DOI:
10.1039/D5QO00509D
(Research Article)
Org. Chem. Front., 2025, Advance Article
Design and synthetic utility of new HAT organocatalysts derived from commercially available diamines†‡
Received
15th March 2025
, Accepted 2nd May 2025
First published on 6th May 2025
Abstract
A series of hydrogen-atom transfer (HAT) organocatalysts were conveniently prepared from commercially available diamine compounds, and their utility in photoinduced HAT catalysis ability was investigated. The combination of these readily available HAT organocatalysts with the Fukuzumi photoredox catalyst enables efficient and site-selective C–H alkylation of various functionalized substrates ranging from simple hydrocarbons to complex molecules. Notably, the sequential one-pot photoinduced dialkylations of bifunctional substrates can be realized. Mechanistic studies suggested that the 1-naphthylmethyl moiety on one nitrogen atom of the diamine compounds plays a crucial role in the reaction by inducing the facile generation of a cationic aminium radical on the other nitrogen of the diamine as an active intermediate for the HAT process.
Introduction
The site-selective functionalization of various C–H bonds in simple and complex organic compounds provides an attractive methodology for the preparation of value-added functionalized products with complex organic structures.1–6 This methodology has high atom and step economy, and allows the facile generation of reactive intermediates for the introduction of specific functional groups, thereby facilitating the straightforward synthesis of pharmaceuticals, agrochemicals and polymer materials.7–12 In recent years, there have been numerous reports on the radical-mediated C–H functionalization of organic molecules.13–18 In these examples, the hydrogen-atom transfer (HAT) process, in which a proton and an electron are transferred from a hydrogen donor to an acceptor in a single step, has attracted attention as a powerful strategy to realize catalytic C–H functionalization via radical intermediates.19–23 Traditional methods for the generation of radical species require the use of toxic and/or hazardous radical initiators such as AIBN24 or Bu3SnH25 under harsh reaction conditions.7 In contrast, recent advances in this exciting field have introduced mild and practical methodologies that use photocatalysis and electrochemistry, whilst catalysts such as transition metal catalysts26–28 and organocatalysts,29–33 which are responsible for the HAT process, have been developed. As early as the 1900s, the generation of an aminium radical cation from N-halogenated amines via thermal or photochemical methods was reported.34,35 These N-radical cations can abstract a hydrogen atom from a C–H bond by an efficient HAT process.36 In addition, many types of organocatalysts for radical HAT processes have been reported. Amine-based organocatalysts are known to be representative models of organocatalysts for the HAT process. For example, catalysts based on quinuclidine,37–41 1,4-diazabicyclo[2.2.2]octane42,43 