DOI:
10.1039/D4RA07274J
(Paper)
RSC Adv., 2025,
15, 6863-6874
Scrutinization of late first-row transition metals decorated octagonal boron (B8) ring complexes as single-atom catalysts for green hydrogen and oxygen production†
Received
10th October 2024
, Accepted 25th February 2025
First published on 3rd March 2025
Abstract
Hydrogen as fuel has gained large interest nowadays as a green energy source. Single-atom catalysis has emerged as a promising strategy for producing hydrogen. Herein, we investigated the late first row transition metals (TM = Co, Cu, Zn, Ni and Fe) adsorbed on eight-membered boron ring (TM@B8) as potential single-atom catalysts (SAC) towards hydrogen evolution reaction (HER) as well as oxygen evolution reaction (OER), aiming to identify less expensive electrocatalysts with high efficiency. Various properties including interaction energy (Eint), energies of frontier molecular orbitals (FMOs), natural bonding orbital (NBO) charges, total density of state (TDOS) spectra and non-covalent interaction (NCI) analyses of considered complexes are explored. These findings demonstrated that both pure TM@B8 and hydrogen-adsorbed TM@B8 complexes have both structural and electronic stability. The Co@B8 complex demonstrated a favorable Gibbs free energy of 0.16 eV toward HER under gaseous conditions. Fe@B8 showed better OER activity having overall ηOER of 1.14 eV. These outcomes show the promising potential of TM@B catalysts for both HER and OER processes.
1 Introduction
Boron's ability to form multiple bonds enables the synthesis of diverse boranes with unique geometric and electronic properties.1,2 Boranes have applications as chemical insulators,3 high-modulus fiber composites,4 semiconductors,5 refractory materials6 and high energy density fuels7 etc. Recently, boron nanoclusters (B6, B8, B38 and B40 etc.) are reported in literature and gained attention due to their thermodynamic stability, electron deficiency, large surface area, low density, high coordination number and covalent nature.8,9 B8 exhibits excellent semiconducting, optical, and catalytic properties. Doping of external metal further enhances the stability, magnetic properties, and electronic transmutation of resultant B8 complexes as evident from various literature reports.10,11
Single atom catalysis (SAC) is a rapidly advancing field across scientific disciplines, where metal atoms are deposited on a chemical surface to enhance catalytic efficiency.12–14 The atomic size of a metal plays a crucial role in determining catalyst performance. As the size of the metal particles decreases, the activity increases, alongside an increase in surface free energy that discourages aggregation into larger clusters.12 Sun et al. used atomic layer deposition (ALD) technique to experimentally synthesize single atom based platinum (Pt) doped graphene sheet and observed better catalytic performance than commercial Pt/C catalyst.15 Shui and coworkers synthesized rare earth metals (yttrium and scandium) doped carbon supported SACs for carbon and nitrogen reduction at room temperature.16
Recently, there has been growing interest toward hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) using single-atom catalysis.17,18 Hydrogen production through HER is an endergonic process, the sustainability of which depends on the suitable catalyst, reactants, and efficient energy sources. Scientists have assigned color codes to classify hydrogen based on the level of sustainability. Currently, small amounts of “low-carbon hydrogen” are produced through water electrolysis using nuclear power (purple hydrogen) or grid electricity (yellow hydrogen).19 The production of truly green hydrogen, however, requires a combination of renewable feedstocks, efficient SACs and efficient energy sources, ideally sunlight.20–23 SACs can lower the overpotential, larger activation barriers and slow kinetics processes. Extensive research has been carried out over the last few years to identify SACs having outstanding efficiency toward green HER. Experimentally, HER electrocatalysis is multi-step process including the transfer of two electrons on metallic surface. The whole reaction occurs under various pH conditions: alkaline,24 acidic25 and neutral media.26 Jousselme and coworkers synthesized non-noble metal single-atom catalysts from carbonized metal-doped ZIFs, exhibiting high activity and stability toward hydrogen evolution achieving −322 mV overpotential at −10 mA cm−2 in acidic media.27 HER under alkaline medium enhances metal stability and address safety and cost concerns associated with acidic conditions.28 Kim et al. described the improvement of HER activity in the presence of alkaline electrolyte.29 Hydrogen production via HER under extreme pH conditions faces severe challenges, and sustainable strategy to overcome those involves utilizing neutral or near-neutral electrolytes for HER. Sun et al. demonstrated theoretically as well as experimentally that nitrogen-doped porous Ni framework exhibited exceptional electrocatalytic performance toward HER under neutral conditions (pH 7) and achieved a current density of 10 mA cm−2 along with low overpotential of 64 mV. These substantial experimental reports demonstrate the effectiveness of transition metal-based SACs toward HER.
Experimental approaches to validate HER activity of catalysts include valence band photoelectron spectroscopy, electrochemistry, and advanced spectroscopic techniques. Theoretical concepts such as adsorption free energy, microkinetic models, volcano plots, and d-band centers are widely employed for the (semi)quantitative evaluation of HER electrocatalysts.30 Pt is the benchmark HER electrocatalyst due to its high exchange current density in one direction (j0) at equilibrium potential and low Tafel slope.31 According to Sabatier principle, the HER free energy diagram evaluates H* adsorption/desorption via ΔGH*,32 which should ideally approach zero for maximum reaction rate (this ideal Tafel step facilitates both adsorption and desorption of hydrogen). More than zero value of ΔGH* represents weak hydrogen adsorption (Volmer step) and less than zero represents strong hydrogen adsorption (Heyrovsky step).33 First-principle calculations allow the construction of HER free-energy diagrams, revealing the thermodynamic favorability of specific reaction pathways on various catalyst surfaces. A correlation between experimental j0 and computed ΔGH* is observed as a “volcano curve,” correlating catalyst surface properties to HER kinetics.34 DFT studies reveal that in MoS2, only the S–Mo–S edge sites are active for H* adsorption, with [1010] Mo edges showing ΔGH* comparable to Pt.35 Electrochemical and STM studies validate this, demonstrating j0 proportionality to edge length rather than basal plane area.36 Similarly, g-C3N4 with nitrogen-doped graphene exhibits higher j0 along with lower overpotential (η) and near-zero ΔGH*, enhancing HER activity.37 In addition to the free energy level of each reaction step (reactant, intermediates, and final product), the possible reaction barriers can also affect the overall reaction rate. The calculation of reaction barriers for the Tafel step at both equilibrium potential and certain overpotential are achievable just by including ne and η for each step (ne denotes the electron numbers carried by the electrode surface and η is the overpotential of the electrode). Such insights are critical for designing and optimizing electrocatalysts by tuning electronic structure and surface chemistry.
Herein, we designed and executed DFT based work to study the late first row transition metals (Zn, Cu, Co, Ni, Fe) doped B8 nanoclusters as single atom catalysts (SACs) toward HER (see Fig. 1). We hope B8 nanocluster with first row transition metals can act as better road map toward SACs for HER. Besides HER, oxygen evolution reaction (OER) potential is also investigated using the same transition metals doped B8 SACs as in literature there are number of SACs are also used to investigate their OER activity.38–40
 |
| | Fig. 1 Schematic diagram showing the proposed mechanism of HER. | |
2 Computational methods
The structures are built in GaussView 5.0 software,41 and subsequently simulation are processed in Gaussian 09 software.42 By building, optimizing, and computing the molecular model, key properties such as energy, bond lengths, charge distribution, atomic and molecular orbitals can be obtained. Gaussian simulations are highly powerful and widely used for calculating molecular electronic structures and spectral characteristics. ωB97XD functional of DFT and Pople's 6-31+G(d,p) basis set are used in these calculations. The energy minima structures of isolated B8 and selected transition metals (Zn, Cu, Co, Ni, Fe) doped B8 complexes are obtained from these calculations both in gas phase and solvent phase (water). Interaction energy (Eint) and Gibbs free energy are also calculated for the validation of thermodynamic stability of the complexes under neutral conditions as well as under aqueous conditions. Eint of all designed complexes is calculated by using the following eqn (1):| | |
Eint = E(B8+TM) − (EB8 + ETM)
| (1) |
Here, E(B8+TM), EB8 and ETM are the notations for the energy of transition metal (TM = Zn, Cu, Co, Ni, Fe) doped B8, the energy of pure B8 and the energy of TM, respectively. The energy gap (EH–L), between the highest occupied and the lowest unoccupied molecular orbitals is calculated by using eqn (2).
The total density of states (TDOS), non-covalent interactions index (NCI) and natural bond orbital (NBO) analyses are studied at ωB97XD/6-31+G(d,p) method. The following half reaction is considered to study the production of hydrogen (eqn (3)).
| |
 | (3) |
Zero-point corrected energies (ΔEZPE) are used for the estimation of change in Gibbs free energy (ΔGH) and change in enthalpies (ΔEH). These parameters (ΔGH, ΔEH ΔEZPE) at absolute temperature (at 298.15 K and 1 atm pressure) are calculated through eqn (4)–(6):
| | |
ΔGH = ΔEH + ΔEZPE − TΔSH
| (4) |
| |
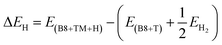 | (5) |
| |
 | (6) |
We also examined the oxygen evolution reaction (OER) through four proton-coupled electron transfer mechanism as shown below as steps (I)–(IV):
| | |
TM@B8 + H2O → TM@B8–OH + H+ + e−
| (I) |
| | |
TM@B8–OH → TM@B8–O + H+ + e−
| (II) |
| | |
TM@B8–O + H2O → TM@B8–OOH + H+ + e−
| (III) |
| | |
TM@B8–OOH → TM@B8 + O2 + H+ + e−
| (IV) |
The variation in Gibb's free energy values of the PCET steps (I)–(IV) is calculated by using eqn (7)–(10) to estimate further the overall OER potential.
| | |
ΔGI = GTM@B8–OH + 1/2GH2 − (GTM@B8 + GH2O)
| (7) |
| | |
ΔGII = GTM@B8–O + 1/2GH2 − GTM@B8–OH
| (8) |
| | |
ΔGIII = GTM@B8–OOH + 1/2GH2 − (GTM@B8–O + GH2O)
| (9) |
| | |
ΔGIV = GTM@B8 + GO2 + 1/2GH2 − GTM@B8–OOH
| (10) |
One of the important steps of the PCET steps is the one having highest Gibb's free energy is known as potential determination step (PDS). The PDS is used to estimate the chemical kinetics of the chemical processing, and it is directly related to overpotential (η). The η of OER is calculated by the below given equation:
EPDS and E° are denoted by electrochemical potential of PDS and standard electrochemical potential of PDS for the total OER at 298.15 K and 1 atm pressure.
3 Results and discussion
3.1 Geometries and stabilities of pure B8 and TM@B8 complexes
The energy minima structure of B8 consists of an eight-member boron ring having 1.55 Å average B–B bond length. We selected three active sites of B8 nanocluster for transition metals doping including top of B8 ring, middle of B–B bond and top of B atom (see Fig. 2).
 |
| | Fig. 2 Optimized geometry of B8 showing the selected sites (B8-top, bond between B–B and top of the B8) for transition metals (TM) doping. | |
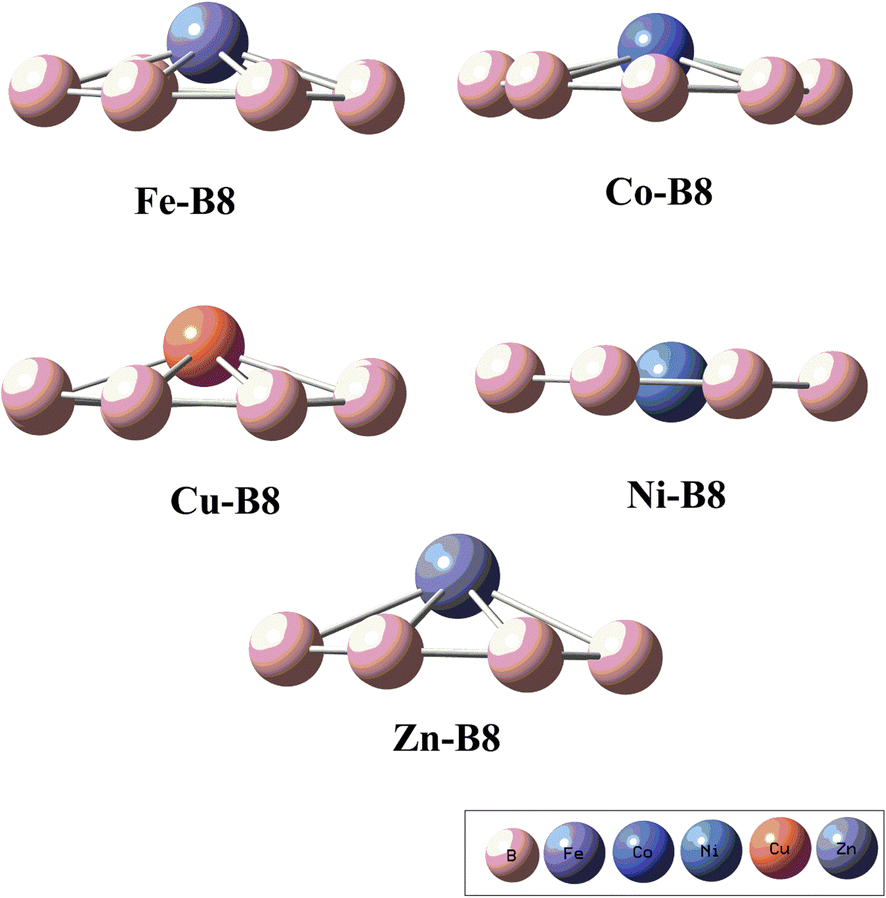
Initially considered TM are doped on all considered sites shown in Fig. 2, but after optimization of TM@B8 complexes, the stable orientation is only obtained is the top central position of B8 nanocluster (B8-top), as given in Fig. 3. The B–B bond length of B8 is changed after interacting with TM. The B–B bond length in Fe@B8, Co@B8, Ni@B8, Cu@B8 and Zn@B8 complexes is 1.56, 1.56, 1.57, 1.55 and 1.57 Å, respectively. We also estimated the interaction distance between boron ring and adsorbed transition metals (DTM–B8). The DTM–B8 in Fe@B8, Co@B8, Ni@B8, Cu@B8 and Zn@B8 is 2.17, 2.09, 2.06, 2.13 and 2.22 Å, respectively. The interaction distance between TM and B8 is noticeably decreased as the interactive forces are operational between the two species.
 |
| | Fig. 3 The optimized geometries of transition metals adsorbed on B8 (TM@B8, TM = Zn, Cu, Co, Ni, Fe). | |
The three lowest spin states of each complex are analyzed to identify the most stable spin multiplicity (see Table 1 for relative energies). The most stable states for Zn@B8 and Fe@B8 are triplet. Singlet is observed as the most stable state for Cu@B8, doublet for Co@B8 and quartet for Ni@B8. Compared to the triplet state of Fe@B8, the quintet and singlet states of this complex have relative energies 15.68 and 17.51 kcal mol−1 less stable than the triplet state, respectively. The most stable spin state for Co@B8 is the doublet, its quartet and sextet states have relative energy of 17.83 and 36.96 kcal mol−1, respectively compared to doublet spin state. For Ni@B8, the quartet state is the most stable one. The sextet and the doublet states have relative energies of 22.91 and 24.66 kcal mol−1 than the quartet one, respectively. In the case of Cu@B8, the singlet spin state is highly stable. Triplet and quintet spin states have relative energies of 13.53 and 63.02 kcal mol−1 as compared to singlet. In Zn@B8 complex, the triplet spin state has the highest stability compared to singlet and quintet spin states, those have higher energies of 15.59 and 57.27 kcal mol−1 than the triplet state.
Table 1 The spin multiplicity and relative energies (Erel, in kcal mol−1) of the different spin states for transition metals (TM) adsorbed on B8
| Properties |
Spin states |
Cu@B8 |
Zn@B8 |
Co@B8 |
Ni@B8 |
Fe@B8 |
| Multiplicities |
Most stable state |
Singlet |
Triplet |
Doublet |
Quartet |
Triplet |
| 2nd stable state |
Triplet |
Singlet |
Quartet |
Sextet |
Quintet |
| 3rd stable state |
Quintet |
Quintet |
Sextet |
Doublet |
Singlet |
| Erel |
Most stable state |
0.00 |
0.00 |
0.00 |
0.00 |
0.00 |
| 2nd stable state |
13.53 |
15.59 |
17.83 |
22.91 |
15.68 |
| 3rd stable state |
63.02 |
57.27 |
36.96 |
24.66 |
17.51 |
Thermodynamic feasibility of all complexes is estimated through interaction energy (Eint) calculation. All TM@B8 complexes have higher Eint, demonstrating the suitability of TM adsorption on the B8. Co@B8 is the most stable (−0.32 eV), followed by Fe@B8 (−0.26 eV), Ni@B8 (−0.25 eV), Cu@B8 (−0.21 eV), and Zn@B8 (−0.15 eV) (see Table 2). The Eint value of complexes under aqueous conditions are −0.30, −10.91, −8.92, −6.29 and −0.17 eV for Fe@B8, Co@B8, Ni@B8, Cu@B8, Zn@B8 respectively (see Table S1, ESI†). The negative value of interaction energy reflects the thermodynamic feasibility of complexation of consider transition metals with B8. The shorter interaction distance between Co and B8 in Co@B8 complex is evident of its highest stability, which is further proved from Eint of this complex (−0.32 eV). The larger interaction distance (2.22 Å) between Zn and B8 is in Zn@B8 complex shows the weak complexation, which is supported by low Eint of this complex. The Eint result of our complexes is similar to the investigation on various 2D nanosheets used for the electrochemical process of the HER.43
Table 2 NBO charge of TM in each complex (QTM in |e|), interaction distance between TM & eight-member boron ring (DTM–B in Å), energy gap (Egap in eV), the energies of LUMOs (ELUMO in eV), the energies of HOMOs (EHOMO in eV), interaction energy of single transition metal adsorbed eight-member boron complexes (Eint in eV) and interaction distance between boron & boron of the boron ring (DB–B in Å) of TM adsorbed eight membered boron complexes (TM@B8, TM = Zn, Cu, Co, Ni, Fe)
| System |
QTM |
DTM–B |
EH–L |
ELUMO |
EHOMO |
Eint |
DB–B |
| B8 |
0 |
— |
8.71 |
0.41 |
−8.29 |
— |
1.55 |
| Fe@B8 |
−0.48 |
2.17 |
5.84 |
−2.61 |
−8.45 |
−0.26 |
1.56 |
| Co@B8 |
0.10 |
2.09 |
5.74 |
−2.68 |
−8.42 |
−0.32 |
1.56 |
| Ni@B8 |
−0.28 |
2.06 |
5.36 |
−2.53 |
−7.89 |
−0.25 |
1.57 |
| Cu@B8 |
0.35 |
2.13 |
5.07 |
−2.93 |
−8.00 |
−0.21 |
1.55 |
| Zn@B8 |
0.74 |
2.22 |
7.32 |
−0.83 |
−8.15 |
−0.15 |
1.57 |
3.2 Electronic properties of pure B8 and TM@B8 complexes
Frontier molecular orbitals (FMOs) analysis is employed to assess the electronic characteristics of B8 and TM@B8 complexes (for values see Table 2). The EH–L of the pure B8 nanocluster is 8.71 eV, which decreases after doping with transition metals. After doping with transition metals, the EH–L for and Zn@B8, Cu@B8, Ni@B8, Co@B8 and Fe@B8 complexes is 7.32, 5.07, 5.36, 5.74 and 5.84 eV, respectively. The EH–L values indicate the semi-conductor nature of TM@B8 complexes and indicate their suitability as electrocatalyst toward HER. The EH–L is decreased due to change in the energies of HOMO and LUMO orbitals. The lowest EH–L of 5.07 eV is seen for Cu@B8 complex, which indicates the increase HOMO energy and decrease of LUMO energy compared to pure B8. Similar behavior is seen for the other complexes except Fe@B8 and Co@B8 complexes, where HOMO energy is increased after complexation. Isodensity in HOMO orbital is located on the bonds between boron atoms and the interaction sites of the boron ring with transition metals, whereas in LUMO orbitals are located primarily on the individual boron atoms of the B8 and to a lesser extent on transition metals (see Fig. S1, ESI†).
Natural bond orbital (NBO) charge analysis revealed the charge transfer between transition metal and B8. NBO charge value on transition metals confirm the direction of charge transfer in B8 complexes (Table 2). NBO charge value on Co, Cu and Zn is 0.10, 0.35 and 0.74|e| in Co@B8, Cu@B8 and Zn@B8 complexes, indicative of transfer of electronic density from metal toward B8. In Fe@B8 and Ni@B8 complexes, the negative charge values on Fe and Ni indicating the shifting of electronic density from B8 surface towards the transition metals. The electronegativity difference of Fe and Co relative to boron is more, which is responsible for withdrawing the electrons from boron and metals act as electron acceptors. Wu and colleagues also observed negative charges during the investigation of HER activity of transition metal doped iron sulphide complexes.44 Janjua also observed the similar trend of NBO charges while working on transition metals adsorbed Mg12O12 nanocages.45
Electron density distribution (EDD) analysis is performed to observe qualitative indication of charge transfer between TM and B8. Two different colors (cyan blue and purple) as shown in Fig. 4 are indicative of charge transfer. In Fe@B8 and Ni@B8 complexes, purple color on B8 represents the charge transfer from the B8 toward the transition metals. The cyan blue color surface on the interacting site of the TM (Fe and Ni) illustrates the acceptance of charge by TM. In Co@B8, Cu@B8 and Zn@B8 complexes, purple color is on TM (Co, Cu and Zn) represent the charge transfer from the transition metals toward the B8. The cyan blue surface on the interacting site of B8 illustrates the acceptance of charge by B8.
 |
| | Fig. 4 Graphics of EDD analysis of TM@B8 (TM = Zn, Cu, Co, Ni, Fe) complexes. | |
3.3 Geometric and electronic properties of hydrogen adsorbed TM@B8 complexes
For analyzing the HER activity atomic hydrogen (H) is adsorbed on each TM@B8 complex and optimized geometries are shown in Fig. 5. Both the transition metal and boron act as cationic species, inducing polarity in nearby atoms which stabilizes the interaction species. The interaction distance between hydrogen and TM is 1.48 Å in H/Fe@B8, 1.44 Å in H/Co@B8, 2.06 Å in H/Ni@B8, 2.09 Å in H/Cu@B8, and 1.69 Å in H/Zn@B8 complexes (see in Table 3). The short bond distance i.e. 1.44, 1.48 and 1.69 Å in H/Co@B8, H/Fe@B8, and H/Zn@B8 complexes confirmed the more effective hydrogen adsorption. In H/Ni@B8 and H/Cu@B8 complexes, hydrogen is adsorbed on boron instead TM having bond distance of 1.35 and 1.19 Å, respectively.
 |
| | Fig. 5 The optimized geometries of hydrogen adsorbed TM@B8 complexes. | |
Table 3 NBO charge value on TM (QTM in |e|), LUMOs energy (ELUMO in eV), HOMOs energy (EHOMO in eV), the HOMO–LUMO gap (EH–L, in eV), NBO charge value on hydrogen (QH in |e|), bond distance between hydrogen & TM (DH–TM in Å) and boron & hydrogen atom (DB–H in Å), adsorption energy of hydrogen in TM@B8 (EH in eV) complexes and Gibb's free energy (ΔGH in eV) of hydrogen adsorbed TM@B8 (TM = Zn, Cu, Co, Ni, Fe) complexes
| System |
QTM |
ELUMO |
EHOMO |
EH–L |
QH |
DH–TM |
DB–H |
DB–B |
EH |
ΔGH |
| H@B8 |
— |
−1.24 |
−7.88 |
6.64 |
0.03 |
— |
1.52 |
1.38 |
−2.37 |
0.76 |
| H/Fe@B8 |
−0.70 |
−1.21 |
−8.56 |
7.35 |
0.14 |
1.48 |
2.67 |
1.56 |
0.02 |
0.42 |
| H/Co@B8 |
−1.34 |
−2.36 |
−8.47 |
6.11 |
0.40 |
1.44 |
2.66 |
1.56 |
−0.01 |
0.16 |
| H/Ni@B8 |
−0.10 |
−2.54 |
−7.96 |
5.42 |
0.20 |
2.06 |
1.35 |
1.56 |
−0.04 |
−1.23 |
| H/Cu@B8 |
0.31 |
−2.45 |
−8.40 |
5.96 |
0.07 |
2.09 |
1.19 |
1.59 |
−0.05 |
−1.13 |
| H/Zn@B8 |
0.57 |
−3.76 |
−8.05 |
4.29 |
−0.32 |
1.69 |
3.35 |
1.56 |
−0.02 |
−0.28 |
The Eint of hydrogen with isolated B8 is −1.82 eV. A variational trend of Eint is observed after H adsorption with TM@B8 (TM = Zn, Cu, Co, Ni, Fe) complexes. The Eint of H atom in H/Fe@B8, H/Co@B8, H/Ni@B8, H/Cu@B8, and H/Zn@B8 complexes is 0.02, −0.01, −0.04, −0.05 and −0.02 eV, respectively (see Table 3). The reasonable thermodynamic stability is seen for H/Cu–B8 complex (EH = −0.05 eV) which is also justified from the less bond distance of 1.19 Å between H and B atoms in respective complex. The interaction energy increases with decreasing interaction distance. You et al. worked on Ni doped phosphide, and they observed almost similar hydrogen adsorption energy trend.46 These results are also similar to results of transition metals adsorbed Mg12O12 complexes which were suggested as potential SACs toward HER.45 Afterward, these complexes are also optimized with hydrogen adsorption under neutral conditions. The EH values of H@B8 (−1.18 eV), H/Ni@B8 (−1.36 eV), H/Cu@B8 (−1.46 eV) and H/Zn@B8 (−0.05 eV) show the high thermodynamically stable of all the catalysts. Meanwhile, the EHOMO of H/Fe@B8 and H/Co@B8 are 0.03 and 0.15 eV, respectively (see Table S1, ESI†).
Variations in energies of frontier molecular orbitals in TM@B8 complexes is observed after hydrogen adsorption. HOMO energy in H/Zn@B8, H/Cu@B8, H/Ni@B8, H/Co@B8 and H/Fe@B8 complexes is −8.05, −8.40, −7.96, −8.47, and −8.56 eV, respectively. The energy of LUMO in H/Zn@B8, H/Cu@B8, H/Ni@B8, H/Co@B8 and H/Fe@B8 complexes is −3.76, −2.45, −2.54, −2.36 and −1.21 eV, respectively. The EH–L is increased after hydrogen adsorption in TM@B8 complexes except H/Zn@B8. The smallest EH–L is observed for H/Zn@B8 (4.29 eV), followed by H/Ni@B8 (5.42 eV), H/Cu@B8 (5.96 eV) and H/Co@B8 (6.11 eV) complexes (see Table 3). The EH–L of H/Fe@B8 is increased to 7.35 eV. A similar trend of change in EH–L gap was observed in TM doped MoSSe complexes by Deng et al.47 which justifies the validity of our results. EH–L g of H/Zn@B8 complex is the lowest one (4.29 eV) as compared to H/B8 complex. The isodensity in HOMO orbitals in H adsorbed complexes is located on the bonds between the boron and the interaction sites of the boron ring with the transition metals. The isodensity in LUMO orbitals is primarily concentrated on the boron atoms of the B8 ring, with small amounts on the transition metals and hydrogen atoms in each complex (see Fig. S2†).
The NBO charge on H in H/Fe@B8, H/Co@B8, H/Ni@B8 and H/Cu@B8 complexes is 0.14, 0.40, 0.20 and 0.07|e|, respectively indicating the shifting of electronic density from hydrogen toward TM@B8 complexes. The highest charge transfer of 0.40|e| is observed in H/Co@B8. In H/Zn@B8 complex, charge on H is −0.32|e|, which indicated the charge transfer toward the hydrogen atom. Specifically, 0.57, 0.31, −0.10, −1.34, and −0.70|e| charges are seen on the TMs of the H/Zn@B8, H/Cu@B8, H/Ni@B8, H/Co@B8, and H/Fe@B8 complexes (see Table 3). Co metal in H/Co@B8 has the highest NBO charge of −1.34|e|, while Ni metal in H/Ni@B8 has the lowest charge transfer (−0.10|e|) in H/Ni@B8. Variation in the NBO charge reflects the interactions between H and designed TM@B8 (T = Zn, Cu, Co, Ni, Fe) complexes. The interaction distance between Co metal and hydrogen is the shortest one compared to other complexes. This represents more interaction, which is also cleared from the highest negative charge on Co (−1.34|e|). The hydrogen attached to the Co has the higher positive charge (0.40|e|) which shows the charge transfer from hydrogen to Co metal in H/Co@B8 complex.
Graphics of EDD analysis showing qualitative indication of charge transfer between TM@B8 and hydrogen are shown in Fig. 4. In H/TM@B8 complexes, purple color on TM@B8 represents the charge transfer from the TM@B8 toward H. The cyan blue color surface on the interacting side of the H which illustrates acceptance of charge by H. particularly, in Fe@B8 Co@B8, Ni@B8 and Cu@B8 complexes, purple color on hydrogen represents the charge transfer from the hydrogen toward the TM@B8. The cyan blue color surface on the interacting side of the TM@B8 which illustrates acceptance of charge by TM@B8 (Fig. S3†).
Total density of states (TDOS) spectral analysis is performed to study the energy states of occupied and unoccupied molecular orbitals. The HOMOs and LUMOs are represented on the right and left sides of each spectrum, respectively (see Fig. 6 and S4†). New energy states are generated in all TM@B8 (TM = Zn, Cu, Co, Ni, Fe) complexes due to charge transfer from TM to B8 complexes. Furthermore, TDOS spectra verified the change in EH–L on the basis of the HOMOs and LUMOs energy states. Prominent variations in energy states of TDOS spectra of hydrogen-adsorbed TM@B8 complexes (H/TM@B8) are seen. Among all these complexes, the H/Zn@B8 complex exhibits a significant variation in the energy states, with the lowest energy gap (EH–L) of 4.29 eV. The peak intensity in spectrum indicates the extent of electronic contribution in each complex. The highest intensity of peaks is seen in H/TM@B8 complexes, followed by TM@B8 complexes and then least in pure B8 spectrum. This analysis depicts the higher extent of electronic contribution in H/TM@B8 complexes. The average electronic contribution is seen in TM@B8 complexes in comparison to H/TM@B8 complexes. The overlapping of the peaks in each spectrum illustrates the stronger electronic interactions between these interacting species in both TM@B8 and H/TM@B8 complexes. These results are very similar to findings of Kosar et al. work on HER of TM doped Si12C12 complexes.48
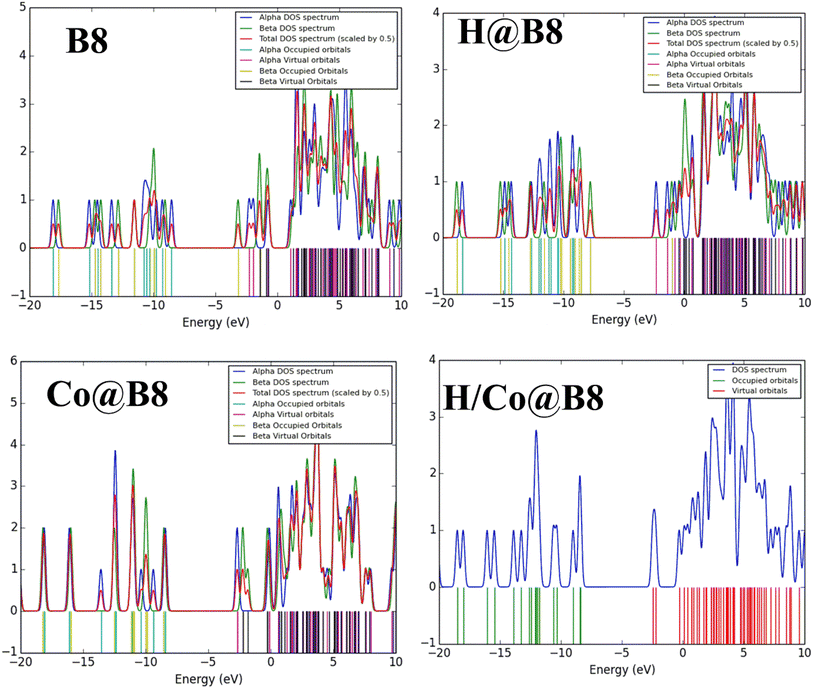 |
| | Fig. 6 Total density of states (DOS) spectra of pure B8 and H@B8, Co@B8 and H/Co@B8 complexes. | |
The nature of interactions is elucidated from non-covalent interaction (NCI) analysis through spotting low and high electronic density regions in complexes. Three-dimensional NCI and two-dimensional reduced density gradient (RDG) graphs of H/TM@B8 complexes, are shown in Fig. 7 and ESI Fig. S5.† 3D-NCI and 2D-RDG graphs interactions are represented by colorful patches and spikes, respectively. These colorful patches and spikes indicate the presence of different types of interactions. Based on this qualitative analysis in both types of NCI and RDG graphs; blue, red and green colors indicate strong hydrogen bonding, repulsive forces, and weak van der Waals forces. In our current study, the red color patches (3D-NCI) and spikes (2D-RDG graphs) show repulsion between the boron atoms of the pure B8 nanocluster. In TM@B8 and H/TM@B8 (TM = Zn, Cu, Co, Ni, Fe) complexes, the dominant factors are the blue and red colors in 3D-NCI and 2D-RDG graphs, respectively. Red color indicates steric repulsion between the boron atoms in the B8 nanocluster in both TM@B8 and H/TM@B8 (T = Zn, Cu, Co, Ni, Fe) complexes. While blue color indicates hydrogen bonding in H/TM@B8 (TM = Zn, Cu, Co, Ni, Fe) complexes. Green reflects the presence of noncovalent interactions in H/TM@B8 (TM = Zn, Cu, Co, Ni, Fe) complexes.
 |
| | Fig. 7 3D-RDG graph (right side) and 2D-NCI plots (left side) and of pure B8, H@B8, Co@B8 and H/Co@B8. | |
For quantitative analysis 2D-RDG graphs are used, the Laplacian of density (∇2ρ) is used to differentiate among various types of bonding. For covalent contacts, negative contributions predominate, resulting in a negative Laplacian value. For weaker noncovalent interactions, positive contribution is dominant in the interatomic region of the Laplacian. The negative sign of λ2 is a key indicator of interactions; thus, the sign of λ2 can distinguish between bonded interactions (λ2 < 0) and non-bonded interactions (λ2 > 0). The λ2 and the density are important parameters to distinguish various kinds of noncovalent interactions. Negative values between −0.020 to −0.035, shown in blue on the left side of the RDG graph, indicate strong hydrogen bonding. Positive values above 0.010, shown in red, indicate repulsive forces. The sign(λ2)ρ values in the range of −0.01 to −0.01, shown in green, indicate weak van der Waals forces in H/TM@B8 complexes. Both blue and red colors are seen in both 3D-NCI figures and 2D-RDG graphs illustrate strong hydrogen bonding and repulsion forces. The green spikes are only seen in the 2D-RDG graphs and represent weak van der Waals interactions in all H/TM@B8 (TM = Zn, Cu, Co, Ni, Fe) complexes. As we moved towards the 3D-RDG graphs of H/TM@B8 complexes, the more prominent blue, green and red spikes are observed indicating the stronger hydrogen bonding, repulsive and van der Waals interactions within each complex after hydrogen adsorption. These results are similar to Kosar et al. work on HER on TM doped Si12C12 complexes.48
3.4 Catalytic activity of TM@B8 complexes toward HER
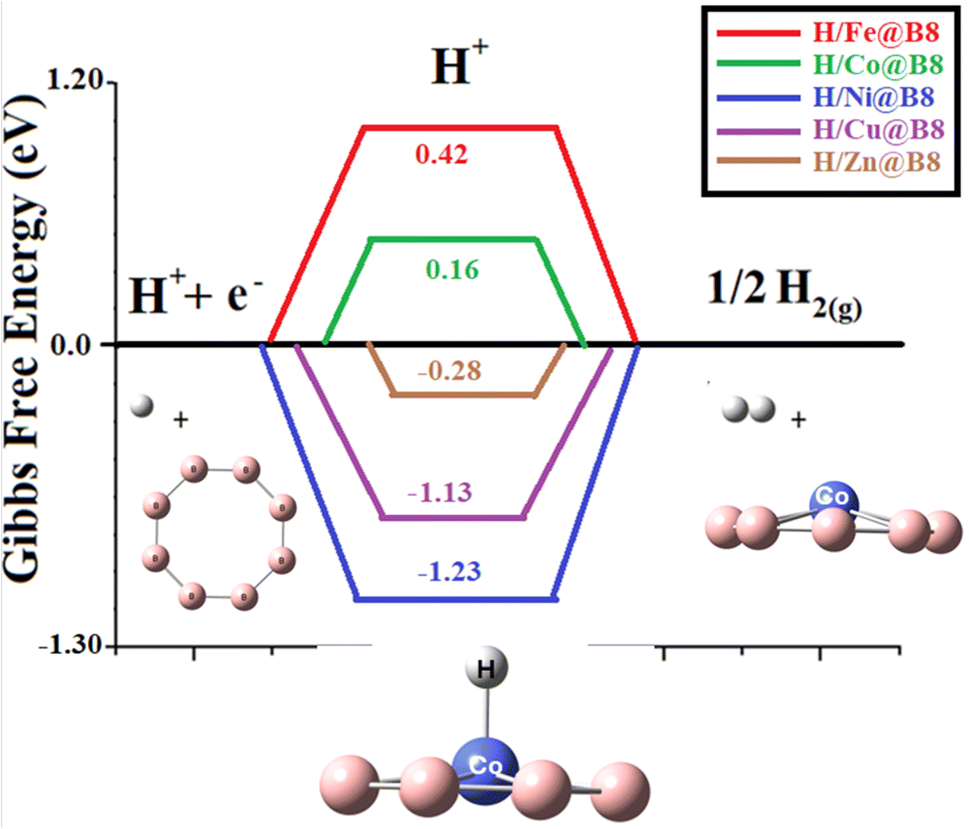
For an ideal HER catalytic activity, the ΔGH value should be closer to zero. The catalytic activity as single-atom catalysts (SACs) is evaluated by calculating the change in Gibbs free energy during HER. Doping of transition metals on the B8 nanocluster is supposed to enhance the catalytic efficiency of B8 toward hydrogen evolution reaction. The graphical comparison of change in Gibbs free energy of all designed catalysts is shown in Fig. 8. ΔGH values near zero indicate excellent performance for HER activity. Table 3 lists the ΔGH values for HER: −0.28, −1.13, −1.23, 0.16, and 0.42 eV for H/Zn@B8, H/Cu@B8, H/Ni@B8, H/Co@B8, H/Fe@B8 complexes, respectively. Overall, Co@B8 exhibits excellent HER activity with a ΔGH value of 0.16 eV, which is near zero. Ni@B8, with a ΔGH value of −1.23 eV, shows the lowest HER activity among the designed TM@B8 complexes.
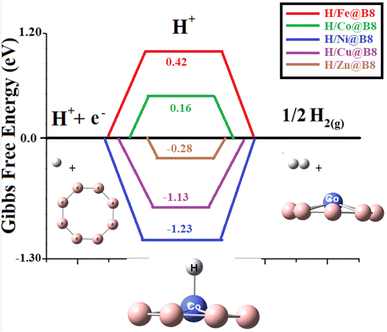 |
| | Fig. 8 Comparative graph of modelled HER catalysts (TM@B8, TM = Zn, Cu, Co, Ni, Fe) with calculated Gibb's free energies. | |
One of the factors that affects the change in Gibbs free energy is the interaction energy of hydrogen. The effective catalyst must have an optimal balance of hydrogen adsorption and desorption (Eint not so much high and not so low). Similarly, ΔGH value should also be optimal near to zero according to Tafel step. This is obeyed by Co@B8 having Eint of 0.01 eV and ΔGH* value of 0.16 eV. The Fe@B8 exhibits weak hydrogen adsorption and has Eint of 0.02 eV. Due to this reason its ΔGH is 0.42 eV which makes the adsorption process energetically unfavorable. Remaining complexes including Ni@B8, Cu@B8 and Zn@B8 show strong hydrogen adsorption as depicted from their Eint values of −0.04, −0.05 and −0.02. Their (Ni@B8, Cu@B8 and Zn@B8) ΔGH are also high and ranges from −0.28 to −1.23 which causes impeding desorption.49 Among all complexes, Co@B8 acts as an effective catalyst for the next generation HER process.50–52 This Gibb's free energy value is comparable to the reported values of various complexes in literature. A detailed comparison of this work is given with the literature in Table 4.
Table 4 Comparative analysis of our best designed single atom catalysts with reported SACs for HER activity
| Complexes |
ΔGH (eV) |
References |
| Fe@GDY |
0.22 |
54 |
| Fe@MO2CO2, Fe@Cr2CO2 |
0.16, −0.12 |
55 |
| Fe@SnO |
−0.44 |
56 |
| 1T-MoS2 |
0.07 |
57 |
| Co@graphene |
0.13 |
58 |
| Re@MoS2 |
−0.43 |
59 |
| Nb@MoS2 |
0.06 |
60 |
| GeP3 |
0.02 |
61 |
| NiPd–gCN |
−0.15 |
62 |
| MoSi2N4/MoSX (X = S, Se) |
∼0 |
63 |
| Ti@MoS2 |
−0.63 |
64 |
| Pt@GDY, Ni@GDY |
0.01, 0.46 |
65 |
| NiCoP on Co, P sites |
0.08, 0.12 |
66 |
| Co@B8 |
0.16 |
Current work |
HER catalytic activity of TM@B8 complexes is also investigated under neutral condition which reported as the best approach both experimentally and theoretically.53 The ΔGH values of HER under neutral conditions are −1.43, 0.82, 0.33, −1.09, −1.16, and 0.76 eV for H@B8, H/Fe@B8, H/Co@B8, H/Ni@B8, H/Cu@B8, H/Zn@B8 (see Table S1, ESI†). Overall, Co@B8 exhibits excellent HER activity with a ΔGH value of 0.33 eV in water as solvent. Cu@B8, with a ΔGH value of −1.16 eV, shows the lowest hydrogen adsorption among the designed TM@B8 catalysts.
3.5 Catalytic activity of TM@B8 toward oxygen evolution reaction (OER)
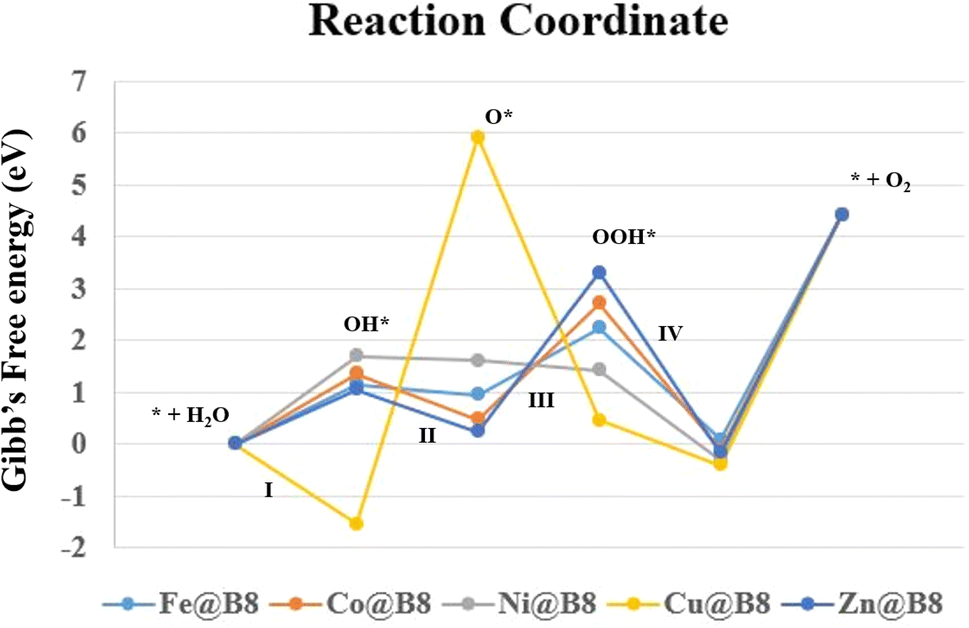
Oxygen evolution reaction (OER) potential of designed complexes is also investigated. OER is an essential process during the electrocatalysis of water splitting. Generally, OER proceeds through four steps proton coupled electron transfer (PCET) process. In PCET, three intermediates play a pivotal role including OH*, O* and OOH* with substrate. All the newly designed intermediates (OH*, O* and OOH* with B8) in this study are optimized at same ωB97XD with 6-31+G(d,p) using three lower spin states starting with default spin state. The most stable spin state of intermediate for each step is given in Table 5, and pictorial representation is given in Fig. 9. The rest of the results are given in Table S2 and Fig. S6 (ESI†). The change in Gibb's free energies of each PCET step (ΔGI − ΔGIV) is calculated to know about the kinetics of the OER. The change in Gibb's free energies is divided into equal values in four PCET steps for OER of an ideal catalyst. The total ΔG of PCET is 4.92 eV where the ΔG of each step is 1.23 eV. The variation in the thermodynamic stabilities between the intermediates of SACs causes variation in the Gibb's free energies and over potential of each PCET steps.67,68 In our current study, total ΔG is 4.42 eV and ΔG ((I)–(IV)) of each step is 1.10 eV. These computational values are in close agreement with experimental values which are accepted in literature.69,70 The change in Gibb's free energies of the total OER is the summation of the four important steps which differ from each other based on thermodynamic stability of the intermediates. Some steps are endergonic in nature and others are exergonic in nature. Although it is desirable to have a reaction with endergonic behavior. Gibb's free energies values are used to calculate the overpotential values (see in Table 5). The higher Gibb's free energies cause increase in overpotential which is the main cause of high potential in OER.65,71 Table 5 illustrates that the desirable potential determination step (PDS) of Fe@B8, Co@B8, and Zn@B8 is step (III). PDS of Ni@B8 and Cu@B8 are (I) and (II), respectively. The best catalytic potential is observed for Fe@B8 with ΔGPDS and ηOER values of 2.24 eV and 1.14 V, respectively. Because the 1.14 eV of Fe@B8 is seen close to the 1.10 eV per step value. The same kind of results are seen in previously published literature.72
Table 5 The most stable spin states for the intermediates of OER, potential determining steps (PDS) along with respective Gibb's free energy change (ΔGmax in eV) and overpotential (ηOER in V)
| Catalysts |
Intermediates with stable spin states |
PDS |
ΔGmax |
ηOER |
| * + OH* |
* + O |
* + OOH |
| Fe@B8 |
Doublet |
Triplet |
Doublet |
(III) |
2.24 |
1.14 |
| Co@B8 |
Triplet |
Sextet |
Triplet |
(III) |
2.71 |
1.60 |
| Ni@B8 |
Quartet |
Quintet |
Quartet |
(I) |
1.69 |
0.59 |
| Cu@B8 |
Singlet |
Sextet |
Triplet |
(II) |
5.91 |
4.80 |
| Zn@B8 |
Doublet |
Quintet |
Doublet |
(III) |
3.30 |
2.20 |
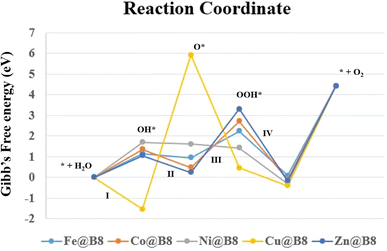 |
| | Fig. 9 Comparative graphs of Modelled OER catalytic activity of (TM@B8, TM = Zn, Cu, Co, Ni, Fe) with calculated Gibb's free energies. | |
Conclusively, Fe@B8 show best performance for OER with ΔGPDS and ηOER values of 2.24 eV and 1.14 V, respectively which is close to the 1.10 eV per step value of PCET. The Co@B8 exhibits excellent HER activity with a ΔGH value of 0.16 eV, which is near zero.
4 Conclusion
Late first row transition metal-adsorbed B8 complexes are investigated as single-atom catalysts (SACs) toward HER application. Various properties including interaction energy (Eint), energies of frontier molecular orbitals (FMOs), natural bonding orbital (NBO) charges, TDOS spectral analysis, and non-covalent interaction (NCI) of considered complexes are explored. These properties helped in describing the thermodynamic stability, electronic reactivity and types of bonding in TM@B8 complexes. The doping of TM with B8 nanocluster and further the H adsorption significantly improved stability of complexes. The Eint. of all H/TM@B8 (T = Zn, Cu, Co, Ni, Fe) complexes are observed in the range of 0.02 to −0.05 eV, indicating thermodynamic feasibility upon complexation. NBO calculations revealed charge transfer from the adsorbed transition metals to the B8 nanocluster. After hydrogen adsorption, NBO charges transfer to hydrogen and vice versa, highlighting the adsorption properties of hydrogen. The HOMO–LUMO energy gap for transition metal-adsorbed complexes ranges from 5.07–7.32 eV, but this gap reduces up to 4.29 eV, after hydrogen adsorption, confirming the improved conductivity of the TM@B8 complex. DOS spectra illustrated the energy states of the molecular orbitals and the extent of electronic contribution in these complexes. NCI plots further explained the existence of hydrogen bonding, noncovalent interactions, and steric repulsion in H/TM@B8 complexes. Gibbs free energy and entropy calculation are instrumental in explaining HER activity. Conclusively, The Co@B8 exhibits excellent HER activity with a ΔGH value of 0.16 eV, which is near zero in gas phase. Fe@B8 show the best performance for OER with ΔGPDS and ηOER values of 2.24 eV and 1.14 V, respectively which is close to the 1.10 eV per step value of PCET. These outcomes show the promising potential of TM@B8 catalysts for both HER and OER process.
Data availability
The data that support the findings of this study are available from the corresponding author, [E-mail: mahmood@cuiatd.edu.pk (T. M.)], upon reasonable request.
Author contributions
Naveen Kosar: simulation, logical methodology, data analysis, writing the original manuscript. Tariq Mahmood: supervision, software, editing, review and finalizing the manuscript. Muhammad Arshad: data analysis, validation. Muhammad Imran: conceptualization, funding acquisition, project administration. Utkirjon Holikulov: resources, project administration, validation.
Conflicts of interest
The authors declare no competing and conflict of interests.
Acknowledgements
The authors acknowledge the Higher Education Commission (NRPU project; 20-16279/NRPU/HEC/2021-2020) of Pakistan. M. I. express appreciation to the Deanship of Scientific Research at King Khalid University Saudi Arabia through the research groups program under grant number R. G. P. 2/608/45. The experiments presented in this paper were carried out using the facilities of the Benefit Advanced AI and Computing Lab at the University of Bahrain {see https://ailab.uob.edu.bh} with support from Benefit Bahrain Company {see https://benefit.bh}.
References
- V. D. Yumatov, E. A. Il'inchik and V. V. Volkov, Russ. Chem. Rev., 2003, 72, 1011–1034 CrossRef CAS.
- R. N. Grimes, J. Chem. Educ., 2004, 81, 657 CrossRef CAS.
- M. Atiş, C. Özdoğan and Z. B. Güvenç, Int. J. Quantum Chem., 2007, 107, 729–744 CrossRef.
- J. Plesek, Chem. Rev., 1992, 92, 269–278 CrossRef CAS.
- O. Mishima, J. Tanaka, S. Yamaoka and O. Fukunaga, Science, 1987, 238, 181–183 CrossRef CAS PubMed.
- I. A. Howard and A. K. Ray, Z. Phys. D:At., Mol. Clusters, 1997, 42, 299–301 CrossRef CAS.
- M. Atis, C. Ozdosan and Z. B. Guvenc, Chin. J. Chem. Phys., 2009, 22, 380–388 CrossRef CAS.
- Z. Cui, C. Chen, Q. Wang, L. Zhao, M. Wang and Y. Ding, New J. Chem., 2020, 44, 17705–17713 RSC.
- W.-L. Li, X. Chen, T. Jian, T.-T. Chen, J. Li and L.-S. Wang, Nat. Rev. Chem, 2017, 1, 0071 CrossRef CAS.
- W.-J. Chen, M. Kulichenko, H. W. Choi, J. Cavanagh, D.-F. Yuan, A. I. Boldyrev and L.-S. Wang, J. Phys. Chem. A, 2021, 125, 6751–6760 CrossRef CAS PubMed.
- Y. Luo, F. Chen, H. Zhang, J. Liu and N. Liu, J. Org. Chem., 2023, 88, 15717–15725 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- Z. Chen, J. Liu and K. P. Loh, Acc. Mater. Res., 2023, 4, 27–41 CrossRef CAS.
- S. Wang, H. Gao, L. Li, K. S. Hui, D. A. Dinh, S. Wu, S. Kumar, F. Chen, Z. Shao and K. N. Hui, Nano Energy, 2022, 100, 107517 CrossRef CAS.
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng, D. Geng, M. N. Banis, R. Li, S. Ye, S. Knights, G. A. Botton, T.-K. Sham and X. Sun, Sci. Rep., 2013, 3, 1775 CrossRef.
- J. Liu, X. Kong, L. Zheng, X. Guo, X. Liu and J. Shui, ACS Nano, 2020, 14, 1093–1101 CrossRef CAS PubMed.
- L. Jin, C. Liu, D. Wang, M. Liu, T. G. Lee, S. G. Peera and X. Qi, Appl. Surf. Sci., 2023, 610, 155580 CrossRef CAS.
- A. Yu, N. Joshi, W. Zhang and Y. Yang, Advanced Sensor and Energy Materials, 2023, 2, 100061 CrossRef.
- S. Griffiths, B. K. Sovacool, J. Kim, M. Bazilian and J. M. Uratani, Energy Res. Soc. Sci., 2021, 80, 102208 CrossRef.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- A. V. Abad and P. E. Dodds, Energy Policy, 2020, 138, 111300 CrossRef.
- F. Zaccaria, G. M. Rodriguez, L. Rocchigiani and A. Macchioni, Front. Catal., 2022, 2, 892183 CrossRef.
- F. Abdelghafar, X. Xu, S. P. Jiang and Z. Shao, Mater. Rep.: Energy, 2022, 2, 100144 CAS.
- H. Meng, W. Zhang, Z. Ma, F. Zhang, B. Tang, J. Li and X. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 2430–2441 CrossRef CAS PubMed.
- A. K. Nayak, M. Verma, Y. Sohn, P. A. Deshpande and D. Pradhan, ACS Omega, 2017, 2, 7039–7047 CrossRef CAS PubMed.
- R. N. Dürr, E. Stéphan, J. Leroy, F. Oswald, B. Verhaeghe and B. Jousselme, ChemElectroChem, 2023, 10, e202300205 CrossRef.
- N. Mahmood, Y. Yao, J. Zhang, L. Pan, X. Zhang and J. Zou, Adv. Sci., 2018, 5, 1700464 CrossRef PubMed.
- J. Kim, H. Kim, W.-J. Lee, B. Ruqia, H. Baik, H.-S. Oh, S.-M. Paek, H.-K. Lim, C. H. Choi and S.-I. Choi, J. Am. Chem. Soc., 2019, 141, 18256–18263 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- B. E. Conway and B. V. Tilak, Electrochim. Acta, 2002, 47, 3571–3594 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, J. Electrochem. Soc., 2005, 152, J23 CrossRef.
- M. Gennero de Chialvo and A. Chialvo, Electrochim. Acta, 1998, 44, 841–851 CrossRef CAS.
- M. Zhang, K. Zhang, X. Ai, X. Liang, Q. Zhang, H. Chen and X. Zou, Chin. J. Catal., 2022, 43, 2987–3018 CrossRef CAS.
- B. Hinnemann, P. G. Moses, J. Bonde, K. P. Jørgensen, J. H. Nielsen, S. Horch, I. Chorkendorff and J. K. Nørskov, J. Am. Chem. Soc., 2005, 127, 5308–5309 CrossRef CAS PubMed.
- T. F. Jaramillo, K. P. Jørgensen, J. Bonde, J. H. Nielsen, S. Horch and I. Chorkendorff, Science, 2007, 317, 100–102 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, L. H. Li, Y. Han, Y. Chen, A. Du, M. Jaroniec and S. Z. Qiao, Nat. Commun., 2014, 5, 3783 CrossRef PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- M. Guo, M. Ji and W. Cui, Appl. Surf. Sci., 2022, 592, 153237 CrossRef CAS.
- X. Zhang, Z. Yang, Z. Lu and W. Wang, Carbon, 2018, 130, 112–119 CrossRef CAS.
- R. Dennington, T. Keith and J. Millam, GaussView 5.0, Semichem Inc., Shawnee Mission KS, 2009 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd Brothers, E. K. N. Kudin, V. N. Staroverov, R. K. J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. C. N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. J. R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz and J. D. J. F. Cioslowski, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- P. Aggarwal, D. Sarkar, K. Awasthi and P. W. Menezes, Coord. Chem. Rev., 2022, 452, 214289 CrossRef CAS.
- Y. Yang, J. Liu, F. Liu, Z. Wang and D. Wu, J. Mater. Chem. A, 2021, 9, 2438–2447 RSC.
- M. R. S. A. Janjua, J. Phys. Chem. Solids, 2022, 167, 110789 CrossRef CAS.
- B. You, Y. Zhang, Y. Jiao, K. Davey and S. Z. Qiao, Angew. Chem., 2019, 131, 11922–11926 CrossRef.
- C. Xiao, R. Sa, Z. Ma, Z. Cui, W. Du, X. Sun, Q. Li and H. Deng, Int. J. Hydrogen Energy, 2021, 46, 10337–10345 CrossRef CAS.
- N. Kosar, S. Rafiq, K. Ayub, M. Imran and T. Mahmood, Int. J. Hydrogen Energy, 2024, 68, 1219–1228 CrossRef CAS.
- M. Ren, X. Guo and S. Huang, Appl. Surf. Sci., 2021, 556, 149801 CrossRef CAS.
- Y. Cheng, X. Fan, F. Liao, S. Lu, Y. Li, L. Liu, Y. Li, H. Lin, M. Shao and S.-T. Lee, Nano Energy, 2017, 39, 284–290 CrossRef CAS.
- D. Chodvadiya, P. K. Jha and B. Chakraborty, Int. J. Hydrogen Energy, 2022, 47, 41733–41747 CrossRef CAS.
- D. Chodvadiya, B. Chakraborty and P. K. Jha, Int. J. Hydrogen Energy, 2023, 48, 18326–18337 CrossRef CAS.
- Z. Zhou, Z. Pei, L. Wei, S. Zhao, X. Jian and Y. Chen, Energy Environ. Sci., 2020, 13, 3185–3206 RSC.
- W. Zhou, Z. Jiang, M. Chen, Z. Li, X. Luo, M. Guo, Y. Yang, T. Yu, C. Yuan and S. Wang, Chem. Eng. J., 2022, 428, 131210 CrossRef CAS.
- L. Sun, B. Wang and Y. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 21808–21817 CrossRef CAS PubMed.
- X. Li, P. Cui, W. Zhong, J. Li, X. Wang, Z. Wang and J. Jiang, Chem. Commun., 2016, 52, 13233–13236 RSC.
- K. Zhang, B. Jin, Y. Gao, S. Zhang, H. Shin, H. Zeng and J. H. Park, Small, 2019, 15, 1804903 CrossRef PubMed.
- M. D. Hossain, Z. Liu, M. Zhuang, X. Yan, G. Xu, C. A. Gadre, A. Tyagi, I. H. Abidi, C. Sun, H. Wong, A. Guda, Y. Hao, X. Pan, K. Amine and Z. Luo, Adv. Energy Mater., 2019, 9, 1803689 CrossRef.
- X. Zhai, L. Li, X. Liu, Y. Li, J. Yang, D. Yang, J. Zhang, H. Yan and G. Ge, Nanoscale, 2020, 12, 10035–10043 RSC.
- K. S. Joseph and S. Dabhi, Electrochim. Acta, 2024, 505, 144968 CrossRef CAS.
- H.-H. Wu, H. Huang, J. Zhong, S. Yu, Q. Zhang and X. C. Zeng, Nanoscale, 2019, 11, 12210–12219 RSC.
- X. Liu, D. K. Hoang, Q. A. T. Nguyen, D. Dinh Phuc, S.-G. Kim, P. C. Nam, A. Kumar, F. Zhang, C. Zhi and V. Q. Bui, Nanoscale, 2024, 16, 13148–13160 RSC.
- A. Jalil, T. Zhao, A. Kanwal and I. Ahmed, Chem. Eng. J., 2023, 470, 144239 CrossRef CAS.
- H. Guo, L. Li, X. Wang, G. Yao, H. Yu, Z. Tian, B. Li and L. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 36506–36514 CrossRef CAS PubMed.
- T. He, S. K. Matta, G. Will and A. Du, Small Methods, 2019, 3, 1800419 CrossRef.
- C. Liu, G. Zhang, L. Yu, J. Qu and H. Liu, Small, 2018, 14, 1800421 CrossRef PubMed.
- E. Sargeant, F. Illas, P. Rodríguez and F. Calle-Vallejo, J. Electroanal. Chem., 2021, 896, 115178 CrossRef CAS.
- S. Tosoni, G. Di Liberto, I. Matanovic and G. Pacchioni, J. Power Sources, 2023, 556, 232492 CrossRef CAS.
- A. Allangawi, T. Mahmood, K. Ayub and M. A. Gilani, Mater. Sci. Semicond. Process., 2023, 153, 107164 CrossRef CAS.
- S. Lu, H. L. Huynh, F. Lou, K. Guo and Z. Yu, Nanoscale, 2021, 13, 12885–12895 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
- S. H. Talib, Z. Lu, X. Yu, K. Ahmad, B. Bashir, Z. Yang and J. Li, ACS Catal., 2021, 11, 8929–8941 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Tariq Mahmood
*a,
Tariq Mahmood