
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntramolecular noncovalent C-bonding driven conformational preference in spiroisatin-based N-acyl hydrazones†‡
Muhammad Imran Ali a,
Javid Hussainb,
Muhammad Usman Anwarb,
Ahmed Al-Harrasic and
Muhammad Moazzam Naseer*a
a,
Javid Hussainb,
Muhammad Usman Anwarb,
Ahmed Al-Harrasic and
Muhammad Moazzam Naseer*a
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan. E-mail: moazzam@qau.edu.pk
bDepartment of Biological Sciences & Chemistry, College of Arts and Sciences, University of Nizwa, Nizwa, Oman
cNatural and Medical Sciences Research Centre, University of Nizwa, Birkat Almouz 616, Oman
First published on 13th January 2025
Abstract
Noncovalent carbon bonding (C-bonding), a recently explored σ-hole interaction, has primarily been characterized through X-ray structural and computational studies. Evidence of C-bonds in solution is scarce, especially in highly polar solvents like DMSO where solvation effects typically overshadow weak non-covalent interactions. In this work, we present three novel spiroisatin-based N-acyl hydrazones (1–3) in which C-bonds play a critical role in stabilizing the cis conformation in solution. Despite the steric preference for the NH–amide bond to adopt the trans geometry (H–N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ≈ 180°), 1H and 13C NMR spectra of compounds 1 and 2 in DMSO-d6 reveal a rotameric mixture with a higher percentage of the cis conformation (82% and 76%, respectively), attributed to the stability provided by intramolecular C-bonding. Compound 3 also predominantly adopts the cis conformation in DMSO but to a lesser extent (60%) than compounds 1 and 2, due to competing intramolecular hydrogen bonding. Single-crystal X-ray analysis of compounds 1 and 2 confirmed the cis conformation, consistent with the solution-state preference. In contrast, compound 3 crystallized in the trans form, likely due to intramolecular hydrogen bonding and solid-state packing effects, which reinforce the steric preference for the trans geometry. Density functional theory (DFT) calculations corroborated the experimental data, predicting greater stability for the cis conformations in compounds 1, 2, and 3 in solution. The ability of intramolecular C-bonding to stabilize the cis conformation, even in highly polar solvents like DMSO, highlights the broader significance of this interaction in supramolecular chemistry and related fields.
O ≈ 180°), 1H and 13C NMR spectra of compounds 1 and 2 in DMSO-d6 reveal a rotameric mixture with a higher percentage of the cis conformation (82% and 76%, respectively), attributed to the stability provided by intramolecular C-bonding. Compound 3 also predominantly adopts the cis conformation in DMSO but to a lesser extent (60%) than compounds 1 and 2, due to competing intramolecular hydrogen bonding. Single-crystal X-ray analysis of compounds 1 and 2 confirmed the cis conformation, consistent with the solution-state preference. In contrast, compound 3 crystallized in the trans form, likely due to intramolecular hydrogen bonding and solid-state packing effects, which reinforce the steric preference for the trans geometry. Density functional theory (DFT) calculations corroborated the experimental data, predicting greater stability for the cis conformations in compounds 1, 2, and 3 in solution. The ability of intramolecular C-bonding to stabilize the cis conformation, even in highly polar solvents like DMSO, highlights the broader significance of this interaction in supramolecular chemistry and related fields.
1. Introduction
Non-covalent interactions are fundamental across chemistry, physics, and biological sciences, playing a key role in maintaining the three-dimensional structures of biomacromolecules such as nucleic acids and proteins, and mediating molecular recognition processes like receptor–ligand binding, enzyme–substrate interactions, and antigen–antibody recognition.1–3 While well-established interactions such as hydrogen4 and halogen bonds5,6 have been extensively characterized, increasing attention has been directed towards weaker non-covalent interactions, particularly in the context of molecular organization in solution (self-assembly)7 and in the solid state (crystal engineering).8,9 Interactions such as C–H⋯O10,11 and C–H⋯π12,13 once considered weak, are now recognized for their importance in stabilizing molecular conformations, promoting crystallization, and influencing selectivity, despite their relatively low energies (3 kcal mol−1 for C–H⋯O14,15 and 0.8 kcal mol−1 for C–H⋯π16 compared to stronger hydrogen bonds). These interactions are also considered crucial in processes like protein folding,17,18 and higher-order molecular assembly.16 Concludingly, the scientific focus has recently been expanded to unravel the behavior of newly discovered non-covalent interactions such as chalcogen,19–21 pnicogen,22–24 aerogen,25,26 and C-bonding.27–29 Among these non-covalent σ-hole interactions (a concept originally introduced by Politzer et al.30,31), C-bonding was first highlighted in 2013 by Mani and Arunan through theoretical calculations on the Ar⋯propargyl alcohol complex,32 and later confirmed experimentally by Guru Row and colleagues via X-ray charge density analysis.33 Although C-bonding has been proposed as a significant interaction with broad implications in supramolecular chemistry, research remains limited in elucidating its role in directing structure in chemical and biological sciences.An analysis of protein structures reveals that, with the exception of proline, all-natural amino acids overwhelmingly favor the trans isomer (>99.9%) due to steric factors.34–37 In contrast, semicarbazones and thiosemicarbazones predominantly adopt the cis conformation for their amide and thioamide groups, respectively, which is attributed to the presence of intramolecular hydrogen bonding (Fig. 1).38 This led to the hypothesis that replacing the NH group in semicarbazones with a CH2 group in acyl hydrazones would result in either a cis conformation stabilized by C-bonding or a trans conformation in the absence of any intramolecular non-covalent interactions (Fig. 1). Based on this hypothesis, we were the first to demonstrate the role of C-bonding in stabilizing the cis conformation in the solid state.39 Later, Sarma and colleagues extended this concept to solution-phase studies specifically in chloroform using N-methyl-N,N-diacylhydrazines as models.40 Mooibroek and co-workers, through X-ray diffraction and DFT studies, further demonstrated the application of C-bonds in crystal engineering.41 Wang et al. later provided substantial evidence of the importance of C-bonds in catalysis through NMR spectroscopy.42 Most notably, Biswal and co-workers underscored the ubiquitous presence of C-bonds in proteins using detailed structural analyses and quantum mechanical calculations to highlight their ubiquity.43
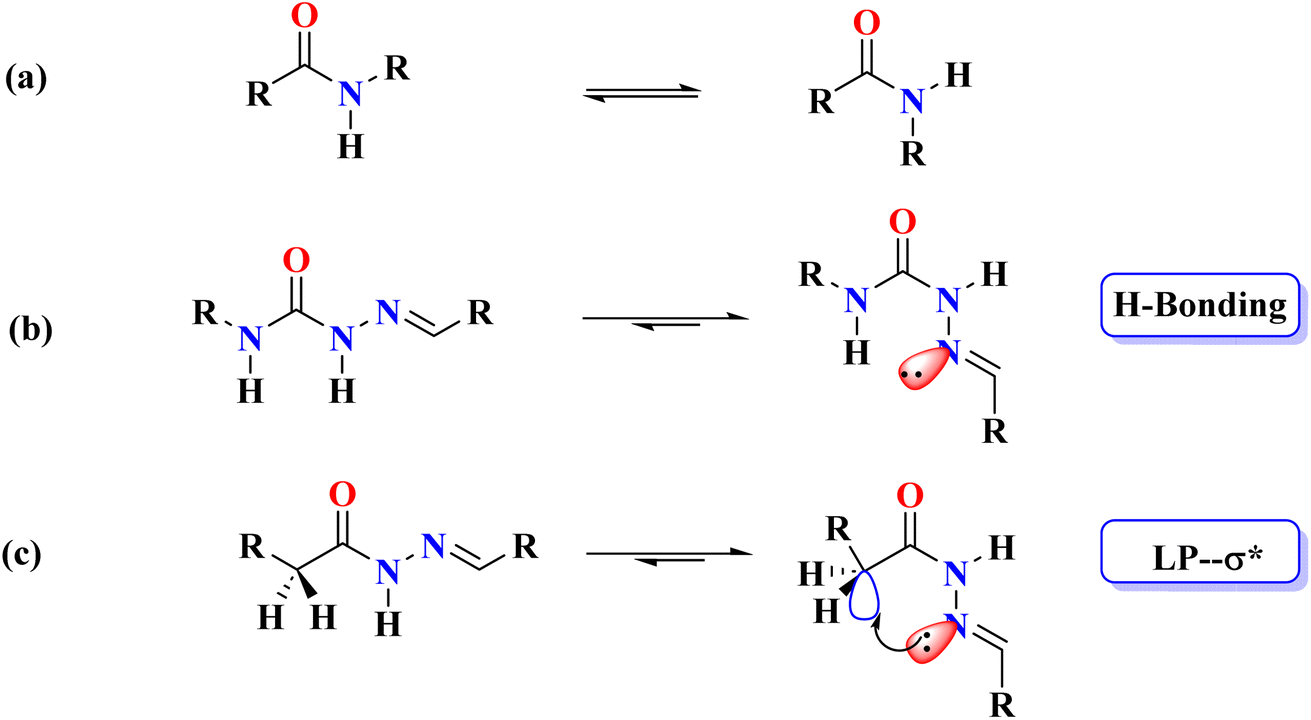 | ||
| Fig. 1 Showing trans (left) and cis (right) conformations, (a) amides; (b) semicarbazones; (c) acylhydrazones. | ||
In this study, we report three novel N-acyl hydrazones 1–3 (Fig. 2) featuring an isatin nucleus, where the nitrogen exhibits increased electronegativity due to lone pair delocalization onto the 2-carbonyl group of the isatin ring. This delocalization enhances the electrophilic character of the adjacent CH2 group, increasing its potential for non-covalent C-bonding interactions. Thus, the 1H and 13C NMR spectra of compounds 1, 2, and 3 in DMSO-d6 reveal a rotameric mixture favoring the cis conformation in 82%, 76%, and 60% respectively due to intramolecular C-bonding. These findings are further supported by X-ray single-crystal analysis and theoretical calculations. This study not only demonstrates the potential of intramolecular C-bonding to stabilize specific conformations but also lays a foundation for further investigation into the nature of this interaction. These insights may have important implications for crystallography, supramolecular chemistry, and related fields of research.
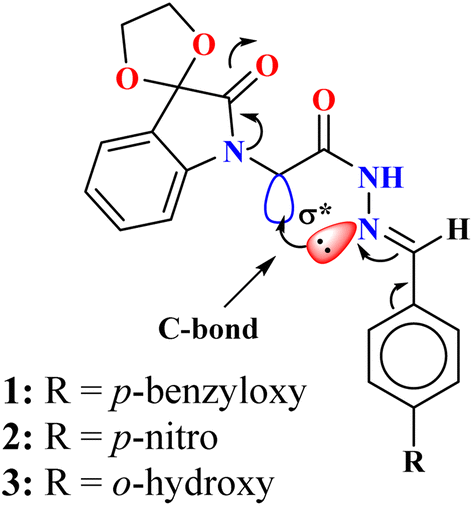 | ||
| Fig. 2 Novel spiroisatin-based N-acyl hydrazones 1–3. | ||
2. Experimental
2.1. Synthesis of ethyl 2-(2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetate (I)
To a round-bottom flask connected to a water separator (Dean–Stark apparatus), commercially available isatin (1 mmol, 147.13 mg) was added in toluene (30 mL) and allowed to react with ethylene glycol (2 mmol, 124.14 mg, 111.83 μL) in the presence of a catalytic amount of p-TsOH. Upon completion of the reaction, the mixture was poured into chilled water. The desired spiroisatin-ketal was extracted with ethyl acetate and recrystallized from ethanol.44 This purified spiroisatin-ketal (1 mmol, 191.19 mg) was then stirred at room temperature with potassium carbonate (1.20 mmol, 165.85 mg) in DMF (20 mL), and ethyl bromoacetate (1.1 mmol, 183.70 mg, 121.66 μL) was added to it dropwise, and the reaction was allowed to continue for 4 hours. Upon completion, the product was collected via solvent extraction and purified through recrystallization from ethanol. The synthesized spiroisatin-based ester I was characterized by comparing its melting point.45,462.2. Synthesis of 2-(2-oxospiro[indoline-3,2′-[1,3]dioxolan]-1-yl)acetohydrazide (II)
In a round-bottom flask, spiroisatin-based ester I (1 mmol, 277.28 mg) was dissolved in ethanol (20 mL), and hydrazine hydrate (3 mmol, 100.12 mg, 97.21 μL) was added to the mixture. The reaction mixture was refluxed with continuous stirring. The resulting hydrazide II was collected by filtration, washed, and recrystallized from ethanol.White solid; yield 90%; Rf 0.3 (EtOAc/n-hexane 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1); m.p. 228–229 °C; FT-IR
:1); m.p. 228–229 °C; FT-IR ![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) (cm−1); 3220 (NH2), 3113 (N–H), 3022 (Csp2–H), 1738 (CO, spiroisatin), 1703 (CO, acetohydrazide), 1274 (C–N, lactam), 1139 (C–O–C).
(cm−1); 3220 (NH2), 3113 (N–H), 3022 (Csp2–H), 1738 (CO, spiroisatin), 1703 (CO, acetohydrazide), 1274 (C–N, lactam), 1139 (C–O–C).
2.3. General procedure for the synthesis of compounds 1–3
In a round-bottom flask, commercially available aromatic aldehyde (1 mmol) was dissolved in methanol (10 mL). A few drops of acetic acid were added as a catalyst, and the reaction mixture was stirred for 15 minutes. Afterward, spiroisatin-bearing N-acetohydrazide II (1 mmol, 263.25 mg) was added, and the reaction mixture was refluxed for 6 hours. The resulting compound precipitated out, were collected by filtration, washed, and recrystallized from ethanol to get pure 1–3. (cm−1); 3144 (N–H), 3079 (Csp2–H), 1734 (CO, spiroisatin), 1678 (CO, acetohydrazide), 1601 (CN imine str.), 1245 (C–N, lactam), 1134 (C–O–C); 1H NMR (300 MHz, DMSO-d6) δ; (cis, 82%) 11.62 (1H, s, N–H), 7.98 (1H, s, NC–H), 4.81 (2H, s, –CH2), (trans, 18%) 11.71 (s, N–H), 8.18 (s, NC–H), 4.41 (s, –CH2), (cis + trans) 4.27–4.39 (∼5H, m, CH2-spiro), 5.16 (3H, s, CH2–O) 6.98–7.89 (∼17H, m, Ar–H); 13C NMR (75 MHz, DMSO-d6) δ; (cis, 82%) 173.3, 167.8, 147.5, 41.1, (trans, 18%) 173.1, 163.0, 144.5, 41.5, (cis + trans) 66.0, 69.8, 101.9, 110.2, 115.6, 123.3, 123.5, 124.2, 124.9, 127.2, 128.2, 128.4, 128.9, 129.0, 129.2, 132.0, 137.2, 144.3, 144.4, 160.3, 160.4; LC-MS (negative mode) [M–H]− = 456.0 m/z. (cm−1); 33154 (N–H), 3089 (Csp2–H), 1731 (CO, spiroisatin), 1694 (CO, acetohydrazide), 1618 (CN imine str.), 1265 (C–N, lactam), 1135 (C–O–C), 1515, 1341 (N–O); 1H NMR (300 MHz, DMSO-d6) δ; (cis, 76%) 12.07 (1H, B, N–H), 8.14 (1H, s, NC–H), 4.90 (2H, s, CH2), (trans, 24%) 12.07 (1H, B, N–H), 8.34 (1H, s, NC–H), 4.47 (s, CH2), (cis + trans) 4.28–4.39 (∼6H, m, CH2-spiro), 7.02–8.31 (∼11H, m, Ar–H); 13C NMR (75 MHz, DMSO-d6) δ; (cis, 76%) 173.3, 168.5, 148.2, 41.2, (trans, 24%) 173.2, 163.8, 144.4, 41.9, (cis + trans) 66.0, 101.9, 110.3, 123.4, 124.2, 128.4, 132.0, 140.6, 142.2, 144.2; LC-MS (negative mode) [M–H]− = 395.1 m/z. (cm−1); 3161 (N–H), 3065 (Csp2–H), 1743 (CO, spiroisatin), 1677 (CO, acetohydrazide), 1687 (CN imine str.), 1264 (C–N, lactam), 1137 (C–O–C); 1H NMR (300 MHz, DMSO-d6) δ: (cis, 60%) 11.67 (s, N–H), 10.06 (s, O–H) 8.35 (s, NC–H), 4.82 (s, CH2), (trans, 40%) 12.05 (1H, s, N–H), 10.94 (1H, s, O–H), 8.45 (1H, s, NC–H), 4.45 (1H, s, CH2) (cis + trans) 4.27–4.41 (∼6H, m, CH2-spiro), 6.84–7.78 (∼13H, m, Ar–H); 13C NMR (75 MHz, DMSO-d6) δ; (cis, 60%) 173.3, 167.7, 147.8, 41.4, (trans, 40%) 173.2, 163.1, 144.5, 41.0, (cis + trans) 66.0, 101.9, 110.2, 110.3, 116.5, 116.8, 119.0, 119.8, 120.5, 123.3, 123.5, 124.2, 124.9, 125.0, 126.7, 129.5, 132.0, 141.9, 144.2; LC-MS (negative mode) [M–H]− = 366.0 m/z.2.4. Quantum-based studies
Non-Covalent Interaction (NCI) analyses were done with Multiwfn47 software. For the NCI plots, we used VMD48 graphical visualization software. Guassian09 suite quantum chemistry program49 was used to optimize the geometry of N-acyl hydrazones. We utilized this basis sets B3LYP/6-311++G (d,p)50 and optimized the structure of our acylhydrazones in DMSO (solution) form to compare the conformations and calculated the difference in their energies. To evaluate the potential energy surface (PES) graph, the Guassian09 suite quantum chemistry program was used with basis sets B3LYP/6-311++G (d,p) on the dihedral angle O28–C23–N27–N29.3. Results and discussion
Spiroisatin-based N-acyl hydrazones 1–3 (Fig. 2) were synthesized in four steps, starting from the inexpensive and readily available isatin (Scheme S1, ESI†). First, the 3-carbonyl group of isatin was protected, followed by a reaction with ethyl bromoacetate in the second step. The resulting intermediate (I) was then treated with hydrazine hydrate to form the corresponding hydrazides (II), which were subsequently reacted with three different aldehydes to produce the target spiroisatin-based N-acyl hydrazones 1–3. Isatin was selected due to its biological relevance and its ability to enhance the electrophilicity of the adjacent CH2 group through nitrogen lone pair delocalization, making it more prone to non-covalent C-bonding interactions. Three aldehydes were chosen based on their electronic characteristics: one with a benzyloxy electron-donating group at the para position, another with a strong electron-withdrawing nitro group, both of which modulate the electron density at the imine nitrogen and influence its involvement in C-bonding. The third aldehyde, featuring an ortho-hydroxy group, was selected to investigate the competition between intramolecular C-bonding and intramolecular hydrogen bonding with imine nitrogen (Fig. 2).Interestingly, the solution-state NMR studies of compound 1 in DMSO-d6 reveal the isomerization of the amide bond, resulting in a rotameric mixture of cis and trans isomers, with the cis conformation being dominant, accounting for 82% of the mixture (Fig. 3). The predominance of the cis isomer can be attributed to intramolecular C-bonding interactions involving the lone pair of the imine nitrogen [N(lp)] and the  orbital, which stabilizes the cis form. Without this stabilization, steric factors would favor the trans isomer.34–37 Similarly, compounds 2 and 3 also exhibit a cis-dominated rotameric mixture, with 76% and 60%, respectively of the cis isomer (Fig. S1 & S2†). The lower percentage of cis conformation of 3 as compared to the compounds 1 and 2 can tentatively attributed to the competing intramolecular H-bonding that exists between the o-hydroxy group and the imine nitrogen, resulting in the relatively lower percentage of cis isomer. The higher percentage of the cis isomer of compound 1 (82%) than the cis isomer of compound 2 (76%) however is credited to electronic factors owing to the presence of two different substituents at the para position of the aryl ring. The electron-donating group like the benzyloxy group present at the para position of the aryl ring of compound 1, makes the imine moiety more electron-rich, and thus a strong C-bond acceptor. On the other hand, the electron-withdrawing nitro group in compound 2 likely withdraws the electron density from the imine moiety, making it relatively less susceptible to make intramolecular C-bond. Notably, the equilibrium showing 40% of the trans isomer for compound 3 on the NMR timescale suggests a low energy barrier between the cis and trans conformers, allowing for rapid interconversion. This low energy barrier and the possibility of rapid conformational exchange are further supported by quantum-based studies (vide infra).
orbital, which stabilizes the cis form. Without this stabilization, steric factors would favor the trans isomer.34–37 Similarly, compounds 2 and 3 also exhibit a cis-dominated rotameric mixture, with 76% and 60%, respectively of the cis isomer (Fig. S1 & S2†). The lower percentage of cis conformation of 3 as compared to the compounds 1 and 2 can tentatively attributed to the competing intramolecular H-bonding that exists between the o-hydroxy group and the imine nitrogen, resulting in the relatively lower percentage of cis isomer. The higher percentage of the cis isomer of compound 1 (82%) than the cis isomer of compound 2 (76%) however is credited to electronic factors owing to the presence of two different substituents at the para position of the aryl ring. The electron-donating group like the benzyloxy group present at the para position of the aryl ring of compound 1, makes the imine moiety more electron-rich, and thus a strong C-bond acceptor. On the other hand, the electron-withdrawing nitro group in compound 2 likely withdraws the electron density from the imine moiety, making it relatively less susceptible to make intramolecular C-bond. Notably, the equilibrium showing 40% of the trans isomer for compound 3 on the NMR timescale suggests a low energy barrier between the cis and trans conformers, allowing for rapid interconversion. This low energy barrier and the possibility of rapid conformational exchange are further supported by quantum-based studies (vide infra).
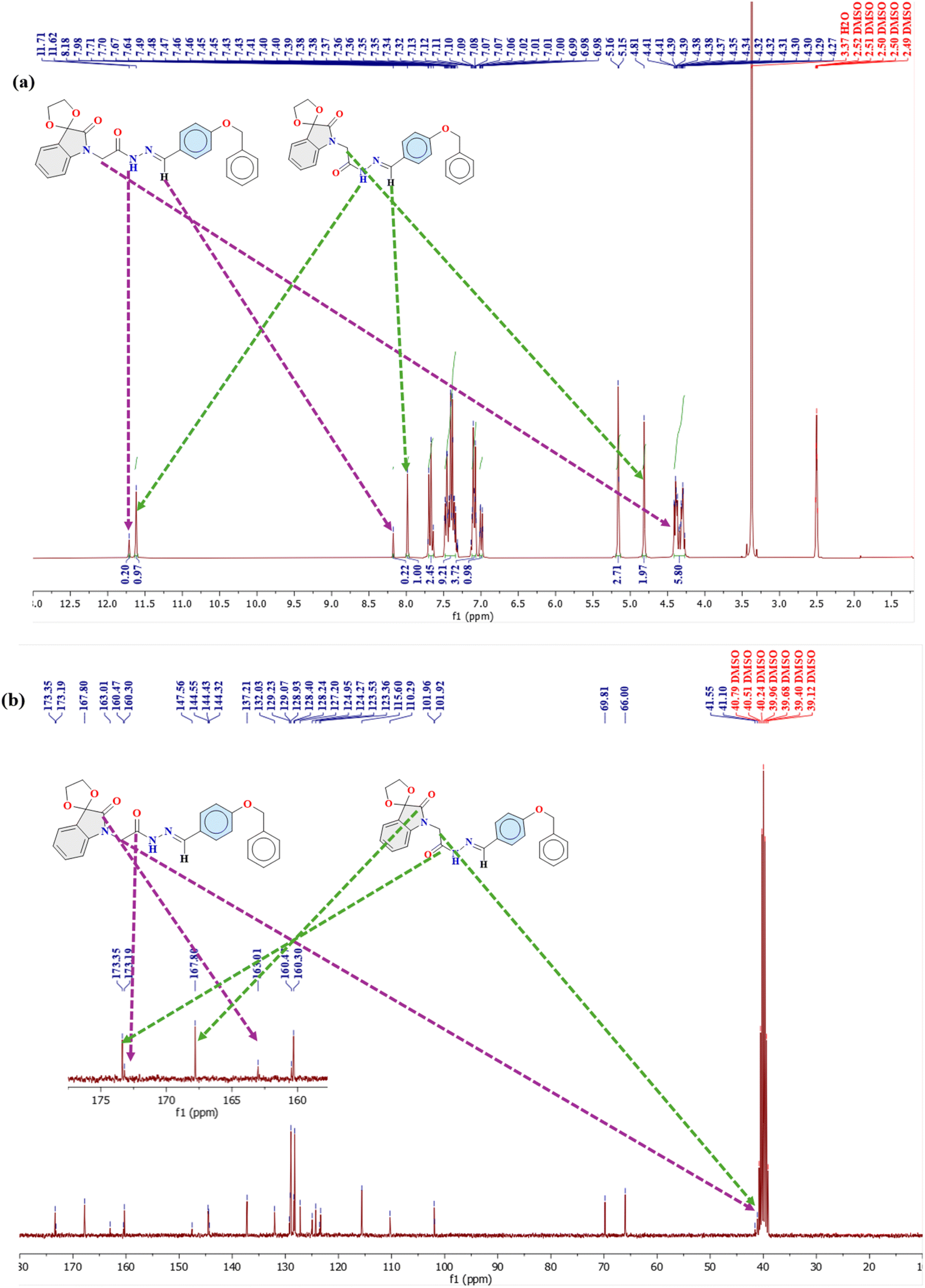 | ||
| Fig. 3 The representative spectra of compound 1; (a) 1H NMR, (b) 13C NMR. | ||
The presence of both isomers in the solution is evident from the distinct proton signals observed in their NMR spectra (Fig. 3a, S1a and S2a†). Generally, the protons of the trans isomer are more deshielded (downfield) compared to those of the cis isomer.40,51 For instance, in compound 1, the NH proton resonates at 11.62 ppm for the cis isomer, while for the trans isomer, it appears downfield at 11.71 ppm (Fig. 3a, Table 1). Similarly, the NC–H proton resonates at 7.98 ppm for the cis isomer and at 8.18 ppm for the trans isomer. The CH2 protons play a crucial role in distinguishing between the cis and trans conformations, as the attached carbon serves as the C-bond donor. In the cis isomer, the lone pair on the imine nitrogen interacts with the 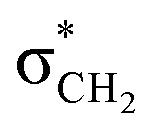 orbital of the CH2 group, leading to a slight elongation of the C–H bond within the methylene group. This interaction results in greater deshielding of the methylene protons, causing a downfield shift in the cis conformation. In contrast, this interaction is absent in the trans isomer, resulting in an upfield shift of these protons. Specifically, in the cis isomer, the methylene protons resonate as a singlet at 4.81 ppm, whereas in the trans conformer, their resonance slightly overlaps with the multiplets of the diastereotopic protons at the 3-position of the isatin ring, appearing at 4.41 ppm (Fig. 3a, Table 1).
orbital of the CH2 group, leading to a slight elongation of the C–H bond within the methylene group. This interaction results in greater deshielding of the methylene protons, causing a downfield shift in the cis conformation. In contrast, this interaction is absent in the trans isomer, resulting in an upfield shift of these protons. Specifically, in the cis isomer, the methylene protons resonate as a singlet at 4.81 ppm, whereas in the trans conformer, their resonance slightly overlaps with the multiplets of the diastereotopic protons at the 3-position of the isatin ring, appearing at 4.41 ppm (Fig. 3a, Table 1).
Likewise, the 13C NMR spectra of compounds 1–3 exhibit two distinct sets of signals corresponding to the cis and trans isomers (Fig. 3b, S1b, S2b† and Table 2). For example, the methylene carbon in compound 1, which participates in C-bonding, resonates at 41.5 ppm for the trans isomer, and a slightly more shielded signal at 41.1 ppm for the cis isomer appeared, indicating intramolecular interactions that shield the methylene group in the cis form through  interactions.40,51 Due to the cyclic nature of isatin, the amide carbonyl of the cyclic amide typically appears more shielded (upfield) than that of the acyclic hydrazone amide. This is evident in the observed carbonyl signals: the amide carbonyl of the hydrazone resonates at 173.1 ppm for the trans isomer and 173.3 ppm for the cis isomer, whereas the cyclic isatin amide appears at 163.0 ppm for the trans isomer and 167.8 ppm for the cis isomer (Fig. 3b, Table 2).
interactions.40,51 Due to the cyclic nature of isatin, the amide carbonyl of the cyclic amide typically appears more shielded (upfield) than that of the acyclic hydrazone amide. This is evident in the observed carbonyl signals: the amide carbonyl of the hydrazone resonates at 173.1 ppm for the trans isomer and 173.3 ppm for the cis isomer, whereas the cyclic isatin amide appears at 163.0 ppm for the trans isomer and 167.8 ppm for the cis isomer (Fig. 3b, Table 2).
| Compounds | Chemical shift δ (ppm) | |||||||
|---|---|---|---|---|---|---|---|---|
| CH2–13C(O)–NH | 13C(O)–N (spiroisatins) | N13C–H |
13CH2–C(O) | |||||
| Trans | Cis | Trans | Cis | Trans | Cis | Trans | Cis | |
| 1 | 173.1 | 173.3 | 163.0 | 167.8 | 144.5 | 147.5 | 41.5 | 41.1 |
| 2 | 173.2 | 173.3 | 163.8 | 168.5 | 144.4 | 148.2 | 41.9 | 41.2 |
| 3 | 173.2 | 173.3 | 163.1 | 166.7 | 144.5 | 147.8 | 41.4 | 41.0 |
As anticipated and consistent with the solution-state results, the solid-state structures (Table S1†) of spiroisatin-based N-acyl hydrazones 1 and 2 are confirmed to adopt a cis conformation (Fig. 4a and b). This preference arises from the alignment of the lone pair on the sp2-hybridized nitrogen atom with the antibonding 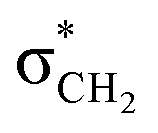 orbital of the CH2(sp3)–N bond, resulting in the formation of non-covalent C-bonding interactions (N⋯C distance 2.740 Å for 1 and 2.707 Å for 2). In contrast, the solid-state structure of compound 3 is found to adopt a trans conformation (Fig. 4c), most probably due to a dominant intramolecular hydrogen bond [O(2)–H(1B)⋯N(2) 1.822 Å] between the imine nitrogen and the hydroxyl group located at the ortho position of the adjacent aromatic ring, and additionally due to packing factors (Fig. S2†). Two previous searches39 in the Cambridge Structural Database (CSD) also support these findings. In search A (Fig. S3†), where both ortho positions of the aromatic ring were unsubstituted, the cis conformation was predominant in 16 out of 20 structures, likely due to the stabilization provided by non-covalent N⋯C, C-bonding interactions. In contrast, in search B (Fig. S3†), where at least one ortho position was substituted by a hydroxyl group, the trans conformation was more common. This trans preference can be collectively attributed to the strong intramolecular O–H⋯N hydrogen bond, steric influence, and the solid state packing effects. The representative structure from search A, TIGNED, exhibits the cis conformation and shows clear evidence of C–bonding interaction (N⋯C distance 2.73 Å) (Fig. S4†). On the other hand, the representative structure EYEKOK from search A, with a trans amide conformation, suggests that other strong non-covalent interactions in the crystal packing (Fig. S4†) stabilize the less favored trans conformation. The predominance of the cis conformation in search A underscores the importance of σ-hole interactions in promoting the cis isomer. In search B, the representative trans conformation structure AYAFAH exhibits a strong intramolecular hydrogen bond that blocks the lone pair on the nitrogen atom, preventing C-bonding. However, in the representative structure YUPSEJ (N⋯C distance 2.75 Å), despite the presence of an ortho OH group, the cis conformation is observed. This is because, in two out of four cases found in the literature, the OH group forms an intramolecular hydrogen bond with a meta-positioned group on the same ring, freeing the nitrogen's lone pair to interact with the carbon atom, thus favoring the cis conformation (Fig. S4†). Additionally, the formation of a stable amide–amide dimer synthon in the solid state further stabilizes the cis arrangement in this case.39
orbital of the CH2(sp3)–N bond, resulting in the formation of non-covalent C-bonding interactions (N⋯C distance 2.740 Å for 1 and 2.707 Å for 2). In contrast, the solid-state structure of compound 3 is found to adopt a trans conformation (Fig. 4c), most probably due to a dominant intramolecular hydrogen bond [O(2)–H(1B)⋯N(2) 1.822 Å] between the imine nitrogen and the hydroxyl group located at the ortho position of the adjacent aromatic ring, and additionally due to packing factors (Fig. S2†). Two previous searches39 in the Cambridge Structural Database (CSD) also support these findings. In search A (Fig. S3†), where both ortho positions of the aromatic ring were unsubstituted, the cis conformation was predominant in 16 out of 20 structures, likely due to the stabilization provided by non-covalent N⋯C, C-bonding interactions. In contrast, in search B (Fig. S3†), where at least one ortho position was substituted by a hydroxyl group, the trans conformation was more common. This trans preference can be collectively attributed to the strong intramolecular O–H⋯N hydrogen bond, steric influence, and the solid state packing effects. The representative structure from search A, TIGNED, exhibits the cis conformation and shows clear evidence of C–bonding interaction (N⋯C distance 2.73 Å) (Fig. S4†). On the other hand, the representative structure EYEKOK from search A, with a trans amide conformation, suggests that other strong non-covalent interactions in the crystal packing (Fig. S4†) stabilize the less favored trans conformation. The predominance of the cis conformation in search A underscores the importance of σ-hole interactions in promoting the cis isomer. In search B, the representative trans conformation structure AYAFAH exhibits a strong intramolecular hydrogen bond that blocks the lone pair on the nitrogen atom, preventing C-bonding. However, in the representative structure YUPSEJ (N⋯C distance 2.75 Å), despite the presence of an ortho OH group, the cis conformation is observed. This is because, in two out of four cases found in the literature, the OH group forms an intramolecular hydrogen bond with a meta-positioned group on the same ring, freeing the nitrogen's lone pair to interact with the carbon atom, thus favoring the cis conformation (Fig. S4†). Additionally, the formation of a stable amide–amide dimer synthon in the solid state further stabilizes the cis arrangement in this case.39
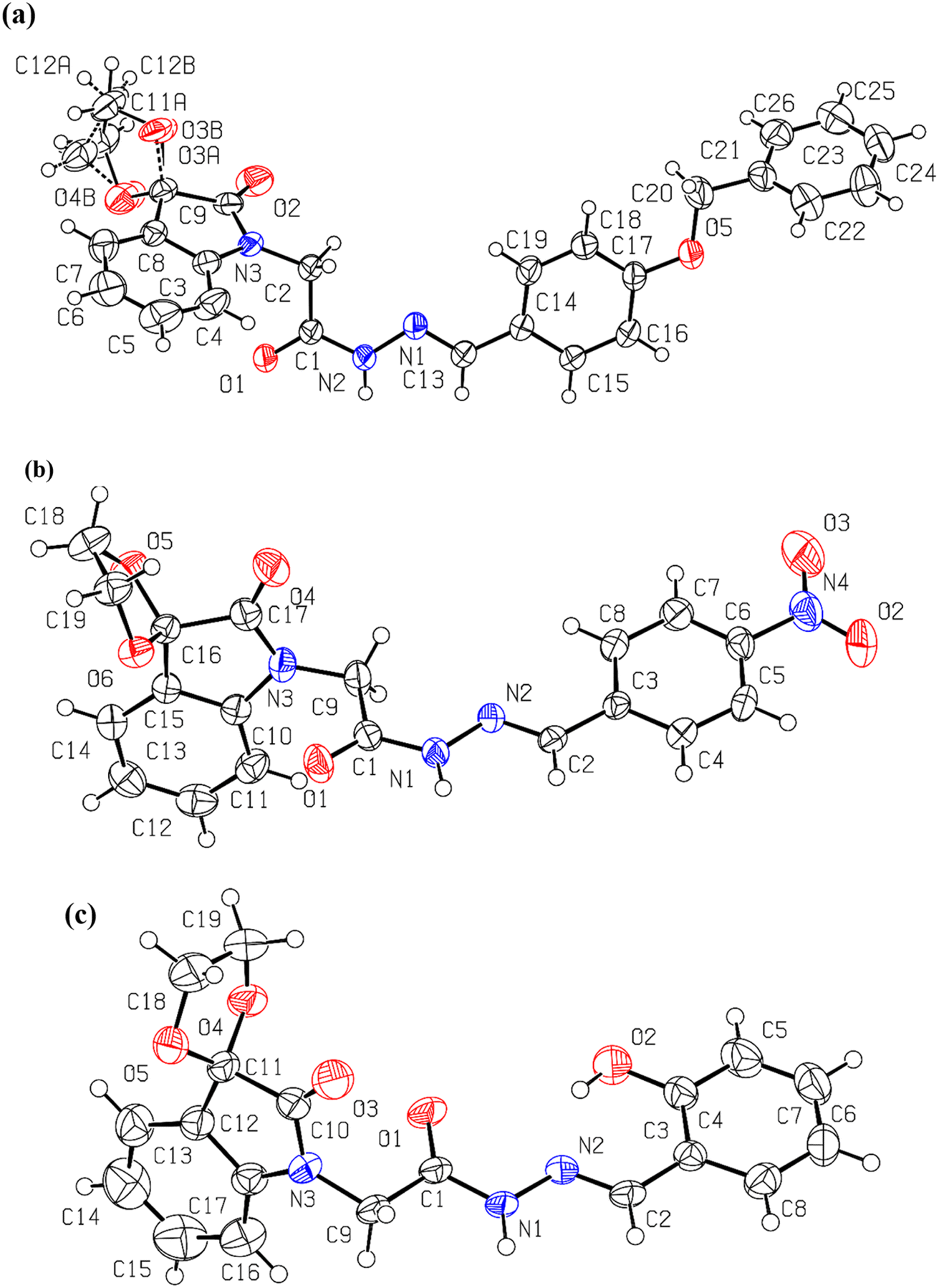 | ||
| Fig. 4 The molecular structure of compounds (a) 1 [C(2)⋯N(1) 2.740 Å, N(3)–C(2)⋯N(1) 171.89°]; (b) 2 [C(9)⋯N(2) 2.707 Å, N(3)–C(9)⋯N(2) 169.31°]; (c) 3. Displacement ellipsoids drawn at the 50% probability level. | ||
The 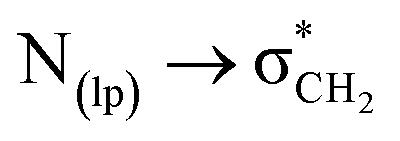 interactions in compounds 1–3 were visualized using the mutliwfn47 to generate non-covalent interaction (NCI) plots.52 NCI plots are advanced visualization tools that go beyond identifying critical points, offering a detailed representation of non-covalent interactions through isosurfaces. These plots are based on electron density and utilize the sign of the second Hessian eigenvalues and color coding to distinguish between favorable and unfavorable interactions. However, the information provided by NCI plots is primarily qualitative, highlighting regions that may be prone to interaction. The color codes represent different types of interactions: red indicates repulsive interactions, blue signals strong attractive interactions, green represents weaker interactions such as C-bonding, and yellow denotes weaker repulsive forces. The NCI plots for spiroisatin-based N-acyl hydrazones 1–3, in solution phase are shown in Fig. 5. The sp2 nitrogen atom participates in C-bonding interactions involving the σ-hole of the CH2 group, as well as weak hydrogen bonding with the ortho hydrogen of the phenyl ring, observed in the cis isomers of compounds 1 and 2 (Fig. 5a and b). In compound 3, strong hydrogen bonding is observed, indicated by the blue color in the NCI plot. In the cis conformer of 3, both C-bonding and hydrogen bonding are present (Fig. 5c), while in the trans conformer, only hydrogen bonding is observed (Fig. 5d).
interactions in compounds 1–3 were visualized using the mutliwfn47 to generate non-covalent interaction (NCI) plots.52 NCI plots are advanced visualization tools that go beyond identifying critical points, offering a detailed representation of non-covalent interactions through isosurfaces. These plots are based on electron density and utilize the sign of the second Hessian eigenvalues and color coding to distinguish between favorable and unfavorable interactions. However, the information provided by NCI plots is primarily qualitative, highlighting regions that may be prone to interaction. The color codes represent different types of interactions: red indicates repulsive interactions, blue signals strong attractive interactions, green represents weaker interactions such as C-bonding, and yellow denotes weaker repulsive forces. The NCI plots for spiroisatin-based N-acyl hydrazones 1–3, in solution phase are shown in Fig. 5. The sp2 nitrogen atom participates in C-bonding interactions involving the σ-hole of the CH2 group, as well as weak hydrogen bonding with the ortho hydrogen of the phenyl ring, observed in the cis isomers of compounds 1 and 2 (Fig. 5a and b). In compound 3, strong hydrogen bonding is observed, indicated by the blue color in the NCI plot. In the cis conformer of 3, both C-bonding and hydrogen bonding are present (Fig. 5c), while in the trans conformer, only hydrogen bonding is observed (Fig. 5d).
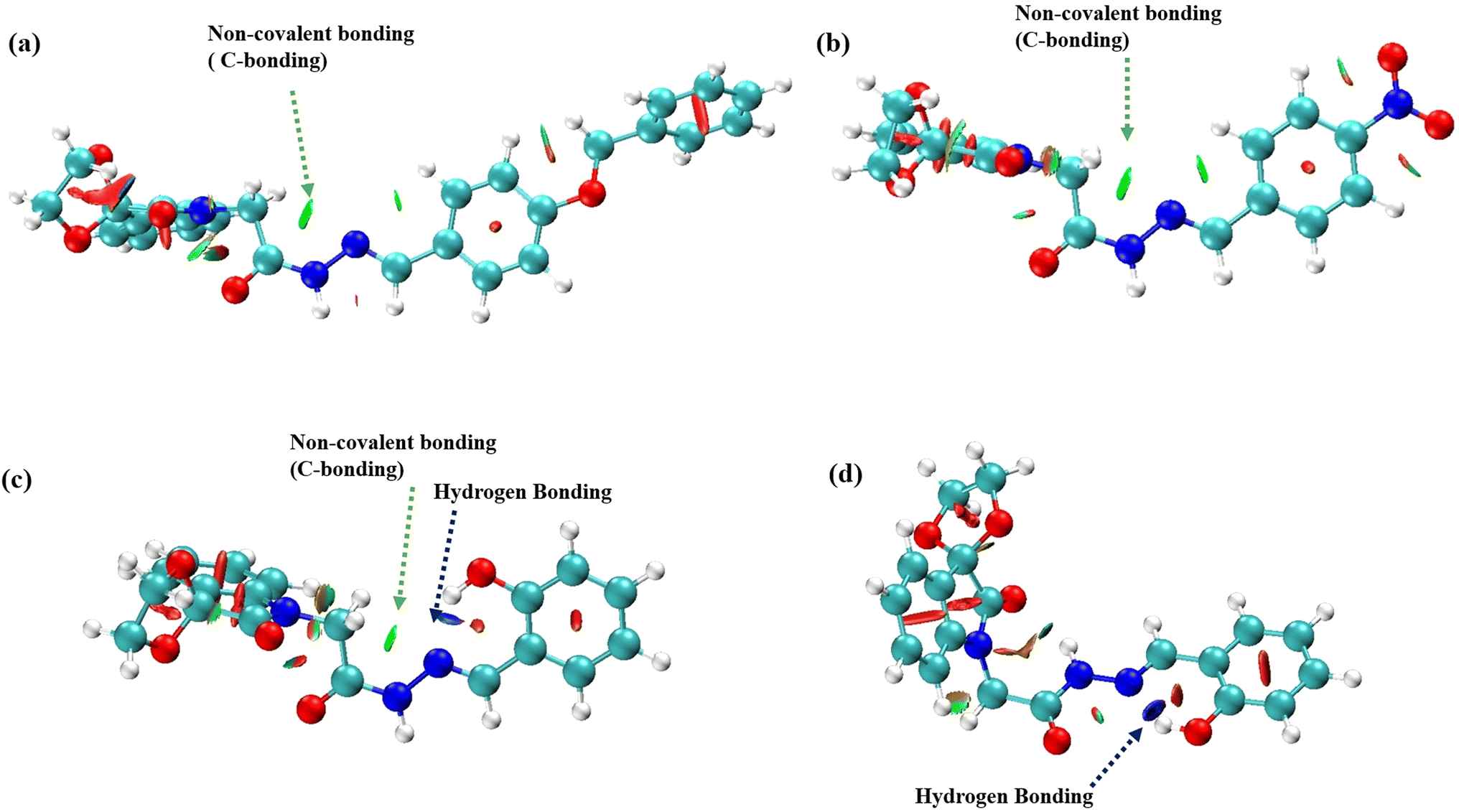 | ||
| Fig. 5 NCI plots of spiroisatin-based N-acyl hydrazones 1–3 showing, (a) C-bonding in 1, (b) C-bonding in 2, C-bonding in 2, (c) C-bonding and H-bonding in 3, (d) intramolecular H-bonding in 3. | ||
The geometry optimization and energy calculations were calculated with natural bond orbital (NBO) analysis utilizing the basis sets B3LYP/6-311++G (d,p)50 at full NBO level. NBO53,54 analysis of representative spiroisatin-based N-acyl hydrazone 2 revealed the existence of 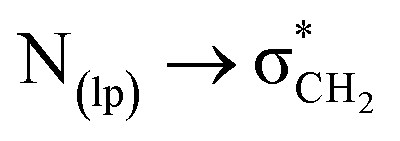 noncovalent interactions between the lone pair of the imine nitrogen and the σ-hole of the CH2 group (Fig. 6). Furthermore, it was determined that the methylene group acts as a C-bond donor, while the nitrogen with a free lone pair serves as the C-bond acceptor.
noncovalent interactions between the lone pair of the imine nitrogen and the σ-hole of the CH2 group (Fig. 6). Furthermore, it was determined that the methylene group acts as a C-bond donor, while the nitrogen with a free lone pair serves as the C-bond acceptor.
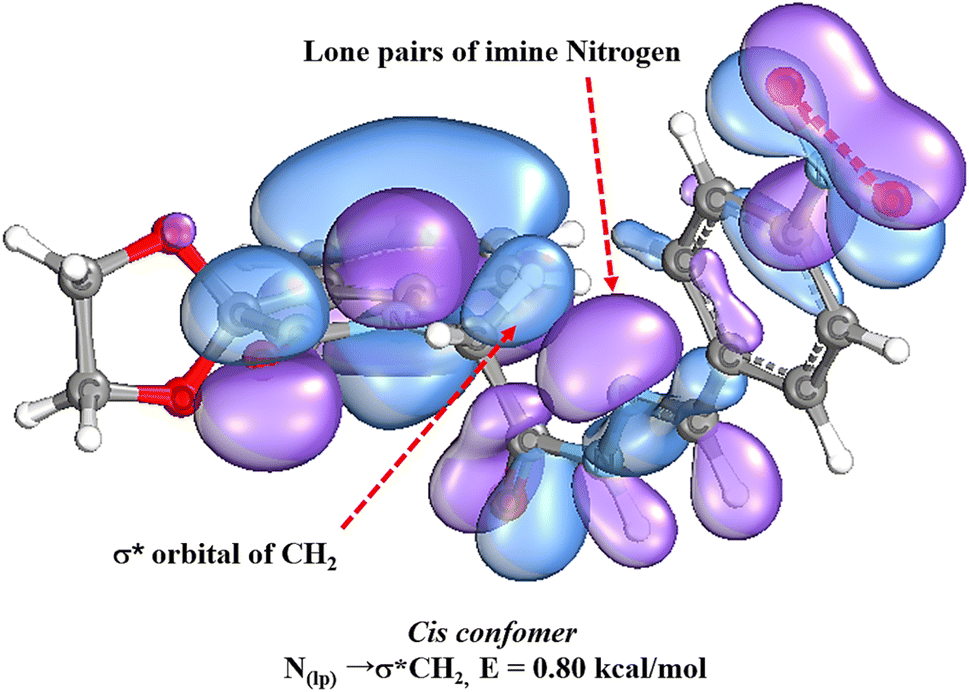 | ||
Fig. 6 NBO analysis showing 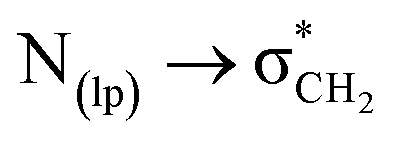 interactions in cis isomer of 2. interactions in cis isomer of 2. | ||
Finally, the quantum chemical calculations55 were performed to validate our solution-state results. The structures of compounds 1–3 were optimized in DMSO to compare their conformations, and the energy differences were calculated using the B3LYP/6-311++G(d,p)50 basis set. The optimized geometries indicated that the structure of the spiroisatin-based N-acylhydrazones is not planar. However, the acyl hydrazone functional group and the phenyl ring of the hydrazone exhibit planarity relative to each other (Fig. 7). For compounds 1 and 2, the trans isomers have higher energies, while the cis isomers exhibit lower energies in DMSO (Table 3). Specifically, for compound 1, the energy difference between the cis and trans isomers is −2.56 kcal mol−1, while for compound 2, the energy barrier is −2.22 kcal mol−1. The subtle energy difference between compounds 1 and 2 suggests that the cis conformer of 1 is slightly more stable than that of 2, likely due to the electron-donating effect of the benzyloxy group. These results also indicate that the cis isomer remains stable even in highly polar solvents with high relative permittivity, as the 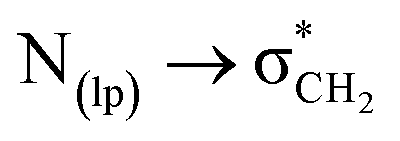 C–bonding interaction can persist in solution. In the case of compound 3, the energy barrier between the cis and trans isomers is reduced to a minimal value of −0.82 kcal mol−1, as both isomers are stabilized by stronger intermolecular hydrogen bonding between the o-hydroxy group and the imine nitrogen atom. Due to this small energy difference, the cis and trans forms of 3 can rapidly interconvert, resulting in a higher proportion of the trans form (40%) observed in the NMR studies compared to the trans isomers of compounds 1 (18%) and 2 (24%) (vide supra).
C–bonding interaction can persist in solution. In the case of compound 3, the energy barrier between the cis and trans isomers is reduced to a minimal value of −0.82 kcal mol−1, as both isomers are stabilized by stronger intermolecular hydrogen bonding between the o-hydroxy group and the imine nitrogen atom. Due to this small energy difference, the cis and trans forms of 3 can rapidly interconvert, resulting in a higher proportion of the trans form (40%) observed in the NMR studies compared to the trans isomers of compounds 1 (18%) and 2 (24%) (vide supra).
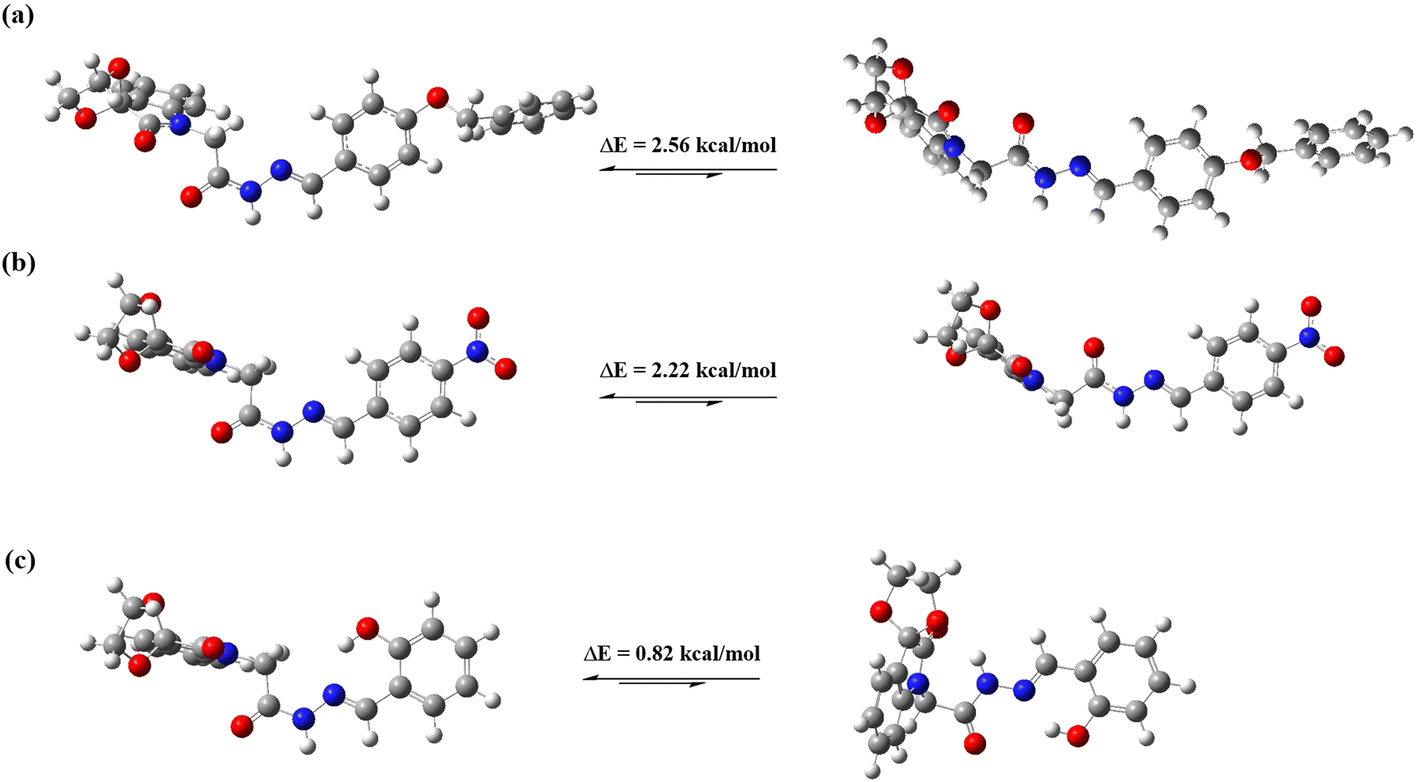 | ||
| Fig. 7 Optimized structures and calculated energy barriers between cis and trans isomers of compounds, (a) 1, (b) 2, (c) 3. | ||
| Energies (Hartree) (trans) | Energies (Hartree) (cis) | ΔE (Hartree) (cis–trans) | ΔE (kcal mol−1) (cis–trans) | |
|---|---|---|---|---|
| 1 | −1527.039505 | −1527.043588 | −0.004083 | −2.56 |
| 2 | −1387.840919 | −1387.844468 | −0.003549 | −2.22 |
| 3 | −1260.000433 | −1260.001748 | −0.001315 | −0.82 |
The conformational and rotational equilibria exhibit significant variation depending on the medium.56,57 It is important to note that the Gibbs free energy differences between conformational or rotational isomers are very small, and the solvation enthalpies must be considerably larger than these free energies in order to induce a substantial shift in the equilibrium position. While highly polar solvents like DMSO can diminish intermolecular interactions, the predominance of cis isomers of compounds 1–3 in solution, driven by intramolecular C-bonding interactions involving 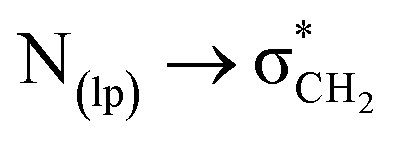 clearly indicate the high polarity of the cis isomer i.e., the isomer with greater dipole moment is more stable in polar solvents like DMSO.56,58,59 Therefore, these quantum-based results are in good agreement with the findings of our solution-phase studies.
clearly indicate the high polarity of the cis isomer i.e., the isomer with greater dipole moment is more stable in polar solvents like DMSO.56,58,59 Therefore, these quantum-based results are in good agreement with the findings of our solution-phase studies.
To determine the stable conformation in DMSO, conformational analysis was performed by sampling points on potential energy. A relaxed potential energy scan was done on the dihedral angle O28–C23–N27–N29 i.e., a standard rotatable bond. The scan was performed from −180° to 180° with every 20° increment. Compound 1 served as a model for these studies. The minimum energy conformation can be found when the θ (dihedral angle) = −0.94°, −179.48°, and 179.48°. Two same conformations, i.e., cis conformer is present when θ = −179.48°, and 179.48°. Likewise, when θ = −0.94°, trans was observed. A minute difference of −0.004083 Hartree (−2.56 kcal mol−1) is found between these two conformers. This subtle energy difference confirms that these two conformers can easily coexist in solution form without any large energy barrier. Moreover, the maxima at the graph indicated the transition geometries between cis and trans-conformers at θ = −0.049° (energy = −1527.04187 Hartree), and 0.107° (energy = −1527.04338 Hartree) (Fig. 8). These sorts of potential energy surface (PES) scans were also undergone for 2 and 3 (Fig. S6 and S7†).
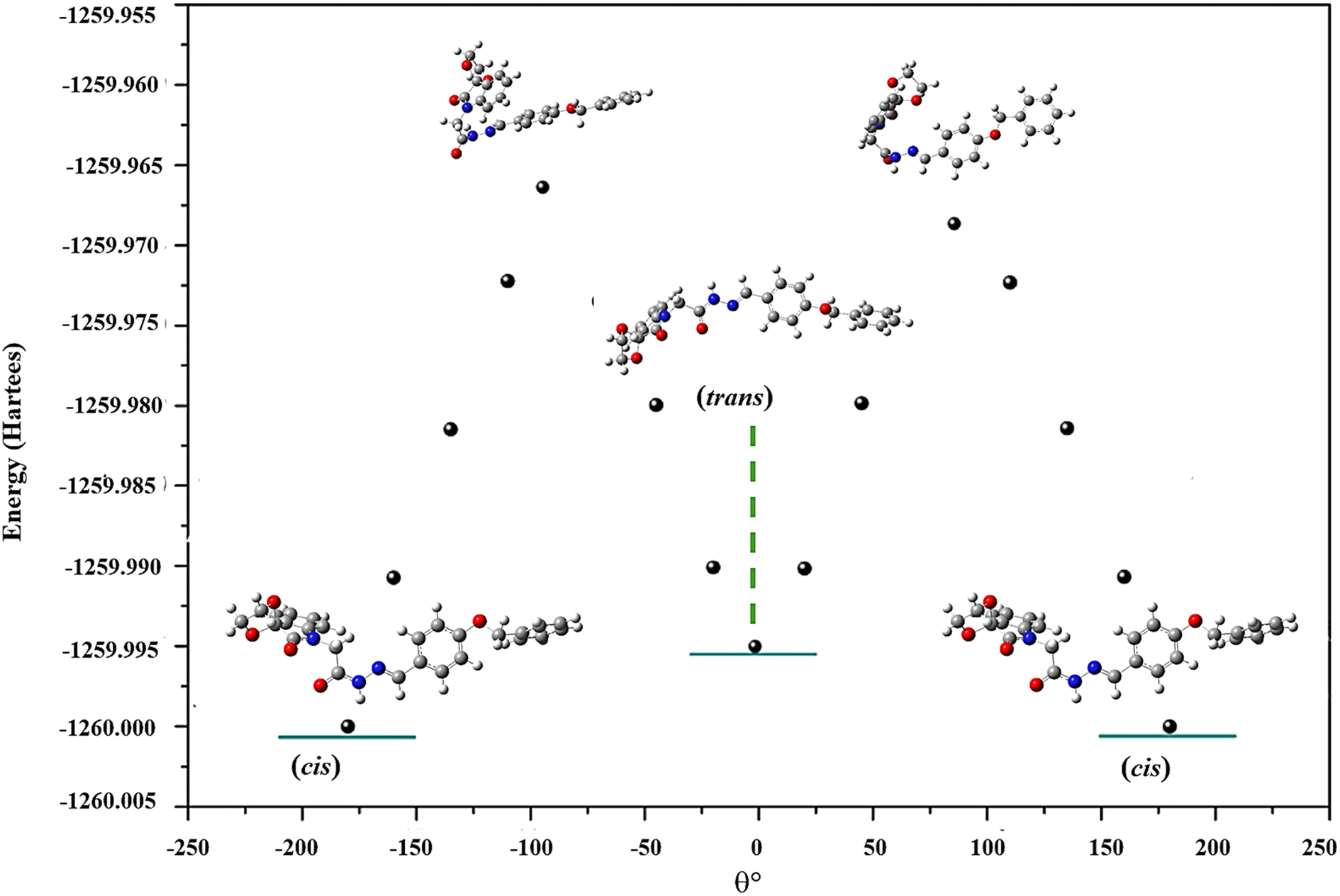 | ||
| Fig. 8 PES-scan of compound 1 with dihedral angle O28–C23–N27–N29. | ||
4. Conclusion
In conclusion, we have highlighted the importance of intramolecular C-bonding in stabilizing a particular conformation in spiroisatin-based N-acyl hydrazones 1–3. Despite the steric preference for a trans geometry around the NH–amide bond, C-bonding (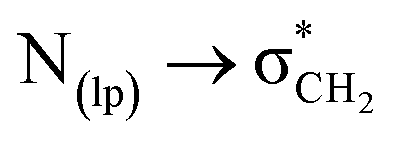 interactions) significantly stabilizes the cis isomer of compounds 1 and 2, as evidenced by 1H and 13C NMR, X-ray diffraction, and density functional theory (DFT) calculations. In DMSO-d6, compounds 1 and 2 predominantly adopt the cis form (82% and 76%, respectively), while compound 3 shows a slightly reduced cis preference (60%) due to competing hydrogen bonding. Single-crystal X-ray studies confirmed cis predominance for compounds 1 and 2, with compound 3 crystallizing in the trans form, likely due to packing and hydrogen bonding effects. Minimal energy barriers between cis and trans forms, supported by DFT calculations and potential energy surface scans, suggest dynamic equilibrium in polar solvents. The stabilization of cis conformation even in a highly polar solvent clearly indicates the broader significance of this interaction in supramolecular chemistry, especially when it is intramolecularly present.
interactions) significantly stabilizes the cis isomer of compounds 1 and 2, as evidenced by 1H and 13C NMR, X-ray diffraction, and density functional theory (DFT) calculations. In DMSO-d6, compounds 1 and 2 predominantly adopt the cis form (82% and 76%, respectively), while compound 3 shows a slightly reduced cis preference (60%) due to competing hydrogen bonding. Single-crystal X-ray studies confirmed cis predominance for compounds 1 and 2, with compound 3 crystallizing in the trans form, likely due to packing and hydrogen bonding effects. Minimal energy barriers between cis and trans forms, supported by DFT calculations and potential energy surface scans, suggest dynamic equilibrium in polar solvents. The stabilization of cis conformation even in a highly polar solvent clearly indicates the broader significance of this interaction in supramolecular chemistry, especially when it is intramolecularly present.
Data availability
The crystallographic data for 1–3 and other data related to this article is available in the ESI.†Author contributions
Muhammad Imran Ali investigation, methodology, formal analysis, writing – original draft writing, Javid Hussain formal analysis, visualization Muhammad Usman Anwar data curation, software, Ahmed Al-Harrasi resources, formal analysis, Muhammad Moazzam Naseer conceptualization, funding acquisition, resources, project administration, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MMN is grateful to The World Academy of Sciences (TWAS) for financial support through Project No. 13-419 RG/PHA/AS_CUNESCO FR: 3240279216.References
- A. S. Mahadevi and G. N. Sastry, Cooperativity in noncovalent interactions, Chem. Rev., 2016, 116, 2775–2825, DOI:10.1021/cr500344ed.
- M. Dubecky, L. Mitas and P. Jurecka, Noncovalent interactions by quantum Monte Carlo, Chem. Rev., 2016, 116, 5188–5215, DOI:10.1021/acs.chemrev.5b00577.
- D. Chatterji, Basics of Molecular Recognition, CRC Press, Boca Raton, 1st edn, 2016, DOI:10.1201/b19645.
- F. Weinhold and R. A. Klein, What is a hydrogen bond? Mutually consistent theoretical and experimental criteria for characterizing H-bonding interactions, Mol. Phys., 2012, 110, 565–579, DOI:10.1080/00268976.2012.661478.
- A. Mukherjee, S. Tothadi and G. R. Desiraju, Halogen bonds in crystal engineering: like hydrogen bonds yet different, Acc. Chem. Res., 2014, 47, 2514–2524, DOI:10.1021/ar5001555.
- B. K. Saha, A. Nangia and M. Jaskólski, Crystal engineering with hydrogen bonds and halogen bonds, CrystEngComm, 2005, 7, 355–358, 10.1039/b501693b.
- D. W. Zhang, J. Tian, L. Chen, L. Zhang and Z. T. Li, Dimerization of Conjugated Radical Cations: An Emerging Non-Covalent Interaction for Self-Assembly, Chem.–Asian J., 2015, 10, 56–68, DOI:10.1002/asia.201402805.
- C. B. Aakeröy, Crystal engineering: strategies and architectures, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 569–586, DOI:10.1107/s0108768197008446.
- A. S. Novikov, Non-covalent interactions in coordination and organometallic chemistry, 2020, DOI:10.3390/books978-3-0365-3287-5.
- Z. Berkovitch-Yellin and L. Leiserowitz, The role played by C–H⋯O and C–H⋯N interactions in determining molecular packing and conformation, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 159–165, DOI:10.1107/s0108768184001919.
- Y. Gu, T. Kar and S. Scheiner, Fundamental properties of the CH⋯O interaction: is it a true hydrogen bond?, J. Am. Chem. Soc., 1999, 121, 9411–9422, DOI:10.1021/ja991795g.
- M. Brandl, M. S. Weiss, A. Jabs, J. Sühnel and R. Hilgenfeld, CH⋯π-interactions in proteins, J. Mol. Biol., 2001, 307, 357–377, DOI:10.1006/jmbi.2000.4473.
- Z. Zhang, Y. Luo, J. Chen, S. Dong, Y. Yu, Z. Ma and F. Huang, Formation of Linear Supramolecular Polymers That Is Driven by C-H⋯π Interactions in Solution and in the Solid State, Angew. Chem., Int. Ed., 2011, 50, 1397, DOI:10.1002/ange.201006693.
- R. Parthasarathi, V. Subramanian and N. Sathyamurthy, Hydrogen bonding without borders: an atoms-in-molecules perspective, J. Phys. Chem. A, 2006, 110, 3349–3351, DOI:10.1021/jp060571z.
- S. Scheiner, T. Kar and Y. Gu, Strength of the CαH⋯O hydrogen bond of amino acid residues, J. Biol. Chem., 2001, 276, 9832–9837, DOI:10.1074/jbc.M010770200.
- M. Nishio, M. Hirota and Y. Umezawa, The CH/π Interaction: Evidence, Nature, and Consequences, John Wiley & Sons, 1st edn, 1998 Search PubMed.
- M. J. Plevin, D. L. Bryce and J. Boisbouvier, Direct Detection of CH/π Interactions in Proteins, Nat. Chem., 2010, 2, 466–471, DOI:10.1038/nchem.650.
- C. A. Sacksteder, S. L. Bender and B. A. Barry, Role for Bound Water and CH–π Aromatic Interactions in Photosynthetic Electron Transfer, J. Am. Chem. Soc., 2005, 127, 7879–7890, DOI:10.1021/ja050659a.
- J. Y. Lim, I. Marques, A. L. Thompson, K. E. Christensen, V. Félix and P. D. Beer, Chalcogen bonding macrocycles and [2] rotaxanes for anion recognition, J. Am. Chem. Soc., 2017, 139, 3122–3133, DOI:10.1021/jacs.6b12745.
- G. E. Garrett, E. I. Carrera, D. S. Seferos and M. S. Taylor, Anion recognition by a bidentate chalcogen bond donor, Chem. Commun., 2016, 52, 9881–9884, 10.1039/c6cc04818h.
- A. Bauzá and A. Frontera, Chalcogen ‘like-like’ interactions involving trisulphide and triselenide compounds: a combined CSD and ab initio study, Molecules, 2018, 23, 699, DOI:10.3390/molecules23030699.
- M. D. Esrafili and A. Sadr-Mousavi, Modulating of the pnicogen-bonding by a H⋯π interaction: an ab initio study, J. Mol. Graphics Modell., 2017, 75, 165–173, DOI:10.1016/j.jmgm.2017.04.017.
- R. Lo, P. Švec, Z. Růžičková, A. Růžička and P. Hobza, On the nature of the stabilisation of the E⋯π pnicogen bond in the SbCl3⋯toluene complex, Chem. Commun., 2016, 52, 3500–3503, 10.1039/c5cc10363k.
- Y. V. Nelyubina, A. A. Korlyukov and K. A. Lyssenko, Experimental charge density evidence for pnicogen bonding in a crystal of ammonium chloride, ChemPhysChem, 2015, 16, 676–681, DOI:10.1002/cphc.201402673.
- M. D. Esrafili and A. Sabouri, Carbene–aerogen bonds: an ab initio study, Mol. Phys., 2017, 115, 971–980, DOI:10.1080/00268976.2017.1297864.
- M. D. Esrafili and F. Mohammadian-Sabet, An ab initio study on anionic aerogen bonds, Chem. Phys. Lett., 2017, 667, 337–344, DOI:10.1016/j.cplett.2016.11.019.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Small Cycloalkane (CN)2C-C(CN)2 Structures Are Highly Directional Non-covalent Carbon-Bond Donors, Chem.–Eur. J., 2014, 20, 10245–10248, DOI:10.1002/chem.201403680.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Non-covalent sp3 carbon bonding with ArCF3 is analogous to CH–π interactions, Chem. Commun., 2014, 50, 12626–12629, 10.1039/c4cc05602g.
- X. García-LLinás, A. Bauzá, S. K. Seth and A. Frontera, Importance of R–CF3⋯O tetrel bonding interactions in biological systems, J. Phys. Chem. A, 2017, 121, 5371–5376, DOI:10.1021/acs.jpca.7b06052.
- P. Politzer, J. S. Murray and T. Clark, in Halogen bonding I: Impact on Materials Chemistry and Life Sciences, Springer, Cham, 2015, pp. 19–42, doi: DOI:10.1007/128_2014_568.
- P. Politzer, J. S. Murray, T. Clark and G. Resnati, The σ-hole revisited, Phys. Chem. Chem. Phys., 2017, 19, 32166–32178, 10.1039/c7cp06793c.
- D. Mani and E. Arunan, The X–C⋯Y (X = O/F, Y = O/S/F/Cl/Br/N/P) ‘carbon bond’ and hydrophobic interactions, Phys. Chem. Chem. Phys., 2013, 15, 14377–14383, 10.1039/c3cp51658j.
- S. P. Thomas, M. S. Pavan and T. G. Row, Experimental evidence for 'carbon bonding' in the solid state from charge density analysis, Chem. Commun., 2014, 50, 49–51, 10.1039/c3cc47226d.
- A. Jabs, M. S. Weiss and R. Hilgenfeld, Non-proline cis peptide bonds in proteins, J. Mol. Biol., 1999, 286, 291–304, DOI:10.1006/jmbi.1998.2459.
- D. E. Stewart, A. Sarkar and J. E. Wampler, Occurrence and role of cis peptide bonds in protein structures, J. Mol. Biol., 1990, 214, 253–260, DOI:10.1016/0022-2836(90)90159-J.
- T. Drakenberg and S. Forsen, Barrier to internal rotation of amides. I. Formamide, J. Phys. Chem., 1970, 74, 1–7, DOI:10.1021/j100696a001.
- T. Drakenberg and S. Forsén, The barrier to internal rotation in monosubstituted amides, J. Chem. Soc. D. Chem. Commun., 1971, 1404–1405, 10.1039/c29710001404.
- R. Jawaria, M. Hussain, Z. Shafiq, H. B. Ahmad, M. N. Tahir, H. A. Shad and M. M. Naseer, Robustness of thioamide dimer synthon, carbon bonding and thioamide–thioamide stacking in ferrocene-based thiosemicarbazones, CrystEngComm, 2015, 17, 2553–2561, 10.1039/c4ce02566k.
- M. M. Naseer, M. Hussain, A. Bauzá, K. M. Lo and A. Frontera, Intramolecular noncovalent carbon bonding interaction stabilizes the cis conformation in acylhydrazones, ChemPlusChem, 2018, 83, 881–885, DOI:10.1002/cplu.201800329.
- J. K. R. Deka, B. Sahariah, K. Baruah, A. K. Bar and B. K. Sarma, Conformational control of N-methyl-N,N′-diacylhydrazines by noncovalent carbon bonding in solution, Chem. Commun., 2020, 56, 4874–4877, 10.1039/d0cc00943a.
- J. J. Roeleveld, S. J. Lekanne Deprez, A. Verhoofstad, A. Frontera, J. I. Van der Vlugt and T. J. Mooibroek, Engineering Crystals Using sp3-C Centred Tetrel Bonding Interactions, Chem.–Eur. J., 2020, 26, 10126–10132, DOI:10.1002/chem.202002613.
- W. Wang, X. Li, P. P. Zhou and Y. Wang, Catalysis with Supramolecular Carbon-Bonding Interactions, Angew. Chem., 2021, 133, 22899–22903, DOI:10.1002/ange.202108973.
- V. R. Mundlapati, D. K. Sahoo, S. Bhaumik, S. Jena, A. Chandrakar and H. S. Biswal, Noncovalent Carbon-Bonding Interactions in Proteins, Angew. Chem., Int. Ed., 2018, 57, 16496–16500, DOI:10.1002/anie.201811171.
- E. V. Zaryanova, A. A. Ignatov and N. A. Lozynskaya, Synthesis and reactivity of new amide-substituted oxindole derivatives, Tetrahedron, 2017, 73, 6887–6893, DOI:10.1016/j.tet.2017.10.049.
- O. Radul, G. Zhungietu, M. Rekhter and S. Bukhanyuk, Simple method for the preparation of 1-substituted isatins, Chem. Heterocycl. Compd., 1983, 19, 286–288, DOI:10.1007/bf00513261.
- S. Gao, J. Zang, Q. Gao, X. Liang, Q. Ding, X. Li, W. Xu, C. J. Chou and Y. Zhang, Design, synthesis and anti-tumor activity study of novel histone deacetylase inhibitors containing isatin-based caps and o-phenylenediamine-based zinc binding groups, Bioorg. Med. Chem., 2017, 25, 2981–2994, DOI:10.1016/j.bmc.2017.03.036.
- T. Lu and F. Chen, Multiwfn: A Multifunctional Wavefunction Analyzer, J. Comput. Chem., 2012, 33, 580–592, DOI:10.1002/jcc.22885.
- W. Humphrey, A. Dalke and K. Schulten, VMD: Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38, DOI:10.1016/0263-7855(96)00018-5.
- A. Frisch, Gaussian 09W Reference, 2009, 470, https://gaussian.com/glossary/g09/ Search PubMed.
- M. P. Andersson and P. Uvdal, New scale factors for harmonic vibrational frequencies using the B3LYP density functional method with the triple-ζ basis set 6-311+G (d,p), J. Phys. Chem. A, 2005, 109, 2937–2941, DOI:10.1021/jp045733a.
- R. Munir, N. Javid, M. Zia-ur-Rehman, M. Zaheer, R. Huma, A. Roohi and M. M. Athar, Synthesis of novel N-acylhydrazones and their CN/NN bond conformational characterization by NMR spectroscopy, Molecules, 2021, 26, 4908, DOI:10.3390/molecules26164908.
- A. D. Isravel, J. K. Jeyaraj, S. Thangasamy, W. J. John and D. F. T. NBO, HOMO-LUMO, NCI, stability, Fukui function and hole–electron analyses of tolcapone, Comput. Theor. Chem., 2021, 1202, 113296, DOI:10.1016/j.comptc.2021.113296.
- E. D. Glendening and F. Weinhold, Natural resonance theory: I. general formalism, J. Comput. Chem., 1998, 19, 593–609, DOI:10.1002/(SICI)1096-987X(19980430)19:6%3C593::AID-JCC3%3E3.0.CO;2-M.
- F. Weinhold, C. Landis and E. Glendening, What is NBO analysis and how is it useful?, Int. Rev. Phys. Chem., 2016, 35, 399–440, DOI:10.1080/0144235X.2016.1192262.
- M. Orio, D. A. Pantazis and F. Neese, Density functional theory, Photosynth. Res., 2009, 102, 443–453, DOI:10.1007/s11120-009-9404-8.
- E. Bretschneider and O. Thomas, Internal Rotation in Molecules, John Wiley & Sons, London-New-York-Sydney-Toronto, 1974, DOI:10.1016/0022-2860(75)85024-1.
- V. Samoshin and N. Zefirov, Conformational transformations of organic-molecules in solutions, Zh. Vses. Khim. O-va. im. D. I. Mendeleeva, 1984, 29, 521–530 CAS.
- J. D. Roberts, Fascination with the conformational analysis of succinic acid, as evaluated by NMR spectroscopy, and why, Acc. Chem. Res., 2006, 39, 889–896, DOI:10.1021/ar050229p.
- O. Tapia and J. Bertrán, Solvent Effects and Chemical Reactivity, Springer, Dordrecht, 1st edn, 2006, DOI:10.1007/0-306-46931-6.
Footnotes |
| † Dedicated to Prof. Dr Klaus Jurkschat of TU Dortmund on occasion of his 73rd birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2401916–2401918. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra08086f |
| This journal is © The Royal Society of Chemistry 2025 |