and piperidine36 that take advantage of the HAT ability of N-radical cations have been developed in recent years. The aminium radical cation is a key species in these catalysts. In this context, we are interested in the possibility of designing new, readily accessible HAT organocatalysts of type 1 (Fig. 1) from commercially available diamine compounds. Our strategy is to introduce a 1-naphthylmethyl moiety onto one nitrogen atom of the diamine compounds, thereby inducing the more favored photooxidation step on the naphthalene ring in 1 first, before the facile electron transfer from a lone pair electron of the other nitrogen atom to the naphthalene ring with radical cation character ([A] to [B] in Fig. 1).42The generated dicationic aminium radical [B] should be able to abstract a hydrogen atom from the C–H bond of a substrate resulting in an efficient HAT process.
 |
| | Fig. 1 Working hypothesis for the design of new HAT organocatalysts from diamines. | |
Results and discussion
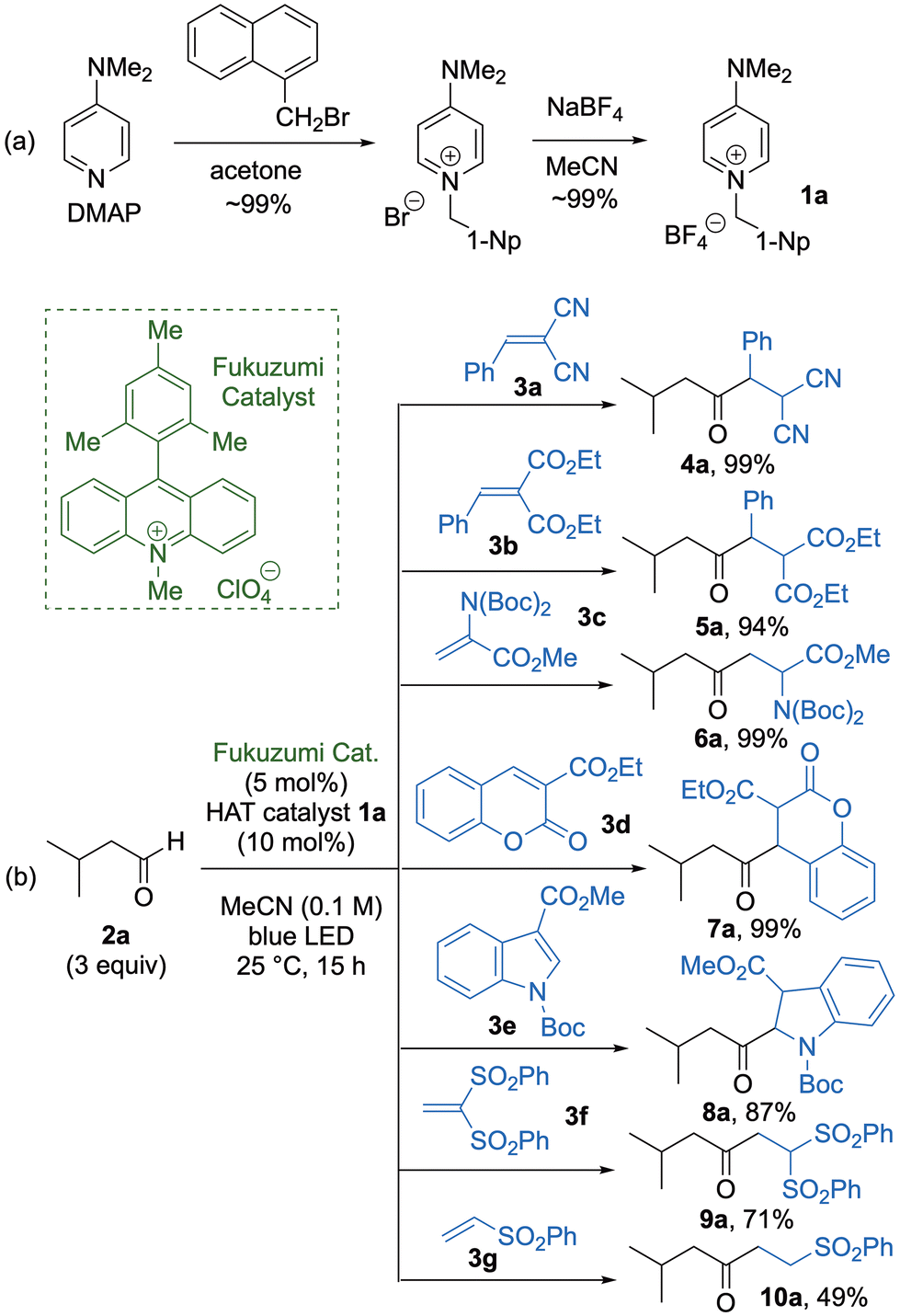
We commenced our study by preparing 1 from a series of commercially available diamine substrates. For example, the reaction of 4-dimethylaminopyridine (DMAP) with 1-naphthylmethyl bromide in acetone furnished the corresponding pyridinium bromide (∼99%), which was treated with sodium tetrafluoroborate (NaBF4) in MeCN to give pyridinium tetrafluoroborate 1a in ∼99% yield (Fig. 2a). The efficiency of 1a as a HAT organocatalyst was then evaluated by applying 1a in the photoinduced C–H alkylation of 3-methylbutanal (2a) with 2-benzylidenemalononitrile (3a) as the radical acceptor in the presence of the Fukuzumi photoredox catalyst in MeCN under irradiation with a blue light-emitting diode (LED) to afford the coupling product 4a in 99% yield (Fig. 2b). Use of other radical acceptors 3b–e furnished the corresponding coupling products 5a–8a, respectively, in high to excellent yields (87–99%). However, when 1,1-ethenediyl bis(sulfone) 3f and vinyl sulfone 3g were used as the radical acceptors, the coupling products 9a and 10a were obtained in moderate to good yields (49–71%).
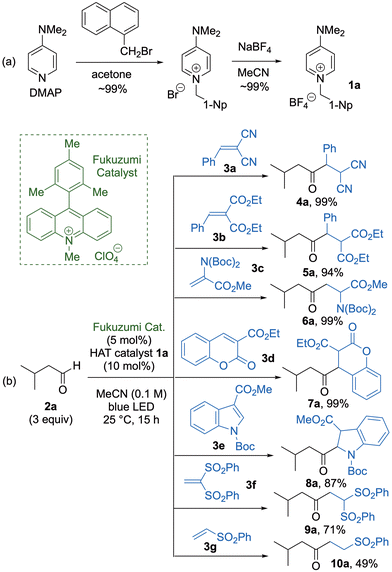 |
| | Fig. 2 (a) Synthesis of HAT organocatalyst 1a. (b) Photoinduced radical reaction of 2a with 3a–3g. | |
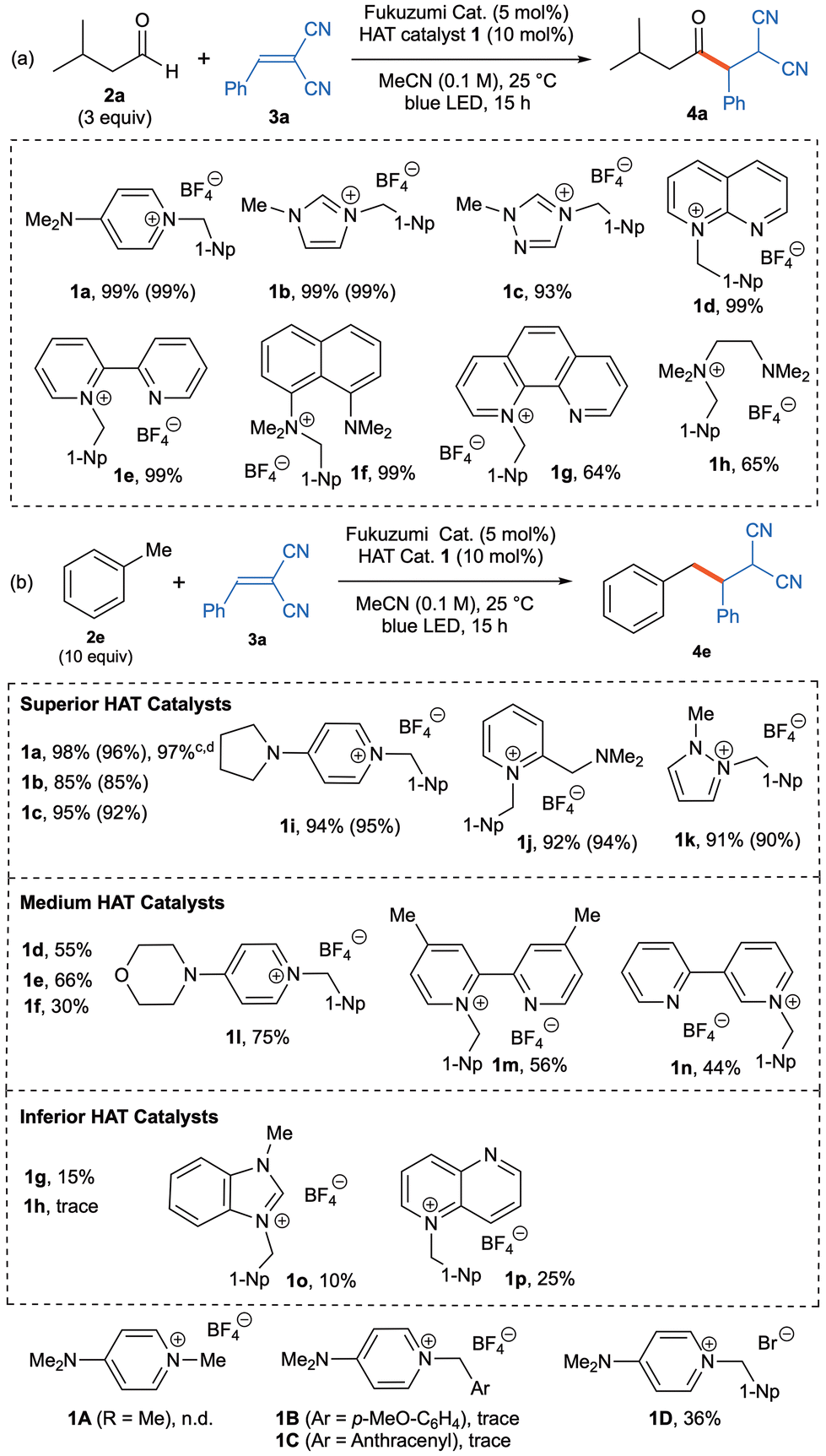
Since the choice of aldehyde substrate 2a generally afforded coupling products 4a–8a in high to excellent yields, this aldehyde substrate did not seem to be appropriate for the examination of the reactivities of other HAT organocatalysts derived from a series of commercially available diamino compounds. Indeed, the attempted use of 5 different HAT organocatalysts 1b–f in the photoinduced C–H alkylation of aldehyde 2a with 3a gave the coupling product 4a in excellent yields as shown in Table 1a. When HAT organocatalysts 1g–h were utilized, moderate yields (64–65%) of 4a were obtained. Accordingly, we selected a toluene (2e) as the substrate as it is less reactive than 2a (Table 1b). Indeed, the attempted reaction of toluene (3 