
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResearch progress on and outlook of direct CO2 thickeners for enhanced oil recovery
Yuxuan Song ab,
Qun Zhang*ab,
Xinyuan Zoub,
Jian Fanb,
Sicai Wangab and
Yan Zhuab
ab,
Qun Zhang*ab,
Xinyuan Zoub,
Jian Fanb,
Sicai Wangab and
Yan Zhuab
aInstitute of Porous Flow and Fluid Mechanics, Chinese Academy of Sciences, Langfang, Hebei 065007, China
bResearch Institute of Petroleum Exploration and Development, Beijing, 100083, China
First published on 9th January 2025
Abstract
Supercritical CO2, as an environmentally friendly and pollution-free fluid, has been applied in various EOR techniques such as CO2 flooding. However, the low viscosity of the gas leads to issues such as early breakthrough, viscous fingering, and gravity override in practical applications. Although effective mobility-control methods, such as CO2 WAG (water alternating gas)–, CO2 foam-, and gel-based methods, have been developed to mitigate these phenomena, they do not fundamentally solve the problem of the high gas–oil mobility ratio, which leads to reduced gas sweep efficiency. Adding CO2 direct thickeners to displacing fluid can increase its viscosity, achieve deeper mobility control, and thus improve the CO2 flooding oil-recovery effect. Unlike other methods, direct thickeners can alter the physical and chemical properties of CO2, making it a fundamentally effective means of achieving mobility control. This approach can be applied in various reservoir environments and formations, or it can assist other methods for more in-depth mobility control. This article reviews the development and application of CO2 direct thickeners and introduces the thickening mechanisms and effects of different types of thickeners as well as their existing problems and future development directions.
1. Introduction
With the development of the global economy, the world's oil consumption has increased significantly, but the production of crude oil is decreasing. The optimization and development of oil extraction technology are therefore urgent priorities.1 Typically, primary and secondary oil recovery can only recover about 40% of a reservoir's reserves; thus, the development of tertiary oil-recovery technology is crucial for maintaining crude oil production and reducing oil imports. CO2 flooding is one of the most promising technologies in enhanced oil recovery (EOR), characterized by its non-toxic and environmentally friendly nature, abundant availability, chemical stability, and ease of miscibility with crude oil. Additionally, CO2 can be stored in subsurface pores, achieving integrated oil displacement and storage during the oil displacement process.2–4 Since the 1960s, the United States has successively carried out a series of studies on CO2 flooding, and there have been more than 130 commercial CO2 flooding projects to date.5 According to statistics, the annual production of CO2 flooding in the U.S. reached 137.1 million tons in 2014, accounting for about 93% of the world's total CO2 flooding production.6 Recently, China proposed the “dual-carbon” goal, where carbon dioxide capture, utilization, and storage (CCUS) is a primary method for end offsetting. CO2 flooding holds significant application value as a vital component of CCUS.7–9Gases have better diffusive capabilities in porous media and tend to enter larger pores first, offering significant permeability at lower gas saturations. However, the high mobility ratio between CO2 and crude oil results in an unstable displacement front, causing early breakthrough, and this problem is exacerbated by increased heterogeneity within the reservoir.10 Moreover, problems such as gravity override and hindered oil–gas mixing and reactivity can significantly reduce sweep efficiency and oil displacement efficiency.11,12 Therefore, reducing the two-phase mobility ratio and strengthening the mobility control of CO2 are critical issues that need to be addressed to improve the oil-recovery rate of CO2 flooding technology.
CO2 thickening is an effective way to reduce the mobility ratio between the oil and gas phases. Due to the low critical temperature (31.1 °C) and critical pressure (7.38 MPa) of CO2, most reservoir temperatures and pressures exceed the critical conditions for CO2, and it often exists in a supercritical state within the reservoir.13 Supercritical carbon dioxide (scCO2) possesses superior heat transfer and diffusive properties, and its density is akin to liquid-state substances, which contribute to its enhanced solvent performance. Dissolving polymers in scCO2 and utilizing the non-bonding forces between solute molecules to form a large molecular network can significantly increase the viscosity of CO2.14
CO2 direct thickeners are not only applicable for EOR processes but also can be widely used in scCO2 fracturing fluids to enhance the sand-carrying capacity of the fracturing fluid, as well as in other related fields, such as CO2 adsorption. Therefore, the development of a high-performance CO2 direct thickener is vital for the entire EOR field. This paper presents a review of CO2 direct thickeners from a molecular perspective, as well as their dissolution thickening mechanisms and application conditions. An outlook on the development prospects of CO2 direct thickeners is also provided as well as insights offering technical support for their future practical application in oil fields.
2. CO2 thickening
2.1 CO2 mobility control and CO2 thickening
It is essential to control the mobility of the gas in the reservoir to alleviate problems such as gas loss and poor sweep efficiency caused by gas channeling. CO2 mobility control can be achieved through three main approaches: First, by reducing the gas saturation and permeability to stabilize the displacement front and alter the flow pattern in the formation. Second, by altering the formation permeability to enhance the control over the flow path of CO2 within the reservoir. Third, by increasing the gas viscosity to reduce the mobility ratio between the oil and gas phases.10,15,16 Based on these principles, CO2-mobility-control methods mainly include water-alternating-gas (WAG), CO2 thickening, CO2 foam, and gel methods, with each method offering certain advantages and disadvantages, as shown in Table 1.17–19| Mobility-control method | Mechanism | Advantages | Shortcomings |
|---|---|---|---|
| CO2-WAG | Alternately injecting water and gas reduces gas permeability and uses the injected water to stabilize the displacement front | High microscopic oil displacement efficiency from gas injection and high swept volume from water injection, while requiring a small amount of gas injection | Water shielding and injection losses, not suitable for oil reservoirs with high heterogeneity, injection and production parameters significantly affect EOR efficiency |
| CO2 foam | Forms a stable foam front at the forefront of displacement fluid, utilizing the high-viscosity characteristic of the foam to reduce the mobility of the gas, with some gas bubbles serving a plugging function | Foam has various forms of mobility-control effects | Injection is difficult, foam stability is poor, and surfactants will adsorb onto the rock surface |
| Gel | Forming a polymer gel in the formation to plug large pores and alter the formation permeability | Repairs and improves the flow channels of displacement fluid and seals off leaks | Plugging stability is poor, not shear-resistant, gel formation exhibits instability, and the effect deteriorates after multiple rounds of profile adjustment |
| CO2 thickening | Polymers are dissolved in supercritical CO2 and increase the viscosity of the gas, thereby reducing the mobility ratio | Water-free systems are easier to inject into the reservoir | High cost |
The factors affecting the efficiency of CO2-WAG flooding include reservoir heterogeneity, wettability, the fluid physicochemical properties, and the injection parameters.17 The water shielding issue caused by the injection of two phases can significantly reduce the recovery rate during the WAG process, especially in formations with high water saturation levels.24,25 Additionally, due to the changes in relative permeability, both water and gas will experience injection losses. This issue can be alleviated by reducing the water slugs and increasing the gas slugs, but at the same time, controlling the gas mobility becomes more difficult.26 The injection parameters of CO2-WAG have a significant impact on the final recovery efficiency. Valeev and Shevelev27 evaluated the effect of the water–gas injection ratio on the final recovery efficiency using a field in the Kogalym region as an example. The addition of water could reduce the mobility of CO2, and at a certain water–gas ratio, the water–oil–gas three-phase system could attain a mixed phase in the porous medium, forming a viscous single phase, which resulted in the optimal displacement efficiency. The miscible process involved the dissolution of CO2 into a mixture of crude oil and water and entailed very complex physicochemical reactions. However, most reservoirs cannot achieve perfect miscible conditions, and the purpose of mobility control can only be achieved through the buffering effect of water between oil and gas with high mobility ratios. To enhance the efficiency of the injection, polymers, surfactants, and nanomaterials are often incorporated to further assist the WAG process. A new technology that has emerged is the chemical enhanced water-alternating-gas (CEWAG) system, where chemical agents are added to enhance the WAG process. These agents are primarily added to the aqueous phase, and there is less research on adding them to the gaseous phase.21
Foam is a non-equilibrium system, and it will gradually collapse and disappear even without disturbance over time. The chemicals used for foaming are typically surfactants, and a continuous injection of surfactant solution is required to stabilize and continuously regenerate the foam in the reservoir.29 CO2 foam flooding has been applied in various field sites. Field experiments have shown that CO2 foam can increase the recovery rate by at least 5%, which is significantly below the results obtained from laboratory tests showing about a 30% increase in recovery rate. The main reason is that surfactants will be extensively adsorbed in the reservoir, causing losses and preventing the continuous and stable formation of the foam.33,35
Based on different thickening mechanisms, CO2 thickeners can be divided into direct thickeners and indirect thickeners. Direct thickeners dissolve in CO2 to form a thermodynamically stable solution, enhancing the viscosity and density of the injected fluid. This process involves only a single phase change of CO2 and does not require water injection, which can prevent issues like water shielding and well acidification. Indirect thickeners thicken the displacement fluid by forming CO2 foams or emulsions with high apparent viscosity. Additionally, there is research on thickening CO2 with nanoparticles, which involves achieving a stable dispersion of nanoparticles in CO2. To achieve the desired viscosity, it is often necessary to modify the nanoparticles or add them together with polymers that are soluble in CO2.
Comparing various mobility-control methods, it could be observed that CO2-WAG, CO2 foam, and gel all aim to control mobility by altering the flow pattern and path of CO2 within the reservoir. These are indirect means of mobility control, as they do not change the physicochemical properties of the supercritical CO2, nor do they fundamentally address the issue of the large viscosity gap between oil and gas. It is also challenging to achieve more precise control over the mobility of CO2 with these methods. Direct thickeners, on the other hand, can increase the viscosity of the CO2 system, altering the fluid's physical and chemical state, and can also assist other methods in achieving further mobility control, making it a fundamentally effective means. Before the study of gas-soluble thickeners, water-soluble thickeners (such as polyacrylamide) had been extensively studied and applied in water flooding. CO2 thickening technology is a challenging and breakthrough technology. The development of high-performance CO2 thickeners represents an innovation in EOR techniques for gas flooding. Regardless of the reservoir's permeability, the saturation of the injected fluid, and the properties of the brine, a thickened CO2 system can always play a role in mobility control.41
2.2 Dissolution and thickening mechanism
Direct CO2 thickeners mainly include polymers, small molecule compounds, and nanoparticles.42–44 The principles of direct thickeners dissolving and thickening in CO2 can mainly be divided into two types. One type is based on the interactions of small molecule compounds with functional groups on the exterior or ends of their molecular chains, which form a network structure in the solution through van der Waals forces and other electrostatic interactions that hinder the flow of CO2 molecules and increase the viscosity of the system (Fig. 1). Most hydrocarbon thickeners and modified copolymers belong to this type. The structure formed by non-covalent interactions is very fragile and can be easily destroyed. The other type involves high-molecular-weight polymers, such as fluoropolymers and siloxane polymers, which can form a large molecular network structure and increase the viscosity by intertwining their macromolecular chains at low concentrations. However, polymers with high molecular weights are difficult to dissolve in CO2, often requiring a significant amount of co-solvents to be added. Nanoparticles primarily increase the viscosity of solutions through surface energy effects and homogeneous dispersion.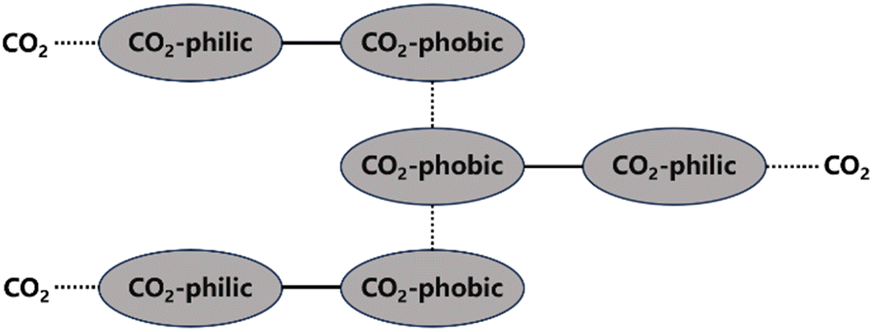 | ||
| Fig. 1 Interactions between polymers and CO2 molecules. | ||
There are three types of forces in a solution system: solute–solute interactions, solute–solvent interactions, and solvent–solvent interactions. The competition and balance among these three determine the final solubility. During dissolution, solute–solvent interactions promote the process, whereas solute–solute and solvent–solvent forces hinder it. Therefore, in polymer–CO2 solutions, the greater the interaction force between the polymer and CO2 molecules, the higher the solubility of the polymer in CO2. Although the solubility of the polymer is increased, the viscosity of the solution may not necessarily be increased because the greater the affinity between the polymer and CO2, the more inclined they are to bind, which can reduce the interactions between the polymer molecules. Similarly, if the polymer has a high cohesive energy and strong intermolecular forces, it will preferentially interact with itself, repelling the binding with CO2 molecules, which would result in reduced solubility. Therefore, it is necessary to ensure that there is a strong interaction between the polymer and CO2 molecules while also maintaining a powerful interaction force between the polymer molecules (or a certain repulsive force between the polymer and CO2) to achieve a good high-viscosity homogeneous CO2 solution.45
According to the requirements of intermolecular forces, direct thickener molecules need to simultaneously possess CO2-philic groups that can increase solubility and CO2-phobic groups that can enhance the interaction forces between polymer molecules. Atactic polymers are more soluble in CO2 than isotactic and syndiotactic polymers.46 Incorporating more short side chains can reduce the polymer's dissolution pressure when it contains the same number of CO2-philic groups. This is because the increase in free volume not only enhances the mixing entropy but also improves the flexibility of the molecular chains.47 Overall, macromolecular chains are more likely to become entangled in a solution, and the thickening ability can be enhanced with increasing the molecular volume, but it is also relatively more difficult for them to stably exist in the solution. Factors that affect the thickening effect mainly include the chemical structure, molecular weight, temperature, and pressure, as shown in Table 2.
| Factor | Solubility | Viscosity |
|---|---|---|
| Concentration | Increase and then decrease | Increase |
| Molecular weight | Decrease | Increase |
| Chain flexibility | Increase | Decrease |
| Atactic structure | Increase | — |
| Branching degree | Increase | Increase |
| Side chain length | Decrease | Increase |
| Temperature | Decrease | Decrease |
| Pressure | Increase | Increase |
| Shear force | Increase | Decrease |
3. CO2 direct thickeners
3.1 Fluorinated polymers
CO2 is a nonpolar molecule and a weak solvent for many polar substances. However, due to the presence of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and a higher quadrupole moment within CO2, there is an electrostatic attraction between CO2 and polar groups, such as fluorocarbons, siloxane, and carbonyl groups. Therefore, compounds with larger polar groups can dissolve in supercritical CO2.48 Research has found that fluorine-containing groups have the best affinity with CO2 and can maintain a certain solubility even at high molecular weights. Dardin49 dissolved n-hexane and perfluorinated n-hexane in supercritical CO2, and compared the chemical shifts of 1H and 19F. The chemical shift of 19F exhibited characteristics related to the density of CO2, with a significant shift greater than that of 1H. This difference originated from the van der Waals forces between the fluorine and CO2 molecules. McHugh50 compared the solubility of poly(vinyl difluoride-co-hexafluoropropylene) (fluorel) and poly(tetrafluoroethylene-co-hexafluoropropylene) (FEP19). It was found that fluorel could dissolve under conditions of 100 °C and 75 MPa, while FEP19, despite the introduction of fluorine-containing groups, lacked polar vinyl groups and tended to precipitate due to strong self-association. It was also observed in fluorinated polyisoprene that polar groups can enhance the interaction between the dipole moment and the quadrupole moment. These studies demonstrated that fluoropolymers are more soluble in CO2 than other compounds containing weakly polar groups.51
O bond and a higher quadrupole moment within CO2, there is an electrostatic attraction between CO2 and polar groups, such as fluorocarbons, siloxane, and carbonyl groups. Therefore, compounds with larger polar groups can dissolve in supercritical CO2.48 Research has found that fluorine-containing groups have the best affinity with CO2 and can maintain a certain solubility even at high molecular weights. Dardin49 dissolved n-hexane and perfluorinated n-hexane in supercritical CO2, and compared the chemical shifts of 1H and 19F. The chemical shift of 19F exhibited characteristics related to the density of CO2, with a significant shift greater than that of 1H. This difference originated from the van der Waals forces between the fluorine and CO2 molecules. McHugh50 compared the solubility of poly(vinyl difluoride-co-hexafluoropropylene) (fluorel) and poly(tetrafluoroethylene-co-hexafluoropropylene) (FEP19). It was found that fluorel could dissolve under conditions of 100 °C and 75 MPa, while FEP19, despite the introduction of fluorine-containing groups, lacked polar vinyl groups and tended to precipitate due to strong self-association. It was also observed in fluorinated polyisoprene that polar groups can enhance the interaction between the dipole moment and the quadrupole moment. These studies demonstrated that fluoropolymers are more soluble in CO2 than other compounds containing weakly polar groups.51
McClain52 demonstrated that high-molecular-weight polyfluoropropyl acrylate (PFOA) could also dissolve in CO2 and increase the viscosity of the system. PFOA is known to be the polymer with the highest affinity for CO2. DeSimone53,54 reported that PFOA with a molecular weight of about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400000 could increase the viscosity from 0.08 cp to 0.2–0.6 cp. Fluorinated polymers can cause environmental pollution, but polymers with fewer than seven fluorocarbons do not have bioaccumulative properties. Consequently, Lemaire55 synthesized PFA with short fluorocarbon chains containing 4 and 6 fluorines, which had almost the same solubility and thickening effect as PFA with long fluorocarbon chains (C8F17) at temperatures between 25–125 °C. Zaberi43 tested the phase behavior and oil displacement efficiency of PFA (Mw = 250000) containing six fluorocarbons in CO2. Although PFA could dissolve well in CO2, it was not soluble in light hydrocarbons. This is very disadvantageous in the mobility-control process because CO2 can extract light hydrocarbons from crude oil during the oil displacement process, and once the light hydrocarbons dissolve in CO2, they can cause the PFA to precipitate. Experiments proved that even at a pressure of 62 MPa, 1 wt% PFA could not dissolve in CO2 containing 30 wt% of mixed light hydrocarbons ranging from C6 to C20. Using Berea sandstone and by injecting 2 PV of pure CO2 and thickened CO2 for comparison, the oil displacement efficiency was found to increase by 16%. Also, the pressure difference increased sharply by 8–9 times after injecting the thickened CO2, which was inconsistent with the viscosity test results showing a 3–4 times increase. The reason for the difference was the blockage of the pore throats caused by polymer precipitation.
400000 could increase the viscosity from 0.08 cp to 0.2–0.6 cp. Fluorinated polymers can cause environmental pollution, but polymers with fewer than seven fluorocarbons do not have bioaccumulative properties. Consequently, Lemaire55 synthesized PFA with short fluorocarbon chains containing 4 and 6 fluorines, which had almost the same solubility and thickening effect as PFA with long fluorocarbon chains (C8F17) at temperatures between 25–125 °C. Zaberi43 tested the phase behavior and oil displacement efficiency of PFA (Mw = 250000) containing six fluorocarbons in CO2. Although PFA could dissolve well in CO2, it was not soluble in light hydrocarbons. This is very disadvantageous in the mobility-control process because CO2 can extract light hydrocarbons from crude oil during the oil displacement process, and once the light hydrocarbons dissolve in CO2, they can cause the PFA to precipitate. Experiments proved that even at a pressure of 62 MPa, 1 wt% PFA could not dissolve in CO2 containing 30 wt% of mixed light hydrocarbons ranging from C6 to C20. Using Berea sandstone and by injecting 2 PV of pure CO2 and thickened CO2 for comparison, the oil displacement efficiency was found to increase by 16%. Also, the pressure difference increased sharply by 8–9 times after injecting the thickened CO2, which was inconsistent with the viscosity test results showing a 3–4 times increase. The reason for the difference was the blockage of the pore throats caused by polymer precipitation.
Xu and Enick56 synthesized a copolymer PolyFAST (Mw = 540000) comprising 71% fluoroacrylate and 29% styrene. PolyFAST with a molecular weight of 540000 had a solubility of 0.25–2 wt% under the conditions of 20 MPa and temperature between 298–373 K. They also found that a thickener concentration of just 1.5 wt% could increase the system's viscosity by 19 times.
Taking advantage of the high solubility of perfluoropolyether in CO2, Enick and Beckman57 synthesized a fluorinated polyurethane disulfonate with a molecular weight of 32500 and that could be mixed with CO2 in any ratio under the conditions of 298 K and 20–34 MPa. Under 34.5 MPa, a thickener concentration of 4 wt% could increase the system viscosity by 2.7 times. However, the high price of the fluorinated ether oil makes it costly to use as a thickener.
Enick58 synthesized a random copolymer of poly(tetrafluoroethylene-vinyl acetate), and it was found that when the content of tetrafluoroethylene (TFE) reached 19 mol%, the polymer had the highest solubility in CO2. When the TFE content in the copolymer was 47 mol%, the solubility was lower than that of PVAc. Molecular dynamics simulation results indicated that when CO2 molecules simultaneously interact with fluorine atoms and hydrogen bonds, the solubility of the copolymer was no longer enhanced by the binding of CO2 to the fluorinated part. This was because the fluorinated units act as Lewis bases and the hydrogen-bonding parts act as Lewis acids during the binding process, leading to a change in the properties of the copolymer.
A. G. Goicochea59 utilized dissipative particle dynamics to simulate the shear viscosity of styrene-fluoroacrylate (HFDA-STY), poly(1-decene) (P1D), and poly(vinyl ethyl ether) (PVEE) in CO2, and their results were within 6% of the experimental outcomes. The findings indicated that the linear PVEE formed a molecular association network, whereas P1D did not associate but rather adsorbed CO2 due to its six branches, thus exhibiting higher solubility. HFDA primarily achieved the best thickening effect through the high affinity of fluorine for CO2 and the π–π stacking between styrene rings, demonstrating a two orders higher magnitude of viscosity than the non-fluorinated solution (Fig. 2).
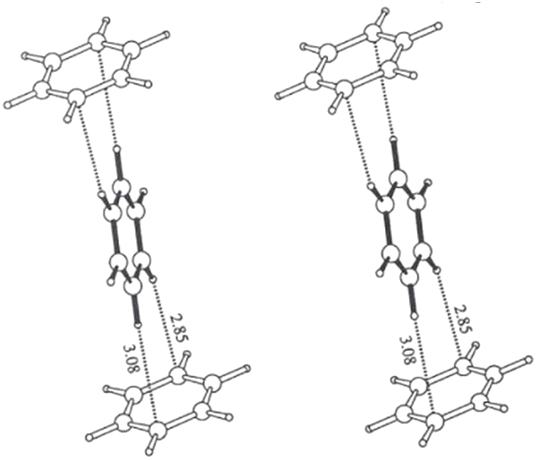 | ||
| Fig. 2 π–π stacking between benzene rings.60 | ||
Dai61,62 synthesized a series of copolymers of heptadecafluorodecyl acrylate and styrene (HFDA-co-STY) with different monomer ratios. As the content of phenyl groups in the copolymer increased, both the solubility pressure and thickening ability of the copolymer increased.
Sun63 synthesized a copolymer of vinyl benzoate and heptadecafluorodecyl acrylate, P(VBe-co-HFDA). Molecular dynamics simulations were used to characterize the solubility and thickening effects of the VBe content in the system, and the results were compared with those of the PHFDA homopolymer. The findings indicated that when the VBe content was 33%, the thickening effect was optimal. At a concentration of 5 wt%, the copolymer could increase the system's viscosity by 438 times, significantly higher than the thickening ability of the PHFDA homopolymer.
Kilic64 investigated the effect of the structure of aromatic acrylate–fluoroacrylate copolymers on the viscosity of CO2, and found that these copolymers could dissolve in CO2 at pressures below 15 MPa and a temperature of 295 K. The solution viscosity increased with the increasing content of aromatic acrylate units in the copolymer. However, beyond a certain content, the phenyl rings shifted from intermolecular association to intramolecular association, leading to a decrease in viscosity. Additionally, higher pressures were found to strengthen the intermolecular association. The experiments demonstrated that an optimal content of 29% aromatic acrylate resulted in the best thickening effect, achieving a dissolution pressure comparable to that of PolyFAST.
Huang65 synthesized a copolymer of fluoroacrylate, styrene, and sulfonated styrene, which increased the solution viscosity through the association of sulfonated styrene. The results showed that this copolymer was less soluble than the non-sulfonated polymer, but the thickening effect was enhanced. The optimal content of styrene was 29%, which obtained the same result as for PolyFAST. The addition of 1–5 wt% copolymer increased the system's viscosity by 3–10 times at 25 °C and 34.48 MPa. Zhang66 further optimized the ratio of copolymer monomers and found that under suitable solubility, a higher content of sulfonated styrene monomer could further enhance the thickening ability. The best ratio was ultimately determined to be 60% PHDA-24% PSt-16% S.
Fluoropolymers do not significantly enhance the viscosity of CO2, but fluorine-containing groups have good affinity with CO2 and high solubility in it. By adding a small amount of fluorine-containing groups to polymers with strong self-association, the originally difficult-to-dissolve molecular chains or groups can become more stable in CO2. The solute molecules can form a network structure in supercritical CO2 without the need for co-solvents, achieving this through strong interactions, such as hydrogen bonding. Although fluoropolymers are costly and have certain biological hazards, their high affinity for CO2, the absence of a need for co-solvents, and their low usage make them advantageous and promising for modification and copolymerization (Table 3).
| Polymer | Structure | Co-solvent | Conditions | Solubility | Viscosity/cp | Reference |
|---|---|---|---|---|---|---|
| PFA | 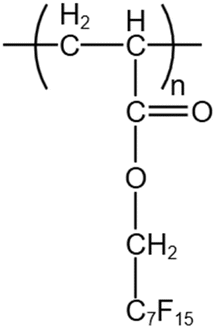 |
— | 50 °C | 6.7 wt% | 0.6 | 43 and 52–55 |
| PolyFAST | 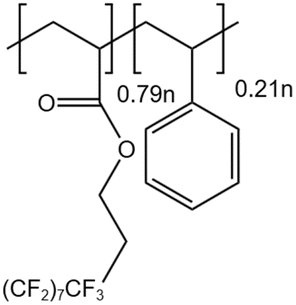 |
— | 373 K 20 MPa | 1.5 wt% | 19 | 56 |
| HFDA-co-STY | 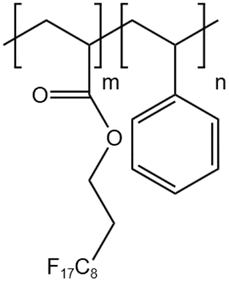 |
— | 35 °C | 5 wt% | 7.06 | 61 and 62 |
| 30 MPa | ||||||
| P(VBe-co-HFDA) | 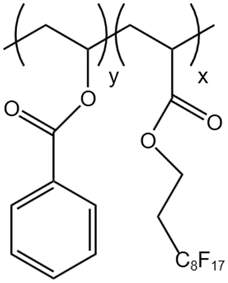 |
— | 308.2 K | 5 wt% | 8.76 | 63 |
| 30 MPa | ||||||
| PEA-FA | 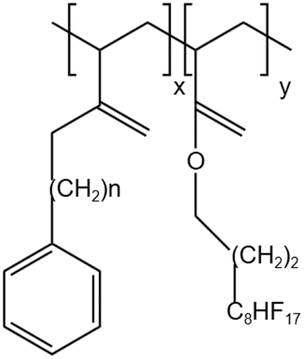 |
— | 295 K | 5 wt% | 2.55 | 64 |
| 41.4 MPa | ||||||
| PHFDA-Pst-S | 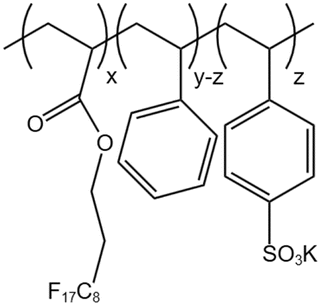 |
— | 273 K | 1 wt% | 2.01 | 65 and 66 |
| 28 MPa |
3.2 Siloxane polymers
Siloxanes are excellent electron-donating groups that can bond with the electron-deficient carbon in CO2. The high flexibility of siloxane chains also promotes the dissolution of molecules in CO2. Siloxane polymers have long been widely applied in the field of CO2 thickeners. Bae and Irani67,68 first discovered that polydimethylsiloxane (PDMS) with a molecular weight of 197000 could dissolve in CO2 and increase the system's viscosity to 1.2 cp, and it effectively delayed gas breakthrough in oil displacement experiments. Du69 also conducted a series of experiments and simulation studies on a PDMS–toluene system as a fracturing fluid, which could thicken CO2 by 40 times at 42 °C and 20 MPa. PDMS remains one of the most effective siloxanes CO2 thickeners, but it requires a large amount of toluene as a co-solvent.
Zhao70 studied the solubility and thickening properties of a series of PDMSs with different end groups. Hydroxyl-end capped PDMS, vinyl-end capped PDMS, and hydrogen-end capped PDMS were compared respectively, and it was found that under the same intrinsic viscosity, the solubility pressure of vinyl-end capped PDMS was the lowest. Further, 8 wt% of 1000 cst vinyl-end capped PDMS achieved a system viscosity of 12.57 mPa s at 313.13 K. Using kerosene as a co-solvent, 5 wt% of 3000 cst vinyl-end capped PDMS and 2 wt% of kerosene achieved a system viscosity of 14.87 mPa s at 313 K and 39.6 MPa.
Enick58 grafted various groups with good affinity for CO2, such as carbonyl, ether, acetate, and trimethyl branched groups, onto siloxanes. The solubility of these groups in CO2 at 295 K was then tested, and it was found that the optimal substitution degree for carbonyl groups was within 4–8%, for ethers it was 8%, and for esters it was 20%. Increasing the degree of substitution increased the solubility pressure, with complete substitution resulting in insolubility in CO2. This was because the addition of side chains increases the rigidity of the molecular chain and the cohesive energy density, and when the substitution degree exceeds a certain critical point, the negative impact on solubility caused by these factors exceeds the Lewis acid–base interaction between the carbonyl group and CO2 molecules. The solubility of trimethyl branched siloxanes was found to be almost the same as that of the PDMS, because the branched substitution increases the free volume of the molecule and enhances the flexibility of the molecular chain, compensating for the increase in cohesive energy density.
Li71 synthesized a siloxane copolymer for fracturing thickening, which could thicken CO2 at 8–12 MPa by 4.1–5.7 times at temperatures between 35–55 °C, with 3 wt% of the copolymer and 7 wt% of the co-solvent toluene. The interaction between the N on the polymer side chain and the C in CO2 increased the solubility, while the O on the siloxane main chain, the C–H bonds in toluene, and the CO double bonds in CO2 worked together to form a stable macromolecular network structure. The team also synthesized siloxane thickeners AOB, BTMT, and PDMS with epoxy groups as end groups. Comparing them with regular PDMS, AOB's thickening effect was 8 times that of PDMS at 310 K and 15 MPa.72 BTMT also showed better resistance to filtration than PDMS in fracturing experiments.73 The solubility pressure of PDMS with epoxy end groups was less than 8 MPa. The thickening effect was 3–4 times higher than that of PDMS at the same molecular weight at temperatures between 20–50 °C.74
O'Brien75 synthesized a series of low-molecular-weight polysiloxanes with aromatic amide groups at the ends, and found that siloxanes with amide-anthraquinone (AQCA) at the ends had a certain thickening effect on CO2 with the co-solvent hexane. A CO2 solution containing 13.3 wt% of the thickener and 26.7 wt% of hexane demonstrated a viscosity 9 times that of the CO2–hexane mixture at 25 °C and 20.7 MPa.
Liu76 synthesized two siloxane-based thickeners for fracturing 1,3,5,7-tetramethyl cyclotetrasiloxane (HBD). The internally branched HBD-2 molecule was found to have a larger free volume and was more prone to self-association, thus having a better thickening effect than HBD-1. They also synthesized a series of HS polymers without a siloxane ring.77 At 298 K and 7.48 MPa, all three thickeners dissolved after being left to stand for 12 h, without a cloud point. Possibly due to CO2 thickening or incomplete dissolution, the solution remained translucent in the reactor. HS-3 performed the best among the three thickeners, which contained more hydroxyl groups and longer side chains. It led to thickening by 151–163 times with the addition of 5 wt% polymer.
Gallo78 utilized the CPCM solvation model and viscosity measurements to compare four different types of siloxane compounds and found that the solubility of polysiloxanes was about 20% higher than that of linear PDMS with similar molecular weights. The polar groups within silsesquioxane molecules could promote solvation, while polar groups on the outer layer and ends of the molecules could lead to strong intermolecular interactions. A mixture of 6.9 wt% silsesquioxanes and nanoparticles with CO2 reached a viscosity of 0.25 cp at 180 bar and 55 °C, showing it was more effective than linear PDMS and it also exhibited greater viscosity stability with temperature changes.
Siloxane polymers have a highly flexible backbone and strong affinity for CO2, making them potentially effective CO2 thickeners. However, siloxane compounds are expensive, and most research has focused on low-molecular-weight siloxanes that do not inherently possess thickening capabilities. Typically, more than 10 wt% co-solvents are required to assist in thickening. In addition to searching for effective molecular structures, it is necessary to further investigate the thickening capabilities of high-molecular-weight siloxane polymers and the mechanisms of co-solvent-assisted thickening. Optimizing the system and selecting the appropriate molecular weight materials are key research focuses for siloxane thickeners (Table 4).
| Polymer | Structure | Co-solvent | Conditions | Solubility | Viscosity/cp | Reference |
|---|---|---|---|---|---|---|
| PDMS | 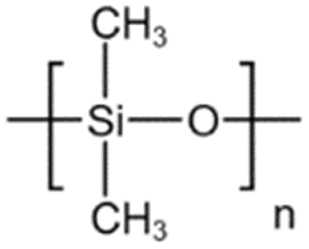 |
Toluene | 54.4 °C | 4 wt% | 1.2 | 67–69 |
| 17.2 MPa | ||||||
| Grafted methylhydrosiloxane–dimethylsiloxane copolymers | 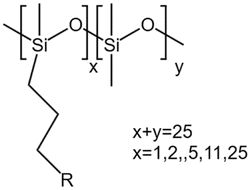 |
— | 295 K | 1–5 wt% | — | 58 |
| 5–45 MPa | ||||||
| Silicone ternary copolymer | 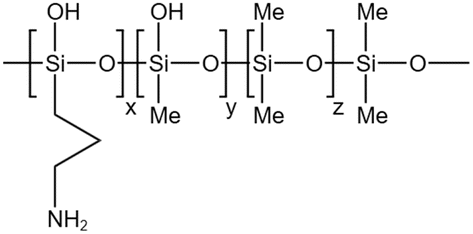 |
Toluene | 55 °C | 3 wt% | 0.15 | 71 |
| 12 MPa | ||||||
| AOB | 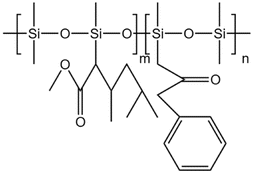 |
Cyclohexane | 310 K | 3 wt% | 1.8 | 72 |
| 15 MPa | ||||||
| Epoxy-terminated PDMS | 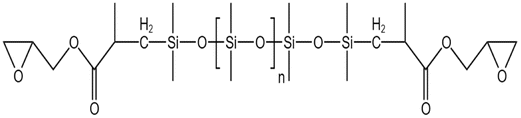 |
Toluene | 303 K | 3 wt% | 0.9 | 74 |
| 14 MPa | ||||||
| AQCA-PDMS | 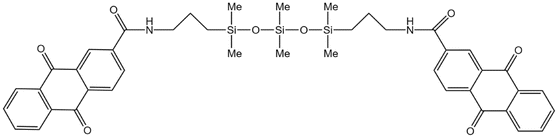 |
Hexane | 25 °C | 13.3 wt% | 0.86 | 75 |
| 20.7 MPa | ||||||
| HBD-1 | 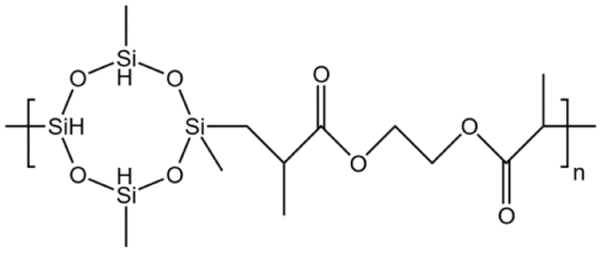 |
— | 32.15 °C | 5 wt% | 3.5 | 76 |
| 10 MPa | ||||||
| HBD-2 | 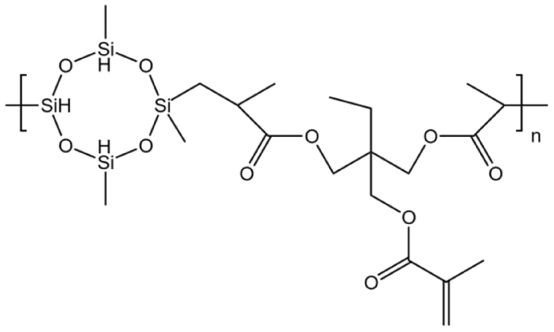 |
— | 32.15 °C | 5 wt% | 4.48 | 76 |
| 10 MPa | ||||||
| HS-1 | 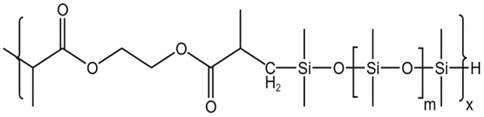 |
— | 16 MPa, 305 K | 5 wt% | 1.02 | 77 |
| HS-2 | 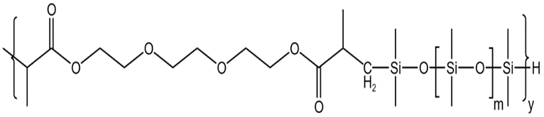 |
— | 16 MPa, 305 K | 5 wt% | 2.78 | 77 |
| HS-3 | 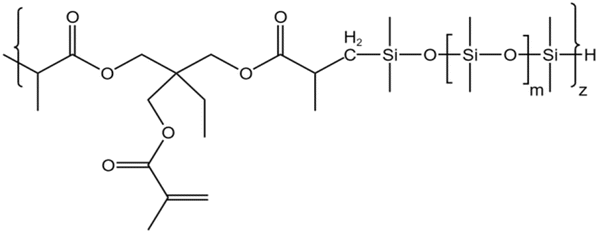 |
— | 16 MPa, 305 K | 5 wt% | 3.26 | 77 |
| Silsesquioxane | 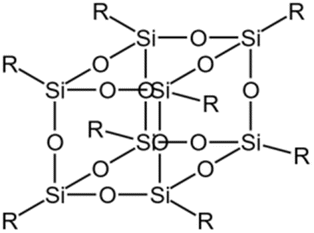 |
— | 180 bar, 55 °C | 6.9 wt% | 0.25 | 78 |
3.3 Hydrocarbon polymers
Due to the lack of highly polar groups in hydrocarbon polymers, their interactions with CO2 molecules are mostly through Lewis acid–base interactions and hydrogen bonding.79 Within the CO2 molecule, the oxygen atoms have a higher electron density than the carbon atom, resulting in a significant charge separation, with the carbon atom bearing a positive charge, acting as a Lewis acid, and the oxygen atoms bearing a negative charge, acting as Lewis bases79 (Fig. 3). The solubility of polymers in CO2 can be enhanced by adding electron-donating groups, such as carbonyl, ether, sulfone, and ester, to the hydrocarbon polymers (Fig. 4), which strengthens the Lewis acid–base and hydrogen-bonding interactions with CO2.81 The acetate group is an excellent electron-donating group, with poly(vinyl acetate) (PVAc) being one of the hydrocarbon polymers with the best solubility in CO2, but its solubility is significantly lower than that of PDMS and PFOA.82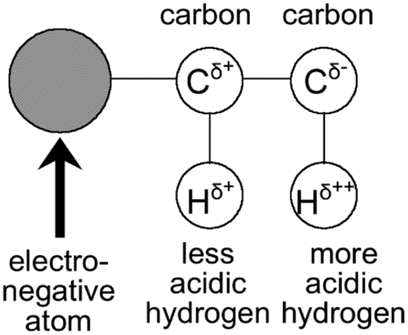 | ||
| Fig. 3 Effect of a highly electronegative atom on charge distribution in a molecule.80 | ||
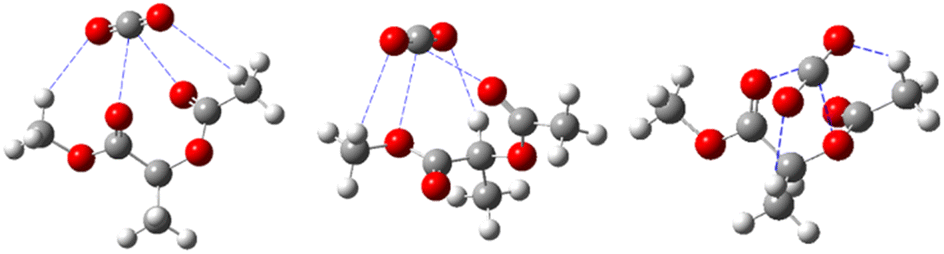 | ||
| Fig. 4 Hydrogen bonding and Lewis acid–base interactions between CO2 and polymers.60 | ||
Khanh83 simulated the interaction between a series of hydrocarbons containing hydroxyl, carbonyl, sulfone, carboxyl, and amide groups with CO2 molecules. The results showed that the stability of the complexes formed by carbonyl and sulfone groups with CO2 was stronger than that of other groups, and the benzene ring also had a certain promoting effect on the stability of the system. It was proven that the stability of the complex was mainly determined by the Lewis acid–base interactions and hydrogen bonding. Apart from the interaction between the carboxyl group and CO2, which was mainly derived from hydrogen bonding, the interaction between other groups and CO2 was mainly determined by the Lewis acid–base interactions.
Polypropylene oxide (PPO) with a molecular weight less than 2000 is more soluble in CO2 than poly(vinyl acetate) (PVAc), but its solubility significantly decreases at higher molecular weights. Therefore, Enick58 attempted to modify PPO by grafting with methyl ether, but this did not increase the solubility. They also investigated the effects of hydroxyl and acetate end groups on PPO and found that only the acetate-terminated PPO showed a decrease in dissolution pressure, by 3.4–6.9 MPa at 295 K. The hydrogen bonds formed by hydroxyl groups increased the intermolecular interactions, reducing the solubility. Then they discovered that low concentrations of poly(ethylene vinyl ether) (PEVE) had nearly the same dissolution pressure as PVAc. Subsequently, Lee84 demonstrated that low-molecular-weight PEVE could dissolve in CO2 but did not exhibit a significant thickening ability.
Xue45 simulated a random copolymer of poly(vinyl acetate–vinyl ether) PVAEE. Under conditions of 308 K and a CO2 density of 0.854 g cm−3, the simulated viscosity of scCO2 was reported to be 0.1268 cp. When the PVAEE content was 1.19 wt% and 2.35 wt%, the viscosity of the system reached 0.2037 cp and 0.4135 cp, respectively showing increases of 2–3 times. They compared the binding energy and dissociation energy of ester groups and ether bonds and found that ester groups could bind more easily to CO2 molecules than ether bonds.
Hu85 synthesized P(VAc-co-VEE) using vinyl acetate (VAc) and vinyl ethyl ether (VEE) groups and found that the random copolymer containing 30% VEE had the highest affinity for CO2. The VEE monomer could also reduce the interactions between polymers.
Wang80 designed a series of polymers, comprising OAO, PVMME, and PVMEE. OAO could dissolve up to 5 wt% at 298 K and 25 MPa, and PVMME could dissolve up to 3 wt% at 120 MPa. Under the same conditions, the solubility of PVMME in CO2 was lower than that of PVAc but better than that of PLA and PMA, because the ether oxygen in the main chain of PVMME had a higher affinity for CO2 than the carbonyl group in PMA.
Heller86 tested several commercially available hydrocarbon polymers soluble in hexane/ethanol, predicting their solubility behavior in CO2 by measuring their solubility in butane. It was ultimately found that poly-alpha-olefins (P1D) and atactic polybutene had the highest solubility, but subsequent research proved that only P-1-D had a very low solubility in CO2.
Tapriyal60 synthesized a non-fluorinated analog of PolyFAST, PolyBOVA, using vinyl acetate and a monomer with a rigid benzene ring side group. PolyBOVA with 5% benzoyl groups could dissolve up to 3 wt% at 298 K with a pressure of 64 MPa required, while it was insoluble in CO2 with 10% benzoyl groups. The viscosity of solutions with 1 wt% and 2 wt% PolyBOVA increased by only 40% and 80%, respectively.
Zhang87 synthesized a polyether-type carbon–hydrogen thickener, poly(ethylene oxide-co-propylene oxide) (PPOGPEAc), and compared it with the non-substituted polymer PPOAc. Under 36.3% PPOGPEAc, the viscosity of the system reached 0.35 mPa s, which was 2.3 times the thickening effect of PPOAc. The copolymer showed the best thickening effect when the phenyl content was 36.3 mol%. It was also found that adding more phenyl groups would increase the intramolecular interactions, affecting the intermolecular associations and reducing the system's solubility.
Raveendran and Wallen88 tested a series of acetylated carbohydrate substances for their solubility in CO2. The large number of hydrogen bonds and Lewis acid–base interactions formed by the carbonyl groups within the sugar molecules made them highly soluble in CO2. Potluri89 found that the higher the content of sugar molecules, the greater the solubility pressure. Cyclodextrin had a cloud point pressure of 25–45 MPa at concentrations of 5–30 wt% at 313 K.
Tapriyal90 synthesized a polymer, PAcGIcVE, with a polyethylene main chain and side chains containing acetylated sugar groups, and compared it with several amorphous polylactic acids (PLAs) with different end groups.91 PLA with end groups containing two ether bonds showed better results than PLA with end groups containing two ether bonds and branched side chains, indicating that linear ether oxygen bonds are more conducive to solubility. The solubility pressure of PAcGIcVE with a molecular weight of 40000 was 65–75 MPa, much lower than that of PLA, but 10–20 MPa higher than that of PVAc.
Due to environmental concerns over fluoropolymers and the high cost of siloxane polymers, which often require large amounts of co-solvents for viscosity enhancement, the development of hydrocarbon polymer thickeners for CO2 is a current research focus. However, hydrocarbon groups do not have a high affinity for CO2, and can only form a stable network structure within the solution through modification of the molecular structure to create Lewis acid–base interactions with CO2, resulting in a solution that is less viscous compared to fluorinated and siloxane polymers (Table 5).
| Polymer | Structure | Co-solvent | Conditions | Solubility | Viscosity/cp | Reference |
|---|---|---|---|---|---|---|
| PVAc | 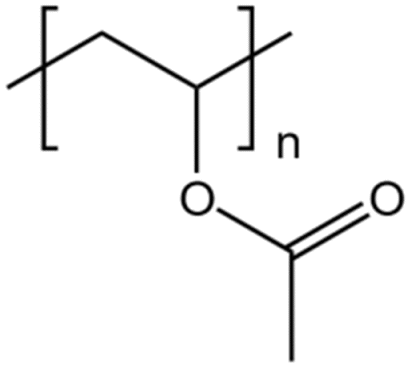 |
— | 308 K, 30.31 MPa | 0.05 wt% | — | 82 |
| PPO | 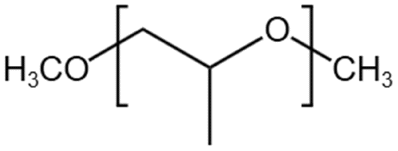 |
— | 295 K, 35 MPa | 3.5 wt% | — | 58 |
| PVAEE |  |
— | 308 K, 18.5 MPa | 2.35 wt% | 0.4135 | 45 |
| P(VAc-co-VEE) | 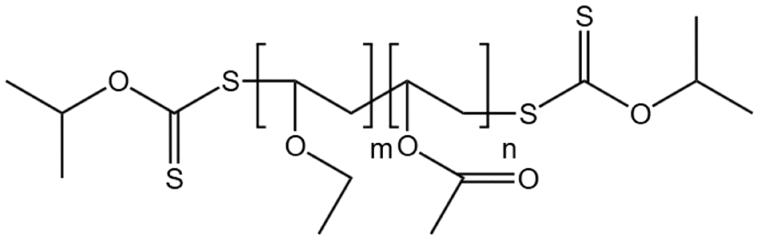 |
— | 35 °C, 24 MPa | 0.2 wt% | — | 85 |
| OAO |  |
— | 298 K, 25 MPa | 5 wt% | — | 80 |
| PVMME | 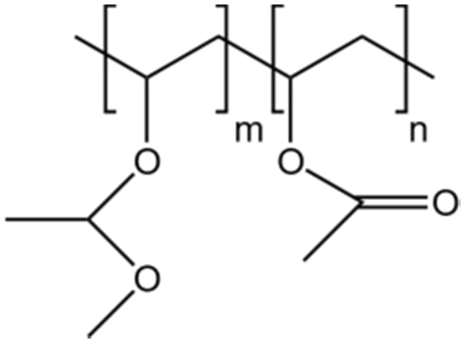 |
— | 298 K, 120 MPa | 3 wt% | — | 80 |
| PolyBOVA | 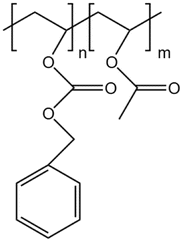 |
— | 298 K, 64 MPa | 2 wt% | 0.27 | 60 |
| PPOGPEAc | 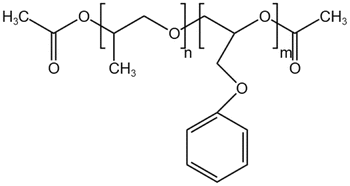 |
— | 30 °C, 35 MPa | 36.3% | 0.35 | 87 |
| Peracetylated sugar derivatives |  |
— | 313 K, 45 MPa | 30 wt% | — | 88 and 89 |
| PAcGIcVE |  |
— | 298 K, 52 MPa | 5 wt% | — | 90 |
| PLA |  |
— | 298 K, 50 MPa | 5 wt% | — | 91 |
| PLA |  |
— | 298 K, 60 MPa | 3 wt% | — | 91 |
| PLA |  |
— | 298 K, 120 MPa | 5 wt% | — | 91 |
3.4 Small molecular compounds
Small molecule compounds have greater solubility and can form rod-like, worm-like, or helical micelles in CO2 through associative interactions, making them potential alternatives to high-molecular-weight polymer thickeners. Similarly, these compounds also need to have a CO2-philic group, which enhances the solubility and a CO2-phobic group, which promotes intermolecular association.Amine-based compounds are considered to have a certain solubility in CO2 due to their weak self-association and low glass transition temperatures, as well as having a cohesive energy density similar to that of siloxanes. Enick58 tested polyvinylethyleneamine (PPEI) and a series of carbonyl-modified compounds, including PPMAEI, PEO, and PDMAA. These compounds showed good miscibility with CO2 in molecular dynamics simulations. However, the experimental results indicated that PPEI, PPMAEI, and PEO were insoluble in CO2 under a high pressure of 45 MPa. Here, the interaction forces between the C atoms and N atoms could not overcome the self-association of amine groups, despite the addition of carbonyl groups. Nevertheless, 0.7 wt% PDMAA swelled in CO2 at 45 MPa, indicating that the N atom, acting as an electron-donating group, enhanced the Lewis acid–base interaction between the carbonyl group and CO2.
J. P. Heller92 found that compounds centered on Sn atoms with three butyl arms and fluorine atoms could increase the viscosity of light hydrocarbons by three orders of magnitude at a low concentration of 1 wt%. Here, the butyl arms could extend into the alkanes to enhance the solubility, while the positively charged Sn atoms could take part in linear associative interactions with the negatively charged F atoms in neighboring molecules. The butyl arms could promote the stability of this associative structure, but these compounds were not soluble in CO2. Therefore, Shi93 synthesized a series of fluorostannanes with three fluoroalkyl arms, among which only the semi-fluorinated hexylfluorostannane increased the viscosity, by 2–3 times. The reason for the relatively low viscosity enhancement is that the fluorine atoms on the semi-fluorinated arms competed with the fluorine atoms bonded to the Sn atoms, disrupting the linear associative structure.
Research has shown that hydroxyl aluminum disoaps can thicken light hydrocarbons, with the ester chains at the molecular ends helping to form cylindrical micelles that can connect with each other, while the straight-chain ends do not have thickening ability. Enick94 attempted to synthesize a series of hydroxyl aluminum disoaps, modifying the end chains to fluorinated chains or branched alkyl chains, but none were soluble in CO2.
12-Hydroxystearic acid (HSA) is considered to be a potential CO2 thickener due to its ability to gel light hydrocarbons,95 However, it is only soluble in CO2 when ethanol is added as a co-solvent. A solution with 3 wt% HSA and 15 wt% ethanol could increase the viscosity of CO2 by 100 times at 28 °C and 12.4 MPa. HSA undergoes a gelation reaction when heated and dissolved in CO2 and then cooled. Hydrogen bonding causes the molecular chains to stack together, forming a fibrous network gel with a high porosity after the solution cools. However, this type of gel can adhere to the rock surface in porous media, making HSA unsuitable for the EOR process.
Zhou96 synthesized a fluorine-containing urea-based tetra-arm oligomer, BPFAUH. After reaching a concentration of 2%, the viscosity of the system increased only slightly, because the formation and rupture of hydrogen bonds reached equilibrium during the formation of the network structure.
Tapriyal60 synthesized a series of self-assembling compounds containing urea and acetate groups derived from acetylated sugars. The carbonyl oxygen in the urea group could form hydrogen bonds with hydrogen atoms, linking molecules to form a two-dimensional sheet-like structure. A bis-arm compound containing one bis-urea and two acetylated sugars displayed much lower solubility compared to a dendritic compound containing one bis-urea and four acetylated sugars under the same conditions.
Doherty97 synthesized three categories of small molecule compounds with amide or urea groups and a siloxane main chain: amide compounds, amides with benzene rings, and ureas with benzene rings. Their experiments showed that compounds containing urea groups were superior to amides and esters in thickening. Highly branched molecules dissolved more easily than linear ones. However, the thickening effect was achieved by adding a large amount of co-solvents. Molecules centered around benzene triurea had very little self-interaction and could not form sheet-like networks or long linear structures.
Small molecular thickeners have shown great potential for viscosity enhancement. Some research has aimed to enhance the solubility and viscosity of CO2 by incorporating nitrogen-containing groups (such as amine and urea groups) that can form weak interactions between N–N atoms and strong interactions between N–C atoms. Extensive experiments have proven that nitrogen-containing groups strengthen the interaction between other CO2-philic groups within the thickener and CO2 molecules, which helps to some extent in the dissolution of polymers in CO2. The weak interactions between N atoms also contribute to the increase in system viscosity. However, these groups only have a certain effect when added to other molecules with good solubility and they are not effective enough to be the main functional groups. Nevertheless, the advantage of forming a significant increase in system viscosity by combining with highly CO2-philic groups still give them a certain application potential (Table 6).
| Polymer | Structure | Co-solvent | Conditions | Solubility | Viscosity/cp | Reference |
|---|---|---|---|---|---|---|
| PDMAA | 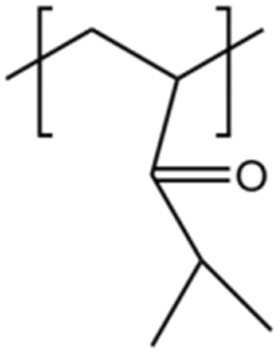 |
— | 298 K, 45 MPa | 0.7 wt% | — | 58 |
| Tri (semi-fluorinated hexyl tin fluoride) | 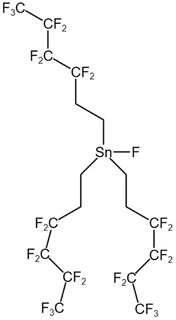 |
— | 297 K, 16.5 MPa | 4 wt% | 0.27 | 93 |
| Hydroxy aluminum disoap | 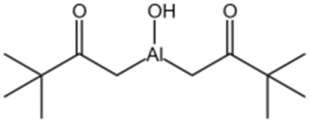 |
— | — | Insoluble | — | 94 |
| 12-HAS | 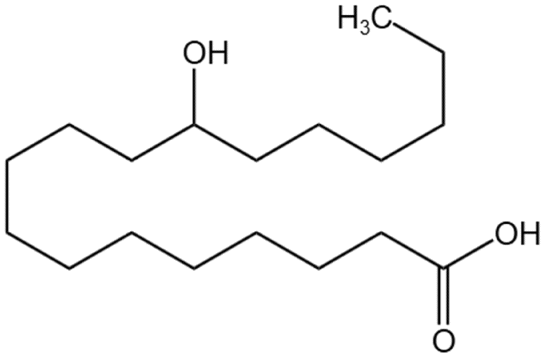 |
Ethanol | 28 °C, 12.4 MPa | 3 wt% | 7.6 | 95 |
| BPFAUH | 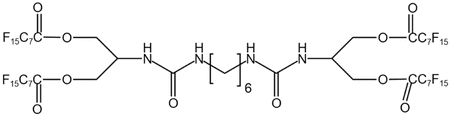 |
— | 313.13 K, 19 MPa | 3 wt% | 19.2 | 96 |
| Bis-urea polymer | 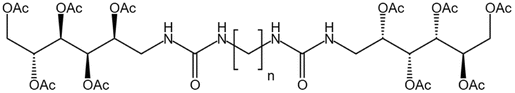 |
— | 298 K, 65 MPa | 5 wt% | — | 60 |
| Bis-urea polymer | 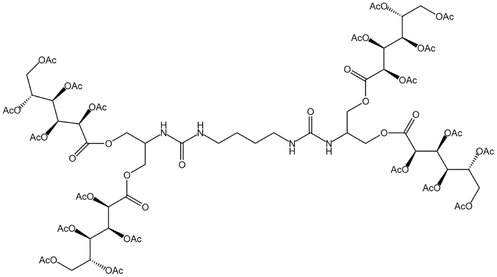 |
— | 298 K, 65 MPa | 1 wt% | — | 60 |
| Benzene trisurea polymer | 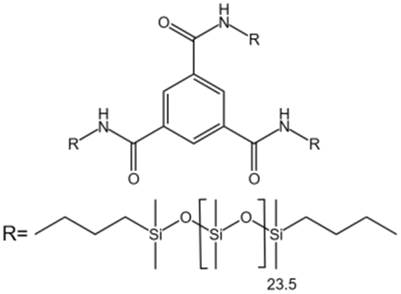 |
Toluene | 25 °C, 5000 psi | 1.5 wt% | 3 | 97 |
| trans-1,2 Cyclohexanedicarboxamides | 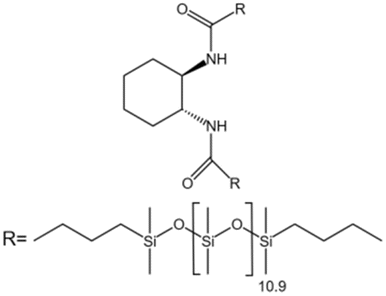 |
Toluene | 25 °C, 1500 psi | 1.6 wt% | 30 | 97 |
3.5 Nanoparticles
Nanomaterials have been applied in various industries for various purposes, such as electrocatalytic CO2 reduction, catalytic conversion of CO2, and the adsorption of CO2 by functionalized composite materials.98–100 Nanoparticles have a high specific surface area, which provides an excellent driving force for diffusion.101 They can travel long distances through the pore channels within the reservoir, interact with the injected fluids, and thus affect the fluid flow characteristics deep within the reservoir.102 Researchers have investigated the relationship between nanomaterials and CO2 across several domains, including the capture of CO2 through carbon nanotubes and the utilization of nanosilica for enhancing CO2 storage within geological formations.103,104 Introducing nanoparticles in the CO2 EOR process can not only alter certain properties, such as the viscosity, density, thermal conductivity, and interfacial tension, of the displacement fluid but also change the reservoir properties, such as the wettability of rock surfaces. The mechanisms of nanomaterials can be divided into two categories in the process of mobility control: first, by stabilizing CO2 foams that are prone to degradation due to rock adsorption and high reservoir temperatures by adsorbing at the two-phase interface; and second, by forming a stable suspension of metal or polymer nanoparticles in CO2 to thicken the fluid.105 The addition of nanoparticles has been proven to be an effective means of mobility control, with studies demonstrating that alternating the injection of thickened CO2 nanofluids and CO2 can achieve higher sweep efficiency.106,107However, the spatial repulsive forces between nanoparticles cannot offset the attractive van der Waals forces, making them prone to flocculation and difficult to stably disperse in CO2. Therefore, it is often necessary to modify the nanoparticles with CO2-philic groups to enhance their stability in solution. There are various types of nanoparticles, including polymer nanoparticles, metal nanoparticles, carbon-based nanoparticles, silicon-based nanoparticles, and Janus nanoparticles, among which SiO2 nanoparticles have been proven to have good thickening effects on displacement fluids.108 Dickson109 modified SiO2 particles with fluoroalkyl ethoxy silane, resulting in a polymer shell that formed a core–shell structure around the particles. This modification allowed SiO2 particles, which were previously insoluble in CO2, to remain stable for up to 30 min at 25 °C and 34 MPa. Visintin110 grafted fluoroalkyl chains onto silica and alumina particles, enabling them to be stably dispersed in CO2. Bell111 used isostearic acid as a ligand to promote the dispersion of silver nanoparticles in CO2, forming a stable solution at 295 K and 13.8 MPa.
Yates112 investigated the effect of the co-solvent hexane on the dispersion of nanoparticles. Similar to the mechanism of polymers dissolving in CO2, nanoparticles could only be stably dispersed when the density of CO2 reached a critical level. The experiment proved that the system could stably exist after adding SiO2 nanoparticles grafted with PDMS and 15 wt% hexane to CO2. The addition of hexane enhanced the interaction between the PDMS side chains and CO2, increasing the number of chains extending into the solvent and weakening the interchain interactions.
Xu113 prepared a type of fluoropolymer-modified nanoparticles for tracing and evaluating CO2 sequestration. These particles did not adsorb on the formation surface and aggregate. The polymer, which contained fluorocarbon, carbonyl oxygen, and benzene rings, was grafted onto Fe3O4 and SiO2 nanoparticles with a diameter of less than 50 nm, increasing their affinity with CO2. The polymer grafting density on the surface of Fe3O4 nanoparticles was higher. It dissolved at 34.5 °C and 16.1 MPa, while SiO2 nanoparticles dissolved at 51 °C and 28.3 MPa.
Zhang114 synthesized a copolymer of fluorinated and sulfonated styrene for fracture thickening. Their experiments proved that 1 wt% of the copolymer could increase the viscosity by 100 times at 333 K and 28 MPa. On this basis, a type of polyester fiber nanoparticle was introduced, and the results showed that this type of nanoparticle had good dispersion in the fluid. At 263 K, 28 MPa, and 5000 s−1, the addition of 0.5 wt% of the nanoparticles could reduce the internal friction by 17%. The system is suitable for the fracturing process, but its ability to pass through core pores is still under investigation.
Khaledialidusti115 used molecular dynamics simulation to investigate the rheological properties of a scCO2–CuO nanoparticle system. They reported that 1 wt% of CuO nanoparticles increased the viscosity of the system by 1.3–2.5 times within the temperature range of 350–410 K at 20 MPa. Su116 conducted a stability analysis of a hydroxylated carbon nanotube (MWCNTs)–scCO2 system. Their results indicated that as the volume concentration and temperature decreased, the stability of the nanofluid continuously improved. The system essentially reached a stable state after 4 min of circulating flow, while the sedimentation density continuously decreased with increasing the volume concentration and circulation flow time.
Wang117 added five types of nanoparticles, namely Cu-BTC, SiO2, MCC, MWCNTs, and TiO2, to the thickener HBD-2 and studied their synergistic effect on the fracturing system. It was found that only the addition of SiO2 could significantly increase the apparent viscosity. A system containing 1 wt% SiO2 nanoparticles and 5 wt% HBD-2 thickener was approximately 1.2–1.7 times more viscous than a system containing pure thickener under the conditions of 305–325 K and 10 MPa. The RDF results proved that the surface energy effect of SiO2 could effectively promote the crosslinking between the polymer molecular chains.
Gandomkar118 synthesized a graphene oxide (GO)/P-1-D nanocomposite polymer, and the results showed that as the content of GO nanoparticles increased, the polymer's dissolution pressure continuously decreased, with the highest dissolution pressure at 100 °C being 24.1 MPa. Compared with pure P-1-D, GO/P-1-D could increase the system's viscosity by more than 5 times, but this thickening system was only tested in light hydrocarbon gases, not CO2.
4. Application of CO2 direct thickeners
CO2-thickening systems are widely used in fracture and EOR processes. Due to the strong heterogeneity, low porosity, and low permeability of unconventional oil and gas reservoirs, it is necessary to rely on reservoir fracturing techniques to further exploit them. Traditional water-based fracturing has certain issues, such as high water consumption and environmental pollution.119 ScCO2 has been used as a fracturing fluid since the 1960s. Compared to water-based fracturing, supercritical CO2 fracturing has the advantages of a higher fracture strength and lower fracture pressure, and it does not clog pores, making it more suitable for the development of unconventional shale oil reservoirs.120 However, CO2 has a low viscosity and poor sand-carrying capacity, which hinders its field application. Adding thickeners to the system to increase the viscosity of CO2 can greatly enhance the effectiveness of CO2 fracturing.Because of the different application scenarios, the requirements for thickeners in CO2 EOR and CO2 fracturing are different. In fracturing experiments, the adsorption amount of thickeners in the formation, shear resistance, and thickening multiple are key to evaluation, as sand-carrying has higher viscosity requirements. Siloxane polymers are commonly used as thickeners in fracturing, while adding silicate or metal nanoparticles can further assist in thickening. To achieve the desired fracturing effect, a significant amount of thickener is required. Similarly, the CO2 EOR process requires great attention to the cost and environmental friendliness of thickeners. EOR requires the thickening system to continuously pass through small pores and maintain displacement pressure, while also considering the temperature and pressure resistance of the thickener and its adsorption properties in the formation. Small molecule thickeners and environmentally friendly hydrocarbon polymers have more application potential in the EOR process.
In practical applications, CO2 direct thickeners face numerous challenges. First, the common polymers available on the market are difficult to dissolve in CO2, and the synthesis process of the thickeners is often complex. The need to add a significant amount of co-solvents leads to higher industrial costs. Second, thickeners tend to adsorb significantly within the formation, especially after coming into contact with oil and water. When the formation conditions are not sufficient to maintain the CO2 thickening system as a stable single phase or at the critical point of a mixed phase, the solute can easily precipitate during flow. Moreover, CO2 can mix with crude oil, and further reactions can occur between the polymer and substances in the crude oil. Although these reactions are mostly detrimental to the dissolution of the polymers in CO2, they can also have certain positive effects. For example, adding PVAc and limonene to scCO2 can prevent the flocculation of non-hydrogen-bonding asphaltenes.121 Considering the current research, while ensuring that functional groups have a high affinity for CO2, the issue of adsorption also needs to be considered. Merely modifying the polymer at the molecular level makes it difficult to further improve the solubility of the polymer. Therefore, optimizing the system is the key to research on CO2 direct thickeners. The polymer modification approach should not be limited to its interactions with CO2; it is also necessary to consider changes in phase behavior after adding other auxiliary thickeners, such as nanoparticles. It is challenging for nanoparticles to achieve a stable dispersed state in CO2; however, if they are added to the CO2–polymer system and the affinity between the polymer and nanoparticles is increased, this can facilitate the uniform dispersion of nanoparticles while also enhancing the viscosity of the CO2 solution. In porous media, nanoparticles preferentially adsorb onto the formation surface, and polymer thickeners can still play a role in increasing viscosity. Based on these challenges, there are currently no field experiment reports on CO2 direct thickeners, and more convincing laboratory experiments are needed to substantiate their effectiveness.
5. Conclusion and outlook
Supercritical CO2 direct thickeners have significant importance for CO2 fracturing and CO2 EOR. The development of thickeners is still mainly at the laboratory research stage, which includes ongoing work on their synthesis, structural characterization, solubility and thickening tests, and exploration of their working mechanisms. Due to high costs and other reasons, there are currently no reports on field applications. Therefore, developing a low-cost, green thickener from a molecular perspective remains the primary research goal in the field of direct CO2 thickeners.In the system, hydrocarbon polymers lack highly polar groups and have poor affinity with CO2, resulting in much lower solubility compared to fluoropolymers and siloxane polymers. The dissolution and thickening effects need to be optimized by modifying the hydrocarbon thickeners. Fluoropolymers are limited by pollution issues and are difficult to widely apply, but fluorine-containing groups have good affinity with CO2 and high solubility in it. Adding a small amount of fluorine-containing groups to polymers with strong self-association can make the originally difficult-to-dissolve molecular chains or groups more stable in CO2. High-molecular-weight siloxane polymers have poor solubility in CO2, while-low-molecular weight siloxanes lack a thickening ability. However, the addition of siloxane chain segments can promote the dissolution of compounds in CO2 and so siloxanes are important materials for modifying hydrocarbon thickeners. For small molecule compounds that are more easily soluble, adjusting the intermolecular interaction forces and forming a stable network structure through intermolecular crosslinking is a key research focus. As research progresses, the addition of nanoparticles has brought new possibilities to the study of thickening agent systems. Nanoparticles can be modified themselves to have similar effects as surfactants, and can also change the properties of the displacing fluid through high surface energy and other microscopic effects. The addition of nanoparticles in CO2–polymer solutions can not only enhance the thickening effect but also reduce the adsorption of polymers onto formations, making it an important research direction worthy of attention. Further studies are needed to understand the mechanisms and factors influencing the uniform dispersion of nanoparticles in the solution, how to strengthen the interaction between the nanoparticles and polymers through modification, and whether the synergistic effect of nanoparticles and polymers can reduce adsorption in porous media.
In characterization methods, most of the characterization techniques for CO2 thickeners are still focused on compound synthesis, phase behavior, solubility pressure, viscosity characterization, and molecular dynamics simulation. The exploration of dissolution mechanisms and solvation patterns is not yet in-depth. It is necessary to start from microscopic factors, such as density, molecular weight, and polymer groups, and combine experiments and simulations to gradually explore the critical points and patterns of the morphological changes in CO2. Characterization often requires high-temperature and high-pressure environments, and the experimental conditions are harsh. There is a need to develop a set of faster, simpler, and standardized characterization methods. The morphological changes of the molecular chains in CO2 can refer to the theoretical studies of polymer chains in dilute solutions, using liquid solvents with solubility parameters close to that of CO2 to replace the CO2 gas, in order to determine the critical association concentration of the molecular chains. Semi-empirical regression equations can then be fitted based on experimental data.
In application, further research is needed on the mechanism of action of CO2 thickening systems. CO2, polymers, and oil–water phases react upon contact in formations, affecting the stability of a single system. Fluoropolymers can precipitate due to repulsive interactions with light hydrocarbons dissolved in CO2, and siloxane polymers can also interact with hydroxyl groups in water molecules. Current displacement experiments have shown that CO2 thickening systems can increase fluid viscosity, thereby increasing displacement pressure and enhancing the oil-recovery rates. However, after phase mixing internally, the main reason for the increased displacement pressure is likely not the increase in fluid viscosity but the precipitation of polymers causing plugging in the pores. Therefore, visualized microscopic displacement experiments are needed to demonstrate the mechanisms of action in the system.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analysed during the current study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the State Key Laboratory of Enhanced Oil and Gas Recovery of the Research Institute of Petroleum Exploration and Development for providing their support.References
- L. Shilun, Y. Tang and H. Chengxi, Reservoir Evaluation and Development, 2019, 9, 1–8 Search PubMed.
- X. Teng, Synthesis and performance evaluation of supercritical carbon dioxide green thickener, Master's thesis, Beijing University of Chemical Technology, 2023, pp. 12–18.
- R. Balch and B. McPherson, in All Days, SPE, Anchorage, Alaska, USA, 2016, p. SPE-180408-MS Search PubMed.
- Y. Hu, M. Hao, G. Chen, R. Sun and S. Li, Pet. Explor. Dev., 2019, 46, 753–766 CrossRef.
- D. Merchant, in All Days, CMTC, Houston, Texas, USA, 2017, p. CMTC-502866-MS Search PubMed.
- J. Qin, H. Han and X. Liu, Pet. Explor. Dev., 2015, 42, 232–240 CrossRef.
- H. Guo, J. Dong, Z. Wang, H. Liu, R. Ma, D. Kong, F. Wang, X. Xin, Y. Li and H. She, OnePetro, 2018, SPE-190286-MS Search PubMed.
- C. Meng, Z. Yang and Z. Ming, Pet. Sci. Technol., 2023, 42, 49–56 Search PubMed.
- W. Qian, Research on the characteristics of CO2 displacement and storage during supercritical CO2 injection in low permeability reservoirs, Doctoral dissertation, China University of Petroleum, Beijing, 2022, pp. 25–29.
- O. Massarweh and A. S. Abushaikha, Petroleum, 2022, 8, 291–317 CrossRef.
- H. F. Al-Riyami, F. Kamali and F. Hussain, in Day 3 Wed, April 26, 2017, SPE, Dammam, Saudi Arabia, 2017, p. D033S046R006 Search PubMed.
- H. Yongmao, W. Zenggui, J. Binshan, C. Yueming, L. Xiangjie and X. Petro, OnePetro, 2004, SPE 88883 Search PubMed.
- P. Nikolai, B. Rabiyat, A. Aslan and A. Ilmutdin, J. Therm. Sci., 2019, 28, 394–430 CrossRef CAS.
- K. Trickett, D. Xing, R. Enick, J. Eastoe, M. J. Hollamby, K. J. Mutch, S. E. Rogers, R. K. Heenan and D. C. Steytler, Langmuir, 2010, 26, 83–88 CrossRef CAS PubMed.
- J. Han, M. Lee, W. Lee, Y. Lee and W. Sung, Appl. Energy, 2016, 161, 85–91 CrossRef CAS.
- Z. Wang, S. Yang, H. Lei, M. Yang, L. Li and S. Yang, J. Pet. Sci. Eng., 2017, 150, 376–385 CrossRef CAS.
- S. Afzali, N. Rezaei and S. Zendehboudi, Fuel, 2018, 227, 218–246 CrossRef CAS.
- B. Brattekås and R. Seright, SPE J., 2023, 1–17 Search PubMed.
- G. Ren, H. Zhang and Q. P. Nguyen, SPE J., 2013, 18, 752–765 CrossRef.
- E. Luo, Y. Hu, B. Zhu, J. Wang and Z. Wang, Oilfield Chem., 2013, 30, 613–619 CAS.
- S. Kumar and A. Mandal, J. Pet. Sci. Eng., 2017, 157, 696–715 CrossRef CAS.
- X. Li, S. Wang, B. Yuan and S. Chen, in Day 4 Tue, April 17, 2018, SPE, Tulsa, Oklahoma, USA, 2018, p. D041S017R006 Search PubMed.
- J. R. Christensen, E. H. Stenby and A. Skauge, SPE Reserv. Eval. Eng., 2001, 4, 97–106 CrossRef CAS.
- D. L. Tiffin and W. F. Yellig, Soc. Pet. Eng. J., 1983, 23, 447–455 CrossRef CAS.
- J.-G. J. Shyeh-Yung, OnePetro, 1991, SPE-22651-MS Search PubMed.
- J. D. Rogers and R. B. Grigg, in All Days, SPE, Tulsa, Oklahoma, 2000, p. SPE-59329-MS Search PubMed.
- A. Valeev and A. Shevelev, in Day 3 Wed, October 18, 2017, SPE, Moscow, Russia, 2017, p. D033S030R001 Search PubMed.
- X. Xu, A. Saeedi, Y. Zhang and K. Liu, in Day 2 Wed, March 23, 2016, OTC, Kuala Lumpur, Malaysia, 2016, p. D022S001R005 Search PubMed.
- M. A. Almobarky, Z. A. Yousef and D. Schechter, OnePetro, 2017, CMTC-486486-MS Search PubMed.
- J. P. Heller, C. L. Lien and M. S. Kuntamukkula, Soc. Pet. Eng. J., 1985, 25, 603–613 CrossRef CAS.
- D. Xing, B. Wei, K. Trickett, A. Mohamed, J. Eastoe, Y. Soong and R. Enick, in All Days, SPE, Tulsa, Oklahoma, USA, 2010, p. SPE-129907-MS Search PubMed.
- S. H. Raza, Soc. Pet. Eng. J., 1970, 10, 328–336 CrossRef CAS.
- J. O. Alvarez, I. W. Saputra and D. S. Schechter, SPE J., 2018, 23, 2103–2117 CrossRef CAS.
- E. Mayoral, J. A. Arcos-Casarrubias and A. Gama Goicochea, Colloids Surf., A, 2021, 615, 126244 CrossRef CAS.
- S. Liang, W. Luo, Z. Luo, W. Wang, X. Xue and B. Dong, Molecules, 2023, 28, 8042 CrossRef CAS PubMed.
- Z. Liu, P. Wei, Y. Qi, X. Huang and Y. Xie, Carbohydr. Polym., 2023, 311, 120759 CrossRef CAS PubMed.
- F. Friedmann, T. L. Hughes, M. E. Smith, G. P. Hild, A. Wilson and S. N. Davies, SPE Reserv. Eval. Eng., 1999, 2, 4–13 CrossRef CAS.
- W.-L. Kang, B.-B. Zhou, M. Issakhov and M. Gabdullin, Pet. Sci., 2022, 19, 1622–1640 CrossRef CAS.
- B. Brattekås, R. Seright and G. Ersland, SPE Prod. Oper., 2020, 35, 202–213 Search PubMed.
- Z. Wang, B. Bai, Y. Long and L. Wang, SPE J., 2019, 24, 2398–2408 CrossRef CAS.
- N. Li, H. Zhang, X. Ren, J. Wang, J. Yu, C. Jiang, H. Zhang and Y. Li, Gas Sci. Eng., 2024, 125, 205312 CrossRef CAS.
- X. Sun, Y. Zhang, G. Chen and Z. Gai, Energies, 2017, 10, 345 CrossRef.
- H. Zaberi, Core flooding study of CO2-soluble polymers for improved mobility control and conformance control, Master's thesis, University of Tulsa, 2011, pp. 28–32.
- P. Raffa, A. A. Broekhuis and F. Picchioni, J. Pet. Sci. Eng., 2016, 145, 723–733 CrossRef CAS.
- P. Xue, J. Shi, X. Cao and S. Yuan, Chem. Phys. Lett., 2018, 706, 658–664 CrossRef CAS.
- J. P. Heller, D. K. Dandge, R. J. Card and L. G. Donaruma, Soc. Pet. Eng. J., 1985, 25, 679–686 CrossRef CAS.
- C. Lepilleur, E. J. Beckman, H. Schonemann and V. J. Krukonis, Fluid Phase Equilib., 1997, 134, 285 CrossRef CAS.
- Y. Qin, X. Yang, Y. Zhu and J. Ping, J. Phys. Chem. C, 2008, 112, 12815–12824 CrossRef CAS.
- A. Dardin, J. M. DeSimone and E. T. Samulski, J. Phys. Chem. B, 1998, 102, 1775–1780 CrossRef CAS.
- M. A. Meilchen, B. M. Hasch and M. A. McHugh, Macromolecules, 1991, 24, 4874–4882 CrossRef CAS.
- T. Huang, J. M. Messman and J. W. Mays, Macromolecules, 2008, 41, 266–268 CrossRef CAS.
- J. B. McClain, D. Londono, J. R. Combes, T. J. Romack, D. A. Canelas, D. E. Betts, G. D. Wignall, E. T. Samulski and J. M. DeSimone, J. Am. Chem. Soc., 1996, 118, 917–918 CrossRef CAS.
- J. M. DeSimone, Z. Guan and C. S. Elsbernd, Science, 1992, 257, 945–947 CrossRef CAS PubMed.
- Y.-L. Hsiao, E. E. Maury, J. M. DeSimone, S. Mawson and K. P. Johnston, Macromolecules, 1995, 28, 8159–8166 CrossRef CAS.
- P. Lemaire, E. Beckman and R. Enick, J. Supercrit. Fluids, 2022, 190, 105728 CrossRef CAS.
- J. Xu and R. M. Enick, OnePetro, 2001, SPE-84949-PA Search PubMed.
- R. Enick, E. Beckman, A. Yazdi, V. Krukonis, H. Schonemann and J. Howell, J. Supercrit. Fluids, 1998, 13, 121–126 CrossRef CAS.
- R. Enick, E. Beckman and A. Hamilton, Oil & Natural Gas Technology, 2005, pp. 68–72 Search PubMed.
- A. G. Goicochea and A. Firoozabadi, J. Phys. Chem. C, 2019, 123, 29461–29467 CrossRef CAS.
- D. Tapriyal, Design of non-fluorous CO2 soluble compounds, Doctoral dissertation, University of Pittsburgh, 2009, pp. 39–70.
- C. Dai, P. Liu, M. Gao, Z. Liu, C. Liu, X. Wang, S. Liu, M. Zhao and H. Yan, J. Mol. Liq., 2022, 119563 CrossRef CAS.
- C. Dai, P. Liu, M. Gao, Z. Liu, C. Liu, Y. Wu, X. Wang, S. Liu, M. Zhao and H. Yan, J. Mol. Liq., 2022, 360, 119563 CrossRef CAS.
- W. Sun, B. Sun, Y. Li, X. Huang, H. Fan, X. Zhao, H. Sun and W. Sun, Polymers, 2018, 10, 268 CrossRef PubMed.
- S. Kilic, R. M. Enick and E. J. Beckman, J. Supercrit. Fluids, 2019, 146, 38–46 CrossRef CAS.
- Z. Huang, C. Shi, J. Xu, S. Kilic, R. M. Enick and E. J. Beckman, Macromolecules, 2000, 33, 5437–5442 CrossRef CAS.
- J. Zhang, L. Q. Rao, S. W. Meng, G. W. Lu, R. Zhang, W. S. Yu and Q. Yang, in Energy and Sustainability V: Special Contributions, ed. H. H. AlKayiem, C. A. Brebbia and S. S. Zubir, Wit Press, Southampton, 2015, pp. 175–182 Search PubMed.
- D. Rousseau, S. Renard, B. Prempain, C. Féjean and S. Betoulle, in All Days, SPE, Tulsa, Oklahoma, USA, 2012, p. SPE-154055-MS Search PubMed.
- J. H. Bae and C. A. Irani, SPE Adv. Technol. Ser., 1993, 1, 166–171 CrossRef.
- M. Du, X. Sun, C. Dai, H. Li, T. Wang, Z. Xu, M. Zhao, B. Guan and P. Liu, J. Pet. Sci. Eng., 2018, 166, 369–374 CrossRef CAS.
- M. Zhao, R. Yan, Y. Li, Y. Wu, C. Dai, H. Yan, Z. Liu, Y. Cheng and X. Guo, Fuel, 2022, 124358 CrossRef CAS.
- Q. Li, Y. Wang, Q. Li, G. Foster and C. Lei, RSC Adv., 2018, 8, 8770–8778 RSC.
- Q. Li, F. Wang, Y. Wang, B. Bai, J. Zhang, C. Lili, Q. Sun, Y. Wang and K. Forson, J. Mol. Liq., 2023, 376, 121394 CrossRef CAS.
- Q. Li, Y. Wang, F. Wang, X. Ning, Z. Chuanbao, J. Zhang and C. Zhang, Environ. Sci. Pollut. Res., 2022, 29, 17682–17694 CrossRef CAS PubMed.
- Y. Wang, Q. Li, W. Dong, Q. Li, F. Wang, H. Bai, R. Zhang and A. B. Owusu, RSC Adv., 2018, 8, 39787–39796 RSC.
- M. J. O'Brien, R. J. Perry, M. D. Doherty, J. J. Lee, A. Dhuwe, E. J. Beckman and R. M. Enick, Energy Fuels, 2016, 30, 5990–5998 CrossRef.
- B. Liu, Y. Wang and L. Liang, Polymers, 2020, 13, 78 CrossRef PubMed.
- B. Liu, Y. Wang, L. Liang and Y. Zeng, RSC Adv., 2021, 11, 17197–17205 RSC.
- G. Gallo, E. Erdmann and C. N. Cavasotto, ACS Omega, 2021, 6, 24803–24813 CrossRef CAS PubMed.
- P. Raveendran and S. L. Wallen, J. Am. Chem. Soc., 2002, 124, 12590–12599 CrossRef PubMed.
- Y. Wang, L. Hong, D. Tapriyal, I. C. Kim, I.-H. Paik, J. M. Crosthwaite, A. D. Hamilton, M. C. Thies, E. J. Beckman, R. M. Enick and J. K. Johnson, J. Phys. Chem. B, 2009, 113, 14971–14980 CrossRef CAS PubMed.
- D. Denney, J. Pet. Technol., 2013, 65, 89–91 CrossRef.
- T. Zhu, H. Gong, M. Dong, Z. Yang, C. Guo and M. Liu, Fluid Phase Equilib., 2018, 458, 264–271 CrossRef CAS.
- P. N. Khanh and N. T. Trung, in Carbon Dioxide Chemistry, Capture and Oil Recovery, ed. I. Karamé, J. Shaya and H. Srour, InTech, 2018 Search PubMed.
- J. J. Lee, S. Cummings, A. Dhuwe, R. M. Enick, E. J. Beckman, R. Perry, M. Doherty and M. O'Brien, in All Days, SPE, Tulsa, Oklahoma, USA, 2014, p. SPE-169039-MS Search PubMed.
- D. Hu, S. Sun, P.-Q. Yuan, L. Zhao and T. Liu, J. Phys. Chem. B, 2015, 119, 12490–12501 CrossRef CAS PubMed.
- D. K. Dandge and J. P. Heller, in All Days, SPE, San Antonio, Texas, 1987, p. SPE-16271-MS Search PubMed.
- Y. Zhang, Z. Zhu and J. Tang, Fluid Phase Equilib., 2021, 532, 112932 CrossRef CAS.
- P. Raveendran and S. L. Wallen, J. Am. Chem. Soc., 2002, 124, 7274–7275 CrossRef CAS PubMed.
- V. K. Potluri, A. D. Hamilton, C. F. Karanikas, S. E. Bane, J. Xu, E. J. Beckman and R. M. Enick, Fluid Phase Equilib., 2003, 211, 211–217 CrossRef CAS.
- D. Tapriyal, Y. Wang, R. M. Enick, J. K. Johnson, J. Crosthwaite, M. C. Thies, I. H. Paik and A. D. Hamilton, J. Supercrit. Fluids, 2008, 46, 252–257 CrossRef CAS.
- S. E. Conway, H. -S. Byun, M. A. McHugh, J. D. Wang and F. S. Mandel, J. Appl. Polym. Sci., 2001, 80, 1155–1161 CrossRef CAS.
- J. P. Heller and D. K. Dandge, US Pat., US4607696A, 1986 Search PubMed.
- C. Shi, Z. Huang, E. J. Beckman, R. M. Enick, S.-Y. Kim and D. P. Curran, Ind. Eng. Chem. Res., 2001, 40, 908–913 CrossRef CAS.
- R. M. Enick, OnePetro, 1991, SPE-21016-MS Search PubMed.
- P. Gullapalli, J. S. Tsau and J. P. Heller, OnePetro, 1995, SPE-28979-MS Search PubMed.
- M. Zhou, H. Tu, Y. He, P. Peng, M. Liao, J. Zhang, X. Xu, W. He, Y. Zhao and X. Guo, J. Mol. Liq., 2020, 312, 113090 CrossRef CAS.
- M. D. Doherty, J. J. Lee, A. Dhuwe, M. J. O'Brien, R. J. Perry, E. J. Beckman and R. M. Enick, Energy Fuels, 2016, 30, 5601–5610 CrossRef CAS.
- L. Xiaoyu, Plasmonic regulation of photo/photothermal CO2 catalytic reaction and mechanism, Doctoral dissertation, Shandong University, 2024, pp. 187–194.
- L. Shuo, Fabrication and performance of polyoxyethlene based membrane for CO2 separation, Doctoral dissertation, Beijing University of Technology, 2023, pp. 85–92.
- D. Tong, Research on regulating electronic structure of copper-sulfur-based electrocatalysts and their performance in carbon dioxide reduction, Doctoral dissertation, Beijing University of Chemical Technology, 2024, pp. 72–79.
- R. D. Shah, in All Days, SPE, New Orleans, Louisiana, 2009, p. SPE-129539-STU Search PubMed.
- M. Al-Shargabi, S. Davoodi, D. A Wood, V. S. Rukavishnikov and K. M. Minaev, ACS Omega, 2022, 12, 9984–9994 CrossRef PubMed.
- N. H. Solangi, R. R. Karri, N. M. Mubarak and S. A. Mazari, Process Saf. Environ. Prot., 2024, 185, 1012–1037 CrossRef CAS.
- O. Olayiwola; N. Coulibaly; N. Liu; B. Guo, OnePetro, 2024, SPE-218367-MS Search PubMed.
- A. U. Rognmo, H. Horjen and M. A. Fernø, Int. J. Greenhouse Gas Control, 2017, 64, 113–118 CrossRef CAS.
- G. Gallo and E. Erdmann, in Day 1 Wed, May 17, 2017, SPE, Buenos Aires, Argentina, 2017, p. D011S001R003 Search PubMed.
- V. Barkhordari and A. Jafari, J. Water Environ. Nanotechnol., 2018, 3, 12–21 CAS.
- S. Davoodi, M. Al-Shargabi, D. A. Wood, V. S. Rukavishnikov and K. M. Minaev, Fuel, 2022, 324, 124669 CrossRef CAS.
- J. L. Dickson, P. S. Shah, B. P. Binks and K. P. Johnston, Langmuir, 2004, 20, 9380–9387 CrossRef CAS PubMed.
- P. M. Visintin, R. G. Carbonell, C. K. Schauer and J. M. DeSimone, Langmuir, 2005, 21, 4816–4823 CrossRef CAS PubMed.
- P. W. Bell, M. Anand, X. Fan, R. M. Enick and C. B. Roberts, Langmuir, 2005, 21, 11608–11613 CrossRef CAS PubMed.
- M. Z. Yates, P. S. Shah, K. P. Johnston, K. T. Lim and S. Webber, J. Colloid Interface Sci., 2000, 227, 176–184 CrossRef CAS PubMed.
- Y. Xu, L. Chen, Y. Zhao, L. M. Cathles and C. K. Ober, Environ. Sci.:Nano, 2015, 2, 288–296 RSC.
- J. Zhang, S. Meng*, H. Liu, X. Lv, R. Zhang and B. Yu, in All Days, SPE, Nusa Dua, Bali, Indonesia, 2015, p. SPE-176221-MS Search PubMed.
- R. Khaledialidusti, A. K. Mishra and A. Barnoush, J. Chem. Phys., 2018, 149, 224702 CrossRef PubMed.
- Z. Su, L. Yang, N. Zhao, J. Song, X. Li and X. Wu, Appl. Therm. Eng., 2024, 241, 122347 CrossRef CAS.
- Y. Wang, B. Liu, D. Li and L. Liang, Energy Sources, Part A, 2022, 44, 6602–6617 CrossRef CAS.
- A. Gandomkar and M. Sharif, J. Pet. Sci. Eng., 2020, 194, 107491 CrossRef CAS.
- L. Thomas, H. Tang, D. M. Kalyon, S. Aktas, J. D. Arthur, J. Blotevogel, J. W. Carey, A. Filshill, P. Fu, G. Hsuan, T. Hu, D. Soeder, S. Shah, R. D. Vidic and M. H. Young, J. Pet. Sci. Eng., 2019, 173, 793–803 CrossRef CAS.
- Y. Feng, K. Haugen and A. Firoozabadi, J. Geophys. Res.:Solid Earth, 2021, 126, e2021JB022509 CrossRef CAS.
- M. Sedghi and L. Goual, OnePetro, 2016, SPE-179040-MS Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |