
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrelithiation strategies for enhancing the performance of lithium-ion batteries
Yiming Zhang,
Huyan Shen,
Yanyu Li,
Yongsheng Hu and
Yao Li *
*
School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai, 200240, China. E-mail: liyaosjtu@sjtu.edu.cn
First published on 15th January 2025
Abstract
During the initial cycling of lithium-ion batteries, the generation of SEI at the electrode–electrolyte interface and the occurrence of irreversible side reactions consume the active lithium, resulting in irreversible loss of volume (ICL), which may also be accompanied by electrode volume changes and structural collapse. Addressing these challenges has become critical, and pre-lithiation with additional lithium has emerged as a key way to improve battery performance. Hence, this review comprehensively analyzes and summarizes the causes of ICL in lithium-ion batteries, and systematically discusses various prelithiation methods and mechanisms of different electrode structures, especially electrodes. Moreover, we discuss the importance of developing effective electrolyte, separator, and binder pre-lithiation technologies to improve ionic conductivity and battery life. The effectiveness of each strategy in improving initial capacity and cycling stability, while addressing compatibility issues and minimizing potential side effects, is evaluated to inform the future development and large-scale application of pre-lithiation technology.
1. Introduction
Given the escalating challenges posed by the energy crisis and environmental pollution, the development of sustainable and clean energy is of paramount importance.1–5 Lithium-ion batteries (LIBs) have emerged as the most prominent energy storage devices owing to their elevated energy density, extended cycle life, absence of memory effect, cost-effectiveness, and eco-friendliness.6–8 During the initial charging and discharging of lithium-ion batteries, a portion of the lithium ions is irreversibly utilized on the surface of the negative electrode to create a solid electrolyte interface (SEI) layer. This process results in a loss of the initial irreversible capacity, causing the actual capacity of the battery to be inferior to the theoretical capacity.9–11 Moreover, owing to significant volume fluctuations of specific electrode materials (P, Si, Sn, etc.), there will be persistent formation and degradation of the SEI, dead Li generation and other side reactions on the electrode surface during subsequent cycles, potentially resulting in substantial active lithium depletion or even structural collapse of the electrode.12,13 The functionality of lithium-ion batteries relies on the intercalation and deintercalation of lithium ions between the anode and cathode; thus, the migration and diffusion of lithium ions during cycling are crucial, directly influencing the battery's capacity. Consequently, pre-lithiation technology aimed at mitigating lithium loss and enhancing battery energy density has been extensively researched.14–19Prelithiation is a technique that uses prelithiation reagents to incorporate active lithium into the electrode material prior to battery assembly. It does not alter the original electrode material or the battery architecture. Pre-introducing lithium ions into the electrode material effectively mitigates irreversible capacity loss during the early charge and discharge cycles, thereby enhancing the battery's initial capacity. Furthermore, prelithiation enhances the structural integrity of electrode materials, mitigates capacity degradation during cycling, and extends the battery's lifespan. To enhance the active lithium content, modifications can be made to the anode, cathode and separator. To date, various prelithiation techniques have been established, which broadly categorized into prelithiation additives, chemical prelithiation, electrochemical prelithiation, and mechanical prelithiation. These approaches can enhance active lithium through diverse mechanisms to augment battery energy density, although they encounter numerous hurdles. Electrochemical prelithiation facilitates meticulous regulation of lithium concentration and distribution inside the electrode, however it may be intricate and labor-intensive. Chemical prelithiation offers ease and scalability, although it may lead to undesirable side reactions and contaminants. Mechanical techniques, including direct interaction with lithium metal, are rapid and uncomplicated but frequently encounter challenges concerning uniformity and regulation. Notwithstanding these gains, other challenges persist, including the stability of prelithiated materials under ambient settings, safety issues related to the handling of reactive lithium, and the scalability of prelithiation techniques for commercial applications.
This review aims to provide a comprehensive analysis of the current status of prelithiation strategies for LIBs, covering the fundamental principles, recent developments, and practical challenges associated with diverse prelithiation methods. We will examine the mechanisms of several prelithiation methods, assess their efficacy in improving battery performance, and investigate their feasibility for large-scale implementation. Furthermore, the review will highlights innovative applications of prelithiation, such as the advancement of next-generation anode materials, optimization of battery components, and enhancement of the safety and longevity of LIBs. Ultimately, we discuss the anticipated the future trajectory of prelithiation research and its potential for large-scale application. By systematically reviewing existing research and identifying future directions, this review aims to enhance the development of efficient and reliable energy storage systems, thereby promoting the broader adoption of high-performance LIBs.
2. Mechanisms of irreversible capacity loss
The three prevalent operational modes of lithium-ion electrodes are intercalation reaction, alloying reaction, and conversion reaction, as illustrated in Fig. 1.20 The initial form of lithium involves intercalation into carbon and lithium titanate. Lithium ions can be reversibly inserted into or detached from the periodic crystal structure, resulting in minimal impact on the anode material's structure. Lithium titanate, known as a 'zero strain material', exhibits excellent structural stability throughout the lithium de-intercalation process; nonetheless, its theoretical specific capacity is limited to 175 mA h g−1, and the lithium intercalation potential is relatively high (1.55 V vs. Li/Li+), resulting in a minimal market share.21 Carbon materials can be classified into graphite and amorphous carbon based on the degree of graphitization, with graphite serving as the primary anode material for LIB due to its low lithium intercalation potential (0.2 V vs. Li/Li+) and cost-effectiveness. Despite graphite's high conductivity, its theoretical capacity is limited to merely 372 mA h g−1.22,23 The rapid advancement of the new energy industry has significantly constrained the potential for enhancing the energy density of traditional graphite rendering it inadequate for further development requirements.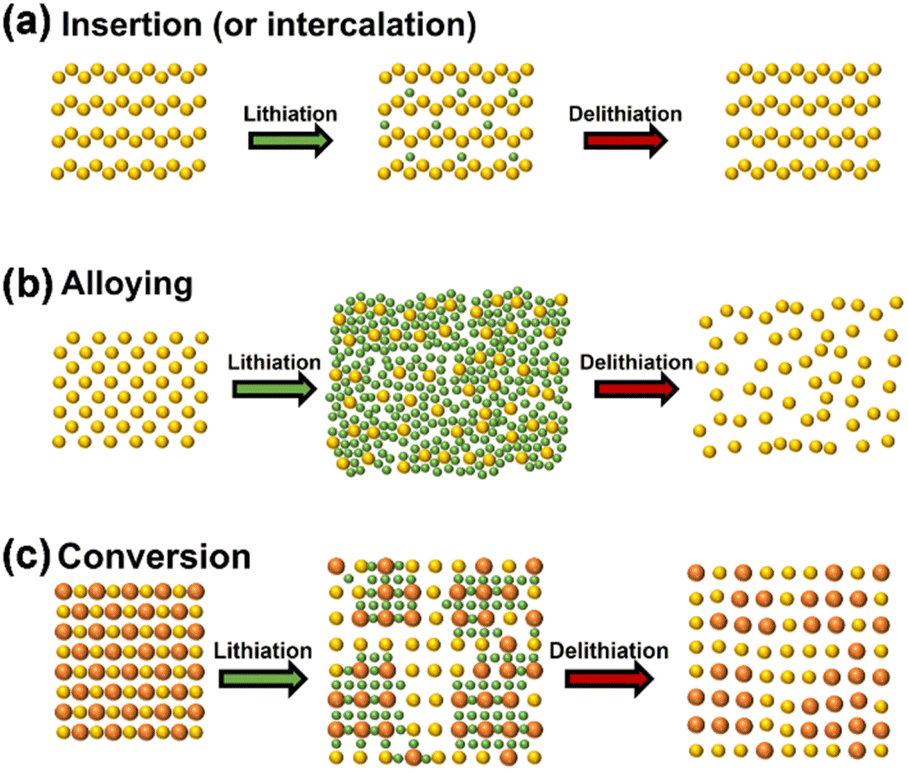 | ||
| Fig. 1 Schematic diagrams of basic lithium storage principles of the anode materials (a) embedding reaction mechanism, (b) alloying reaction mechanism, (c) transformation reaction mechanism (reprinted from ref. 20. Copyright 2020, with permission from MDPI). | ||
The second anode material comprises Si, Sn and P alloys, which undergo a chemical reaction with lithium for lithium storage. These materials possess a high theoretical specific capacity, nevertheless, they experience more significant side reactions and greater initial active lithium loss compared to graphite.30–35 The significant volume change of this alloy material during charging and discharging will result in the collapse of its electrode structure, subsequently causing the fracture and pulverization of the electrode material. The SEI film is concurrently degraded alongside the electrode damage, subsequently reacting with the electrolyte to generate a new SEI film that envelops the electrode material. This cycle may repeat multiple times, leading to significant lithium loss. The redox potential, ICE, and electronic/ionic conductivity of anode materials significantly influence the electrochemical parameters of LIBs, including energy density, power density, and cycling stability. The side reaction between the electrode material and the electrolyte results in the formation of a SEI film on the surface of the anode, depleting active lithium from the lithium oxide or phosphate in the cathode during the battery's initial charging, significantly diminishing the capacity and energy density of the lithium-ion battery.
The third is a conversion reaction characterized by multi-electron processes. Unlike intercalation or alloying reactions, where lithium ions insert into or combine with the metal structure, the conversion reaction is more complex. Certain transition metal compounds undergo reduction or oxidation by lithium ions during charging and discharging, resulting in the formation of diminutive particles comprised of metal and lithium anion complexes.24–29 The conversion reaction is attractive due to its potential for much higher specific capacities compared to traditional intercalation materials. This is because the reaction involves the transfer of multiple electrons per lithium ion, allowing for a significantly greater number of lithium ions to be stored in the material. For example, a material like iron oxide (Fe2O3) can achieve a theoretical capacity much higher than graphite, which is the anode material commonly used in lithium-ion batteries. This anode material exhibits a superior specific capacity, yet, it is plagued by significant volumetric fluctuations, inadequate reaction reversibility, and elevated redox potential.
2.1 Formation of SEI films
The high electrochemical activity of lithium metal leads to a reduction reaction with the electrolyte, resulting in the formation of a passivation film. This phenomenon was initially identified by Dey in 1970 and subsequently termed SEI film by Peled in 1979.36 Fig. 2 illustrates the development process of the SEI membrane, wherein the ongoing decomposition of the electrolyte leads to the progressive thickening of the SEI membrane, potentially resulting in a mosaic or a layered structure.37 In the mosaic configuration, inorganic nanocrystals are randomly distributed inside an organic substrate;38–40 In the hierarchical structure, the inner layer primarily consists of inorganic substances, whereas the outer layer is predominated constituted of organic matter.41,42 The ideal SEI membrane should exhibit low electron conductivity and high ion diffusion capacity, insolubility in electrolytes, and stability through a broad spectrum of operating temperatures and voltages.43 The predominant model is the rapid migration of lithium ions via the mosaic model. Fig. 2 illustrates that the SEI layer is mainly composed of an inorganic layer and an organic layer. The region adjacent to graphite anode typically consists of an inorganic layer, mainly of LiF, Li2O, Li2CO3, Li3N, LiOH, etc., which offers strength support for SEI. The outer layer consists of an organic layer that mainly forms an organic lithium salt, such as COO2Li, HOOCLi and other substances with high oxidation state.44 This bilayer structure may lead to the electrolyte being coated with organic components on the electrode's surface after the electrode reacts, while the inner layer gradually transforms into inorganic substances such as LiF.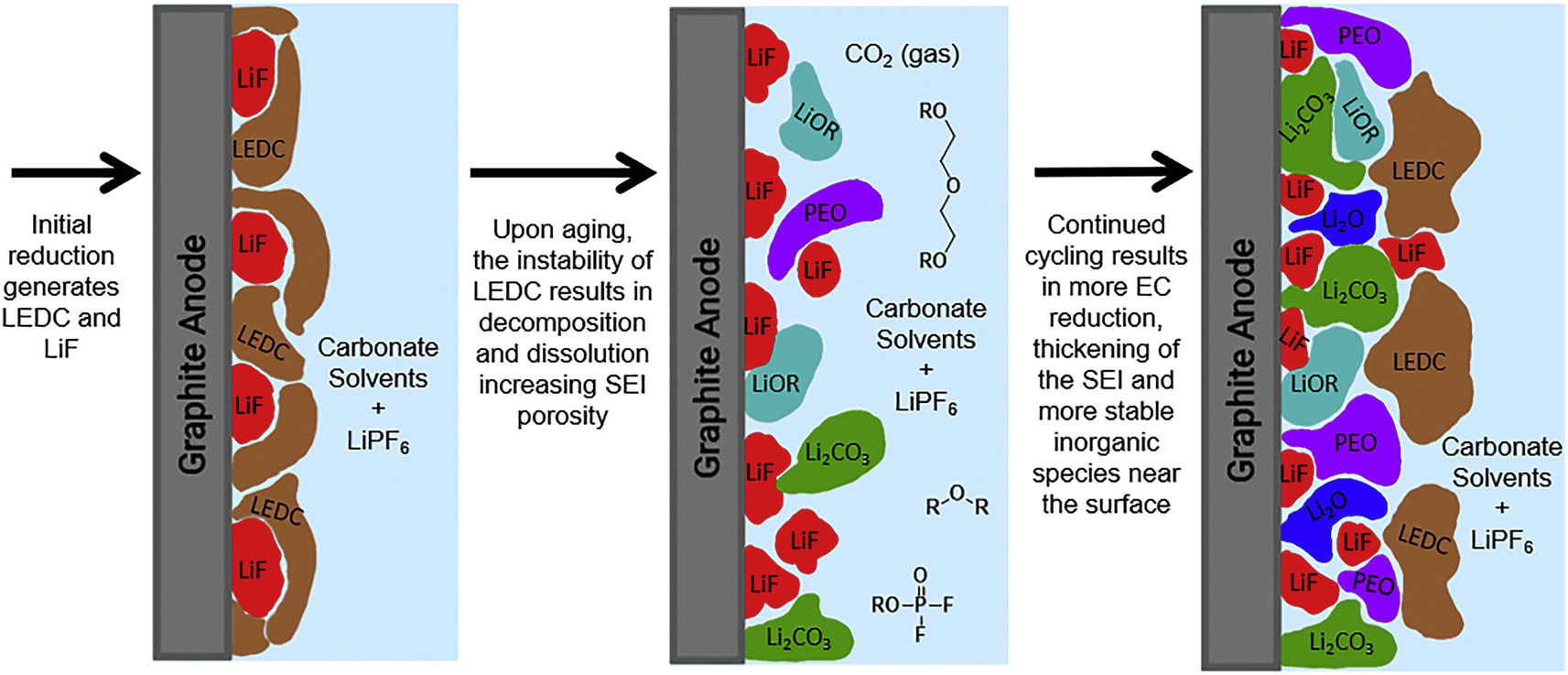 | ||
| Fig. 2 Schematic diagram of the formation of an SEI film on a graphite anode (reprinted from ref. 37. Copyright 2019, with permission from Elsevier). | ||
Goodenough posited that the formation of the SEI layer on the electrode surface is associated with the energy levels of the HOMO (orbital with the highest energy level that has occupied electrons)–LUMO (the orbital with the lowest energy level that has not occupied electrons)of the electrolyte.45 Fig. 3 illustrates the comparative electron energies of the cathode and anode and electrolyte redox pairs in LIBs. When the electrochemical potential μA of the anode exceeds the minimum energy of the unoccupied molecular orbital (LUMO) of the electrolyte, a reduction reaction will occur on the anode's surface. Likewise, when μC is beneath the energy of the highest occupied molecular orbital (HOMO) of the electrolyte, the electrolyte experiences an oxidation process at the cathode surface. To enhance the energy density of the redox pair in lithium-ion batteries, it is essential to achieve the maximum energy differential between the cathode and anode, which corresponds to an elevated operating voltage. The oxidation potential of typical organic electrolytes is approximately 4.6 V (versus. Li+/Li), while the reduction potential is nearly 1.0 V (vs. Li+/Li). The reduction potential of Li+ embedded in graphite ranges from 0 to 0.25 V (vs. Li+/Li), which is inferior to that of the electrolyte.46 Consequently, when the potential of the graphite anode falls below the stable range of the electrolyte, the electrolyte decomposes on the graphite surface, resulting in the formation of a solid electrolyte interphase (SEI) film during the charging of the graphite anode (lithium intercalation reaction at the graphite anode).47
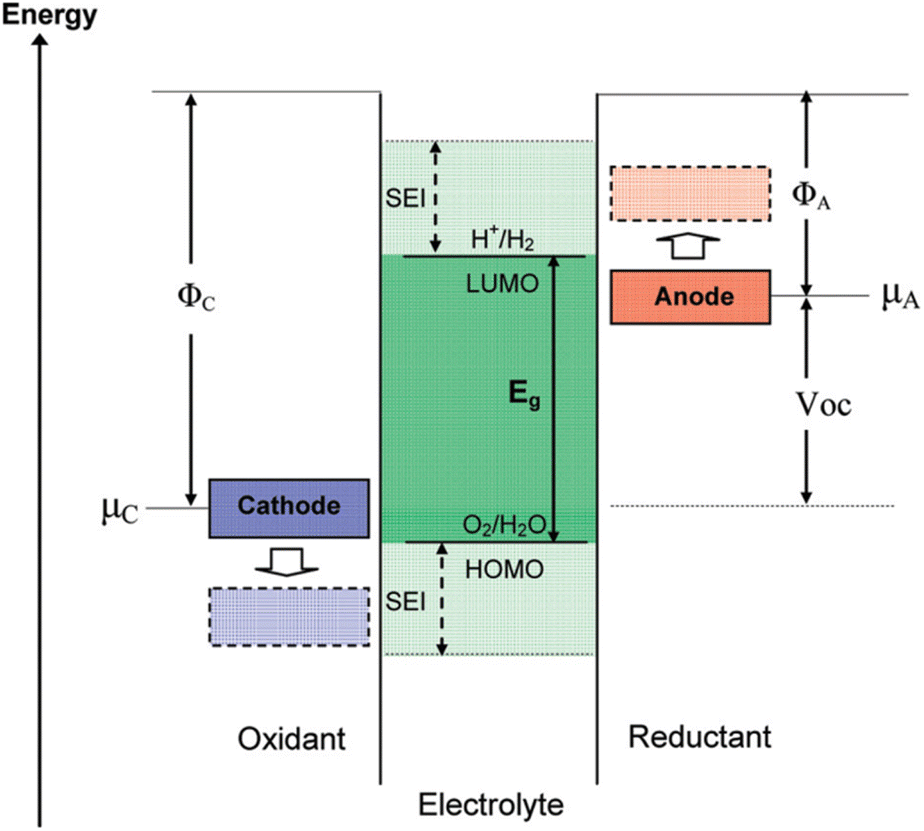 | ||
| Fig. 3 Schematic open-circuit energy diagram of an aqueous electrolyte. ΦA and ΦC are the anode and cathode work functions. Eg is the window of the electrolyte for thermodynamic stability (reprinted from ref. 45. Copyright 2009, with permission from ACS Publications). | ||
2.2 Electrolyte breakdown
The electrolyte in lithium-ion batteries (LIBs) generally comprises a lithium salt, like lithium hexafluorophosphate (LiPF6), dissolved in an organic solvent, such as ethylene carbonate (EC) or dimethyl carbonate (DMC).47 Under conditions of elevated pressure or temperature, both solvents and salts may undergo decomposition.48 This decomposition produces gaseous by-products and solid residues that negatively impact the battery's performance and safety. Using the common EC solvent as an example, during battery operation, especially at elevated temperatures or under high voltage circumstances, EC experiences reductive breakdown on the anode surface. This process produces ethylene gas (C2H4), which increases the internal gas pressure in the battery.49 Furthermore, any residual water (H2O) in the electrolyte may breakdown during the charging process, generating hydrogen gas (H2). The buildup of these gasses results in elevated internal pressure, leading to the battery's expansion and deformation. This distortion may compromise the precise alignment of the battery's electrodes and other components, potentially resulting in problems such as electrode delamination, loss of electrical connectivity, and heightened risk of electrolyte leakage or cell rupture. Furthermore, the solid residues generated from electrolyte breakdown accumulate on the surfaces of the electrodes. These residues may additionally augment the thickness of the solid electrolyte interphase (SEI) layer or generate wholly novel interfacial layers. The stability of the SEI layer is essential for safeguarding the anode and maintaining long-term cycle stability; nevertheless, the persistent generation of new SEI from continuing breakdown reactions results in several issues. The augmented SEI layer elevates the internal resistance of the battery, thus diminishing its overall conductivity and efficiency. The creation of these solid residues also depletes active lithium ions, leading to irreversible capacity loss (ICL) and reducing the battery's energy density over time.2.3 Lithium deposition
Due to the larger potential of lithium-ion embedding in graphite (0.1 V vs. Li/Li+) compared to lithium deposition (<0 V vs. Li/Li+), lithium insertion into graphite is thermodynamically favored over deposition. Nonetheless, the lithium insertion rate in the anode surpasses the solid-phase diffusion rate of lithium ions within the anode at elevated charging rates or low temperatures, resulting in the facile deposition of lithium ions on the anode surface, leading to the formation of dendritic or needle-like structures. Fig. 4 demonstrates that dendritic development will destabilize the electrode and current interface during the cycling of lithium-ion batteries, compromising the integrity of the formed SEI layer.50–52 Simultaneously, during the growing phase, lithium dendrites will persist in depleting the electrolyte, resulting in the irreversible deposition of metallic lithium, hence forming dead lithium and inducing low coulombic efficiency. Lithium dendrites can penetrate the separator, causing internal short circuits in lithium-ion batteries, leading to thermal runaway, ignition, combustion, and explosion.53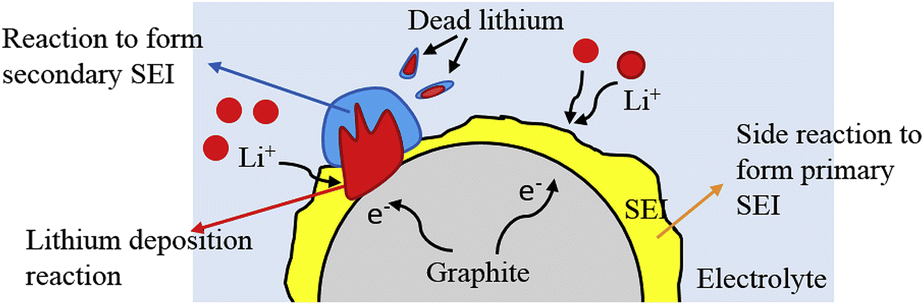 | ||
| Fig. 4 Schematic diagram of degradation mechanisms at low temperature (reprinted from ref. 50. Copyright 2019, with permission from Elsevier). | ||
2.4 Transition metal dissolution
The dissolution of transition metals in lithium-ion batteries (LIBs) significantly impacts battery performance and longevity. The dissolving process is intensified by elevated operating voltages, high temperatures, and the chemical instability of the cathode material. Transition metal ions (e.g., Mn, Co, Ni) in the cathode material may dissolve into the electrolyte during cycling, particularly at elevated voltages.54 The dissolved metal may migrate to the anode and be deposited on the solid electrolyte interphase (SEI) or within the anode material, so promoting the breakdown of the electrolyte and resulting in the formation of further SEI films. The presence of these metal ions at the anode–electrolyte interface destabilizes the solid electrolyte interface (SEI), a protective layer essential for sustaining battery performance. The continuous formation and decomposition of SEI films lead to increased lithium-ion consumption, resulting in irreversible capacity loss (ICL). The migration and deposition of dissolved transition metals on the anode can inflict structural damage to the anode material. Shin et al. included Mn(PF6)2 into the ester electrolyte, demonstrating that the deposition of dissolved Mn ions on the graphite anode results in the thickening of the SEI layer and an increase in the charge transfer impedance of the battery (Fig. 5).55 This leads to increased resistance, diminished conductivity, and reduced overall cell efficiency.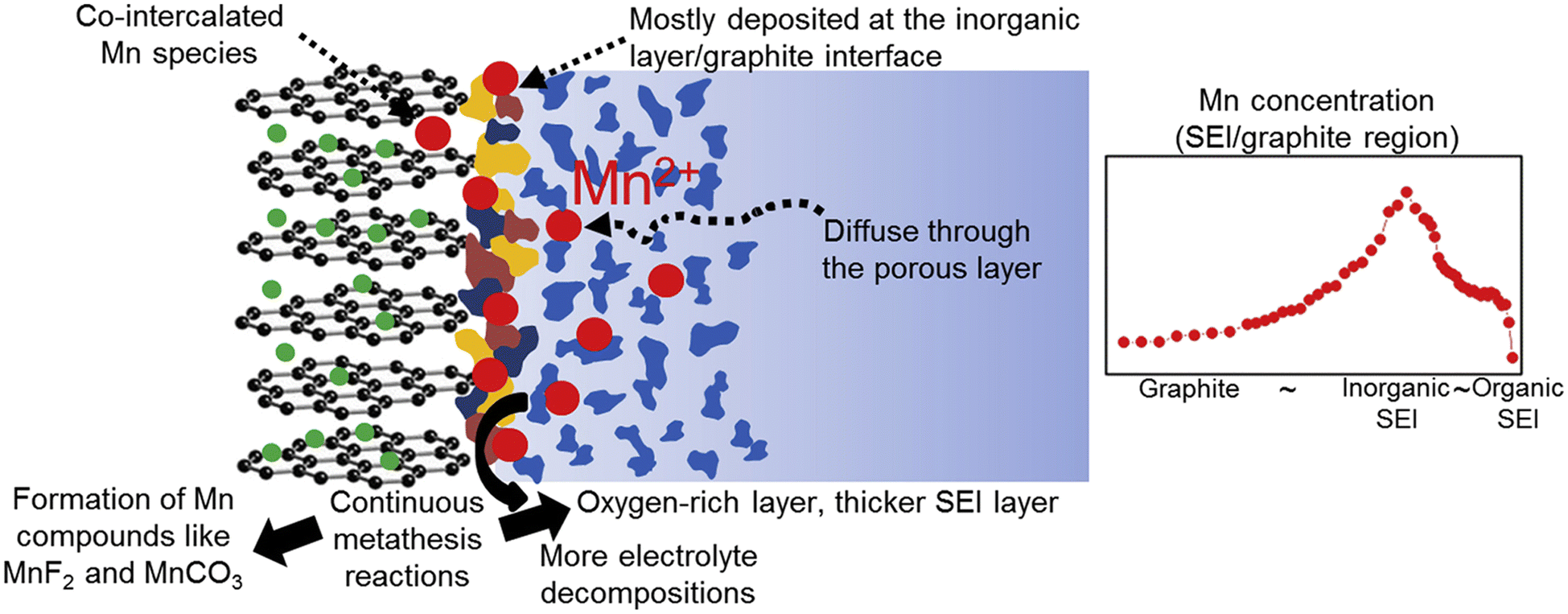 | ||
| Fig. 5 The spatial distribution of Mn deposited at the SEI/graphite interface (reprinted from ref. 55. Copyright 2015, with permission from Elsevier). | ||
2.5 Volume changes and structural degradation of electrodes
High-capacity anode materials, including silicon, tin, and phosphorus, have attracted much attention for their potential to substantially enhance the energy density of lithium-ion batteries (LIBs). Nonetheless, these materials encounter considerable hurdles, primarily due to the enormous volume alterations they experience during lithiation and delithiation processes.56,57 Silicon, which can theoretically accommodate ten times more lithium than graphite, undergoes a volume increase of up to 300% upon full lithiation. Likewise, tin and phosphorus experience significant volume alterations, albeit to a somewhat smaller degree. The significant expansions and contractions during charge and discharge cycles induce substantial mechanical stress within the electrode material (Fig. 6).58–61 This stress can induce the fragmentation and fissuring of anode particles, causing the active material to detach from the surrounding binder and disassociate from the current collector. Structural degradation caused by these volume variations adversely impacts battery performance and cycle stability. The development and spread of fissures in the anode material compromise the electrode's integrity, resulting in diminished electrical contact and reduced conductivity.62–65 This degradation can segregate sections of the active material, rendering them inactive and thereby diminishing the overall capacity of the battery. Furthermore, the persistent disruption and reformation of the solid electrolyte interphase (SEI) layer on the anode surface intensify the issue. The SEI is a passivation layer that develops during the early cycles, stabilizing the electrode–electrolyte interface by inhibiting additional electrolyte breakdown. Nonetheless, with each cycle, when the anode material undergoes expansion and contraction, the solid electrolyte interphase (SEI) layer may fracture and then regenerate. This process depletes additional lithium ions, resulting in irreversible capacity loss (ICL), while promoting the growth of thicker and less stable solid electrolyte interphase (SEI) layers, hence elevating internal resistance and diminishing cell efficiency.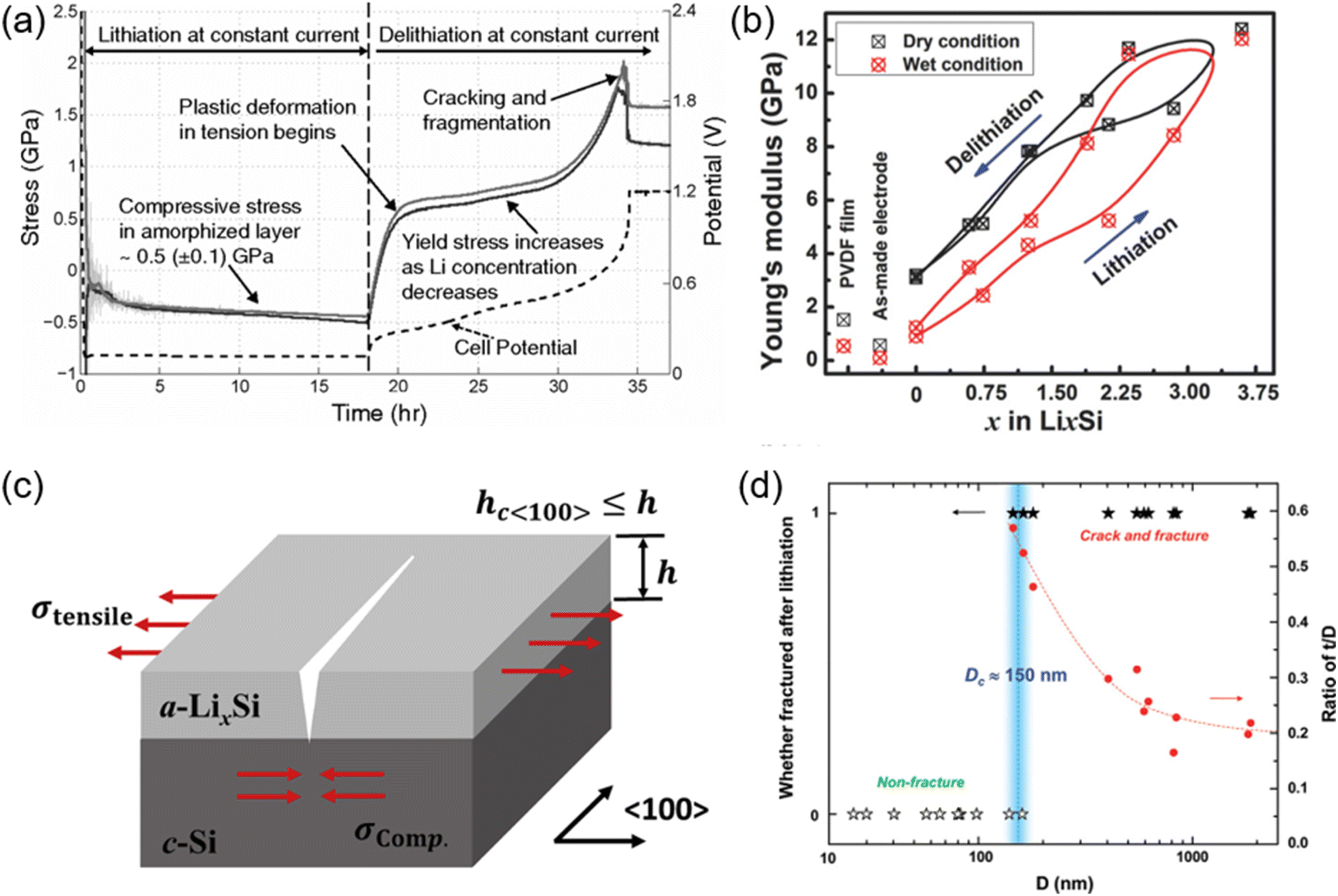 | ||
| Fig. 6 (a) Evolution of stress in amorphized layer as a function of time during lithiation and delithiation of a crystalline Si film (two solid lines corresponding to two orthogonal directions on the wafer) (reprinted from ref. 58. Copyright 2011, with permission from American Physical Society). (b) The variation of Young's modulus of the composite electrode with Li concentration (x in LixSi) during the lithiation/delithiation process (reprinted from ref. 61. Copyright 2018, with permission from Wiley-VCH). (c) Schematic illustration of 〈100〉-oriented crack (reprinted from ref. 62. Copyright 2019, with permission from Elsevier). (d) Statistics showing the critical size (Dc) around 150 nm (reprinted from ref. 60. Copyright 2012, with permission from ACS Publications). | ||
3. Current status of prelithiation research
The creation of the SEI layer and the occurrence of irreversible side reactions lead to significant consumption of active lithium, a reduction in battery energy density, and degradation of the electrode structure, ultimately shortening cycle life of lithium-ion batteries. Consequently, the supplementary adjustment of active lithium through pre-lithiation has emerged as a pivotal subject for scientific researchers. Pre-lithiation is conceptually distinct from the pretreatment of battery materials or electrodes; it aims to supply supplementary active lithium to lithium-ion batteries through specific pre-lithiation reagents, materials, or treatments prior to battery cycling. This process increases the lithium content prior to charging and discharging (Fig. 7).19 The pre-lithiation effect can facilitate the pre-regulation of SEI composition, stimulate the synergistic coexistence of many components, and enhance SEI stability. In conclusion, prelithiation techniques are essential for enhancing the performance and efficiency of lithium-ion batteries. By tackling lithium depletion and improving the stability of anode materials, these methods are crucial in promoting the advancement of high-energy-density lithium-ion batteries. Ongoing research and innovation in this field are crucial for overcoming existing limits and realizing the complete potential of lithium-ion battery technology.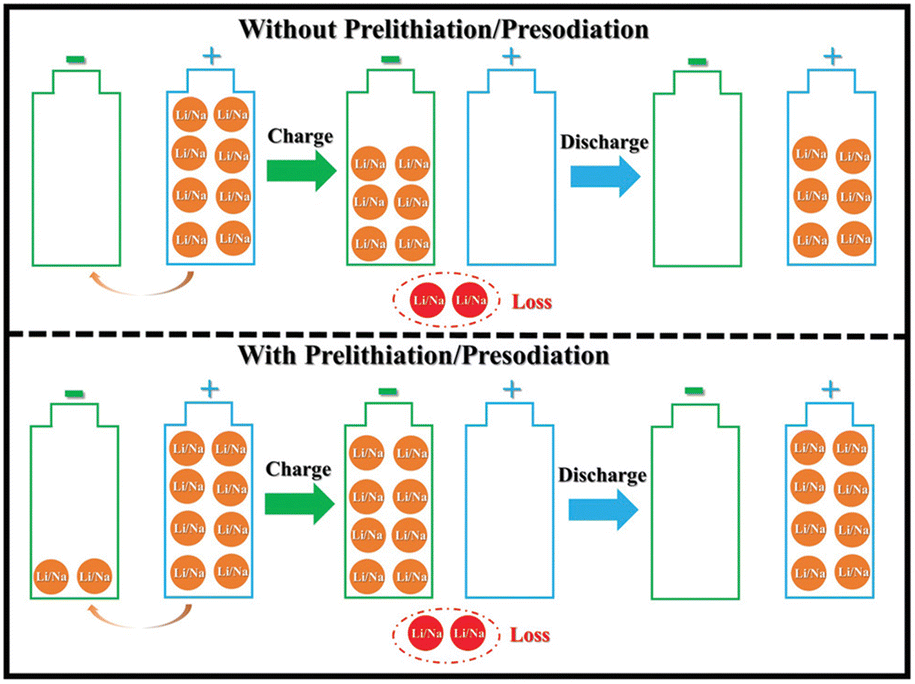 | ||
| Fig. 7 Schematic of the influence of prelithiation/presodiation in LIBs and SIBs (reprinted from ref. 19. Copyright 2020, with permission from Wiley-VCH). | ||
Prelithiation can be executed on various battery components, including cathodes and anodes, through methods such as electrochemical prelithiation at the electrode level, chemical prelithiation at the material or electrode level, the use of prelithiation additives at the cathode, and direct contact or short circuiting of the anode with lithium metal. Fig. 8 illustrates pre-lithiation at both the material and electrode levels, encompassing the synthesis and utilization of pre-lithiation materials, electrochemical pre-lithiation, and chemical pre-lithiation at the electrode level.66 Various pre-lithiation techniques, employing distinct mechanisms, have demonstrated varying efficiencies in enhancing the energy density and other performance metrics of lithium-ion batteries (LIBs). Five prevalent pre-lithiation techniques are as follows: first, active lithium is electrochemically deposited onto the electrode; second, a lithium sheet is mechanically pressed onto the electrode material to provide lithium; third, lithium-containing additives are utilized; fourth, lithium metal is combined with an organic reagent to create a slurry that coats the electrode surface via solid reaction; fifth, the electrode material is treated through chemical immersion. The cathode is typically stable, but the anode is the primary site of active lithium loss, prompting extensive research on the surface engineering of the anode. This paper will systematically present the research on pre-lithiation in recent years, beginning with the fundamental structure of the battery and addressing the electrode, electrolyte, separator, and binder levels, in accordance with the reaction mechanisms of various components to compensate for lithium. The electrode-level lithium source replenishment can be categorized into four methods of pre-lithiation technology: electrode pre-lithiation additives, chemical pre-lithiation, electrochemical pre-lithiation, and mechanical pre-lithiation.