equiv.) with 2-benzylidenemalononitrile (3a) in the presence of HAT organocatalyst 1a and the Fukuzumi photocatalyst under similar conditions with longer reaction time afforded the coupling product 4e in 97% yield. The use of an excess of toluene (10 equiv.) enhanced the yield of 4e to 98% under similar conditions with shorter reaction time (15 h). Now using an excess of toluene (10 equiv.) as standard, the reactivities of various HAT organocatalysts in the radical-promoted alkylation to 2-benzylidenemalononitrile (3a) was thoroughly investigated (Table 1b). Among these, HAT organocatalysts 1b–c and 1i–k derived from 1-methyl-1H-imidazole, 1-methyl-1H-1,2,4-triazol, 4-(pyrrolidin-1-yl)pyridine, N,N-dimethyl-1-(pyridin-2-yl)methanamine and 1-methyl-1H-pyrazol, respectively, also demonstrate excellent catalytic performance (85–94% yields) in the photoinduced C–H alkylation of toluene (2e) with 2-benzylidenemalononitrile (3a). It should be noted that the use of 4-(dimethylamino)-1-methylpyridin-1-ium tetrafluoroborate (1A) in place of the 1-(1-naphthylmethyl) pyridin-1-ium salt 1a gave none of the desired product 4e under the standard conditions. In addition, replacement of the 1-naphthylmethyl moiety in 1a with p-methoxybenzyl or anthracen-9-ylmethyl moieties (i.e., catalysts 1B or 1C) resulted in the production of only a trace amount of 4e. The choice of counter anion is crucially important, and the use of 4-(dimethylamino)-1-(naphthalen-1-ylmethyl)pyridin-1-ium bromide (1D) gave a low yield (36%) of the desired 4e. Next, we investigated HAT organocatalysts 1d–f and 1l–n derived from 1,8-naphthyridine, 2,2′-bipyridine, 1,8-bis(dimethylamino)naphthalene (proton sponge), 4-(pyridin-4-yl)morpholine, 4,4′-dimethyl-2,2′-bipyridine and 2,3′-bipyridine, respectively. These HAT organocatalysts exhibited moderate to good performance (30–75% yield of 4e) in the photoinduced C–H alkylation of toluene (2e) with 2-benzylidenemalononitrile (3a). Considering the high oxidation potential of pyridine moiety on HAT catalyst 1e, 1m and 1n, these catalysts might generate a pyridyl-radical cation, which contributes to their HAT ability. Accordingly, 1-methyl substituted 2,2′-bipyridine HAT catalyst 1E was synthesized. However, product 4e is not detected in the 1E-catalyzed C–H alkylation of toluene with 3a. Use of simple 1-naphthylmethylpyridinium salt 1F gave coupling product 4e in lower yield (45% vs. 66% with 1e). These results indicated that the presence of 1-naphthylmethyl moiety is crucially important, and another pyridyl moiety showed some positive results in this transformation. The similar tendency is also observed with HAT catalyst 1d in comparison with 1G and 1H. (for details, see Scheme S1 in the ESI‡). Furthermore, HAT organocatalysts 1g–h and 1o–p derived from 1,10-phenanthroline, N,1N1,N2,N2-tetramethylethylenediamine (TMEDA), 1-methyl-1H-benzo[d]imidazole and 1,5-naphthyridine, respectively, exhibited the low reactivities (trace ∼25% yields of 4e) in the same photoinduced C–H alkylation of toluene with 3a.
