
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular complexation of C60 and C70 by helical nanographene incorporating N-heterotriangulene and hexabenzocoronene subunits†
Marina Kinzelmann a,
Nina Fröhlichb,
Frederik Gnanntb,
Jan Borstelmannf,
Stefan Frühwaldc,
Christoph Oleszake,
Norbert Juxe,
Andreas Görlingcd,
Milan Kivalaf and
Thomas Drewello*a
a,
Nina Fröhlichb,
Frederik Gnanntb,
Jan Borstelmannf,
Stefan Frühwaldc,
Christoph Oleszake,
Norbert Juxe,
Andreas Görlingcd,
Milan Kivalaf and
Thomas Drewello*a
aPhysical Chemistry I, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany. E-mail: thomas.drewello@fau.de
bOrganic Chemistry I, Friedrich-Alexander-Universität Erlangen-Nürnberg, Nikolaus-Fiebiger-Straße 10, 91058 Erlangen, Germany
cTheoretical Chemistry, Friedrich-Alexander-Universität Erlangen-Nürnberg, Egerlandstraße 3, 91058 Erlangen, Germany
dErlangen National High Performance Computing Center (NHR@FAU), Martensstr. 1, 91058 Erlangen, Germany
eOrganic Chemistry II, Friedrich-Alexander-Universität Erlangen-Nürnberg, Nikolaus-Fiebiger-Straße 10, 91058 Erlangen, Germany
fInstitute of Organic Chemistry, Ruprecht-Karls-Universität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany
First published on 31st January 2025
Abstract
Supramolecular host–guest complexes are studied in the gas-phase evaluating a new host molecule for fullerenes (C60 and C70). The new host molecule is a double N-heterotriangulene-[5]helicene (NTH), consisting of two N-heterotriangulene (N-HTA) blades embedded into a hexabenzocoronene-like backbone with helically curved topology. Host–guest complexes of [1:1]+˙/2+, [1:2]+˙/2+, [2:1]2+ and [2:3]2+ stoichiometry and charge state are formed by electrospray ionization-mass spectrometry (ESI-MS). Ion formation occurs through electrochemical oxidation of the N-HTA moieties. Energy-resolved collision-induced dissociation (ER-CID) experiments reveal the noncovalent binding of the fullerenes to the NTH molecule and provide an order of stability for the complexes. Density-functional theory (DFT) calculations establish the lowest energy geometries of the complexes.
Introduction
In recent years, tremendous progress has been made in the area of molecular nanographenes.1–26 Synthetic efforts were directed towards bottom-up approaches in order to gain control over size, structure and properties of the nanographenes.27,28 A major motivation behind these efforts lies in their potential applications in photovoltaic, molecular electronics and sensing.2,29–32 Nanographenes are particularly suited to overcome the zero band gap problem2 of pristine single-layer graphene, which poses severe limitations on electronic applications of graphene.33 Crucial to improving the applicability of molecular nanographenes is the detailed understanding of electron and energy transfer processes34 and connected with this, insight into the interaction of nanographenes with other molecules. Along those lines, the host–guest chemistry of nanomaterials with fullerenes (C60 and C70) has been of prime interest. Fullerenes are widely established as electron acceptors in electronic applications.35 But also their unique spherical structure has an impact on their host–guest chemistry. The importance of shape complementarity to the host–guest chemistry of nanomaterials with fullerenes has been the subject of several reviews.36–39 Crucial to the formation and stability of such complexes is thus the precise fit of host and guest molecule. Noncovalent interaction and supramolecular complex formation with the convex fullerene surface will be enhanced through a complementary concave surface of the nanographene. In other words, for the perfect fit with the fullerene, the nanomaterial should be curved. Curvature can be induced into the two-dimensional (2D) honey-comb lattice by introducing non-hexagonal rings.40 The implementation of a five-membered ring (or lower) will lead to positive Gaussian curvature (bowl shape structure) and introducing a seven-membered ring (or higher)41,42 results in negative Gaussian curvature (saddle shape structure). There are even examples of nanographenes possessing both curvature motifs simultaneously.43–48 Retaining the six-membered rings, curvature can be created by introducing a helical twist to the molecule.40A helical twist can be imposed on a system by intentionally inducing steric repulsion between different, overlapping parts of the molecule. This is the case in the title molecule of this study shown in Fig. 1a. In the double N-heterotriangulene-[5]helicene (NTH) studied here, two N-heterotriangulene (N-HTA) units constitute the terminal parts of a [5]helicene. The [5]helicene is part of a π-extension which is reminiscent of the hexa-peri-hexabenzocoronene (HBC) molecule (Fig. 1c). The dimethylmethylene bridges of the N-HTA moieties protrude out of the molecular plane, causing steric repulsion of the two units, ultimately leading to a distortion of the π-system from planarity. This curvature in the NTH structure offers potentially a good interaction area for the formation of supramolecular complexes with C60 and C70. Fig. 1 also depicts the synthetic precursor of the NTH molecule (Fig. 1b), which turns into the NTH molecule by cyclodehydrogenation.
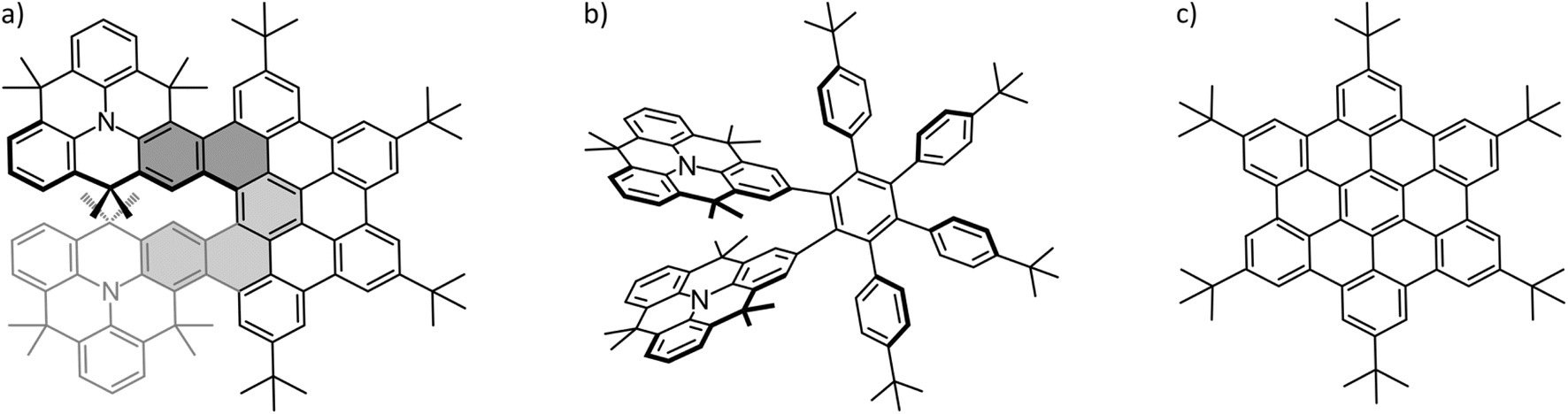 | ||
| Fig. 1 Structure of the investigated molecules: (a) NTH, (b) the NTH precursor and (c) tbutyl-decorated hexabenzocoronene (HB-HBC). | ||
The gas-phase host–guest chemistry of single-core N-HTA hosts with C60 has been the topic of an earlier investigation by us.49 In the present study, we employ electrospray ionization-(tandem) mass spectrometry (ESI-MS(/MS)) to ionize the NTH molecule and characterise the complexes formed with C60 and C70. ESI is a soft ionization method, which may allow both the direct transfer of intact noncovalent complexes from solution into the gas-phase, as well as their formation through aggregation phenomena during the spraying process. The mass spectrometry-based gas-phase experiment on solitary ions enables the investigation of intrinsic properties within complexes without solvent effects or the influence of counter ions.50 Hosseini et al. showed that the fullerene solubility has an inverse correlation on the binding strength of host–guest complexes.51 Therefore, association constants measured in solution will be dependent upon the employed solvent. Energy-resolved collision-induced dissociation experiments provide insight into the fragmentation dynamics of noncovalent complexes and allow the establishment of relative stability orders. Thus, gas-phase studies of host–guest systems provide insight into intermolecular interactions, representing a unique complementary to condensed phase or solid state investigations. DFT calculations accompany the experimental findings and are used to establish the geometries of the complexes.
Experimental
The racemic NTH was synthesized following a reported procedure.52 A dedicated publication detailing the NTH synthesis and its photophysical and redox properties will be published separately.53 The 1H- and 13C-NMR spectra of NTH, as well as a link to the crystal structure are provided in the ESI.† The fullerenes (C60 and C70) and trifluoroacetic acid (TFA) were purchased from Merck. The solvents dichloromethane (DCM), acetonitrile (ACN) and toluene were purchased from VWR chemicals in HPLC grade purity. The stock solutions of NTH and the fullerenes were prepared in DCM (0.5 g L−1) and toluene (1.0 g L−1), respectively. The final concentration for the ESI measurements were 1 × 10−5 M and 5 × 10−5 M for the NTH and fullerenes, respectively, in a mixture of DCM and ACN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1; v:v) to which a small amount of TFA (2 μL) was added.
:1; v:v) to which a small amount of TFA (2 μL) was added.
All MS1 and MS2 measurements were performed with a quadrupole time-of-flight (qToF) instrument (micrOTOF-QII, Bruker Daltonics, Bremen, Germany). The analyte solution was directly injected into the ESI source with a flow rate of 3.0 μL min−1; the temperature of the nitrogen counter flow was set to 180 °C. All measurements were performed in the positive ion mode, whereby the capillary voltage was set to 4.5 kV and the endplate offset to −0.5 kV. For the MS2 experiments, the precursor ions were selected by the mass analyzer quadrupole and accelerated into the collision cell quadrupole. A Parker LCMS64 nitrogen generator provided the nitrogen, used as a collision gas, with a purity of 99.999% and a flow rate of 0.5 L min−1. The instrument parameters were optimized to obtain good intensities for each experiment.
The survival yield (SY)54,55 of the precursor ions was calculated as the intensity ratio of the ion of interest to all observed ions and was recorded as a function of the collision energy in the centre-of-mass frame (Ecom).
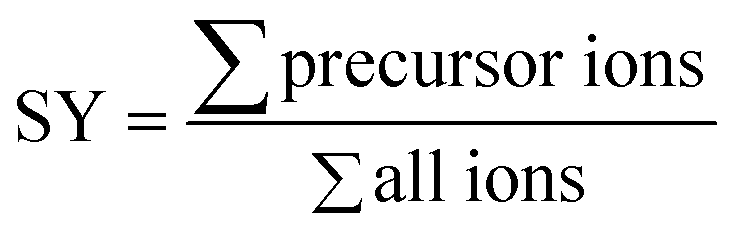
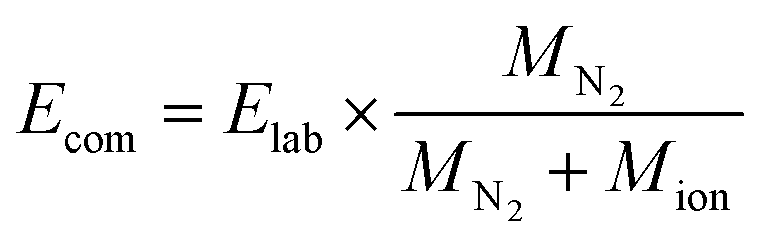
The breakdown graphs were obtained under multiple collision conditions and fitted with a sigmoid function. The collision energy, E50, at witch 50% of the precursor ions is dissociated is used as a measure of its relative stability.
DFT calculations on the formed complexes were performed with the TURBOMOLE program package (Version 7.3).56 Because of the increased size of the fullerene aggregates the GGA functional PBE was used in combination with the basis set def2-TZVP57 and the D3 dispersion correction to correctly account for the dispersion interactions between NTH and fullerene.58 Fragmentation energies were computed using fully relaxed structures of the complexes and fragments, respectively. An estimation of the BSSE error can be found in the ESI.† For the visualization of these interactions the program NCIPLOT (Version 3.0) was used.59 Areas of noncovalent interactions were determined by calculating and visualizing contour plots of the reduced density gradient given by
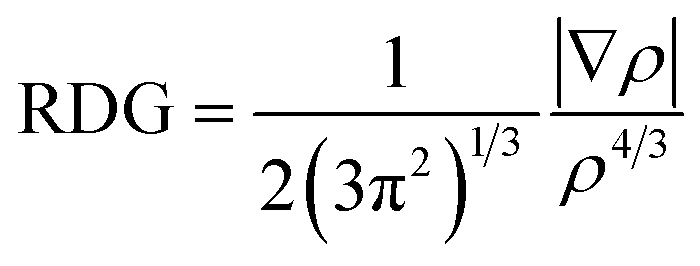
The contour plots were visualized at areas of low electron density below a chosen threshold, for which ρ = 0.2 was selected. In order to identify the attractive or repulsive nature of the noncovalent interaction the expression sign(λ2)ρ was evaluated, which contains the electron density ρ and the second largest eigenvalue of the electron density Hessian matrix which changes its sign according to the nature of the noncovalent interaction. For the visualization isosurfaces of the RDG at 0.3 arbitrary units were used and colored according to a color scale of −0.05 < ρ < 0.05.
Results and discussion
Electrospraying a NTH/C60 analyte solution results in the positive-ion ESI mass spectrum displayed in Fig. 2. The most abundant ion corresponds to the molecular dication NTH2+ (m/z 661.9). The other intense signals are assigned to the molecular ion NTH+˙ (m/z 1323.7) as well as to the doubly and singly charged [1:1] NTH–fullerene complexes at m/z 1021.9 and 2043.7, respectively. Less intense are the signals for the [1:2] NTH(C60)2+˙, [1:2] NTH(C60)22+, [2:1] NTH2(C60)2+ and the [2:3] NTH2(C60)32+ complexes at m/z 2764.8, m/z 1382.4, m/z 1683.7 and m/z 2404.8, respectively. The molecular ions of the bare NTH host are the most abundant species observed, followed by the [1:1] complexes with C60 and eventually the larger complexes with more than only one host and/or guest molecule incorporated. This product distribution is fully in line with a plausible formation scenario in which the [1:1] complex is initially formed with further coordination to it by NTH and/or C60 towards the larger complexes. All ions observed are either radical cations or dications and as such the result of single or double electron transfer reactions. Considering that the ion formation by ESI is commonly based on acid/base chemistry, the essential lack of protonation is truly remarkable. However, the trialkylamine building block as being present in the N-HTA units is amendable to facile electrochemical oxidation. Redox reactions may occur as intrinsic electrochemical processes within the ESI source.60–64 Previous ESI studies on azatriangulenes confirmed their facile electrochemical oxidation.49,54,65 Therefore, one can confidently conclude that oxidation occurs at the N-HTA unit(s). This means for the complexes observed in Fig. 2 that the NTH molecule carries the charge and C60 is present as a neutral molecule.49 This conclusion is also corroborated by the results of the dissociation experiments of these complexes (vide infra).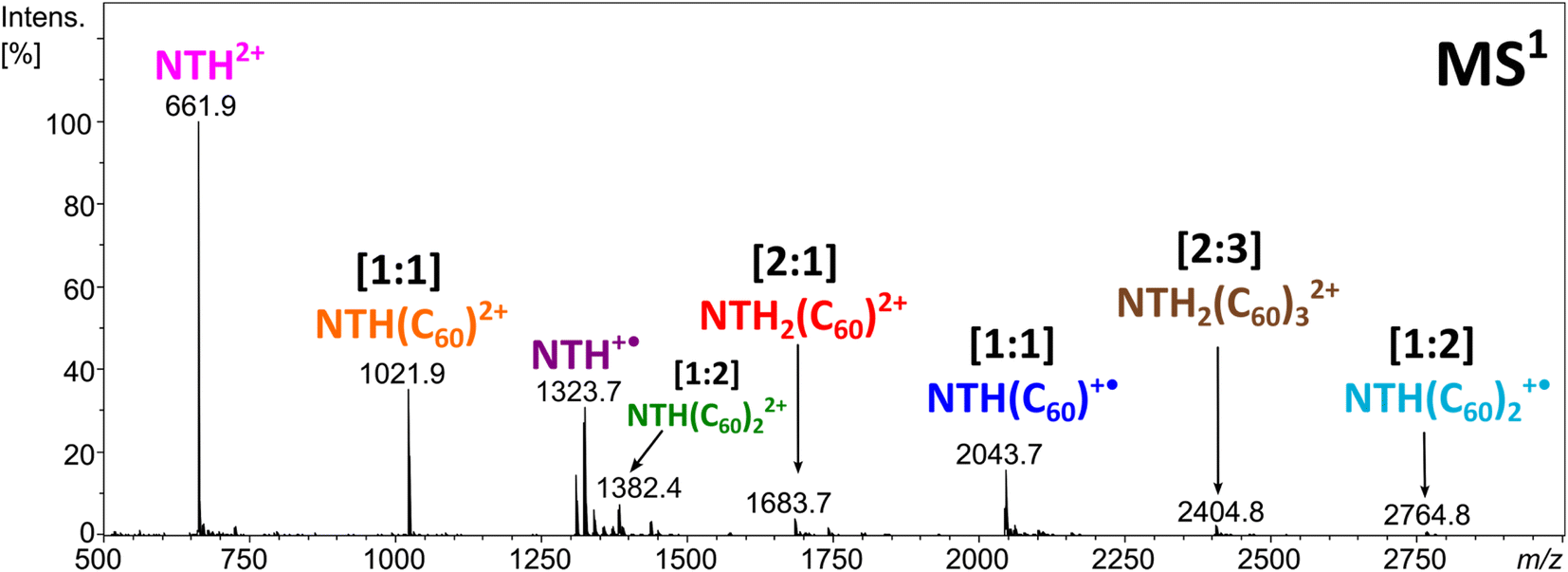 | ||
| Fig. 2 Positive ion-mode ESI-MS of the NTH/C60 analyte solution. | ||
Clearly, the MS1 experiment provides evidence of the formation of multiple host–guest complexes with NTH and C60, featuring the prominent formation of singly and doubly ionised [1:1] complexes and – to a lesser extent also – larger complexes. The question arises as to whether or not the observed aggregation extent is indicative of a particularly favourable interaction within the host–guest complex or if similar results would also be obtained with other host molecules of comparable size but different structure. Moreover, due to the helical structure of the NTH molecule, there are two potential binding sites for complex formation. This refers to either interaction of the guest with both of the N-HTA units in a tweezer-like fashion. On the other hand, the fullerene could interact with the backbone of the molecule. In order to answer these questions, we performed experiments with two different host molecules containing the relevant structural elements and allowing decisive insight.
Firstly, we applied the synthetic uncyclized precursor of NTH as a host (Fig. 1b), for which the resulting mass spectrum shows no complex formation with C60 at all. The NTH precursor still features the tweezer-like arrangement of the two N-HTA moieties, although the cyclodehydrogenation to form the HBC-backbone has not yet occurred. Instead, the phenyl groups are arranged in a propeller-shaped fashion to avoid steric repulsion (see Fig. S2†). The cavity between the two N-HTA moieties is thus available in both hosts, NTH (Fig. 1a) and its precursor (Fig. 1b) and constitutes a highly attractive binding site for metal cations. Such a tweezer-like binding site is well established for all-carbon helicenes.66 The fact that the NTH precursor shows no complexation with C60 has a structural implication for the observed [1:1] complex of C60 with NTH. As a matter of fact, we assume that the cavity of the N-HTA tweezer is not well suited to form stable complexes with the fullerene, simply because the fullerene is too big for a favourable interaction. This should be even more the case for the NTH host where movements are more restricted because of the ridged backbone. Thus, the tweezer binding motif is not operative in this system. A bigger host system based on a porphyrin-decorated HBC has been able to effectively attach up to eight C60 molecules.67 As a conclusion, in the observed complexes with NTH, the C60 must be associated with the “outside” of the NTH host.
Secondly, since the HBC-backbone appears to be an essential requirement for the complexation, we tested hexa-tert-butyl-hexa-peri-hexabenzocoronene (HB-HBC) as a host (Fig. 1c). The resulting mass spectra (Fig. S3†) are characterized by intense signals of mono- and dimeric structures of the HB-HBC molecule. The mass spectra reveal the formation of only very minor amounts of an HB-HBC fullerene complex of [2:1] composition. CID experiments show that the fullerene is only loosely bound to an HB-HBC dimeric structure (Fig. S4†). The complex dissociates into C60 and the dimer cation of HB-HBC and this occurs at much lower collision energies than the dissociation of the [1:1] complex of C60 with NTH+˙. The signal of a [1:1] C60:HB-HBC complex can only just be identified, but is too weak to be examined further. The low abundance of the [1:1] and [2:1] complexes, together with the low bond strength between C60 and HB-HBC dimer ion, indicate that complex formation is possible but not efficient. This can be attributed to a poor shape-complementarity of the planar HB-HBC and the convex surface of the fullerene.26
The experiments with the three different host molecules indicate that the observed extent of complexation of NTH with C60 is significant and that the curved structure of the HBC-backbone is clearly the decisive factor for the successful complex formation with C60. Interaction of the fullerene with the curved HBC-backbone, perhaps even including one of the N-HTA units, would result in a convex–concave binding motif, for which many examples are reported in the literature.36–39 This proposed binding motif will be reconsidered in the discussion of the DFT calculations (vide infra).
In order to obtain further insight into the composition of the complexes and the charge distribution within the complexes, collision-induced dissociation (CID) of the ions was studied in MS2 experiments. The singly and doubly charged [1:1] complexes both fragment by the loss of a neutral fullerene, leading to NTH+˙ and NTH2+, respectively (Fig. 3a and b). Also, the [1:2] complex (Fig. 3c) shows the successive release of two C60s. The two doubly charged complexes, [2:1]2+ (Fig. 3d) and [2:3]2+ (Fig. 3e), dissociate by a reaction which is known as Coulomb explosion,55,68,69 which refers to the dissociation of a dication into two singly charged fragment ions. NTH2(C60)2+ dissociates into NTH+˙ and (C60)NTH+˙ and NTH2(C60)32+ decomposes into NTH(C60)+˙ and NTH(C60)2+˙. These reactions suggest that the two positive charges are located on each of the two NTH molecules in the complexes. There is no indication of a reaction into a smaller fragment complex with only one doubly charged NTH unit which would have supported the alternative initial charge distribution of one NTH2+ dication and one neutral NTH molecule in the complex. All dissociation reactions confirm that NTH was charged while C60 is the neutral component in the complex.
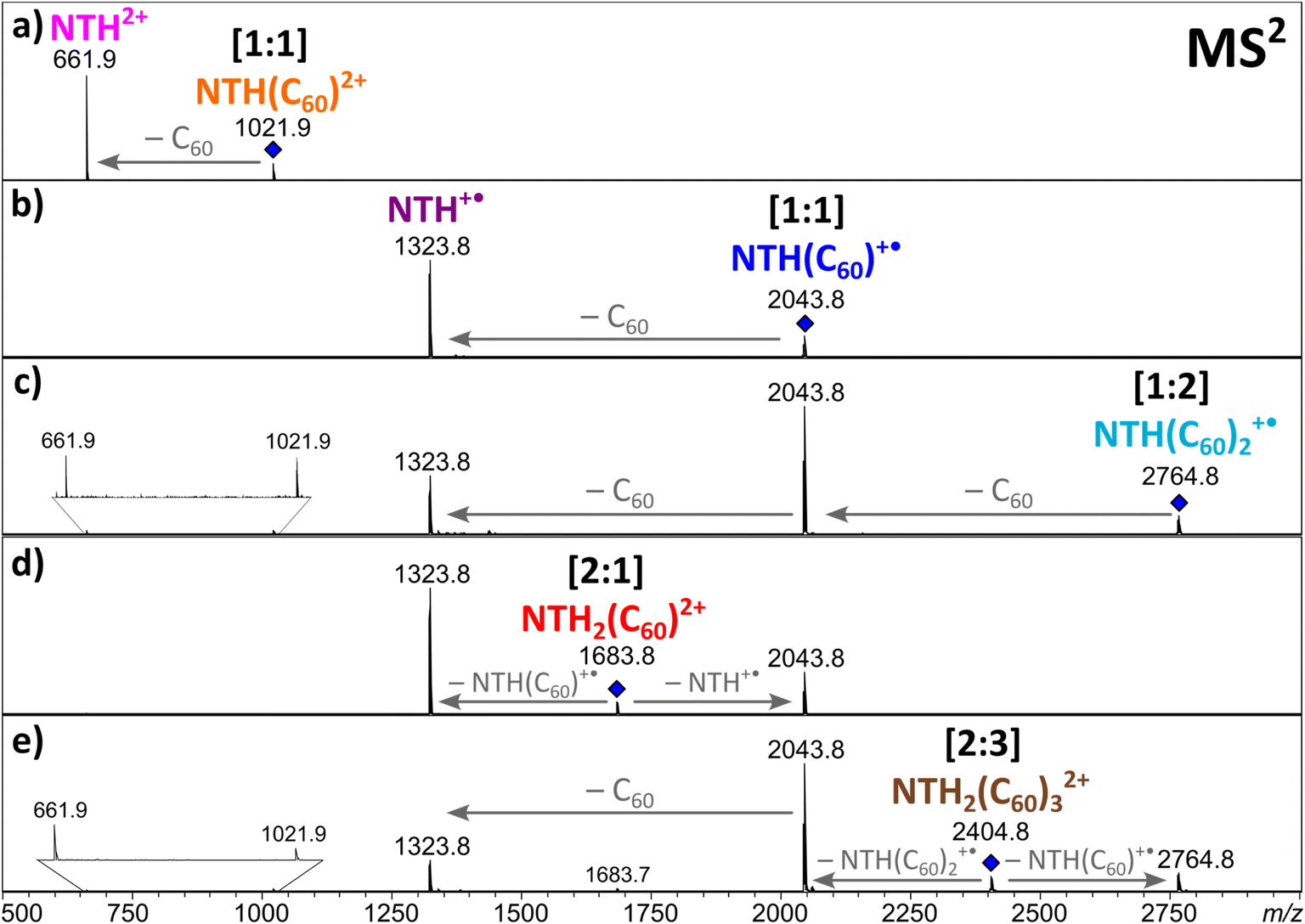 | ||
| Fig. 3 MS2 spectra of the selected NTH/C60 complexes. | ||
In the following, the stability of the complexes is evaluated by energy-dependent CID measurements. In these experiments, a selected complex is collisionally activated to induce decomposition. By increasing the collision energy, a breakdown graph is obtained. The E50 value represents the collision energy at which half of the population of the selected complex is decomposed and is taken as a measure of stability. By comparing the breakdown graphs of the singly and doubly charged [1:1] complexes (Fig. 4a and b), one notices that the doubly charged complex decomposes at a slightly higher collision energy. NTH(C60)2+ has an E50 value of 0.31 eV, and NTH(C60)+˙ possesses an E50 value of 0.28 eV. The dicationic complex is thus more stable than the singly charged complex. At first sight this finding appears somewhat puzzling since charge repulsion should weaken the dication and allow for more facile dissociation.70 However, in the host–guest complex, the Coulomb repulsion within the NTH unit does not affect the complex stability, as the charge repulsion is only restricted to the host entity and does not affect the guest molecule. In contrast, the two charges may even enhance the polarization of the C60 guest and lead to a more firmly connected complex. Our DFT calculations (vide infra) also confirm that the dissociation of dicationic NTH(C60)2+ into NTH2+ and neutral C60 (Fig. 3a) requires more energy than the fragmentation of the monocationic NTH(C60)+˙ into NTH+˙ and neutral C60 (Fig. 3b).
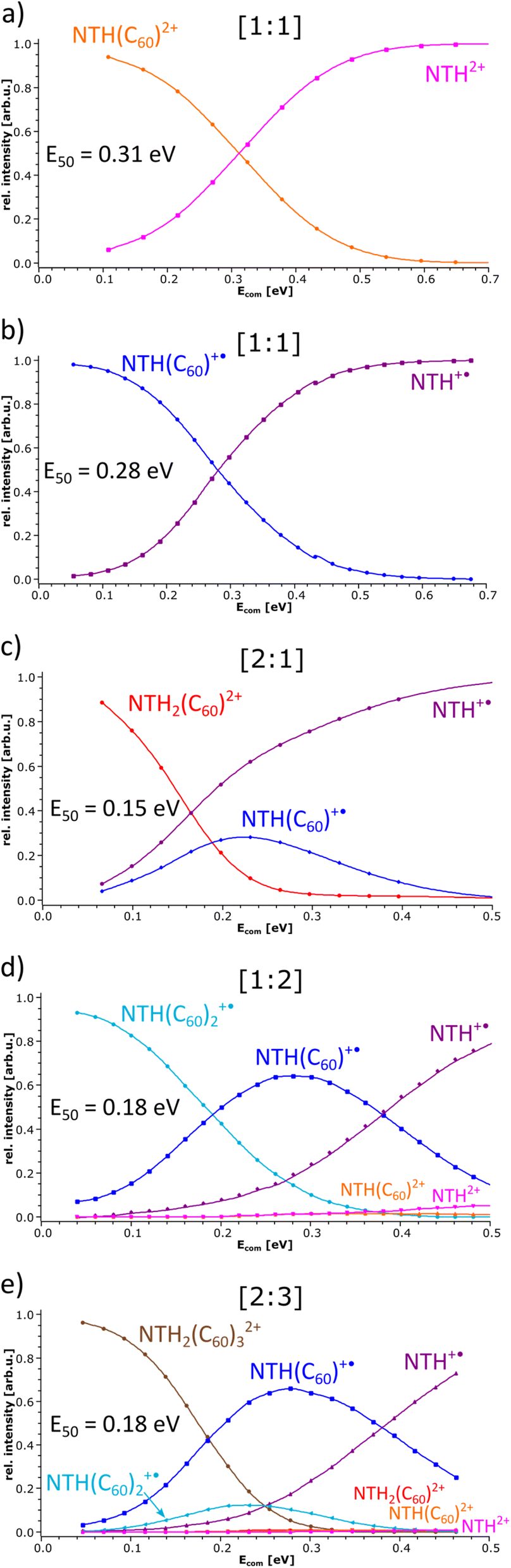 | ||
| Fig. 4 Breakdown graphs of the selected NTH/C60 complexes. | ||
The breakdown graphs of the larger complexes beyond the [1:1] composition (Fig. 4c–e) reveal a clearly reduced complex stability. The larger complexes possess approximately only half of the stability seen for the [1:1] complexes. The E50 values were obtained as 0.15 eV for NTH2(C60)2+ (Fig. 4c), 0.18 eV for NTH(C60)2+˙ (Fig. 4d) and again 0.18 eV for the largest complex NTH2(C60)32+ (Fig. 4e). The lower stability confirms the assumption that larger entities are generated by the addition of further building blocks to the initial [1:1] complex. Amongst the larger complexes, NTH2(C60)2+ (Fig. 4c) is by a small margin the weakest complex. This may be caused by a somewhat more pronounced repulsion of the two positive charges in this complex. In NTH2(C60)2+ (Fig. 4c) there is only one C60 to accommodate the two positively charged NTH+˙ cations, while in NTH2(C60)32+ (Fig. 4e) two more C60s may stabilise the complex. The other larger complex NTH(C60)2+˙ (Fig. 4d) is just singly charged and therefore not affected by Coulombic repulsion. The breakdown graphs also reveal the fragmentation dynamics of the larger complexes. NTH2(C60)2+ (Fig. 4c) with two NTH+˙ cations attached to one neutral C60 decomposes almost directly into NTH+˙ and the intermediate NTH(C60)+˙ is not as abundantly formed as for the other larger complexes. On the one hand, NTH+˙ is already formed in the Coulomb explosion of NTH2(C60)2+ into NTH+˙ and NTH(C60) +˙. But also, the resulting NTH(C60)+˙ seems to dissociate much more easily than in Fig. 4d when it is formed from NTH(C60)2+˙ by a simple loss of neutral C60. We assume that the Coulomb explosion reaction leads to an excitation of the NTH(C60)+˙ intermediate, which decomposes more easily. NTH(C60)2+˙ (Fig. 4d) shows after the loss of one C60 the pronounced formation of an intermediate [1:1] NTH(C60)+˙ complex, which eventually undergoes a second C60 loss to result in the formation of bare NTH+˙. Despite its different composition, NTH2(C60)32+ (Fig. 4e) shows almost exactly the same breakdown behaviour as NTH(C60)2+˙ (Fig. 4d). This is caused by the fact that NTH2(C60)32+ (Fig. 4e) decomposes by a Coulomb explosion reaction efficiently into NTH(C60)+˙, the central [1:1] intermediate fragment ion and into NTH(C60)2+ which also easily decomposes into NTH(C60)+˙ by C60 loss. The breakdown graphs of the NTH(C60)2+ complex (Fig. 4d) and the NTH2(C60)32+ complex (Fig. 4e) show minute signals for the doubly charged ions: NTH2+, NTH(C60)2+ and NTH2(C60)2+ (only observed for NTH2(C60)32+) in the MS2 spectra at elevated collision energies (Ecom > 0.32 eV). These doubly charged ion signals appear only after complete decomposition of the [2:3] complex and are therefore not connected to direct dissociations of the selected precursor complex. In fact, a likely source of these unwanted, interfering ions are even larger multiply charged complexes of the same m/z value (isobaric ions) as the mass-selected ions of interest.
Unfortunately, the [1:2] complex ion NTH(C60)22+ could not be isolated from a protonated species which interfered with the ion of interest, so that no reliable E50 value could be obtained. However, the dissociation pattern shows as expected the consecutive loss of two C60s into NTH2+ as the final ion.
The experiments were also performed with C70. Two different binding modes result from the oval shape of C70: “end-on” binding, which resembles the binding of C60, and “side-on” binding. Which binding motif is preferred depends on the structural arrangement of the molecule. In the absence of steric hindrance, side-on binding is commonly preferred because the interaction area is larger than it is for end-on binding.71 Our experiments show (Fig. S5†) that more NTH–fullerene complexes are formed for C60 (e.g. [2:1] and [2:3]) than for C70. Indicating that complex formation with C60 is more efficient. However, those C70 complexes that were observed are all more stable compared to the same complexes with C60 (Fig. S6†). Which clearly indicates that there is more binding interaction with C70. The increased complex stability is most evident for the mono and dicationic [1:1] complexes. For the [2:1] complex, only a minor increase of the complex stability could be determined. This agrees with the assumption that the larger complexes are the result of lose coordination of additional NTH molecules or fullerenes to an already existing [1:1] complex. Therefore, we do not expect the same interaction strength as for the [1:1] complexes. A list with all determined E50 values for comparison of complex stabilities can be found in the ESI (Table S1†).
DFT calculations were performed in support of the experimental findings. The purpose of these calculations is it to identify the lowest energy structures of the host–guest complexes and to establish their fragmentation energies. Due to the large system size, only the [1:1], [1:2] and [2:1] NTH/C60 complexes were calculated in the gas phase. The most likely position of the fullerene guest with respect to the NTH host, as well as the energy required for complex dissociation were calculated. Fig. 5 displays the most stable complex geometries. The other structures as well as a table of all fragmentation energies can be found in the ESI (Fig. S8 and Tables S2–S4†). For the [1:1] complex, three complex geometries were considered (see ESI†). The fullerene was either placed between both N-HTA units (I-hta), between HBC-backbone and the backside of an N-HTA unit (I-hbc-in) or above the HBC with no further interaction (I-hbc-out). The least stable structure could be assigned to the tweezer-like I-hta conformation. The N-HTA units apparently cannot open wide enough to properly enclose the fullerene. This is in agreement with the experimental results on the basis of which a bonding to the N-HTA tweezer could be discounted. The two geometries involving interaction with the HBC backbone form more strongly coordinated complexes. However, the I-hbc-out geometry lacks the additional N-HTA binding and is, therefore, slightly less stable than the I-hbc-in geometry. We therefore assume that the [1:1] complexes observed in the experiments adopt the I-hbc-in conformation. For the [1:2] complex, a second fullerene was added to the most stable [1:1] complex. Two structures were considered. The tweezer-like position between the N-HTA units (II-hta) as well as the position over the other unoccupied side of the HBC-backbone (II-hbc). Again, the HBC coordination is favoured, leading to the complex depicted in Fig. 5. For the [2:1] complex, the fullerene was either encapsulated by the N-HTA units (III-hta) or placed between the HBC-backbones (III-hbc) of both NTH molecules. The III-hbc position is much more stable, and therefore, we assign this geometry to the [2:1] complex observed in the experiment.
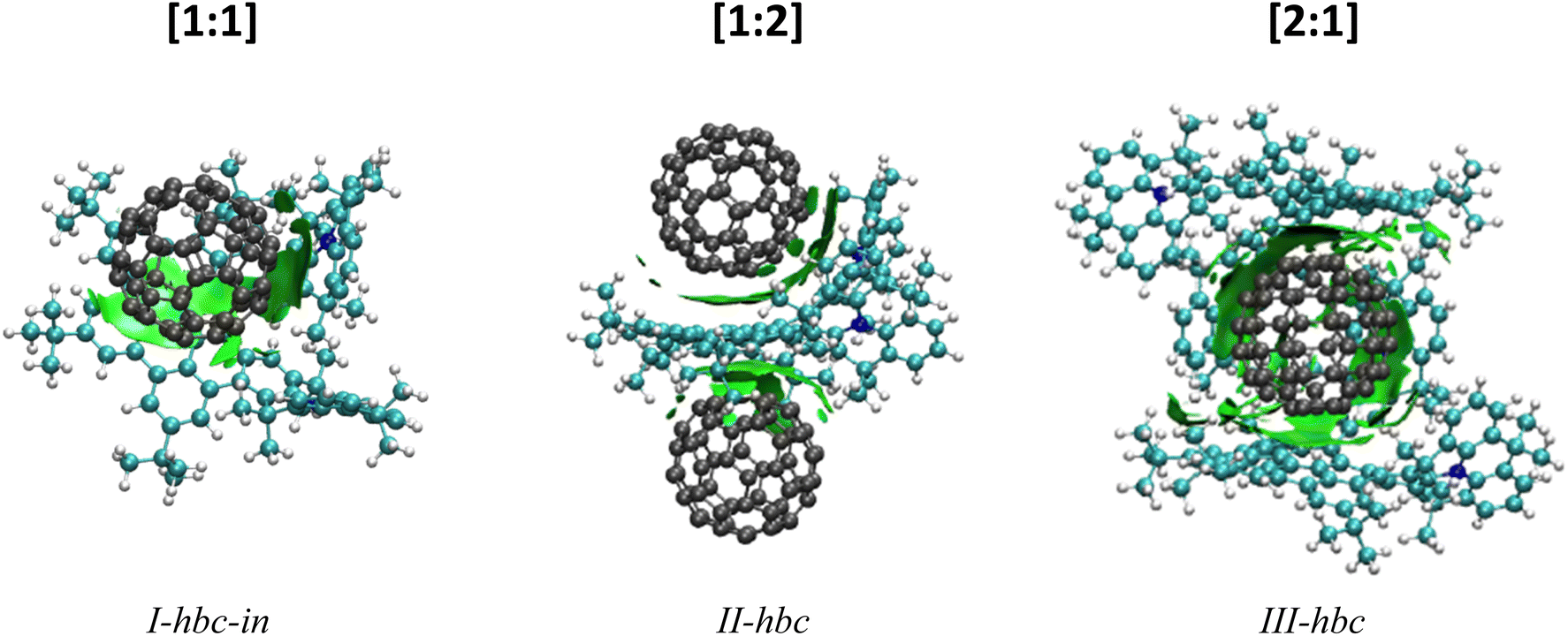 | ||
| Fig. 5 DFT-optimized geometries of the NTH/C60 complexes with visualization of long-range interactions. | ||
For the three different complex compositions studied here, the fragmentation energies were calculated for the neutral, singly and doubly charged complexes. The fragmentation energy is obtained as the difference in energy of the noncovalent complex to the separated components of the complex (no reverse activation barrier assumed) and – represents the binding energy within the complex. While a list of all fragmentation energies can be found in the ESI (Tables S2–S4†), we will discuss in the following the complexes that were actually observed in the experiments.
On the whole, the fragmentation energies confirm the experimentally observed charge distributions upon dissociation of the complexes. For all singly charged complexes, the charge resides on the NTH rather than on C60, which is a consequence of the lower ionization energy of the NTH molecule. This is confirmed even though the ionization energy of C60 was calculated as 7.32 eV, which is clearly lower than the well-established experimental value of 7.6 eV,72,73 representing a considerable discrepancy. The first and second IE of the NTH molecule were obtained as 1IENTH = 5.33 eV and 2IENTH = 7.49 eV, respectively. There is no experimental gas-phase data available for comparison. For the doubly charged NTH2(C60)2+ complexes with two NTH units the calculations confirm the experimental observation and predict a more favourable charge distribution of NTH+˙/NTH+˙ rather than NTH2+/NTH0. For the doubly charged [1:1] complex NTH(C60)2+ theory predicts the feasibility of electron transfer within the complex from C60 (IP = 7.32 eV) to NTH2+ (2IENTH = 7.49 eV). Therefore, the fragmentation into C60+˙ and NTH+˙ (fragmentation energy = 1.05 eV, ESI Table S2†) should be favoured over the experimentally observed dissociation into a neutral fullerene and the doubly charged molecule NTH2+ (fragmentation energy = 1.22 eV, ESI, Table S2†). Even when C60 is replaced by C70, no indication of this reaction could be observed. C70 has a more favourable thermochemistry towards electron transfer to NTH2+, because of its slightly lower IP of 7.47 eV.74 Only when C60 was replaced by C78 in the complex, which possesses an experimental IP of 7.0 eV,75 that is 0.6 eV lower than the IP of C60, the electron transfer followed by dissociation into NTH+˙ and C78+˙ was abundantly observed. However, the formation of NTH2+ and C78 was still the more abundantly occurring dissociation reaction (see Fig. S7†). Unfortunately, it is not clear at this point as to whether the thermochemistry of the charge transfer within the NTH2+(fullerene) complexes is adequately described at this level of theory. It is also possible that the collision experiment is affected by a kinetic shift. In which case the electron transfer within the complex would not take place if the difference of the second IP of the NTH and the first IP of the fullerene is not sufficiently large.
Conclusion
The host–guest chemistry of the helical NTH molecule comprising N-HTA and HBC moieties with the fullerenes C60 and C70 has been explored in gas-phase experiments accompanied by DFT analysis of the geometries and fragmentation energies. The NTH molecule is identified as a new host for the fullerenes. The concave–convex shape complementarity is established as the essential prerequisite for complex formation between the helical nanographene host and the spherical fullerene guest. Convex C60 binds to the concave helicene-like backbone of NTH, which also involves the terminal N-HTA moiety. Since the respective binding site is present twice in both the host and the guest molecule, complex formation beyond the [1:1] stoichiometry occurs leading to [2:1], [1:2] and even bigger complexes. However, complexes beyond the [1:1] composition show less stability. Unfavorable shape matching may prevent the efficient formation of larger complexes with C70. However, the larger surface area of C70 promotes the development of dispersion forces between NTH and fullerenes, which explains the higher stability of the [1:1] complexes with C70 compared to those with C60. The findings of this investigation contribute to a better understanding of the noncovalent bonding within complexes of NTH and fullerenes and may aid efforts to enhance the applicability of nanographenes in areas such as photovoltaic, molecular electronics and sensing.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
We are grateful to the DFG for their funding through SFB953 “Synthetic Carbon Allotropes”, Projektnummer 182849149. We are indebted to Dr Christian Neiß (FAU, Theoretical Chemistry) for his supporting help.References
- Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed.
- A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616–6643 RSC.
- P. J. Evans and N. Martín, Chem, 2016, 1, 16–31 CAS.
- Y. Cao, V. Fatemi, S. Fang, K. Watanabe, T. Taniguchi, E. Kaxiras and P. Jarillo-Herrero, Nature, 2018, 556, 43–50 CrossRef CAS PubMed.
- N. J. Schuster, R. Hernández Sánchez, D. Bukharina, N. A. Kotov, N. Berova, F. Ng, M. L. Steigerwald and C. Nuckolls, J. Am. Chem. Soc., 2018, 140, 6235–6239 CrossRef CAS PubMed.
- P. J. Evans, J. Ouyang, L. Favereau, J. Crassous, I. Fernández, J. Perles and N. Martín, Angew. Chem., Int. Ed., 2018, 57, 6774–6779 CrossRef CAS PubMed.
- K. Kato, Y. Segawa, L. T. Scott and K. Itami, Angew. Chem., Int. Ed., 2018, 57, 1337–1341 CrossRef CAS PubMed.
- S. H. Pun and Q. Miao, Acc. Chem. Res., 2018, 51, 1630–1642 CrossRef CAS PubMed.
- K. Y. Cheung, S. Gui, C. Deng, H. Liang, Z. Xia, Z. Liu, L. Chi and Q. Miao, Chem, 2019, 5, 838–847 CAS.
- F. Zhang, E. Michail, F. Saal, A.-M. Krause and P. Ravat, Chem.–Eur. J., 2019, 25, 16241–16245 CrossRef CAS PubMed.
- Y. Zhu, X. Guo, Y. Li and J. Wang, J. Am. Chem. Soc., 2019, 141, 5511–5517 CrossRef CAS PubMed.
- X.-Y. Wang, X. Yao, A. Narita and K. Müllen, Acc. Chem. Res., 2019, 52, 2491–2505 CrossRef CAS PubMed.
- C. M. Cruz, I. R. Márquez, S. Castro-Fernández, J. M. Cuerva, E. Maçôas and A. G. Campaña, Angew. Chem., Int. Ed., 2019, 58, 8068–8072 CrossRef CAS PubMed.
- J. M. Farrell, V. Grande, D. Schmidt and F. Würthner, Angew. Chem., Int. Ed., 2019, 58, 16504–16507 CrossRef CAS PubMed.
- K. Y. Cheung, Y. Segawa and K. Itami, Chem.–Eur. J., 2020, 26, 14791–14801 CrossRef CAS PubMed.
- S. Ma, J. Gu, C. Lin, Z. Luo, Y. Zhu and J. Wang, J. Am. Chem. Soc., 2020, 142, 16887–16893 CrossRef CAS PubMed.
- M. M. Martin, F. Hampel and N. Jux, Chem.–Eur. J., 2020, 26, 10210–10212 CrossRef CAS PubMed.
- L. Moshniaha, M. Żyła-Karwowska, P. J. Chmielewski, T. Lis, J. Cybińska, E. Gońka, J. Oschwald, T. Drewello, S. M. Rivero, J. Casado and M. Stępień, J. Am. Chem. Soc., 2020, 142, 3626–3635 CrossRef CAS PubMed.
- J. Urieta-Mora, M. Krug, W. Alex, J. Perles, I. Fernández, A. Molina-Ontoria, D. M. Guldi and N. Martín, J. Am. Chem. Soc., 2020, 142, 4162–4172 CrossRef CAS PubMed.
- S. Matsubara, Y. Koga, Y. Segawa, K. Murakami and K. Itami, Nat. Catal., 2020, 3, 710–718 CrossRef CAS.
- C. Zhu, K. Shoyama and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 21505–21509 CrossRef CAS PubMed.
- M. A. Medel, R. Tapia, V. Blanco, D. Miguel, S. P. Morcillo and A. G. Campaña, Angew. Chem., Int. Ed., 2021, 60, 6094–6100 CrossRef CAS PubMed.
- P. Izquierdo-García, J. M. Fernández-García, I. Fernández, J. Perles and N. Martín, J. Am. Chem. Soc., 2021, 143, 11864–11870 CrossRef PubMed.
- S. Escayola, A. Poater, A. Muñoz-Castro and M. Solà, Chem. Commun., 2021, 57, 3087–3090 RSC.
- C. Zhu, K. Shoyama, M. A. Niyas and F. Würthner, J. Am. Chem. Soc., 2022, 144, 16282–16286 CrossRef CAS PubMed.
- H. He, Y. J. Lee, Z. Zong, N. Liu, V. M. Lynch, J. Kim, J. Oh, D. Kim, J. L. Sessler and X.-S. Ke, J. Am. Chem. Soc., 2024, 146, 543–551 CrossRef CAS PubMed.
- X.-Y. Wang, A. Narita and K. Müllen, Nat. Rev. Chem., 2018, 2, 0100 CrossRef.
- J. M. Fernández-García, P. J. Evans, S. Filippone, M. Á. Herranz and N. Martín, Acc. Chem. Res., 2019, 52, 1565–1574 CrossRef PubMed.
- Y. Gu, Z. Qiu and K. Müllen, J. Am. Chem. Soc., 2022, 144, 11499–11524 CrossRef CAS PubMed.
- A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Chem. Rev., 2014, 114, 7150–7188 CrossRef CAS PubMed.
- Z. Liu, S. Fu, X. Liu, A. Narita, P. Samorì, M. Bonn and H. I. Wang, Adv. Sci., 2022, 9, 2106055 CrossRef PubMed.
- J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718–747 CrossRef CAS PubMed.
- F. Schwierz, Nat. Nanotechnol., 2010, 5, 487–496 CrossRef CAS PubMed.
- G. M. Beneventi, M. Krug, D. Reger, N. Jux and D. M. Guldi, J. Photochem. Photobiol., C, 2023, 56, 100602 CrossRef.
- T. Liu and A. Troisi, Adv. Mater., 2013, 25, 1038–1041 CrossRef CAS PubMed.
- E. M. Pérez and N. Martín, Chem. Soc. Rev., 2008, 37, 1512–1519 RSC.
- E. M. Pérez and N. Martín, Chem. Soc. Rev., 2015, 44, 6425–6433 RSC.
- S. Selmani and D. J. Schipper, Chem.–Eur. J., 2019, 25, 6673–6692 CrossRef CAS PubMed.
- X. Chang, Y. Xu and M. von Delius, Chem. Soc. Rev., 2024, 53, 47–83 RSC.
- M. Rickhaus, M. Mayor and M. Juríček, Chem. Soc. Rev., 2017, 46, 1643–1660 RSC.
- J. Borstelmann, J. Bergner, F. Rominger and M. Kivala, Angew. Chem., Int. Ed., 2023, 135, e202312740 CrossRef.
- J. Borstelmann, L. Schneider, F. Rominger, F. Deschler and M. Kivala, Angew. Chem., Int. Ed., 2024, 136, e202405570 CrossRef.
- K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739–744 CrossRef CAS PubMed.
- K. Kato, Y. Segawa, L. T. Scott and K. Itami, Chem.–Asian J., 2015, 10, 1635–1639 CrossRef CAS PubMed.
- J. M. Fernández-García, P. J. Evans, S. Medina Rivero, I. Fernández, D. García-Fresnadillo, J. Perles, J. Casado and N. Martín, J. Am. Chem. Soc., 2018, 140, 17188–17196 CrossRef PubMed.
- J. Liu, S. Mishra, C. A. Pignedoli, D. Passerone, J. I. Urgel, A. Fabrizio, T. G. Lohr, J. Ma, H. Komber, M. Baumgarten, C. Corminboeuf, R. Berger, P. Ruffieux, K. Müllen, R. Fasel and X. Feng, J. Am. Chem. Soc., 2019, 141, 12011–12020 CrossRef CAS PubMed.
- Y. Fei, Y. Fu, X. Bai, L. Du, Z. Li, H. Komber, K.-H. Low, S. Zhou, D. L. Phillips, X. Feng and J. Liu, J. Am. Chem. Soc., 2021, 143, 2353–2360 CrossRef CAS PubMed.
- S. Zank, J. M. Fernández-García, A. J. Stasyuk, A. A. Voityuk, M. Krug, M. Solà, D. M. Guldi and N. Martín, Angew. Chem., Int. Ed., 2022, 61, e202112834 CrossRef CAS PubMed.
- B. D. Gliemann, V. Strauss, J. F. Hitzenberger, P. O. Dral, F. Hampel, J.-P. Gisselbrecht, T. Drewello, W. Thiel, D. M. Guldi and M. Kivala, Chem.–Eur. J., 2017, 23, 12353–12362 CrossRef CAS PubMed.
- L. Cera and C. A. Schalley, Chem. Soc. Rev., 2014, 43, 1800–1812 RSC.
- A. Hosseini, S. Taylor, G. Accorsi, N. Armaroli, C. A. Reed and P. D. W. Boyd, J. Am. Chem. Soc., 2006, 128, 15903–15913 CrossRef CAS PubMed.
- N. Fröhlich, It's All a Matter of Proper Communication, PhD thesis, Friedrich-Alexander-Universitär Erlangen-Nürnberg, 2020.
- J. Borstelmann, S. Zank, M. Krug, G. Berger, N. Fröhlich, G. Glotz, F. Gnannt, L. Schneider, F. Deschler, T. Clark, G. Gescheidt, D. M. Guldi and M. Kivala, in preparation.
- J. F. Hitzenberger, P. O. Dral, U. Meinhardt, T. Clark, W. Thiel, M. Kivala and T. Drewello, ChemPlusChem, 2017, 82, 204–211 CrossRef CAS PubMed.
- R. W. Kirschbaum, M. Hausmann, O. V. Boltalina, S. H. Strauss and T. Drewello, Phys. Chem. Chem. Phys., 2015, 17, 23052–23058 RSC.
- TURBOMOLE V7.3 2018, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, TURBOMOLE GmbH, since 2007, available from http://www.turbomole.com.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- G. J. Van Berkel and V. Kertesz, Anal. Chem., 2007, 79, 5510–5520 Search PubMed.
- M. Schäfer, M. Drayß, A. Springer, P. Zacharias and K. Meerholz, Eur. J. Org Chem., 2007, 2007, 5162–5174 CrossRef.
- R. Vessecchi, A. E. M. Crotti, T. Guaratini, P. Colepicolo, S. E. Galembeck and N. P. Lopes, Mini-Rev. Org. Chem., 2007, 4, 75–87 CrossRef CAS.
- T. Cai, X.-Y. Xu, D.-M. Fang, H.-Y. Qi, Y. Jiang, G.-L. Zhang and Z.-J. Wu, Int. J. Mass Spectrom., 2014, 373, 39–42 CrossRef CAS.
- T. Cai, X.-Y. Xu and Z.-J. Wu, Analyst, 2015, 140, 7864–7867 RSC.
- M. Kinzelmann, N. Fröhlich, A. Vogel, M. Kivala and T. Drewello, J. Mass Spectrom., 2024, 59, e5079 CrossRef CAS PubMed.
- J. Oschwald, D. Reger, S. Frühwald, V. Warmbrunn, A. Görling, N. Jux and T. Drewello, ChemPhysChem, 2023, 24, e202300496 CrossRef CAS PubMed.
- P. Haines, R. Kaur, M. M. Martin, M. B. Minameyer, S. Frühwald, S. Bönisch, D. Lungerich, F. Hampel, A. Görling, T. Drewello, N. Jux and D. M. Guldi, Adv. Energy Mater., 2021, 11, 2100158 CrossRef CAS.
- J. Li, W. Wei, L. C. Nye, P. S. Schulz, P. Wasserscheid, I. Ivanović-Burmazović and T. Drewello, Phys. Chem. Chem. Phys., 2012, 14, 5115–5121 RSC.
- T. Drewello, C. B. Lebrilla, H. Schwarz and T. Ast, J. Organomet. Chem., 1988, 339, 333–338 CrossRef CAS.
- P. Scheier, A. Stamatovic and T. D. Märk, Chem. Phys. Lett., 1988, 144, 119–124 CrossRef CAS.
- S. Jung, J. Seo and J. Seo, J. Phys. Chem. A, 2010, 114(43), 11376–11385 CrossRef CAS PubMed.
- D. L. Lichtenberger, K. W. Nebesny, C. D. Ray, D. R. Huffman and L. D. Lamb, Chem. Phys. Lett., 1991, 176, 203–208 CrossRef CAS.
- J. de Vries, H. Steger, B. Kamke, C. Menzel, B. Weisser, W. Kamke and I. V. Hertel, Chem. Phys. Lett., 1992, 188, 159–162 CrossRef CAS.
- D. L. Lichtenberger, M. E. Rempe and S. B. Gogosha, Chem. Phys. Lett., 1992, 198, 454–460 CrossRef CAS.
- H. Steger, J. Holzapfel, A. Hielscher, W. Kamke and I. V. Hertel, Chem. Phys. Lett., 1995, 234, 455–459 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07837c |
| This journal is © The Royal Society of Chemistry 2025 |