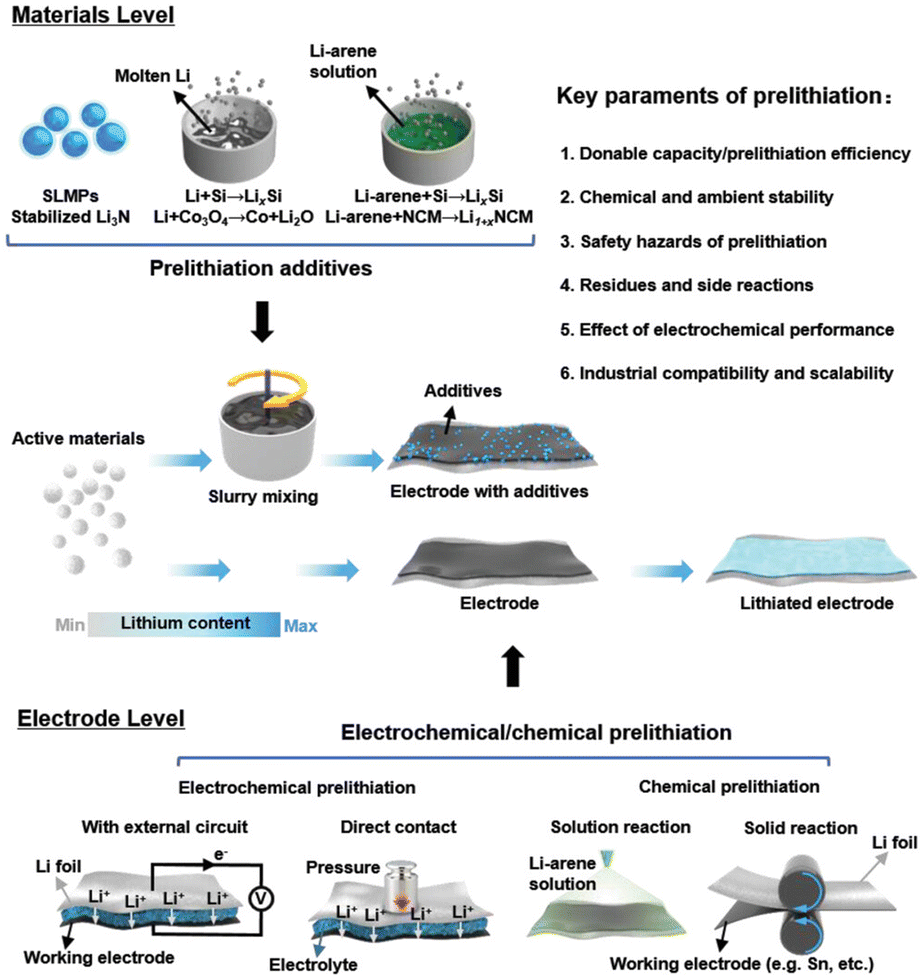 | ||
| Fig. 8 Materials, methods, and key parameters for battery prelithiation (reprinted from ref. 66. Copyright 2021, with permission from Wiley-VCH). | ||
3.1 Electrode prelithiation
The first cyclic irreversible lithium loss of the electrode material mostly results from the solid electrolyte interface (SEI) established at the electrode/electrolyte interface and irreversible lithium-ion intercalation. Prelithiation at the electrode level primarily involves the incorporation of supplementary lithium sources into the positive and negative electrode materials to enhance the initial coulombic efficiency of the battery and mitigate the irreversible lithium loss resulting from the electrode/electrolyte interface reaction and structural alterations during the initial cycle. This section will explore the existing mechanisms of action of pre-lithiation additives, electrochemical prelithiation, chemical prelithiation, and mechanical prelithiation, along with their present problems.Lithium alloys exhibit a reduced lithium intercalation potential and can discharge lithium ions during the initial cycle, compensating for lithium loss in the anode material. LixSi nanoparticles exhibit a low lithiation potential of 10 mV and a large theoretical specific capacity of 2000 mA h g−1, facilitating efficient and rapid prelithiation of the negative electrode. Zhao et al. documented LixSi–Li2O core–shell nanoparticles exhibiting high specific capacity as an effective pre-lithiation agent to mitigate first-pass capacity loss (Fig. 9a).69 LixSi–Li2O core–shell nanoparticles are amenable to processing in a one-step thermal alloying process in slurries, demonstrate excellent capacity in dry air environments, and are safeguarded by Li2O passivated shells. This method is facile and suitable for mass-producible. Both the silicon and graphite anodes were effectively prelithiated with these nanoparticles, attaining elevated first-pass coulombic efficiencies ranging from 94% to over 100%. LixSi can benefit from the mature manufacturing infrastructure of the Si industry for scale-up and low-cost manufacturing. However, this material is unstable in air with low relative humidity and has a limited lifespan; therefore, it is essential to produce pre-lithiation additives with enhanced air stability (Fig. 9b). Yang proposed a straightforward, economical, and regulated approach to synthesis of environmentally stable LixSi-based prelithiation reagents.71 A chemical reaction between aluminum isopropanoxide and LixSi results in the formation of a LixAlySiOz/Li2O protective layer on the surface, endowing the LixSi (A-LixSi) covered with this layer with chemical stability and robust environmental adaptability (Fig. 9c). Utilizing A-LixSi as a lithium supplement for the graphite anode enhances the anode's initial coulombic efficiency while also imparting stable chemical qualities and robust environmental adaptability. Zhao et al. devised a comprehensive one-pot metallurgical method to prelithiate group IV elements and their respective oxides, yielding electrode materials that approach theoretical specific capacity (Fig. 10d).73 The synthesized Li22Z5 alloy and Li22Z5–Li2O composites (Z = Si, Ge, Sn, etc.) served as prelithiation agents to enhance the initial coulombic efficiency of graphite and alloy-type anode materials. LixGe demonstrates superior stability in air because of the substantial binding energy between Li and Ge atoms in the Li22Ge5 crystal (Fig. 10d). The uniformly distributed reactive LixZ nanodomain is consistently integrated inside a durable, highly crystalline Li2O matrix.
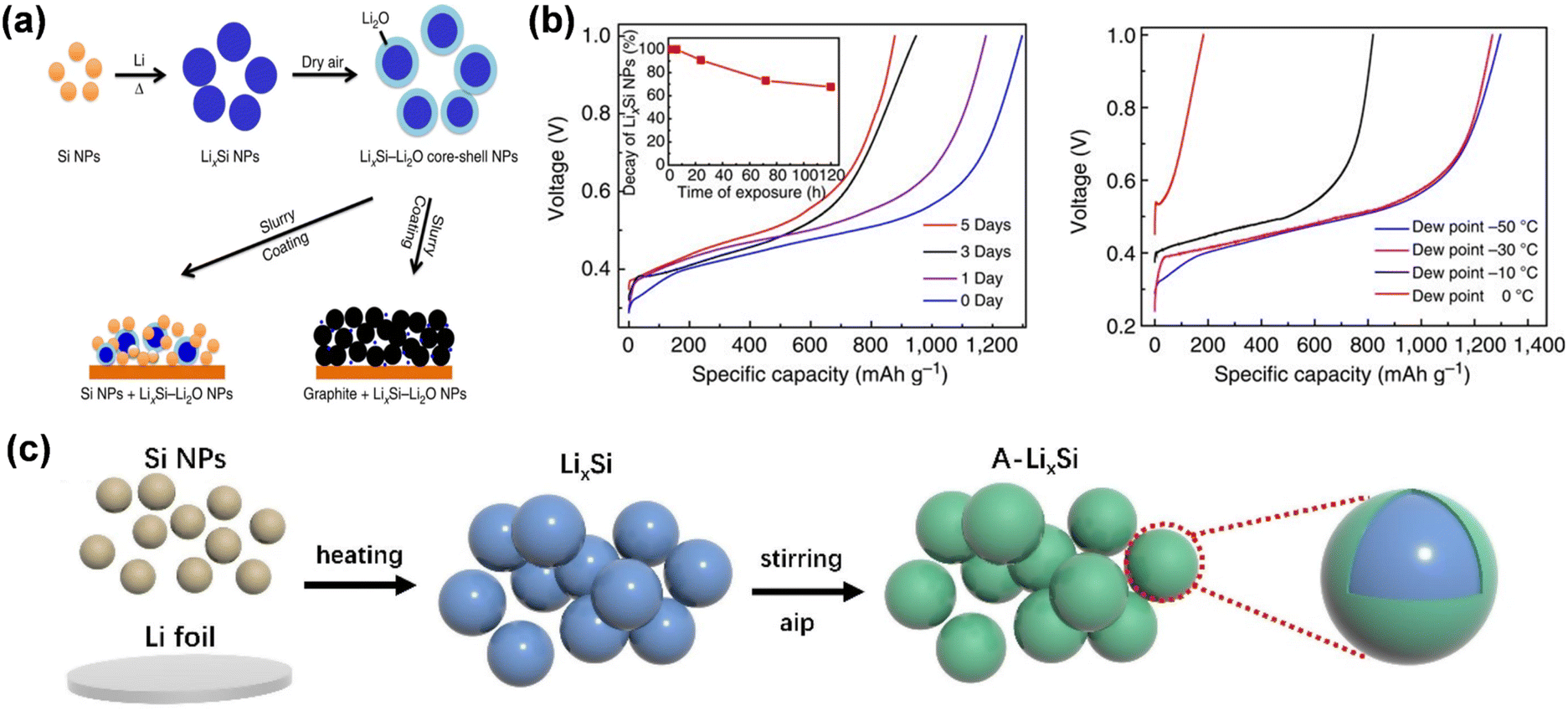 | ||
| Fig. 9 (a) Schematic diagrams showing Si NPs react with melted Li to form LixSi NPs. (b) The capacity retention of LixSi–Li2O NPs exposed to dry air with varying durations and air at different humidity levels (reprinted from ref. 69. Copyright 2014, with permission from Springer Nature). (c) Synthesis process for the LixAlySiOz/Li2O-coated LixSi (A-LixSi). | ||
 | ||
| Fig. 10 (a) Images of galvanostatic discharge/charge profiles of A-LixSi NPs at 0.05 A g−1 in the first cycle. (b) First-cycle voltage profiles of graphite/A-LixSi (blue line), graphite/Si (purple line) and graphite (red line) (reprinted from ref. 71. Copyright 2022, with permission from Elsevier). (c) The Li8Si10 pseudo-tetrahedron from cubic Li22Si5, Li5Ge10 pseudo-tetrahedron from cubic Li22Ge5, and Li4Sn6 octahedron from cubic Li22Sn5 were selected as the simulation units to calculate the binding energy (Eb) of Li with Z (Z = Si, Ge and Sn). (d) Schematic diagram showing a one-pot metallurgical process to synthesize Li22Z5 alloys and Li22Z5-Li2O composites by using Z and ZO2 as the starting materials, respectively (reprinted from ref. 73. Copyright 2018, with permission from Elsevier). | ||
The utilization of lithium metal as a pre-lithiation additive for the anode prevents the incorporation of extraneous contaminants, while the Li2CO3 nanoshell on the surface of SLMPs offers effective protection to maintain air stability. SLMPs, when prelithiated with slurry, exhibit incompatibility with polar solvents; therefore, they must be combined with styrene-butadiene rubber (SBR) and toluene (SST) to ensure stable lithium supplementation (Fig. 11a). The quantity of ICE and SST is roughly linear, allowing for the adjustment of ICE to 60–120% by varying the volume of SST for different levels of prelithiation.67 Upon calendering and prelithiation of SLMPs, the Li2CO3 protective layer fractures at an external pressure of 15 MPa, thereby exposing the fresh lithium metal to graphite.74 Upon the addition of the electrolyte to moisten the electrode, a spontaneous electrochemical reaction transpires between the exposed lithium and graphite particles, yielding a partially lithiated graphite anode. Furthermore, Liu et al. synthesized stable nanoscale lithium powders (<500 nm) from lithium foil using ball milling high melting point ionic liquids ([P4444]TFSI) at low temperatures, in conjunction with employing Li2CO3 nanoshell protection (Fig. 11b).75 The solid [P4444]TFSI ionic liquid at ambient temperature serves as a dispersion for the preparation of fine lithium powder particles and functions as a protection agent for newly exposed lithium surfaces. Utilizing it as a pre-lithiation reagent for lithium-free anode materials (Si, SiO, SnO), the initial coulombic efficiency of the constructed half-cell improved from 48.4%, 44.2%, and 52.9% to 93.2%, 93.7%, and 92.8%. Furthermore, Huang et al. synthesized ultrafine air-stabilized lithium spheres (ASLSs) consisting of metal lithium cores and LiF/Li2O shells using electrochemical plating (Fig. 11c).76 The LiF/Li2O shell efficiently safeguards ASLSs in atmospheric conditions and remains stable for 30 days. ASLS has superior pre-lithiation performance relative to coarse-grained Li powder, owing to its consistent small particle size and thin layer protection. Prelithiation with ASLSs increased the CE of graphite electrodes rose from 77.4% to 101.9%, and the energy density of the entire cell constructed with LiCoO2 was markedly enhanced.
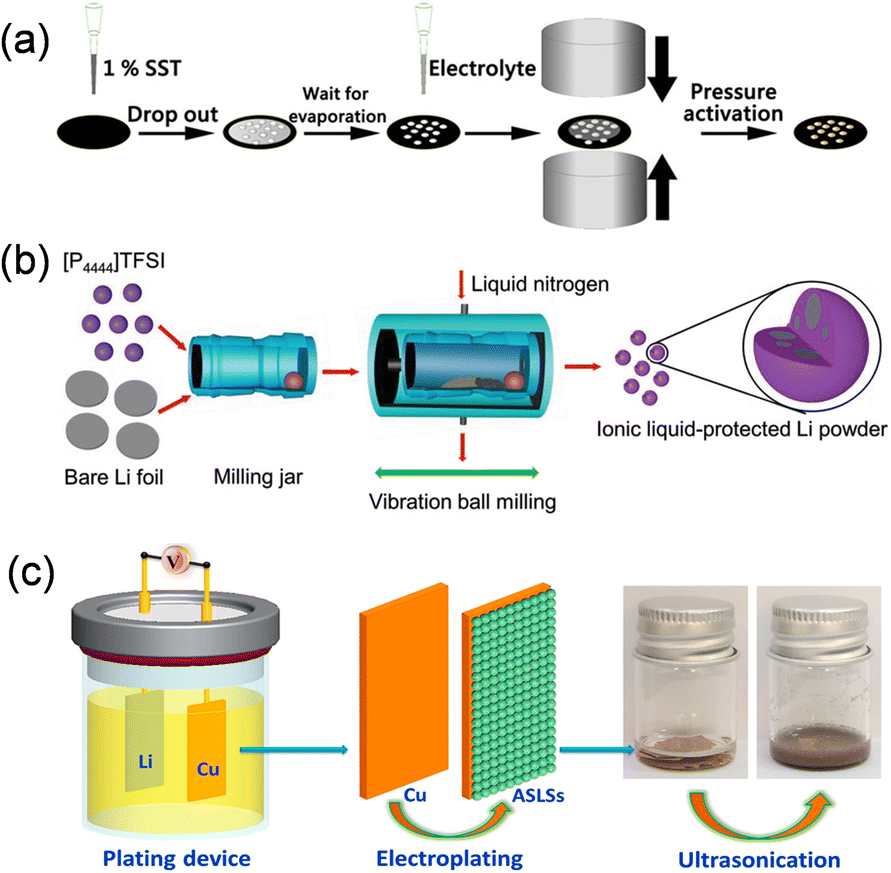 | ||
| Fig. 11 (a) Schematic illustration of pre-lithiation process on an SiO electrode (reprinted from ref. 67. Copyright 2021, with permission from ACS Publications). (b) Schematic illustration of the ionic liquid-assisted cryomilling preparation process for nanoscaled Li powders (reprinted from ref. 75. Copyright 2019, with permission from Wiley-VCH). (c) Schematic of the fabrication process of the ASLSs (reprinted from ref. 76. Copyright 2021, with permission from Elsevier). | ||
The primary objective of cathode prelithiation is to mitigate irreversible capacity loss by using lithium-rich additives that possess a greater lithium-ion supply capacity, hence releasing supplementary lithium ions during the initial charge–discharge cycle. To compensate for lithium on the cathode side, lithium carbon-nitrogen oxide LiX (where X represents C, N, O, P, etc.), lithium-rich ternary compounds LixMyOz (with M as Ni, Co, Fe, etc.), and metal/Li2O nanocomposites can offer substantial lithium-ion capacity beneath the charge cut-off potential window of the current cathode.77–91
Upon charging the lithium-rich ternary material LixMyOz to the lithium extraction potential at the negative electrode, lithium ions will be transported from the additives and incorporated into the negative lattice or used to form a solid electrolyte interphase (SEI) film to compensate for the consumption of active lithium. The lithium-rich compound Li8ZrO6 (LZO) contains 8 lithium ions per formula unit, enabling a substantial release of lithium ions at reduced mass loads to offset the irreversible capacity loss observed in the initial week. LZO is integrated with the cathode material LiNi0.5Mn1.5O4 (LNMO) to construct a button battery, with graphite as the anode, exhibiting a reversible specific capacity improvement of 15–18% and a capacity retention rate of 30% after 50 charge–discharge cycles (Fig. 12a).81 Furthermore, electrochemical impedance spectroscopy indicated a substantial reduction in the recombination resistance of the LNMO half-cell with LZO prelithiation (Fig. 12b). Li6CoO4 possesses an initial charge capacity of up to 573.6 mA h g−1 and a discharge capacity of merely 25.6 mA h g−1, yielding a supplemental lithium capacity of 548 mA h g−1. Moreover, at 50% relative humidity (RH), Li6CoO4 can preserve its structure integrity, morphology, and electrochemical stability for one hour. Li et al. effectively synthesized an air-stable Li6CoO4@Li5FeO4 (LCO@LFO) composite prelithiation reagent by meticulously encapsulating Li5FeO4 with Li6CoO4 (Fig. 12c).89 The Li6CoO4@Li5FeO4 demonstrated exceptional resistance to water and oxygen, maintaining stability in air for one hour (Fig. 12d).