Table 1 Reactivity of various new HAT organocatalysts derived from commercially available diaminesa,b
| Condition: 3-Methylbutanal (2a, 0.6 mmol) or toluene (2e, 2.0 mmol), acceptor 3a (0.20 mmol), Fukuzumi catalyst (5.0 mol%), and HAT catalyst 1 (10 mol%) in MeCN (2.0 mL) under light irradiation (blue LED, 448 nm) at 25 °C for 15 h. NMR yield using 1,1,2,2-tetrachloroethane as internal standerard; isolated yeilds are indicated in parentheses. n.d. = not detected. Trace = <5% yield. With 3 equiv. of toluene. For 36 h. |
 |
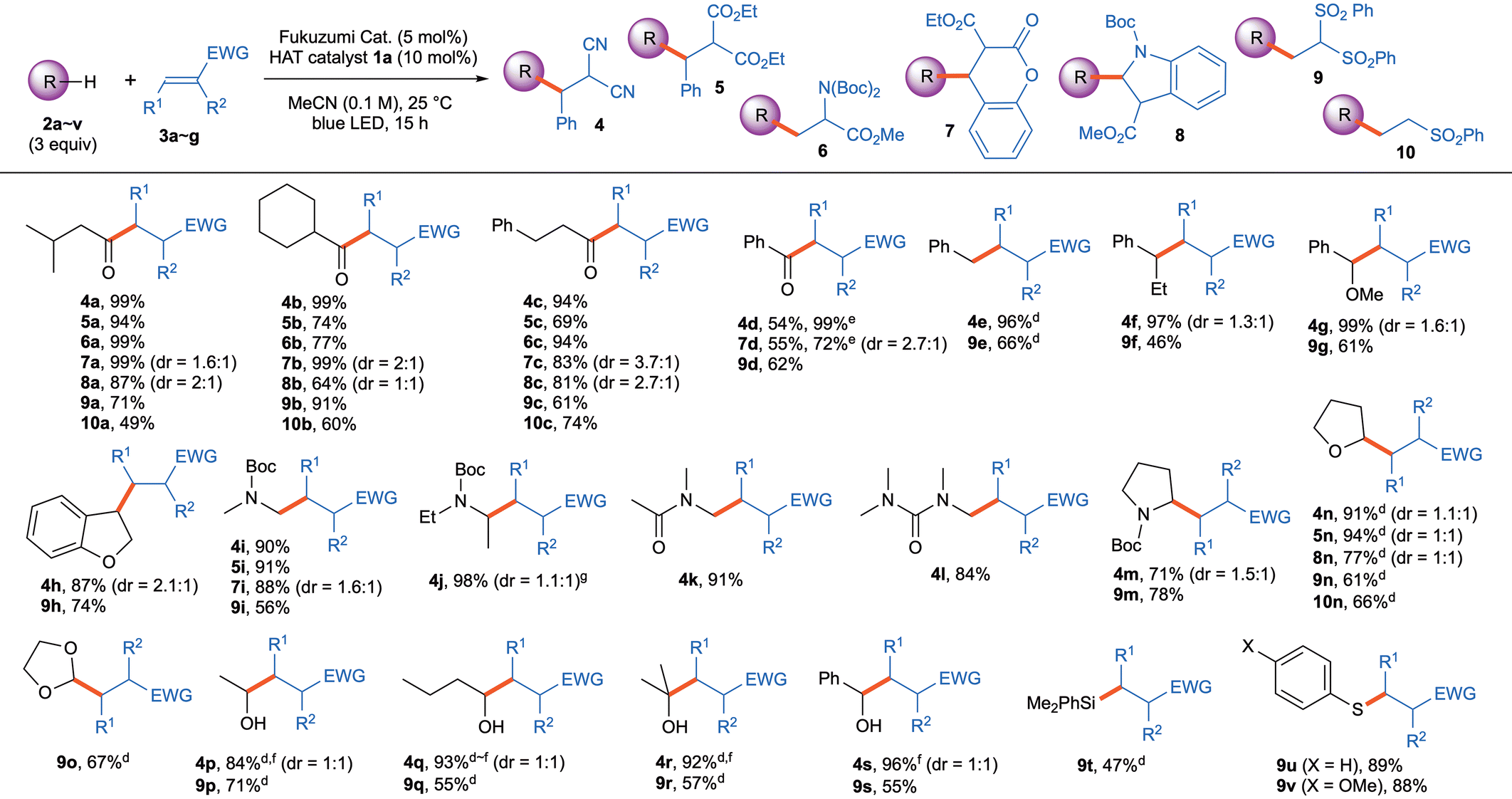
With this information in hand, we examined the substrate scope of the various functionalized compounds in the presence of HAT catalyst 1a, as shown in Table 2. Of the aldehyde substrates 2a–d, the aliphatic aldehydes 2a–c reacted with various radical acceptors 3a–g smoothly to furnish the corresponding coupling products 4–10 in good to excellent yields. However, aromatic aldehyde 2d was found to be less reactive. The in situ-generated benzoyl radical smoothly reacted with acceptors 3a,d,f to give products 4d, 7d, and 9d in good to excellent yields, while the reaction with acceptors 3b,c,e,g proceeded sluggishly to give low yields (17–38%) of products 5d, 6d, 8d, and 10d, respectively (for details see Table S4 in the ESI‡). The benzylic radicals generated in situ from benzylic substrates 2e–h, only underwent conjugate addition with reactive acceptors 3a and 3f, whilst the other acceptors (i.e., 3b–e and 3g) reacted very sluggishly under the standard conditions (for details see Table S4 in the ESI‡). Among amido substrates, the α-amido carbon radical generated from N-Boc-dimethylamine is the more reactive, and the radical coupling took place smoothly with acceptors 3a,b,d,f to afford the corresponding products 4i, 5i, 7i, and 9i, respectively, in good to high yields. It was found that N-Boc-pyrrolidine (2m) also worked well. However, the other amido substrates 2j–l would only react with 2-benzylidenemalononitrile (3a) to give 4j–l in good to excellent yields. The tetrahydrofuran-2-yl radical, generated from THF (2n), can be coupled easily with various radical acceptors 3a,b,e–g to furnish coupling products 4n, 5n, 8n, 9n, and 10n, respectively, in good to high yields. A carbon radical derived from 1,3-dioxolane (2o) added to reactive acceptor 3f to give coupling product 9o in good yield. We also examined several alcohol substrates 2p–s, and products with α-hydroxy C–C bonds 4p–s and 9p–s, respectively, were formed in moderate to high yields. Dimethylphenylsilane (2t) and aromatic thiols 2u–v only reacted with the reactive acceptor 3f to give coupling products 9t–v, respectively, in moderate to high yields.
