, H. Knust and R. Britton, Direct Photocatalytic Fluorination of Benzylic C–H Bonds with N-Fluorobenzenesulfonimide, Chem. Commun., 2015, 51, 11783–11786 RSC.
- (a) C. L. Hill, D. A. Bouchard, M. Kadkhodayan, M. M. Williamson, J. A. Schmidt and E. F. Hilinski, Catalytic Photochemical Oxidation of Organic Substrates by Polyoxometalates. Picosecond Spectroscopy, Photochemistry, and Structural Properties of Charge-Transfer Complexes between Heteropolytungstic Acids and Dipolar Organic Compounds, J. Am. Chem. Soc., 1988, 110, 5471–5479 CrossRef CAS; (b) D. M. Schultz, F. Lévesque, D. A. DiRocco, M. Reibarkh, Y. Ji, L. A. Joyce, J. F. Dropinski, H. Sheng, B. D. Sherry and I. W. Davies, Oxyfunctionalization of the Remote C−H Bonds of Aliphatic Amines by Decatungstate Photocatalysis, Angew. Chem., Int. Ed., 2017, 56, 15274–15278 CrossRef CAS PubMed; (c) G. Laudadio, S. Govaerts, Y. Wang, D. Ravelli, H. F. Koolman, M. Fagnoni, S. W. Djuric and T. Noël, Selective C(sp3)−H Aerobic Oxidation Enabled by Decatungstate Photocatalysis in Flow, Angew. Chem., Int. Ed., 2018, 57, 4078–4082 CrossRef CAS PubMed; (d) W. Wu, Z. Fu, S. Tang, S. Zou, X. Wen, Y. Meng, S. Sun, J. Deng, Y. Liu and D. Yin, (nBu4N)4W10O32-Catalyzed Selective Oxygenation of Cyclohexane by Molecular Oxygen under Visible Light Irradiation, Appl. Catal., B, 2015, 164, 113–119 CrossRef CAS.
- (a) R. F. Renneke and C. L. Hill, Selective Photochemical Dehydrogenation of Saturated Hydrocarbons with Quantum Yields Approaching Unity, Angew. Chem., Int. Ed. Engl., 1988, 27, 1526–1527 CrossRef; (b) J. G. West, D. Huang and E. J. Sorensen, Acceptorless Dehydrogenation of Small Molecules through Cooperative Base Metal Catalysis, Nat. Commun., 2015, 6, 10093 CrossRef PubMed; (c) H. Cao, Y. Kuang, X. Shi, K. L. Wong, B. B. Tan, J. M. C. Kwan, X. Liu and J. Wu, Photoinduced Site-Selective Alkenylation of Alkanes and Aldehydes with Aryl Alkenes, Nat. Commun., 2020, 11, 1956 CrossRef CAS PubMed.
- (a) M. Okada, T. Fukuyama, K. Yamada, I. Ryu, D. Ravelli and M. Fagnoni, Sunlight Photocatalyzed Regioselective β-Alkylation and Acylation of Cyclopentanones, Chem. Sci., 2014, 5, 2893–2898 RSC; (b) K. Yamada, T. Fukuyama, S. Fujii, D. Ravelli, M. Fagnoni and I. Ryu, Cooperative Polar/Steric Strategy in Achieving Site–Selective Photocatalyzed C(sp3)−H Functionalization, Chem. – Eur. J., 2017, 23, 8615–8618 CrossRef CAS PubMed; (c) D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa and I. Ryu, Site-Selective C–H Functionalization by Decatungstate Anion Photocatalysis: Synergistic Control by Polar and Steric Effects Expands the Reaction Scope, ACS Catal., 2018, 8, 701–713 CrossRef CAS.
- P. Bellotti, H.-M. Huang, T. Faber and F. Glorius, Photocatalytic Late-Stage C–H Functionalization, Chem. Rev., 2023, 123, 4237–4352 CrossRef CAS PubMed.
- V. D. Waele, O. Poizat, M. Fagnoni, A. Bagno and D. Ravelli, Unraveling the Key Features of the Reactive State of Decatungstate Anion in Hydrogen Atom Transfer (HAT) Photocatalysis, ACS Catal., 2016, 6, 7174–7182 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (b) N. A. Romero and D. A. Nicewicz, Organic Photoredox Catalysis, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- D. C. Duncan and M. A. Fox, Early Events in Decatungstate Photocatalyzed Oxidations: A Nanosecond Laser Transient Absorbance Reinvestigation, J. Phys. Chem. A, 1998, 102, 4559–4567 CrossRef CAS.
- (a) S. Montanaro, D. Ravelli, D. Merli, M. Fagnoni and A. Albini, Decatungstate as Photoredox Catalyst: Benzylation of Electron-Poor Olefins, Org. Lett., 2012, 14, 4218–4221 CrossRef CAS PubMed; (b) L. Capaldo, R. Riccardi, D. Ravelli and M. Fagnoni, Acyl Radicals from Acylsilanes: Photoredox-Catalyzed Synthesis of Unsymmetrical Ketones, ACS Catal., 2018, 8, 304–309 CrossRef CAS.
- (a) L. Capaldo, L. Buzzetti, D. Merli, M. Fagnoni and D. Ravelli, Smooth Photocatalyzed Benzylation of Electrophilic Olefins via Decarboxylation of Arylacetic Acids, J. Org. Chem., 2016, 81, 7102–7109 CrossRef CAS PubMed; (b) A. Krech, V. Yakimchyk, T. Jarg, D. Kananovich and M. Ošeka, Ring–Opening Coupling Reaction of Cyclopropanols with Electrophilic Alkenes Enabled by Decatungstate as Photoredox Catalyst, Adv. Synth. Catal., 2024, 366, 91–100 CrossRef CAS.
- S. Angioni, D. Ravelli, D. Emma, D. Dondi, M. Fagnoni and A. Albini, Tetrabutylammonium Decatungstate (Chemo)Selective Photocatalyzed, Radical C-H Functionalization in Amides, Adv. Synth. Catal., 2008, 350, 2209–2214 CrossRef CAS.
- (a) R. F. Renneke, M. Pasquali and C. L. Hill, Polyoxometalate Systems for the Catalytic Selective Production of Nonthermodynamic Alkenes from Alkanes. Nature of Excited-State Deactivation Processes and Control of Subsequent Thermal Processes in Polyoxometalate Photoredox Chemistry, J. Am. Chem. Soc., 1990, 112, 6585–6594 CrossRef CAS; (b) P. J. Sarver, N. B. Bissonnette and D. W. C. MacMillan, Decatungstate-Catalyzed C(sp3)–H Sulfinylation: Rapid Access to Diverse Organosulfur Functionality, J. Am. Chem. Soc., 2021, 143, 9737–9743 CrossRef CAS PubMed.
- I. B. Perry, T. F. Brewer, P. J. Sarver, D. M. Schultz, D. A. DiRocco and D. W. C. MacMillan, Direct Arylation of Strong Aliphatic C–H Bonds, Nature, 2018, 560, 70–75 CrossRef CAS PubMed.
- (a) D. Mazzarella, A. Pulcinella, L. Bovy, R. Broersma and T. Noël, Rapid and Direct Photocatalytic C(sp3)−H Acylation and Arylation in Flow, Angew. Chem., Int. Ed., 2021, 60, 21277–21282 CrossRef CAS PubMed; (b) P. Fan, Y. Lan, C. Zhang and C. Wang, Nickel/Photo-Cocatalyzed Asymmetric Acyl-Carbamoylation of Alkenes, J. Am. Chem. Soc., 2020, 142, 2180–2186 CrossRef CAS PubMed; (c) Y. Jin, E. W. H. Ng, T. Fan, H. Hirao and L.-Z. Gong, Photochemical Allylation of Alkanes Enabled by Nickel Catalysis, ACS Catal., 2022, 12, 10039–10046 CrossRef CAS; (d) P. Martínez-Balart, B. L. Tóth, Á. Velasco-Rubio and M. Fañanás-Mastral, Direct C–H Allylation of Unactivated Alkanes by Cooperative W/Cu Photocatalysis, Org. Lett., 2022, 24, 6874–6879 CrossRef PubMed; (e) S. Xu, H. Chen, Z. Zhou and W. Kong, Three–Component Alkene Difunctionalization by Direct and Selective Activation of Aliphatic C−H Bonds, Angew. Chem., Int. Ed., 2021, 60, 7405–7411 CrossRef CAS PubMed; (f) V. Murugesan, A. Ganguly, A. Karthika and R. Rasappan, C–H Alkylation of Aldehydes by Merging TBADT Hydrogen Atom Transfer with Nickel Catalysis, Org. Lett., 2021, 23, 5389–5393 CrossRef CAS PubMed; (g) P. Fan, Z. Chen and C. Wang, Nickel/Photo-Cocatalyzed Three-Component Alkyl-Acylation of Aryl-Activated Alkenes, Org. Lett., 2023, 25, 8877–8882 CrossRef CAS PubMed.
- (a) Q. Wang, S. Ni, X. Wang, Y. Wang and Y. Pan, Visible-Light-Mediated Tungsten-Catalyzed C-H Amination of Unactivated Alkanes with Nitroarenes, Sci. China: Chem., 2022, 65, 678–685 CrossRef CAS; (b) E. Mao and D. W. C. MacMillan, Late-Stage C(sp3)–H Methylation of Drug Molecules, J. Am. Chem. Soc., 2023, 145, 2787–2793 CrossRef CAS PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Direct Photocatalyzed Hydrogen Atom Transfer (HAT) for Aliphatic C–H Bonds Elaboration, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Mechanistic Studies in Photocatalysis, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- (a) L. Li, Y. Yao and N. Fu, Free Carboxylic Acids: The Trend of Radical Decarboxylative Functionalization, Eur. J. Org. Chem., 2023, e202300166 CrossRef CAS; (b) C. Anyaegbu, G. Vidali, D. Haridas and J. F. Hooper, Radical Decarboxylation: An Emerging Tool in Polymer Synthesis, Polym. Chem., 2024, 15, 2537–2547 RSC; (c) S. Bonciolini, A. Pulcinella, M. Leone, D. Schiroli, A. L. Ruiz, A. Sorato, M. A. J. Dubois, R. Gopalakrishnan, G. Masson, N. Della Ca’, S. Protti, M. Fagnoni, E. Zysman-Colman, M. Johansson and T. Noël, Metal-Free Photocatalytic Cross-Electrophile Coupling Enables C1 Homologation and Alkylation of Carboxylic Acids with Aldehydes, Nat. Commun., 2024, 15, 1509 CrossRef CAS PubMed.
- (a) K. Okada, K. Okamoto, N. Morita, K. Okubo and M. Oda, Photosensitized Decarboxylative Michael Addition through N-(Acyloxy)Phthalimides via an Electron-Transfer Mechanism, J. Am. Chem. Soc., 1991, 113, 9401–9402 CrossRef CAS; (b) S. K. Parida, T. Mandal, S. Das, S. K. Hota, S. De Sarkar and S. Murarka, Single Electron Transfer-Induced Redox Processes Involving N-(Acyloxy)Phthalimides, ACS Catal., 2021, 11, 1640–1683 CrossRef CAS.
- (a) J. Schwarz and B. König, Metal-Free, Visible-Light-Mediated, Decarboxylative Alkylation of Biomass-Derived Compounds, Green Chem., 2016, 18, 4743–4749 RSC; (b) A. Claraz, C. Allain and G. Masson, Electroreductive Cross–Coupling of Trifluoromethyl Alkenes and Redox Active Esters for the Synthesis of Gem–Difluoroalkenes, Chem. – Eur. J., 2022, 28, e202103337 CrossRef CAS PubMed.
- (a) G. Pratsch, G. L. Lackner and L. E. Overman, Constructing Quaternary Carbons from N -(Acyloxy)phthalimide Precursors of Tertiary Radicals Using Visible-Light Photocatalysis, J. Org. Chem., 2015, 80, 6025–6036 CrossRef CAS PubMed; (b) X. Lu, B. Xiao, L. Liu and Y. Fu, Formation of C(sp3)–C(sp3) Bonds through Nickel–Catalyzed Decarboxylative Olefin Hydroalkylation Reactions, Chem. – Eur. J., 2016, 22, 11161–11164 CrossRef CAS PubMed; (c) Y. Zhao, J.-R. Chen and W.-J. Xiao, Visible-Light Photocatalytic Decarboxylative Alkyl Radical Addition Cascade for Synthesis of Benzazepine Derivatives, Org. Lett., 2018, 20, 224–227 CrossRef CAS PubMed; (d) C. Zheng, G. Wang and R. Shang, Catalyst–free Decarboxylation and Decarboxylative Giese Additions of Alkyl Carboxylates through Photoactivation of Electron Donor–Acceptor Complex, Adv. Synth. Catal., 2019, 361, 4500–4505 CrossRef CAS; (e) L.-Y. Zeng, P.-Z. Qu, M. Tao, G. Pu, J. Jia, P. Wang, M. Shang, X. Li and C.-Y. He, Synthesis of Alkylated Polyfluorobenzenes through Decarboxylative Giese Addition of Aliphatic N -Hydroxyphthalimide Esters with Polyfluorostyrene, J. Org. Chem., 2023, 88, 14105–14114 CrossRef CAS PubMed; (f) D. M. Kitcatt, E. Pogacar, L. Mi, S. Nicolle and A.-L. Lee, Light-Mediated Direct Decarboxylative Giese Aroylations without a Photocatalyst, J. Org. Chem., 2024, 89, 16055–16059 CrossRef CAS PubMed; (g) D. M. Kitcatt, K. A. Scott, E. Rongione, S. Nicolle and A.-L. Lee, Direct decarboxylative Giese amidations: photocatalytic vs. metal- and light-free, Chem. Sci., 2023, 14, 9806–9813 RSC; (h) X. Jin and L. Zhang, Expedient access to N-alkylphthalimides via redox-neutral photocatalysed Giese-type reactions, Org. Biomol. Chem., 2022, 20, 5377–5382 RSC; (i) C. S. Batsika, N. A. Stini and C. G. Kokotos, Visible Light–Mediated Decarboxylative Giese Addition Utilizing Thioxanthone or Thioxanthone–TfOH Complex as the Photocatalyst, Chem. – Eur. J., 2025, 31, e202404483 CrossRef CAS PubMed.
- (a) S. Garbarino, S. Protti, S. Gabrielli, M. Fagnoni, A. Palmieri and D. Ravelli, Multi-Step Continuous Flow Synthesis of β/γ-Substituted Ketones, ChemPhotoChem, 2018, 2, 847–850 CrossRef CAS; (b) T. Fukuyama, K. Yamada, T. Nishikawa, D. Ravelli, M. Fagnoni and I. Ryu, Site-selectivity in TBADT-photocatalyzed C(sp3)–H Functionalization of Saturated Alcohols and Alkanes, Chem. Lett., 2018, 47, 207–209 CrossRef CAS; (c) C. Raviola, S. Protti, D. Ravelli and M. Fagnoni, Photogenerated acyl/alkoxycarbonyl/carbamoyl radicals for sustainable synthesis, Green Chem., 2019, 21, 748–764 RSC.
- (a) Y. Fu, Z. Wang, Y. Zhang, G. Shen and X. Zhu, Quantitative Evaluation of the Hydrogen–Donating Abilities of Amines and Amides in Acetonitrile, ChemistrySelect, 2022, 7, e202202625 CrossRef CAS; (b) W. Tantawy and H. Zipse, Hydroxylic Solvents as Hydrogen Atom Donors in Radical Reactions, Eur. J. Org. Chem., 2007, 5817–5820 CrossRef CAS; (c) A. Gansäuer, L. Shi, M. Otte, I. Huth, A. Rosales, I. Sancho-Sanz, N. M. Padial and J. E. Oltra, Hydrogen Atom Donors: Recent Developments, in Radicals in Synthesis III, ed. M. Heinrich and A. Gansäuer, Springer Berlin Heidelberg, Berlin, Heidelberg, 2011, vol. 320, pp. 93–120 Search PubMed.
- (a) M. Salamone, M. Galeotti, E. Romero-Montalvo, J. A. Van Santen, B. D. Groff, J. M. Mayer, G. A. DiLabio and M. Bietti, Bimodal Evans–Polanyi Relationships in Hydrogen Atom Transfer from C(sp3)–H Bonds to the Cumyloxyl Radical. A Combined Time-Resolved Kinetic and Computational Study, J. Am. Chem. Soc., 2021, 143, 11759–11776 CrossRef CAS PubMed; (b) Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, 1st edn, CRC Press, 2007 CrossRef.
- B. Taleb, R. Jahjah, D. Cornu, M. Bechelany, M. Al Ajami, G. Kataya, A. Hijazi and M. H. El-Dakdouki, Exploring Hydrogen Sources in Catalytic Transfer Hydrogenation: A Review of Unsaturated Compound Reduction, Molecules, 2023, 28, 7541 CrossRef CAS PubMed.
- (a) D. J. Van Hoomissen and S. Vyas, Impact of Conjugation and Hyperconjugation on the Radical Stability of Allylic and Benzylic Systems: A Theoretical Study, J. Org. Chem., 2017, 82, 5731–5742 CrossRef CAS PubMed; (b) F. S. Sharopov, M. Wink and W. N. Setzer, Radical Scavenging and Antioxidant Activities of Essential Oil Components – An Experimental and Computational Investigation, Nat. Prod. Commun., 2015, 10, 153–156 CrossRef PubMed.
- S. M. Nabavi, A. Marchese, M. Izadi, V. Curti, M. Daglia and S. F. Nabavi, Plants Belonging to the Genus Thymus as Antibacterial Agents: From Farm to Pharmacy, Food Chem., 2015, 173, 339–347 CrossRef CAS PubMed.
- https://www.sigmaaldrich.com/IT/it [accessed on 13/01/2025].
- C. S. Funari, R. L. Carneiro, M. M. Khandagale, A. J. Cavalheiro and E. F. Hilder, Acetone as a Greener Alternative to Acetonitrile in Liquid Chromatographic Fingerprinting, J. Sep. Sci., 2015, 38, 1458–1465 CrossRef CAS PubMed.
- (a) A. A. Zavitsas, D. W. Rogers and N. Matsunaga, Shortcomings of Basing Radical Stabilization Energies on Bond Dissociation Energies of Alkyl Groups to Hydrogen, J. Org. Chem., 2010, 75, 5697–5700 CrossRef CAS PubMed; (b) M. D. Wodrich, W. C. McKee and P. V. R. Schleyer, On the Advantages of Hydrocarbon Radical Stabilization Energies Based on R−H Bond Dissociation Energies, J. Org. Chem., 2011, 76, 2439–2447 CrossRef CAS PubMed.
- (a) L. Capaldo, D. Merli, M. Fagnoni and D. Ravelli, Visible Light Uranyl Photocatalysis: Direct C–H to C–C Bond Conversion, ACS Catal., 2019, 9, 3054–3058 CrossRef CAS; (b) R. Rutkauskaite, X. Zhang, A. W. Woodward, Y. Liu, G. Herrera, J. Purkis, S. D. Woodall, M. Sarsfield, G. Schreckenbach, L. S. Natrajan and P. L. Arnold, The effect of ancillary ligands on hydrocarbon C–H bond functionalization by uranyl photocatalysts, Chem. Sci., 2024, 15, 6965–6978 RSC; (c) D. Liu, A. Hazra, X. Liu, R. Maity, T. Tan and L. Luo, CdS Quantum Dot Gels as a Direct Hydrogen Atom Transfer Photocatalyst for C−H Activation, Angew. Chem., Int. Ed., 2024, 63, e202403186 CrossRef CAS PubMed.
- R. O. Loutfy, C. K. Hsiao, B. S. Ong and B. Keoshkerian, Electrochemical evaluation of electron acceptor materials, Can. J. Chem., 1984, 62, 1877–1885 CrossRef CAS.
- R. Salgado, A. Oehmen, G. Carvalho, J. P. Noronha and M. A. M. Reis, Biodegradation of Clofibric Acid and Identification of Its Metabolites, J. Hazard. Mater., 2012, 241–242, 182–189 CrossRef CAS PubMed.
- (a) A. D. Farahani, A. D. Martin, H. Iranmanesh, M. M. Bhadbhade, J. E. Beves and P. Thordarson, Gel– and Solid–State–Structure of Dialanine and Diphenylalanine Amphiphiles: Importance of C-H Interactions in Gelation, ChemPhysChem, 2019, 20, 972–983 CrossRef CAS PubMed; (b) I. W. Hamley, Small Bioactive Peptides for Biomaterials Design and Therapeutics, Chem. Rev., 2017, 117, 14015–14041 CrossRef CAS PubMed.
- (a) Unnatural Amino Acids: Methods and Protocols, ed. L. Pollegioni and S. Servi, Humana Press, Totowa, NJ, 2012, vol. 794 Search PubMed; (b) T. Narancic, S. A. Almahboub and K. E. O'Connor, Unnatural Amino Acids: Production and Biotechnological Potential, World J. Microbiol. Biotechnol., 2019, 35, 67 CrossRef PubMed.
- (a) C. Che, Y. Li, X. Cheng, Y. Lu and C. Wang, Visible–Light–Enabled Enantioconvergent Synthesis of α–Amino Acid Derivatives via Synergistic Brønsted Acid/Photoredox Catalysis, Angew. Chem., Int. Ed., 2021, 60, 4698–4704 CrossRef CAS PubMed; (b) R. Qi, C. Wang, Y. Huo, H. Chai, H. Wang, Z. Ma, L. Liu, R. Wang and Z. Xu, Visible Light Induced Cu-Catalyzed Asymmetric C(sp3)–H Alkylation, J. Am. Chem. Soc., 2021, 143, 12777–12783 CrossRef CAS PubMed; (c) Z. Gu, L. Zhang, H. Li, S. Cao, Y. Yin, X. Zhao, X. Ban and Z. Jiang, Deracemization through Sequential Photoredox–Neutral and Chiral Brønsted Acid Catalysis, Angew. Chem., Int. Ed., 2022, 61, e202211241 CrossRef CAS PubMed; (d) Y. Cui, J. Ai, Y. Duan, M. Jia, T. Ouyang, A. Liu, L. Yu, J. Liu, X. Liu, C. Chu, Y. Li, Y. Ma, L. Chen, L. Han, J. Chen, C. Tian, S. Che and Y. Fang, Enantioselective synthesis of amino acids by photocatalytic reduction of CO2 on chiral mesostructured ZnS, Chem, 2025, 11, 102390 CrossRef.
- F. Albericio, Orthogonal Protecting Groups for Nα-Amino and C-Terminal Carboxyl Functions in Solid-Phase Peptide Synthesis, Biopolymers, 2000, 55, 123–139 CrossRef CAS PubMed.
- S. Protti, D. Ravelli, M. Fagnoni and A. Albini, Solar Light-Driven Photocatalyzed Alkylations. Chemistry on the Window Ledge, Chem. Commun., 2009, 7351 RSC.
- (a) B. Schweitzer-Chaput, M. A. Horwitz, E. de Pedro Beato and P. Melchiorre, Photochemical Generation of Radicals from Alkyl Electrophiles Using a Nucleophilic Organic Catalyst, Nat. Chem., 2019, 11, 129–135 CrossRef CAS PubMed; (b) J. Davies, T. D. Svejstrup, D. Fernandez Reina, N. S. Sheikh and D. Leonori, Visible-Light-Mediated Synthesis of Amidyl Radicals: Transition-Metal-Free Hydroamination and N -Arylation Reactions, J. Am. Chem. Soc., 2016, 138, 8092–8095 CrossRef CAS PubMed.
- A. Molinari, R. Amadelli, V. Carassiti and A. Maldotti, Photocatalyzed Oxidation of Cyclohexene and Cyclooctene with (nBu4N)4W10O32 and (nBu4N)4W10O32/FeIII[Meso-Tetrakis(2,6-Dichlorophenyl)Porphyrin] in Homogeneous and Heterogeneous Systems, Eur. J. Inorg. Chem., 2000, 2000, 91–96 CrossRef.
- S. Didarataee, A. Suprun, N. Joshi and J. C. Scaiano, NIR phosphorescence from decatungstate anions allows the conclusive characterization of its elusive excited triplet behaviour and kinetics, Chem. Commun., 2024, 60, 1896–1899 RSC.
- S. M. Aschmann, J. Arey and R. Atkinson, Formation of P-Cymene from OH + γ-Terpinene: H-Atom Abstraction from the Cyclohexadiene Ring Structure, Atmos. Environ., 2011, 45, 4408–4411 CrossRef CAS.
- For selected examples see: (a) J.-Q. Chen, R. Chang, J.-B. Lin, Y.-C. Luo and P.-F. Xu, Photoredox-Induced Intramolecular 1,5-H Transfer Reaction of Aryl Iodides for the Synthesis of Spirocyclic γ-Lactams, Org. Lett., 2018, 20, 2395–2398 CrossRef CAS PubMed; (b) W. Zhou, I. A. Dmitriev and P. Melchiorre, Reductive Cross-Coupling of Olefins via a Radical Pathway, J. Am. Chem. Soc., 2023, 145, 25098–25102 CrossRef CAS PubMed.
- (a) J. C. Walton and A. Studer, Evolution of Functional Cyclohexadiene-Based Synthetic Reagents: The Importance of Becoming Aromatic, Acc. Chem. Res., 2005, 38, 794–802 CrossRef CAS PubMed; (b) A. Bhunia and A. Studer, Recent Advances in Radical Chemistry Proceeding through Pro-Aromatic Radicals, Chem, 2021, 7, 2060–2100 CrossRef CAS; (c) S. Didarataee, J. Ong, A. Suprun, N. Joshi and J. C. Scaiano, Kinetics, quantum yield and mechanism of the decatungstate-catalyzed photooxidation of C–H hydrogen donors: role of the persistent radical effect, Catal. Sci. Technol., 2025, 15, 1149–1156 RSC.
- M. C. Foti, C. Rocco, Z. Jin and R. Amorati, Rate Constants for H-Atom Abstraction by HOO˙ from H-Donor Compounds of Antioxidant Relevance, New J. Chem., 2024, 48, 16047–16056 RSC.
- (a) J. A. Hawari, P. S. Engel and D. Griller, Rate Constants for the Reactions of Alkyl Radicals with 1,4−cyclohexadiene, Int. J. Chem. Kinet., 1985, 17, 1215–1219 CrossRef CAS; (b) M. Newcomb and S. Un. Park, N-Hydroxypyridine-2-Thione Esters as Radical Precursors in Kinetic Studies. Measurements of Rate Constants for Hydrogen-Atom-Abstraction Reactions, J. Am. Chem. Soc., 1986, 108, 4132–4134 CrossRef CAS.
- (a) L. Jackson and J. C. Walton, The Efficiency of Alkyl Radical Generation and Hydrogen Transfer from 1-Alkylcyclohexa-2,5-Diene-1-Carboxylic Acids, Tetrahedron Lett., 1999, 40, 7019–7021 CrossRef CAS; (b) H. Togo, R. Taguchi, K. Yamaguchi and M. Yokoyama, Reactivity of [Bis(1-Adamantylcarbonyloxy)Iodo]Arenes in Substitution and Addition Reactions, J. Chem. Soc., Perkin Trans. 1, 1995, 2135 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, characterization data, and original 1H and 13C NMR spectra. See DOI: https://doi.org/10.1039/d5qo00399g |
| This journal is © the Partner Organisations 2025 |