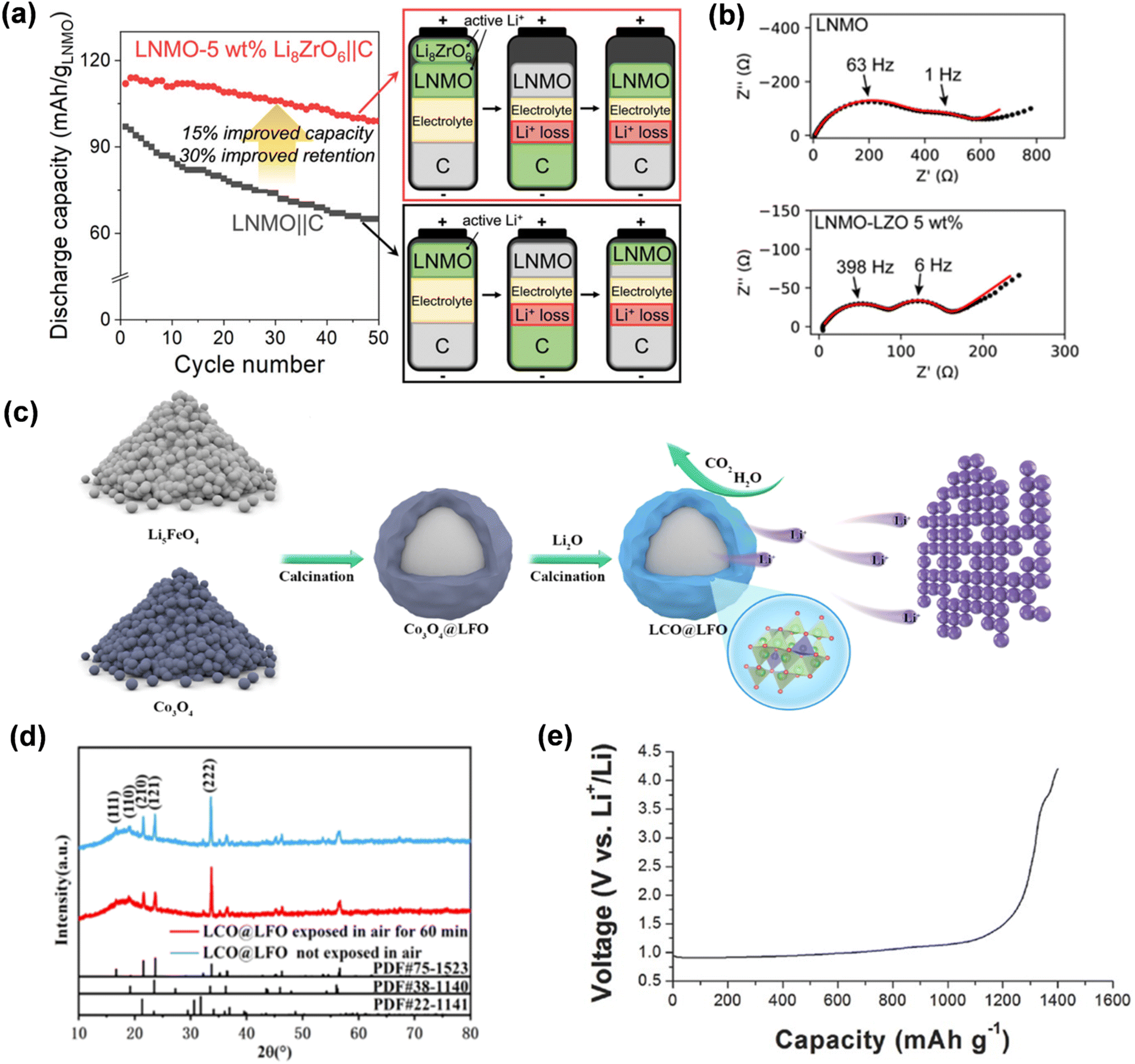 | ||
| Fig. 12 (a) The discharge specific capacity of the cathode material lithium nickel manganate (LNMO) before and after the combination with the prelithiation additive LZO. (b) Nyquist plots for the impedance spectra of LNMO and LNMO–LZO half-cells after charging to 4.9 V at 0.2C with the indicated LZO loadings (reprinted from ref. 81. Copyright 2022, with permission from ACS Publications). (c) The preparation process of LCO@LFO pre-lithiation reagent. (d) XRD of LCO@LFO and LCO@LFO exposed in air for 60 min (reprinted from ref. 89. Copyright 2021, with permission from IOP Publishing). (e) Initial voltage curve of the ground Li3N during charging to 4.2 V versus Li+/Li (reprinted from ref. 78. Copyright 2016, with permission from Wiley-VCH). | ||
Nonetheless, the majority of ternary pre-lithiation additions exhibit poor theoretical specific capacity, leading to a restricted enhancement in battery energy density. Li3N is a widely utilized self-sacrificing pre-lithiation additive with a substantial theoretical specific capacity of 2308.5 mA h g−1. It decomposes at voltages exceeding 0.44 V (vs. Li+/Li) and remains unoxidized by the discharge cathode during charging, thereby serving as a sacrificial additive to augment lithium for the discharge cathode. Park et al. employed it as an addition to the cathode material LiCoO2 and discovered that only the small particles with a pristine surface of Li3N exhibited electrochemical activity for prelithiation of the cathode.78 It exhibits chemical stability in a dry environment, enhancing the reversible capacity of the entire cell without compromising rate performance. In the initial cycle of electrochemical oxidation of the half-cell, the voltage plateau at 0.9 V (versus Li+/Li) signifies the generation of N2 and active lithium. Upon charging to 4.2 V, a substantial capacity of 1399.3 mA h g−1 is achieved, equating to 1.8 Li in Li3N (Fig. 12e). Substituting 2wt% of LiCoO2 with Li3N, the charging capacity rose from 149.7 mA h g−1 to 178.4 mA h g−1, representing an increase of 28.7 mA h g−1 over LiCoO2, equivalent to 1584.2 mA h g−1 (≈2.1Li) of Li3N. Although LiN3 is stable in air, it thermally decomposes between 115 and 298 °C. In general, battery electrodes are dried at 120 °C or higher, so their practical application may be limited.
Alongside Li3N, the self-sacrificial prelithiation additions will generate gas, while Li2S, Li3P, and Li2Se will yield S, P, and Se residues post-delithiation. Li2S possesses a substantial theoretical capacity (1167 mA h g−1); however, the delithiation process generates sulfur compounds with inadequate conductivity and polysulfide intermediates that are incompatible with ester electrolytes. Consequently, to utilize it for cathode prelithiating agents, modifications are necessary. The core–shell Li2S/KB/PVP nanocomposites developed by Zhan et al. have an irreversible specific capacity of 1084 mA h g−1. The capacity retention rate of the entire cell, comprising a Si–C anode and LiFePO4, improved from 53% to nearly 100% after 200 cycles (Fig. 13b).92 They synthesized M/Li2O (M: Co, Fe, Ni, etc.) nanocomposites in a one-step method, which is versatile and widely generalized for industrial production. In addition, its Li-extraction specific capacity was maintained at a certain level, only 51 mA h g−1 lower than the initial value after eight hours of exposure to the environment, indicating compatibility with conventional lithium-ion battery manufacturing environments.
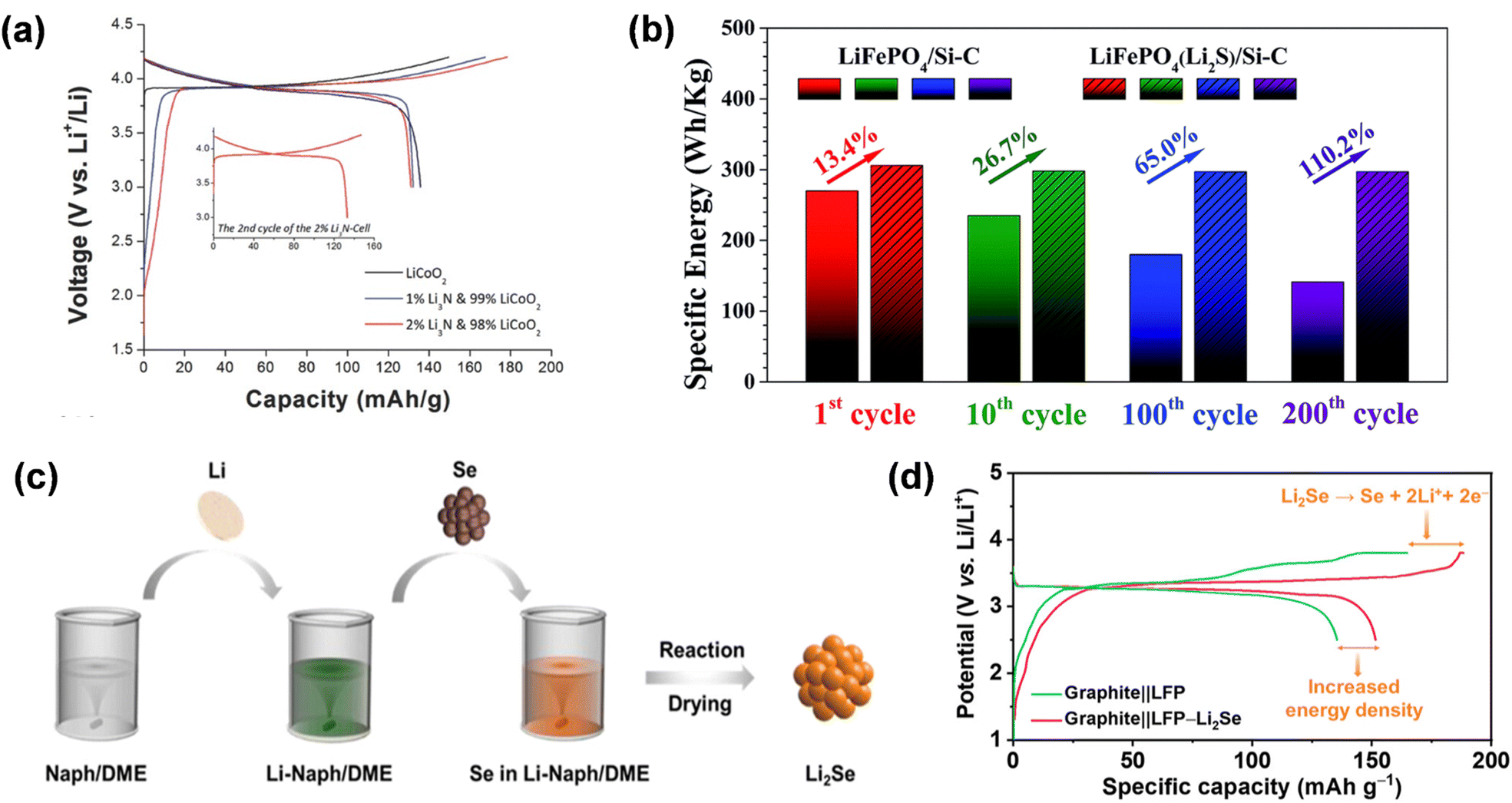 | ||
| Fig. 13 (a) The initial charge/discharge voltage curves of LiCoO2 and mixed electrodes doped with 1 wt% or 2 wt% Li3N (reprinted from ref. 78. Copyright 2016, with permission from Wiley-VCH). (b) Comparison of the specific energy for the LiFePO4/Si–C with LiFePO4(Li2S)/Si–C full cells (reprinted from ref. 92. Copyright 2018, with permission from Royal Society of Chemistry). (c) Schematic diagram of the synthesis procedure of Li2Se powder. (d) Initial charge/discharge profiles at 0.05C (reprinted from ref. 80. Copyright 2023, with permission from ACS Publications). | ||
Lithium selenide (Li2Se) exhibits superior theoretical specific capacity and conductivity compared to the previously documented Li6CoO4, Li2O, and Li2S, and can be permanently transformed into selenium (Se) at 2.5–3.8 V, hence supplying extra lithium. Significantly, Li2Se prevents gas evolution during electrochemical decomposition and is compatible with carbonate electrolytes. Pan et al. included 6 wt% Li2Se into the LiFePO4 (LFP) cathode, resulting in a 9% enhancement in the initial specific capacity of the Li//LFP half-cell and a 19.8% increase in energy density.80 The discharge specific capacity of the Gr//LFP complete battery increased from 135 mA h g−1 to 151 mA h g−1 during the initial cycle, hence enhancing the energy density of lithium-ion batteries (Fig. 13d).
Furthermore, the cathode prelithiation additives derived from the amalgamation of metals with binary or ternary materials exhibit elevated prelithiation efficiency and excellent compatibility, enhance the capacity of lithium-ion batteries, and prevent the emission of deleterious gases. Sun described a cathode additive M/Li2O composed of nanoscale mixes of transition metals and lithium oxide, synthesized through conversion processes of metal oxide and lithium (MxOy + 2yLi + 2ye+− → xM + yLi2O) (Fig. 14a).93 The theoretical specific capacity of M/Li2O nanocomposites surpasses that of current cathode materials, with values of 724 mA h g−1 for Co/Li2O (molar ratio = 3/4), 799 mA h g−1 for Fe/Li2O (molar ratio = 2/3), and 935 mA h g−1 for Mn/Li2O (molar ratio = 1/2). The research demonstrated that the overall capacity of the LiFePO4 electrode containing 4.8% Co/Li2O was enhanced by 11% relative to the original LiFePO4 electrode in the complete cell (Fig. 14b). Besides employing various metals, pre-lithiation can also be executed with binary compounds produced through chemical processes involving distinct oxides, sulfides, fluorides, and lithium. Li2S/Co nanocomposites were suggested as a solution to the issue of pre-lithiation. A comprehensive technique has been suggested, wherein the lithium source in the composite can be electrochemically extracted during the charging process through a reverse electrochemical conversion reaction in the initial cycle (Fig. 14c).94 Li2S/Co nanocomposites can deliver a substantial specific capacity of 670 mA h g−1 to offset lithium depletion in the anode. Furthermore, multi-component Fe/LiF/Li2O nanocomposites have been engineered for cathode prelithiation, capable of delivering up to 550 mA h g−1 of lithium-ion capacity during the initial charging phase by a multi-electron reverse conversion event.95 Importantly, such prelithiation additive showed good compatibility with the existing battery fabrication process. Fe/LiF/Li2O nanocomposites provide superior lithium compensation effects as additions for diverse cathode materials, including LiCoO2, LiFePO4, and LiNi1−x−yCoxMnyO2 (15% increase in reversible capacity using 4.8 wt% of the additives).
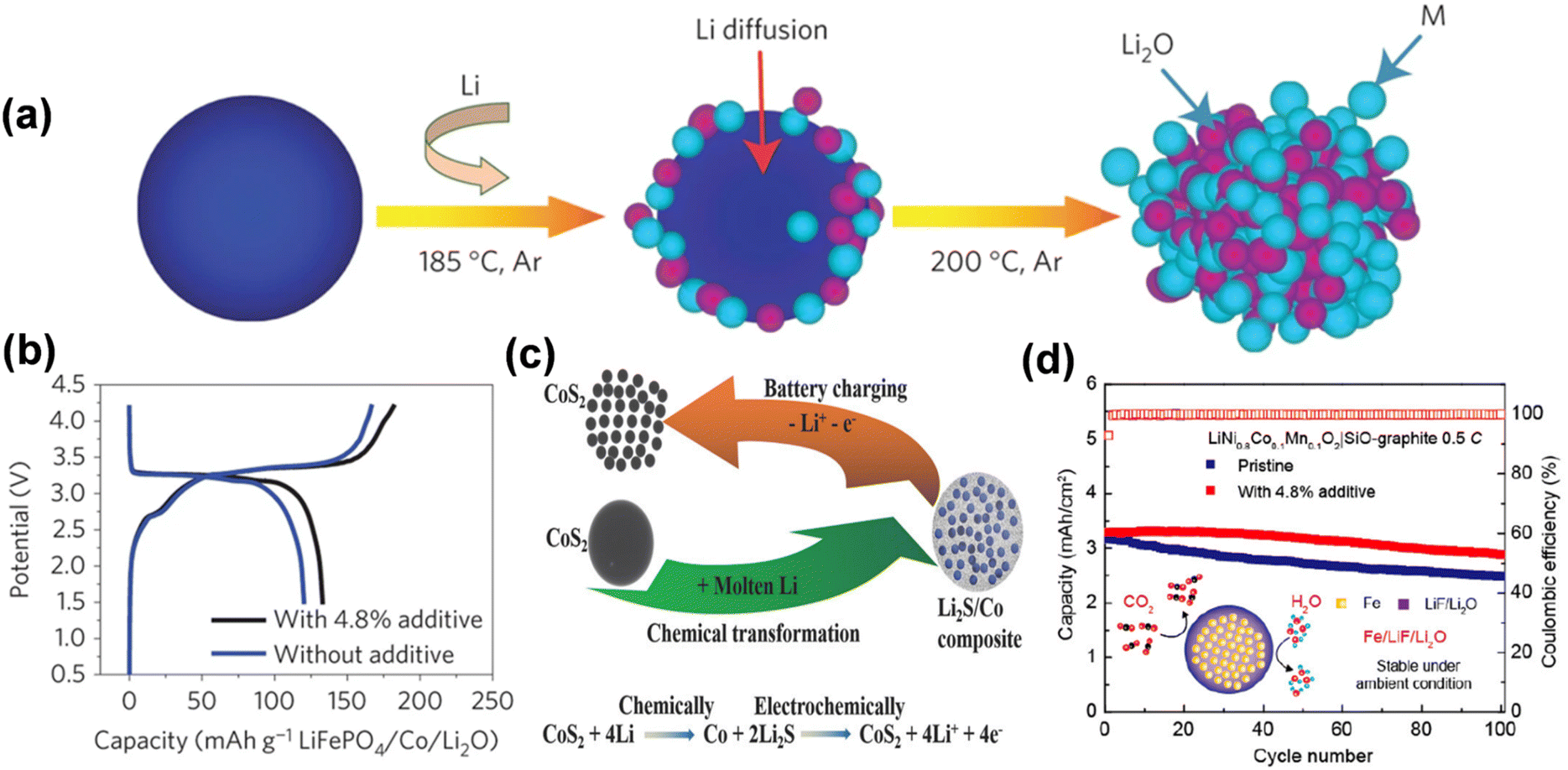 | ||
| Fig. 14 (a) Schematic of the fabrication process of the N–M/N–Li2O composites. (b) The initial charge/discharge potential profiles of LiFePO4/graphite full cells with and without the N–Co/N–Li2O additive (reprinted from ref. 93. Copyright 2016, with permission from Springer Nature). (c) Schematic of the chemical synthesis of Li2S/metal (e.g., Li2S/Co) composite and the electrochemical extraction of lithium during the charge process (reprinted from ref. 94. Copyright 2016, with permission from Wiley-VCH). (d) Cycling stability of LiNi0.8Co0.1Mn0.1O2|SiO–graphite full cells with and without Fe/LiF/Li2O additive (reprinted from ref. 95. Copyright 2020, with permission from ACS Publications). | ||
An overview of representative anode prelithiation strategies is shown in Table 1. Most prelithiation additives have significant advantages in lithium-ion batteries, as they can provide a high specific capacity, which significantly increases the capacity of the battery. In addition, pre-lithiation additives can be mixed directly with the electrode material, which is simple and easy to implement without changing the basic structure of the electrode material. However, when selecting electrode prelithiation additives, we must consider their compatibility with electrode materials, but this has considerable limitations for a wide range of applications. Second, some additives may undergo undesirable side reactions with the electrolyte or electrodes, or they may decompose or fail during long charge–discharge cycles, affecting the life of the battery. In addition, the application of pre-lithiation additives may increase the production cost of batteries, especially in the industrial production process. Balancing improved battery performance with cost control remains an important issue. Finally, the environmental impact and health risks of pre-lithiation additives also need to be further evaluated, especially for additives that may contain harmful ingredients, and their safety and environmental friendliness still need to be rigorously tested in practical applications. Positive and negative prelithiation additives play an important role in improving battery performance, but their application still faces challenges in terms of technology, cost and long-term stability.
| Prelithiation material | Targeted substance | Cell configuration | ICE | Performance | Refs |
|---|---|---|---|---|---|
| SLMP | SiO | NCM622//SiO | 70% | 40 cycles, 50% capacity retention | 67 |
| SLMP | Si-CNT | NCA//Si-CNT | 79% | 50 cycles, 93% capacity retention | 68 |
| LixSi-Li2O | Graphite | Li//Gr | 104% | 50 cycles | 69 |
| Si | Li//Si | 94% | 20 cycles | ||
| Li foil | Sn@PPy | LiFePO4//LixSn@PPy | — | 80 cycles, 83% capacity retention | 70 |
| Li22Z5 alloys/Li22Z5-Li2O composites | Gr | Li//Gr | 100% | 100 cycles | 73 |
| Li microspheres | SiO@C | LiCoO2//SiO@C | 95% | 100 cycles, 81.5% capacity retention | 76 |
| Li3N | LiCoO2 | LiCoO2//SiOx/C@Si | 78.1% | 100 cycles | 78 |
| Li2Se | LiFePO4 | LiFePO4//Gr | — | 100 cycles, 89.4% capacity retention | 80 |
| Li2C4O4 | NCM622 | NCM622//Si/Gr | 71.4% | 138 cycles, 60% capacity retention | 83 |
| Li2C2O4/NiO | HC | LiCoO2//HC | 53.8% | 200 cycles, 77% capacity retention | 84 |
| Li2O2 | NCM | NCM//Gr | — | 55 cycles | 85 |
| Al2O3@Li2NiO2 | LiCoO2 | LiCoO2//Gr | 100% | 250 cycles, 87% capacity retention | 86 |
| CS-LiCoO2 | LiCoO2 | CS-LiCoO2//Gr-SiO | — | 100 cycles, 80% capacity retention | 88 |
| Li6CoO4@Li5FeO4 | NCM811 | NCM811//Gr | 63.1% | 50 cycles | 89 |
| Li2S | Si–C | LiFePO4//Si–C | 100% | 200 cycles, 100% capacity retention | 92 |
| Co/Li2O | LiFePO4 | LiFePO4//Gr | — | 100 cycles | 93 |
| Li2S/metal composite | LiFePO4 | Li//LiFePO4 | — | 100 cycles | 94 |
| Fe/LiF/Li2O | Gr | NCM811//SiO-Gr | 80% | 100 cycles, 88.4% capacity retention | 95 |
With the continuous research and development of new pre-lithiation materials (such as organic lithium compounds and inorganic lithium salts), pre-lithiation additives will be able to achieve large-scale production at lower cost and higher performance in the future. In addition, the optimization of formulations and the development of novel additives will further promote their application in large-scale production. Depending on market demand, the production cost and environmental impact of pre-lithiation additives are expected to be further reduced.