Table 2 Substrate scope of photocatalyzed C–H alkylations of 2 and 3 with HAT catalyst 1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a,b,c
a,b,c
| Conditions: 2 (0.6 mmol), 3 (0.20 mmol), Fukuzumi cat. (5.0 mol%), and HAT catalyst 1a (10 mol%) in MeCN (2.0 mL) under light irradiation (blue LED, 448 nm) at 25 °C for 15 h. Yeilds are isolated yields. Diastereomer ratio (dr) were determined by 1H NMR of the crude product. With 10 equiv. of C–H substrate. Reaction time is 36 hours. Cyclized product was isolated after silica-gel column chromatography (see the ESI‡). Diatereoisomeric ratio (dr) was determined by the yields of isolated products. |
 |
A characteristic feature of our approach is that it can be utilized in late-stage functionalization reactions, and this has been successfully demonstrated via the site-selective C–H alkylation of bifunctional molecules (Table 3). Thus, p- and m-methylbenzaldehyde 2w,x generated p- and m-methylbenzoyl radicals solely, without the formation of any benzylic radicals, in a site-selective manner under photoinduced conditions in the presence of 2-benzylidenemalononitrile (3a) to furnish the coupling products 4w and 4x, respectively, in excellent yields. In isochromane substrate 2y, the preferential generation of an α-oxy carbon radical took place to afford the coupling product 4y in excellent yield. With butyl methyl ether (2z), only coupling product 4z was obtained in excellent yield with a small amount of methyleneoxy radical formation. Amide substrates 2aa–ad were examined and N-Ac-morpholine (2aa) was converted to the corresponding α-amido carbon radical in a site-selective manner without formation of the α-oxy carbon radical to provide coupling product 4aa in high yield. In the cases of N-Boc-isopropylmethylamine (2ab) and N-Boc-methylpropylamine (2ac), the α-amido methyl radicals can be generated preferentially tofurnish coupling products 4ab and 4ac with high to excellent site-selectivity. An α-amido methylene radical is preferentially formed from the unsymmetric urea derivative 2ad to give coupling product 4ad in high yield. When benzo[d][1,3]dioxole-5-carbaldehyde (2ae) was treated with HAT catalyst 1a, coupling product 4ae was obtained as a major product in high yield with a small amount of 4ae′ via acyl radical formation. In marked contrast, however, 4ae′ was produced almost exclusively in excellent yield with HAT catalyst 1e (for more information on the site-selectivity of various HAT catalyst, see Tables S5 and S6 of the ESI‡).44 Based on the calculation, the bond dissociation energies (BDEs) for the CH2 and aldehyde C–H positions in piperonyl aldehyde (2ae) are similar, the CH2 position (90.8 kcal mol−1) is slightly higher than aldehyde C–H position (89.1 kcal mol−1). The difference in HAT site-selectivity between the catalysts 1a and 1e in the reaction using 2ae more likely arises from π–π interaction of catalyst/substrate (Fig. 3). When the naphthyl group of the catalyst 1a forms a π interaction with the substrate 2ae, the hydrogen abstraction from the CH2 group occurs more easily as shown in [C] in comparison with the case [D]. In contrast, when the naphthyl group of the catalyst 1e forms a π interaction with the substrate 2ae, the hydrogen abstraction from the aldehyde C–H position occurs more easily as shown in [E] (for details of calculation method and the optimized structures, see section 8 in the ESI‡). Next, the site-selective C–H alkylation of natural products and biologically active compounds was investigated (Table 3). Dihydrocitronellal (2af) was compatible with this approach and 4af, 6af, and 7af were delivered in excellent yields with rigorous site-selectivity. Geraniol (2ag) was also successfully reacted without the protection of the hydroxy group, providing the coupling product 4ag with rigorous regioselectivity. In the multi-functional molecules 2ah and 2ai, which have multiple reactive C–H bonds, the present approach afforded 4ah and 4ai, respectively, in a highly site-selective manner.
 |
| | Fig. 3 Site-selective C–H alkylations of 2ae with HAT catalyst 1a or 1e. | |
Table 3 Substrate scope of various site-selective photocatalyzed C–H alkylations with HAT catalyst 1aa,b,c
| Conditions: 2 (0.6 mmol), 3a (0.20 mmol), Fukuzumi cat. (5 mol%), and HAT catalyst 1a (10 mol%) in MeCN (2.0 mL) under light irradiation (blue LED, 448 nm) at 25 °C for 15 h. Combined isolated yields of all isomers. Diastereomer ratio (dr) were determined by 1H NMR of the crude product. Regioisomeric ratio (rr) was determined by 1H NMR of crude or isolated products. Reaction time is 36 hours. Reaction time is 48 hours. |
 |
Our approach was then applied to the sequential, one-pot photoinduced C–H dialkylations of bifunctional substrate 3h with the Fukuzumi/HAT catalysts. Thus, the initial reaction of 3h with toluene in the presence of the Fukuzumi catalyst (5 mol%) and HAT catalyst 1a (10 mol%) in MeCN under blue LED irradiation at 25 °C for 24 h was carried out to furnish monoalkylation product 11 in almost quantitative yield with rigorous site-selectivity. After removal of the solvent and toluene via vacuum evaporation, the second reaction was then executed by adding 3-methylbutanal (2a) (1 mmol) in the presence of the Fukuzumi catalyst (5 mol%) and HAT catalyst 1a (10 mol%) in MeCN under blue LED irradiation at 25 °C for 15 h to afford only dialkylation product 12 in 81% yield with rigorous site-selectivity (Fig. 4a). In a similar manner, the first-step reaction of 3h with toluene and the subsequent reaction with THF were accomplished under similar photoinduced conditions to give the dialkylation product 13 in 66% yield (Fig. 4b). In addition, the initial photoinduced reaction of 3h was conducted with ethanol to furnish the monoalkylation product 14 in 74% yield, and the subsequent reaction with 3-methylbutanal (2a) afforded the dialkylation product 15 in 65% yield (Fig. 4c).
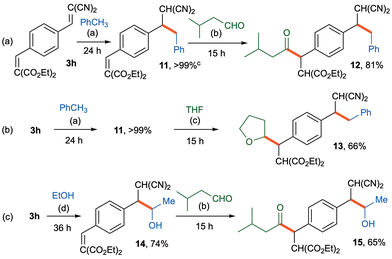 |
| | Fig. 4 Sequential one-pot photocatalyzed C–H alkylations of 3h with HAT catalyst 1a. (a) 3h (0.2 mmol), toluene (2 mmol), Fukuzumi cat. (5 mol%), and HAT catalyst 1a (10 mol%) in MeCN (2.0 mL) under light irradiation (blue LED, 448 nm) at 25 °C for 24 h. (b) 3-Methylbutanal (1 mmol), Fukuzumi cat. (5 mol%), and HAT catalyst 1a (10 mol%) in MeCN (2.0 mL) under light irradiation (blue LED, 448 nm) at 25 °C for 15 h. (c) THF (2 mmol), Fukuzumi cat. (5 mol%), and HAT catalyst 1a (10 mol%) in MeCN (2.0 mL) under light irradiation (blue LED, 448 nm) at 25 °C for 15 h. (d) EtOH (1 mmol), Fukuzumi cat. (5 mol%), and HAT catalyst 1a (10 mol%) in MeCN (2.0 mL) under light irradiation (blue LED, 448 nm) at 25 °C for 36 h. | |
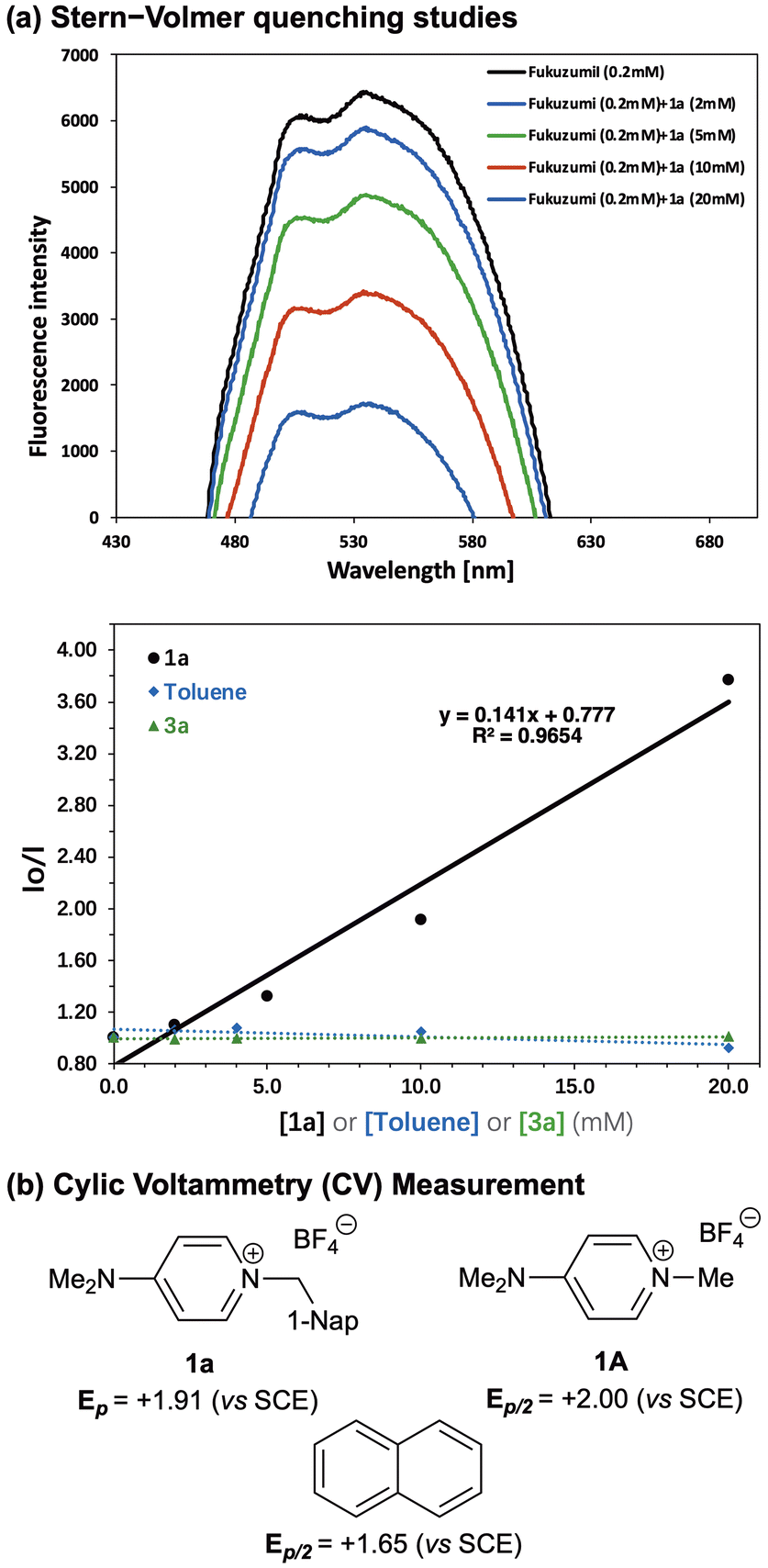
In order to thoroughly understand the reaction mechanism, we performed Stern–Volmer fluorescence quenching and cyclic voltammetry (CV) studies (Fig. 5). The Stern–Volmer fluorescence quenching studies indicate that HAT catalyst 1a quenches the excited state of the Fukuzumi catalyst, while neither the C–H substrate, toluene, nor the 2-benzylidenemalononitrile (3a) as acceptor quenched the Fukuzumi catalyst (Fig. 5a). Cyclic voltammetry (CV) analysis of 1a revealed a peak at +1.91 V vs. the saturated calomel electrode (SCE). In comparison, naphthalene and the N-methyl-substituted catalyst 1A showed half-peak potentials (Ep/2) of +1.65 V vs. SCE and +2.00 V vs. SCE, respectively (Fig. 5b). These observations imply that the excited state of the Fukuzumi catalyst (with a reductive quenching potential of *Ered1/2 = +2.09 V vs. SCE42) can oxidize both the tertiary amine and naphthalene groups in 1a, though oxidation of the naphthalene group occurs more readily than that of the tertiary amine. These results show that the N-tethered naphthalene moiety would be oxidized by the photoredox catalyst and assist the generation of an aminium radical cation on the other nitrogen atom of the diamine catalyst via intramolecular electron transfer. The generated active aminium radical cation species then proceed to abstract the hydrogen atom from the C–H bond of the substrate.
 |
| | Fig. 5 Control experiments. | |
In addition, the radical clock experiments using citronellal (2aj) as substrate in the presence or absence of electron-deficient alkene (3a) were carried out. In the presence of electron-deficient alkene (3a), neither the coupling product nor the radical cyclization-addition product was detected, and the desired cyclization product, menthone was obtained in 3% yield. In the absence of electron-deficient alkene (3a), the desired cyclization product, menthone was obtained in 8% yield. These results indicated the participation of radical species under this reaction condition (for detail, see ESI, Schemes S3 and S4‡).
Conclusions
In summary, we have designed and synthesized various types of hydrogen-atom transfer (HAT) organocatalysts from commercially available diamine compounds. Some of the best HAT organocatalysts demonstrated here, in combination with a photoredox organocatalyst allowed efficient and site-selective C–H alkylation of various functionalized organic substrates, ranging from simple hydrocarbons to complex molecules. This study should encourage further development of readily available, new HAT organocatalysts for the site-selective C–H alkylation of multifunctional molecules. The application of chiral HAT organocatalysts derived from commercially available chiral diamine substrates for asymmetric C–H alkylation is ongoing in our laboratory.
Author contributions
K. M. conceptualized the research. J. J. performed the experiments. K. M. prepared the manuscript. J. J. and T. K. prepared and edited the ESI.‡ K. M. supervised the project and edited the manuscript.
Data availability
The data supporting this article have been included as part of the ESI.‡
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the JSPS KAKENHI Grant no. 21H05026 and 23H04910 (Green Catalysis Science).
References
- M. S. Chen and N. C. White, Combined effects on selectivity in Fe-catalyzed methylene oxidation, Science, 2010, 327, 566–571 CrossRef PubMed.
- T. Newhouse and P. S. Baran, If C–H bonds could talk: selective C–H bond oxidation, Angew. Chem., Int. Ed., 2011, 50, 3362–3374 CrossRef CAS PubMed.
- S. R. Neufeldt and M. S. Sanford, Controlling site selectivity in palladium-catalyzed C–H bond functionalization, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS.
- H. M. Daviesand and D. Morton, Recent advances in C–H functionalization, J. Org. Chem., 2016, 81, 343–350 CrossRef PubMed.
- K.-J. Jiao, Y.-K. Xing, Q.-L. Yang, H. Qiu and T.-S. Mei, Site-selective C–H functionalization via synergistic use of electrochemistry and transition metal catalysis, Acc. Chem. Res., 2020, 53, 300–310 CrossRef CAS.
- B. Prabagar, Y. Yang and Z. Shi, Site-selective C–H functionalization to access the arene backbone of indoles and quinolines, Chem. Soc. Rev., 2021, 50, 11249–11269 RSC.
- T. G. Frihed, M. Bols and C. M. Pedersen, C–H functionalization on carbohydrates, Eur. J. Org. Chem., 2016, 2740–2756 CrossRef CAS.
- X.-Q. Chu, D. Ge, Z.-L. Shen and T.-P. Loh, Recent advances in radical-initiated C (sp3)–H bond oxidative functionalization of alkyl nitriles, ACS Catal., 2018, 8, 258–271 CrossRef CAS.
- O. P. Patel, N. K. Nandwana, L. J. Legoabe, B. C. Das and A. Kumar, Recent advances in radical C–H bond functionalization of imidazoheterocycles, Adv. Synth. Catal., 2020, 362, 4226–4255 CrossRef CAS.
- R. Cannalire, S. Pelliccia, L. Sancineto, E. Novellino, G. C. Tron and M. Giustiniano, Visible light photocatalysis in the late-stage functionalization of pharmaceutically relevant compounds, Chem. Soc. Rev., 2021, 50, 766–897 RSC.
- P. Bellotti, H.-M. Huang, T. Faber and F. Glorius, Photocatalytic late-stage C–H functionalization, Chem. Rev., 2023, 123, 4237–4352 CrossRef CAS PubMed.
- M. Sadeghi, C(sp3)–H functionalization using chlorine radicals, Adv. Synth. Catal., 2024, 366, 2898–2918 CrossRef CAS.
- M. Kim, E. You, J. Kim and S. Hong, Site-selective pyridylic C–H functionalization by photocatalytic radical cascades, Angew. Chem., Int. Ed., 2022, 61, e202204217 CrossRef PubMed.
- Z. Lu, M. Ju, Y. Wang, J. M. Meinhardt, J. I. Martinez Alvarado, E. Villemure, J. A. Terrett and S. Lin, Regioselective aliphatic C–H functionalization using frustrated radical pairs, Nature, 2023, 619, 514–520 CrossRef.
- R. T. Simons, M. Nandakumar, K. Kwon, S. K. Ayer, N. M. Venneti and J. L. Roizen, Directed photochemically mediated nickel-catalyzed (hetero) arylation of aliphatic C–H bonds, J. Am. Chem. Soc., 2023, 145, 3882–3890 CrossRef.
- H. T. Ang, Y. Miao, D. Ravelli and J. Wu, Pyridine N-oxides as hydrogen atom transfer reagents for site-selective photoinduced C(sp3)–H functionalization, Nat. Synth., 2024, 3, 568–575 CrossRef.
- X. He, Y. Zhang, S. Liu, W. Zhang, Z. Liu, Y. Zhao and X. Shen, Photocatalytic site-selective radical C(sp3)–H aminoalkylation, alkylation and arylation of silanes, Nat. Synth., 2025, 4, 188–195 CrossRef.
- Z. Li, X. Chen, C. Peng, Y. Xu, H. Wang, S. Liu and X. Shen, Merging radical brook rearrangement and 1,5-hydrigen atom transfer: Facile synthesis of ketone-containing α-fluoroalkyl alcohols, ChemCatChem, 2024, 16, e202301075 CrossRef.
- J. M. Mayer, Understanding hydrogen atom transfer: from bond strengths to Marcus theory, Acc. Chem. Res., 2011, 44, 36–46 CrossRef PubMed.
- N. Hoffmann, Photochemical electron and hydrogen transfer in organic synthesis: The control of selectivity, Synthesis, 2016, 1782–1802 CrossRef.
- L. Capaldo and D. Ravelli, Photochemical electron and hydrogen transfer in organic synthesis: The control of selectivity, Eur. J. Org. Chem., 2017, 2056–2071 CrossRef PubMed.
- H. Cao, X. Tang, H. Tang, Y. Yuan and J. Wu, Photoinduced intermolecular hydrogen atom transfer reactions in organic synthesis, Chem. Catal., 2021, 1, 523–598 Search PubMed.
- L. Capaldo, D. Ravelli and M. Fagnoni, Direct photocatalyzed hydrogen atom transfer (HAT) for aliphatic C–H bonds elaboration, Chem. Rev., 2021, 122, 1875–1924 CrossRef PubMed.
- L. Androvič, J. Bartáček and M. Sedlák, Recent advances in the synthesis and applications of azo initiators, Res. Chem. Intermed., 2016, 42, 5133–5145 Search PubMed.
- A. Studer and S. Amrein, Tin hydride substitutes in reductive radical chain reactions, Synthesis, 2002, 835–849 CrossRef.
- S. W. Crossley, C. Obradors, R. M. Martinez and R. A. Shenvi, Mn-, Fe-, and Co-catalyzed radical hydrofunctionalizations of olefins, Chem. Rev., 2016, 116, 8912–9000 CrossRef PubMed.
- S. A. Green, S. W. Crossley, J. L. M. Matos, S. Vasquez-Cespedes and R. A. Shenvi, The high chemofidelity of metal-catalyzed hydrogen atom transfer, Acc. Chem. Res., 2018, 51, 2628–2640 CrossRef CAS PubMed.
- P. P. Singh, S. Sinha, P. Gahtori, S. Tivari and V. Srivastava, Recent advances of decatungstate photocatalyst in HAT process, Org. Biomol. Chem., 2024, 22, 2523–2538 RSC.
- X.-Z. Fan, J.-W. Rong, H.-L. Wu, Q. Zhou, H.-P. Deng, J. D. Tan, C.-W. Xue, L.-Z. Wu, H.-R. Tao and J. Wu, Eosin Y as a Direct hydrogen atom transfer photocatalyst for the functionalization of C–H bonds, Angew. Chem., Int. Ed., 2018, 57, 8514–8518 CrossRef CAS PubMed.
- Z.-X. Wu, G.-W. Hu and Y.-X. Luan, Development of N-hydroxy catalysts for C–H functionalization via hydrogen atom transfer: challenges and opportunities, ACS Catal., 2022, 12, 11716–11733 CrossRef CAS.
- A. Matsumoto and K. Maruoka, Design of organic radical cations as potent hydrogen–atom transfer catalysts for C–H functionalization, Asian J. Org. Chem., 2024, 13, e202300580 CrossRef CAS.
- S. P. Singh, V. Srivastava, P. K. Singh and P. P. Singh, Visible-light induced eosin Y catalysed C(sp2)–H alkylation of carbonyl substrates via direct HAT, Tetrahedron, 2023, 132, 133245 CrossRef CAS.
- J. Xu, R. Li, Y. Ma, J. Zhu, C. Shen and H. Jiang, Site-selective α-C (sp3)–H arylation of dialkylamines via hydrogen atom transfer catalysis-enabled radical aryl migration, Nat. Commun., 2024, 15, 6791 CrossRef CAS.
- M. E. Wolff, Cyclization of N-Halogenated amines (The Hofmann-Löffler Reaction), Chem. Rev., 1963, 63, 55–64 CrossRef CAS.
- M. S. Chen and M. C. White, Combined effects on selectivity in Fe-catalyzed methylene oxidation, Science, 2010, 327, 566–571 CrossRef CAS PubMed.
- A. J. McMillan, M. Sieńkowska, P. D. Lorenzo, G. K. Gransbury, N. F. Chilton, M. Salamone, A. Ruffoni, M. Bietti and D. Leonori, Practical and selective sp3 C–H bond chlorination via aminium radicals, Angew. Chem., Int. Ed., 2021, 60, 7132–7139 CrossRef CAS.
- J. L. Jeffrey, J. A. Terrett and D. W. MacMillan, O–H hydrogen bonding promotes H-atom transfer from α C–H bonds for C-alkylation of alcohols, Science, 2015, 349, 1532–1536 CrossRef CAS.
- C. Le, Y. Liang, R. W. Evans, X. Li and D. W. MacMillan, Selective sp3 C–H alkylation via polarity-match-based cross-coupling, Nature, 2017, 547, 79–83 CrossRef CAS PubMed.
- W. Xiao, X. Wang, R. Liu and J. Wu, Quinuclidine and its derivatives as hydrogen-atom-transfer catalysts in photoinduced reactions, Chin. Chem. Lett., 2021, 32, 1847–1856 CrossRef CAS.
- L. Yi, C. Zhu, X. Chen, H. Yue, T. Ji, Y. Ma, Y. Cao and R. Kancherla, O–H bond activation of β, γ-unsaturated oximes via hydrogen atom transfer (HAT) and photoredox dual catalysis, Chem. Sci., 2023, 14, 14271–14279 RSC.
- H. Hu, Z. Shi, X. Guo, F.-H. Zhang and Z. A. Wang, A Radical approach for asymmetric α-C–H addition of N-sulfonyl benzylamines to aldehydes, J. Am. Chem. Soc., 2024, 146, 5316–5323 CrossRef CAS PubMed.
- A. Matsumoto, M. Yamamoto and K. Maruoka, Cationic DABCO-based catalyst for site-selective C–H alkylation via photoinduced hydrogen-atom transfer, ACS Catal., 2022, 12, 2045–2051 CrossRef.
- J. Caner, A. Matsumoto and K. Maruoka, Facile synthesis of 1, 2-aminoalcohols via α-C–H aminoalkylation of alcohols by photoinduced hydrogen-atom transfer catalysis, Chem. Sci., 2023, 14, 13879–13884 RSC.
- The site-selectivity of HAT catalyst 1a and our previously reported DABCO-Nap HAT catalyst is compared by doing several bifunctional molecules. The newly designed DMAP-based HAT catalyst 1a showed excellent potential to achieve site-selective alkylation of multifunctional molecules (for details, see Table S7 of the ESI‡).
Footnotes |
| † Dedicated to Professor S. Chandrasekaran on his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures and characterization for all relevant compounds. See DOI: https://doi.org/10.1039/d5qo00509d |
|
| This journal is © the Partner Organisations 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab