
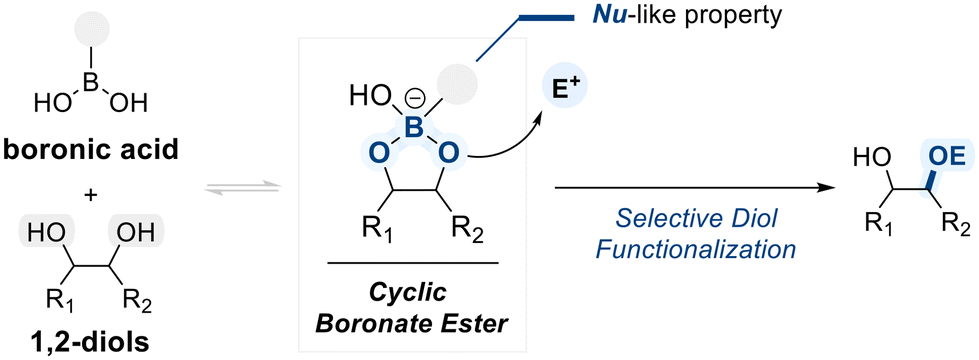
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress in selective functionalization of diols via organocatalysis
Liaba Niaz†
,
Sojeong Bang† and
Jeonghyo Lee*
and
Jeonghyo Lee*
Department of Chemistry, Hanyang University, 222 Wangsimni-ro, Seongdong-gu, Seoul 04763, Korea. E-mail: jeonghyolee@hanyang.ac.kr
First published on 23rd May 2025
Abstract
Polyols bearing multiple hydroxyl groups present persistent challenges for site-selective functionalization due to their inherent reactivity similarity. As minimal polyol systems, diols offer a practical and conceptually rich platform for developing regioselective catalytic strategies. This review highlights recent progress in organocatalyzed diol functionalization, with a survey of organocatalysts incorporating boron, nitrogen, and phosphorus-based motifs, as well as emerging photoredox methodologies. These systems enable selective transformation under mild conditions, avoiding stoichiometric activation and minimizing reaction complexity. Steric and electronic effects, along with noncovalent interactions, are examined in detail to rationalize the observed selectivity and guide the rational design of catalysts. This review offers a conceptual foundation for advancing sustainable, selective methods in diol derivatization.
1. Introduction
Hydroxyl groups are among the most fundamental and ubiquitous functional groups in organic chemistry, frequently found in biologically important molecules.1,2 Their chemical versatility allows transformations via oxidation, reduction, and substitution, however, achieving regio- and stereoselective functionalization in highly functionalized environments remains a major challenge. A seminal contribution in this field came from the Miller group, who developed small-peptide catalysts for the enantioselective kinetic resolution of alcohols via hydrogen bonding, selectively acylating hydroxyl groups over amides.3 This work exemplified the potential of non-enzymatic catalysts for selective hydroxyl functionalization in complex settings.Polyols, which contain multiple hydroxyl groups, are widespread in both natural and synthetic compounds, including carbohydrates,4 glycosides,5 and other biomolecules,6 and are key intermediates in pharmaceutical,7 agrochemical,8 and materials science.9 The dense presence of hydroxyl groups often necessitates complex protecting group strategies to achieve site selectivity.10 To address this, direct and site-selective functionalization approaches have emerged. In another pioneering example, the Miller group achieved desymmetrization of myo-inositol through site-selective phosphorylation,11,12 and later extended this concept to the selective modification of erythromycin A, a complex natural product polyol.13,14 These studies placed a premium on selective catalysis and functional group tolerance in high-complexity environments, setting the stage for today's late-stage functionalization strategies. More recent approaches further advance selectivity through the strategic use of non-covalent interactions,15 minimizing undesired over-functionalization.
Diols represent an essential subclass within the broader polyol family and serve as a critical platform for advancing selective functionalization methodologies.16 Despite possessing fewer hydroxyl groups compared to higher-order polyols, diols still pose the fundamental challenge of distinguishing between two closely similar OH functionalities. Consequently, diols act as practical model systems for developing innovative site-selective transformations, potentially extendable to more structurally complex polyols. Modern catalytic strategies are designed to achieve not only regioselectivity, by selectively modifying one hydroxyl group over the other, but also stereoselectivity and chemoselectivity, thereby broadening the synthetic versatility and application potential of diol-based transformations.17
From a synthetic standpoint, diols are easily accessible via well-established methods, including the dihydroxylation of alkenes,18 hydrolysis of epoxides,19 carbonyl reduction,20 and cascade aldol/reduction sequences,21 rendering them both cost-effective and widely available building blocks (Fig. 1a). Beyond their synthetic accessibility, diols possess notable biological significance due to their widespread occurrence in natural products and bioactive molecules (Fig. 1b). Prominent examples include (1R,2S)-Goniodiol, known for its antibacterial activity; Zephyranthine, utilized in respiratory treatments; and Fluvastatin, an anti-hypercholesterolemic agent.22–25 Targeted diol manipulation via functional group interconversion, ring formation, or derivatization can dramatically alter biological activity or streamline synthetic pathways. These attributes highlight the critical importance of selective diol functionalization in contemporary organic synthesis.
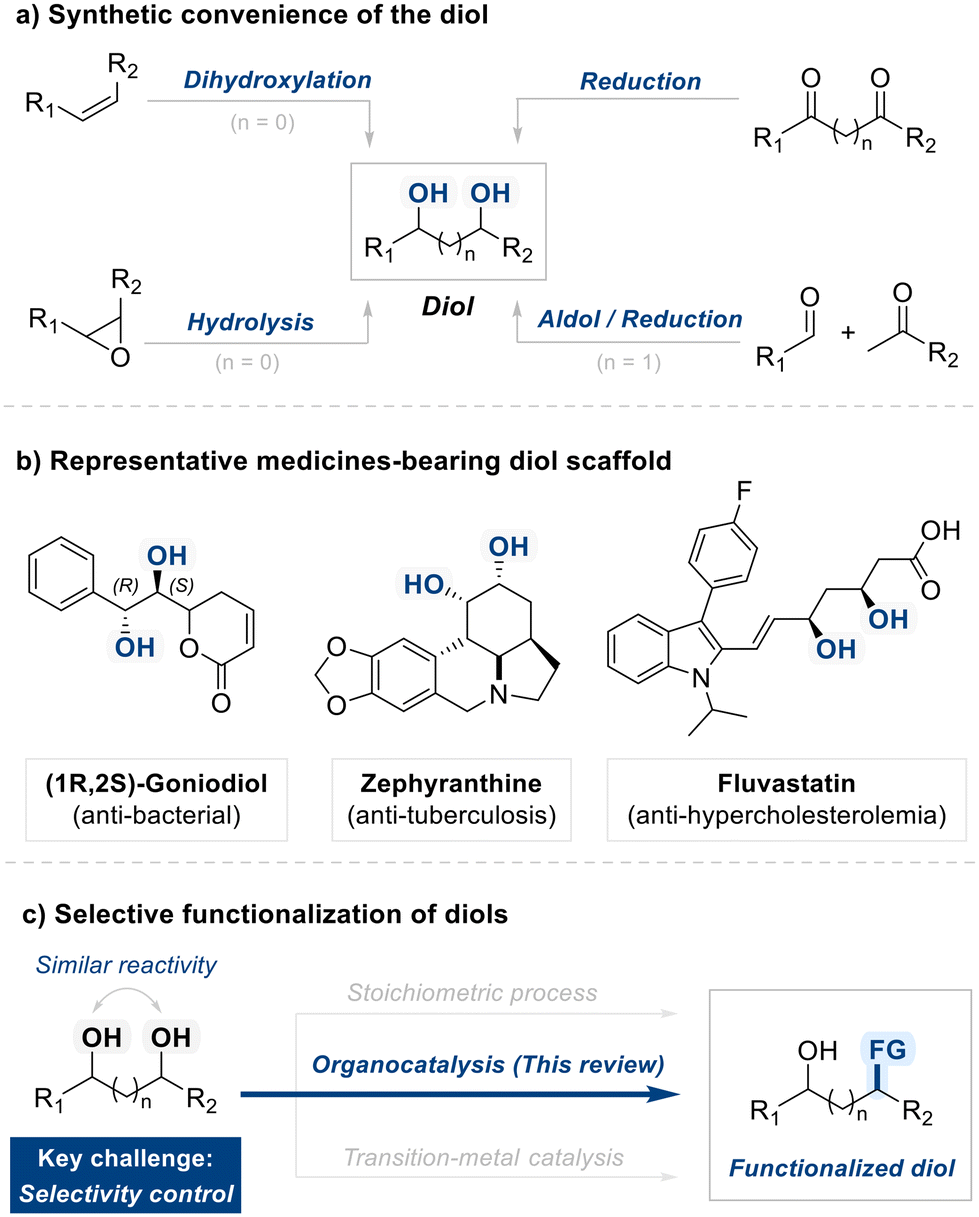 | ||
| Fig. 1 Overview of diol substrate: synthetic accessibility, pharmaceutical relevance, and the thematic focus of this review. | ||
However, these manipulations demand precise control, and achieving regioselective functionalization of diol remains a persistent challenge due to the similar reactivity of hydroxyl groups (Fig. 1c). Traditional solutions often involve multi-step protection and stoichiometric activation, which can inflate the length and cost of a synthetic sequence while generating undesirable waste.26,27 Over the past few decades, researchers have made substantial progress in developing more direct and sustainable processes.28 Regiodivergent reactions, for instance, allow a single diol to give different regioisomers by modulating reaction parameters, thereby offering a potent means to generate structural diversity in fewer steps.29
A broad array of catalytic paradigms has been developed to realize this objective. Early efforts predominantly utilized organometallic or tin-based reagents,30,31 whereas enzymatic approaches32 leveraged the remarkable selectivity of biological catalysts. Transition-metal complexes, with palladium,33 ruthenium,34 and copper35 among popular choices, continue to demonstrate impressive regio- and stereoselectivities in diol functionalization. More recently, organocatalysts have gained prominence owing to their mild conditions, minimal toxicity, and straightforward reaction setups. Through the integration of hydrogen-bond donor frameworks (e.g., peptides, thioureas), Lewis and Brønsted acid catalysis (e.g., boron-based species, phosphoric acids), and photoredox platforms, organocatalytic methods have achieved high selectivity in the direct modification of diols, significantly reducing the need for protective groups and harsh reagents.
While the selective functionalization of carbohydrate systems has been extensively explored and reviewed,28,36–46 contemporary research increasingly turns to non-carbohydrate diols as platforms for testing new catalytic concepts. These investigations have not only yielded streamlined synthetic pathways, but also critical mechanistic insights, revealing how subtle variations in sterics, electronics, and reaction conditions can bias reactivity toward one hydroxyl group over another. A detailed understanding of these control factors provides the foundation for designing next-generation catalysts that enable highly selective functionalization of diols and structurally complex polyols.
This review provides an overview of recent advancements in the organocatalyzed selective functionalization of diol substrates. Selective transformations of carbohydrates are deliberately excluded, as they have been comprehensively covered in previous reviews.28,36–46 Likewise, transition metal-catalyzed approaches to diol functionalization are beyond the scope of this discussion.31,47–52 Our focus is confined to organocatalytic strategies, reflecting their significance in selective diol derivatization. The literature is systematically categorized based on the nature of the organocatalyst, with particular emphasis on systems, incorporating nitrogen, boron, or phosphorus centers, as well as emerging photoredox methodologies. Throughout, we highlight key mechanistic insights and showcase representative synthetic applications that underscore the potential and versatility of these catalytic platforms (Fig. 2). By contextualizing these recent advances within a broader historical and conceptual framework, this review aims to guide practitioners toward innovative and sustainable solutions for this enduring challenge in organic synthesis.
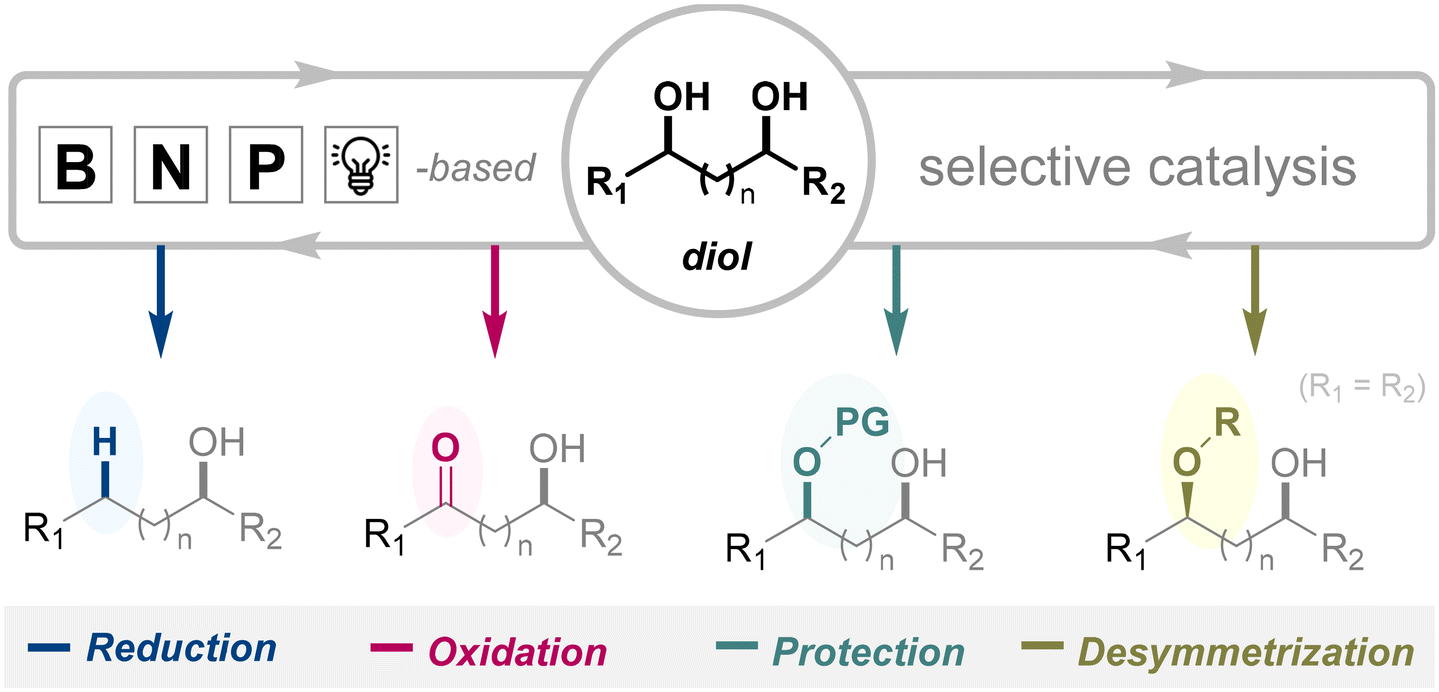 | ||
| Fig. 2 Selective functionalization of diol via organocatalysis. | ||
2. Organoboron catalytic systems
Boron is a main-group element uniquely characterized by its electron deficiency, possessing only three valence electrons and commonly adopting sp2 and sp3 hybridizations. These features give rise to tricoordinate, planar geometries with an empty p-orbital, allowing boron compounds to act as Lewis acids capable of engaging nucleophiles (Fig. 3a).53 Upon coordination, tetracoordinate boron species are formed, which often serve as key intermediates in organic transformations.54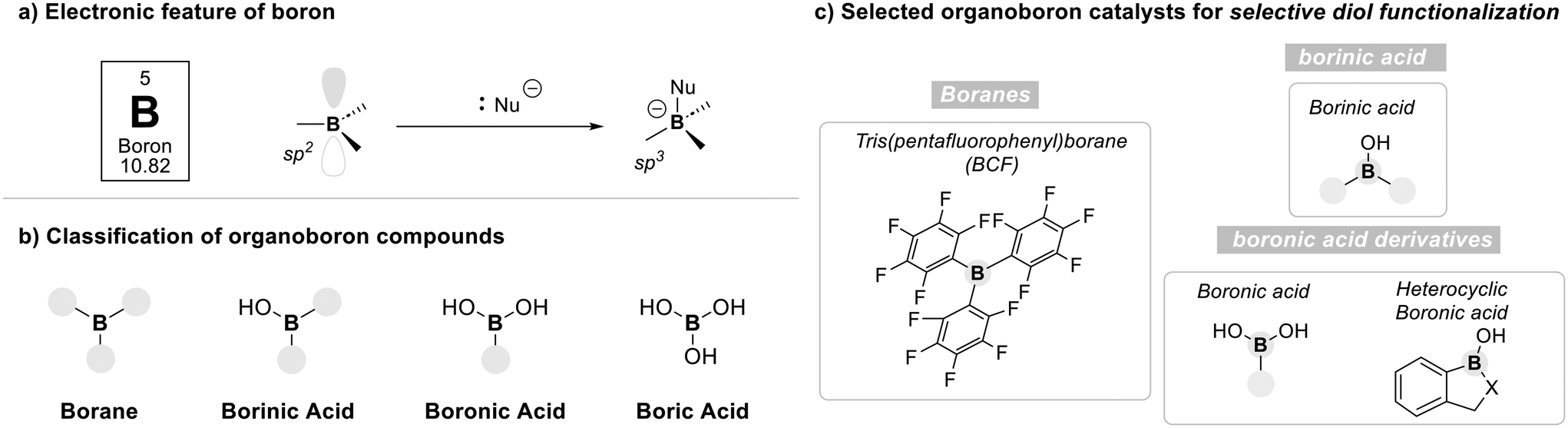 | ||
| Fig. 3 Overview of boron-based organocatalyst. | ||
Organoboron compounds are classified into borane, borinic acids, boronic acids, and boric acid according to their oxidation states55 (Fig. 3b). While each of these boron species has demonstrated substantial utility in organic synthesis, their differing oxidation states impart distinct electronic properties. These variations in electronic nature strongly influence their reaction mechanisms and acidity profiles.56 Among them, boranes, borinic acids, and boronic acids possess at least one organic substituent, allowing for structural and electronic tunability.57,58 In contrast, boric acid consists solely of hydroxyl groups, offering limited flexibility for such modifications, and is therefore excluded from further discussion in this review.
In the following sections, four representative classes of organo-boron catalysts, such as boranes, borinic acids, boronic acids, and heterocylic boronic acid derivatives, are introduced with a focus on their characteristic activation modes and their application to the selective functionalization of diol substrates (Fig. 3c).
2.1. Borane catalysis
Boranes have a trivalent boron center bonded to three organic groups and no hydroxyl groups, which makes them highly electron-deficient and Lewis acidic. To further enhance their electrophilicity, strong electron-withdrawing groups are introduced onto the borane framework. A prominent example is tris(pentafluorophenyl)borane (BCF), an exceptionally electron-deficient Lewis acid owing to the strong inductive and resonance effects of the pentafluorophenyl groups. BCF has found widespread utility in organic synthesis, particularly in combination with hydrosilanes for the deoxygenation of alcohols (Scheme 1).59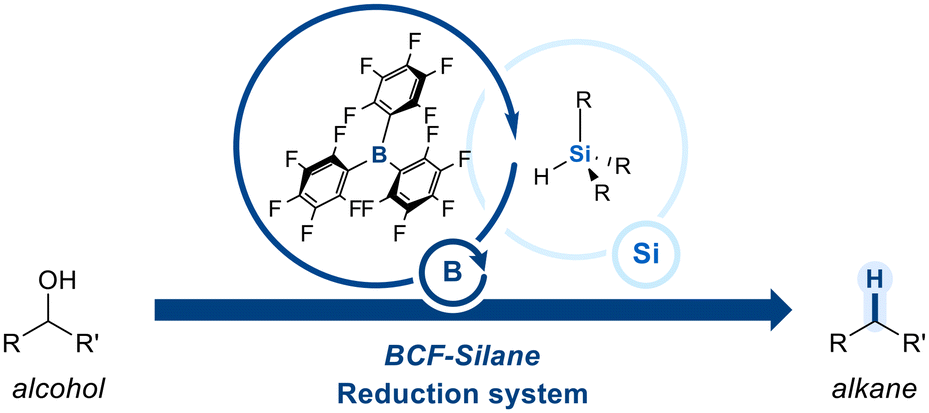 | ||
| Scheme 1 The BCF-silane catalytic reduction system. | ||
The deoxygenation mechanism initiates when BCF interacts with a hydrosilane, leading to polarization of the Si–H bond (Scheme 2a).60 The electron-deficient boron center withdraws electron density from silicon, resulting in partial positive charge on silicon and partial negative charge on boron. Next, oxygen atom of an alcohol coordinate to the electrophilic silicon center, forming a transient oxonium ion. Concurrently, this interaction facilitates the transfer of a hydride ion (H−) from the silane to the boron center, resulting in the formation of a reactive boron–hydride species. The resultant hydride is then delivered to the α-carbon to substitute the oxonium leaving group, leading to cleavage of the C–O bond.
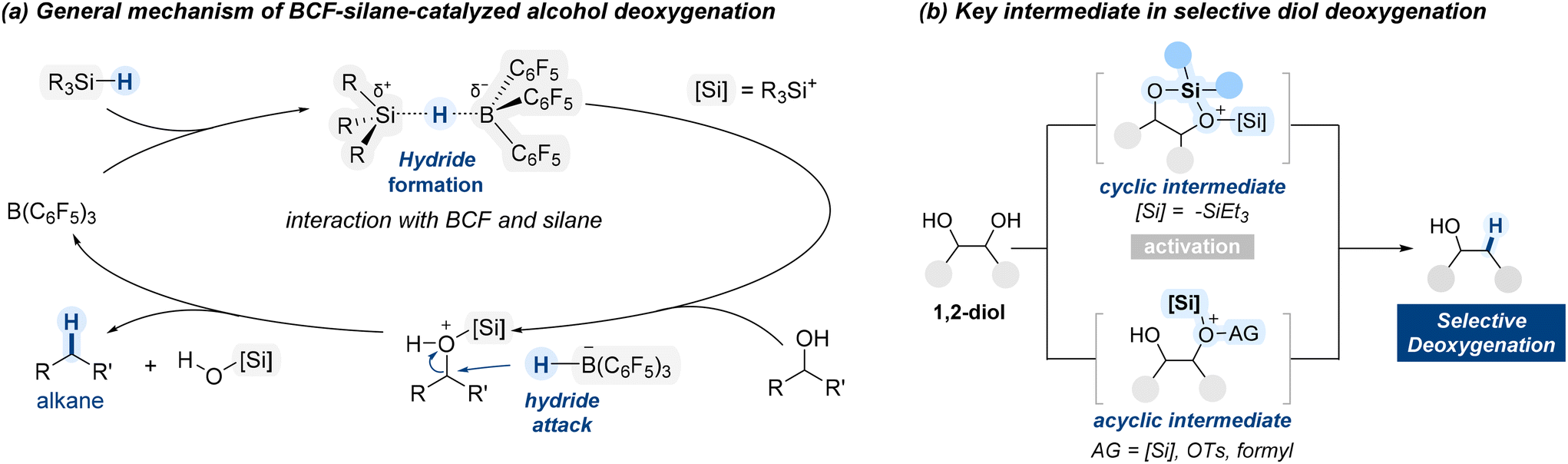 | ||
| Scheme 2 General mechanism for BCF-catalysed deoxygenation of alcohols. | ||
When diol substrates are treated with the BCF/hydrosilane system, selective deoxygenation can proceed through either cyclic or acyclic intermediates, as illustrated in Scheme 2b. The nature of the intermediate, cyclic or acyclic, depends on the substrate's structure and the reaction conditions, influencing both selectivity and efficiency. In the following section, we will explore examples demonstrating how the BCF/silane system enables selective deoxygenation of diol substrates.
In a 2015 study, Morandi demonstrated a BCF (OC-1)-catalyzed regioselective deoxygenation of terminal 1,2-diols with a preference on 1°-hydroxyl group reduction (Scheme 3).61 In this reaction, two types of silane sources were employed. The first, Ph2SiH2, which contains two equivalents of hydride, is sacrificially consumed to generate the key cyclic siloxane intermediate. In the subsequent addition of the second silane, Et3SiH, the more sterically accessible primary alcohol coordinates to the electrophilic silicon center, forming an oxonium intermediate. Subsequently, the hydride was selectively delivered to the activated carbon center resulting in high primary regioselectivity. From their DFT calculation study, the authors proposed that the cyclic siloxane pathway is energetically favored, providing a rationale for the observed selectivity.62
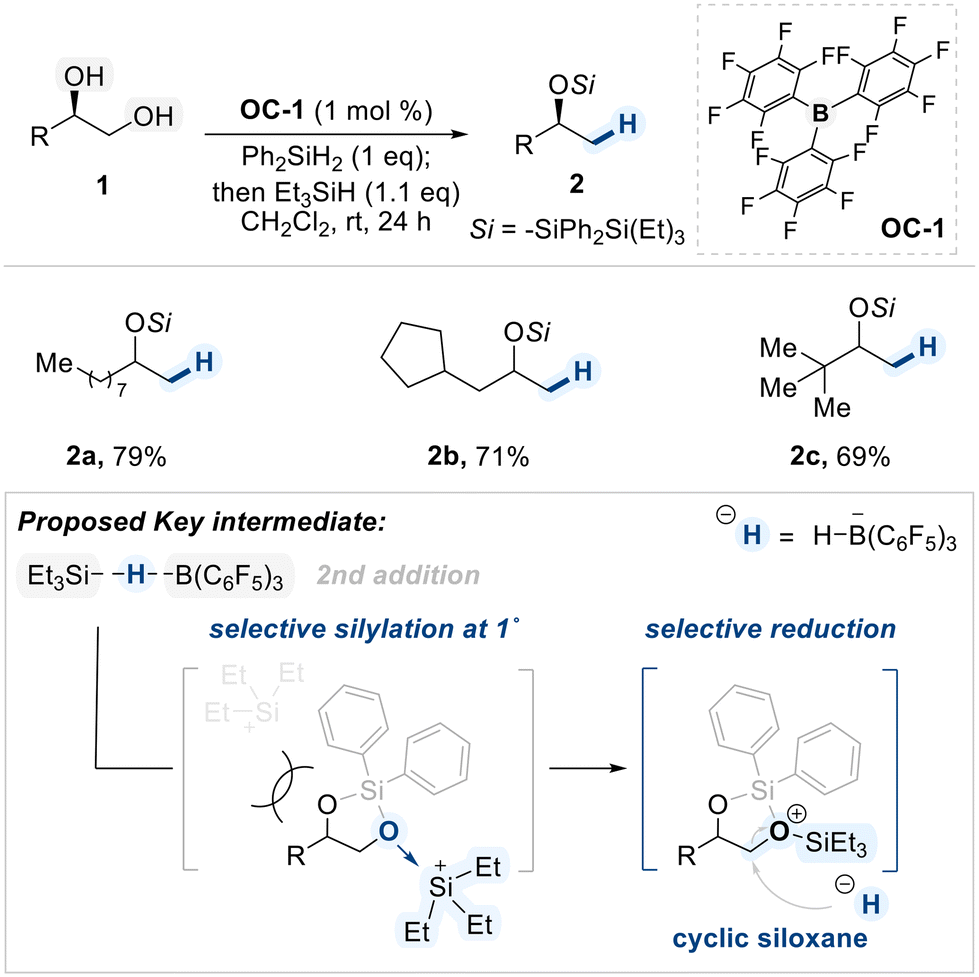 | ||
| Scheme 3 BCF-catalyzed regioselective deoxygenation of terminal 1,2-diols. | ||
Extending their earlier findings, the Morandi group applied the previously established B(C6F5)3/silane conditions to internal 1,2-diols, rather than terminal ones, and observed distinct reactivity, leading to a catalytic reductive pinacol-type rearrangement (Scheme 4a).63 This transformation enabled stereoinvertive migration of alkyl groups from secondary–secondary diols (Scheme 4b), efficiently affording rearranged alcohols. Computational studies indicated that steric hinderance between BCF scaffold and alkyl substituents of a cyclic siloxane intermediate prohibited direct hydride insertion (TS-A, Scheme 4c) and that hyperconjugative effects from the migrating group stabilize the transition state, thereby favoring rearrangement over direct hydride deoxygenation (TS-B, Scheme 4c).
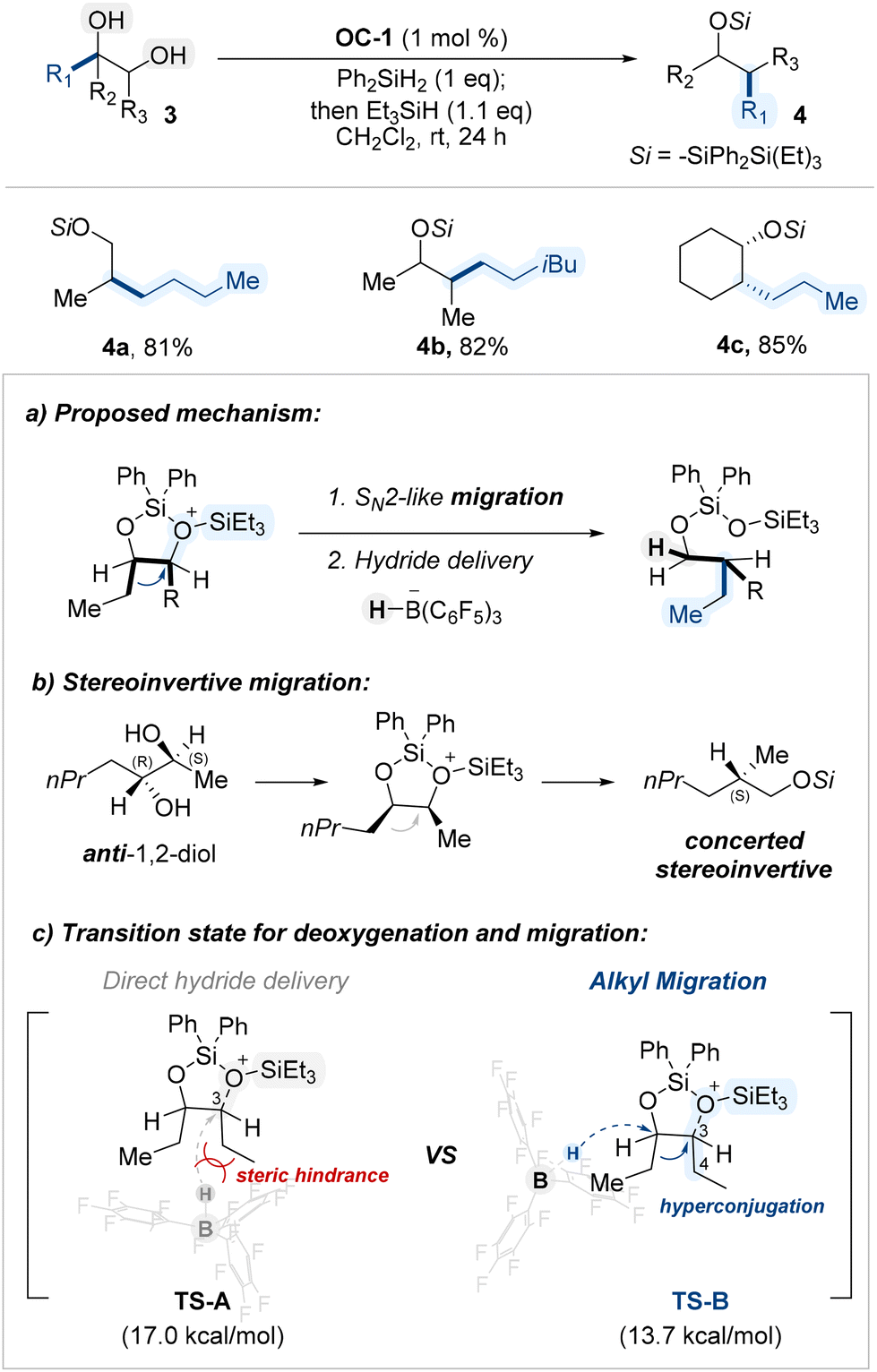 | ||
| Scheme 4 BCF-catalyzed reductive pinacol-type rearrangement of internal 1,2-diols. | ||
Oestreich and co-workers reported a B(C6F5)3-catalyzed chemoselective reduction of primary alkyl tosylates under mild conditions (Scheme 5).64 In this study, remote diols, in which one hydroxyl group was converted into a tert-butyldimethylsilyl (TBDMS) ether and the other into a tosylate, were examined for evaluating the leaving group aptitudes of these two different groups. Upon addition of BCF/hydrosilane, in situ generated boron–hydride species was found to be preferentially delivered to tosylated position for the chemoselective deoxygenation (Scheme 5a).
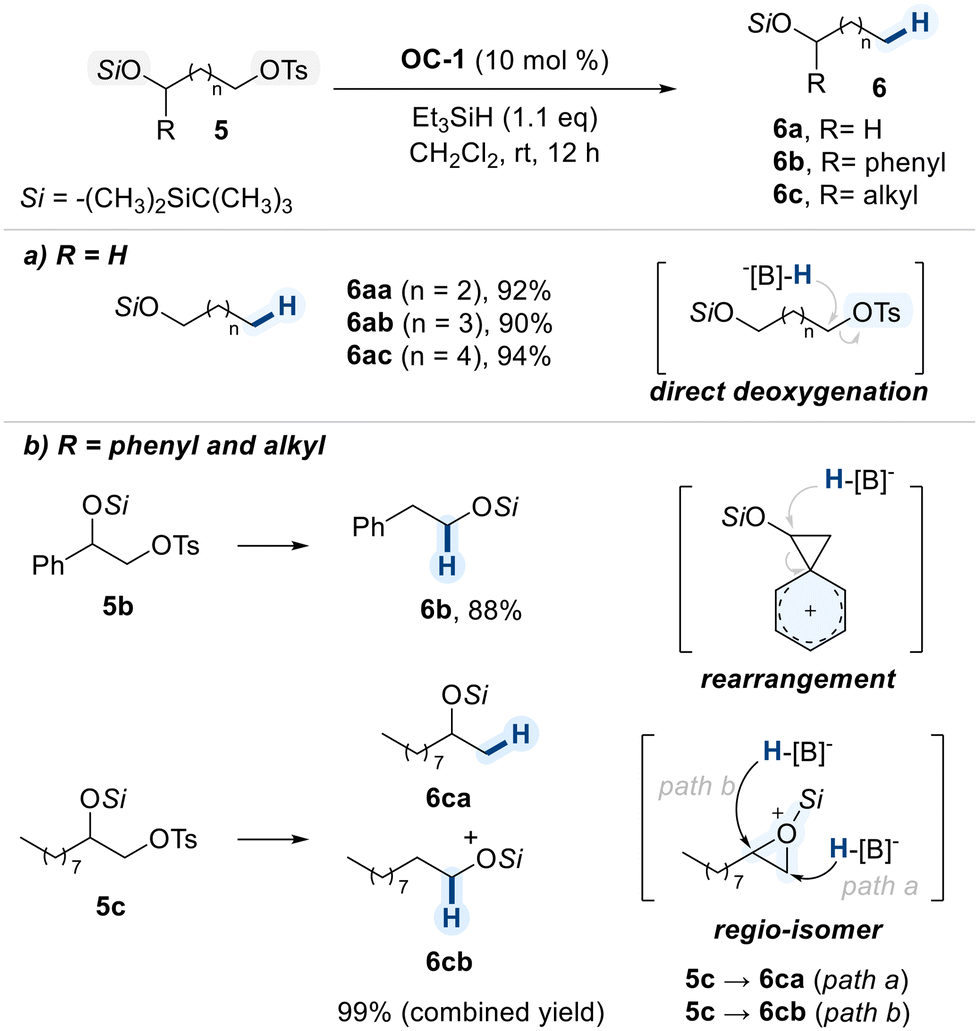 | ||
| Scheme 5 Chemoselective deoxygenation of tosylated diols. | ||
This strategy was further applied to terminal 1,2-diols bearing a primary tosylate (Scheme 5b). Notably, the product outcome varied significantly depending on the nature of the secondary substituent. In aryl-substituted substrates (5b), the benzene π-system participates as a neighboring group, facilitating the formation of a spirocyclic phenonium ion intermediate. The resulting three-membered ring was then opened by hydride delivery from the boron species, affording the rearranged product (6b).
In contrast, alkyl-substituted diols (5c) underwent intramolecular attack of the tosyl group by the oxygen atom on vicinal silyl ether to form a silyl oxonium ion. This intermediate was reduced by hydride attack at either ring carbon (path a or path b), resulting in a nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of regioisomeric products (6ca and 6cb). These results demonstrate that distinct reaction pathways can emerge from subtle changes in substrate structure, highlighting the mechanistic flexibility of this catalytic system.
:1 mixture of regioisomeric products (6ca and 6cb). These results demonstrate that distinct reaction pathways can emerge from subtle changes in substrate structure, highlighting the mechanistic flexibility of this catalytic system.
Following this investigation, the same group presented a BCF/silane-stimulated selective deoxygenation, particularly designed for secondary benzylic alcohols from remote diol substrates (Scheme 6).65 The process is associated with a tandem formylation and deoxygenation sequence. The prior formylation reactions take place only for primary or secondary alcohol substrates due to steric accessibility to formyl group, and the latter deoxygenation proceeds via presupposed carbocation formation followed by hydride delivery. In the second step, secondary benzylic selectivity was achieved given that carbocation is more stable in secondary position than primary position. This result implied that the rationale design of substrates with proper understanding of reaction mechanism would facilitate the development intriguing selective functionalization on unsymmetrical diol substrates.
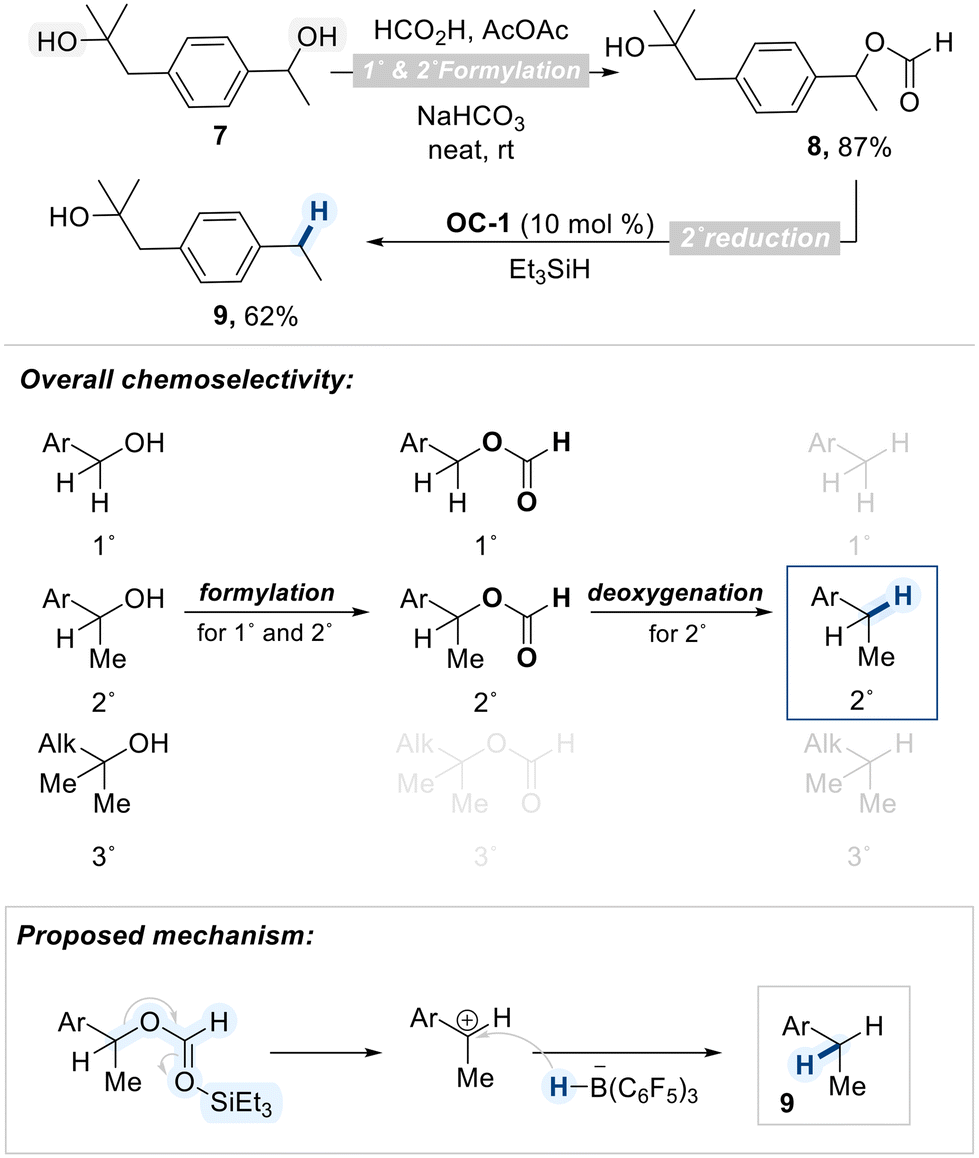 | ||
| Scheme 6 BCF-catalyzed tandem activation–reduction sequences for selective deoxygenation of remote diols. | ||
In light of the research by Song and Lu, a B(C6F5)3-catalyzed deoxygenation protocol was developed for mono-protected diols using a uniquely structured bis(silyl) reductant, (HMe2SiCH2)2 (Scheme 7).66 The reaction displayed high selectivity for free hydroxyl groups over aryl and silyl ethers, as well as other functional groups. The remarkable chemoselectivity is attributed to the dual coordination mode of the bis(silyl) reagent. (HMe2SiCH2)2 features two atom-centered silicon units capable of simultaneously coordinating to a single hydroxyl group, thereby enhancing the leaving group ability of the oxygen and facilitating hydride transfer from the in situ generated boron–hydride species. This mode of activation preferentially engages naked hydroxyl groups over sterically hindered silyl ethers, allowing for selective deoxygenation even in the presence of multiple oxygen-containing functionalities. Notably, the method was also extended to more hindered secondary and tertiary alcohols, which were efficiently converted to the corresponding hydrocarbons, 11c and 11d, respectively.
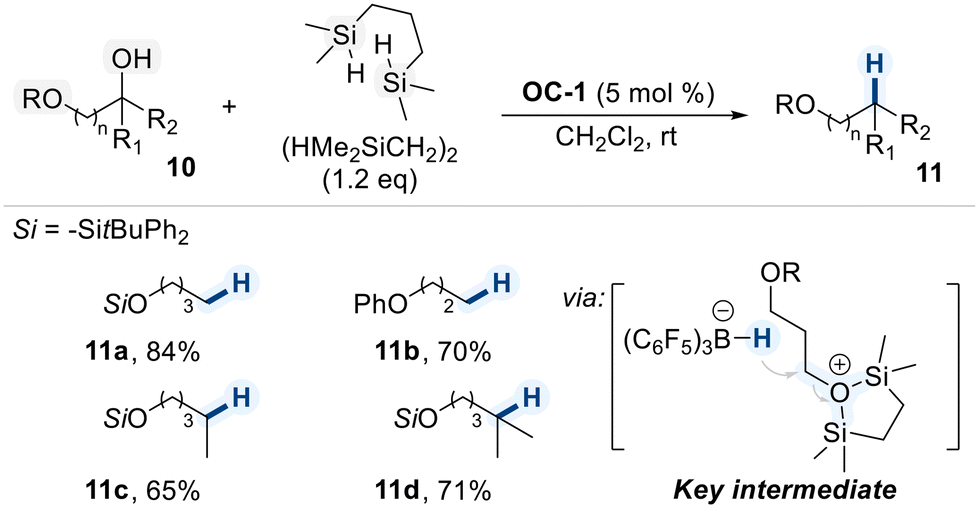 | ||
| Scheme 7 BCF-catalyzed chemoselective deoxygenation of naked hydroxyl group in diols containing diverse ether functionalities. | ||
2.2. Borinic acid catalysis
Borinic acids, featuring one hydroxyl group and two organic substituents bound to a trivalent boron center, possess a vacant p-orbital, rendering them moderately Lewis acidic. The electrophilic boron center of borinic acids can reversibly interact with the electron-rich hydroxyl groups of diols. This interaction facilitates a reversible alcoholysis, leading to the formation of a key cyclic borinate ester intermediate (Scheme 8a).67 The formation of this cyclic borinate intermediate not only enhances the nucleophilicity of the coordinated oxygen, but also creates a tunable spatial and electronic environment that allows for precise control over site-selectivity in subsequent transformations.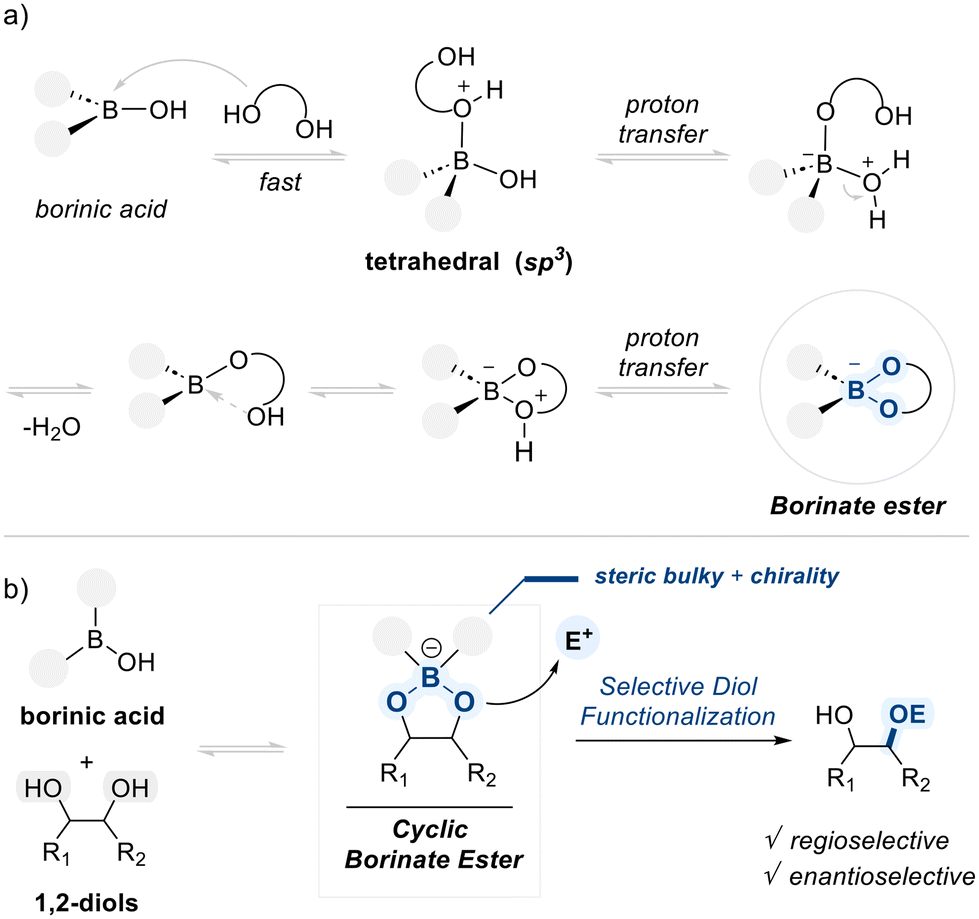 | ||
| Scheme 8 (a) Formation of borinate ester, (b) borinic acid-mediated selectivity control. | ||
The selectivity of borinic acid-mediated transformations can be modulated by altering the nature of the substituents on the boron center (Scheme 8b). When sterically bulky groups are introduced, the reaction is directed toward the more accessible hydroxyl group in unsymmetric diols. Alternatively, attaching chiral substituents can restrict the spatial approach of electrophiles, thereby promoting functionalization in an enantioselective manner. In this section, we introduce examples of diol functionalization using borinic acid catalysts, focusing on how steric and electronic features of the catalyst structure influence selectivity.
Taylor and coworkers employed a diphenylborinic acid ethanolamine complex (OC-2) as a catalyst for the monobenzoylation of vicinal-1,2-diol substrates (Scheme 9a).68 The catalyst OC-2 was turned out to form the key cyclic borinate intermediate preferentially with cis-1,2-diols over trans-diols (cis/trans = 11:1), which was owing to the less angle and torsional strain in cis-intermediate.
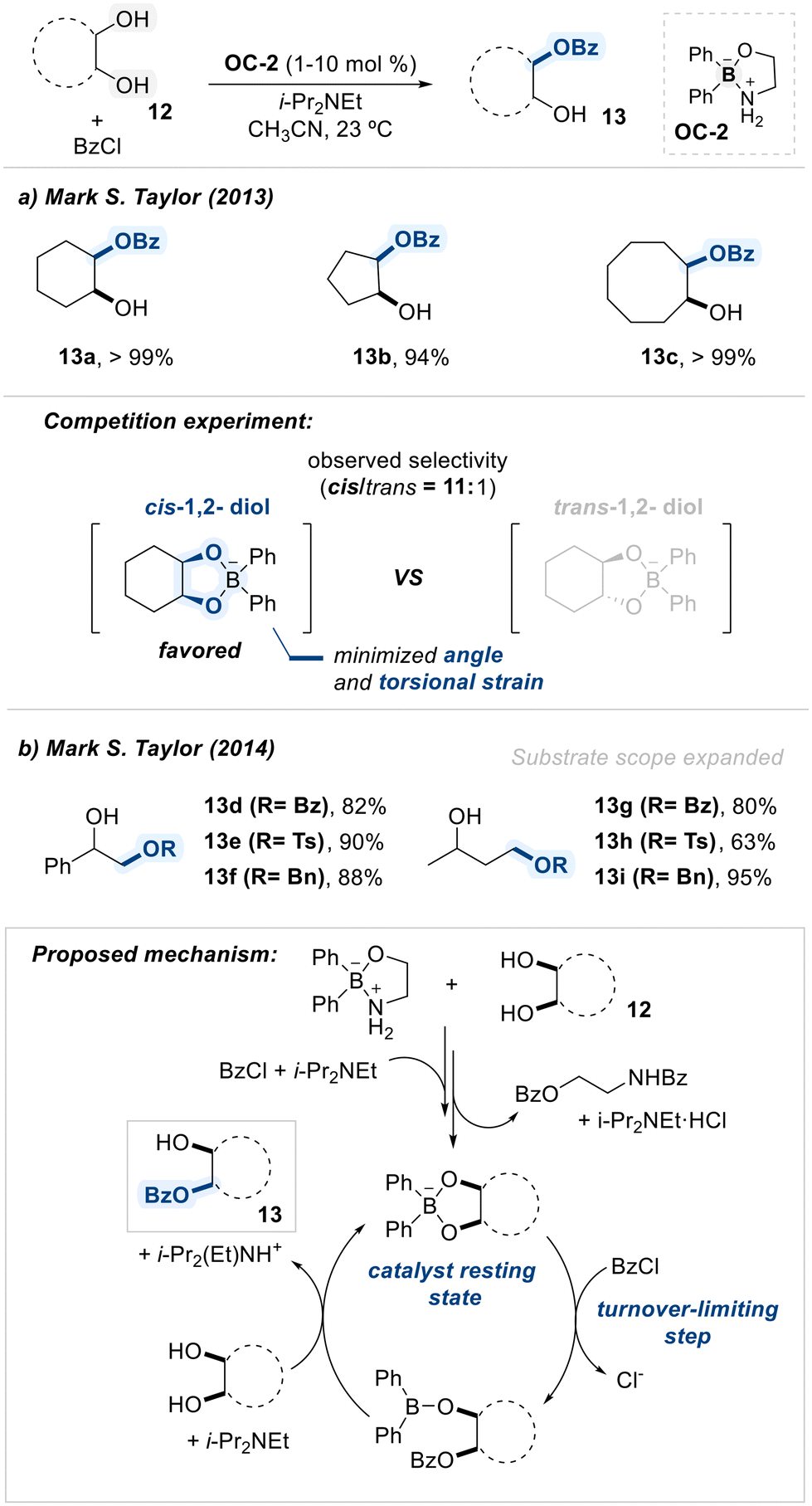 | ||
| Scheme 9 Borinic acid-catalyzed regioselective acylation of diols. | ||
Diphenylborinic acid ethanolamine catalyst (OC-2) forms a reversible complex with the diol substrate, which subsequently undergoes nucleophilic attack on benzoyl chloride to furnish the monobenzoylated products with high selectivity. Importantly, the reaction exhibits a strong preference for cis-vicinal diols, whose adjacent hydroxyl groups adopt a chelating geometry conducive to the formation of a stable five-membered borinate ring. This preference is attributed to the reduced dihedral angle and torsional strain associated with the cis conformation.
Building on this strategy, Taylor and co-workers expanded the scope of the borinic acid-catalyzed monofunctionalization to include sulfonylation and alkylation of non-carbohydrate diols (Scheme 9b). The proposed catalytic cycle begins with the activation of catalyst (OC-2) via irreversible bis-benzoylation of the ethanolamine ligand, followed by coordination of the resulting boron center with the diol substrate to form a stable borinate adduct. The nucleophilic intermediate then undergoes a turnover-limiting benzoylation step, forming a monobenzoylated intermediate. This species is readily displaced by another diol substrate, yielding the desired product and regenerating the resting-state borinate complex.
As a part of their ongoing studies on borinic acid catalysis, the Taylor group introduced a heteroboraanthracene-derived catalyst (OC-3) for the 1°-alcohol selective mono-functionalization of terminal 1,2-diols (Scheme 10).69 The system exhibited high selectivity for primary alcohols, which was attributed to both electronic and steric factors favoring activation at the less hindered hydroxyl group. Mechanistically, the catalyst forms a borinate intermediate with the diol substrate, which then undergoes nucleophilic attack on the electrophile to furnish the functionalized product. Although the interaction between the catalyst and diol is relatively weak, the resulting borinate species is sufficiently nucleophilic to ensure efficient transformation and high selectivity.
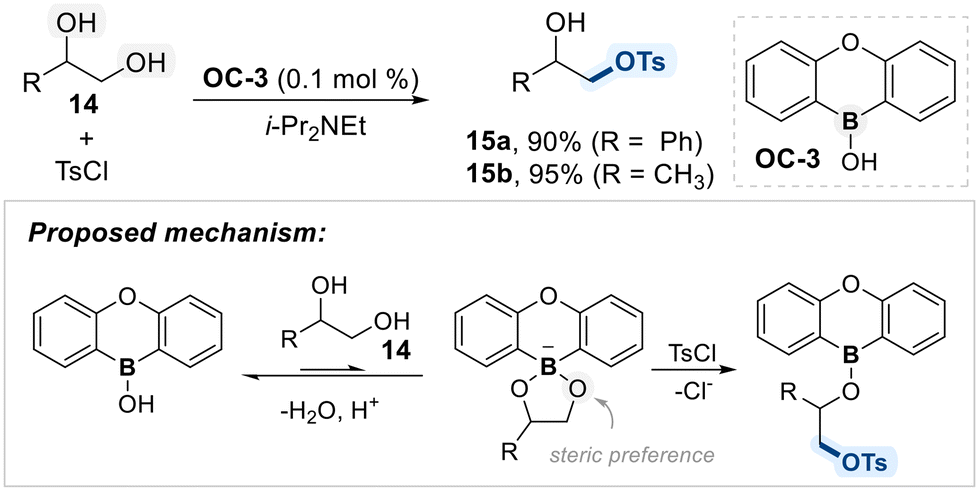 | ||
| Scheme 10 Borinic acid (OC-3)-catalyzed regioselective tosylation of terminal 1,2-diol. | ||
Maruoka and Hashimoto demonstrated that a combination of a borinic acid catalyst (OC-4) and a chiral ammonium salt (Co-cat-1) can induce an enantioselective benzylation of terminal vicinal diols selectively targeting the primary hydroxyl group (Scheme 11).70 In this catalytic process, a borinate ester intermediate, formed between catalyst OC-4 and the diol substrate, interacts with the chiral ammonium salt, generating ion-pair complex labeled, IPS and IPR. Stoichiometric experiments indicated that the formation of the IPS intermediate is kinetically favored; however, the subsequent benzylation step was proposed as the enantio-determining event. While nucleophilic attack by IPS on benzyl chloride is kinetically accessible, nucleophilic substitution involving the IPR intermediate is kinetically disfavored. This kinetic difference results in an effective kinetic resolution of the racemic diol.
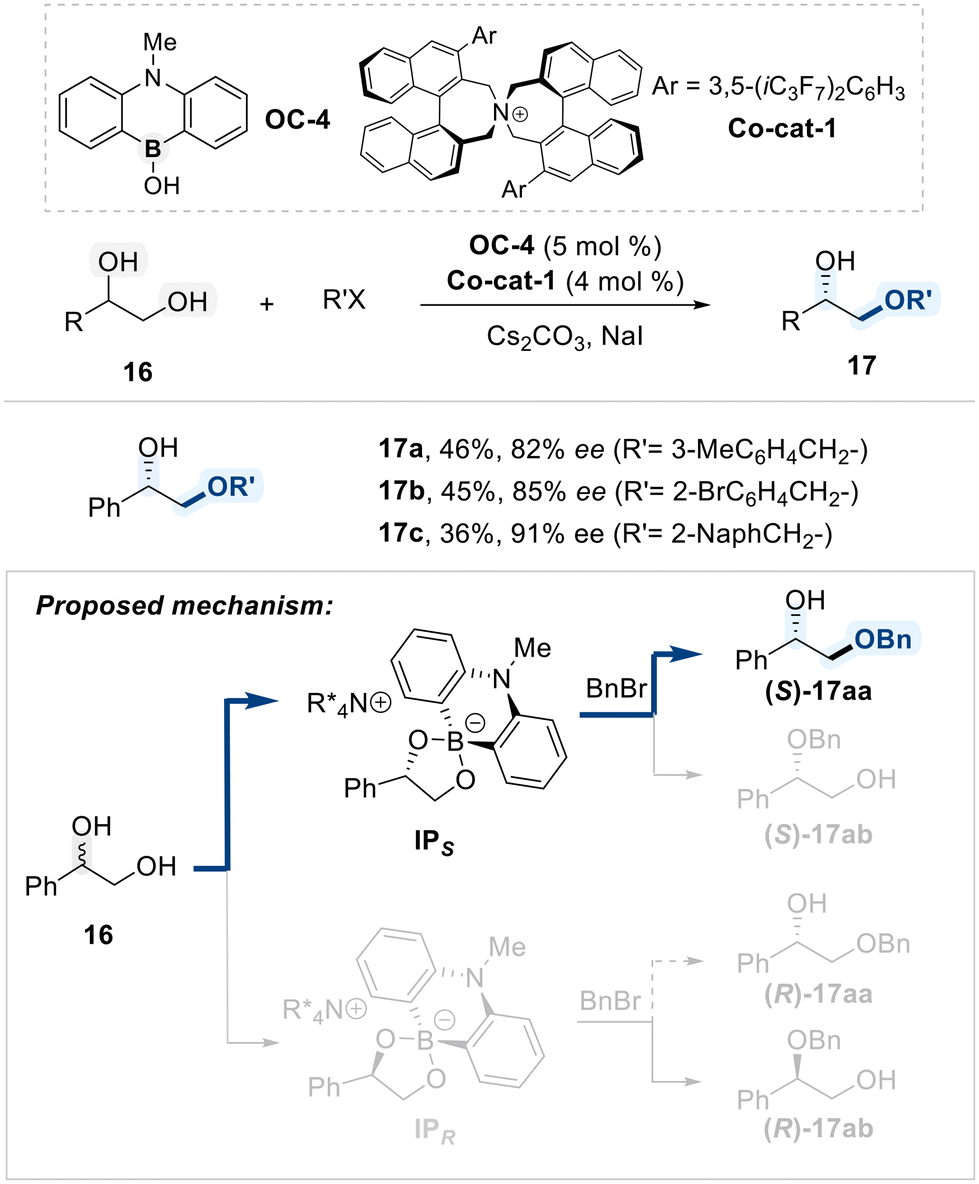 | ||
| Scheme 11 Alkylative functionalization of vicinal diols catalyzed by a chiral ammonium borinate. | ||
Zheng designed a C2-symmetric chiral borinic acid catalyst (OC-5) for the enantioselective desymmetrization of 2,2-disubstituted-1,3-propanediols (Scheme 12).71 The catalyst features a rigid oxaboraanthracene core with two chiral side arms, which adopt a twisted C2-symmetric conformation upon diol binding. This conformation generates a well-defined asymmetric cleft that enforces a fixed geometry on the bound diol. Within this cleft, the two enantiotopic hydroxyl groups are differentiated by their spatial environments, one is deeply buried and sterically shielded by the catalyst framework, while the other remains accessible for reaction. As a result, nucleophilic attack occurs selectively at the exposed site, enabling high levels of enantioselectivity. This cleft-based differentiation is key to the catalyst's ability to control stereochemical outcome, even with sterically hindered diol substrates bearing quaternary centers.
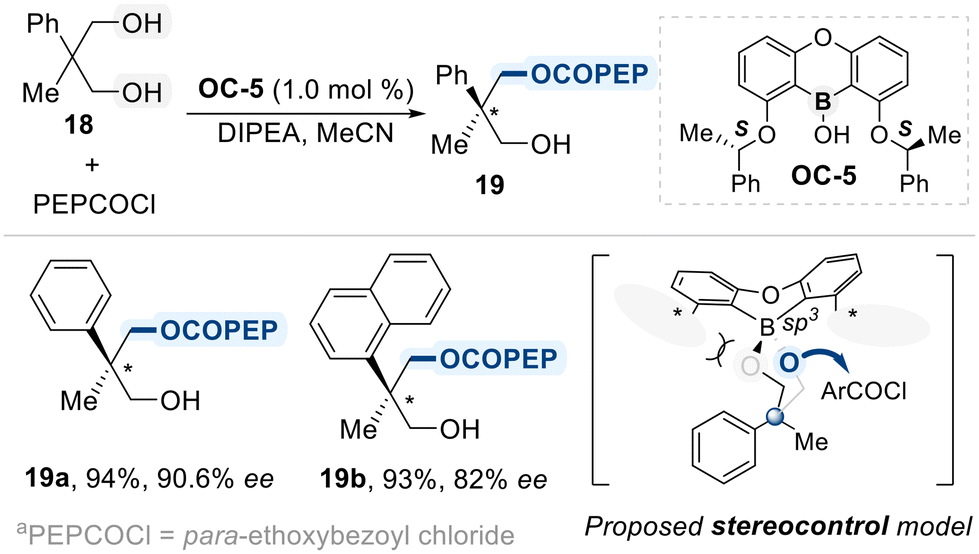 | ||
| Scheme 12 Asymmetric desymmetrization using chiral borinic acid. | ||
Arisawa and Sako presented an enantioselective alkylative desymmetrization of meso-1,2-diols (20) using an axially chiral boronic acid catalyst (OC-6) (Scheme 13).72 The catalyst featured a C2-symmetric biaryl scaffold that established a chiral environment upon coordination with the diol substrate. This interaction generated a cyclic borinate intermediate, positioning the two enantiotopic hydroxyl groups unsymmetrically within the catalyst's architecture. Consequently, one hydroxyl group became sterically shielded, while the other remained accessible for nucleophilic attack, enabling asymmetric induction.
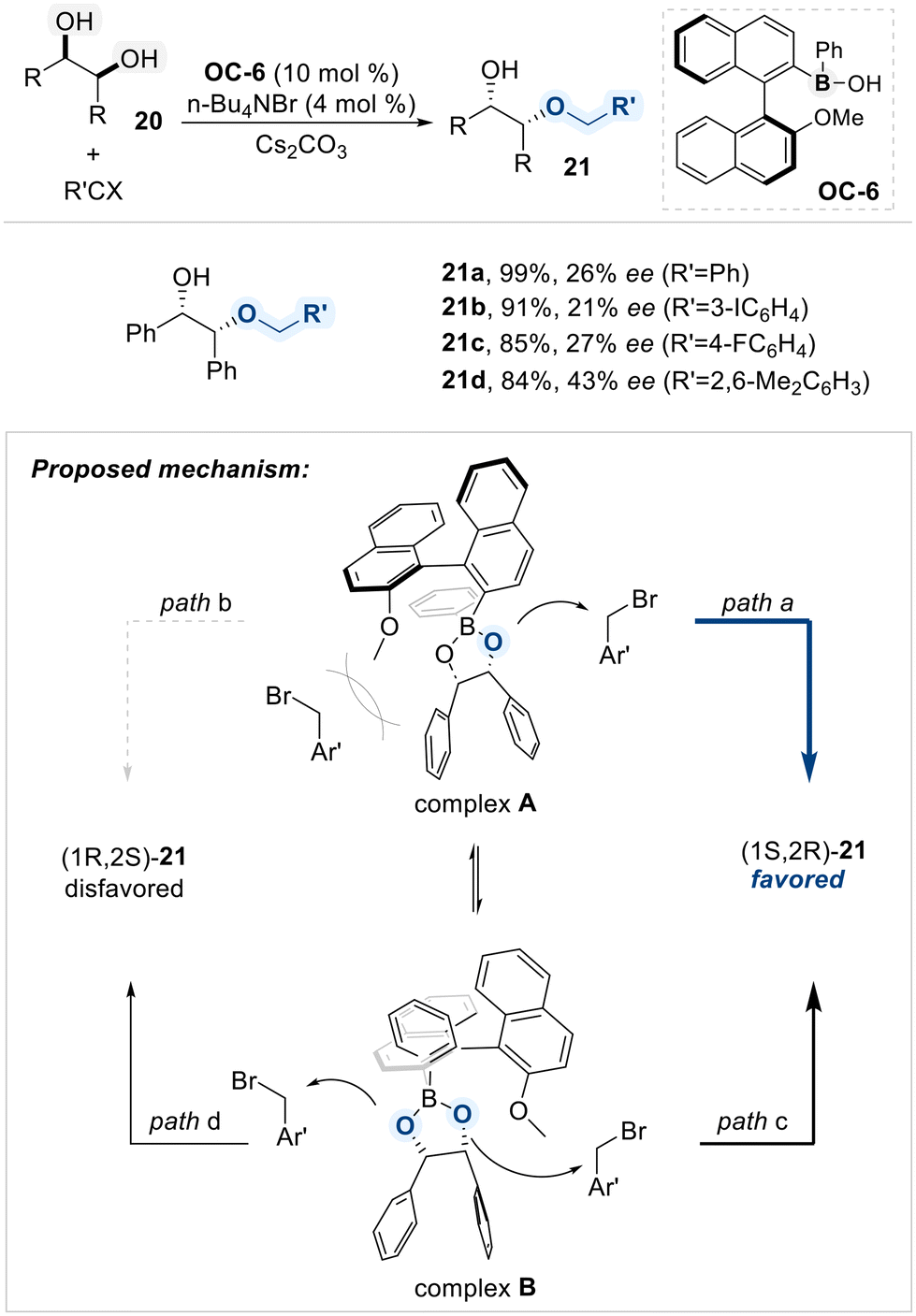 | ||
| Scheme 13 Alkylative desymmetrization of 1,2-diols catalyzed by chiral borinic acid. | ||
According to their mechanistic study via DFT calculations using (R,R)-butane-2,3-diol ((1R,2R)-20), coordination to the chiral boronic acid catalyst OC-6 generates two borinate complexes (complex A and B), resulting in a total of four possible transition states (paths a–d). For complex A, path a is energetically favored, as minimal steric interference between the oxygen and the electrophile leads to the (1S,2R)-product. In contrast, path b is disfavored due to steric congestion from the catalyst's methoxy group. The other conformational complex B proceeds through paths c and d, which compete and reduce overall enantioselectivity. Path c, despite nearby steric bulk, benefits from a twisted geometry that enables effective nucleophilic attack, forming the major enantiomer, (1S,2R)-product. Path d, though free from steric hindrance, suffers from a stereoelectronic mismatch and leads to the minor (1R,2S)-product. These features collectively account for the modest enantioselectivity observed.
2.3. Boronic acid catalysis
Boronic acids are well known for their utility as synthetic building blocks in a variety of bond-forming reactions, such as the Suzuki–Miyaura coupling,73 Petasis reaction,74 and Chan–Lam coupling,75 where they typically function in a stoichiometric manner. However, their role as catalytic reagents, particularly in the context of diol functionalization, is comparatively less developed and remains distinct from that of other boron-based species such as borinic acids, which offer greater structural tunability and catalytic versatility.This limitation is closely linked to the structural simplicity of boronic acids, which possess a trivalent boron center bound to two hydroxyl groups and a single organic substituent. While this architecture is well suited for reversible covalent interactions with diol substrates, a key feature in dynamic covalent catalysis, it inherently restricts opportunities for fine-tuning steric and electronic environments. As a result, in boronic acid-catalyzed selective functionalization of diols, regio- or chemoselectivity is rarely dictated by the catalyst itself and instead arises from the intrinsic electronic or steric bias within unsymmetric diol substrates (Scheme 14).
 | ||
| Scheme 14 Boronic acid-catalyzed selective functionalization of diol. | ||
While numerous methods employing boronic acids have been developed for diol modification, this review focuses specifically on catalytic approaches. Accordingly, systems requiring stoichiometric or excess amounts of boronic acids are excluded.76,77 In the following section, we highlight representative examples in which boronic acid-derived catalysts have enabled selective diol functionalization, with particular attention to the underlying reaction mechanisms.
Onomura's group reported that a boronic acid catalyst (OC-7) efficiently facilitates the selective oxidation of vicinal-diols to α-hydroxyketones under aqueous conditions, performing in both chemical and electrochemical reaction systems (Scheme 15).78 According to their proposed mechanism, the 1,2-diol substrate is first activated by the boronic acid catalyst OC-7 via formation of a boronate ester intermediate. In the chemical pathway, the cyclic boronate undergoes nucleophilic attack on an Br+ species, generated in situ from DBI. This transformation ultimately affords the ketone product 23 through an elimination process. The authors further demonstrated that a combination of KBrO3 and KHSO4 can also serve as an electrophilic bromine source for this transformation.79 In the electrochemical pathway, Br+ is produced at the anode from KBr, while OH− ions formed at the cathode and subsequently act as the base in the elimination step.
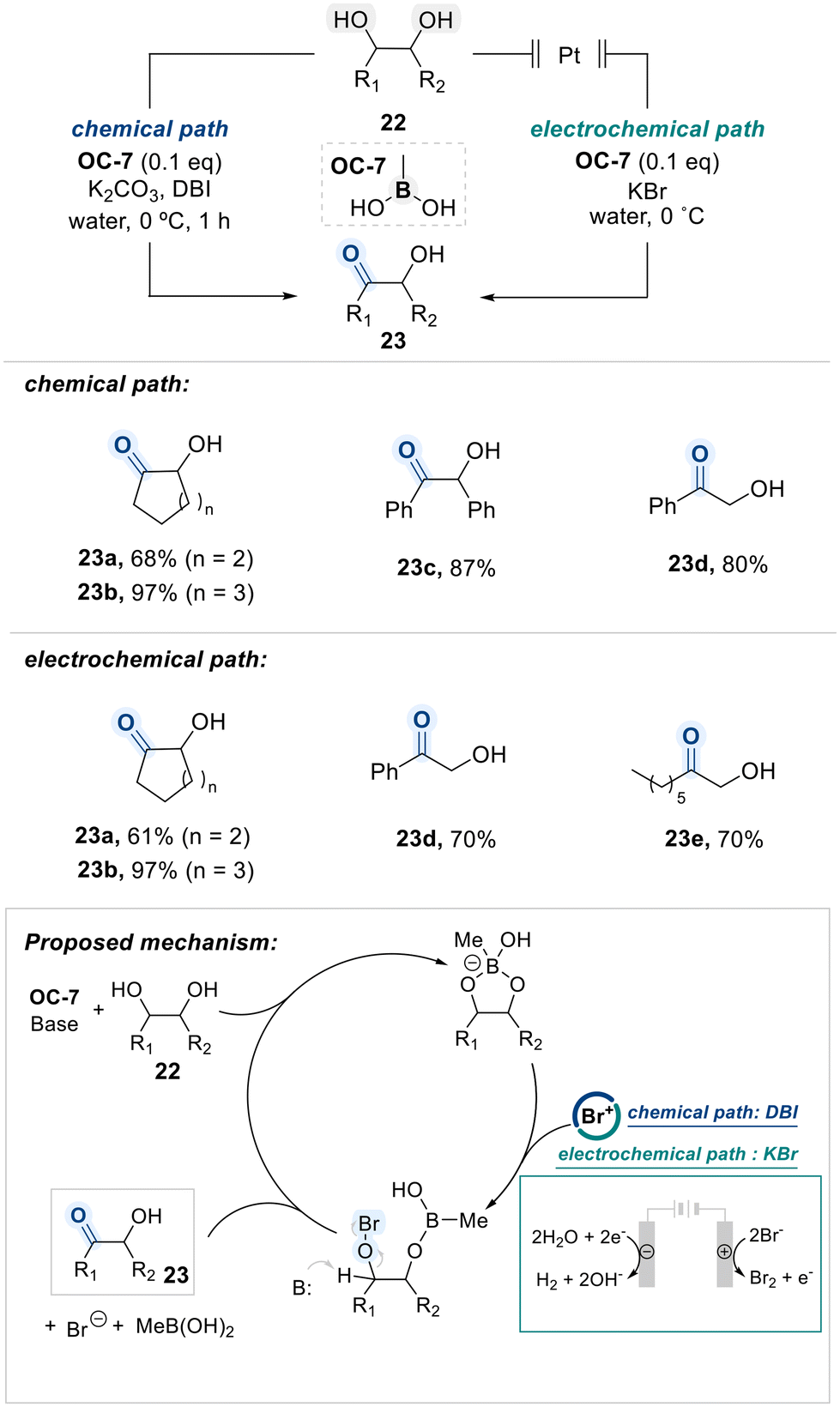 | ||
| Scheme 15 Boronic acid-catalyzed selective oxidation of 1,2-diols to α-hydroxyl ketones. | ||
In this context unsymmetrical terminal diol substrates exhibited selective oxidation at the secondary hydroxyl group, highlighting a preference over the primary site under the optimized conditions.
In 2021, Taylor and co-workers reported a site-selective intramolecular cyclization of remote diols utilizing a pentafluorophenylboronic acid catalyst (OC-8) and oxalic acid co-catalyst (Co-cat-2) (Scheme 16).80 The reaction proceeds via the formation of a Brønsted acid-like boron species (I), generated through dynamic association between OC-8 and Co-cat-2, which acts as the active catalyst. Upon exposure to the diol substrate, species (I) functions as a proton donor toward the benzylic hydroxyl group, thereby promoting the elimination of water and generating a benzylic carbocation intermediate. This intermediate subsequently undergoes intramolecular nucleophilic attack by the pendant hydroxyl group, resulting in formation of the cyclic ether. Concurrently, the conjugate base boron species (II), generated in the initial proton transfer step, accepts the proton released during ether formation to afford the protonated boron species (III). Rehydration of (III) under aqueous conditions restores the active species (I), thus completing the catalytic cycle. The observed regioselectivity is attributed to the involvement of a carbocation intermediate, as benzylic positions provide sufficient stabilization to allow efficient cyclization.
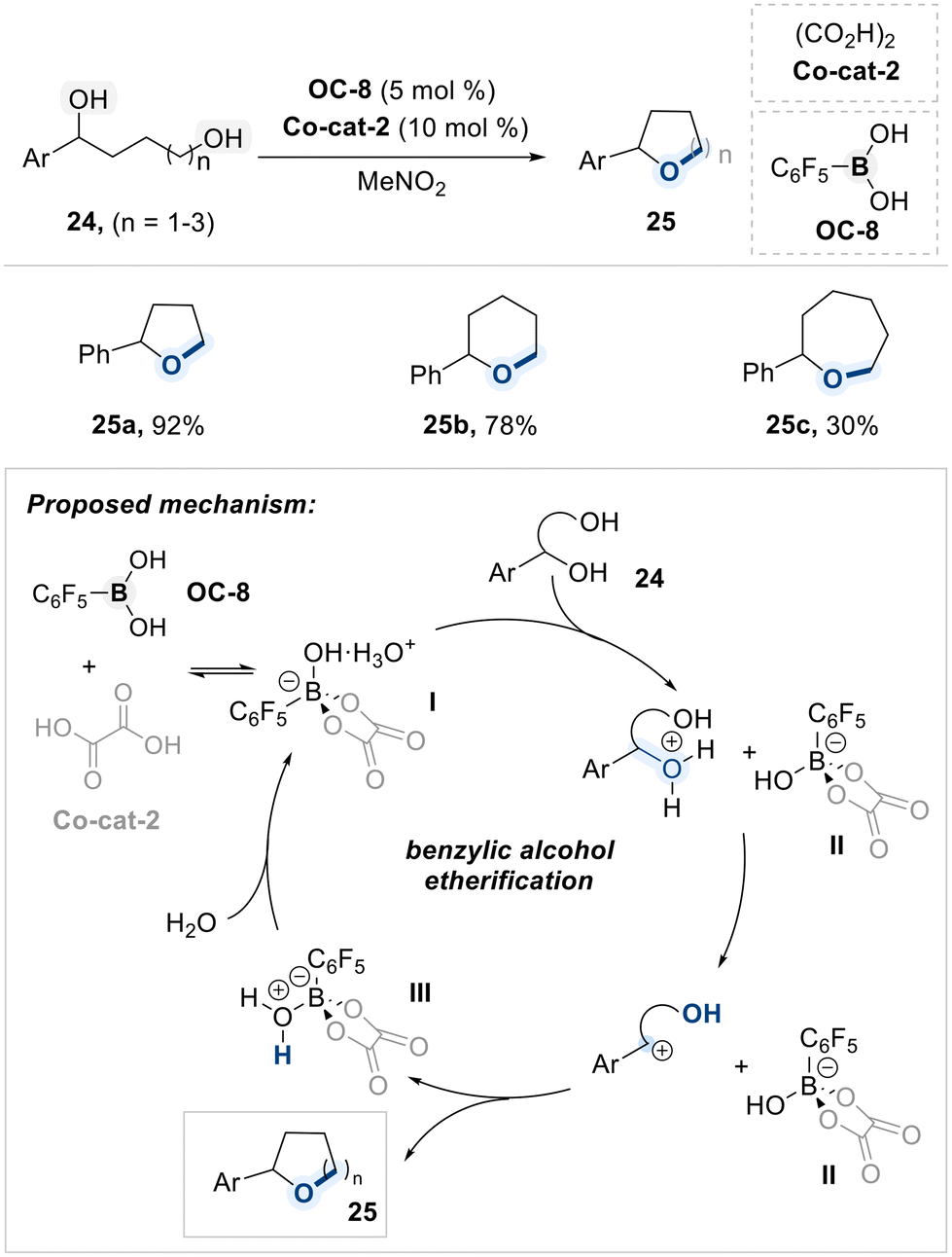 | ||
| Scheme 16 Intramolecular cyclization of remote diols. | ||
2.4. Catalysis by heterocyclic boronic acid derivatives
Heterocyclic boronic acid derivatives have gained attention as promising catalysts for selective polyol functionalization due to their customizable properties in catalyst desgin.81 A subset of these catalysts possesses boron-containing heterocycles incorporating oxygen or nitrogen atoms within the ring, which provide stable rigidity and sufficient Lewis acidity for effective diol binding (Scheme 17).82 These heterocyclic frameworks often feature unsymmetric geometries distinct from classical boronic or borinic acids, allowing differentiation between the two hydroxyl groups in diol substrates during complexation.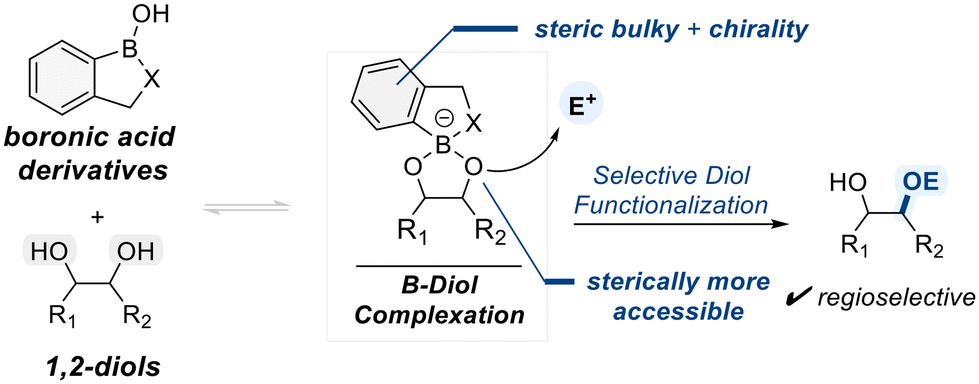 | ||
| Scheme 17 General mechanism for heterocyclic boronic acid-catalyzed selective functionalization of diols. | ||
Incorporating steric hindrance or chiral elements into the heterocyclic backbone can further enhance the selectivity of electrophilic reactions mediated by such boron–diol complexes. The following section highlights representative examples of selective diol functionalization, with a particular focus on catalysts based on benzoxaborole scaffolds. This section presents various examples of selective diol functionalization, focusing on catalysts derived from heterocyclic boron-based scaffolds.
Hall and Rygus developed a neutral benzoxazaborine-based organocatalyst (OC-9) for the regioselective functionalization of vicinal-diols (Scheme 18).83 Upon reversible formation of a tetracoordinate boronate species (IV), the sterically accessible oxygen, originating from the primary alcohol, selectively attacks chlorophosphate electrophiles, affording the desired products in high yields with excellent site-selectivity.
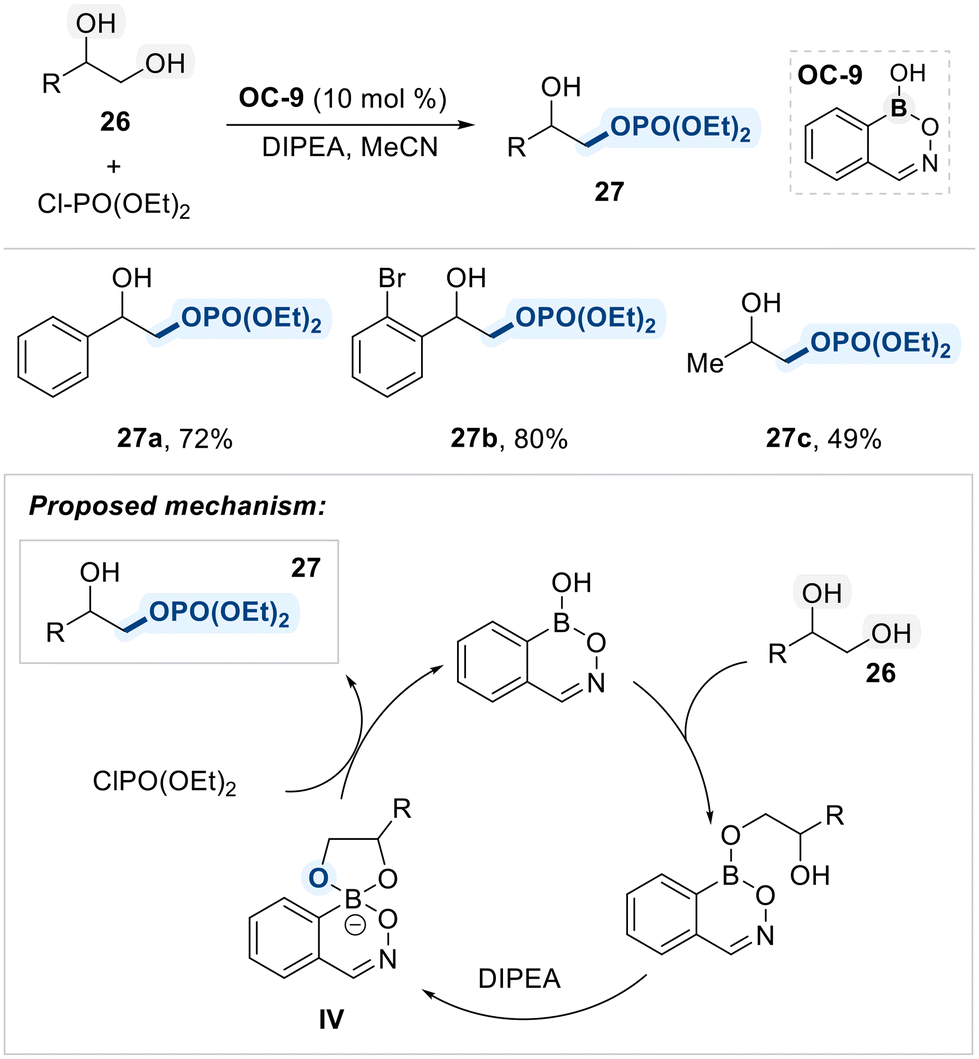 | ||
| Scheme 18 Monophosphorylation reaction of vicinal diols catalyzed by benzoxazaborine scaffold. | ||
Kuwano and Arai unveiled that a chiral benzazaborole catalyst (OC-10), in combination with an N-methylimidazole (NMI) co-cataylst (Co-cat-3), effectively induced the enantio-selective sulfonylation of cis-1,2-diols (Scheme 19).84 The benzazaborole scaffold of OC-10 facilitates the stepwise formation of a key boronate intermediate upon binding to the cis-diol substrate. Meanwhile, the NMI co-catalyst activates sulfonyl chloride by generating a tosyl imidazolium species, which serves as the actual electrophile. The conformationally more accessible oxygen atom within the cyclic boronate intermediate then performs a nucleophilic attack on the activated tosyl electrophile, providing the enantioriched tosylated product. Finally, the boronate ester is regenerated as the catalytic boron center is displaced by another incoming diol substrate, thus completing the catalytic cycle.
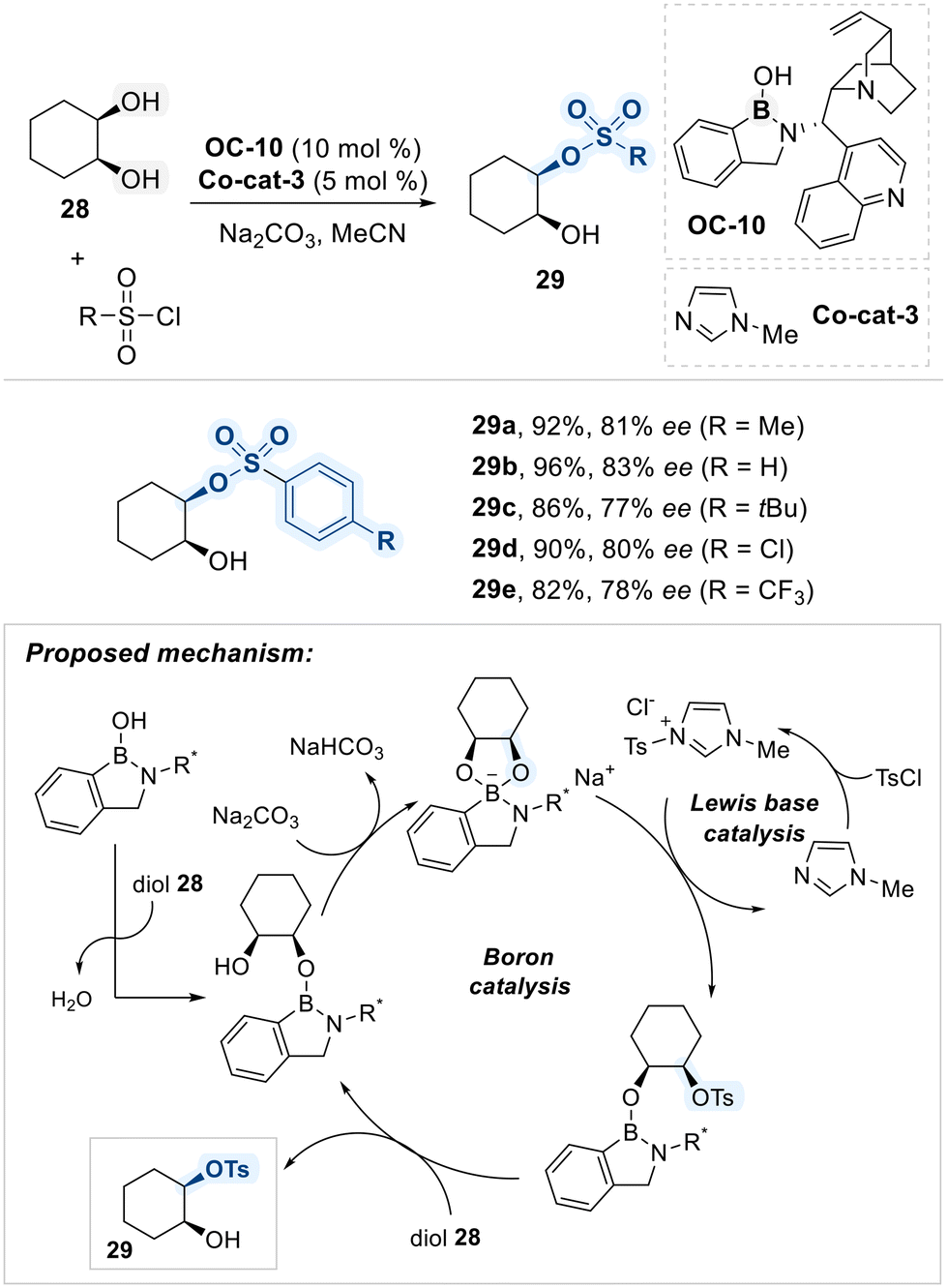 | ||
| Scheme 19 Chiral benzazaborole-catalyzed enantioselective sulfonylation of meso-1,2-diol. | ||
The Kusano research group presented a benzoxaborole-based catalytic platform for the site-selective functionalization of 1,2-diols (Scheme 20).81 The catalyst features a benzoxaborole core bearing electron-withdrawing aryl substituents, which increase the binding affinity toward diol substrates and promote the formation of catalytically active boronate intermediates. This conformational advantage was experimentally shown to confer high selectivity for cis-1,2-diols over their trans-isomers, as evidenced by competitive benzoylation reactions in which preferential functionalization of the cis-isomer was observed. In substrates bearing both primary and secondary hydroxyl groups, functionalization predominantly occurs at the primary site, likely due to its lower steric hindrance.
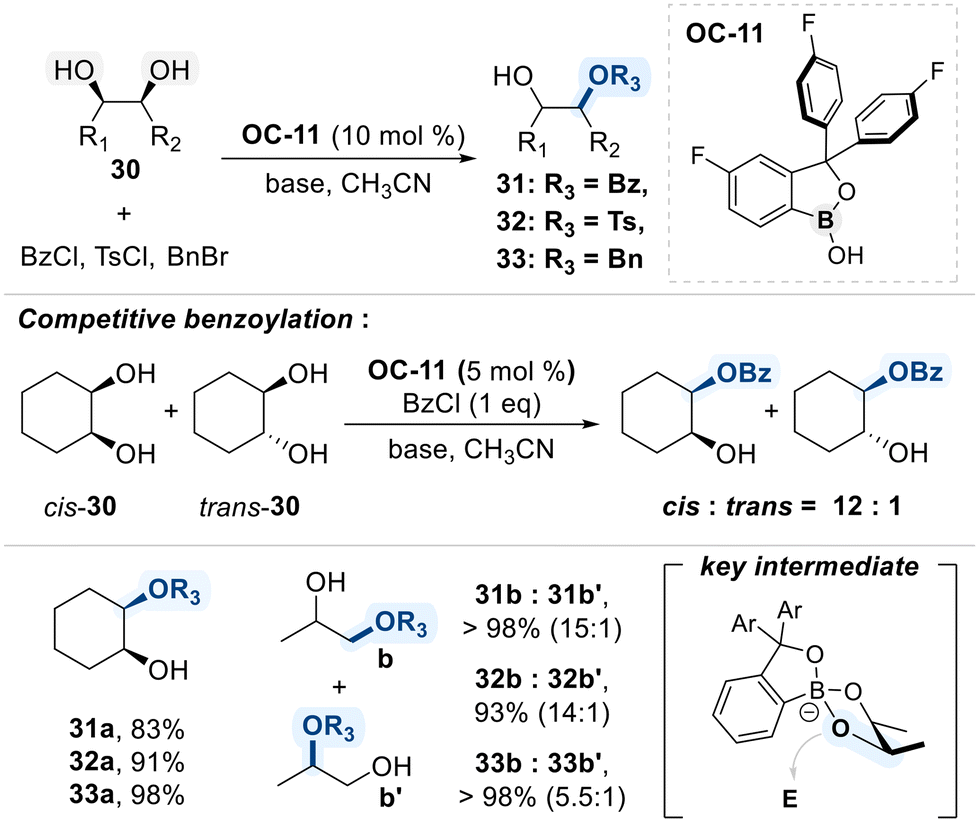 | ||
| Scheme 20 Site-selective modification of diols catalyzed by OC-11. | ||
In a follow-up study, the same research group developed an improved bifunctional benzoxaborole catalyst (OC-12) bearing a pendant N,N-dimethylamino group for the regioselective benzoylation of 1,2-diols (Scheme 21).85 The catalyst engages in bifunctional activation, wherein the benzoxaborole moiety selectively forms a tetrahedral boronate intermediate with cis-1,2-diols due to their favorable geometry for cyclic ester formation, while the pendant N,N-dimethylamino group simultaneously performs nucleophilic activation of the benzoyl chloride, enabling an intramolecular acyl transfer to the coordinated diol. This cooperative mechanism not only enhances catalytic efficiency but also confers high regioselectivity, favoring monobenzoylation at the less hindered primary alcohol of the terminal 1,2-diol substrate.
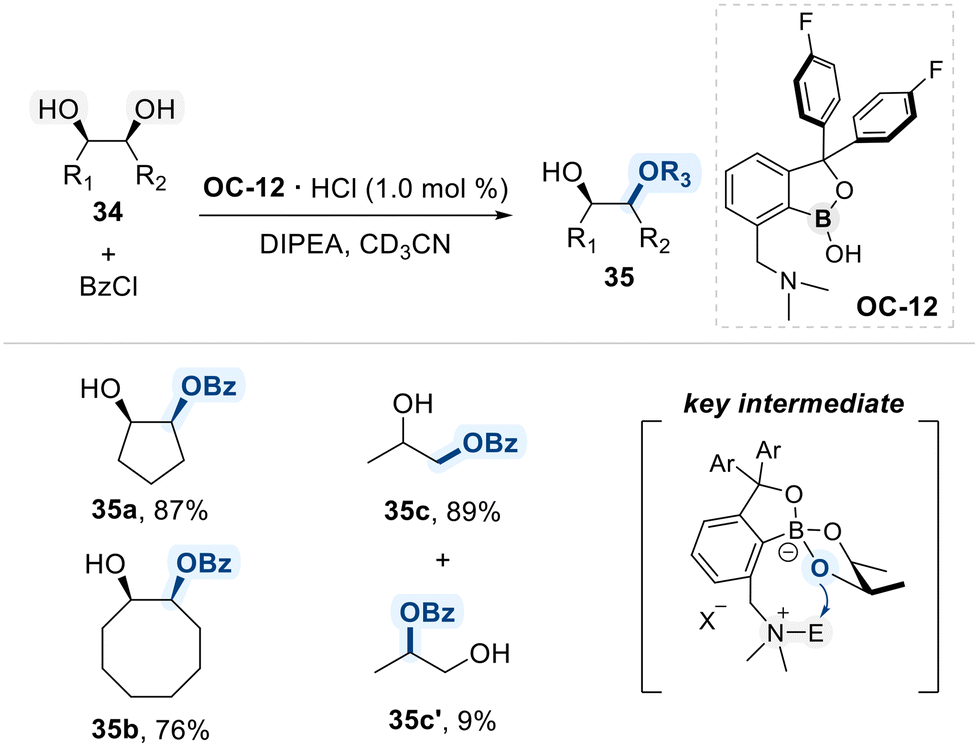 | ||
| Scheme 21 Selective cis-1,2-diol modification using the benzoxaborole catalyst embedded with Lewis base. | ||
Hall and co-workers described a method for the desymmetrization of meso-1,3-diols via an enantioselective alkylation, employing a chiral hemiboronic acid catalyst (OC-13) (Scheme 22).86 A boroxarophenanthrene core from a BINOL-derived backbone, along with steric elements such as a trityl ether and two methyl groups, confers the catalyst (OC-13) for having conformational-biased environment when reactants approach to them. Complexation of OC-13 catalyst with the 1,3-diol forms a six-membered borinate ester, positioning the two hydroxyl groups in distinct spatial orientations. Selective alkylation occurs at the less hindered hydroxyl group, located away from the bulky trityl substituent, furnishing the enantioriched alkylated product.
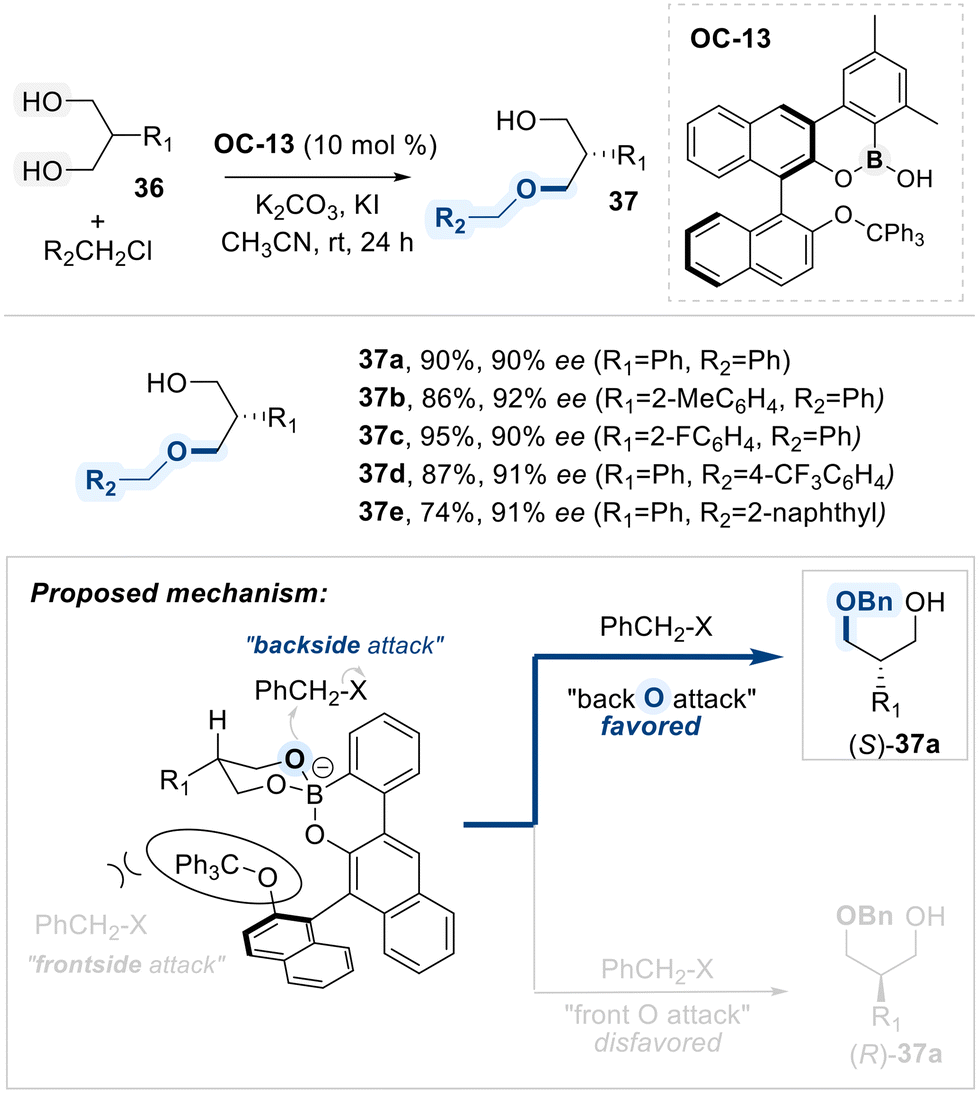 | ||
| Scheme 22 Enantioselective alkylation of 1,3-diols with a chiral hemiboronic acid catalyst. | ||
3. Nitrogen-based organocatalytic systems
In recent years, nitrogen-containing organocatalysts have played a crucial role in modern organic synthesis, offering versatile reactivity, favorable electronic properties, and enhanced catalytic efficiency.87 In the context of selective diol functionalization, various types of nitrogen-containing catalytic systems have been employed, including N-heterocycles, diamines, peptides and N-heterocyclic carbenes (NHCs), and others, each operating through distinct mechanistic pathways. The continued application of these catalysts to a broader range of diol substrates has enabled value-added structural modifications with high selectivity.883.1. N-Heterocycle catalysis
Aromatic N-heterocycles are widely recognized as efficient catalysts for the functionalization of alcohols.89 The mechanism underlying their catalytic activity in hydroxyl group functionalization is well understood (Scheme 23).90 It begins with the activation of the N-heterocycle by an electrophilic species, leading to the formation of a key ammonium intermediate (V), which acts as a new electrophile, for the subsequent nucleophilic attack by the alcohol. This nucleophilic attack leads to the functionalization of the alcohol, forming a desired product with a second ammonium intermediate (VI). Consequently, intermediate VI undergoes deprotonation by an external base, regenerating the catalyst and completing the catalytic cycle.91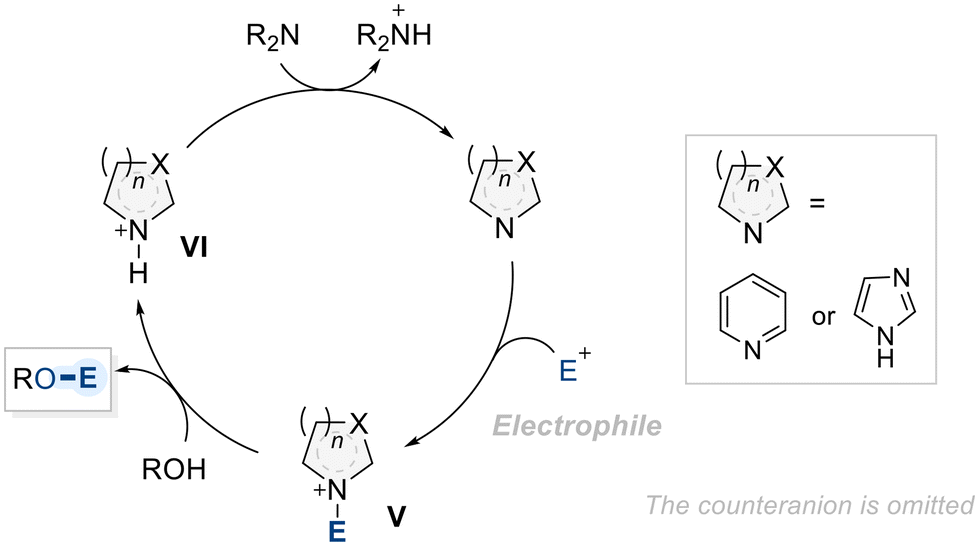 | ||
| Scheme 23 General mechanism of N-heteroycle-catalyzed alcohol functionalization. | ||
With these mechanistic features in hand, researchers have explored the potential of N-heterocycles in selective catalysis for diol functionalization. Notably, chiral 4-amino pyridine structures have been widely considered as an asymmetric catalyst in the context of enantioselective acylation of diol substrates. In 2003, Kawabata et al. introduced a chiral 4-pyrrolidinopyridine catalyst (OC-14), featuring two distinct functional side chains at C2 and C4 of the pyrrolidine ring, to perform enantioselective isopropanoylation of meso-1,2-cyclohexane diol (Scheme 24).92 As outlined in the general mechanism (Scheme 23), forming a chiral isopropyl acyl-pyridinium intermediate was identified as a key step in achieving enantioselectivity. Despite moderate yield (61%) and enantioselectivity (65% ee), the successful sterochemical induction achieved by introducing chiral substituents onto the N-heterocycle offers pioneering insights and establishes a foundation for advancing chiral pyridine-based catalysts in the selective functionalization of diols.
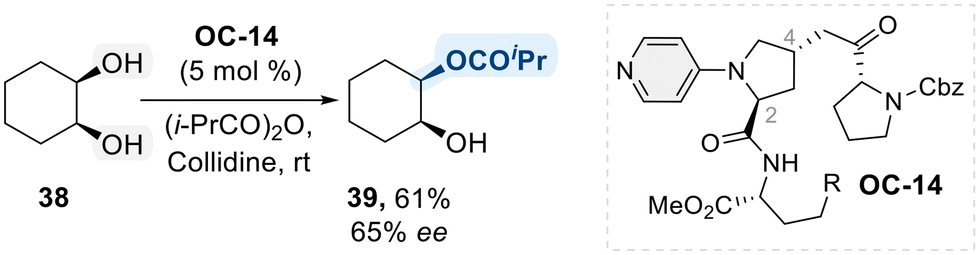 | ||
| Scheme 24 Desymmetrization of meso-1,2-cyclohexane diol using organo-catalyst (OC-14). | ||
Building on these insights, Yamada and co-workers designed a novel class of chiral 4-(dimethylamino)pyridine (DMAP) catalyst (OC-15) for the asymmetric isopropanoylation of meso-diols (Scheme 25).93 A distinctive feature of this catalyst is its unique conformation switch system, which is based on the interconversion between the uncomplexed form (VII) and the self-complexed form (VIII), induced by N-acylation. The proposed mechanistic model suggests that self-complexation, driven by an intramolecular cation–π interaction between the pyridinium ring and a thiocarbonyl group, creates a chiral environment around the active site, which is used for discriminating symmetric diols.
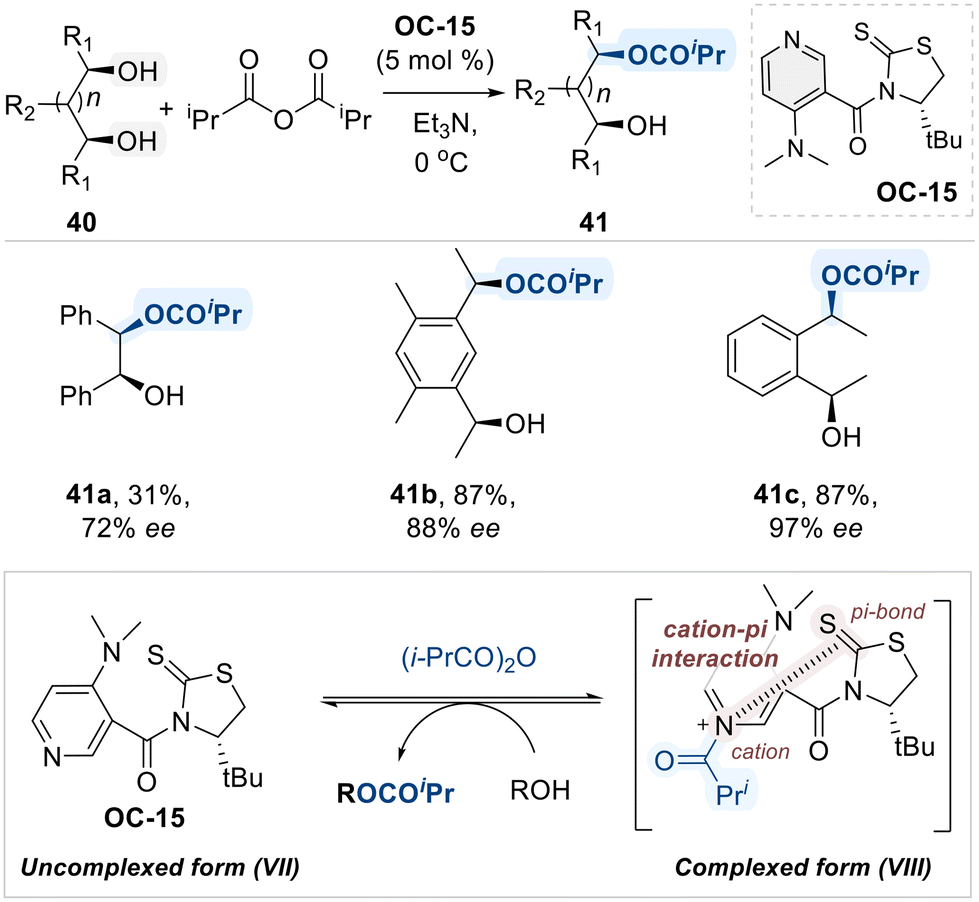 | ||
| Scheme 25 Enantioselective isopropanoylation of meso-diols using chiral-DMAP catalyst (OC-15). | ||
Furthermore, a study by Mandai et al. made a significant contribution to the chiral 4-amino pyridine catalytic system by introducing a 1,1′-binaphthyl containing two 3° hydroxyl units (OC-16) (Scheme 26).94 The study highlighted that the two 3° hydroxyl units were crucial for achieving high catalytic activity and enantioselectivity in desymmetrization of 1,3-diols, though their specific role in the reaction mechanism has not yet elucidated.
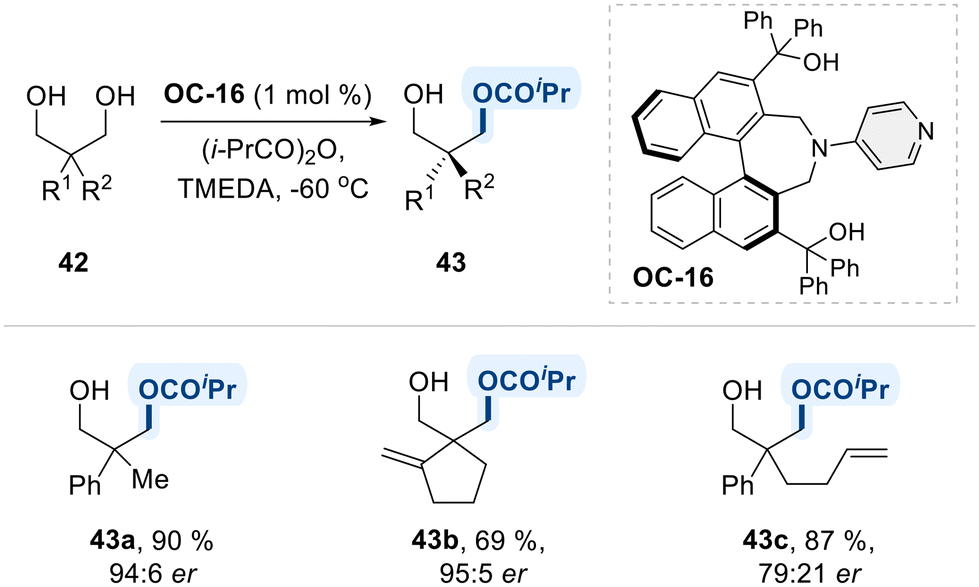 | ||
| Scheme 26 Enantioselective isopropylation of 1,3-diols via chiral-DMAP catalyst (OC-16). | ||
In 2018, Takata and his colleagues disclosed a novel rotaxane-type catalyst (OC-17) to undergo O-benzoylative asymmetric desymmetrization of meso-1,2-diol (Scheme 27).95 The rotaxane-type catalyst OC-17 exhibited a cooperative effect between its crown ether wheel bearing (R)-binaphthyl group (back structure of OC-17, Scheme 27) and its axle containing N-pyridylmethyl substituent (front structure of OC-17, Scheme 27). As described in Scheme 23, the reaction initiated with the formation of an N-acylpyridinium intermediate, followed by the enantioselective approach of the diol substrate. The proximity effect, driven by the localization of the chiral wheel around the N-benzoyl pyridinium intermediate, facilitated this selectivity.
 | ||
| Scheme 27 Rotaxane-based catalyst (OC-17) for asymmetric regioselective benzoylation of meso-1,2-diol. | ||
As a further contribution, Lian et al. recently developed a chiral C2-substituted aminopyridine-N-oxide (OC-18) and a chiral C3-substituted arylpyridine-N-oxide (OC-19), enabling enantio-divergent benzoyl transfer in the desymmetrization of meso-1,2-diols (Scheme 28).96 Depending on the choice of chiral catalyst (OC-18 or OC-19), benzoylation of meso-diol 46 proceeded selectively to afford either enantiomer, 47 or 48. DFT calculations further elucidated the reaction mechanism, revealing that it proceeded through a bifunctional activation, where the benzoyl-activated N-oxide and the proton of the amides played pivotal roles in both catalytic reactivity and enantio-control.
 | ||
| Scheme 28 Desymmetrization of meso-diols catalyzed by chiral pyridine-N-oxide (OC-18 and OC-19). | ||
Selective functionalization has been achieved using not only pyridine-based organocatalysts but also imidazole-derived catalysts. In the context of imidazole-based selective organocatalysis, Tan and coworkers demonstrated that an imidazole bearing a chiral substituent at the C2-position (OC-20) effectively catalyzed the enantioselective silylation of meso-1,2-diols (Scheme 29).97 This catalyst exhibits bifunctional properties: a 2-methoxy oxazolidine moiety functions as the diol-binding site, while the imidazole group serves as the silyl-binding site. This arrangement enables efficient and selective transfer of silyl groups to diols in an enantioselective manner.
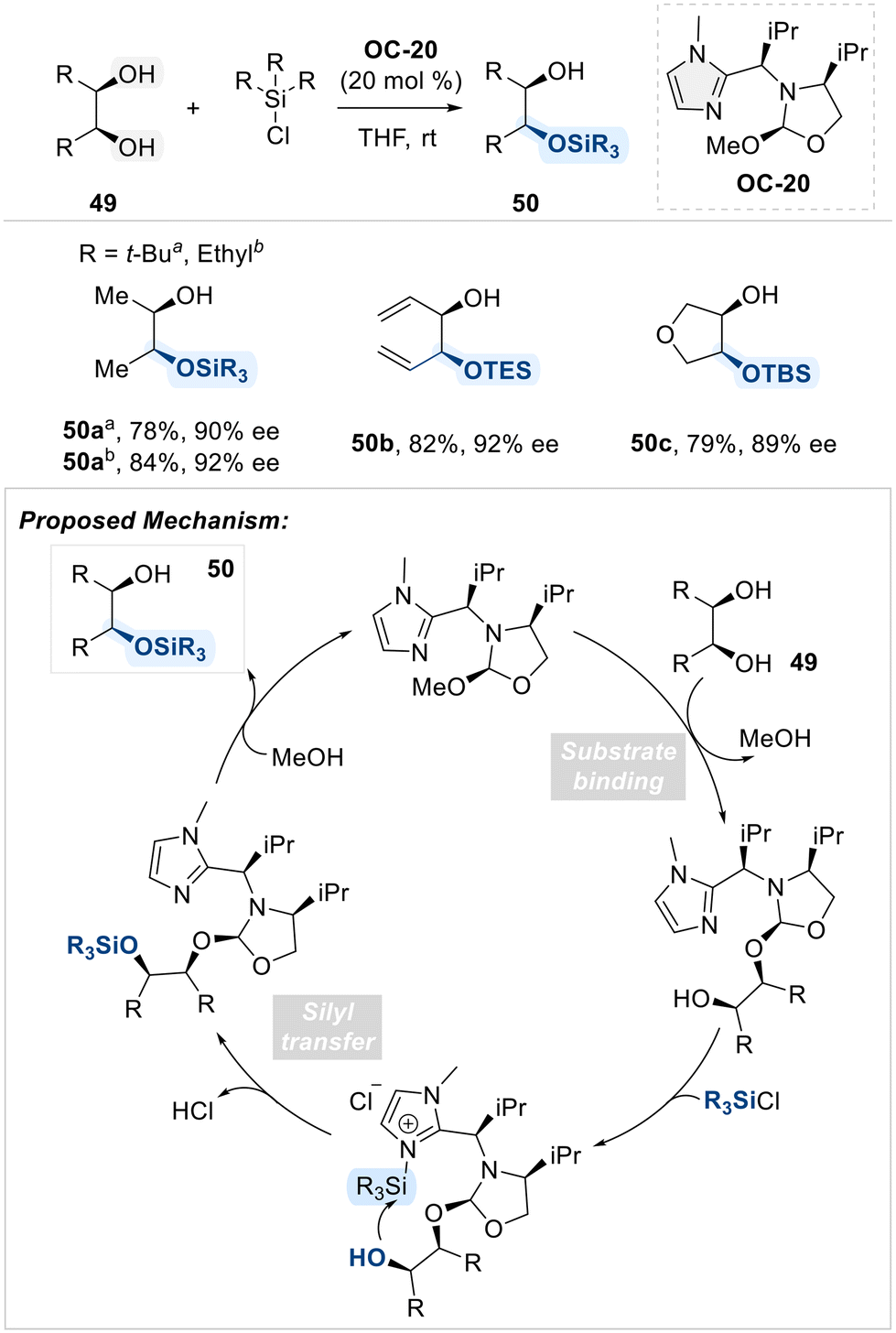 | ||
| Scheme 29 Enantioselective silylation of meso-1,2-diols employing bifunctional imidazole catalyst (OC-20). | ||
In the following year, the same group presents a regiodivergent kinetic resolution of terminal 1,2-diols through asymmetric silyl transfer, utilizing the same chiral catalyst OC-20 (Scheme 30).98 This strategy enables selective silylation of either the primary or secondary hydroxyl group in each enantiomer, thereby achieving both chemical differentiation and enantiomeric resolution. The key to this transformation lies in the reversible covalent bonding between the catalyst and substrate, which plays a pivotal role in directing regio- and enantioselectivity. Initially, the primary alcohol reacts more rapidly due to its higher intrinsic reactivity, leading to preferential formation of the silylated product from one enantiomer. As the reaction proceeds, acid byproducts promote dynamic exchange between catalyst and substrate, which accelerates the silylation of the less reactive enantiomer. By fine-tuning the reaction parameters, such as the rate of silyl chloride addition and reaction temperature, the authors achieved high enantioselectivity and synthetically useful yields. This regiodivergent resolution offers an efficient approach to access enantiopure, mono-protected diols.
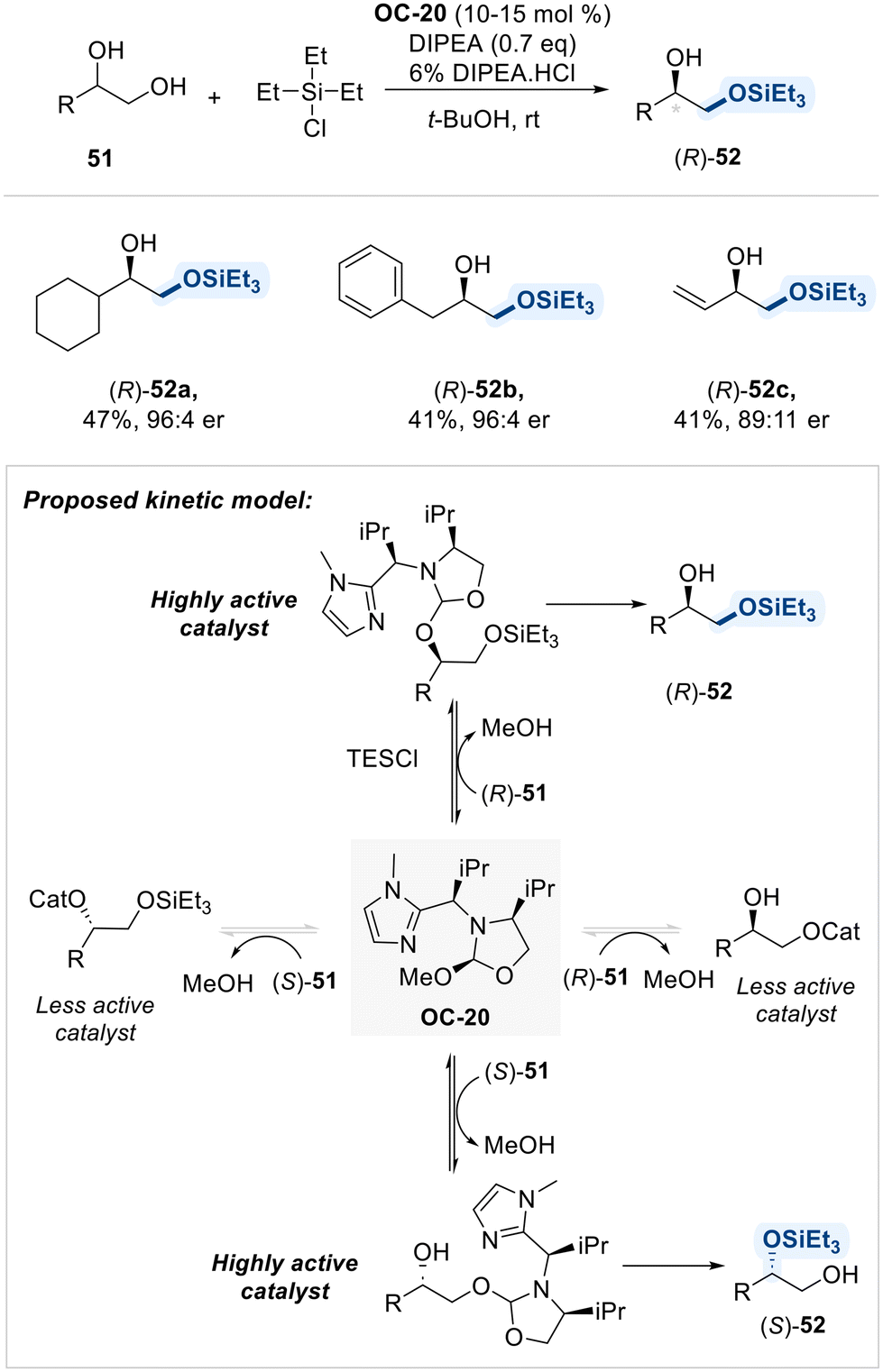 | ||
| Scheme 30 Regiodivergent kinetic resolution of terminal 1,2-diols. | ||
Subsequently, the Ishihara group developed a novel bifunctional organocatalyst (OC-21), which integrates a chiral sulfonamide moiety as a hydrogen-bond donor and an imidazole unit as an electrophilic acceptor (Scheme 31).99 Leveraging its potent hydrogen-bond donating capability, catalyst OC-21 effectively promoted the enantioselective acylation of meso-1,3-diols bearing a hydrogen-bond-accepting 3-pyrroline-1-carbonyl (Pyroc) group. The established hydrogen-bond network between sulfonamide and Pyroc group creates a chiral environment that facilitates acyl transfer from the acyl ammonium intermediate to the diol substrate, achieving enantioselectivity.
 | ||
| Scheme 31 Enantioselective acylation of meso-1,3-diols using OC-21. | ||
Moreover, like pyridine and imidazole, isothioureas are widely recognized as highly effective nucleophilic organocatalysts, owing to the presence of an electron-rich nitrogen atom adjacent to a thiocarbonyl group. This structural feature endows isothioureas with strong nucleophilic character, enabling them to efficiently activate electrophilic centers, particularly carbonyl-containing compounds. Considering this intrinsic nucleophilic reactivity of isothioureas, Bressy group developed a highly enantioselective method for the desymmetrization of acyclic meso-1,3-diols via acylation, utilizing a chiral isothiourea-based organocatalyst (OC-22) (Scheme 32).100 Mechanistically, the reaction proceeds via a nucleophilic attack by the electron-rich nitrogen atom of the catalyst (OC-22) on the anhydride electrophile, generating a chiral N-acyl isothiouronium intermediate. This intermediate acts as an efficient acyl transfer agent, enabling the enantioselective acylation of the meso-diol with high stereocontrol.
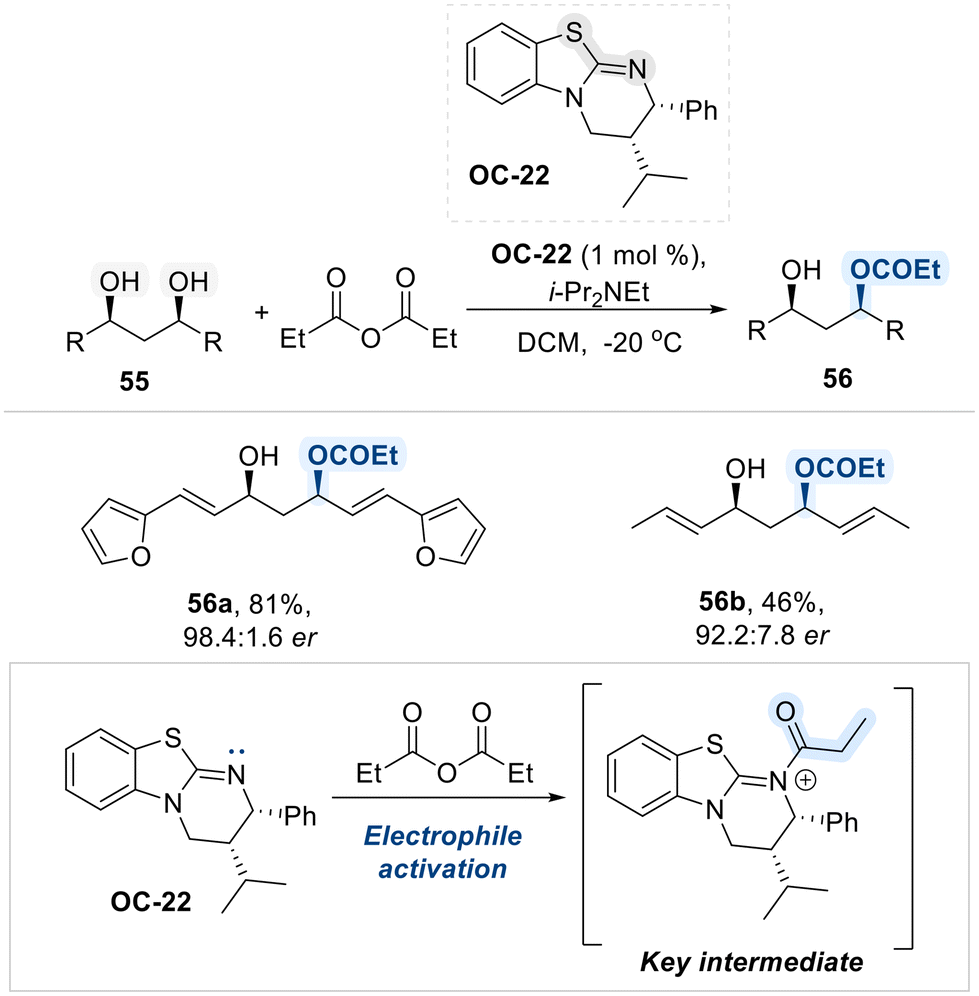 | ||
| Scheme 32 Isothiourea catalyzed desymmetrization of meso 1,3-diols. | ||
Expanding upon this strategy, Birman et al. employed an isothiourea-based organocatalyst (OC-23) for the enantio-selective acylation of lobelanidine (57), enabling the asymmetric synthesis of lobeline under ambient conditions using propionic anhydride as the acyl donor (Scheme 33).101 The reaction proceeds via formation of a chiral N-isopropanoyl isothiouronium intermediate, which acts as an electrophilic acyl transfer agent, delivering the acyl group with high regio- and enantioselectivity.
 | ||
| Scheme 33 Synthesis of lobeline via regioselective acylation of lobelanidine (57). | ||
Building on the nucleophilic character of N-heterocycles in facilitating selective diol functionalization, Ren and co-workers subsequently reported a cost-effective and highly efficient method for the regioselective acylation of diols using 1,5-diazabicyclo[4.3.0]non-5-ene (DBN, OC-24) as an organo-catalyst (Scheme 34).102 The authors propose that the bifunctional character of DBN arises from its ability to simultaneously engage in dual hydrogen-bonding interactions with the diol substrate and promote acyl transfer through the formation of a reactive acyl–ammonium electrophilic intermediate. This dual activation mode facilitates regioselective acylation at the sterically less hindered hydroxyl group, ultimately affording the monoacylated product 60.
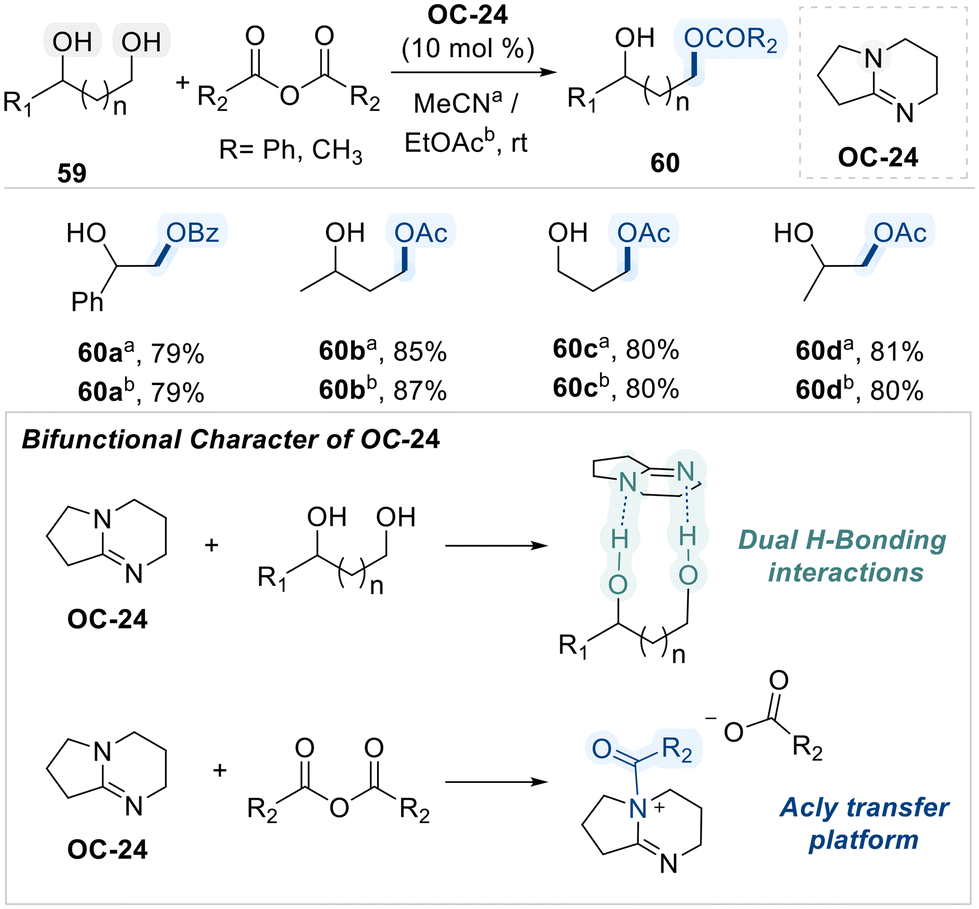 | ||
| Scheme 34 DBN (OC-24) catalyzed regioselective acylation of diols. | ||
3.2. Diamine catalysis
Diamine catalytic system features two nitrogen atoms whose electron-rich character imparts strong nucleophilic and Brønsted basic properties, enabling activation of electrophilic substrates or acidic protons.103 Leveraging their inherent nucleophilic character, diamines have been employed as effective catalysts in acyl transfer reactions. Notably, chiral ethylenediamine frameworks have attracted considerable attention as asymmetric catalysts in the enantioselective acylation of diol substrates. A plausible mechanistic pathway involves the bidentate coordination of the diamine catalyst to the carbonyl carbon of the acylating agent, thereby enhancing the electrophilicity of the acyl donor and facilitating regioselective acylation at a specific hydroxyl group of the diol substrate (Scheme 35).104 The resulting regioselectivity is governed by the steric and electronic properties of the diamine catalyst, providing a strategic platform for the rational design of highly selective diamine-based catalytic systems for diol functionalization. | ||
| Scheme 35 General mechanism of 1,2-diamine-catalyzed regioselective acylation of diols. | ||
A study conducted by Oriyama and colleagues demonstrated the enantioselective benzoylation of meso-diols utilizing the chiral 1,2-diamine reagent OC-25, which was synthesized from (S)-proline (Scheme 36).105 Although this method requires a stoichiometric amount (1.0 equivalent) of OC-25, it illustrates that diamine-based systems serve as effective platforms for the acylation of diol substrates, and that high enatioselectivity can be achieved through the rational design of the diamine catalyst. To enhance economic feasibility, the same research group subsequently developed a structurally optimized chiral 1,2-diamine catalyst (OC-26), which exhibited significantly enhanced catalytic efficiency in the enantioselective benzoylation reaction when employed alongside triethylamine as a base.106 Remarkably, utilizing only 0.5 mol% of OC-26 resulted in benzoylation of meso-diols with selectivity comparable to the stoichiometric OC-25 system, while simultaneously increasing reaction efficiency and sustainability through substantial catalyst load reduction.
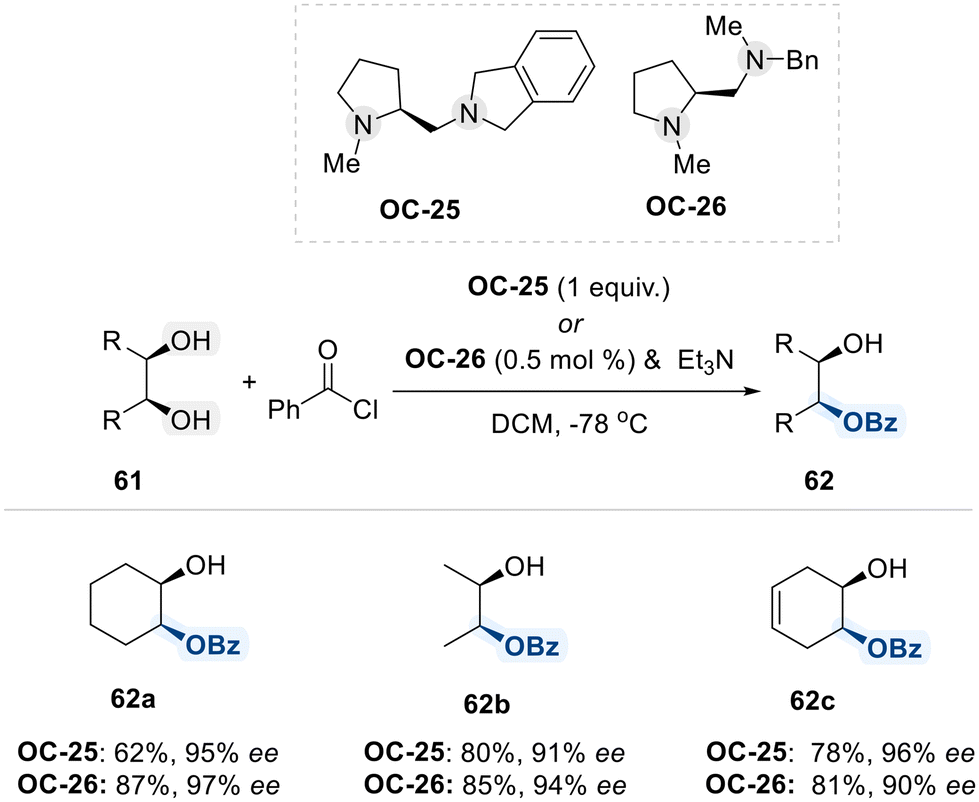 | ||
| Scheme 36 Enantioselective benzoylation of meso-diol using chiral diamines (OC-25 and OC-26). | ||
Next, Kundig group explored their application in the desymmetrization of meso-1,4-diol complexes derived from [Cr(CO)3(naphthoquinone)] while preserving metal coordination (Scheme 37).107 This transformation was achieved through enantioselective monobenzoylation, facilitated by a new quinidine-derived chiral diamine catalyst (OC-27). As illustrated in the proposed mechanism in Scheme 35, OC-27 interacts with the carbonyl carbon of benzoyl chloride, increasing its electrophilicity and enabling transfer of the benzoyl group to meso-1,4-diol complexes in an enantioselective manner. This strategy enables access to highly enantioenriched planar chiral chromium complexes 64, which serve as valuable chiral building blocks for highly diastereoselective transformations, such as chromium-mediated dearomatization reactions.108
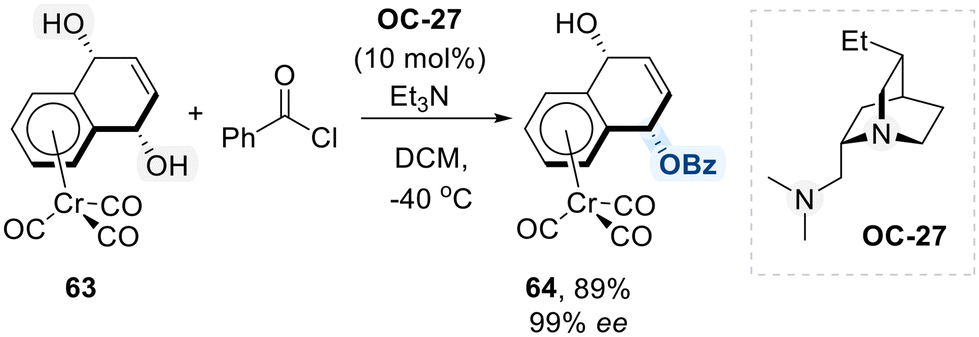 | ||
| Scheme 37 Desymmetrization of meso-1,4-diol chromium complex. | ||
To further explore the applicability of the chiral diamine catalyst OC-27, the same group extended its use to commonly encountered meso-1,2-diols (Scheme 38).109 Employing OC-27, they successfully achieved the desymmetrization of various cyclic and acyclic meso-diols through benzoylation, obtaining monobenzoylated products with good to excellent yields and high enantioselectivities. This group study underscored the versatility of quinidine-derived organocatalyst (OC-27) in enantioselective benzoylation of meso diols.
 | ||
| Scheme 38 Enantioselective benzoylation of meso-1,2-diols using cinchona alkaloid-derived catalyst (OC-27). | ||
3.3. Peptide catalysis
Peptides have emerged as powerful catalysts in regioselective transformation of organic compounds due to their inherent structural flexibility, precise molecular recognition, and ability to engage in non-covalent interactions, such as hydrogen bonding and electrostatic forces.110 In the context of selective functionalization of diols, these properties enable peptide-based catalysts to distinguish between two hydroxyl groups, thereby facilitating regio- and enantioselective transformations, including acylation and silylation reactions.Particularly, short peptide sequences containing proline, histidine, or other nucleophilic residues have been employed to catalyze diol desymmetrization, yielding regioselectively modified products with high enantiocontrol. Their catalytic activity primarily stems from functional groups, bearing amines, carboxylates, and hydroxyls, which participate in hydrogen bonding, Brønsted acid–base interactions, and steric control.111 These interactions play a crucial role in stabilizing key transition states and reactive intermediates, thereby minimizing activation energy barriers and precisely directing selective transformations at a single hydroxyl site. Such cooperative effects not only enhance reaction efficiency but also expand the scope of peptide catalysis in asymmetric diol transformation.
In 2005, Lewis et al. disclosed desymmetrizative acylation of meso-glycerol derivatives 67, employing a chiral histidine-derived peptide-based organocatalyst (OC-28) (Scheme 39).112 The catalyst combines an imidazole moiety, which activates the acyl group, with a peptide chain that accepts a hydrogen bond from one hydroxyl group of the diol substrate. This interaction spatially orients the second hydroxyl group, guiding highly enantioselective acylation. Collectively, this approach enables access to enantioenriched products with excellent selectivity.
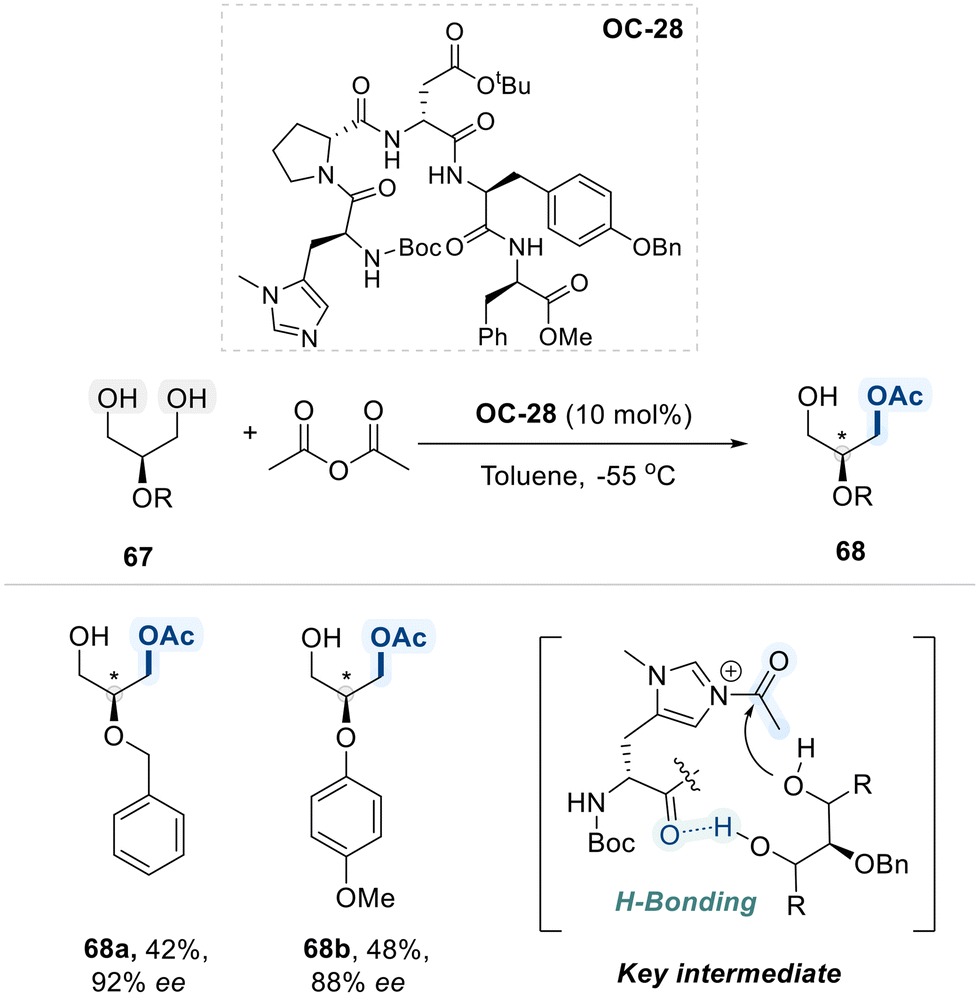 | ||
| Scheme 39 Selective acylation of glycerol derivatives. | ||
Building on their earlier work, the same research group developed OC-29, a synthetic, miniaturized peptide-based organocatalyst that mimics enzyme function, for the desymmetrization of sterically and structurally challenging remote meso-diols 69 (Scheme 40).113 This transformation proceeds via an enantioselective desymmetrizing acylation reaction. Although the precise mode of asymmetric induction remains ambiguous, the authors propose that undefined, yet functionally significant, non-covalent interactions between the catalyst and substrate contribute to the observed enantioselectivity. Notably, reducing the steric bulk of the alkyl substituents on diol substrate 69 led to a marked decrease in enantioselectivity, underscoring the reaction's sensitivity to substrate sterics.
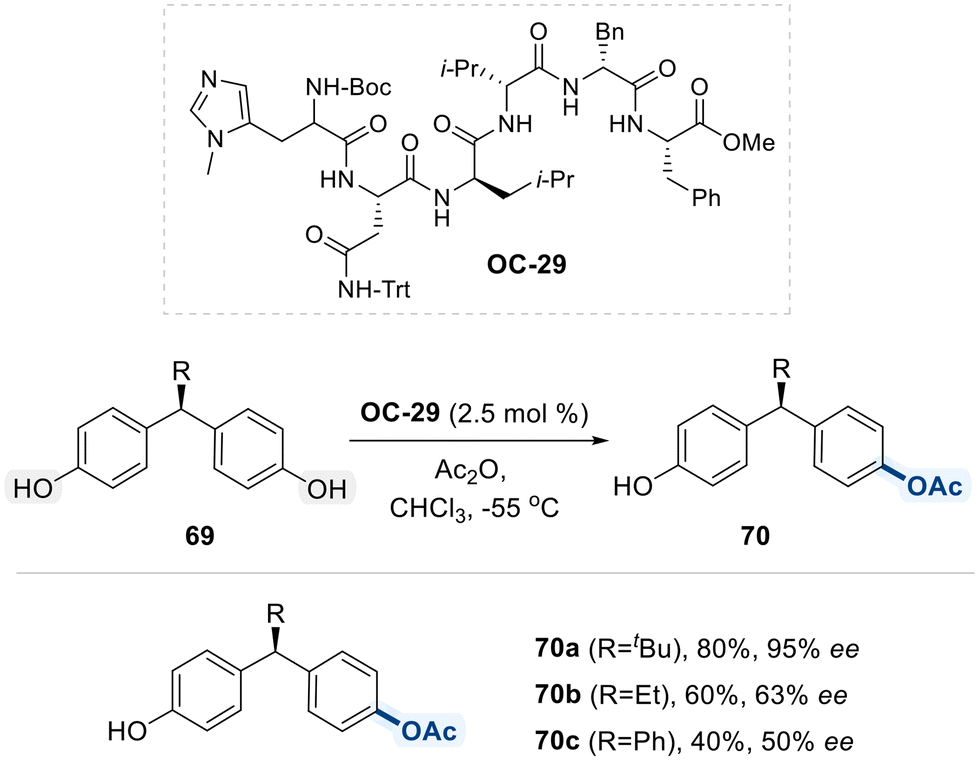 | ||
| Scheme 40 Desymmetrization of remote meso-diol via enantioselective acylation. | ||
Schreiner et al. have introduced a novel approach for desymmetrizing meso-diols through enantioselective acylation, coupled with an in situ oxidation step to protect the valuable product from racemization (Scheme 41).114 This strategy employs a highly lipophilic, histidine-derived peptide catalyst (OC-30), incorporating an adamantane-based amino acid motif. Their study demonstrated that the lipophilic peptide catalyst OC-30 effectively facilitates the desymmetrization of vicinal meso-diols via enatioselective acyl transfer from a chiral imidazolium intermediate. Furthermore, the subsequent TEMPO-mediated oxidation of the monoacylated product provided the corresponding α-acetoxy ketones in high isolated yields, with no loss of enantioselectivity.
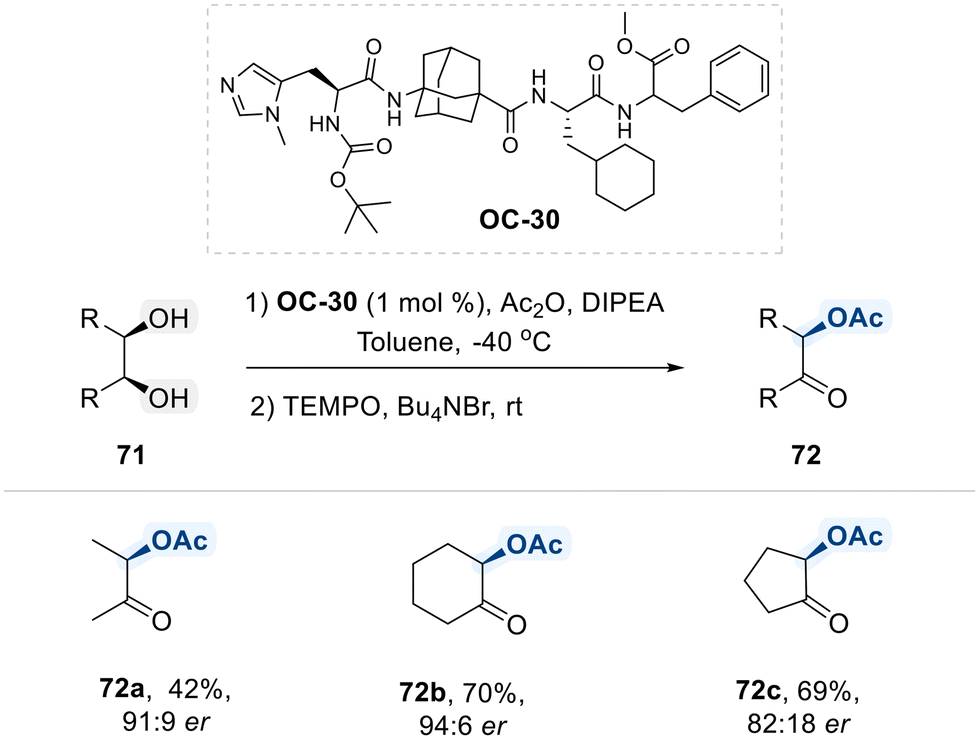 | ||
| Scheme 41 Enantioselective acylation of meso-1,2-diols using OC-30. | ||
Building on their earlier work, the same group further advanced the field by reporting an enantioselective kinetic resolution of trans-cycloalkane-1,2-diols through a monoacylation process, employing their previously developed peptide-based organocatalyst OC-30 (Scheme 42).115 The reaction proceeds via a selective acyl transfer from a chiral imidazolium intermediate, wherein one hydroxyl group of the diol undergoes acylation while the other engages in hydrogen bonding with the peptide backbone, an interaction essential for precise chiral recognition. Notably, when the reaction was performed in more polar solvents such as acetonitrile, dichloromethane (DCM), or trifluoromethylbenzene, both enantioselectivity and reaction rate declined significantly. These findings highlight the pivotal role of solvent polarity in maintaining the non-covalent interactions necessary for effective substrate orientation and high stereocontrol within the catalytic system.
 | ||
| Scheme 42 Kinetic resolution of trans-cycloalkane-1,2-diols employing OC-30. | ||
Subsequently, the Snapper group designed a simple, amino-acid-based small molecule as a chiral organocatalyst (OC-31) for the enantioselective silylation of meso-diols (Scheme 43).116,117 Structurally, OC-31 incorporates a Lewis basic imidazole group, which enhances the electrophilicity of the silyl halide reagent, along with a chiral amino acid moiety that engages in hydrogen bonding interactions with the diol substrate. These interactions facilitate the selective activation of one hydroxyl group, thereby enabling enantioselective silylation. In their initial investigation of meso-diol desymmetrization via silyl group transfer, the reaction required a high catalyst loading (30 mol%) and prolonged reaction times of several days.116 To enhance the efficiency of this catalytic system, the authors utilzed an achiral co-catalyst (Co-cat-4) as a nucleophilic promotor alongside a chiral catalyst (OC-31) as Brønsted base.117 This dual-catalyst system effectively accelerated the reaction, enabling completion within one hour while delivering the desired products in high yields and enantiomeric ratios with a reduced catalyst loading of just 5 mol% catalyst (OC-31).
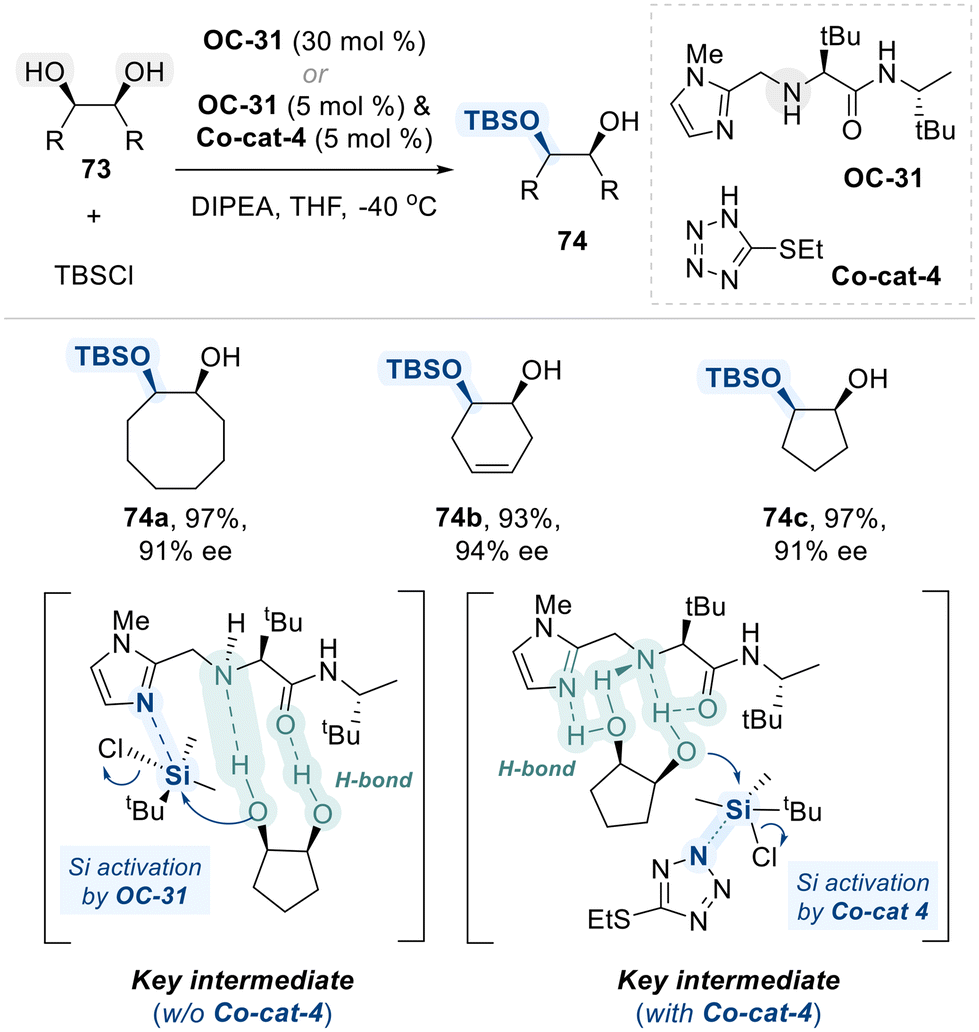 | ||
| Scheme 43 Enantioselective silylation of meso-1,2-diols utilizing amino-acid-based organocatalyst (OC-31). | ||
A landmark contribution from the Miller group introduced a pioneering strategy for the oxidative desymmetrization of meso-1,4-diols using a novel aminoxyl-functionalized, peptide-based organocatalyst (OC-32). This elegant approach enables the enantioselective synthesis of enantioenriched lactones with remarkable efficiency and selectivity (Scheme 44).118
 | ||
| Scheme 44 Oxidative desymmetrization of meso-1,4-diols using OC-32. | ||
Central to this transformation is OC-32, a bifunctional catalyst that integrates an oxidizing oxoammonium species with a chiral peptide backbone. The oxoammonium group serves as an oxidizing agent, while the peptide framework provides a well-organized hydrogen-bonding network that reinforces chiral induction. Mechanistically, the reaction begins by forming a stabalized hydrogen bond of one of the hydroxyl groups of meso diol with the peptide backbone, anchoring the substrate within a chiral environment followed by the site-selective oxidation of free hydroxyl group of meso-diol substrate, generating a transient aldehyde intermediate. Following initial oxidation, intramolecular cyclization occurs via nucleophilic attack of the unoxidized hydroxyl group on the newly formed aldehyde, resulting in the formation of a chiral lactol. This intermediate is then subjected to a second oxidation step, typically mediated by trichloroisocyanuric acid (TCCA), to furnish the corresponding enantioenriched lactone product.
Recently, the same research group reported a regiodivergent oxidation of unsymmetrical diols (79) using structurally tailored, aminoxyl-containing peptide-based organocatalysts (OC-33 and OC-34) (Scheme 45).119 This approach enabled catalyst-controlled selective oxidation of either the less hindered or the more hindered hydroxyl group with high levels of regioselectivity. According to their findings, OC-34 favors oxidation at the less hindered hydroxyl group. This preference arises because a nearby methyl group at the aminoxyl-core in the catalyst (OC-34) structure sterically blocks access to the more hindered hydroxyl group, making the less hindered site more accessible for oxidation. In contrast, OC-33 features a bulkier, more sterically crowded peptide core. This design encourages binding of the less hindered hydroxyl group within a confined “pocket” of the peptide, thereby positioning the more hindered hydroxyl group near the reactive oxoammonium center. As a result, oxidation occurs preferentially at the more hindered site. As outlined in Scheme 45, initial oxidation leads to the formation of a chiral lactol via intramolecular cyclization, which undergoes further oxidation with TCCA to afford the final chiral lactone products.
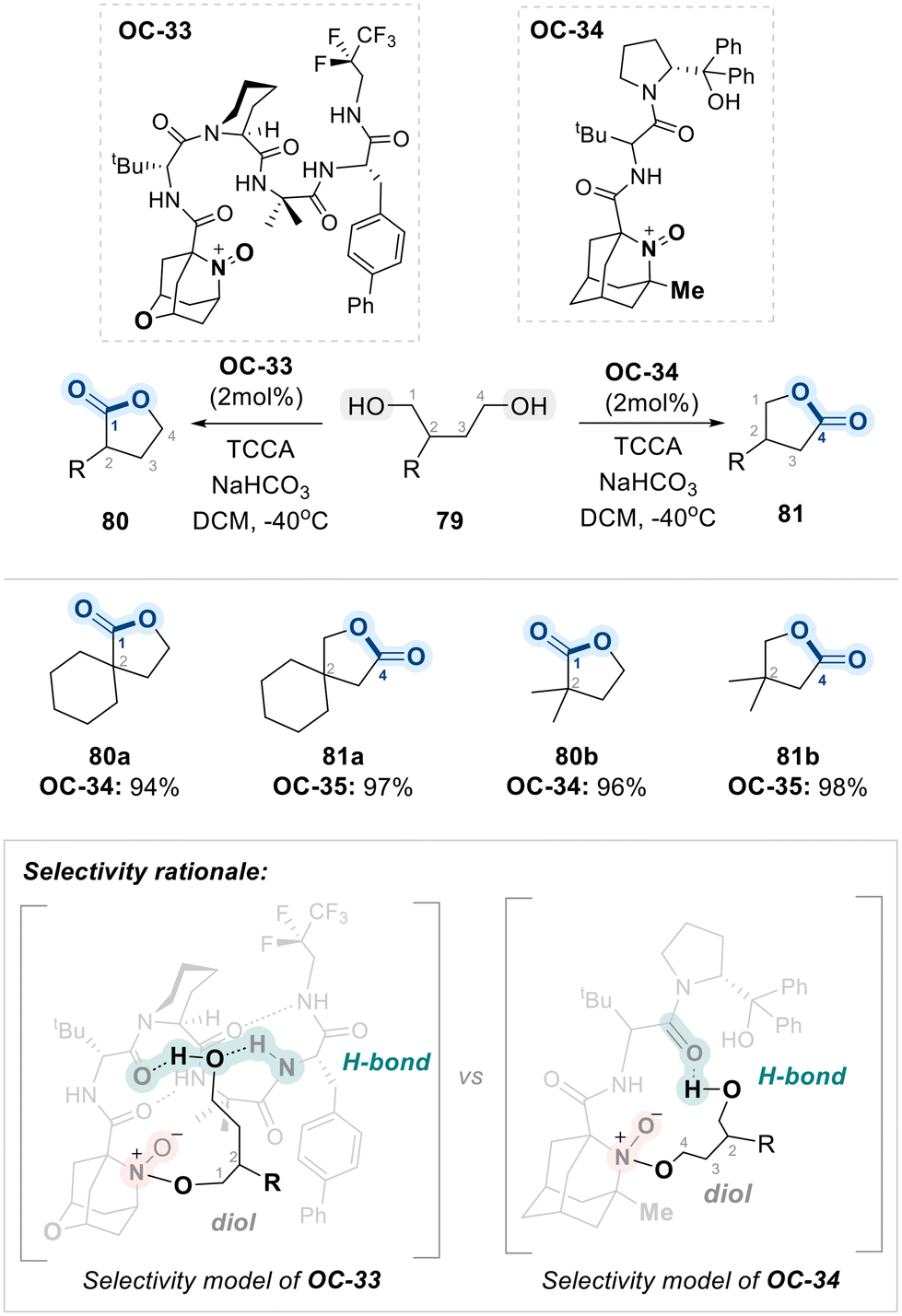 | ||
| Scheme 45 Regiodivergent oxidation of unsymmetrical-1,4-enabled by OC-33 and OC-34. | ||
Inspired by the participation of the peptide's carbonyl group in hydrogen bonding and secondary interactions, the Yeung group utilized a chiral amino-thiocarbamate catalyst (OC-35), featuring a thiocarbamate moiety analogous to the amide functional group, to promote an intriguing bromocyclization of diolefinic diols (82) with high diastereo- and enantioselectivity (Scheme 46).120 The selectivity observed in this transformation is governed by a combination of structural features of the 1,3-diol substrate and noncovalent interactions induced by the catalyst. Specifically, an internal hydrogen-bonding network within the 1,3-diol enforces a pseudo-chair conformation resembling a six-membered ring. This conformation is further stabilized by a bulky substituent adopting a pseudo-equatorial orientation, thereby minimizing steric repulsion.
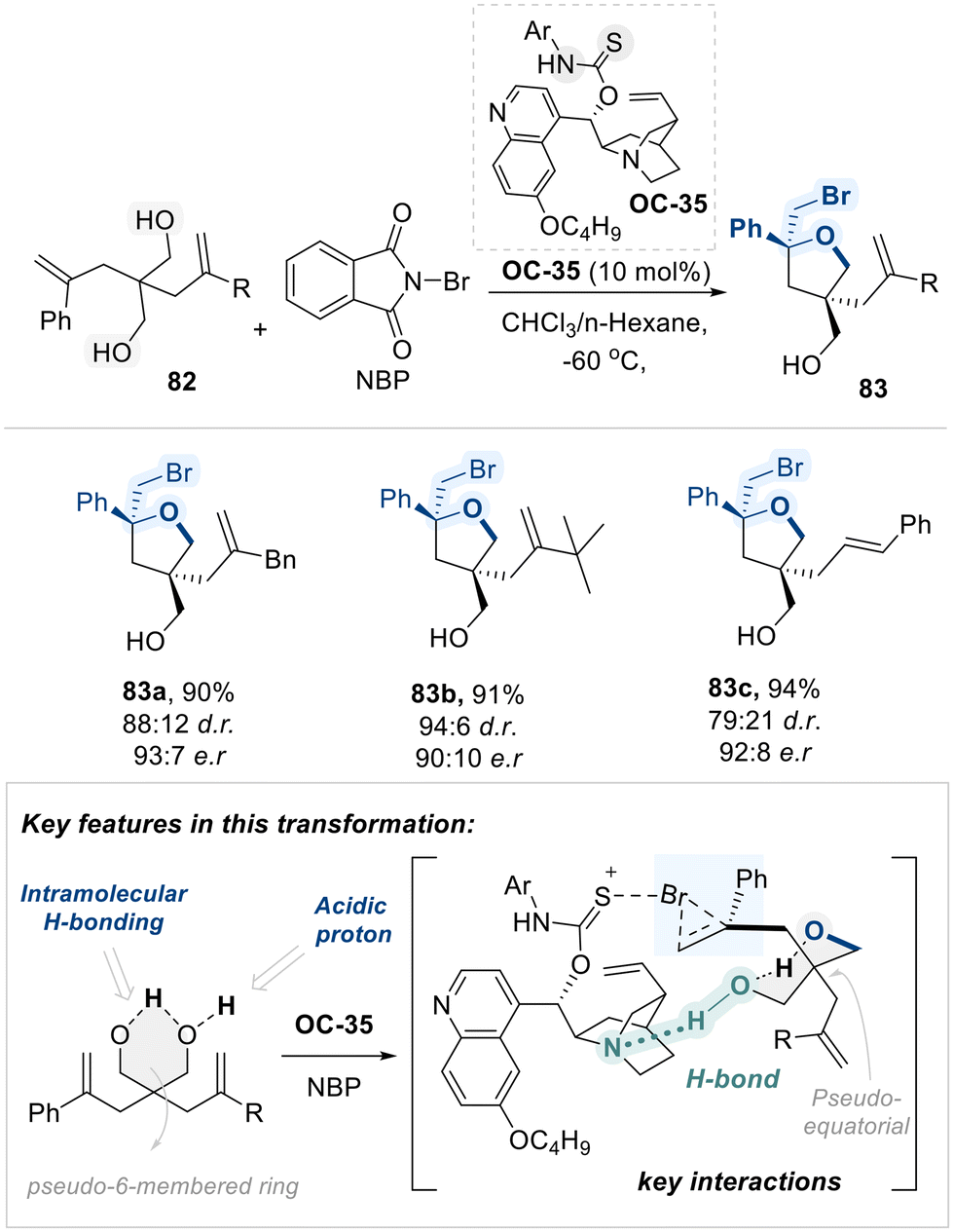 | ||
| Scheme 46 Amino thiocarbamate (OC-35)-catalyzed bromocycliazation of diolefinic diols. | ||
Concurrently, the amino-thiocarbamate catalyst (OC-35) forms a bifunctional catalytic pocket. First, the quinuclidine moiety acts as a hydrogen bond acceptor, interacting with the acidic proton of the 1,3-diol. Second, it serves as a bromine transfer platform: the catalyst abstracts bromine from N-bromophthalimide (NBP) and delivers it to the alkene substrate. This controlled delivery imposes a geometrical constraint that promotes selective bromonium ion formation. Subsequent intramolecular nucleophilic attack by the free hydroxyl group of the 1,3-diol onto the bromonium intermediate affords the cyclic ether product with high diastereo- and stereoselectivity.
3.4. N-Heterocyclic carbenes (NHCs) catalysis
N-Heterocyclic carbenes (NHCs) represent a distinct class of stable carbenes, characterized by a divalent carbon atom embedded within a nitrogen-containing heterocyclic ring.121 Unlike conventional carbenes, which are typically highly reactive and short-lived, NHCs exhibit remarkable stability due to electron donation from adjacent nitrogen atoms and steric protection provided by the ring system. Owing to their strong electron-donating properties, NHCs play a pivotal role in catalyzing the acylation of alcohols by facilitating the activation of aldehydes as acyl anion equivalents. Mechanistically, the reaction commences with the generation of an NHC carbene from the deprotonation of its corresponding azolium salt using base (Scheme 47).122 The carbene, functioning as a strong nucleophile, attacks the electrophilic carbonyl carbon of the aldehyde to generate a tetrahedral intermediate (IX), which undergoes proton transfer to yield the corresponding enaminol (X). This is followed by an internal redox process that generates the NHC–acyl intermediate (XI), which exhibits enhanced electrophilicity at the carbonyl center. This activation facilitates nucleophilic attack by the alcohol, leading to the formation of the ester product while simultaneously regenerating the NHC catalyst.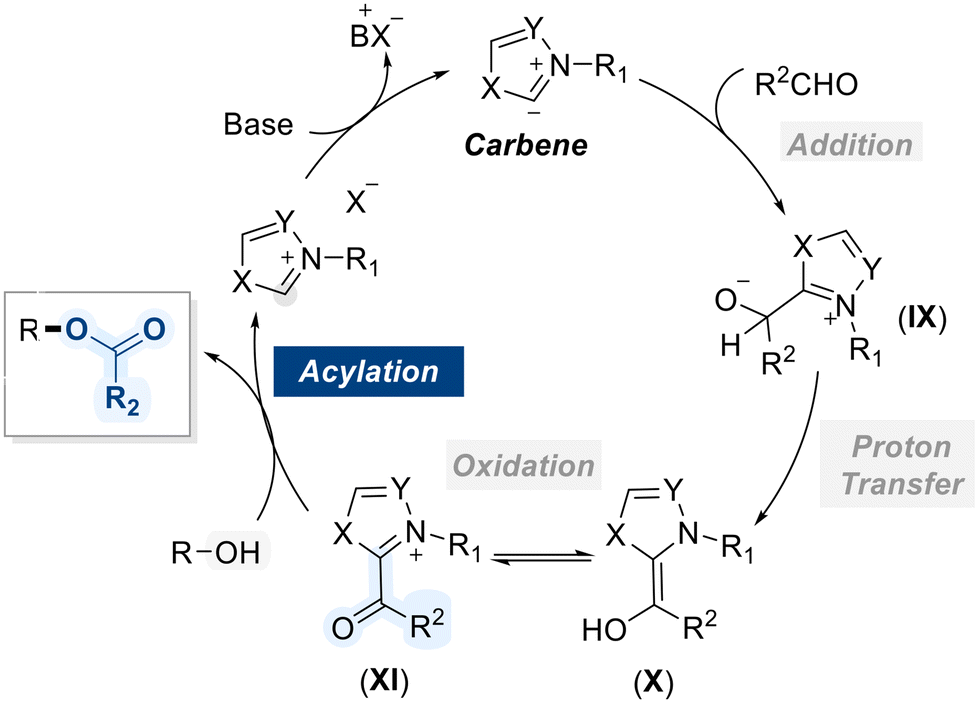 | ||
| Scheme 47 General mechanism of NHC-catalyzed acylation of alcohols. | ||
Considering these mechanistic insights, the Rovis group investigated the enantioselective acylation of meso-hydrobenzoin 84 using a chiral nucleophilic carbene catalyst (OC-36) and α-bromo-cyclohexane carboxaldehyde as an acylating source (Scheme 48).123 The catalytic cycle begins with the nucleophilic attack of the carbene on the aldehyde, followed by hydrogen transfer to generate an α-bromo enol intermediate (XIII) which undergoes leaving group elimination to form an enol intermediate (XIV), which tautomerizes to yield an active acylating species (XV) for the selective acylation of the diol. This approach offers a unique and efficient strategy for the enantioselective functionalization of diols, thereby expanding the scope of chiral carbene-catalyzed transformations in asymmetric synthesis.
 | ||
| Scheme 48 Enantioselective acylation of meso-hydrobenzoin using chiral carbene catalyst (OC-36). | ||
In the following year, Scheidt et al. developed the enantioselective monoacylation of cis-cyclohexane-1,2-diol with cinnamaldehyde using a chiral triazolium salt (OC-37) as an N-heterocyclic carbene (NHC) catalyst (Scheme 49).124 The transformation initiates with NHC-catalyzed activation of cinnamaldehyde, yielding addition intermediate XVI, which undergoes oxidation to furnish the key α,β-unsaturated carbonyl–NHC species XVII. This chiral acyl transfer platform subsequently facilitates the enantioselective acylation of diol substrates.
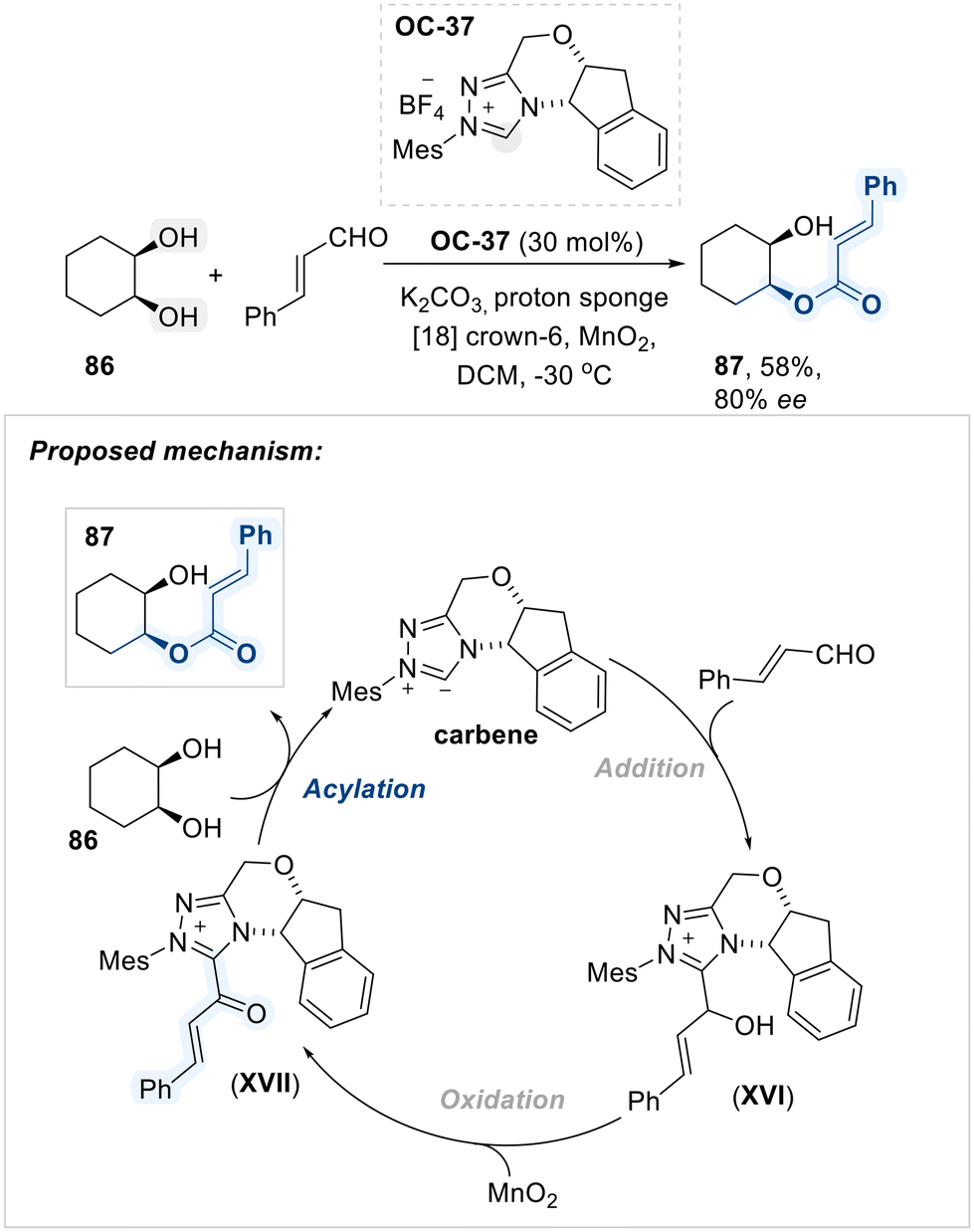 | ||
| Scheme 49 Asymmetric monoacylation of cis-cyclohexane-1,2-diol using OC-37. | ||
Subsequent study by Iwahana and his team disclosed a combination of a chiral N-heterocyclic carbene (OC-38) and a redox-active flavin (Co-cat-5) as an effective system for promoting the enantioselective benzoylation of cis-cyclohexane-1,2-diol with benzaldehyde under ambient air conditions (Scheme 50).125 The reaction mechanism involves the generation of enol intermediate (XIX) by the nucleophilic addition of carbene to benzaldehyde. The riboflavin derivative (Co-cat-5) serves as the oxidant, facilitating the oxidation of the enol intermediate (XIX) to an NHC-acyl intermediate (XX), thereby directing enantioselective benzoylation of diol. This approach provides a straightforward and effective method for asymmetric functionalization under mild, air-tolerant conditions.
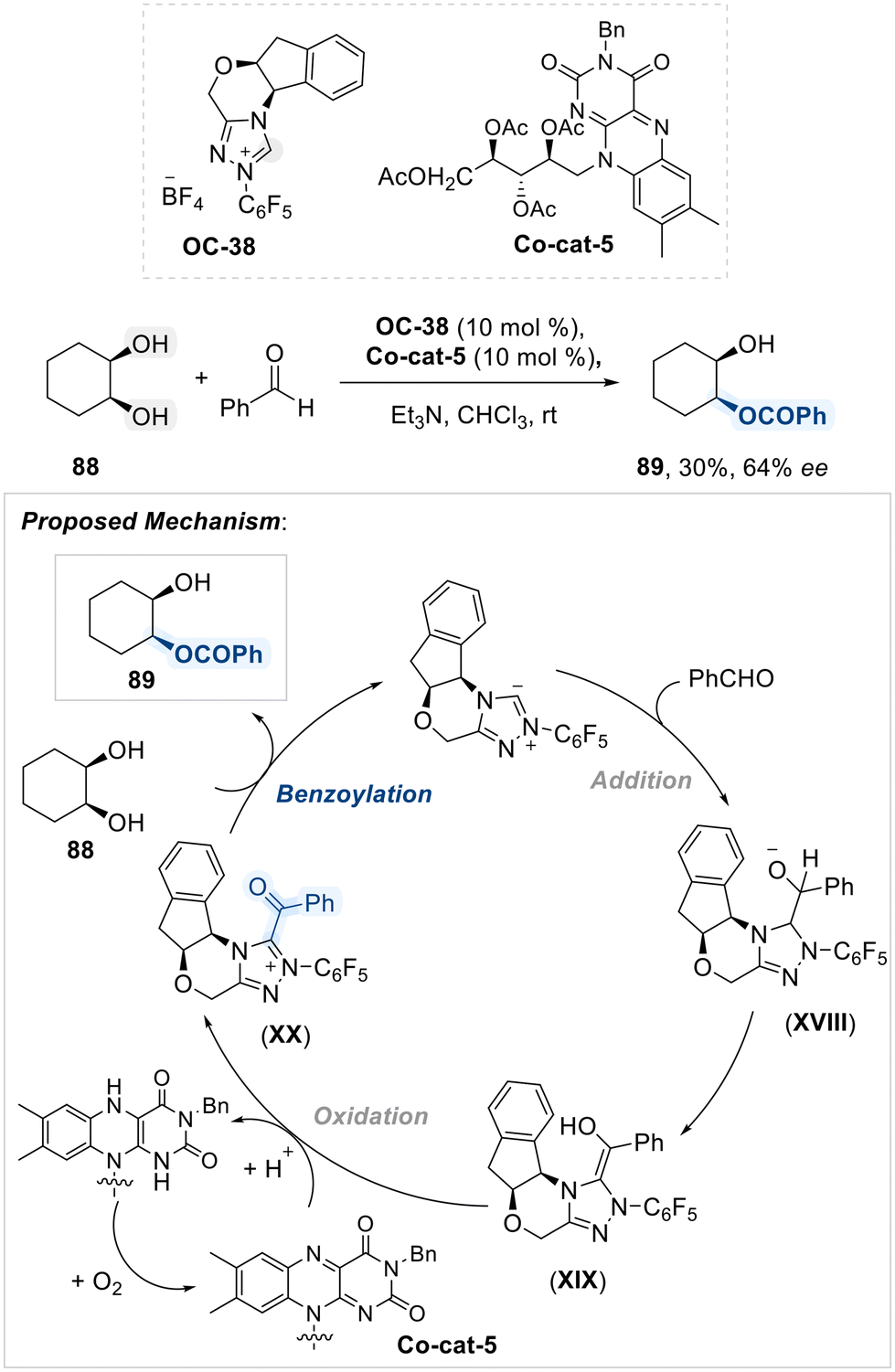 | ||
| Scheme 50 Desymmetrization of 1,2-diol using chiral N-heterocyclic carbene (OC-38) and a redox-active flavin (Co-cat-5). | ||
4. Phosphorus-based organocatalytic systems
Phosphorus-based organocatalysts play a crucial role in modern organic synthesis, owing to their versatile reactivity profiles that encompass nucleophilic activation, Brønsted acid catalysis, and Lewis's base interactions.126 This intrinsic tunability renders them particularly valuable in the selective functionalization of diols. Among the commonly employed phosphorus-containing catalysts are chiral phosphines, chiral phosphoric acids (CPAs), and phosphonium salts, each leveraging distinct mechanistic pathways to promote selective transformations of diols.127 Like chiral phosphines act as nucleophilic catalysts, enabling the activation of electrophiles for regioselective modifications of diol substrates. On the other hand, CPAs, typically derived from BINOL or SPINOL frameworks, serve as Brønsted acid catalysts, promoting transformations through hydrogen bonding and ion-pair interactions.128 The versatility of phosphorus-based catalysts underscores their pivotal role in the development of selective diol functionalization strategies.In this contribution, Vedejs and co-workers initially developed a chiral phosphine organocatalyst (OC-39) catalyzed monobenzoylation of meso-hydrobenzoin (90), achieving moderate levels of conversion and enantioselectivity (Scheme 51).129 To improve these outcomes, the same group subsequently developed a structurally refined phosphine catalyst (OC-40), which exhibited markedly enhanced reactivity and enantioselectivity in the functionalization of diol (90).130 It is assumed that chiral phosphines promote acylation through nucleophilic activation of the acyl donor, generating a reactive chiral P-acyl phosphonium intermediate. This intermediate facilitates enantioselective acyl transfer to the hydroxyl group, enabling high levels of stereocontrol during the transformation.
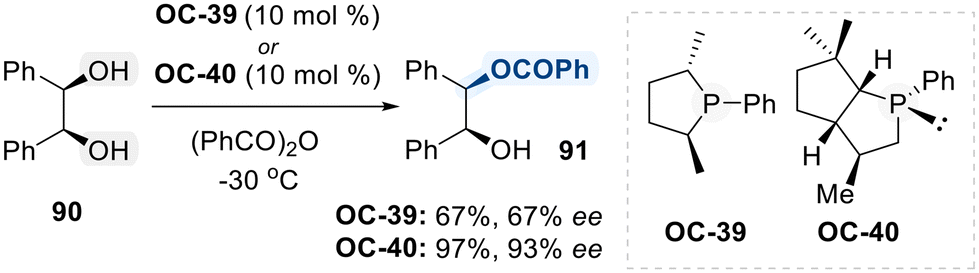 | ||
| Scheme 51 Phosphines catalyzed regioselective reaction of cis-cyclohexane-1,2-diol. | ||
As a further advancement, the Fujimoto group developed a bifunctional phosphinite catalyst (OC-41), derived from cinchona alkaloids, which integrates a Lewis-basic phosphinite moiety with a Brønsted-basic tertiary amine to achieve the enantioselective desymmetrization of meso-1,2-diols via benzoylation (Scheme 52).131 Despite its effectiveness, this system required over 30 mol% catalyst loading. To make it more economical, subsequently the same group introduced a more practical and readily accessible amino-phosphinite catalyst (OC-42), synthesized from cis-aminoindanol.132 This improved catalyst enabled the enantioselective benzoylation of meso-1,2-diols using substantially lower loadings (2.5 mol%). Although the detailed reaction mechanism remains unelucidated, the authors propose that benzoyl chloride activation likely proceeds through coordination to the phosphinite Lewis base.
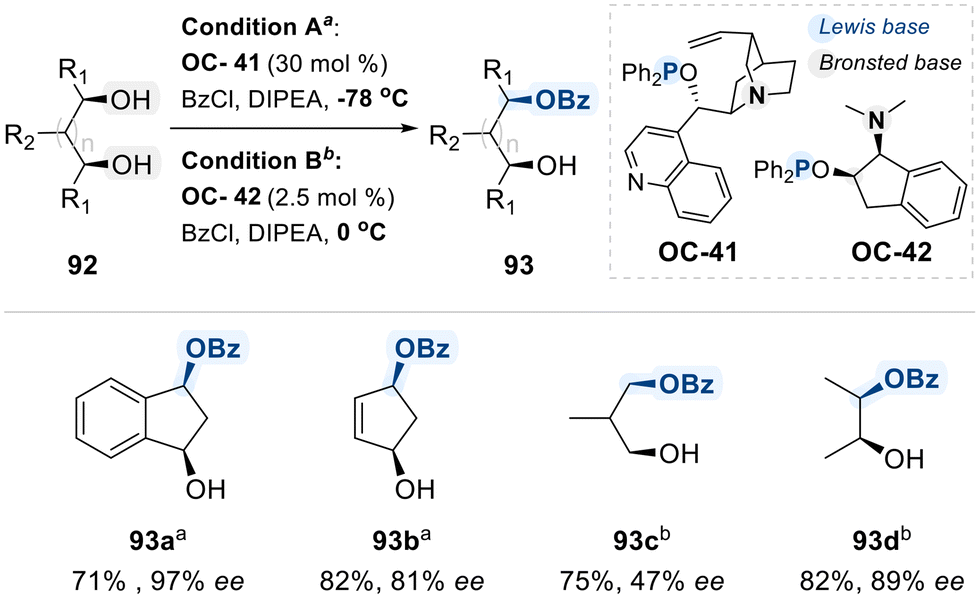 | ||
| Scheme 52 Enantioselective monobenzoylation of meso-diols via phosphinite derivatives (OC-41 or OC-42). | ||
In 2014, Zheng and co-workers reported a noteworthy application of a BINOL-based chiral phosphoric acid catalyst (OC-43) for the highly enantioselective desymmetrization of 2-substituted 1,3-diols (Scheme 53).133 The reaction begins with dimethyldioxirane (DMDO)-promoted oxidation of a 1,3-diol benzylidene acetal 94, generating acetone and an ortho-ester intermediate. This intermediate subsequently undergoes chiral phosphoric acid (OC-43) mediated proton transfer reaction, delivering the final product 95 with efficient yield and selectivities. DFT calculations reveal that the oxidation of the acetal by DMDO is the rate-determining step, while the exceptional enantioselectivity arises from favorable aryl–aryl interactions between the substrate and catalyst. In subsequent studies, the authors demonstrated that this asymmetric desymmetrization strategy could be further utilized for the highly enantioselective synthesis of acyclic α-tertiary amines.134
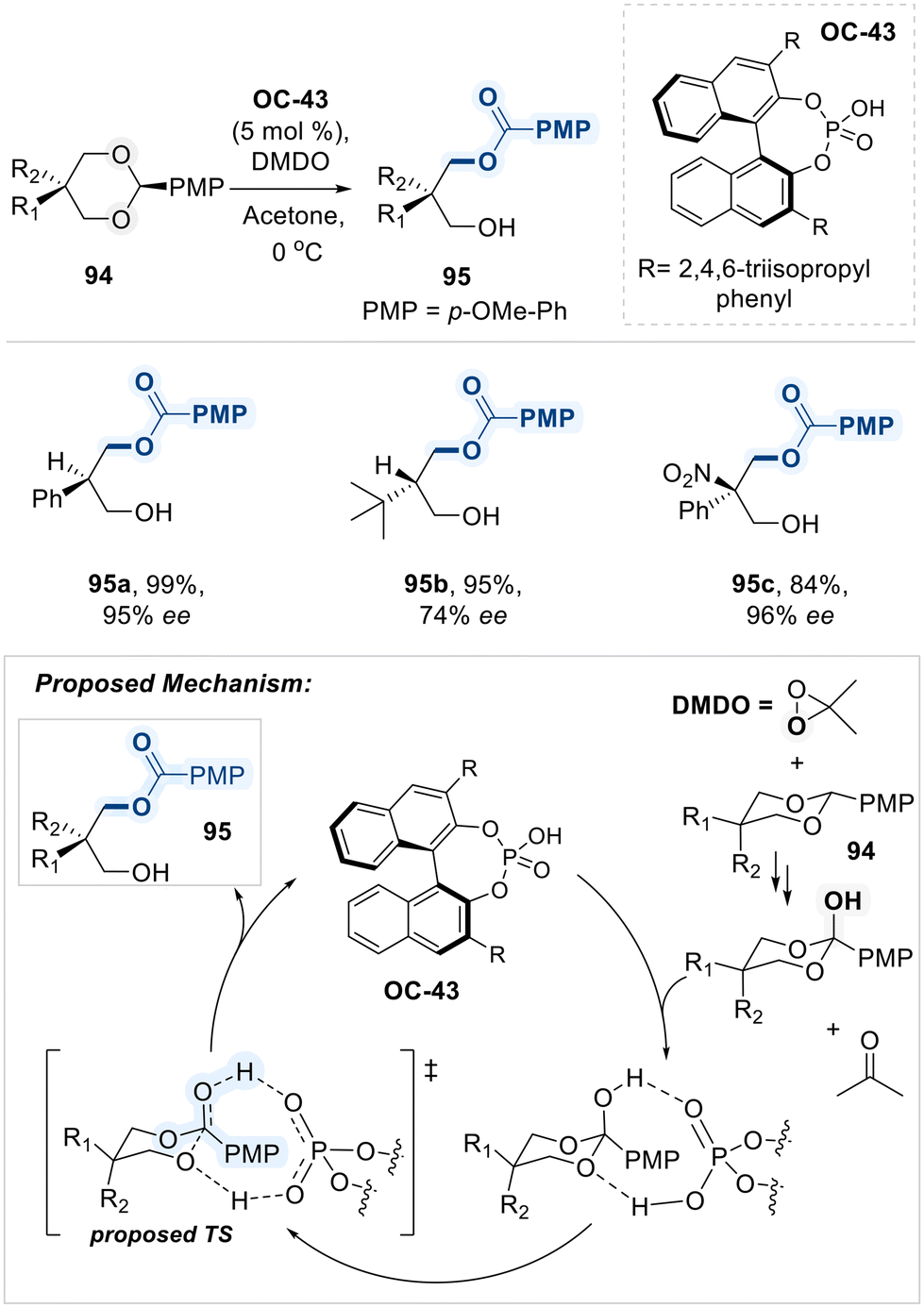 | ||
| Scheme 53 Desymmetrization of 1,3-diols employing chiral phosphoric acid (OC-43). | ||
Recognizing the potential of chiral phosphoric acids as Brønsted acids, Cui and co-workers developed a highly enantioselective desymmetrization of meso-1,3-diol bearing ynamide scaffold 96, employing a BINOL-derived CPA catalyst (OC-44) (Scheme 54).135 The reaction begins with protonation of the ynamide 96 by the catalyst OC-44, generating a keteniminium intermediate 97. This intermediate then undergoes intramolecular hydroalkoxylation to afford a chiral oxazolidine intermediate 98, which serves as the enantio-determining step. A subsequent proton transfer from the CPA (OC-44) to intermediate 98 generates a cyclic iminium species (98), which undergoes hydrolysis to deliver the chiral β-amino alcohol 100 with regeneration of the catalyst. The observed enantioselectivity arises from well-defined hydrogen-bonding and ion-pairing interactions between the keteniminium intermediate 97 and the chiral Brønsted acid catalyst.
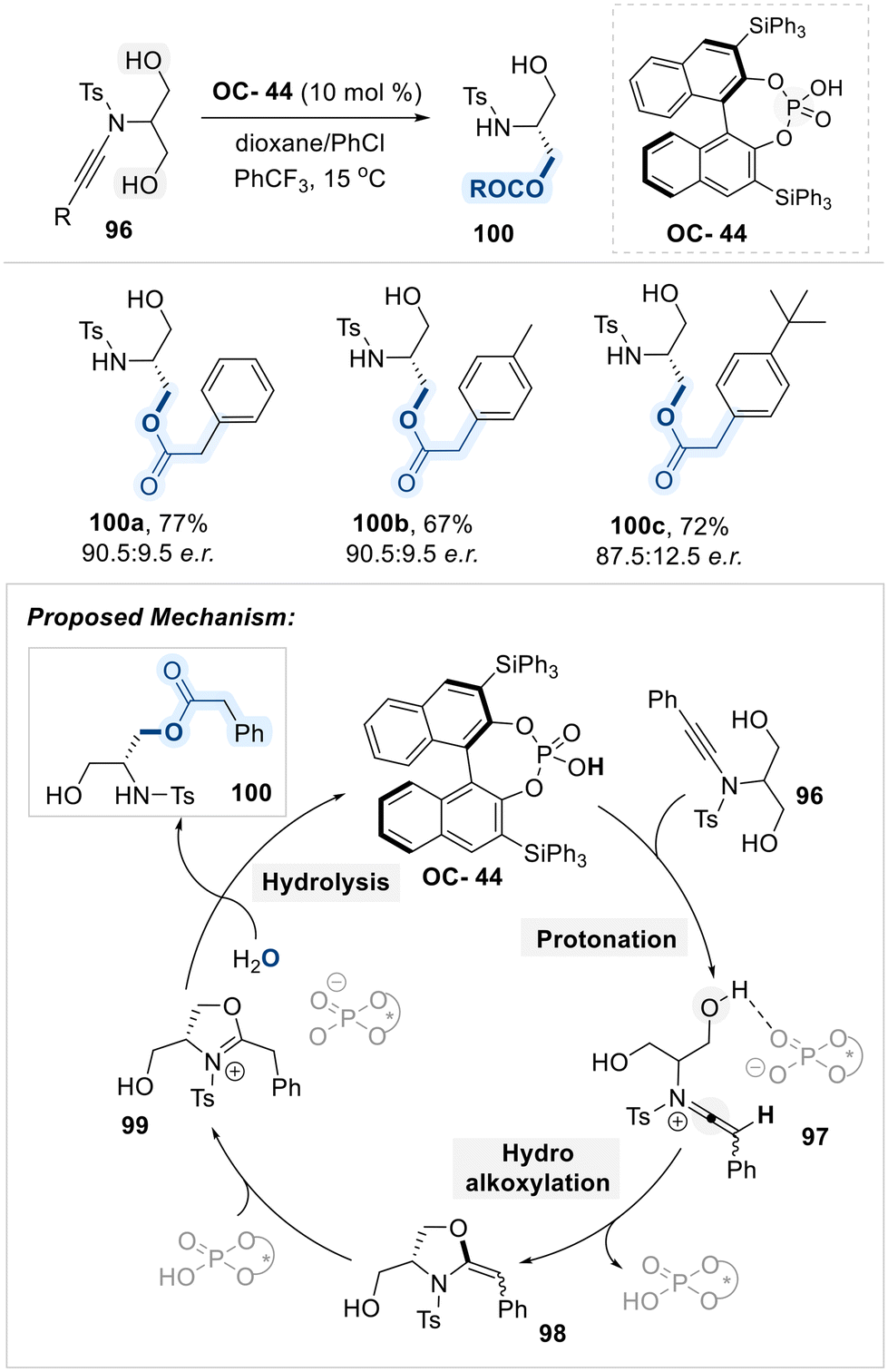 | ||
| Scheme 54 Desymmetrization of serinol-derived ynamides using chiral phosphoric acid catalyst (OC-44). | ||
Notably, Suga group employed a novel carbonyl-stabilized phosphonium ylide (OC-45) as an ionic nucleophilic catalyst for the selective acylation of primary alcohols in terminal diols (Scheme 55).136 The proposed mechanism initiates with the nucleophilic attack of the phosphonium ylide (OC-45) on the electrophilic carbon of isopropyl anhydride, resulting in the formation of an O-isopropylated intermediate is activated species subsequently undergoes acyl transfer to the diol substrate, selectively targeting the primary hydroxyl group. The observed regioselectivity is attributed to steric hindrance imposed by the bulky triphenylphosphine moiety, which disfavors nucleophilic attack at the more hindered secondary alcohol, thereby favoring acylation at the less hindered primary site.
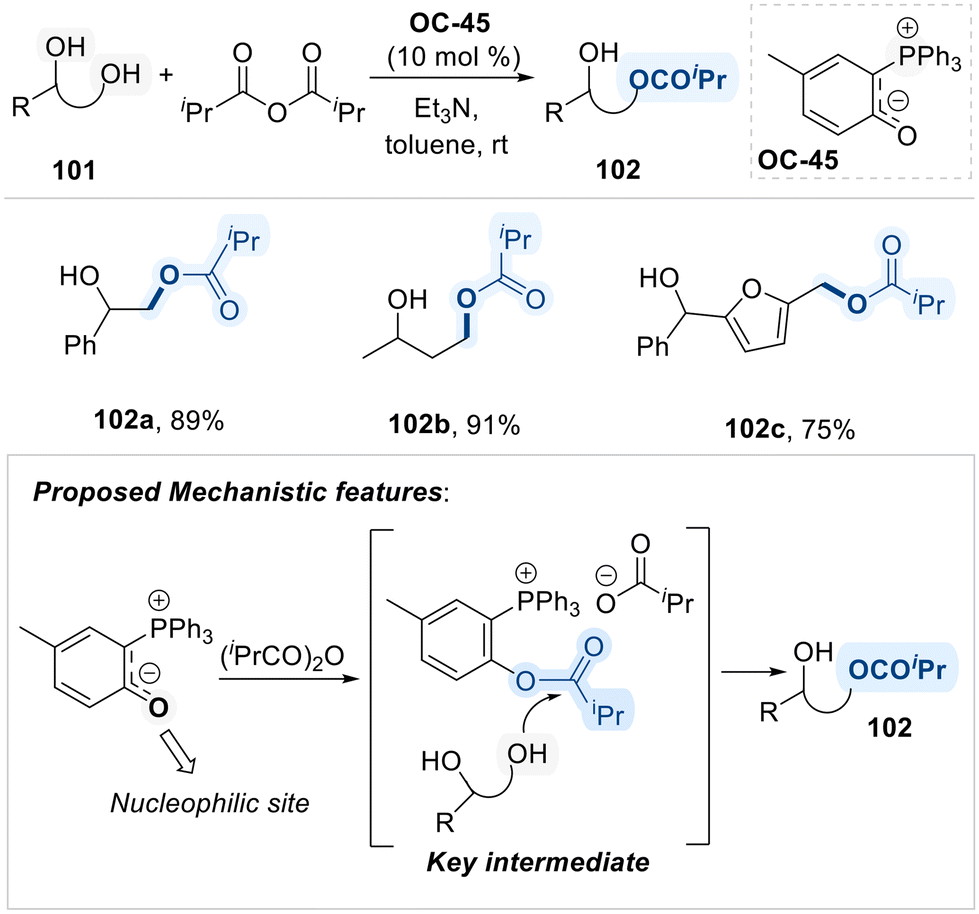 | ||
| Scheme 55 Phosphonium ylide (OC-45) catalyzed regioselective acylation of diol. | ||
5. Organophoto-catalytic systems
Traditional approaches to site-selective diol functionalization have primarily relied on two-electron pathways. In these systems, selectivity is often induced by the structural features of the catalyst, particularly in enantioselective transformations where chiral architectures play a central role. Alternatively, selectivity may result from the intrinsic steric or electronic properties of the substrate.More recently, organophotocatalysis has emerged as a mechanistically distinct strategy, enabling site-selective modification of diol substrates through single-electron processes initiated by visible light. Upon photoexcitation, the excited photocatalyst (PC*) engages in a single-electron transfer with a redox-active additive (A), generating a radical species (˙A) (Scheme 56). This additive radical species subsequently reacts with the diol substrate through hydrogen atom transfer (HAT), forming reactive intermediates, such as alkoxy, benzylic, or ketyl radical species. These reactive intermediates serve as branching points for various downstream reactions, including selective oxidation at specific hydroxyl groups and oxidative cleavage of vicinal C–C bonds. This section examines how such photochemical radical-based strategies have been harnessed to achieve site-selective transformations of diols through well-controlled single-electron pathways.
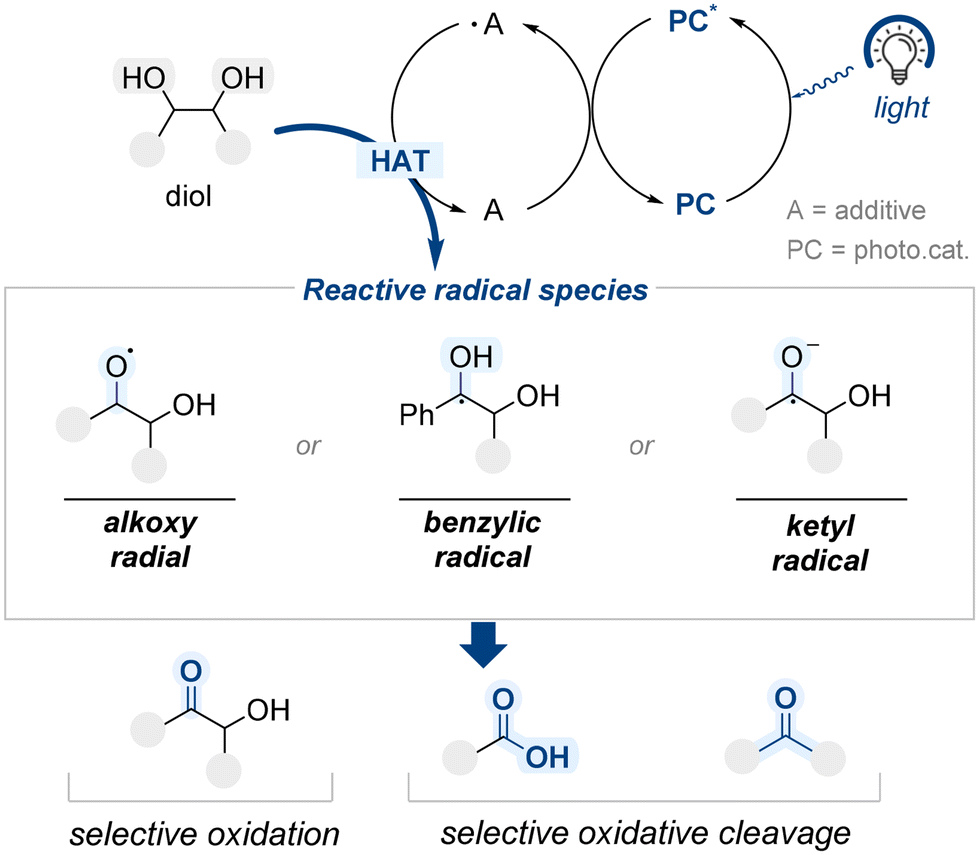 | ||
| Scheme 56 General mechanism of selective oxidation of diol via photocatalysis. | ||
A regioselective oxidation protocol targeting benzylic hydroxyl groups within diol substrates was developed by Rizvi, and Shah, employing Eosin Y (OC-46) as an organophotocatalyst and tert-butyl hydroperoxide (TBHP) as the oxidant under blue light irradiation (Scheme 57).137 Underlying this transformation is a mechanism in which photoexcited Eosin Y initiates homolytic cleavage of TBHP to generate tert-butoxy and hydroxyl radicals. The former abstracts a hydrogen atom adjacent to the aromatic ring, forming a stabilized benzylic radical, which is subsequently oxidized by the latter to yield the carbonyl product. The observed selectivity stems from the lower bond dissociation energy and enhanced stability of the benzylic radical. In substrates such as 1,3-diol, oxidation occurs exclusively at the benzylic hydroxyl group, leaving the primary alcohol unmodified.
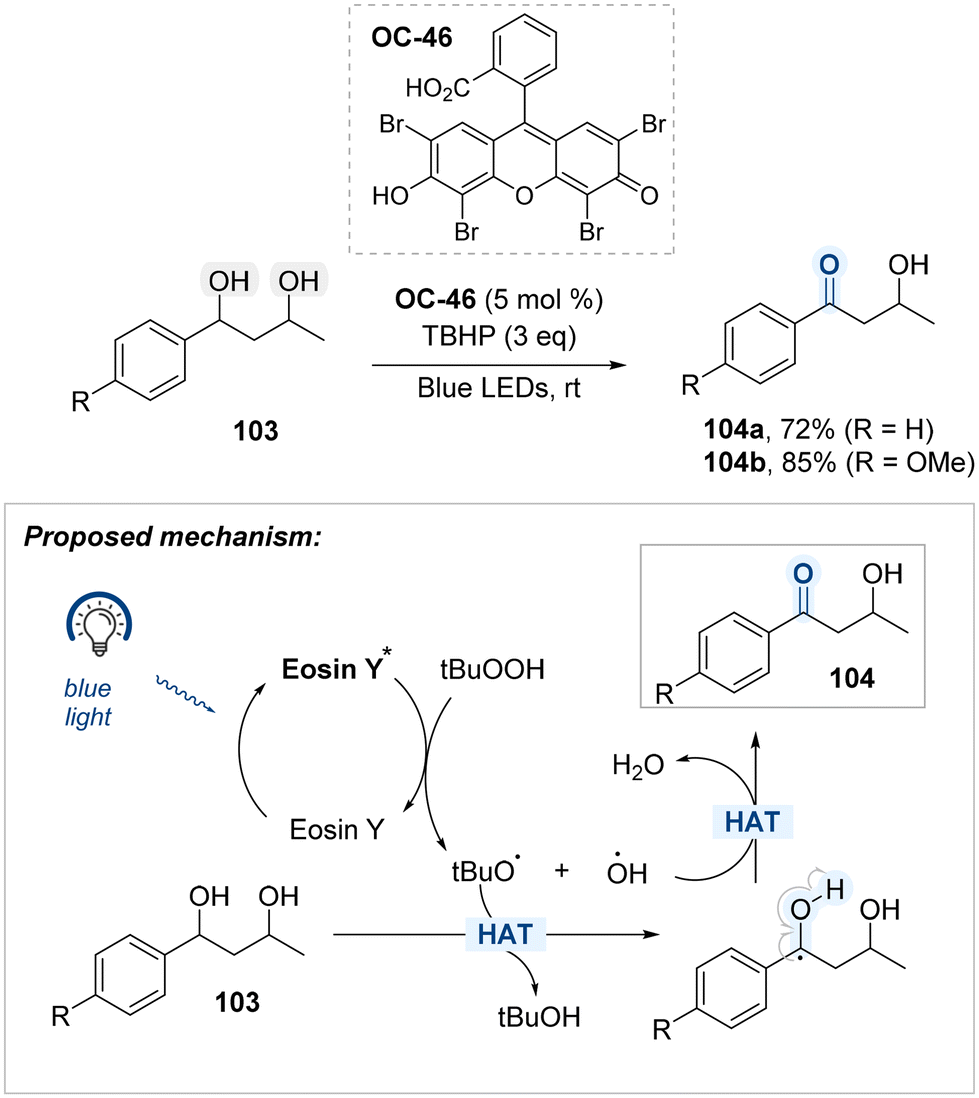 | ||
| Scheme 57 Blue light-mediated chemoselective oxidation of 1,3-diol. | ||
Building on the utility of Eosin Y (OC-46) as an organo-photocatalyst, Song, Xu, and Gao developed a blue-light-mediated protocol for the selective oxidation of hydroxyl groups located at benzylic positions, employing molecular oxygen as the terminal oxidant (Scheme 58).138 OC-46 has been shown to promote aerobic oxidation at benzylic positions via a hydrogen atom transfer (HAT) mechanism under visible light irradiation. Upon photoexcitation, the Eosin Y catalyst reaches its excited state and abstracts a hydrogen atom from the benzylic C–H bond, generating a benzylic radical and the reduced form of the catalyst (Eosin Y–H). The benzylic radical is subsequently oxidized to furnish a benzyl carbocation, which undergoes deprotonation to yield the corresponding carbonyl product 106. Under blue light irradiation, the reduced photocatalyst (Eosin Y–H) is reoxidized by molecular oxygen, thereby regenerating Eosin Y and closing the catalytic cycle.
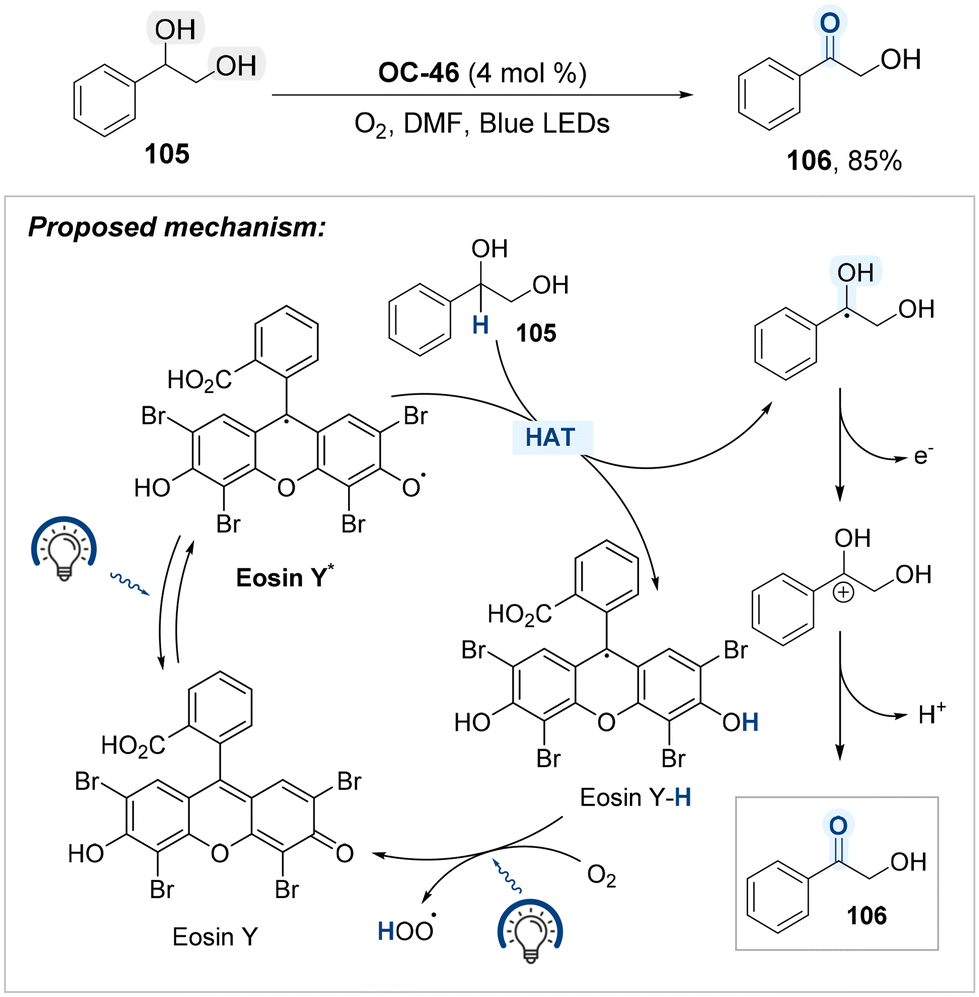 | ||
| Scheme 58 Photocatalyzed selective oxidation of 1,2-diols. | ||
Ananthakrishnan and co-workers also developed a photoinduced oxidation protocol for benzylic hydroxyl groups using bromodimethylsulfonium bromide (BDMS, OC-47) and molecular oxygen, enabling the site-selective conversion of secondary alcohols to ketones (Scheme 59).139 In the proposed mechanism, OC-47 undergoes photoactivation to generate bromine radicals, which initiate hydrogen atom abstraction from the benzylic C–H bond of diol 107, forming benzylic radical intermediates 108. These radicals subsequently react with molecular oxygen to produce a diol–peroxy radical adduct 109. The resulting peroxy radicals abstract hydrogen atoms from in situ generated HBr, thereby regenerating bromine radicals and forming hydroperoxide species 110,140 which are believed to be intermediates en route to the formation of ketone 111. Notably, overoxidation beyond the ketone stage is not observed under the reaction conditions.
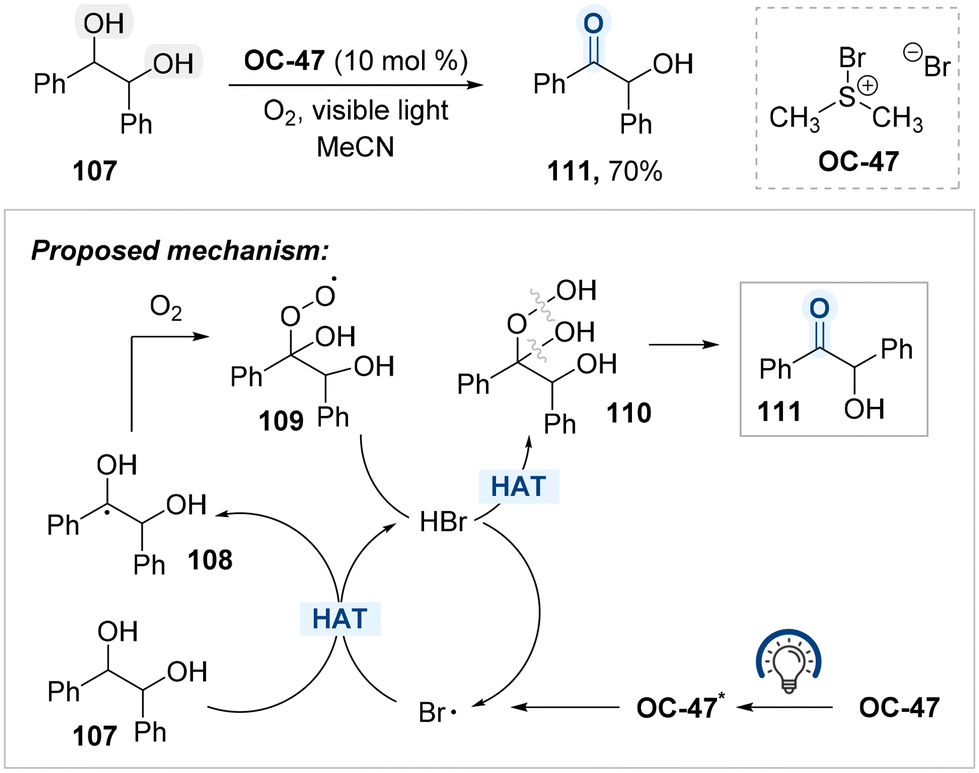 | ||
| Scheme 59 OC-47-catalyzed selective benzylic oxidation under visible light. | ||
Phipps and colleagues reported a catalytic system for the enantioselective oxidation of meso-diols via visible-light-driven radical generation (Scheme 60).141 This transformation utilizes 4CzIPN (OC-48) as an organophotocatalyst in combination with a cinchona alkaloid-derived co-catalyst (Co-cat-6), which collectively enable the stereocontrolled formation of radical intermediates under mild conditions. Upon photoexcitation, OC-48 undergoes oxidative quenching with diisopropyl azodicarboxylate (DIAD), a sacrificial oxidant that generates the radical ion pair [OC-48]˙+ and DIAD˙−. The latter is protonated to afford a persistent DIAD˙ species capable of participating in downstream radical coupling. Meanwhile, the oxidized photocatalyst facilitates single-electron oxidation of quinuclidine moiety of Co-cat-6, producing a chiral quinuclidinium radical cation XXI that selectively abstracts a hydrogen atom from the meso-diol substrate 112. This enantio-determining step breaks molecular symmetry to yield a stereodefined ketyl radical intermediate 113, which subsequently undergoes radical–radical coupling with DIAD˙ to form intermediate 114. Elimination of this adduct furnishes the enantioenriched hydroxyketone product 115. This strategy represents a mechanistically distinct approach for the de-symmetrization of meso-diols, achieving high enantioselectivity through controlled generation and selective capture of ketyl radical intermediates.
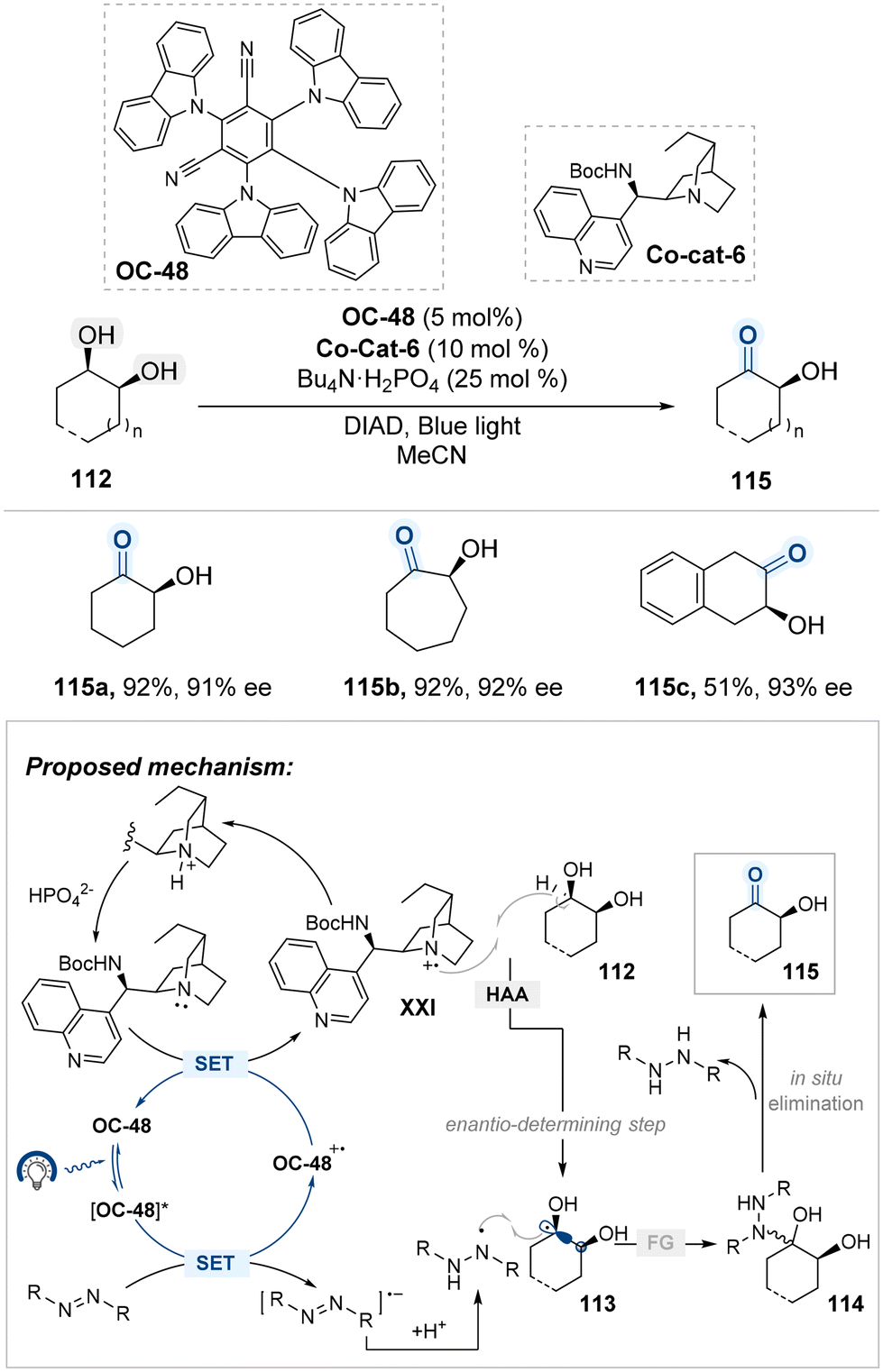 | ||
| Scheme 60 Photocatalyzed oxidative desymmetrization of meso-1,2-diols. | ||
Kundu and co-workers reported a site-selective oxidative C–C bond cleavage of diol substrates under blue light irradiation, employing an acridinium-based photocatalyst (OC-50), Selectfluor as a HAT agent, and DMAP as a base (Scheme 61).142 This transformation operates through a radical mechanism involving sequential single-electron transfer (SET) and hydrogen atom transfer (HAT) steps. In this system, photo-excited photocatalyst (OC-50*) facilitate a SET to oxidize the base-activated form of Selectfluor (XXII), generating the electrophilic radical cation species N-(chloromethyl)triethylene-diamine (TED˙2+, [XXII]˙2+). This radical cation serves as an effective hydrogen atom abstractor, selectively removing a hydrogen atom from the benzylic position of the diol 116 to form a resonance-stabilized benzylic radical intermediate 117. The benzylic radical species goes through oxidation by superoxide radical (O2˙−), forming a hydroperoxide intermediate 118. This species is subsequently converted into an α-hydroxy ketone intermediate 119, which is further transformed into the corresponding benzoic acid 120 via a well-established oxidative pathway143,144 The reaction showcases the role of [XXII]˙2+ as a potent photogenerated HAT species, driving selective benzylic oxidation through a multi-step cascade involving radical formation, oxygenation, and hydrolysis.
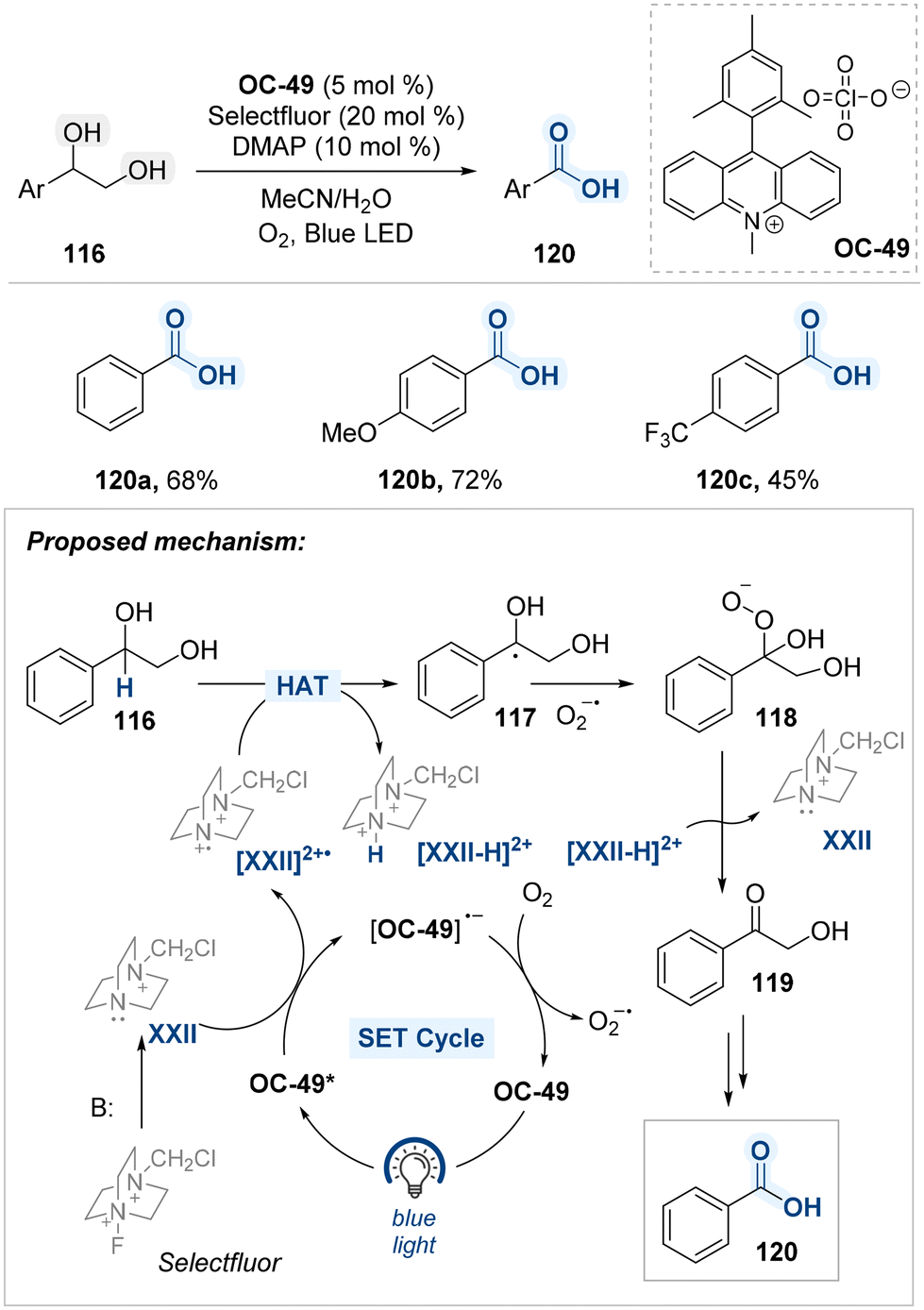 | ||
| Scheme 61 Blue light-mediated oxidative cleavage of terminal 1,2-diols. | ||
Fu and co-workers demonstrated that mesoporous graphitic carbon nitride (mpg-C3N4, OC-50), a visible-light-responsive organic semiconductor with a high surface area and tunable nitrogen-rich structure, can efficiently promote the aerobic oxidative cleavage of 1,2-diols under mild conditions (Scheme 62).145 Under the visible light, the OC-50 promotes a photoredox-mediated activation of molecular oxygen. During this process, charge separation occurs to generate holes (h+) in the valence band and electrons (e−) in the conduction band. The photoexcited electron in conduction band reduces molecular oxygen to produce reactive oxygen species, such as superoxide radicals (O2˙− or HOO˙). Concurrently, the photogenerated hole facilitates hydrogen atom transfer from benzylic alcohol moiety of the diol substrate 121, furnishing an alkoxy radical intermediate. This radical undergoes β-scission,146 yielding a benzaldehyde 122′ with benzylic radical. The resulting benzylic radical can go through a second hydrogen atom transfer or couple with reactive oxygen species, ultimately forming an additional equivalent of aldehyde. In subsequent steps, aldehydes are proposed to be oxidized to carboxylic acids 122 through a well-established radical-mediated pathway.
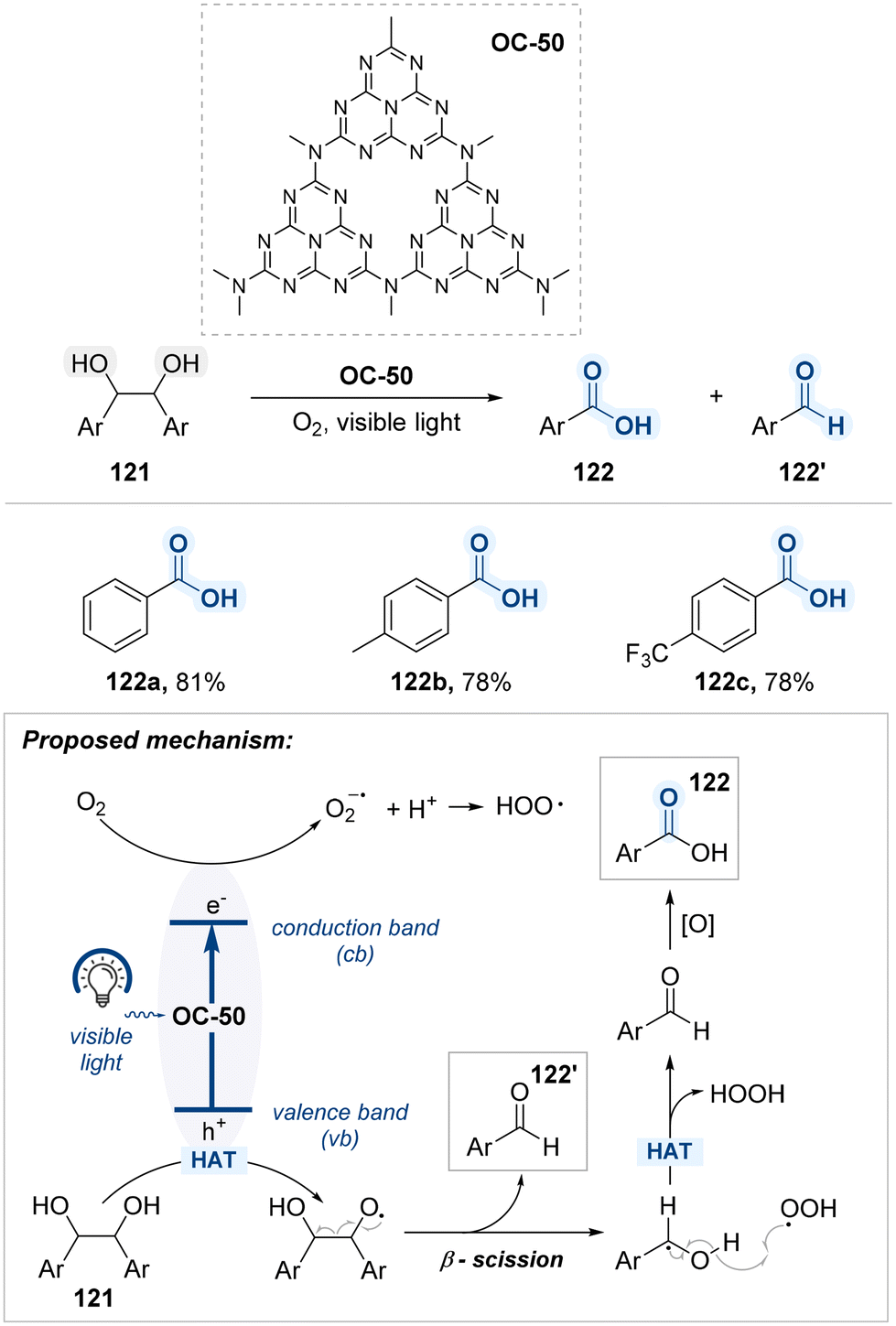 | ||
| Scheme 62 Photocatalyzed oxidative cleavage of C–C bonds in vicinal diol substrates. | ||
As part of ongoing efforts to design photocatalytic systems operable in aqueous media, a method employing nitrogen-deficient carbon nitride (CN620, OC-51) was reported by Niu and Ni for the oxidative cleavage of 1,2-diols under visible light (Scheme 63).147 Photoexcitation of OC-51 generates electron–hole pairs, initiating redox processes in which the photogenerated electrons reduce molecular oxygen to superoxide radicals (O2˙−), while the holes oxidize 1,2-diols to form alkoxy radical intermediates. From this point, two distinct mechanistic pathways are proposed, depending on the nature of the substituent (R) on the diol substrate 123: pathway A operates when R is a non-hydrogen substituent, while pathway B proceeds when R is a hydrogen atom. In pathway A, the alkoxy radical undergoes β-scission to cleave the adjacent C–C bond, generating a α-hydroxy radical and a carbonyl compound 124. The resulting carbon-centered radical is then oxidized via hydrogen atom transfer (HAT) from either the solvent or another alkoxy radical, affording a second aldehyde or ketone product 124′.148 In contrast, pathway B involves direct oxidation of the alkoxy radical by a putative superoxide radical species, generating a peroxy anion intermediate and water. This intermediate undergoes hydrolysis to form a gem-diol and desired aldehyde 124 which subsequently dehydrates to afford the carbonyl product 124′.
 | ||
| Scheme 63 Photocatalytic aerobic oxidative cleavage of C–C bonds in 1,2-diols. | ||
6. Conclusions and future outlook
This review provides a comprehensive overview of recent advances in the organocatalytic functionalization of diols, with particular emphasis on methodologies that enable precise regio- and stereochemical control. The literature is categorized based on the nature of the organocatalyst, with particular attention to boron, nitrogen, and phosphorus-based systems, alongside emerging photoredox-catalyzed methodologies. Throughout, key mechanistic insights were highlighted, emphasizing their impact on reactivity and regio- and stereoselective outcomes.In the domain of boron-based organocatalysis, representative classes such as boranes, borinic acids, boronic acids, and their heterocyclic derivatives (e.g., benzoxazaborine, benzazaborole and benzoxaborole) were introduced, with a focus on their characteristic activation modes in diol functionalization. For borane catalysts, the selective deoxygenation of diols using the BCF/hydrosilane system has been discussed, proceeding via either cyclic or acyclic intermediates. The nature of the intermediate is highly dependent on both substrate structure and reaction conditions, thereby influencing the transformation's efficiency and selectivity. In the case of borinic acids, boronic acids, and their derivatives, the electrophilic boron center engages in reversible interactions with the hydroxyl groups of diols, forming cyclic borinate or boronate ester intermediates. In this scenario, the selective transformation of the diol was achieved through careful tuning of the catalyst's steric and electronic environment.
In the context of nitrogen-based organocatalytic functionalization of diols, this review encompassed a diverse range of catalytic systems, including N-heterocycles, diamines, peptides, and N-heterocyclic carbenes (NHCs). These catalysts operate via diverse mechanistic pathways, such as electrophile activation, hydrogen bonding, Brønsted acid–base interactions, and steric modulation. Such interactions facilitate differentiation between hydroxyl groups and stabilize key transition states, ultimately enabling high levels of regioselectivity.
For phosphorus-containing organocatalysts, systems such as chiral phosphines, chiral phosphoric acids, and phosphonium salts have been reviewed for their ability to promote the selective transformation of diols via mechanisms involving nucleophilic activation, Brønsted acidity, or Lewis base catalysis.
The last section highlighted recent advances in diol-selective organophotocatalytic oxidation, which typically proceed via redox-additive mediated photoinduced single-electron transfer (SET) process that generates radical species. These radical species react with diol through hydrogen atom transfer (HAT), forming reactive intermediates that drive site-selective oxidation. Crucially, the selective generation of these intermediates is governed by differences in C–H bond dissociation energies, radical stability, and steric accessibility factors.
Although selective transformations of hydroxyl groups in diols have been pursued for more than a century, several key challenges persist, offering compelling avenues for future research. A major limitation lies in the relatively narrow substrate generality, as most strategies discussed in this review have predominantly been developed for meso-diols or secondary-primary diols, underscoring a constrained chemical space. In contrast, the regioselective functionalization of more challenging motifs, such as unsymmetrical secondary–secondary diols, remote diols, and tertiary–secondary diols, remains particularly demanding due to their comparable reactivity profiles and lack of inherent differentiation.
Another significant challenge is the development of more economical and finely tunable catalytic platforms that minimize the use of sacrificial reagents, override steric biases, and enable reversals of inherent selectivity trend. The design of such catalysts may benefit from leveraging novel synergistic non-covalent interactions, which can provide the level of control required for broad substrate applicability and enhanced selectivity. Given that diols represent a valuable model system for developing functionalization strategies applicable to more complex polyol frameworks, advances in catalyst design for diol substrates are likely to translate effectively to polyols, governed by similar selectivity-controlling principles. Ultimately, such developments will open new avenues for the regioselective transformation of complex polyol architectures, thereby accelerating innovation in polyol building block synthesis.
We believe that the organocatalyzed methods for selective transformation of diol discussed in this review will serve as an attractive catalytic platform to achieve further exciting breakthroughs. We hope this contribution provides a succinct summary of the field and encourages exploration of related synthetic methodologies.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare that they have no conflicts of interest.Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. RS-2023-00239404). This work was supported by the research fund of Hanyang University (HY-202300000001161).References
- D. J. Trader and E. E. Carlson, Chemoselective hydroxyl group transformation: an elusive target, Mol. Biosyst., 2012, 8, 2484–2493 Search PubMed.
- J. Cramer, C. P. Sager and B. Ernst, Hydroxyl Groups in Synthetic and Natural-Product-Derived Therapeutics: A Perspective on a Common Functional Group, J. Med. Chem., 2019, 62, 8915–8930 Search PubMed.
- S. J. Miller, G. T. Copeland, N. Papaioannou, T. E. Horstmann and E. M. Ruel, Kinetic Resolution of Alcohols Catalyzed by Tripeptides Containing the N-Alkylimidazole Substructure, J. Am. Chem. Soc., 1998, 120, 1629–1630 Search PubMed.
- A. C. Weymouth-Wilson, The Role of Carbohydrates in Biologically Active Natural Products, Nat. Prod. Rep., 1997, 14, 99–110 Search PubMed.
- K. Vladimir and M. Ludmila, Glycosides in Medicine: The Role of Glycosidic Residue in Biological Activity, Curr. Med. Chem., 2001, 8, 1303–1328 CrossRef PubMed.
- G. A. Evrendilek, in Sweeteners: Nutritional Aspects, Applications, and Production Technology, ed. T. Varzakas, A. Labropoulos and S. Anestis, CRC Press, Boca Raton, 1st edn, 2012, vol. 14, ch. 3, pp. 56–60 Search PubMed.
- D. B. Werz and P. H. Seeberger, Carbohydrates as the Next Frontier in Pharmaceutical Research, Chem. – Eur. J., 2005, 11, 3194–3206 Search PubMed.
- P. Mulqueen, Recent advances in agrochemical formulation, Adv. Colloid Interface Sci., 2003, 106, 83–107 CrossRef CAS PubMed.
- C. Feldmann, Polyol-mediated synthesis of nanoscale functional materials, Solid State Sci., 2005, 7, 868–873 CrossRef CAS.
- S. S. Kulkarni, C.-C. Wang, N. M. Sabbavarapu, A. R. Podilapu, P.-H. Liao and S.-C. Hung, “One-Pot” Protection, Glycosylation, and Protection–Glycosylation Strategies of Carbohydrates, Chem. Rev., 2018, 118, 8025–8104 CrossRef CAS PubMed.
- B. R. Sculimbrene and S. J. Miller, Discovery of a Catalytic Asymmetric Phosphorylation through Selection of a Minimal Kinase Mimic: A Concise Total Synthesis of d-myo-Inositol-1-Phosphate, J. Am. Chem. Soc., 2001, 123, 10125–10126 Search PubMed.
- B. R. Sculimbrene, A. J. Morgan and S. J. Miller, Nonenzymatic peptide-based catalytic asymmetric phosphorylation of inositol derivatives, Chem. Commun., 2003, 1781–1785 Search PubMed.
- C. A. Lewis and S. J. Miller, Site-Selective Derivatization and Remodeling of Erythromycin A by Using Simple Peptide-Based Chiral Catalysts, Angew. Chem., Int. Ed., 2006, 45, 5616–5619 CrossRef CAS PubMed.
- J.-H. Tay, A. J. Argüelles, M. D. DeMars II, P. M. Zimmerman, D. H. Sherman and P. Nagorny, Regiodivergent Glycosylations of 6-Deoxy-erythronolide B and Oleandomycin-Derived Macrolactones Enabled by Chiral Acid Catalysis, J. Am. Chem. Soc., 2017, 139, 8570–8578 CrossRef CAS PubMed.
- R. R. Knowles and E. N. Jacobsen, Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20678–20685 Search PubMed.
- L. Bergelson, V. Vaver, N. Prokazova, A. Ushakov and G. Popkova, Diol Lipids, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 1966, 116, 511–520 CrossRef CAS PubMed.
- O. Robles and D. Romo, Chemo- and Site-Selective Derivatizations of Natural Products Enabling Biological Studies, Nat. Prod. Rep., 2014, 31, 318–334 Search PubMed.
- M. C. Noe, M. A. Letavic and S. L. Snow, Asymmetric Dihydroxylation of Alkenes, Org. React., 2004, 66, 109–625 Search PubMed.
- M. Thirumalaikumar, Ring-Opening Reactions of Epoxides: A Review, Org. Prep. Proced. Int., 2022, 54, 1–39 CrossRef CAS.
- R. Thompson, The Reduction of α-Diketones, J. Am. Chem. Soc., 1939, 61, 1281–1283 CrossRef CAS.
- S. E. Bode, M. Wolberg and M. Müller, Stereoselective Synthesis of 1,3-Diols, Synthesis, 2006, 557–588 CAS.
- M. S. Butler, Natural Products to Drugs: Natural Product-Derived Compounds in Clinical Trials, Nat. Prod. Rep., 2008, 25, 475–516 Search PubMed.
- G. R. Pettit, G. M. Cragg, S. B. Singh, J. A. Duke and D. L. Doubek, Antineoplastic Agents, 162. Zephyranthes Candida, J. Nat. Prod., 1990, 53, 176–178 Search PubMed.
- Y.-C. Wu, C.-Y. Duh, F.-R. Chang, G.-Y. Chang, S.-K. Wang, J.-J. Chang, D. R. McPhail, A. T. McPhail and K.-H. Lee, The Crystal Structure and Cytotoxicity of Goniodiol-7-monoacetate from Goniothalamus amuyon, J. Nat. Prod., 1991, 54, 1077–1081 CrossRef CAS PubMed.
- L.-J. Yang, X.-Y. Peng, Y.-H. Zhang, Z.-Q. Liu, X. Li, Y.-C. Gu, C.-L. Shao, Z. Han and C.-Y. Wang, Antimicrobial and Antioxidant Polyketides from a Deep-Sea-Derived Fungus Aspergillus versicolor SH0105, Mar. Drugs, 2020, 18, 636 CrossRef CAS PubMed.
- X. Luo, Y. Li, N. K. Gupta, B. Sels, J. Ralph and L. Shuai, Protection Strategies Enable Selective Conversion of Biomass, Angew. Chem., 2020, 132, 11800–11812 CrossRef.
- T. Wang and A. V. Demchenko, Synthesis of Carbohydrate Building Blocks via Regioselective Uniform Protection/Deprotection Strategies, Org. Biomol. Chem., 2019, 17, 4934–4950 RSC.
- V. Dimakos and M. S. Taylor, Site-Selective Functionalization of Hydroxyl Groups in Carbohydrate Derivatives, Chem. Rev., 2018, 118, 11457–11517 Search PubMed.
- N. Funken, Y. Q. Zhang and A. Gansäuer, Regiodivergent Catalysis: A Powerful Tool for Selective Catalysis, Chem. – Eur. J., 2017, 23, 19–32 Search PubMed.
- W. Muramatsu, S. Tanigawa, Y. Takemoto, H. Yoshimatsu and O. Onomura, Organotin-Catalyzed Highly Regioselective Thiocarbonylation of Nonprotected Carbohydrates and Synthesis of Deoxy Carbohydrates in a Minimum Number of Steps, Chem. – Eur. J., 2012, 18, 4850–4853 CrossRef CAS PubMed.
- M. Guillaume and Y. Lang, Further Improvements of the Dibutyl Tin Oxide-Catalyzed Regioselective Diol Tosylation, Tetrahedron Lett., 2010, 51, 579–582 Search PubMed.
- M. Therisod and A. M. Klibanov, Regioselective Acylation of Secondary Hydroxyl Groups in Sugars Catalyzed by Lipases in Organic Solvents, J. Am. Chem. Soc., 1987, 109, 3977–3981 CrossRef CAS.
- K. Chung, S. M. Banik, A. G. De Crisci, D. M. Pearson, T. R. Blake, J. V. Olsson, A. J. Ingram, R. N. Zare and R. M. Waymouth, Chemoselective Pd-Catalyzed Oxidation of Polyols: Synthetic Scope and Mechanistic Studies, J. Am. Chem. Soc., 2013, 135, 7593–7602 CrossRef CAS PubMed.
- C. K. Hill and J. F. Hartwig, Site-Selective Oxidation, Amination, and Epimerization Reactions of Complex Polyols Enabled by Transfer Hydrogenation, Nat. Chem., 2017, 9, 1213–1221 CrossRef CAS PubMed.
- R. Ramírez-Contreras and B. Morandi, Chemo-and Regioselective Functionalization of Polyols through Catalytic C (sp3)–C (sp3) Kumada-Type Coupling of Cyclic Sulfate Esters, Org. Lett., 2016, 18, 3718–3721 CrossRef PubMed.
- L. Xia, Q. Huang and L. Dai, Site-Selective Editing of Carbohydrate Scaffolds to Access Rare Sugars Enabled by Photoinduced Radical Processes, Org. Chem. Front., 2024, 11, 4926–4933 RSC.
- K. Yamatsugu and M. Kanai, Catalytic Approaches to Chemo- and Site-Selective Transformation of Carbohydrates, Chem. Rev., 2023, 123, 6793–6838 CrossRef CAS PubMed.
- A. Shatskiy, E. V. Stepanova and M. D. Kärkäs, Exploiting Photoredox Catalysis for Carbohydrate Modification through C–H and C–C Bond Activation, Nat. Rev. Chem., 2022, 6, 782–805 CrossRef CAS PubMed.
- C. E. Suh, H. M. Carder and A. E. Wendlandt, Selective Transformations of Carbohydrates Inspired by Radical-Based Enzymatic Mechanisms, ACS Chem. Biol., 2021, 16, 1814–1828 CrossRef CAS PubMed.
- D. J. Gorelik, V. Dimakos, T. Adrianov and M. S. Taylor, Photocatalytic, Site-Selective Oxidations of Carbohydrates, Chem. Commun., 2021, 57, 12135–12138 RSC.
- V. Dimakos and M. S. Taylor, Recent Advances in the Direct O-Arylation of Carbohydrates, Org. Biomol. Chem., 2021, 19, 514–524 RSC.
- S. A. Blaszczyk, T. C. Homan and W. Tang, Recent Advances in Site-Selective Functionalization of Carbohydrates Mediated by Organocatalysts, Carbohydr. Res., 2019, 471, 64–77 CrossRef CAS PubMed.
- H.-Y. Wang, S. A. Blaszczyk, G. Xiao and W. Tang, Chiral Reagents in Glycosylation and Modification of Carbohydrates, Chem. Soc. Rev., 2018, 47, 681–701 RSC.
- Y. Ueda and T. Kawabata, in Site-Selective Catalysis, ed. T. Kawabata, Springer Chem, Cham, 1st edn, 2016, ch. 8, pp. 203–231 Search PubMed.
- W. Muramatsu, Recent Advances in the Regioselective Functionalization of Carbohydrates Using Non-Enzymatic Catalysts, Trends Glycosci. Glycotechnol., 2016, 28, E1–E11 CrossRef.
- D. Lee and M. S. Taylor, Catalyst-Controlled Regioselective Reactions of Carbohydrate Derivatives, Synthesis, 2012, 3421–3431 CAS.
- Y. Zhou, K.-S. Liao, T.-Y. Chen, Y. S. Hsieh and C.-H. Wong, Effective Organotin-Mediated Regioselective Functionalization of Unprotected Carbohydrates, J. Org. Chem., 2023, 88, 7141–7151 CrossRef CAS PubMed.
- D. J. Gorelik, J. A. Turner, T. S. Virk, D. A. Foucher and M. S. Taylor, Site- and Stereoselective C–H Alkylations of Carbohydrates Enabled by Cooperative Photoredox, Hydrogen Atom Transfer, and Organotin Catalysis, Org. Lett., 2021, 23, 5180–5185 CrossRef CAS PubMed.
- B. Ren, N. Yan and L. Gan, Regioselective Alkylation of Carbohydrates and Diols: A Cheaper Iron Catalyst, New Applications, and Mechanism, RSC Adv., 2017, 7, 46257–46262 RSC.
- V. Saikam, S. Dara, M. Yadav, P. P. Singh and R. A. Vishwakarma, Dimethyltin Dichloride Catalyzed Regioselective Alkylation of cis-1,2-Diols at Room Temperature, J. Org. Chem., 2015, 80, 11916–11925 CrossRef CAS PubMed.
- Y. Zhou, O. Ramström and H. Dong, Organosilicon-Mediated Regioselective Acetylation of Carbohydrates, Chem. Commun., 2012, 48, 5370–5372 RSC.
- A. Bouzide and G. Sauve, Highly Selective Silver(I) oxide Mediated Monoprotection of Symmetrical Diols, Tetrahedron Lett., 1997, 38, 5945–5948 CrossRef CAS.
- G.-W. Yang, Y.-Y. Zhang and G.-P. Wu, Modular Organoboron Catalysts Enable Transformations with Unprecedented Reactivity, Acc. Chem. Res., 2021, 54, 4434–4448 CrossRef CAS PubMed.
- R. S. Mancini, J. B. Lee and M. S. Taylor, Boronic Esters as Protective Groups in Carbohydrate Chemistry: Processes for Acylation, Silylation, and Alkylation of Glycoside-Derived Boronates, Org. Biomol. Chem., 2017, 15, 132–143 RSC.
- D. G. Hall, in Boronic Acids: Preparation, Applications in Organic Synthesis and Medicine, ed. and D. G. Hall, John Wiley & Sons, Hoboken, New Jersey, United States, 1st edn, 2006, ch. 1, pp. 1–3 Search PubMed.
- B. J. Graham and R. T. Raines, Emergent Organoboron Acid Catalysts, J. Org. Chem., 2024, 89, 2069–2089 CrossRef CAS PubMed.
- M. Boyet, L. Chabaud and M. Pucheault, Recent Advances in the Synthesis of Borinic Acid Derivatives, Molecules, 2023, 28, 2660 CrossRef CAS PubMed.
- G. Springsteen and B. Wang, A Detailed Examination of Boronic Acid–Diol Complexation, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS.
- H. Fang and M. Oestreich, Defunctionalisation Catalysed by Boron Lewis Acids, Chem. Sci., 2020, 11, 12604–12615 RSC.
- L. L. Adduci, T. A. Bender, J. A. Dabrowski and M. R. Gagné, Chemoselective Conversion of Biologically Sourced Polyols into Chiral Synthons, Nat. Chem., 2015, 7, 576–581 Search PubMed.
- N. Drosos and B. Morandi, Boron–Catalyzed Regioselective Deoxygenation of Terminal 1, 2-Diols to 2-Alkanols Enabled by the Strategic Formation of a Cyclic Siloxane Intermediate, Angew. Chem., Int. Ed., 2015, 54, 8814–8818 Search PubMed.
- G.-J. Cheng, N. Drosos, B. Morandi and W. Thiel, Computational Study of B(C6F5)3-Catalyzed Selective Deoxygenation of 1,2-Diols: Cyclic and Noncyclic Pathways, ACS Catal., 2018, 8, 1697–1702 CrossRef CAS.
- N. Drosos, G. J. Cheng, E. Ozkal, B. Cacherat, W. Thiel and B. Morandi, Catalytic Reductive Pinacol–Type Rearrangement of Unactivated 1, 2-Diols through a Concerted, Stereoinvertive Mechanism, Angew. Chem., Int. Ed., 2017, 56, 13377–13381 Search PubMed.
- I. Chatterjee, D. Porwal and M. Oestreich, B(C6F5)3-Catalyzed Chemoselective Defunctionalization of Ether-Containing Primary Alkyl Tosylates with Hydrosilanes, Angew. Chem., Int. Ed., 2017, 56, 3389–3391 CrossRef CAS PubMed.
- S. C. Richter and M. Oestreich, Chemoselective Deoxygenation of 2° Benzylic Alcohols through a Sequence of Formylation and B(C6F5)3-Catalyzed Reduction, Eur. J. Org. Chem., 2021, 2103–2106 CrossRef CAS.
- W. Yang, L. Gao, J. Lu and Z. Song, Chemoselective Deoxygenation of Ether-Substituted Alcohols and Carbonyl Compounds by B(C6F5)3-Catalyzed Reduction with (HMe2SiCH2)2, Chem. Commun., 2018, 54, 4834–4837 RSC.
- M. G. Chudzinski, Y. Chi and M. S. Taylor, Borinic Acids: A Neglected Class of Organoboron Compounds for Recognition of Diols in Aqueous Solution, Aust. J. Chem., 2011, 64, 1466–1469 CrossRef CAS.
- D. Lee and M. S. Taylor, Borinic Acid-Catalyzed Regioselective Acylation of Carbohydrate Derivatives, J. Am. Chem. Soc., 2011, 133, 3724–3727 CrossRef CAS PubMed.
- E. Dimitrijević and M. S. Taylor, 9-Hetero-10-Boraanthracene-Derived Borinic Acid Catalysts for Regioselective Activation of Polyols, Chem. Sci., 2013, 4, 3298–3303 Search PubMed.
- M. Pawliczek, T. Hashimoto and K. Maruoka, Alkylative Kinetic Resolution of Vicinal Diols under Phase-Transfer Conditions: A Chiral Ammonium Borinate Catalysis, Chem. Sci., 2018, 9, 1231–1235 Search PubMed.
- J. Song and W.-H. Zheng, Synthesis of a C2-Symmetric Chiral Borinic Acid and Its Application in Catalytic Desymmetrization of 2,2-Disubstituted-1,3-Propanediols, J. Am. Chem. Soc., 2023, 145, 8338–8343 CrossRef CAS PubMed.
- Y. Chikashige, T. Takehara, T. Matsuzaki, T. Suzuki, K. Murai, M. Arisawa and M. Sako, Axially Chiral Borinic Acid Catalysts: Design, Synthesis, and Application in Alkylative Desymmetrization of 1, 2-Diols, J. Org. Chem., 2023, 88, 14178–14183 Search PubMed.
- A. Suzuki, Cross-Coupling Reactions of Organoboranes: An Easy Way to Construct C–C Bonds (Nobel Lecture), Angew. Chem., Int. Ed., 2011, 50, 6722–6737 Search PubMed.
- N. A. Petasis and I. Akritopoulou, The Boronic Acid Mannich Reaction: A New Method for the Synthesis of Geometrically Pure Allylamines, Tetrahedron Lett., 1993, 34, 583–586 Search PubMed.
- P. Y. S. Lam, C. G. Clark, S. Saubern, J. Adams, M. P. Winters, D. M. T. Chan and A. Combs, New Aryl/Heteroaryl C–N Bond Cross-Coupling Reactions via Arylboronic Acid/Cupric Acetate Arylation, Tetrahedron Lett., 1998, 39, 2941–2944 Search PubMed.
- V. Ortega, E. del Castillo and A. G. Csákÿ, Transition-Metal-Free Stereocomplementary Cross-Coupling of Diols with Boronic Acids as Nucleophiles, Org. Lett., 2017, 19, 6236–6239 Search PubMed.
- H. Yu, R. Lee, H. Kim and D. Lee, Lewis Acid-Promoted Regio- and Diastereoselective Cross-Coupling of Aryl-Substituted 1,2-Diols and Boronic Acids, J. Org. Chem., 2019, 84, 3566–3578 CrossRef CAS PubMed.
- J. M. William, M. Kuriyama and O. Onomura, Boronic Acid–Catalyzed Selective Oxidation of 1, 2-Diols to α–Hydroxy Ketones in Water, Adv. Synth. Catal., 2014, 356, 934–940 Search PubMed.
- J. M. William, M. Kuriyama and O. Onomura, Simple Method for Selective Oxidation of 1,2-Diols in Water with KBrO3/KHSO4, Tetrahedron Lett., 2014, 55, 6589–6592 Search PubMed.
- S. Estopiñá-Durán, L. J. Donnelly, E. B. Mclean, B. M. Hockin, A. M. Slawin and J. E. Taylor, Aryl Boronic Acid-Catalyzed Dehydrative Substitution of Benzylic Alcohols for C–O Bond Formation, Chem. – Eur. J., 2019, 25, 3950–3956 Search PubMed.
- S. Kusano, S. Miyamoto, A. Matsuoka, Y. Yamada, R. Ishikawa and O. Hayashida, Benzoxaborole Catalyst for Site–Selective Modification of Polyols, Eur. J. Org. Chem., 2020, 2020, 1598–1602 Search PubMed.
- S. Vshyvenko, M. L. Clapson, I. Suzuki and D. G. Hall, Characterization of the Dynamic Equilibrium between Closed and Open Forms of the Benzoxaborole Pharmacophore, ACS Med. Chem. Lett., 2016, 7, 1097–1101 CrossRef CAS PubMed.
- J. P. Rygus and D. G. Hall, Direct Nucleophilic and Electrophilic Activation of Alcohols Using a Unified Boron-Based Organocatalyst Scaffold, Nat. Commun., 2023, 14, 2563 Search PubMed.
- S. Kuwano, Y. Hosaka and T. Arai, Chiral Benzazaboroles as Catalysts for Enantioselective Sulfonylation of cis-1,2-Diols, Org. Biomol. Chem., 2019, 17, 4475–4482 Search PubMed.
- S. Kusano, Y. Yamada and S. Hagihara, Benzoxaborole Catalyst Embedded with a Lewis Base: A Highly Active and Selective Catalyst for cis-1, 2-diol Modification, J. Org. Chem., 2024, 89, 6714–6722 Search PubMed.
- C. D. Estrada, H. T. Ang, K.-M. Vetter, A. A. Ponich and D. G. Hall, Enantioselective Desymmetrization of 2-Aryl-1,3-Propanediols by Direct O-Alkylation with a Rationally Designed Chiral Hemiboronic Acid Catalyst That Mitigates Substrate Conformational Poisoning, J. Am. Chem. Soc., 2021, 143, 4162–4167 Search PubMed.
- S. E. Denmark and G. L. Beutner, Lewis Base Catalysis in Organic Synthesis, Angew. Chem., Int. Ed., 2008, 47, 1560–1638 CrossRef CAS PubMed.
- M. Hossain and A. K. Nanda, A Review on Heterocyclics: Synthesis and Their Application in Medicinal Chemistry of Imidazole Moiety, Science, 2018, 6, 83–94 CAS.
- R. Gujjarappa, N. Vodnala and C. Malakar, Recent Advances in Pyridine–Based Organocatalysis and its Application towards Valuable Chemical Transformations, ChemistrySelect, 2020, 5, 8745–8758 CrossRef CAS.
- S. K. Verma, B. Acharya and M. Kaushik, Imidazole-Catalyzed Monoacylation of Symmetrical Diamines, Org. Lett., 2010, 12, 4232–4235 Search PubMed.
- E. F. Scriven, 4-Dialkylaminopyridines: Super Acylation and Alkylation Catalysts, Chem. Soc. Rev., 1983, 12, 129–161 Search PubMed.
- T. Kawabata, R. Stragies, T. Fukaya, Y. Nagaoka, H. Schedel and K. Fuji, Preparation and Properties of Chiral 4-Pyrrolidinopyridine (PPY) Analogues with Dual Functional Side Chains, Tetrahedron Lett., 2003, 44, 1545–1548 CrossRef CAS.
- S. Yamada, T. Misono, Y. Iwai, A. Masumizu and Y. Akiyama, New Class of Pyridine Catalyst Having a Conformation Switch System: Asymmetric Acylation of Various sec-Alcohols, J. Org. Chem., 2006, 71, 6872–6880 CrossRef CAS PubMed.
- H. Mandai, K. Ashihara, K. Mitsudo and S. Suga, Enantioselective Desymmetrization of 1,3-Diols by a Chiral DMAP Derivative, Chem. Lett., 2018, 47, 1360–1363 CrossRef CAS.
- K. Xu, K. Nakazono and T. Takata, Design of Rotaxane Catalyst for O-Acylative Asymmetric Desymmetrization of meso-1,2-Diol Utilizing the Cooperative Effect of the Components, Chem. Lett., 2016, 45, 1274–1276 CrossRef CAS.
- S.-Y. Lian, N. Li, Y. Tian, C. Peng, M.-S. Xie and H.-M. Guo, Reversal of Enantioselectivity for the Desymmetrization of meso-1, 2-Diols Catalyzed by Pyridine-N-oxides, J. Org. Chem., 2023, 88, 13771–13781 CrossRef CAS PubMed.
- X. Sun, A. D. Worthy and K. L. Tan, Application of Scaffolding Catalysts in a Highly Enantioselective Desymmetrization Reaction, Angew. Chem., Int. Ed., 2011, 50, 8167 CrossRef CAS PubMed.
- X. Sun, A. D. Worthy and K. L. Tan, Resolution of Terminal 1,2-Diols via Silyl Transfer, J. Org. Chem., 2013, 78, 10494–10499 CrossRef CAS PubMed.
- A. Sakakura, S. Umemura and K. Ishihara, Desymmetrization of meso–Glycerol Derivatives Induced by L–Histidine–Derived Acylation Catalysts, Adv. Synth. Catal., 2011, 353, 1938–1942 CrossRef CAS.
- J. Merad, P. Borkar, T. Bouyon Yenda, C. Roux, J.-M. Pons, J.-L. Parrain, O. Chuzel and C. Bressy, Highly Enantioselective Acylation of Acyclic Meso 1, 3-Diols through Synergistic Isothiourea-Catalyzed Desymmetrization/Chiroablative Kinetic Resolution, Org. Lett., 2015, 17, 2118–2121 Search PubMed.
- V. B. Birman, H. Jiang and X. Li, Enantioselective Synthesis of Lobeline via Nonenzymatic Desymmetrization, Org. Lett., 2007, 9, 3237–3240 CrossRef CAS PubMed.
- B. Ren, M. Zhang, S. Xu, L. Gan, L. Zhang and L. Tang, DBN–Catalyzed Regioselective Acylation of Carbohydrates and Diols in Ethyl Acetate, Eur. J. Org. Chem., 2019, 4757–4762 CrossRef CAS.
- J.-C. Kizirian, Chiral Tertiary Diamines in Asymmetric Synthesis, Chem. Rev., 2008, 108, 140–205 CrossRef CAS PubMed.
- S. Vera, A. Landa, A. Mielgo, I. Ganboa, M. Oiarbide and V. Soloshonok, Catalytic Asymmetric α-Functionalization of α-Branched Aldehydes, Molecules, 2023, 28, 2694 CrossRef CAS PubMed.
- T. Oriyama, K. Imai, T. Hosoya and T. Sano, Asymmetric Acylation of meso-Diols with Benzoyl Halide in the Presence of a Chiral Diamine, Tetrahedron Lett., 1998, 39, 397–400 CrossRef CAS.
- T. Oriyama, K. Imai, T. Sano and T. Hosoya, Highly Efficient Catalytic Asymmetric Acylation of meso-1,2-Diols with Benzoyl Chloride in the Presence of a Chiral Diamine Combined with Et3N, Tetrahedron Lett., 1998, 39, 3529–3532 CrossRef CAS.
- E. P. Kündig, T. Lomberget, R. Bragg, C. Poulard and G. Bernardinelli, Desymmetrization of a meso-Diol Complex Derived from [Cr(CO)3(η6–5,8-Naphthoquinone)]: Use of New Diamine Acylation Catalysts, Chem. Commun., 2004, 1548–1549 RSC.
- A. R. Pape, K. P. Kaliappan and E. P. Kündig, Transition-Metal-Mediated Dearomatization Reactions, Chem. Rev., 2000, 100, 2917–2940 CrossRef CAS PubMed.
- E. P. Kündig, A. E. Garcia, T. Lomberget, P. P. Garcia and P. Romanens, Truncated Cinchona Alkaloids as Catalysts in Enantioselective Monobenzoylation of meso-1,2-Diols, Chem. Commun., 2008, 3519–3521 RSC.
- D. de Wied, Peptides and Behavior, Life Sci., 1977, 20, 195–204 CrossRef CAS PubMed.
- E. R. Jarvo and S. J. Miller, Amino Acids and Peptides as Asymmetric Organocatalysts, Tetrahedron, 2002, 58, 2481–2495 CrossRef CAS.
- C. A. Lewis, B. R. Sculimbrene, Y. Xu and S. J. Miller, Desymmetrization of Glycerol Derivatives with Peptide-based Acylation Catalysts, Org. Lett., 2005, 7, 3021–3023 CrossRef CAS PubMed.
- C. A. Lewis, A. Chiu, M. Kubryk, J. Balsells, D. Pollard, C. K. Esser, J. Murry, R. A. Reamer, K. B. Hansen and S. J. Miller, Remote Desymmetrization at Near-Nanometer Group Separation Catalyzed by a Miniaturized Enzyme Mimic, J. Am. Chem. Soc., 2006, 128, 16454–16455 CrossRef CAS PubMed.
- C. E. Müller, D. Zell and P. R. Schreiner, One–Pot Desymmetrization of meso–1, 2-Hydrocarbon Diols through Acylation and Oxidation, Chem. – Eur. J., 2009, 15, 9647–9650 CrossRef PubMed.
- C. E. Müller, L. Wanka, K. Jewell and P. R. Schreiner, Enantioselective Kinetic Resolution of trans-Cycloalkane-1,2-diols, Angew. Chem., Int. Ed., 2008, 47, 6180–6183 CrossRef PubMed.
- Y. Zhao, J. Rodrigo, A. H. Hoveyda and M. L. Snapper, Enantioselective Silyl Protection of Alcohols Catalysed by an Amino-Acid-Based Small Molecule, Nature, 2006, 443, 67–70 CrossRef CAS PubMed.
- N. Manville, H. Alite, F. Haeffner, A. H. Hoveyda and M. L. Snapper, Enantioselective Silyl Protection of Alcohols Promoted by a Combination of Chiral and Achiral Lewis Basic Catalysts, Nat. Chem., 2013, 5, 768–774 CrossRef CAS PubMed.
- J. Rein, S. D. Rozema, O. C. Langner, S. B. Zacate, M. A. Hardy, J. C. Siu, B. Q. Mercado, M. S. Sigman, S. J. Miller and S. Lin, Generality-Oriented Optimization of Enantioselective Aminoxyl Radical Catalysis, Science, 2023, 380, 706–712 CrossRef CAS PubMed.
- S. B. Zacate, J. Rein, S. D. Rozema, R. A. Annor, S. J. Miller and S. Lin, Catalyst-Controlled Regiodivergent Oxidation of Unsymmetrical Diols, J. Am. Chem. Soc., 2025, 147, 8118–8124 CrossRef CAS PubMed.
- D. W. Tay, G. Y. Leung and Y. Y. Yeung, Desymmetrization of Diolefinic Diols by Enantioselective Amino–thiocarbamate–Catalyzed Bromoetherification: Synthesis of Chiral Spirocycles, Angew. Chem., Int. Ed., 2014, 53, 5161–5164 Search PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, An Overview of N-Heterocyclic Carbenes, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- K. Zeitler, Extending Mechanistic Routes in Heterazolium Catalysis – Promising Concepts for Versatile Synthetic Methods, Angew. Chem., Int. Ed., 2005, 44, 7506–7510 CrossRef CAS PubMed.
- N. T. Reynolds, J. Read de Alaniz and T. Rovis, Conversion of α-Haloaldehydes into Acylating Agents by an Internal Redox Reaction Catalyzed by Nucleophilic Carbenes, J. Am. Chem. Soc., 2004, 126, 9518–9519 CrossRef CAS PubMed.
- B. E. Maki, A. Chan, E. M. Phillips and K. A. Scheidt, Tandem oxidation of allylic and benzylic alcohols to esters catalyzed by N-heterocyclic carbenes, Org. Lett., 2007, 9, 371–374 Search PubMed.
- S. Iwahana, H. Iida and E. Yashima, Oxidative Esterification, Thioesterification, and Amidation of Aldehydes by a Two–Component Organocatalyst System Using a Chiral N–Heterocyclic Carbene and Redox–Active Riboflavin, Chem. – Eur. J., 2011, 17, 8009–8013 Search PubMed.
- E. Maciá, The Role of Phosphorus in Chemical Evolution, Chem. Soc. Rev., 2005, 34, 691–701 RSC.
- K. Fourmy, D. H. Nguyen, O. Dechy-Cabaret and M. Gouygou, Phosphole-Based Ligands in Catalysis, Catal. Sci. Technol., 2015, 5, 4289–4323 RSC.
- J. L. Methot and W. R. Roush, Nucleophilic Phosphine Organocatalysis, Adv. Synth. Catal., 2004, 346, 1035–1050 CrossRef CAS.
- E. Vedejs, O. Daugulis and S. Diver, Enantioselective Acylations Catalyzed by Chiral Phosphines, J. Org. Chem., 1996, 61, 430–431 CrossRef CAS PubMed.
- E. Vedejs, O. Daugulis and N. Tuttle, Desymmetrization of meso-Hydrobenzoin Using Chiral, Nucleophilic Phosphine Catalysts, J. Org. Chem., 2004, 69, 1389–1392 CrossRef CAS PubMed.
- S. Mizuta, M. Sadamori, T. Fujimoto and I. Yamamoto, Asymmetric Desymmetrization of meso–1, 2-Diols by Phosphinite Derivatives of Cinchona Alkaloids, Angew. Chem., 2003, 115, 3505–3507 CrossRef.
- H. Aida, K. Mori, Y. Yamaguchi, S. Mizuta, T. Moriyama, I. Yamamoto and T. Fujimoto, Enantioselective Acylation of 1,2- and 1,3-Diols Catalyzed by Aminophosphinite Derivatives of (1S,2R)-1-Amino-2-Indanol, Org. Lett., 2012, 14, 812–815 CrossRef CAS PubMed.
- S.-S. Meng, Y. Liang, K.-S. Cao, L. Zou, X.-B. Lin, H. Yang, K. Houk and W.-H. Zheng, Chiral Phosphoric Acid-Catalyzed Enantioselective Desymmetrization of 2-Substituted and 2,2-Disubstituted 1,3-Diols via Oxidative Cleavage of Benzylidene Acetals, J. Am. Chem. Soc., 2014, 136, 12249–12252 CrossRef CAS PubMed.
- S.-S. Meng, W.-B. Tang and W.-H. Zheng, Catalytic Enantioselective Synthesis of Acyclic α-Tertiary Amines via Desymmetrization of 2-Substituted 2-Nitro-1,3-Diols, Org. Lett., 2018, 20, 518–521 CrossRef CAS PubMed.
- D.-Q. Cui, Y.-Q. Wang, B. Zhou and L.-W. Ye, Brønsted-Acid-Catalyzed Enantioselective Desymmetrization of 1, 3-Diols: Access to Chiral β-Amino Alcohol Derivatives, Org. Lett., 2023, 25, 9130–9135 CrossRef CAS PubMed.
- Y. Toda, T. Sakamoto, Y. Komiyama, A. Kikuchi and H. Suga, A Phosphonium Ylide as an Ionic Nucleophilic Catalyst for Primary Hydroxyl Group-Selective Acylation of Diols, ACS Catal., 2017, 7, 6150–6154 CrossRef CAS.
- S. Devari, M. A. Rizvi and B. A. Shah, Visible-Light-Mediated Chemoselective Oxidation of Benzylic Alcohols, Tetrahedron Lett., 2016, 57, 3294–3297 CrossRef CAS.
- Z.-X. He, B. Yin, X.-H. Li, X.-L. Zhou, H.-N. Song, J.-B. Xu and F. Gao, Photochemical Selective Oxidation of Benzyl Alcohols to Aldehydes or Ketones, J. Org. Chem., 2023, 88, 4765–4769 CrossRef CAS PubMed.
- S. Gazi and R. Ananthakrishnan, Bromodimethylsulfonium Bromide: A Potential Candidate for Photocatalytic Selective Oxidation of Benzylic Alcohols Using Oxygen and Visible Light, RSC Adv., 2012, 2, 7781–7787 RSC.
- Y.-M. Zhong, H.-C. Ma, J.-X. Wang, X.-J. Jia, W.-F. Li and Z.-Q. Lei, AlBr3·6H2O-Catalyzed Oxidation of Benzylic Alcohols, Catal. Sci. Technol., 2011, 1, 927–931 RSC.
- N. Y. S. Lam, J. Dhankhar, A. S. K. Lahdenperä and R. J. Phipps, Catalytic Enantioselective Hydrogen Atom Abstraction Enables the Asymmetric Oxidation of Meso Diols, J. Am. Chem. Soc., 2024, 146, 33302–33308 CrossRef CAS PubMed.
- I. Borthakur, A. Joshi, S. Kumari and S. Kundu, Metal-Free Visible-Light-Induced Oxidative Cleavage of C(sp3)–C and C(sp3)–N Bonds in Nitriles, Alcohols, and Amines, Chem. – Eur. J., 2024, 30, e202303295 CrossRef CAS PubMed.
- H. Luo, L. Wang, S. Shang, J. Niu and S. Gao, Aerobic Oxidative Cleavage of 1,2-Diols Catalyzed by an Atomic-Scale Cobalt-Based Heterogeneous Catalyst, Commun. Chem., 2019, 2, 17 CrossRef CAS.
- Y. Matsusaki, T. Yamaguchi, N. Tada, T. Miura and A. Itoh, Aerobic Photooxidative Cleavage of Vicinal Diols to Carboxylic Acids Using 2-Chloroanthraquinone, Synlett, 2012, 23, 2059–2062 CrossRef CAS.
- R. Zhu, G. Zhou, J. N. Teng, X. Li and Y. Fu, Metal–free Photocatalytic Aerobic Oxidative Cleavage of C-C Bonds in 1, 2-Diols, ChemSusChem, 2020, 13, 5248–5255 CrossRef CAS PubMed.
- A. Hu, Y. Chen, J.-J. Guo, N. Yu, Q. An and Z. Zuo, Cerium-Catalyzed Formal Cycloaddition of Cycloalkanols with Alkenes through Dual Photoexcitation, J. Am. Chem. Soc., 2018, 140, 13580–13585 CrossRef CAS PubMed.
- T. Niu, S. Chen, M. Hong, T. Zhang, J. Chen, X. Dong and B. Ni, Heterogeneous Carbon Nitride Photocatalyst for C–C Bond Oxidative Cleavage of Vicinal Diols in an Aerobic Micellar Medium, Green Chem., 2020, 22, 5042–5049 RSC.
- J. Schwarz and B. König, Visible-Light-Mediated C–C Bond Cleavage of 1,2-Diols to Carbonyls by Cerium Photocatalysis, Chem. Commun., 2019, 55, 486–488 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © the Partner Organisations 2025 |