A prevalent chemical prelithiation method involves submerging the electrode in an electrochemically active lithium-aromatic-ether solution and infusing lithium ions from the prelithiation system into the active material, therefore enhancing the initial coulombic efficiency (ICE). The primary mechanism involves utilizing the electrophilicity of aromatic hydrocarbons to react with lithium metal in ether solvents, resulting in the formation of lithium-containing aromatic radical compounds (LACs), followed by the transport of dissolved lithium ions to the negative electrode via the potential difference. The extensively researched organic reagents, including lithium naphthalene (Li-NaPh) and lithium biphenyl (Li-Bp), have low chemical potentials (about 0.33 and 0.35 V versus Li+/Li), facilitating the lithiation of carbon-based anodes. The excellent wettability of ether solvents allows for complete infiltration of the electrode, ensuring homogeneous lithiation of the active substance. The sensitivity of lithium-aromatic-ether-based solutions to air or moisture is less than that of metallic lithium foils or powders, hence enhancing the operational scope of pre-lithiation investigations. Solvents and organic reagents are inherently poisonous and volatile, necessitating their use in an inert atmosphere. Consequently, researchers have investigated the pre-lithiation of hard carbon, phosphorus, tin oxide, and sulfur electrodes utilizing lithium–naphthalene and lithium–biphenyl systems.
Abe employed naphthalene as the solvent for lithium to investigate the electrochemical properties of natural graphite flakes subjected to various solvent treatments.96 The solvents include tetrahydrofuran (THF), 2-methyltetrahydrofuran (MeTHF), 2,5-dimethyltetrahydrofuran (di-Me-THF), 1,2-dimethoxyethane (DME), 1,2-diethoxyethane (DEE), 1-methoxypropane (MP), 1-methoxybutane (MB), and ether (Et2O) (Fig. 15a). Li co-intercalated with THF, DME, and DEE formed ternary Li-solvent graphite intercalation compounds (GICs), while solvent-free binary Li-GICs were achieved using MeTHF, diMeTHF, MP, MB, and Et2O. Shen, drawing on ionic solvation and coordination chemistry, introduced an innovative design strategy for pre-lithiation solutions, effectively achieving the pre-lithiation of the Gr anode by selecting a solvent characterized by strong electron donation, steric hindrance, and chemical stability to modulate the redox potential of the pre-lithiation reagent and inhibit solvent co-embedding during the pre-lithiation process.97 The Gr anode may be accurately prelithiated to the necessary state within minutes using lithium biphenyl/2-methyltetrahydrofuran (Li-BP/2-MeTHF), without compromising its lattice structure (Fig. 15b). When the prelithiated graphite negative electrode (pGr) is coupled with the positive electrode, the entire cell demonstrates markedly enhanced initial coulombic efficiency (ICE) and increased energy density relative to the original graphite anode. Likewise, the pre-lithiation of the graphite anode utilizing lithium naphthalene as a lithiation agent and 2-methyltetrahydrofuran as a reductive solvent exhibits a minimal redox potential of 0.19 V, enabling the controlled and efficient prelithiation of the graphite anode to the desired level within a few minutes, without requiring solvent co-intercalation.98 The prelithiated Gr anode for 3 minutes demonstrated an initial coulombic efficiency of 100%, superior rate capability, and enhanced cycle capacity retention. By managing the time and temperature of the immersion process, the degree of pre-lithiation and spatial uniformity of the active lithium throughout the electrode can be finely controlled, thus ensuring process fidelity and low cost for large-scale integration.
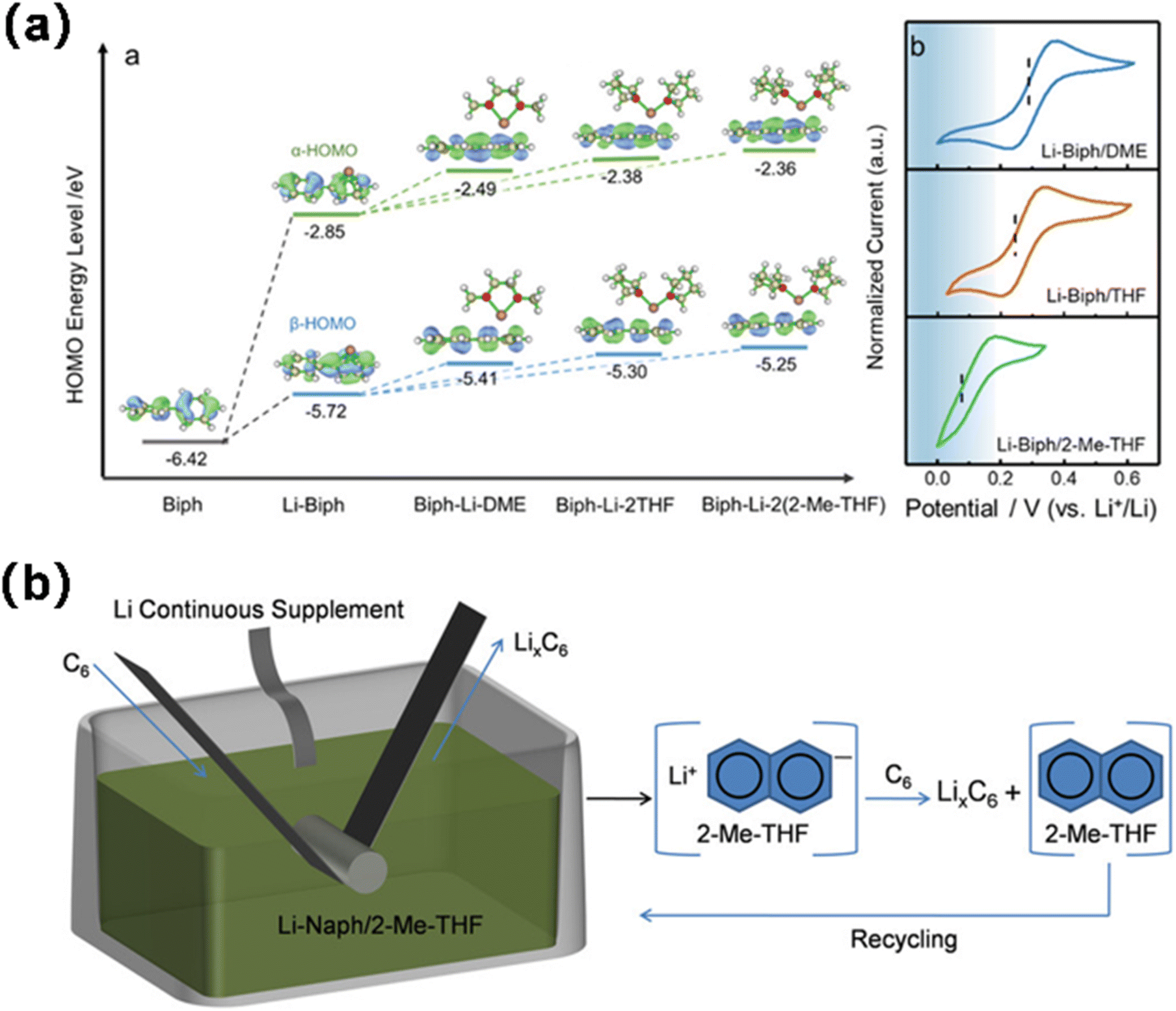 | ||
| Fig. 15 (a) Geometrical configurations and HOMO energy levels (reprinted from ref. 97. Copyright 2021, with permission from Wiley-VCH). (b) Schematic illustration for the prelithiation reaction of Gr with Naph-Li/2-Me-THF solution (reprinted from ref. 98. Copyright 2022, with permission from Wiley-VCH). | ||
Chemical pre-lithiation with reducing chemical solutions has been shown to inhibit the direct incorporation of active lithium into the silicon-based anode. The reducible reagent on the silicon anode surface preferentially reacts with the electrolyte components or the silicon surface, facilitating the formation of a solid electrolyte interface (SEI) coating. The creation of this SEI film can stabilize the anode surface to some degree and mitigate later irreversible lithium loss; nevertheless, it obstructs further lithium-ion intercalation and prevents effective lithium alloying with silicon. Conversely, the reducibility of lithium–naphthalene, lithium–biphenyl, and similar systems is insufficient to prelithiate anode materials with low lithiation potential, such as silicon, graphite, and silicon oxide. The incorporation of diverse electron-donor functional groups (CH3, CH2CH3, CH(CH3)2, and OH) into aromatic hydrocarbon molecules significantly enhances electron density, consequently diminishing electron affinity and reduction potential, which further augments the reducing capability of prelithiation reagents. The pre-lithiation potential of SiO in methyl butyl ether solvents for silicon-based anodes is 0.21 V.99 According to the electron donor group theory, Lee et al. indicated that the reduction potential of LAC was modified to below 0.2 V by molecular engineering of BP derivatives, and the active lithium doping in silicon-based anodes was demonstrated for the first time via a uniform and scalable chemical reaction.100 The insertion of methyl groups on the benzene ring elevates the lowest unoccupied molecular orbital (LUMO) level of BP derivatives. In comparison to the neighboring methyl group (2-Me), there exists a more significant potential shift concerning the intermediate position (3,3-2Me) and para position (4,4-2Me). Conversely, for the 4 substituents in 3,3,4,4-4MeBP, the redox potential of LACs diminishes to 0.129 V, which is inferior to the redox potential of SiOx, thereby instigating the regulation of active lithium in SiOx and elevating the initial coulombic efficiency to over 100%. This indicates that the electronic influence of the substituents correlates with the elevation of the molecular orbital energy level, addressing the issue of the SiOx anode's inability to undergo prelithiation with BP, hence facilitating the practical use of this technology.
Chemical pre-lithiation can form a stable artificial SEI film through the electrode material and pre-lithiation reagent in advance, which can reduce the direct contact between the electrolyte and the anode material and reduce the occurrence of side reactions. Stability is enhanced by precise control of composition and structure, so as to better cope with the volume changes of the anode material during charging and discharging, especially in high-capacity anode materials such as silicon. Sun et al. proposed a novel strategy to construct an artificial hybrid SEI layer composed of LiF and Li3Sb in situ at the anode by the spontaneous chemical reaction of prelithiated SiOx anode with SbF3.101 The prefabricated artificial SEI layer with long-term cycling stability and enhanced Li transmission capacity can inhibit the volume change of SiOx anode, and significantly improve the capacity retention and rate performance of the modified SiOx+, and increase the ICE to 94.5%. In addition, the whole cell assembled with NCM811 and modified SiOx exhibited high ICE (86.0%) and capacity retention (86.6%) after 100 cycles at 0.5C.
To attain elevated energy density, the positive and negative electrode materials may be prelithiated concurrently. Shen et al. devised a straightforward prelithiation method employing Li-Naph reagents to fully prelithiate sulfur-polyacrylonitrile (S-PAN) composites into a Li2S-PAN cathode and partially prelithiate nanosilicon into a LixSi anode, thereby creating an innovative variant of silicon/sulfur lithium-ion batteries.102 The LixSi/Li2S-PAN battery demonstrates a high specific energy of 710 W h kg−1, an initial coulombic efficiency of 93.5%, and substantial cycling performance.
Chemical pre-lithiation is an effective strategy for enhancing the performance and stability of lithium-ion batteries, especially in high-capacity anode materials like silicon. By pre-lithiating the anode material with a pre-lithiation reagent, a stable artificial solid electrolyte interphase (SEI) film is formed. This pre-formed SEI layer reduces the direct interaction between the electrolyte and the anode material, minimizing undesirable side reactions during battery cycling. The stability of the SEI film can be further improved by precisely controlling its composition and structure, enabling it to better accommodate the volume expansion and contraction of the anode material during charging and discharging cycles. Furthermore, the fundamental principle of chemical pre-lithiation is straightforward; it circumvents slurry processing and facilitates swift lithium replenishment, hence enhancing the practical applicability of certain unstable pre-lithiation materials. This chemical pre-lithiation approach is mild, effective, and broadly applicable to various lithium-deficient electrodes, creating new opportunities for the advancement of cost-effective, eco-friendly, and high-capacity lithium-ion batteries. However, the chemical pre-lithiation technology consumes a large amount of lithium source, and it is impractical to apply it commercially. In the future, this technology will be optimized with catalysts, environmentally friendly lithium sources, and more efficient reaction paths, making it more suitable for large-scale applications.
ISC refers to a self-discharge phenomenon in the electrolyte, wherein a potential difference arises between the negative electrode and lithium metal due to short circuit contact, facilitating electron transfer. During this process, lithium ions are released into the electrolyte through electron transfer and subsequently migrate from the electrolyte to the negative electrode to maintain charge equilibrium. Liu et al. introduced a technique for prelithiating silicon nanowire (SiNW) anodes by a straightforward self-discharge mechanism.103 In the pre-lithiation technique, silicon nanowires (SiNWs) grown on stainless steel (SS) are immediately affixed to a sheet of lithium metal foil in the presence of an electrolyte, which is caused by pressure from an electrical short circuit for electrochemical lithiation. Silicon nanowires (SiNWs) undergo two distinct lithiation processes: when the SiNW contacts the lithium foil (left side of Fig. 16a), electrons traverse through the wire's tip; conversely, when the SiNW does not contact the lithium foil (right side of Fig. 16a), electrons flow through the interface between the lithium foil and the stainless steel substrate, subsequently ascending through the base of the SiNW. The extent of pre-lithiation can be readily regulated by adjusting the duration of pre-lithiation. The study revealed that following 20 minutes of pre-lithiation, SiNW maintains its nanostructure, attaining a substantial pre-stored lithium capacity of around 2000 mA h g−1, demonstrating the efficacy of this direct contact electrochemical pre-lithiation approach (Fig. 16b). Electrolyte-mediated electrochemical prelithiation necessitates significantly lower applied pressure than mechanical roll-to-roll processes (approximately 30 MPa for mechanical methods versus approximately 60 kPa for electrolyte-mediated electrochemical prelithiation), making it suitable for electrodes with low mechanical stability.
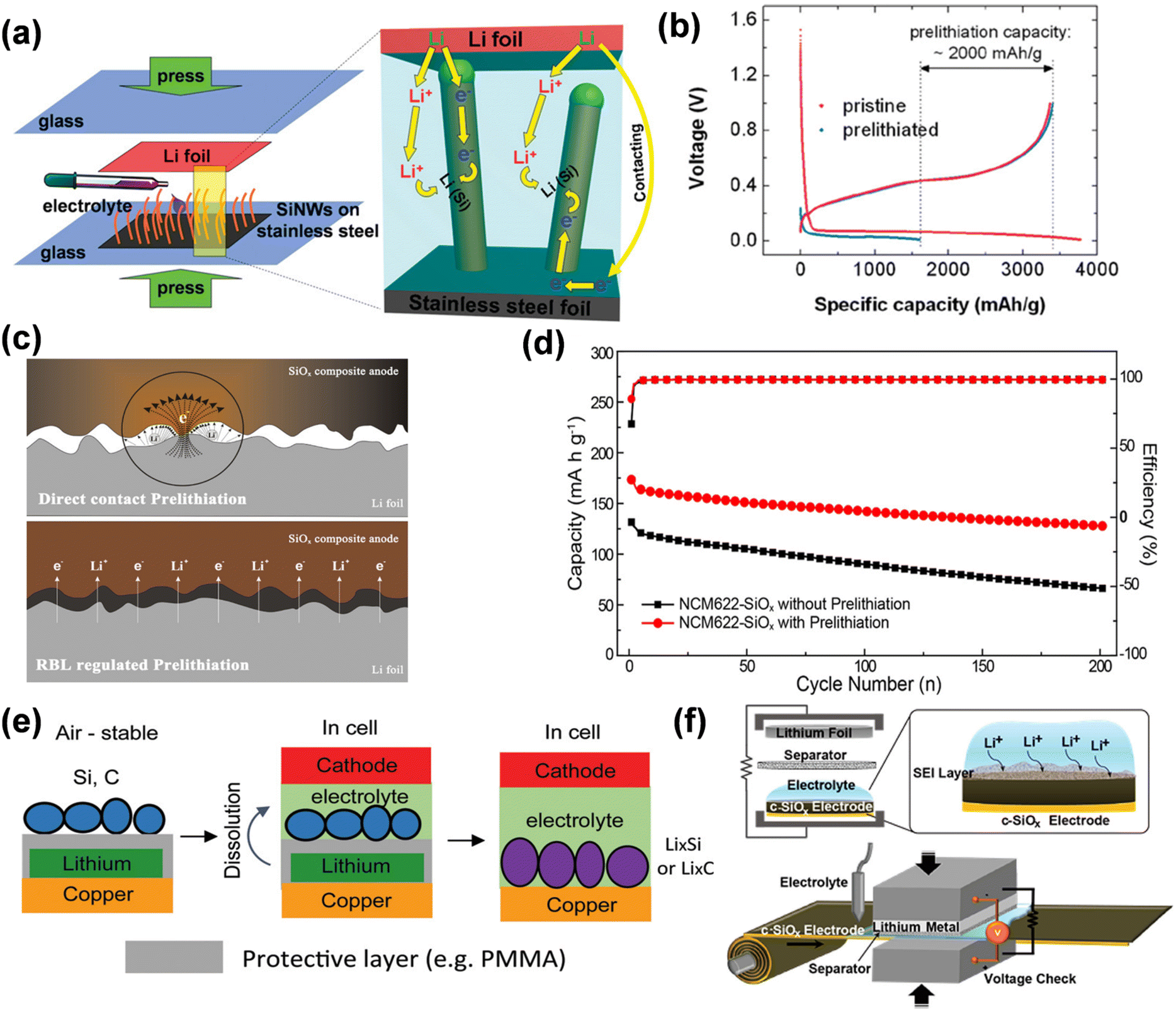 | ||
| Fig. 16 (a) Schematic diagrams showing the prelithiation of SiNWs on stainless steel (SS) foil, and the internal electron and Li+ pathways during the prelithiation. (b) Comparison of first cycle voltage profiles of pristine and 20 min prelithiated SiNWs with Li metal as the counter electrode (reprinted from ref. 103. Copyright 2011, with permission from ACS Publications). (c) Illustration of Li-ion and electron transfer in the direct contact and RBL-regulated prelithiation process. (d) Cycling performance and coulombic efficiency of NCM622-SiOx full cell with and without prelithiation (reprinted from ref. 104. Copyright 2019, with permission from ACS Publications). (e) Schematic of the process to prepare ambient-air-stable lithiated anode (reprinted from ref. 105. Copyright 2016, with permission from ACS Publications). (f) Graphical illustration of prelithiation process of c-SiOx electrode and its scalable roll-to-roll process scheme (reprinted from ref. 106. Copyright 2016, with permission from ACS Publications). | ||
Meng modified the speed and extent of lithiation by incorporating a resistive buffer layer (RBL) between the SiOx anode and the lithium foil, while optimizing the pre-embedded lithium intercalation technique in direct contact with the anode and the lithium foil (Fig. 16c).104 This method is controllable and efficient, but it requires the press of a 2 kg weight and dropping of electrolytes in a vacuum condition, so it is currently only in the small-scale laboratory stage. The porous architecture and elevated conductivity of RBL enhance Li+ transport and electron transfer, while its pliable characteristics guarantee intimate contact between the Li foil and the negative electrode for consistent pre-lithiation. The prelithiated SiOx anode has elevated initial coulombic efficiency and specific capacity in both half-cells, ensuring steady performance for 200 cycles in the NCM622//SiOx whole cell (Fig. 16d). Internal short-circuit pre-lithiation employs unstable lithium metal in atmospheric conditions, impeding large-scale industrial applications. Cao et al. delineated a tri-layer configuration of active material/polymer/lithium anode that exhibits stability in ambient air for a duration of time.105 The polymer layer safeguards lithium from oxygen and moisture while providing a stable coating for the active ingredient. The polymer layer is progressively dissolved in the battery electrolyte, allowing the active material to interact with lithium and create a lithiated anode. This tri-layer configuration not only maintains the electrode's stability in ambient air but also facilitates uniform lithiation. Utilizing this method, an initial coulombic efficiency of 99.7% was achieved in the graphite anode, surpassing 100%.
In the presence of an electrolyte, ISC-based pre-lithiation facilitates direct contact between the electrode material and lithium foil, simplifying operation and eliminating the need for battery disassembly and reassembly. The extent of prelithiation can be modified by regulating the contact duration and the quantity of lithium exposed; nevertheless, achieving precise control remains challenging. By constructing a lithium-metal half-cell and establishing an external circuit discharge (ESC), the cut-off voltage can be meticulously regulated to introduce a precise quantity of active lithium, thereby controlling the extent of prelithiation, regulating the formation of the SEI film, and enhancing the ICE of lithium-ion batteries. Kim et al. proposed a pre-lithiation method utilizing an electro-short circuit in lithium metal foil, which generates an external short circuit when electrolyte and separator are present.106 This pre-lithiation solution is suitable for conventional roll-to-roll battery manufacturing methods. Spontaneous prelithiation can be commenced by utilizing the potential difference between the two electrodes (Fig. 16f) and by optimizing circuit resistance for accurate short-circuit duration and voltage monitoring, facilitating the precise adjustment of the prelithiation level of carbon-coated SiOx (c-SiOx) electrodes without lithium plating. The lithiation of the c-SiOx electrode is facilitated by the stream of Li ions and does not involve direct contact with the Li metal. Consequently, the deposition of metallic lithium can be circumvented, allowing for the formation of the same solid electrolyte interphase (SEI) layer generated during the electrochemical cycle, which consists of lithium carbonate and alkyl lithium carbonate, along with additional inorganic constituents such as LiF, Li2O, and LixSiOy. Throughout the entire cycle, the ICE of the half-cell reaches 94.9% while maintaining the structural integrity of the SiOx. In comparison to 73.6% of the pristine battery, the initial charge efficiency (ICE) of the entire cell, which consists of nickel-rich layered cathode material Li(Ni0.8Co0.15Al0.05)O2, was markedly enhanced from 58.85% to 85.34%.
This poses a safety concern, as the ESC approach necessitates the utilization of lithium metal for electrochemical prelithiation. Zhou et al. presented a unique electrolytic cell including a Cu pitting anode half-cell in a 0.5 M Li2SO4 aqueous solution electrolyte and a Si lithiated cathode half-cell in a gelatinous polymer electrolyte (saturated with 1 M LiPF6 at EC/DEC = 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7).107 The two half-cells are divided by a thin layer of lithium super-ion conductors, and the silicon electrode facilitates precise pre-lithiation from an aqueous solution containing lithium without the presence of lithium. MnOx/Si and S/Si lithium-ion batteries were constructed with prelithiated Si anodes, achieving high specific energies of 349 and 732 W h kg−1, respectively. Despite a high specific power of 1710 W kg−1, the MnOx/Si complete cell maintains a capacity of 138 W h kg−1. This marks the first report of a complete technique for fabricating an all-lithium-ion battery utilizing two lithium-deficient electrodes, without employing lithium metal as a lithium supply. This unique pre-lithiation method, characterized by great controllability, absence of short circuits, and ample lithium resources, is anticipated to significantly advance the development of safe, environmentally friendly, and durable lithium-ion batteries.
:7).107 The two half-cells are divided by a thin layer of lithium super-ion conductors, and the silicon electrode facilitates precise pre-lithiation from an aqueous solution containing lithium without the presence of lithium. MnOx/Si and S/Si lithium-ion batteries were constructed with prelithiated Si anodes, achieving high specific energies of 349 and 732 W h kg−1, respectively. Despite a high specific power of 1710 W kg−1, the MnOx/Si complete cell maintains a capacity of 138 W h kg−1. This marks the first report of a complete technique for fabricating an all-lithium-ion battery utilizing two lithium-deficient electrodes, without employing lithium metal as a lithium supply. This unique pre-lithiation method, characterized by great controllability, absence of short circuits, and ample lithium resources, is anticipated to significantly advance the development of safe, environmentally friendly, and durable lithium-ion batteries.
Electrochemical prelithiation enables efficient lithium embedding through electrochemical reactions, which enables precise control of lithium injection in a short period of time, thereby improving the initial coulombic efficiency and cycle life of the battery. By modulating the current and voltage, electrochemical prelithiation may accurately regulate the extent of prelithiation, prevent both excessive and insufficient prelithiation, and guarantee uniform incorporation of lithium ions within the anode. Furthermore, the electrochemical prelithiation process is environmentally friendly and does not rely on toxic or polluting chemicals, aligning with green environmental protection and sustainable development. At the same time, electrochemical pre-lithiation eliminates the need for chemical additives, avoiding the impurities that can be introduced by pre-lithiation additives, which is essential for the purity and stability of battery materials. However, additional side reactions, including electrolyte breakdown, may transpire. Consequently, meticulous regulation of electrochemical parameters is essential to guarantee that the primary reaction is lithium intercalation. The electrochemical prelithiation technology is based on the conventional charge–discharge mode of the battery, and optimizes the interface reaction between the electrode and the electrolyte. But this strategy necessitates supplementary equipment and procedures, including auxiliary electrodes and meticulous regulation of current and voltage, hence increasing complexity and expense in battery production. In the future, the pre-lithiation efficiency will be further improved and the cost will be reduced through a more efficient battery management system, intelligent control and adjustable current mode.
Li et al. synthesized three variants of LixSi (x = 4.4, 3.75, and 2.33) using high-energy ball milling of lithium metal and silicon powder.108 All three LixSi phases demonstrated superior performance compared to unmodified Si, with Li4.4Si exhibiting the most exceptional results, achieving a first discharge capacity of 3306 mA h g−1 at a current density of 358 mA g−1, a capacity exceeding 2100 mA h g−1 after 30 cycles, and a capacity surpassing 1200 mA h g−1 after 60 cycles. The formation of a nitride layer (LixNySiz) on the surface of Li4.4Si enhances cycling performance, maintaining a capacity above 1200 mA h g−1 after 80 cycles. Enhancing the nitriding degree markedly improves the capacity retention rate, decreasing from an average of 1.06% each cycle to 0.15% per cycle, however the initial discharge capacity diminishes due to the inactivation of Si in the LixNySiz layer. The initial coulombic efficiency of all LixSi-based electrodes was markedly higher than that of Si electrodes (about 90% compared to 40–70%).
The anode tin foil of lithium-ion batteries exhibits a substantial specific capacity of 993 mA h g−1; yet, its initial coulombic efficiency (ICE) ranges merely from 10% to 20%, significantly lower than that of silicon or SnO2 nanoparticles. Xu et al. discovered that bare Sn facilitates the dissolution of liquid electrolytes at intermediate voltages, resulting in bubble formation that obstructs lithium ion transport and degrades the SEI layer.109 They synthesized LixSn foil through metallurgical pre-alloying of Li, resulting in a reduced initial anode potential that concurrently mitigates outgassing and facilitates the development of a cohesive passivated solid electrolyte interphase (SEI). A one-step mechanical prelithiation approach was employed to address the issue of extremely poor initial coulombic efficiency associated with the self-supporting Sn-based foil anode. The roll-to-roll calendering of stacked Li and Sn foils induces a spontaneous reaction at the interface, resulting in the formation of a LixSn layer on the Sn foil's surface, thereby creating a LixSn/Sn electrode, with the un-lithiated Sn foil substrate serving as a current collector. Roll-to-coil rolling is now being used in industrial production, so this method is expected to be commercially applied. A lithium-ion capacity over 3 mA h cm−2 is incorporated into the Sn foil electrode. At a current density of 2.65 mA h cm−2, the initial coulombic efficiency (ICE) of the LiFePO4//LixSn full cell rose from 20% to 94% and attained 200 stable cycles. The moisture resistance of the pre-lithiated Sn anode is excellent; even after 12 hours of exposure, the samples show an industrially acceptable capacity retention rate with no observed capacity decay after exposure to ambient air of various humidities.
Mechanical prelithiation provides significant advantages for improving the performance of anode materials in lithium-ion batteries. It enhances the initial coulombic efficiency (ICE) by directly incorporating lithium into the active material, reducing the formation of a resistive solid electrolyte interphase (SEI) and ensuring better charge–discharge efficiency. Additionally, mechanical prelithiation enhances cycling stability, as demonstrated by LixSi and LixSn electrodes, which maintain high capacities over extended cycles. This approach also improves safety by mitigating issues such as gas formation and electrode degradation commonly associated with bare lithium or poor initial performance. However, challenges remain in optimizing the process, including balancing lithium content and structural integrity, preventing inactivation of active materials like Si, and addressing the stability of the pre-lithiated layer in long-term cycling. Further advancements are required to refine the process and ensure consistency for large-scale applications. With the development of 3D printing and advanced manufacturing technologies, mechanical pre-lithiation can more precisely control the distribution of lithium, thereby improving the energy density and long-life performance of batteries. In the future, mechanical pre-lithiation technology is expected to achieve economic large-scale application through intelligent manufacturing and automated production lines.
Table 2 shows three other methods in electrode prelithiation, which are primarily prelithiation of graphite and silicon compounds, significantly increasing their initial coulombic efficiency and cycle capacity. However, their cycle life needs to be further improved, and the commercial application of prelithiation technology still faces substantial challenges.
| Prelithiation method | Prelithiation material | Targeted substance | Cell configuration | ICE | Performance | Refs |
|---|---|---|---|---|---|---|
| Chemical prelithiation | Li-Biph/2-Me-THF | Gr | LiFePO4//Gr | 99.6% | 60 cycles | 97 |
| LiCoO2//Gr | 96.5% | 60 cycles | ||||
| NCM811//Gr | 90.8% | 60 cycles | ||||
| Li-Naph/2-Me-THF | Gr | LiCoO2//Gr | 96.1% | 50 cycles | 98 | |
| Li–arene | SiOx | NMC532//SiOx | 86.4% | 116% energy density | 100 | |
| Li–Np | Si | Si//Li2S-PAN | 93.5% | 50 cycles, 82.6% capacity retention | 102 | |
| Electrochemical prelithiation | Li foil | SiNW | Sulfur/mesoporous carbon//SiNW | — | 10 cycles, 80% capacity retention | 103 |
| Li foil | Si | Li4Ti5O12//Gr/PMAA/Li | — | 50 cycles, 84% capacity retention | 105 | |
| Li foil | c-SiOx | NCA//c-SiOx | 85.34% | 100 cycles | 106 | |
| Mechanical prelithiation | Li | Si | Li//Li4.4Si@LixNySiz-1 | 89.2% | Capacity decay of 2.6% per cycle in 20 cycles | 108 |
| Li foil | Sn | LFP//Sn | 94% | 200 cycles, 94.5% capacity retention | 109 |
3.2 Other prelithiation
Li developed a pre-lithiation binder approach utilizing polyacrylic acid (LixPAA) for SiOx anodes.110 By modifying the molar ratio of the carboxyl group and LiOH to meticulously regulate the lithiation degree of the PAA binder, the initial coulombic efficiencies and cycle life of the SiOx anode were markedly enhanced. When Li0.75PAA binder substitutes PAA binder, the interchain entanglement diminishes binder aggregation and enhances mechanical strength. The pre-reaction with the –COOH group of the PAA polymer concurrently diminishes irreversible side reactions during cycling, while the substantial quantity of –COOLi groups produced by the reaction serves as an auxiliary lithium source, facilitating lithium insertion and extraction. The ICE of the heavily loaded (3 mg cm−2) SiOx electrode utilizing this binder exceeds 70%, significantly surpassing the 57.4% of the SiOx@PAA electrode, and it remains stable cycling performance, delivering over 2.7 mA h cm−2 at 0.2C for 170 cycles. Furthermore, it is suggested that this method is broadly applicable to various silicon anodes, including silicon nanoparticles and silicon/graphite composites. This universal binder method and proposed operational mechanism provide valuable insights for the development of silicon-based anodes with elevated initial coulombic efficiency, higher energy density, and extended lifespan.
Throughout the cycling process of lithium batteries, the silicon-based anode experiences significant volume fluctuations due to lithium alloying and dealloying, leading to diminished cycling stability. Commonly utilized PVDF is incapable of alleviating the tension induced by volumetric expansion due to its insufficient van der Waals contact with silicon. Zhang et al. examined a water-soluble rigid rod polymer, poly(2,2′-disulfonyl-4,4′-benzidine terephthalamide) (PBDT), utilized as a binder for silica-based electrodes.111 The amino, carbonyl, and sulfonic acid groups of the PBDT bundle can establish hydrogen bonds with the surface of silicon nanoparticles (SiNPs), providing strong adhesion to preserve the integrity of the silicon electrode. Furthermore, PBDT adhesives can create highly organized nematic stiff rod bundles (double helix chains), enhancing their strength and toughness to mitigate the volumetric expansion of SiNPs. The lithiated PBDT binder has a high ionic conductivity (3 × 10−4 S cm−1), which partially replenishes the active lithium utilized during SEI formation and facilitate the establishment of a stable solid electrolyte interface (SEI). The silicon-based electrode half-cell utilizing PBDT binder sustains a substantial capacity of 2163 mA h g−1 at the C/4 rate, although the particular capacity of Si@PVDF significantly diminishes to 111 mA h g−1. The pre-lithiated PBDT binder can effectively improve ICE and specific capacity, but it is only suitable for active materials with large volume expansion, and lacks universal for practical applications.
Silicon exhibits relatively low ionic conductivity when employed as the anode material in lithium batteries. Low ionic conductivity results in the sluggish diffusion of lithium ions within silicon, hence diminishing ion transmission efficiency during charging and discharging, which adversely impacts both rate performance and total battery performance. The ion transport performance and overall efficacy of lithium batteries can be substantially enhanced through tactics including nanomaterialization, composite materials, surface modification, and structural design. Li presented a binder pre-lithiation approach for silicon electrodes.112 A trifunctional network binder (N–P–LiPN) was synthesized using hydrogen bonding, utilizing partially lithiated rigid polyacrylic acid as the backbone and partially lithiated soft Nafion as the buffer layer. The fabrication process of N–P–LiPN binder is aqueous, enabling low cost and environmental friendliness in scale-up applications. N–P–LiPN exhibits robust adhesion and mechanical capabilities, enabling it to accommodate the significant volume fluctuations of silicon anodes. Furthermore, lithium ions are conveyed via the lithiation group of N–P–LiPN, which markedly improves the ionic conductivity of the silicon anode. The Si@N–P–LiPN electrode demonstrated an initial coulombic efficiency of 93.18% and maintained consistent cycling performance for 500 cycles in the half-cell.
Besides volume expansion and low ionic conductivity, silicon anodes also face the issue of exothermic reactions with lithium, necessitating the investigation of heat-resistant binders. Zhu et al. introduced a water-soluble poly(amic acid)-derived binder exhibiting superior mechanical strength, solubility, and adhesive properties.113 It not only endures the elevated temperatures of the pre-lithiation process but also proficiently maintains the cohesion of the active components over successive charge–discharge cycles. This lithium-substituted polyamic acid binder (Li–Pa) provides exceptional thermal stability and mechanical flexibility, making it suitable for large-scale, eco-friendly electrode production methods.
Binder pre-lithiation enhances the initial coulombic efficiency (ICE) and overall battery performance while circumventing the intricacies associated with including additional pre-lithiation materials directly into the electrode. Furthermore, pre-lithiation with binders maintains the integrity of the electrode material and structure without compromising the conductivity and mechanical stability of the electrode. Binder pre-lithiation enhances battery efficiency and cycle longevity by substituting carboxyl-based protons with lithium ions, hence establishing an effective pre-lithiation mechanism. Nonetheless, binder pre-lithiation has several obstacles, including the necessity of maintaining lithium stability and appropriate release inside the binder, as well as preventing detrimental reactions during electrode fabrication and battery operation. Consequently, selecting the appropriate binder type, optimizing lithium incorporation process, and maximizing the extent of pre-lithiation are essential for effective binder pre-lithiation. Binder pre-lithiation enhances the initial efficiency and performance of lithium-ion batteries without substantially escalating process complexity and expense, presenting great application possibilities in battery manufacturing and performance tuning.
:EMC = 3:7, with a coulombic efficiency of 95–96%. During the experiment, a large amount of the gaseous product Cl2 was dissolved in the electrolyte, but the Si active material and SEI membrane in contact with it did not degrade significantly. In the NCM811//Si full cell, the capacity retention rate for the 100th cycle can be significantly improved from 54% to 78% by electrolytic prelithiation compared to the reference cell without Si prelithiation.The utilization of electrolyte is essential for the pre-lithiation of the anode material by direct contact. Nevertheless, a significant limitation of this method is the inadequate utilization efficiency of the lithium supply (<65.0%), as the inert “dead lithium” produced obstructs the diffusion and migration of lithium ions within the battery. Consequently, to augment the efficacy of this contact prelithiation, the electrolyte can be optimally tailored. Yue et al. examined contact prelithiation in traditional ethylene carbonate (EC)-based electrolytes with monosolvent dimethyl carbonate (DMC) electrolytes to analyze the impact of solid electrolyte interphase (SEI) development at the interface of dense primordial electrolytes with low electron conductivity on the transformation of lithium sources.116 The findings indicated that the limited usage of lithium sources is due to the fast obstruction of electron channels by the dense REI formed from the electrolyte composition. DMC can produce a mixed ionic/electron conductor interface (MCI) that is irregular on the anode surface and devoid of organic material, allowing the REI-wrapped lithium metal to be perpetually transformed throughout the pre-lithiation process, resulting in a 92.8% utilization of the lithium supply. The pouch cell with the NCM811 cathode had an impressive capacity retention rate of 94.9% over 210 cycles, attributed to the efficient pre-lithiation enabled by DMC electrolyte.
Qiao et al. optimized the electrolyte composition by substituting the liquid electrolyte with a carbon-coated lithium phosphate oxide (LiCPON) solid electrolyte, thereby preventing various side reactions associated with lithium metal and forming an ideal interface with the decomposition products of LiCPON.117 The initial coulombic efficiency of a prelithiated electrode with a solid electrolyte can be enhanced by around 10% relative to the original electrode, reaching 98.6% in half cells and 88.9% in full cells. Prelithiation anodes exhibiting solid-state corrosion demonstrate superior prelithiation efficiency compared to prelithiated electrodes utilizing liquid electrolytes.
In comparison to alternative pre-lithiation techniques, electrolyte lithium salts exhibit stability and cost-effectiveness in atmospheric conditions, with no breakdown or side reactions occurring during the electrolysis process, thereby substantially minimizing economic expenses and safety hazards. Additionally, it is highly versatile and can be seamlessly incorporated into current battery manufacturing processes with minimal alterations to the production workflow. Excessive electrolyte consumption during electrolysis will elevate the internal resistance of the battery, hinder lithium ion migration, compromise safety protections, and diminish battery lifespan. Consequently, choosing the appropriate electrolyte salt and optimizing its quantity is crucial for the effective implementation of electrolyte pre-lithiation to prevent adverse impacts on the cycling performance and stability of the battery.
Cao et al. recently produced a two-dimensional lithiated covalent organic framework nanosheet (Li–CON) layer including organized lithiation sites within the intrinsic nanopores of Li–CON (Fig. 17a).118 The low diffusion barrier of these lithiation sites enables them to function as mediators for lithium ion transfer, thereby influencing the diffusion behavior of lithium ions and markedly enhancing their transport. Furthermore, the nitrogen in the pyridine of the triazole unit serves as an adsorption site to capture polysulfides via dipole–dipole interactions on the Li–CON surface. The adsorption site and lithiation site function synergistically, with Li–CON facilitating the breaking of Li–S bonds at relatively low activation energy, accelerating polysulfide conversion rates. The Li–CON modified separator demonstrates elevated lithium-ion conductivity and transfer number, while preventing polysulfide shuttling and diminishing charge overpotential. The lithium-sulfur battery exhibits an initial capacity of 982 mA h g−1 at 1C and demonstrates a capacity degradation of merely 0.057% per cycle after 600 cycles, indicating exceptional long-cycle performance (Fig. 17b).
 | ||
| Fig. 17 (a) Synthetic scheme of the Li–CON. (b) Long-term cycling performance of the Li–CON@GN and CON@GN cells at 1C, respectively (reprinted from ref. 118. Copyright 2020, with permission from Elsevier). | ||
Meng introduced a novel functional separator prelithiation (FSP, Fig. 18a) that enhances a conventional polymer separator by applying a coating of pre-lithiated antifluorite (LFO) through an industry-adaptive spread coating process, facilitating swift detection and reaction to first dendrite growth.119 It prevents the incorporation of extremely alkaline chemicals into the cathode slurry, mitigates the risk of slurry gelation, safeguards the cathode electrode from degradation, and preserves the stability of the electrode–electrolyte interface. The industrial preparation of the separator paves the way for its further expansion of production, which can be applied to electrode preparation, battery manufacturing, and formation. By lowering the discharge cut-off voltage, it reduces lithium loss from the positive electrode, reversibly absorbs lithium from the negative electrode, and prevents perlithiation and dendrite formation on the negative electrode. The 3 A h lithium-ion pouch battery, utilizing a functional separator, a silicon-based anode, and a high-nickel layered oxide cathode, demonstrated a consistent energy density of 330 W h kg−1 and significantly enhanced cycling performance.
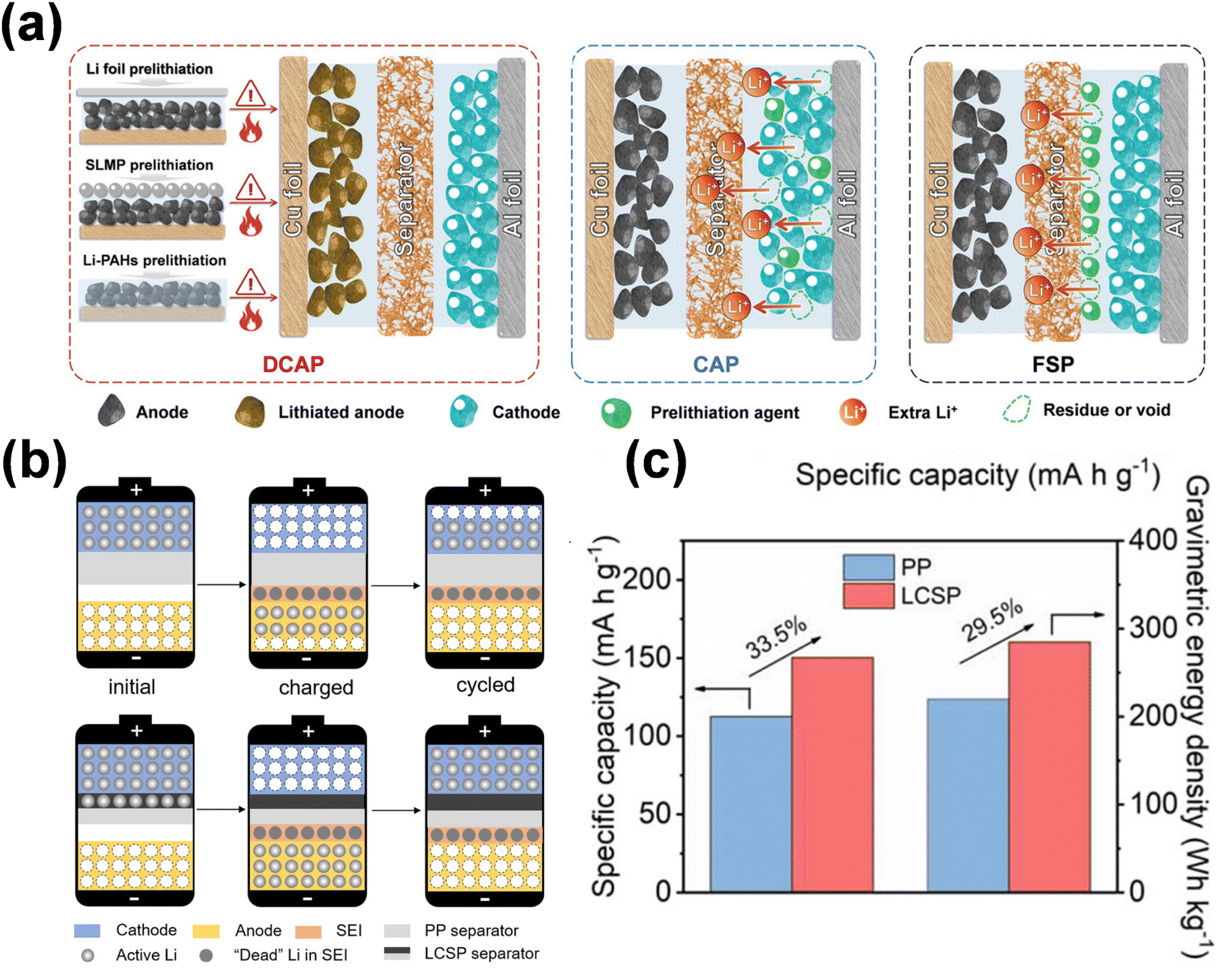 | ||
| Fig. 18 (a) Schematics of the three prelithiation strategies (reprinted from ref. 119. Copyright 2023, with permission from Wiley-VCH). (b) Schematic illustration of Li-ion full cells and the LCSP separator compensating for the initial Li loss. (c) Comparison of the initial specific capacity and energy density for the LiFePO4|graphite full cells with different separators (reprinted from ref. 120. Copyright 2021, with permission from ACS Publications). | ||
Rao et al. similarly presented a prelithiated separator to mitigate initial capacity loss by altering a commercial polypropylene separator (CSP) with Li2S/Co nanocomposites.120 The Li2S/Co coating facilitates the extraction of active lithium ions during charging, demonstrating a delithiation capacity of 993 mA h g−1. The assembly of the entire battery utilizing LiFePO4 and graphite resulted in an increase in reversible capacity from 112.6 mA h g−1 to 150.3 mA h g−1, an enhancement in capacity retention rate from 73% to 84.4% over 100 cycles, and a 29.5% rise in energy density (Fig. 18c). Post-delithiation, the LCSP separator maintains electrochemical stability and does not compromise cathode or overall battery performance. The fabricated pouch cells demonstrated consistent cycling performance. The superior electrochemical properties of the prelithiated separator, together with the attributes of the roll-to-roll production process, indicate its potential for industrial-scale lithium-ion battery manufacturing.
The uniform distribution of the lithium source within the separator facilitates an accurate release of lithium ions, enhancing battery performance and longevity. Furthermore, the pre-lithiation of the separator prevents direct contact between the lithium source and the cathode or anode materials, mitigates the production of lithium dendrites, diminishes the likelihood of potential side reactions, and enhances the safety of the battery.121 Separator pre-lithiation is relatively straightforward comparing to alternative pre-lithiation techniques and may be seamlessly incorporated into current battery production processes. This technology must be meticulously engineered and optimized to guarantee a consistent application of the lithium source on the separator and an adequate lithium release rate, while preserving the mechanical integrity and stability of the separator to enhance battery performance without jeopardizing overall safety. In addition, the pre-lithiation process may lead to changes in separator properties, such as changes in porosity, which in turn can affect the overall performance of the battery. Therefore, how to optimize the pre-lithiation process to ensure the stability of the lithiation layer and the long-term effect of the separator is the key to realizing the wide application of this technology.
4. Effect of prelithiation
Prelithiation, the procedure of incorporating lithium ions into the electrode material of lithium-ion batteries before assembly, significantly influences battery performance in various aspects, notably in augmenting capacity, prolonging cycle life, reducing initial capacity fade rate, enhancing rate performance, and increasing energy density. Prelithiation markedly improves the battery's capacity by augmenting the electrode material with more lithium ions. The increase in the lithium reservoir immediately results in enhanced energy density, allowing the battery to accumulate more charge. As a result, equipment utilizing prelithiated batteries can function for extended durations between charges, which is particularly advantageous for electric vehicles and portable electronics. Furthermore, prelithiation is essential for prolonging the cycle life of batteries. The technique promotes the development of a more stable solid electrolyte interface (SEI) layer, serving as a protective barrier between the electrode materials and the electrolyte. This SEI layer aids in preventing the deterioration of the electrode structure over successive charge–discharge cycles. Prelithiation preserves the integrity of the electrode materials, enabling the battery to endure a higher number of cycles with minimal performance degradation. The prolonged cycle life reduces the frequency of battery replacements, providing both economic and environmental benefits. Simultaneously, the initial rate of capacity degradation in batteries was enhanced. The early capacity fade denotes the swift decline in capacity that generally transpires during the initial charge–discharge cycles of a new battery. Prelithiation facilitates the early formation of a stable solid electrolyte interphase (SEI) layer in the battery's life cycle, hence averting irreversible interactions between the electrode materials and the electrolyte. This stabilization reduces initial capacity loss, guaranteeing that the battery maintains a greater proportion of its original capacity from the beginning. Consequently, the battery demonstrates enhanced performance and reliability from the outset, offering a more stable and trustworthy power supply. Moreover, prelithiation improves the rate performance of lithium-ion batteries. The rate performance, defined as a battery's capacity to charge and discharge rapidly, is essential for applications demanding high power output, including electric vehicles and grid energy storage. Pre-lithiation enhances the kinetics of charge–discharge reactions, facilitating quicker response times and more efficient energy transfer. This results in enhanced power delivery and performance in high-demand scenarios, rendering prelithiated batteries appropriate for a broader spectrum of applications. Ultimately, prelithiation enhances the energy density of lithium-ion batteries, which dictates the amount of energy a battery can store in relation to its weight or volume. The pre-lithiation treatment supplies extra active lithium to offset the Initial coulombic Loss (ICL) and enhances the Initial coulombic Efficiency (ICE), hence elevating the operational voltage. Consequently, it directly improves energy density, facilitating the development of lighter and more compact batteries without sacrificing capacity or performance. This is especially beneficial for portable devices and electric vehicles, where space and weight are crucial factors.5. Conclusions and perspective
This review systematically examines pre-lithiation techniques categorized by electrode architecture, emphasizing the prelithiation of electrodes, electrolytes, separators, and binders. Each category offers distinct opportunities and challenges that are essential for enhancing the performance of lithium-ion batteries. We examined four primary techniques of electrode prelithiation: prelithiation additives, electrochemical prelithiation, chemical prelithiation, and mechanical prelithiation. Pre-lithiation additives demonstrate significant potential in enhancing initial capacity and cycling stability; nonetheless, their compatibility with various electrode materials continues to be a critical research focus. Electrochemical prelithiation provides substantial performance enhancements, however it may be constrained by process intricacy and expense. Chemical prelithiation exhibits a high lithium consumption rate; nonetheless, it poses issues regarding material compatibility and may result in side reactions. Mechanical prelithiation, albeit simple to execute, raises difficulties regarding the structural stability of the electrode material. The emphasis should be on advancing pre-lithiation methods to enhance ionic conductivity and ensure stability in electrolytes and separators. Advancements in these domains can enhance the overall efficiency and longevity of the battery. Several critical research avenues are essential for the advancement of pre-lithiation technology in the future. Initially, enhancing the compatibility and performance of pre-lithiation additives across various electrode materials is essential for maximizing their advantages. Secondly, the simplification and cost reduction of electrochemical and chemical prelithiation processes will be crucial for their commercial feasibility. A hybrid method that integrates various pre-lithiation techniques may yield unique solutions by leveraging the benefits of each method.With the growing demand for efficient, sustainable lithium ion batteries, it is necessary to improve prelithiation techniques to address issues related to battery performance and resource constraints. By compensating for the irreversible lithium loss during the initial charge cycle, prelithiation technology has positively influenced battery performance by augmenting capacity, extending cycle life, reducing initial capacity degradation, and improving power performance and safety. With the ongoing advancements in pre-lithiation technology research and development, the prospects for enhancements in battery technology are increasingly optimistic, facilitating the emergence of more potent, resilient, and efficient energy storage systems and electric vehicles. Prelithiation has emerged as a promising strategies to enhance the performance of lithium-ion batteries (LIBs), particularly for next-generation high-capacity anodes like Si-based and SiOx-based materials. Apart from lithium-ion batteries, prelithiation will also become an indispensable process for lithium–sulfur, lithium–oxygen, and sodium-ion batteries, which face similar challenges with low initial coulombic efficiency (ICE) and energy density.
Although pre-lithiation technology has achieved remarkable results in laboratory and small-scale production, there are still some issues to consider before the industrialization of prelithiation technologies. While techniques such as mechanical rolling have demonstrated good lithiation efficiency and industrial compatibility, challenges remain in controlling lithiation uniformity and degree, which can result in localized side reactions and the formation of lithium dendrites, posing risks to battery safety and cycle stability. Additionally, the high reactivity of certain prelithiation materials, such as SLMPs and lithium alloys, presents serious safety concerns during manufacturing. These materials require careful handling and processing to avoid explosions or other hazardous events.
Addressing the challenges of prelithiation technology requires several strategic approaches. Precise control over the prelithiation process is essential to minimize issues like localized over-lithiation and dendrite formation. The development of advanced real-time monitoring systems and feedback mechanisms could provide the necessary precision. Additionally, the exploration of novel prelithiation methods, such as electrospray and plasma spray, which allow for more uniform and controlled lithium distribution, could offer solutions to current limitations in existing techniques. Another pivotal challenge is the moisture sensitivity and environmental instability of many prelithiation materials. Developing more stable materials that are compatible with aqueous binders and polar solvents commonly used in electrode manufacturing will be crucial for scaling prelithiation and making it environmentally friendly. Surface passivation strategies, such as using Li2CO3 coatings on SLMPs or F-based layers for Si-based anodes, could help mitigate the reactivity of these materials. Moreover, finding cost-effective, safe, and greener prelithiation reagents that are compatible with existing industrial production lines remains a major goal. In addition to improving prelithiation methods, it is essential to develop new battery management systems (BMS) and cell design principles to accommodate the unique electrochemical behaviors of prelithiated cells. For example, adjusting charge/discharge protocols to prevent issues like over-lithiation or metallic lithium plating during cycling will be key to maintaining safety and cycle life. Furthermore, the recycling of prelithiated batteries presents a unique challenge, as the high reactivity of prelithiated anodes could pose risks during battery disassembly. The development of safe and efficient recycling techniques for these batteries will be crucial for ensuring the sustainability of prelithiation technologies.
For large-scale industrial applications, cost-effectiveness and scalability are paramount. Methods like in situ electrochemical prelithiation using three-electrode designs or roll-to-roll processing offer promising routes for improving the efficiency and economic viability of prelithiation. However, the large-area Li-foil counter electrode required for some of these methods still adds to the cost and affects the thermal safety of the cells. Using through-hole structured electrodes could reduce the amount of Li foil needed, but the current high cost of laser-drilled electrodes remains a barrier. At the same time, the technology of extracting lithium from waste lithium batteries and other lithium resources can effectively realize the reuse of lithium resources, reduce environmental pollution, reduce dependence on primary lithium resources, and promote economic benefits.
In conclusion, prelithiation technologies offer substantial potential to improve the performance of lithium-ion batteries, especially with the advent of high-capacity anode materials and next-generation battery systems. However, realizing their full potential requires overcoming challenges related to control, safety, cost, and scalability. Future prelithiation innovations must focus on developing precise, safe, and cost-efficient methods that are environmentally friendly and compatible with large-scale industrial applications. With continued research and development, prelithiation will play a pivotal role in the advancement of high-performance energy storage technologies for a wide range of applications, from electric vehicles to renewable energy storage systems.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Yiming Zhang: conception, investigation, writing-review, and editing; Yao Li: methodology, review and editing; Huyan Shen: investigation and methodology; Yanyu Li: investigation and methodology; Yongsheng Hu: investigation and methodology. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Yuan Chuang Fund of Shanghai Institute of Space Power-Sources, YF07050118F3537.References
- A. A. Kebede, T. Kalogiannis, J. Van Mierlo and M. Berecibar, Renewable Sustainable Energy Rev., 2022, 159, 112213 CrossRef CAS.
- D. M. Davies, M. G. Verde, O. Mnyshenko, Y. R. Chen, R. Rajeev, Y. S. Meng and G. Elliott, Nat. Energy, 2018, 4, 42–50 CrossRef.
- A. G. Olabi, Energy, 2017, 136, 1–6 CrossRef.
- Y. Tong, J. Liang, H. K. Liu and S. X. Dou, Energy Storage Mater., 2019, 20, 176–187 CrossRef.
- J. Liu, Z. Huang, M. Fan, J. Yang, J. Xiao and Y. Wang, Nano Energy, 2022, 104, 107915 CrossRef CAS.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef PubMed.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- G. E. Blomgren, J. Electrochem. Soc., 2017, 164, A5019 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS.
- Y. Matsumura, S. Wang and J. Mondori, J. Electrochem. Soc., 1995, 142, 2914–2918 CrossRef CAS.
- C. Yan, R. Xu, Y. Xiao, J. Ding, L. Xu, B. Li and J. Huang, Adv. Funct. Mater., 2020, 30, 1909887 CrossRef CAS.
- J. Lu, Z. Chen, F. Pan, Y. Cui and K. Amine, Electrochem. Energy Rev., 2018, 1, 35–53 CrossRef CAS.
- L. Y. Beaulieu, K. W. Eberman, R. L. Turner, L. J. Krause and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A137 CrossRef CAS.
- Z. Huang, Z. Deng, Y. Zhong, M. Xu, S. Li, X. Liu, Y. Zhou, K. Huang, Y. Shen and Y. Huang, Carbon Energy, 2022, 4, 1107–1132 CrossRef CAS.
- L. Jin, C. Shen, Q. Wu, A. Shellikeri, J. Zheng, C. Zhang and J. P. Zheng, Adv. Sci., 2021, 8, 2005031 CrossRef CAS PubMed.
- R. Ding, S. Tian, K. Zhang, J. Cao, Y. Zheng, W. Tian, X. Wang, L. Wen, L. Wang and G. Liang, J. Electroanal. Chem., 2021, 893, 115325 CrossRef CAS.
- C. Sun and X. Zhang, Energy Storage Mater., 2020, 32, 497–516 CrossRef.
- F. Wang, B. Wang, J. Li, B. Wang, Y. Zhou, D. Wang, H. Liu and S. Dou, ACS Nano, 2021, 15, 2197–2218 CrossRef CAS PubMed.
- K. Zou, W. Deng, P. Cai, X. Deng, B. Wang, C. Liu, J. Li, H. Hou, G. Zou and X. Ji, Adv. Funct. Mater., 2021, 31, 2005581 CrossRef CAS.
- B.-S. Lee, Polymers, 2020, 12, 2035 CrossRef CAS.
- H. Ge, N. Li, D. Li, C. Dai and D. Wang, J. Phys. Chem. C, 2009, 113, 6324–6326 CrossRef CAS.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4, 5387–5416 RSC.
- K. Sawai, Y. Iwakoshi and T. Ohzuku, Solid State Ionics, 1994, 69, 273–283 CrossRef CAS.
- B. Liu, X. Hu, H. Xu, W. Luo, Y. Sun and Y. Huang, Sci. Rep., 2014, 4, 4229 CrossRef PubMed.
- B. Joshi, E. Samuel, H. S. Jo, Y.-I. Kim, S. Park, M. T. Swihart, W. Y. Yoon and S. S. Yoon, Electrochim. Acta, 2017, 253, 479–488 CrossRef CAS.
- X. Wei Kong, R. Liang Zhang, S. Kui Zhong and L. Wu, Mater. Sci., 2016, 34, 227–232 Search PubMed.
- J. Wang, N. Du, H. Wu, H. Zhang, J. Yu and D. Yang, J. Power Sources, 2013, 222, 32–37 CrossRef CAS.
- L. Wang, Y. Yu, P. C. Chen, D. W. Zhang and C. H. Chen, J. Power Sources, 2008, 183, 717–723 CrossRef CAS.
- L. Fan, W. Zhang, S. Zhu and Y. Lu, Ind. Eng. Chem. Res., 2017, 56, 2046–2053 CrossRef CAS.
- J. R. Szczech and S. Jin, Energy Environ. Sci., 2011, 4, 56–72 RSC.
- B.-S. Lee, J. Yoon, C. Jung, D. Y. Kim, S.-Y. Jeon, K.-H. Kim, J.-H. Park, H. Park, K. H. Lee, Y.-S. Kang, J.-H. Park, H. Jung, W.-R. Yu and S.-G. Doo, ACS Nano, 2016, 10, 2617–2627 CrossRef CAS PubMed.
- Y. Jin, B. Zhu, Z. Lu, N. Liu and J. Zhu, Adv. Energy Mater., 2017, 7, 1700715 CrossRef.
- H. Liu, S. Wang, L. Liu, J. Zhao, W. Zhang, R. Bao, L. Wang, J. Yang, Y. Li and Z. Jing, Chem. Eng. J., 2024, 495, 152444 CrossRef CAS.
- S. Liang, Y. Cheng, J. Zhu, Y. Xia and P. Müller-Buschbaum, Small Methods, 2020, 4, 2000218 CrossRef CAS.
- R. Mo, D. Rooney, K. Sun and H. Y. Yang, Nat. Commun., 2017, 8, 13949 CrossRef CAS.
- E. Paled, J. Electrochem. Soc., 1979, 126, 2047 CrossRef.
- S. K. Heiskanen, Joule, 2019, 3, 2322–2333 CrossRef CAS.
- E. Peled, D. Golodnitsky and G. Ardel, J. Electrochem. Soc., 1997, 144, L208–L210 CrossRef CAS.
- D. Chen, M. A. Mahmoud, J.-H. Wang, G. H. Waller, B. Zhao, C. Qu, M. A. El-Sayed and M. Liu, Nano Lett., 2019, 19, 2037–2043 CrossRef CAS PubMed.
- S. Jurng, Z. L. Brown, J. Kim and B. L. Lucht, Energy Environ. Sci., 2018, 11, 2600–2608 RSC.
- J. Christensen and J. Newman, J. Electrochem. Soc., 2004, 151, A1977 CrossRef CAS.
- D. Aurbach, J. Power Sources, 2000, 89, 206–218 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- Y. Zhou, M. Su, X. Yu, Y. Zhang, J.-G. Wang, X. Ren, R. Cao, W. Xu, D. R. Baer, Y. Du, O. Borodin, Y. Wang, X.-L. Wang, K. Xu, Z. Xu, C. Wang and Z. Zhu, Nat. Nanotechnol., 2020, 15, 224–230 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- G. Wang, W. Brown and M. Kvetny, Curr. Opin. Electrochem., 2019, 13, 112–118 CrossRef CAS.
- G. Ramos-Sanchez, F. A. Soto, J. M. Martinez De La Hoz, Z. Liu, P. P. Mukherjee, F. El-Mellouhi, J. M. Seminario and P. B. Balbuena, J. Electrochem. Energy Convers. Storage, 2016, 13, 031002 CrossRef.
- E. W. C. Spotte-Smith, T. B. Petrocelli, H. D. Patel, S. M. Blau and K. A. Persson, ACS Energy Lett., 2023, 8, 347–355 CrossRef CAS.
- K. Leung, Y. Qi, K. R. Zavadil, Y. S. Jung, A. C. Dillon, A. S. Cavanagh, S.-H. Lee and S. M. George, J. Am. Chem. Soc., 2011, 133, 14741–14754 CrossRef CAS PubMed.
- X. Zhao, Y. Yin, Y. Hu and S.-Y. Choe, J. Power Sources, 2019, 418, 61–73 CrossRef CAS.
- G. Yang, S. Zhang, S. Weng, X. Li, X. Wang, Z. Wang and L. Chen, Nano Lett., 2021, 21, 5316–5323 CrossRef CAS PubMed.
- F. Hao, J. Mater. Chem. A, 2018, 6, 19664 RSC.
- W. Cai and Y. Yao, Chem. Soc. Rev., 2020, 49, 3806 RSC.
- D. P. Abraham, T. Spila, M. M. Furczon and E. Sammann, Electrochem. Solid-State Lett., 2008, 11, A226 CrossRef CAS.
- H. Shin, J. Park, A. M. Sastry and W. Lu, J. Power Sources, 2015, 284, 416–427 CrossRef CAS.
- M. Winter, Electrochim. Acta, 1999, 45, 31–50 CrossRef CAS.
- M. T. McDowell, S. W. Lee, W. D. Nix and Y. Cui, Adv. Mater., 2013, 25, 4966–4985 CrossRef CAS PubMed.
- M. J. Chon, V. A. Sethuraman, A. McCormick, V. Srinivasan and P. R. Guduru, Phys. Rev. Lett., 2011, 107, 045503 CrossRef CAS PubMed.
- V. A. Sethuraman, M. J. Chon, M. Shimshak, V. Srinivasan and P. R. Guduru, J. Power Sources, 2010, 195, 5062–5066 CrossRef CAS.
- X. H. Liu, L. Zhong, S. Huang, S. X. Mao, T. Zhu and J. Y. Huang, ACS Nano, 2012, 6, 1522–1531 CrossRef CAS PubMed.
- Y. Wang, Q. Zhang, D. Li, J. Hu, J. Xu, D. Dang, X. Xiao and Y.-T. Cheng, Adv. Energy Mater., 2018, 8, 1702578 CrossRef.
- T. Yoon, C. Xiao, J. Liu, Y. Wang, S. Son, A. Burrell and C. Ban, J. Power Sources, 2019, 425, 44–49 CrossRef CAS.
- M. Pharr, Z. Suo and J. J. Vlassak, Nano Lett., 2013, 13, 5570–5577 CrossRef CAS PubMed.
- C. S. Kang, J. Power Sources, 2014, 267, 739–743 CrossRef CAS.
- Y. S. Choi, M. Pharr, C. S. Kang, S.-B. Son, S. C. Kim, K.-B. Kim, H. Roh, S.-H. Lee, K. H. Oh and J. J. Vlassak, J. Power Sources, 2014, 265, 160–165 CrossRef CAS.
- R. Zhan, X. Wang, Z. Chen, Z. W. Seh, L. Wang and Y. Sun, Adv. Energy Mater., 2021, 11, 2101565 CrossRef CAS.
- B. Huang, T. Huang, L. Wan and A. Yu, ACS Sustain. Chem. Eng., 2021, 9, 648–657 CrossRef CAS.
- M. W. Forney, M. J. Ganter, J. W. Staub, R. D. Ridgley and B. J. Landi, Nano Lett., 2013, 13, 4158–4163 CrossRef CAS PubMed.
- J. Zhao, Z. Lu, N. Liu, H.-W. Lee, M. T. McDowell and Y. Cui, Nat. Commun., 2014, 5, 5088 CrossRef CAS PubMed.
- S. Li, C. Wang, J. Yu, Y. Han and Z. Lu, Energy Storage Mater., 2019, 20, 7–13 CrossRef.
- Z. Yang, Electrochem. Commun., 2022, 138, 107272 CrossRef CAS.
- H. Xu, S. Li, X. Chen, C. Zhang, Z. Tang, H. Fan, Y. Yu, W. Liu, N. Liang, Y. Huang and J. Li, Nano Energy, 2020, 74, 104815 CrossRef CAS.
- J. Zhao, J. Sun, A. Pei, G. Zhou, K. Yan, Y. Liu, D. Lin and Y. Cui, Energy Storage Mater., 2018, 10, 275–281 CrossRef.
- B. Xiang, L. Wang, G. Liu and A. M. Minor, J. Electrochem. Soc., 2013, 160, A415–A419 CrossRef CAS.
- K. Pu, X. Qu, X. Zhang, J. Hu, C. Gu, Y. Wu, M. Gao, H. Pan and Y. Liu, Adv. Sci., 2019, 6, 1901776 CrossRef CAS PubMed.
- X. Li, J. Power Sources, 2021, 496, 229868 CrossRef CAS.
- J. Guo, S. Li, B. Zhu, H. Zhang, X. Gao, Y. Wu, S. Zhang, N. Wen, X. Wang, Y. Lai and Z. Zhang, Chem. Eng. J., 2023, 471, 144744 CrossRef CAS.
- K. Park, B. Yu and J. B. Goodenough, Adv. Energy Mater., 2016, 6, 1502534 CrossRef.
- Y. Liu, X. Meng, Y. Shi, J. Qiu and Z. Wang, Adv. Mater., 2023, 35, 2305386 CrossRef CAS PubMed.
- Y. Pan, X. Qi, H. Du, Y. Ji, D. Yang, Z. Zhu, Y. Yang, L. Qie and Y. Huang, ACS Appl. Mater. Interfaces, 2023, 15, 18763–18770 CrossRef CAS PubMed.
- M. Kim, B. D. Spindler, L. Dong and A. Stein, ACS Appl. Energy Mater., 2022, 5, 14433–14444 CrossRef CAS.
- D. Shanmukaraj, S. Grugeon, S. Laruelle, G. Douglade, J.-M. Tarascon and M. Armand, Electrochem. Commun., 2010, 12, 1344–1347 CrossRef CAS.
- A. Gomez-Martin, M. M. Gnutzmann, E. Adhitama, L. Frankenstein, B. Heidrich, M. Winter and T. Placke, Adv. Sci., 2022, 9, 2201742 CrossRef CAS PubMed.
- G. Huang, J. Liang, X. Zhong, H. Liang, C. Cui, C. Zeng, S. Wang, M. Liao, Y. Shen, T. Zhai, Y. Ma and H. Li, Nano Res., 2023, 16, 3872–3878 CrossRef CAS.
- Y. Bie, J. Yang, J. Wang, J. Zhou and Y. Nuli, Chem. Commun., 2017, 53, 8324–8327 RSC.
- M. G. Kim and J. Cho, J. Mater. Chem., 2008, 18, 5880 RSC.
- M. Noh and J. Cho, J. Electrochem. Soc., 2012, 159, A1329–A1334 CrossRef CAS.
- X. Liu, Y. Tan, W. Wang, C. Li, Z. W. Seh, L. Wang and Y. Sun, Nano Lett., 2020, 20, 4558–4565 CrossRef CAS PubMed.
- J. Li, B. Zhu, S. Li, D. Wang, W. Zhang, Y. Xie, J. Fang, B. Hong, Y. Lai and Z. Zhang, J. Electrochem. Soc., 2021, 168, 080510 CrossRef CAS.
- K.-S. Park, D. Im, A. Benayad, A. Dylla, K. J. Stevenson and J. B. Goodenough, Chem. Mater., 2012, 24, 2673–2683 CrossRef CAS.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacín, Adv. Mater., 2010, 22 CrossRef PubMed.
- Y. Zhan, H. Yu, L. Ben, B. Liu, Y. Chen, Y. Wu, H. Li, W. Zhao and X. Huang, J. Mater. Chem. A, 2018, 6, 6206–6211 RSC.
- Y. Sun, H.-W. Lee, Z. W. Seh, N. Liu, J. Sun, Y. Li and Y. Cui, Nat. Energy, 2016, 1, 15008 CrossRef CAS.
- Y. Sun, H. Lee, Z. W. Seh, G. Zheng, J. Sun, Y. Li and Y. Cui, Adv. Energy Mater., 2016, 6, 1600154 CrossRef.
- J. Du, W. Wang, A. Y. Sheng Eng, X. Liu, M. Wan, Z. W. Seh and Y. Sun, Nano Lett., 2020, 20, 546–552 CrossRef CAS PubMed.
- T. Abe, Y. Mizutani, T. Tabuchi, K. Ikeda, M. Asano, T. Harada, M. Inaba and Z. Ogumi, J. Power Sources, 1997, 68, 216–220 CrossRef CAS.
- Y. Shen, X. Shen, M. Yang, J. Qian, Y. Cao, H. Yang, Y. Luo and X. Ai, Adv. Funct. Mater., 2021, 31, 2101181 CrossRef CAS.
- Y. Luo, Y. Deng, Y. Shen, H. Li, Y. Cao and X. Ai, Energy Technol., 2022, 10, 2200269 CrossRef CAS.
- T. Tabuchi, H. Yasuda and M. Yamachi, J. Power Sources, 2005, 146, 507–509 CrossRef CAS.
- J. Jang, I. Kang, J. Choi, H. Jeong, K. Yi, J. Hong and M. Lee, Angew. Chem., Int. Ed., 2020, 59, 14473–14480 CrossRef CAS PubMed.
- Y. Sun, K. Zhang, R. Chai, Y. Wang, X. Rui, K. Wang, H. Deng and H. Xiang, Adv. Funct. Mater., 2023, 33, 2303020 CrossRef CAS.
- Y. Shen, J. Zhang, Y. Pu, H. Wang, B. Wang, J. Qian, Y. Cao, F. Zhong, X. Ai and H. Yang, ACS Energy Lett., 2019, 4, 1717–1724 CrossRef CAS.
- N. Liu, L. Hu, M. T. McDowell, A. Jackson and Y. Cui, ACS Nano, 2011, 5, 6487–6493 CrossRef CAS PubMed.
- Q. Meng, G. Li, J. Yue, Q. Xu, Y.-X. Yin and Y.-G. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 32062–32068 CrossRef CAS PubMed.
- Z. Cao, P. Xu, H. Zhai, S. Du, J. Mandal, M. Dontigny, K. Zaghib and Y. Yang, Nano Lett., 2016, 16, 7235–7240 CrossRef CAS PubMed.
- H. J. Kim, S. Choi, S. J. Lee, M. W. Seo, J. G. Lee, E. Deniz, Y. J. Lee, E. K. Kim and J. W. Choi, Nano Lett., 2016, 16, 282–288 CrossRef CAS PubMed.
- H. Zhou, X. Wang and D. Chen, ChemSusChem, 2015, 8, 2737–2744 CrossRef CAS PubMed.
- X. Li, F. E. Kersey-Bronec, J. Ke, J. E. Cloud, Y. Wang, C. Ngo, S. Pylypenko and Y. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 16071–16080 CrossRef CAS PubMed.
- H. Xu, S. Li, C. Zhang, X. Chen, W. Liu, Y. Zheng, Y. Xie, Y. Huang and J. Li, Energy Environ. Sci., 2019, 12, 2991–3000 RSC.
- Z. Li, W. Tang, Y. Yang, G. Lai, Z. Lin, H. Xiao, J. Qiu, X. Wei, S. Wu and Z. Lin, Adv. Funct. Mater., 2022, 32, 2206615 CrossRef CAS.
- J. Zhang, J. Sun, Y. Zhao, Y. Su, X. Meng, L. Yan and T. Ma, J. Colloid Interface Sci., 2023, 649, 977–985 CrossRef CAS PubMed.
- Z. Li, Y. Zhang, T. Liu, X. Gao, S. Li, M. Ling, C. Liang, J. Zheng and Z. Lin, Adv. Energy Mater., 2020, 10, 1903110 CrossRef CAS.
- T. Zhu, T.-N. Tran, C. Fang, D. Liu, S. P. Herle, J. Guan, G. Gopal, A. Joshi, J. Cushing, A. M. Minor and G. Liu, J. Power Sources, 2022, 521, 230889 CrossRef CAS.
- H. Zhou, X. Wang and D. Chen, ChemSusChem, 2015, 8, 2737–2744 CrossRef CAS PubMed.
- L. Haneke, F. Pfeiffer, P. Bärmann, J. Wrogemann, C. Peschel, J. Neumann, F. Kux, S. Nowak, M. Winter and T. Placke, Small, 2023, 19, 2206092 CrossRef CAS PubMed.
- X. Yue, Y. Yao, J. Zhang, Z. Li, S. Yang, X. Li, C. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202205697 CrossRef CAS PubMed.
- Y. Qiao, S. Yang, Z. Ma, Y. Yang, X. Hong and Z. Fu, Nano Res., 2023, 16, 8394–8404 CrossRef CAS.
- Y. Cao, C. Liu, M. Wang, H. Yang, S. Liu, H. Wang, Z. Yang, F. Pan, Z. Jiang and J. Sun, Energy Storage Mater., 2020, 29, 207–215 CrossRef.
- Q. Meng, M. Fan, X. Chang, H. Li, W. Wang, Y. Zhu, J. Wan, Y. Zhao, F. Wang, R. Wen, S. Xin and Y. Guo, Adv. Energy Mater., 2023, 13, 2300507 CrossRef CAS.
- Z. Rao, J. Wu, B. He, W. Chen, H. Wang, Q. Fu and Y. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 38194–38201 CrossRef CAS PubMed.
- X. Dai, X. Zhang, J. Wen, C. Wang, X. Ma, Y. Yang, G. Huang, H.-M. Ye and S. Xu, Energy Storage Mater., 2022, 51, 638–659 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |