
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, biological and pharmacokinetic characterization of a novel leucine ureido derivative as a multi-target anticancer agent†
Fangyuan Shia,
Qifu Xub,
Yingjie Zhang b,
Jiangying Cao*c,
Chunxi Liu*a and
Anchang Liu*a
b,
Jiangying Cao*c,
Chunxi Liu*a and
Anchang Liu*a
aDepartment of Pharmacy, Qilu Hospital of Shandong University, Jinan, Shandong 250012, China. E-mail: acleu@126.com; liuchunxi1985@163.com
bDepartment of Medicinal Chemistry, Key Laboratory of Chemical Biology (Ministry of Education), School of Pharmaceutical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, P. R. China
cSchool of Pharmacy, Shandong University of Traditional Chinese Medicine, Jinan, Shandong 250355, P. R. China. E-mail: 60030103@sdutcm.edu.cn
First published on 10th February 2025
Abstract
Previously, a novel series of leucine ureido derivatives containing the 1,2,3-triazole moiety were identified and validated as potent aminopeptidase N inhibitors with marked in vitro and in vivo antitumor potencies. Moreover, synergistic anti-proliferation effects against tumor cells were found when used in combination with 5-Fluorouracil (5-FU). Herein, a novel leucine ureido derivative (compound 3) was synthesized by coupling cytotoxic agent 5-FU with leucine ureido derivatives containing the 1,2,3-triazole moiety via esterification. The biological activity evaluation showed that compound 3 exhibited more potent in vitro anti-proliferative, anti-metastatic, anti-angiogenic activities than the positive control bestatin. Furthermore, it was observed that compound 3 was very stable in simulated gastric fluid, while slowly cleaved in simulated intestinal fluid. In vivo pharmacokinetic study displayed that compound 3 was absorbed quickly after oral administration in rats and maintained in vivo for a long time, but exhibited poor oral bioavailability. Generally speaking, compound 3 is a promising lead for further development of more potent analogs as anticancer agents.
1. Introduction
Aminopeptidase N (APN; CD13; EC 3.4.11.2) belonging to the M1 family of the MA clan of peptidase is a Zn2+ dependent membrane-bound exopeptidase,1,2 which consist of a short cytoplasmic domain, a single transmembrane part and a large extra-cellular domain.3 As a moonlighting protein, APN presents multiple biological functions, such as enzymatic activity, antigen presentation and the receptor for some viruses.4 It was reported that APN was associated with the tumor migration, invasion, metastasis and angiogenesis.5–7 Moreover, APN is a functional marker of semi-dormant liver cancer cells which are responsible for chemotherapy resistance and cancer relapse.8Due to the important role of APN in tumors, APN inhibitors (APNIs) had caught the attention of scientists. So far, many APN inhibitors have been reported.9,10 Bestatin, containing an AHPA skeleton, has displayed diverse biological activities, such as anti-angiogenic, anti-metastatic, and immunomodulatory effects.11,12 5-Fluorouracil (5-FU, compound 1), a pyrimidine fluoride derivative, presents inhibitory activities against various carcinomas. However, it is limited in clinical application due to the side effects and shortcomings, such as marrow toxicity, short plasma half-life, and poor tumor selectivity.13 Therefore, the N1 and N3 positions of 5-FU were usually modified with biodegradable linkers to generate prodrugs to improve the efficacy and reduce toxicity.14,15
In the previous work of our group, we synthesized a number of different series of APN inhibitors, of which the leucine ureido derivatives with the 1,2,3-triazole moiety (compound 2) exhibited excellent APN inhibitory potency and promising in vitro and in vivo anti-angiogenic and anti-metastatic effects.16,17 Notably, when combined with 5-fluorouracil (5-FU), synergistic anti-proliferation effects against several tumor cell lines were exhibited and no significant systemic toxicity was found in a mouse hepatoma H22 tumor transplant model.16
According to the multi-target drug design approach,18,19 we designed a new leucine ureido derivative (compound 3, Fig. 1) by coupling 5-FU moiety with the carboxyl group of leucine ureido derivatives containing the 1,2,3-triazole group. In detail, the ester linker was liable and could be cleaved by the esterase enzyme. Moreover, the hydroxymethyl-5-FU (compound 4) could be converted to 5-FU via an extremely fast process. Therefore, the hybrid drug was supposed to release cytotoxic agent 5-FU and compound 5 by the enzyme-catalyzed metabolic reactions. In addition, compound 3 might have enhanced antitumor activity due to the aforementioned synergistic anti-proliferation effects of 5-FU combined with the leucine ureido derivatives against tumor cells.16 Thereby, the in vitro anti-proliferative activities, stability and in vivo pharmacokinetic properties of compound 3 are reported in this paper.
 | ||
| Fig. 1 Design strategy and proposed degradation pathway for the target compound. | ||
2. Results and discussion
2.1. Chemistry
The target compound 3 is synthesized following the routes in Schemes 1 and 2. As shown in Scheme 1, compound 6 reacted with triphosgene to generate isocyanate, which was then immediately reacted with propargylamine to yield the ureido derivative 7. Then, NH2OK transformed the methyl ester group of 7 to hydroxamate moiety to give the key intermediate 8. | ||
| Scheme 1 Reagents and conditions: (a) triphosgene, NaHCO3, DCM/H2O, 0 °C, 1.5 h; (b) propargylamine, TEA, DCM, 25 °C, 12 h; (c) NH2OK, MeOH, 25 °C, 0.5 h. | ||
 | ||
Scheme 2 Reagents and conditions: (a(i)) 10% HCl, NaNO2, 0 °C, 0.5 h, (ii) NaN3, 0 °C, 0.5 h; (b) NaOH, H2O, MeOH, 25 °C, 3 h; (c) 37% oxymethylene, 70 °C, 2 h; (d) PyBOP, Et3N, DCM, THF, 24 h; (e) 8, CuSO4·5H2O, sodium ascorbate, DMSO/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 25 °C, 5 h. :1), 25 °C, 5 h. | ||
As shown in Scheme 2, activation of the amino groups of the aromatic amine 9 with sodium nitrite in 10% hydrochloric acid and then reacted with sodium azide led to the azide derivative 10, which was hydrolyzed with sodium hydroxide to obtain carboxyl derivative 11. 5-Fluorouracil (1) was reacted with 37% oxymethylene to give the hydroxymethylated intermediate 4, which was coupled with 11 via esterification of carboxyl and hydroxyl group to give intermediate 12. Finally, the target compound 3 was generated by coupling 12 with 8 via click chemistry.
2.2. Enzyme inhibitory activity of the target compound against porcine APN
The target compound 3 synthesized was firstly evaluated for the inhibitory activity against porcine APN with bestatin as the positive control. The results in Table 1 showed that the APN inhibitory activity of compound 3, with the IC50 value of 0.50 ± 0.2 μM, was over 8-fold more potent than the positive control bestatin (APN IC50 = 4.08 ± 0.35 μM).2.3. In vitro anti-proliferative activities of the target compound against tumor cells
Compound 3 was further evaluated in the anti-proliferation assay against six tumor cell lines (MDA-MB-231, HEL, K562, HuH7, PLC/PRF/5 and HepG2). The results are listed in Table 2. Compared with other tumor cells, HEL and K562 cell lines were more sensitive to compound 3, with the IC50 values of 47.18 ± 19.29 μM and 25.58 ± 12.31 μM, respectively. Moreover, compound 3 presented better anti-proliferative potencies than bestatin against HEL and K562 cell lines.2.4. In vitro and ex vivo anti-angiogenesis assays
To evaluate the in vitro anti-angiogenic potency of compound 3, the human umbilical vein endothelial cells (HUVECs) tubular structure formation assay was performed. The results in Fig. 2 showed that compound 3 could inhibit the capillary tube formation at the concentration of 5 μM. Moreover, compared with 10 μM of bestatin, 5 μM of compound 3 presented less tubular structure formed by HUVECs, indicating the better anti-angiogenesis activity. | ||
| Fig. 2 Representative images of compound 3 on the formation of HUVECs capillary tube-like structure. | ||
It is well-known that the rat aortic ring model could simulate the in vivo angiogenesis environment better than the HUVECs tube formation model. The ex vivo rat thoracic aorta ring assay was used to further evaluate the anti-angiogenic activity of compound 3. The results are shown in Fig. 3. Compared with the control group, compound 3 could prevent the micro-vessel growth at the concentration of 5 μM. Moreover, similar with the results shown in the HUVECs tubular structure formation assay, 5 μM of compound 3 demonstrated much better inhibitory potency against micro-vessel growth relative to 10 μM of bestatin.
 | ||
| Fig. 3 Representative images of compound 3 on the micro-vessels growth of the rat aortic ring. | ||
2.5. Stability of the test compound in vitro
The stability of compound 3 in simulated gastric fluid and simulated intestinal fluid was conducted using a LC/MS/MS method. Briefly, the compound 3 was incubated at 37 °C for the appointed time. At predetermined time points, a sample of the mixture was removed, mixed, centrifuged and analyzed by LC/MS/MS. It was observed that compound 3 was very stable in simulated gastric fluid, while slowly cleaved in simulated intestinal fluid (Fig. 4). The results showed that compound 3 could release significant levels of compound 5 and 5-FU in vitro. Therefore, the anti-proliferative activities of compound 3 against tumor cells might be partly due to the synergistic effect of compound 5 and 5-FU.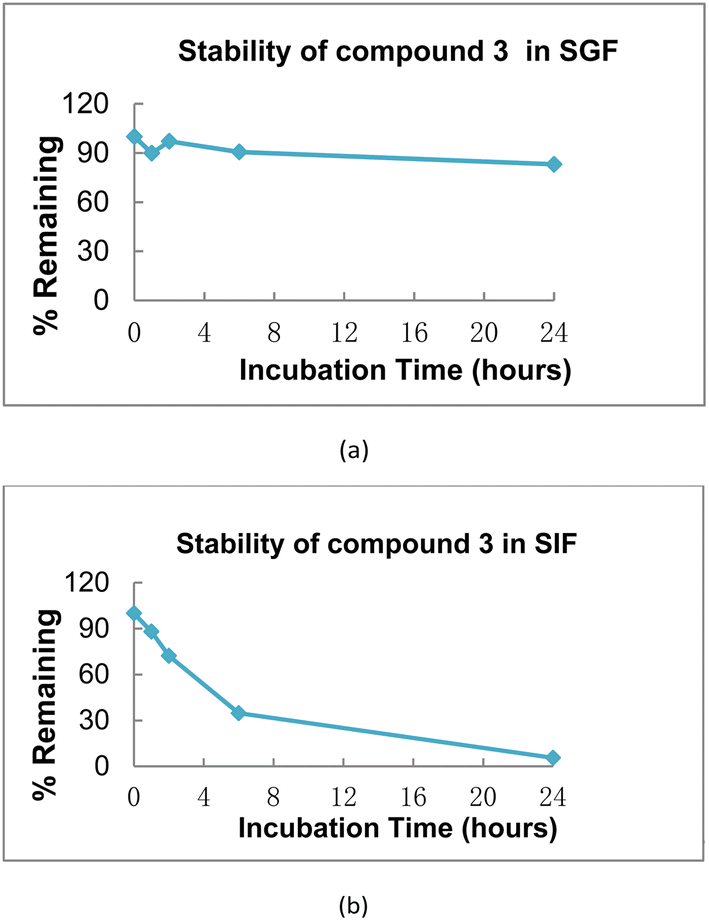 | ||
| Fig. 4 (a) Stability of compound 3 in simulated gastric fluid, points were achieved after 0, 1, 2, 6, 24 h, respectively; (b) stability of compound 3 in simulated intestinal fluid, points were achieved after 0, 1, 2, 6, 24 h, respectively. | ||
2.6. Metabolic stability in mouse liver microsomes (MLMs)
Metabolic stability assay of compound 3 with testosterone as the positive control, was performed in liver microsomes of mouse at the concentration of 1 μM. The metabolism of compound 3 in mouse liver microsomes was determined for phase I oxidative reactions. The results were listed in Table 3. Compound 3 was stable in MLMs with T1/2 = 94.9 min and CLint(liver) = 57.8 mL min−1 kg−1. The remaining amount of compound 3 at 60 min was 69.4% in MLMs, comparing with the group with no co-factor, which indicated that NADPH was involved in the metabolism of the target compound.2.7. In vivo PK studies
Given its promising results in the in vitro studies, the in vivo pharmacokinetic properties of compound 3 were assayed after a single intravenous (i.v.) or oral (p.o.) administration, respectively, which could be beneficial for the further preclinical studies.The degradation kinetics of compound 3 in vivo was evaluated in Sprague Dawley rats (Table 4 and Fig. 5), and LC-MS/MS analysis was conducted to quantitatively determine the plasma concentration of compound 3 and the corresponding metabolites. As shown in Table 4, it was revealed that the mean values of the maximum plasma concentration (Cmax) of compound 3 after i.v. and p.o. administration were 47400 ± 15838 ng mL−1 and 337 ± 158 ng mL−1, respectively. The areas under the plasma concentration–time curve extrapolated to the last time point (AUC0–t) after i.v. and p.o. administration were 16059 ± 6837 ng h mL−1 and 484 ± 119 ng h mL−1, respectively. Moreover, the plasma T1/2 values of compound 3 were 0.867 ± 0.387 h (i.v.) and 4.44 ± 4.1 h (p.o.), respectively. The time to Cmax (Tmax) was 0.417 h, indicating that compound 3 was absorbed rapidly after oral administration.
| PK parameters | Dosed (mg kg−1) | Cmax (ng mL−1) | Tmax (h) | AUC0–t (ng h mL−1) | AUC0–∞ (ng h mL−1) | CL (mL h−1 kg−1) | T1/2 (h) | F (%) |
|---|---|---|---|---|---|---|---|---|
| i.v. | 10 | 47400 ± 15838 |
0.0833 ± 0 | 16059 ± 6837 |
16067 ± 6840 |
737 ± 407 | 0.867 ± 0.387 | NA |
| p.o. | 50 | 337 ± 158 | 0.417 ± 0.144 | 484 ± 119 | 510 ± 99.3 | NA | 4.44 ± 4.1 | 0.64 |
 | ||
| Fig. 5 (a) Concentration–time curve of compound 3 after i.v. administration in rats; (b) concentration–time curve of compound 3 after p.o. administration in rats; (c) concentration–time curve of compound 5 after i.v. administration in rats; (d) concentration–time curve of compound 5 after p.o. administration in rats; (e) concentration–time curve of compound 5-FU after i.v. administration in rats; (f) concentration–time curve of compound 5-FU after p.o. administration in rats. | ||
As shown in Fig. 5, after administration of compound 3, its degradation products 5-FU and compound 5 were founded almost simultaneously. Therefore, it was speculated that compound 3 was rapidly metabolized to release 5-FU and compound 5. Although absorbed quickly in rats and maintained in vivo for a long time, the absolute oral bioavailability of compound 3 was determined to be 0.64%. Therefore, the attention could be focused on the optimal delivery system to increase the oral bioavailability of compound 3 and enhance the pharmacological efficacy.
3. Experimental
3.1. Chemistry: general procedures
All the commercially available materials were used without further purification otherwise noted. All reactions were monitored by thin-layer chromatography (TLC) on 0.25 mm silica gel plates (60 GF-254). The product spots were visualized by UV light, ferric chloride and iodine vapor. The purification of products was performed via column chromatography and recrystallization. Melting points were determined on an electrothermal melting point apparatus without correction. 1H NMR and 13C NMR spectra were determined on a Brucker DRX spectrometer with TMS as an internal standard. Chemical shifts were described as δ in parts per million and J in hertz. HRMS and ESI-MS were conducted by Shandong Analysis and Test Center.:2:40) to give compound 12 as a white solid (2.25 g, yield: 41%). mp: 168.4–169.8 °C. 1H NMR (400 MHz, DMSO-d6): δ 12.03 (s, 1H), 8.26 (d, J = 6.5 Hz, 1H), 7.82 (d, J = 7.8 Hz, 1H), 7.68 (t, J = 7.8 Hz, 1H), 7.44 (d, J = 8.1 Hz, 1H), 7.31 (t, J = 7.8 Hz, 1H), 5.80 (s, 2H).:10) to give compound 3 as a white solid (0.31 g, yield: 25%). mp: 124.3–125.8 °C. 1H NMR (400 MHz, DMSO-d6): δ 11.97 (s, 1H), 10.68 (s, 1H), 8.80 (s, 1H), 8.31 (s, 1H), 8.00–7.95 (m, 2H), 7.83 (t, J = 7.7 Hz, 1H), 7.72 (t, J = 7.7 Hz, 1H), 7.65 (d, J = 7.7 Hz, 1H), 6.44 (t, J = 5.7 Hz, 1H), 6.21 (d, J = 8.8 Hz, 1H), 5.63 (s, 2H), 4.35–4.24 (m, 2H), 4.11–4.05 (m, 1H), 1.58–1.50 (m, 1H), 1.35 (t, J = 7.2 Hz, 2H), 0.89–0.85 (m, 6H); 13C NMR (100 MHz, DMSO-d6): δ 169.9, 165.1, 157.7, 149.5, 146.7, 141.1, 138.8, 135.7, 133.9, 131.2, 130.3, 129.7, 129.4, 126.5, 124.1, 71.3, 49.6, 42.8, 35.2, 24.6, 23.2, 22.6. Retention time: 36.03 min, eluted with MeOH/water (0.1% formic acid), purity: 97.16%.3.2. Biological evaluation
000–12000 cells per well. After incubation for 4 h, compound sample (100 μL) which were previously diluted to the working concentrations (0.16/0.8/4/20/100 μmol L−1) with complete medium were added to the tested 96-well plates and incubated for a further 48 h. Subsequently, MTT (20 μL, 5 mg mL−1) solution was added into the test well, followed by the incubation for another 4 h. The plates were centrifuged at 800 rpm for 3 min. The supernatant was poured off and DMSO (200 μL) was added to dissolve the formed formazan. Finally, the mixture was shaken for 15 min and the absorbance values were measured using a plate reader at 570 nm (Varioskan, Thermo Fisher Scientific, Waltham, MA, USA).Simulated intestinal fluid was prepared as follows. Dissolved 0.136 g KH2PO4 and 0.2 g pancreatin and added sufficient H2O to make the total volume of 20 mL. The pH value of the test solution was adjusted to 6.8 ± 0.05.
To the stock solution of compound 3 was added simulated gastric fluid and simulated intestinal fluid, of which the final test concentration was 2 μM. The mixture was incubated at 37 °C and 600 rpm for the appointed time. After incubation of 60 min, 120 min, 360 min and 1440 min, respectively, to the test samples were added 400 μL of cold acetonitrile containing 200 ng per mL tolbutamide and labetalol (internal standard) immediately. Then, 200 μL of suspension was separated and mixed with 400 μL of cold acetonitrile containing 200 ng per mL tolbutamide and labetalol again. Subsequently, samples were subjected to centrifuge at 4000 rpm, 4 °C for 20 min. 200 μL of supernatant was used for the LC/MS/MS analysis to determine the remaining amount of test compound based on peak area ratio of analyte/IS. LC/MS/MS analysis was using a ACQUITY UPLC BEH C18 column (1.7 μm 2.1 × 50 mm). Compounds were eluted with water (containing 0.1% formic acid)/acetonitrile (containing 0.1% formic acid) over 60 min. The absorbance was measured at 282 nm, the flow rate was 0.5 mL min−1 and the quantity of injection was 20 μL.
| F (%) = (doseiv × AUCoral(0–∞))/(doseoral × AUCiv(0–∞)) × 100% |
4. Conclusions
In summary, according to the previous series of leucine ureido derivatives containing the 1,2,3-triazole moiety as APN inhibitors, we designed and synthesized the conjugated compound 3 following the multi-target drug design approach. Compound 3 exhibited more potent anti-proliferative activities against two human leukemic cell lines and anti-angiogenesis activity compared with the positive control bestatin. Furthermore, the preliminary stability of compound 3 revealed that the hybrid could release significant level of compound 5 and 5-FU in vitro, while NADPH was involved in the metabolism. Moreover, in vivo PK studies revealed that compound 3 was absorbed rapidly after oral administration, but the absolute oral bioavailability was approximately 0.64% in rats. Therefore, further attention can be focused on increasing the oral bioavailability and enhancing the pharmacological efficacy.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Conceptualization, Jiangying Cao, Chunxi Liu and Anchang Liu; data curation, Fangyuan Shi, Yingjie Zhang and Qifu Xu; formal analysis, Fangyuan Shi, Yingjie Zhang and Qifu Xu; investigation, Fangyuan Shi and Qifu Xu; methodology, Jiangying Cao, Chunxi Liu and Anchang Liu; project administration, Jiangying Cao, Chunxi Liu and Anchang Liu; supervision, Jiangying Cao, Chunxi Liu and Anchang Liu; writing – original draft, Fangyuan Shi; writing – review & editing, Jiangying Cao, Chunxi Liu and Anchang Liu.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 82003578) and Shandong Provincial Natural Science Foundation, China (No. ZR2020QH341).Notes and references
- N. D. Rawlings and A. J. Barrett, MEROPS: the peptidase database, Nucleic Acids Res., 1999, 27(1), 325–331 CrossRef CAS PubMed.
- C. Antczak, I. De Meester and B. Bauvois, Ectopeptidases in pathophysiology, Bioessays, 2001, 23(3), 251–260 CrossRef CAS PubMed.
- Y. Luan and W. Xu, The structure and main functions of aminopeptidase N, Curr. Med. Chem., 2007, 14(6), 639–647 CrossRef CAS PubMed.
- P. Mina-Osorio, The moonlighting enzyme CD13: old and new functions to target, Trends Mol. Med., 2008, 14(8), 361–371 Search PubMed.
- T. Tokuhara, N. Hattori, H. Ishida, T. Hirai, M. Higashiyama, K. Kodama and M. Miyake, Clinical significance of aminopeptidase N in non-small cell lung cancer, Clin. Cancer Res., 2006, 12(13), 3971–3978 Search PubMed.
- Y. Aozuka, K. Koizumi, Y. Saitoh, Y. Ueda, H. Sakurai and I. Saiki, Anti-tumor angiogenesis effect of aminopeptidase inhibitor bestatin against B16-BL6 melanoma cells orthotopically implanted into syngeneic mice, Cancer Lett., 2004, 216(1), 35–42 CrossRef CAS PubMed.
- L. Guzman-Rojas, R. Rangel, A. Salameh, J. K. Edwards, E. Dondossola, Y. G. Kim, A. Saghatelian, R. J. Giordano, M. G. Kolonin and F. I. Staquicini, et al., Cooperative effects of aminopeptidase N (CD13) expressed by nonmalignant and cancer cells within the tumor microenvironment, Proc. Natl. Acad. Sci. U. S. A., 2012, 109(5), 1637–1642 Search PubMed.
- N. Haraguchi, H. Ishii, K. Mimori, F. Tanaka, M. Ohkuma, H. M. Kim, H. Akita, D. Takiuchi, H. Hatano and H. Nagano, et al., CD13 is a therapeutic target in human liver cancer stem cells, J. Clin. Invest., 2010, 120(9), 3326–3339 CrossRef CAS PubMed.
- A. Mucha, M. Drag, J. P. Dalton and P. Kafarski, Metallo-aminopeptidase inhibitors, Biochimie, 2010, 92(11), 1509–1529 CrossRef CAS PubMed.
- S. A. Amin, N. Adhikari and T. Jha, Design of Aminopeptidase N Inhibitors as Anti-cancer Agents, J. Med. Chem., 2018, 61(15), 6468–6490 CrossRef PubMed.
- Y. Mishima, Y. Terui, N. Sugimura, Y. Matsumoto-Mishima, A. Rokudai, R. Kuniyoshi and K. Hatake, Continuous treatment of bestatin induces anti-angiogenic property in endothelial cells, Cancer Sci., 2007, 98(3), 364–372 CrossRef CAS PubMed.
- M. Lis, M. Szczypka, A. Suszko and B. Obminska-Mrukowicz, Influence of bestatin, an inhibitor of aminopeptidases, on T and B lymphocyte subsets in mice, Pol. J. Vet. Sci., 2011, 14(3), 393–403 CAS.
- Z. Y. Tian, G. J. Du, S. Q. Xie, J. Zhao, W. Y. Gao and C. J. Wang, Synthesis and bioevaluation of 5-fluorouracil derivatives, Molecules, 2007, 12(11), 2450–2457 CrossRef CAS PubMed.
- X. Pan, C. Wang, F. Wang, P. Li, Z. Hu, Y. Shan and J. Zhang, Development of 5-Fluorouracil derivatives as anticancer agents, Curr. Med. Chem., 2011, 18(29), 4538–4556 CrossRef CAS PubMed.
- N. Shimma, I. Umeda, M. Arasaki, C. Murasaki, K. Masubuchi, Y. Kohchi, M. Miwa, M. Ura, N. Sawada and H. Tahara, et al., The design and synthesis of a new tumor-selective fluoropyrimidine carbamate, capecitabine, Bioorg. Med. Chem., 2000, 8(7), 1697–1706 Search PubMed.
- J. Cao, J. Zang, X. Kong, C. Zhao, T. Chen, Y. Ran, H. Dong, W. Xu and Y. Zhang, Leucine ureido derivatives as aminopeptidase N inhibitors using click chemistry. Part II, Bioorg. Med. Chem., 2019, 27(6), 978–990 CrossRef CAS PubMed.
- J. Cao, C. Ma, J. Zang, S. Gao, Q. Gao, X. Kong, Y. Yan, X. Liang, Q. Ding and C. Zhao, et al., Novel leucine ureido derivatives as aminopeptidase N inhibitors using click chemistry, Bioorg. Med. Chem., 2018, 26(12), 3145–3157 CrossRef CAS PubMed.
- Z. Chen, L. Han, M. Xu, Y. Xu and X. Qian, Rationally designed multitarget anticancer agents, Curr. Med. Chem., 2013, 20(13), 1694–1714 CrossRef CAS PubMed.
- L. M. Espinoza-Fonseca, The benefits of the multi-target approach in drug design and discovery, Bioorg. Med. Chem., 2006, 14(4), 896–897 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03200d |
| This journal is © The Royal Society of Chemistry 2025 |