DOI:
10.1039/D4RA08514K
(Paper)
RSC Adv., 2025,
15, 6742-6752
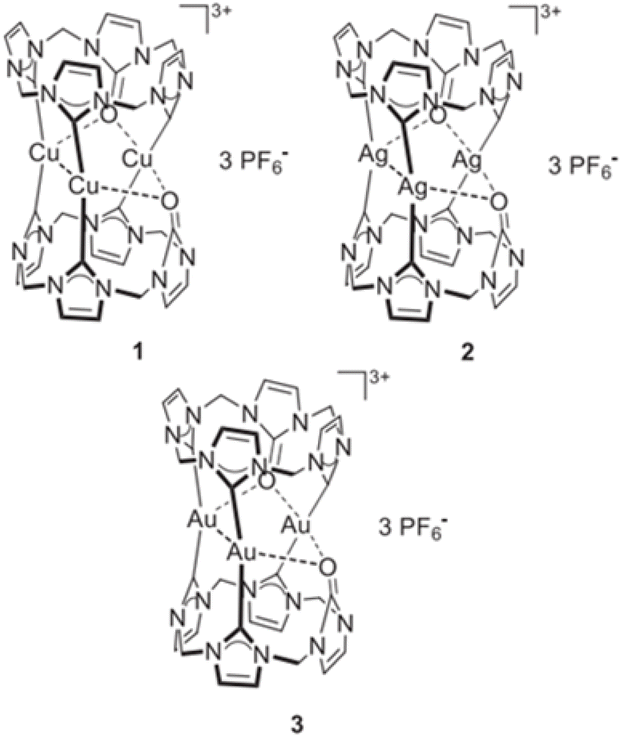
Trinuclear cylinder-like potential anticancer and antibacterial Cu(I), Ag(I) and Au(I) nano-sized cationic complexes with tris-NHC ligands: cationic M3 metal cluster displaying positive or negative cooperativity in triad [L2(R)6→M3]3+ complexes?†
Received
3rd December 2024
, Accepted 24th February 2025
First published on 28th February 2025
Abstract
N-Heterocyclic carbenes (NHCs) are a class of organic molecules containing a divalent carbon atom, known as a carbene, within a heterocyclic (ring) structure where nitrogen atoms (N) form part of the ring. These molecules have garnered significant attention in coordination chemistry due to their unique bonding properties, particularly as strong σ-donor ligands that facilitate the formation of stable complexes. A theoretical study was conducted to investigate the structural and bonding characteristics of M←C bonds in trinuclear, nano-sized Cu(I), Ag(I), and Au(I) cations with two tris-NHC ligands, which exhibit promising anti-cancer and antibacterial potential. The study employed natural bond orbital (NBO) techniques, energy decomposition analysis (EDA), and extended transition-state natural orbital for chemical valence (ETS-NOCV) methods to analyze the bonding interactions. The cooperativity values between bonds were also examined, revealing positive values indicative of anti-cooperativity within the complexes. The results further demonstrated that the M←C interactions are predominantly electrostatic in nature. These findings highlight the unique structural and electronic properties of the complexes, suggesting their potential as candidates for anti-cancer and antibacterial applications.
Introduction
N-Heterocyclic carbenes (NHCs) have emerged as versatile ligands in both organometallic and inorganic chemistry. Owing to their strong σ-donating properties, NHCs form more stable bonds with metals than phosphines,1 with the lone pair of electrons on the carbene carbon stabilized by adjacent nitrogen atoms through an inductive effect.2 Additionally, metal–NHC complexes have attracted significant attention due to their wide range of catalytic3,4 and pharmacological5–7 properties. They are extensively used as ligands in inorganic and organometallic chemistry for two primary reasons: first, their strong metal coordination, and second, their resistance to moisture and air.2,8 As a result, NHCs are preferred as substitutes for phosphine ligands. Modifications at the (N1) position of the NHC ligand significantly impact its reactivity and binding affinity. Transition metal–NHC complexes have been employed in medicinal chemistry due to their remarkable biological properties.9 N-heterocyclic carbene–metal complexes have gained significance in fields like organometallic chemistry, catalysis and bioorganic/bioinorganic chemistry. Among these, coinage metal–NHCs are particularly noteworthy due to their varied biological properties and applications.10,11 The NHC copper complexes have the potential to generate reactive oxygen species (ROS), which can cause DNA strand breaks and contribute to the observed cytotoxicity.12 Metallic silver was recognized by the Chaldeans as early as 4000 B.C.E., making it the third metal used by ancient civilizations, following gold and copper.13 Although most organometallic pharmaceutical research has focused on platinum and gold, the medicinal applications of silver are well-documented.14 Multinuclear silver N-heterocyclic carbene (Ag-NHC) complexes have garnered interest in medicinal chemistry due to their unique properties and potential applications. These complexes represent a promising area in medicinal chemistry, with potential applications in antimicrobial and anticancer therapies.15–19 Gold has long been essential in human history. Likely the first metal to be discovered, it has been utilized in medicinal treatments since ancient times.20 Multinuclear gold-N-heterocyclic carbene (Au-NHC) complexes are an evolving area of research in medicinal chemistry, showing promise for various therapeutic applications. These complexes are emerging as a promising class of compounds in medicinal chemistry. They hold potential for applications in both cancer therapy and antimicrobial treatments.21–26 In 2021, Jacob reported the synthesis and identification of the first large cyclic trinuclear, tri-carbene complexes Cu(I), Ag(I), and Au(I). The ability of these complexes to inhibit the growth of bacteria (S. aureus, E. coli) and cancer cells (HeLa, MCF7) was investigated. The copper and silver complexes demonstrated antiproliferative activity in both cell lines, ranging from 3.03 μM to 25.01 μM (ref. 27) (see Fig. 1).27 Compared to mononuclear complexes, multinuclear complexes have more active sites that can coordinate with substituents possessing biological properties, and as a result, they may exhibit stronger biological activities.28
 |
| | Fig. 1 The structure of the pharmaceutical complexes studied by Jacob. | |
Recent studies have investigated the structure and nature of metal–NHC bonds in some pharmaceutical coinage metal ion complexes.29–33 Cooperativity in bonds refers to the phenomenon where the formation, breaking, or alteration of one bond influences the behavior of nearby bonds. This interaction can enhance or inhibit subsequent bonding events, depending on the system. Cooperativity is widely observed in physical, chemical, and biological systems, and it is particularly important in stabilizing molecular structures and driving complex reactions. Herein, a comparative theoretical investigation into the nature of the metal–NHC bonds of some cylindrical trinuclear clusters of group 11 in coordination with two symmetrical tri-N-heterocyclic carbene (NHC) ligand was reported. The effect of substituents, C2H5, CH3, H, F, Cl, Br, Ph, and SiH3 on six NHC rings of cationic three-metal clusters of copper (1), silver (1) and gold (1) are also investigated. It is worth noting that the X-ray crystal structures of three metal clusters, copper, silver and gold, substituted with C2H5 were synthesized by Rit in 2010 (ref. 34) and 2011.35 Our research group has used the crystal structure of these clusters as input files for the calculations in this study Fig. 2.
 |
| | Fig. 2 Schematic representation of the [L2(R)6→M3]3+ nano-sized cationic complexes. | |
Computational details
The [L2(C2H5)6→Au3]3+ complex is analysed using eight density functional methods: Cam-B3LYP, M05-2X, M06-2X, B3LYP, PBE,35 BLYP, BP86 and M06, in conjunction with D3 dispersion corrections. Additionally, the structural results obtained were compared with the corresponding experimental values derived from X-ray crystallography.34,35 The results obtained from the RMS methodological findings indicate that PBE-D3,36 the most relevant functional among the methods mentioned, shows the strongest correlation between quantitative and experimental structural data in Table 1.
Table 1 RMS calculation of [L2(C2H5)6→Au3]3+ complex
| Method |
Au–C (Å) |
Au–C (Å) |
Au–C (Å) |
RMS |
| B3LYP-D3 |
2.06 |
2.06 |
2.06 |
0.09 |
| BLYP-D3 |
2.06 |
2.06 |
2.06 |
0.09 |
| BP86-D3 |
2.04 |
2.04 |
2.04 |
0.06 |
| CAM-B3LYP-D3 |
2.05 |
2.05 |
2.05 |
0.08 |
| M05-2X-D3 |
2.05 |
2.05 |
2.05 |
0.08 |
| M06-D3 |
2.06 |
2.06 |
2.06 |
0.09 |
| M06-2X-D3 |
2.05 |
2.05 |
2.05 |
0.08 |
| PBE-D3 |
2.04 |
2.04 |
2.04 |
0.06 |
| Exp |
2.00 |
2.01 |
2.00 |
— |
In this article, the cationic parts of the complexes with the general formula [L2(R)6→M3]3+ (where M = Cu(I), Ag(I), Au(I); R = C2H5, CH3, H, F, Cl, Br, Ph, and SiH3) were investigated. All calculations (optimized structures and single points) were performed at the PBE-D3/def2-TZVP37,38 level of theory using Gaussian 09 software.39 All structures were optimized and their energies were obtained in gas phase using PBE-D3/def2-TZVP level of theory. The vibrational frequency analysis indicates that the optimized structures at stationary points, obtained at the same theoretical level, correspond to local minima, as evidenced by the absence of imaginary frequencies. NBO analysis calculations were performed to investigate the nature of the
C(tris-NHC)→M3 bonds using the Gaussian 09 at the BP86/def2-TZVP theoretical level.40,41 Energy decomposition analysis (EDA) was conducted to assess the nature of the C(tris-NHC)→M3 bonds at the BP86-D3/TZ2P level of theory, utilizing the ADF 2013.01 software package.
Result and discussion
Structural studies
The mentioned complexes optimized on PBE-D3/def2TZVP level of theory in the gas phase. The optimized structures and length of the corresponding of [L2(R)6→M3]3+ complexes; M = Cu(I), Ag(I), Au(I); R = C2H5 and CH3 are shown in Fig. 3. The optimized structure of the other related complexes has been reported in the Tables S3 and S4.†
 |
| | Fig. 3 The optimized structures and length of [L2(R)6→M3]3 complexes; M = Cu(I), Ag(I), Au(I); R = C2H5 and CH3. All hydrogen atoms were removed to enhance clarity. | |
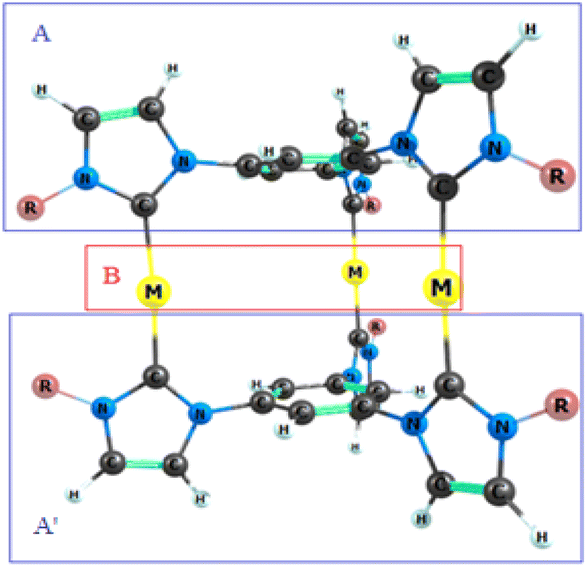
To measure the length of complexes, [L2(R)6→M3]3+; M = Cu(I), Ag(I), Au(I); R = H, F, Cl, Br, CH3 and SiH3, the distance between two atoms H, F, Cl, Br, C, and Si located on reciprocal R's was calculated. Similarly, for complexes [L2(R)6→M3]3+; M = Cu(I), Ag(I), Au(I); R = C2H5 and Ph, the distance between two distant carbon atoms located on R was measured.
Structural data confirmed that this series of complexes has a nanostructure sized (see Fig. 3–5). All coordination details of the Au(I); R = C2H5, CH3, H, F, Cl, Br, Ph, and SiH3, were 1.90 Å for information. Optimized complexes mentioned are provided in the supporting copper complexes and 2.08 Å for silver complexes. Also, C(tris-NHC)→M3 bond lengths for the gold complexes are approximately 2.03 Å. According to the results, a reverse V-shaped trend is observed. With the change in substituents, no significant changes are observed. Due to the symmetrical nature of the studied complexes, the bond length of each of the six C(tris-NHC)→M3 bonds in each complex remains the same. Therefore, only one bond length for the C(tris-NHC)→M3 bond is reported. The bond lengths of the C(tris-NHC)→M3 bonds in the mentioned complexes are listed in the ESI, Table S1.†
 |
| | Fig. 4 Fragments analysis of [L2(R)6→M3]3+ complexes investigated here. | |
 |
| | Fig. 5 Contour plots of NOCV deformation densities Δρ and associated energies ΔE(ρ), computed at the BP86-D3/TZ2P level of theory. The corresponding deformation electron densities were depicted by the direction of charge transitioning from red to blue. The eigenvalues (ν) give the size of the charge migration. | |
Interaction energy
The interaction energies between the examined M33+ ions and the ligand fragments in the optimized structures of the nano-sized complexes were calculated using the PBE/def2-TZVP level of theory. The results are shown in Table 2. To calculate the interaction energy in the complexes [L2(R)6→M3]3+, three fragments A, A′, and B were defined as follows Fig. 4. In this study, the interaction energy between fragments A and B, as well as A and A′B, was investigated. Single point (SP) energy of all fragments, (A, B, A′, AB and A′B) were calculated using the PBE/def2-TZVP level of theory. Due to the symmetry of the complex, the interaction energy was analysed for only one side of the complex. The interaction energy was measured according to the following equations.42 The total interaction energy was calculated using two formulas, eqn (1) and eqn (2). The total interaction energies obtained from both formulas are same.| |
 | (1) |
| | |
IEBTotal = EABC − (EABCA + EABCB + EABCC)
| (2) |
Table 2 Interaction energy (IE) (kcal mol−1) of [L2(R)6→M3]3+ complexes
| M |
R |
IEABA′A−B |
IEABA′B−A′ |
IEABA′A−BA′ |
IEABA'AB−A′ |
IEtotal(eqn (1)) |
IEtotal(eqn (2)) |
| Cu |
C2H5 |
−360.60 |
−360.60 |
−272.42 |
−272.42 |
−633.02 |
−633.02 |
| CH3 |
−353.73 |
−353.73 |
−270.31 |
−270.31 |
−624.04 |
−624.04 |
| H |
−341.57 |
−341.56 |
−267.69 |
−267.68 |
−609.25 |
−609.25 |
| F |
−311.61 |
−311.62 |
−251.54 |
−251.55 |
−563.16 |
−563.16 |
| Cl |
−330.02 |
−330.02 |
−259.21 |
−259.21 |
−589.23 |
−589.23 |
| Br |
−336.10 |
−336.10 |
−261.11 |
−261.11 |
−598.11 |
−598.11 |
| SiH3 |
−358.89 |
−358.89 |
−271.66 |
−271.66 |
−630.55 |
−630.55 |
| Ph |
−373.52 |
−373.09 |
−277.89 |
−277.46 |
−650.98 |
−650.98 |
| Ag |
C2H5 |
−318.43 |
−318.43 |
−244.87 |
−244.87 |
−563.30 |
−563.30 |
| CH3 |
−308.74 |
−308.74 |
−240.28 |
−240.27 |
−549.01 |
−549.01 |
| H |
−305.49 |
−305.49 |
−247.25 |
−247.25 |
−552.74 |
−552.74 |
| F |
−268.23 |
−268.23 |
−222.27 |
−222.27 |
−490.50 |
−490.50 |
| Cl |
−289.92 |
−289.93 |
−232.30 |
−232.32 |
−522.24 |
−522.24 |
| Br |
−293.81 |
−293.80 |
−232.19 |
−232.18 |
−525.99 |
−525.99 |
| SiH3 |
−312.10 |
−312.10 |
−242.03 |
−242.02 |
−554.12 |
−554.12 |
| Ph |
−328.82 |
−328.82 |
−246.73 |
−246.73 |
−575.55 |
−575.55 |
| Au |
C2H5 |
−439.04 |
−438.66 |
−304.27 |
−303.89 |
−742.93 |
−742.93 |
| CH3 |
−429.20 |
−429.21 |
−301.16 |
−301.17 |
−730.37 |
−730.37 |
| H |
−415.94 |
−415.95 |
−300.52 |
−300.53 |
−716.48 |
−716.48 |
| F |
−380.23 |
−380.23 |
−284.79 |
−284.79 |
−665.02 |
−665.02 |
| Cl |
−402.31 |
−402.32 |
−291.90 |
−291.90 |
−694.21 |
−694.21 |
| Br |
−411.16 |
−411.16 |
−293.81 |
−293.81 |
−704.97 |
−704.97 |
| SiH3 |
−434.76 |
−434.79 |
−303.19 |
−303.22 |
−737.98 |
−737.98 |
| Ph |
−449.84 |
−449.30 |
−307.12 |
−306.57 |
−756.41 |
−756.41 |
Results indicated that the interaction energy values for Cu(I), Ag(I) Au(I); in [L2(R)6→M3]3+ complexes were in the ranges of (−563.16 to −650.98) kcal mol−1, (−490.50 to −575.55) kcal mol−1 and (−665.02 to −756.41) kcal mol−1 respectively. Assuming R is constant, the trend of total interaction energy for group 11 metals is V-shaped, IE Au(1) > IE Cu(1) > IE Ag(1). For example, the total interaction energy for the complexes [L2(C2H5)6→Au3]3+, [L2(RC2H5)6→Ag3]3+ and [L2(C2H5)6→Cu3]3+ were −742.93 kcal mol−1, −563.30 kcal mol−1 and −633.02 kcal mol−1, respectively. Changing the substituent on the NHC rings alters the interaction energy. The total interaction energies for electron-donating substituents, such as Ph, C2H5, and CH3, are higher than those for electron-withdrawing substituents such as F, Cl, and Br. This trend was observed across all three metal clusters investigated. Additionally, the highest and lowest total interaction energies were associated with the complexes [L2(Ph)6→Au3]3+, which has an interaction energy of −756.41 kcal mol−1, and [L2(F)6→Ag3]3+, which has an interaction energy of −490.50 kcal mol−1.
Cooperativity
In 1957 Frank and Wen43 qualitatively described the presence of cooperative effects among hydrogen bonds as a nonadditive enhancement of interactions through the formation of additional bonds. Significant attention has been focused on cooperativity within a triad formed by noncovalent interactions among three connected monomers44 Hankins45 and colleagues proposed a characteristic feature of the cooperative effect in a triadic configuration of H2O molecules, treating it as a noncyclic A–B–C system based on the concept of “pairwise nonadditivity” represented by the following equation: eqn (3)| | |
Ecoop = SEABC − SEAB − SEBC – SE(AC, T)
| (3) |
SEABC denotes the stabilization energy of the triad, while SEAB and SEBC represent the stabilization energies of the isolated dyads within their respective minima configurations. The term SE(AC, T) also signifies the stabilization energy of molecules A and C in the triad configuration. However, while several theoretical studies46–51 have calculated the cooperative energy using eqn (3) and other studies have employed eqn (4), which does not take SE(AC, T) into account.52–56
| | |
Ecoop = SEABC − SEAB − SEBC
| (4) |
Cooperativity refers to the phenomenon where multiple interactions within a system influence each other, causing the system to behave differently than would be expected from the individual interactions acting alone. This coupling can result in either positive cooperativity (where one interaction enhances another) or negative cooperativity (where one interaction reduces another). It is a fundamental aspect of systems chemistry that produces collective properties not found in the individual components.57 One example of cooperativity is hemoglobin, where the binding of oxygen to one site increases the affinity of the other sites for oxygen.58 In 2024, Sanei Movafagh and co-workers introduced a new equation eqn (5) for calculating cooperativity based on bond interaction energy in three-component systems.55
| |
 | (5) |
The calculated values of the cooperativity energy based on eqn (5) for the nano-sized complexes [L2(R)6→M3]3+; M = Cu(I), Ag(I), Au(I) and R = C2H5, CH3, H, F, Cl, Br, Ph, and SiH3) are summarized in Table 3. Based on the results, the cooperativity values for all complexes are positive, indicating the presence of anti-cooperativity. The trend of cooperativity for group 11 metals, similar to the interaction energy, exhibits a V-shaped pattern and follows the order, Ag(I) < Cu(I) < Au(I). For example, cooperativity energy for the complexes [L2(F)6→Cu3]3+, [L2(F)6→Ag3]3+ and [L2(F)6→Au3]3+ were 73.88 kcal mol−1, 58.24 kcal mol−1 and 115.42 kcal mol−1, respectively.
Table 3 Cooperativity energy (kcal mol−1) of [L2(R)6→M3]3+ complexes
| M |
R |
IEABA′A−B |
IEABA′A−BA′ |
Ecoop |
| Cu |
C2H5 |
−360.60 |
−272.42 |
88.18 |
| |
CH3 |
−353.73 |
−270.31 |
83.42 |
| |
H |
−341.57 |
−267.69 |
73.88 |
| |
F |
−311.61 |
−251.54 |
60.07 |
| |
Cl |
−330.02 |
−259.21 |
70.81 |
| |
Br |
−336.1 |
−261.11 |
74.99 |
| |
SiH3 |
−358.89 |
−271.66 |
87.23 |
| |
Ph |
−373.52 |
−277.89 |
95.63 |
| Ag |
C2H5 |
−318.43 |
−244.87 |
73.56 |
| |
CH3 |
−308.74 |
−240.28 |
68.46 |
| |
H |
−305.49 |
−247.25 |
58.24 |
| |
F |
−268.23 |
−222.27 |
45.96 |
| |
Cl |
−289.92 |
−232.30 |
57.62 |
| |
Br |
−293.81 |
−232.19 |
61.62 |
| |
SiH3 |
−312.1 |
−242.03 |
70.07 |
| |
Ph |
−328.82 |
−246.73 |
82.09 |
| Au |
C2H5 |
−439.04 |
−304.27 |
134.77 |
| |
CH3 |
−429.2 |
−301.16 |
128.04 |
| |
H |
−415.94 |
−300.52 |
115.42 |
| |
F |
−380.23 |
−284.79 |
95.44 |
| |
Cl |
−402.31 |
−291.9 |
110.41 |
| |
Br |
−411.16 |
−293.81 |
117.35 |
| |
SiH3 |
−434.76 |
−303.19 |
131.57 |
| |
Ph |
−449.84 |
−307.12 |
142.72 |
NBO analysis
In this study, we used Gaussian 09 software for NBO analysis. NBO analysis was utilized to quantify various bond parameters and to characterize the metal–ligand interactions. Based on NBO calculation at the PBE-D3/def2-TZVP level of theory, the nature of the C(tris-NHC)→M3 bonds in the mentioned nano-sized complexes were analysed.
Natural charge and charge transfer
The natural charge on M3 atoms of the complexes [L2(R)6→M3]3+; M = Cu(I), Ag(I), Au(I); R = C2H5, CH3, H, F, Cl, Br, Ph, and SiH3 at the PBE-D3/def2-TZVP level of theory were collected. The results were shown in Table 4. In the complexes [L2(R)6→M3]3+, the formal charges of the M3 and L2(R6) fragments are +3 and 0, respectively. The results indicate that charge transfer occurs from the L2(R)6 fragment to the M3. While the R substituent is constant, a decreasing trend in the natural charge was observed by changing the M from Cu(I) to Au(I). For example, the natural charges of the M3 metal cluster in the complexes [L2(Br)6→Cu3]3+, [L2(Br)6→Ag3]3+ and [L2(Br)6→Au3]3+ were found to be 1.17e, 1.04e, and 0.65e, respectively. Additionally, the natural charge of the M3 metal cluster is generally higher in the presence of electron-withdrawing substituents compared to electron-donating substituents. For example, the natural charges of complexes [L2(F)6→Ag3]3+ and [L2(C2H5)6→Ag3]3+ were 1.21 e and 1.04 respectively. The highest and lowest natural charges of the mentioned complexes correspond to the [L2(F)6→Cu3]3+ and [L2(C2H5)6→Au3]3+, with values of 1.25 e and 0.61 e, respectively. The amount of charge transfer from L2(R6) fragment to M3 was also calculated. The results indicate an increasing trend in charge transfer when changing the M3 from Cu(I) to Au(I) while keeping the same R substituent. For instance, the charge transfer amounts of [L2(Br)6→Cu3]3+, [L2(Br)6→Ag3]3+, and [L2(Br)6→Au3]3+ complexes are −1.83e, −1.96e and −2.35e, respectively.
Table 4 Natural charge and the amount of charge transfer L2(R)6→M3 for [L2(R)6→M3]3+ complexes
| M |
R |
Natural charge |
Charge transfer |
| M3 |
L2(R)6 |
L2(R)6→Cu3 |
| Cu |
C2H5 |
1.09 |
1.91 |
−1.91 |
| CH3 |
1.09 |
1.91 |
−1.91 |
| H |
1.14 |
1.86 |
−1.86 |
| F |
1.25 |
1.75 |
−1.75 |
| Cl |
1.20 |
1.80 |
−1.80 |
| Br |
1.17 |
1.83 |
−1.83 |
| SiH3 |
1.15 |
1.85 |
−1.85 |
| Ph |
1.16 |
1.84 |
−1.84 |
| Ag |
C2H5 |
1.05 |
1.95 |
−1.95 |
| CH3 |
1.05 |
1.95 |
−1.95 |
| H |
1.11 |
1.89 |
−1.89 |
| F |
1.21 |
1.79 |
−1.79 |
| Cl |
1.10 |
1.90 |
−1.90 |
| Br |
1.04 |
1.96 |
−1.96 |
| SiH3 |
1.09 |
1.91 |
−1.91 |
| Ph |
1.09 |
1.91 |
−1.91 |
| Au |
C2H5 |
0.61 |
2.39 |
−2.39 |
| CH3 |
0.66 |
2.34 |
−2.34 |
| H |
0.75 |
2.25 |
−2.25 |
| F |
0.82 |
2.18 |
−2.18 |
| Cl |
0.71 |
2.29 |
−2.29 |
| Br |
0.65 |
2.35 |
−2.35 |
| SiH3 |
0.68 |
2.32 |
−2.32 |
| Ph |
0.67 |
2.33 |
−2.33 |
The results confirm that changing the metal from Cu(I) to Au(I) leads to a reduction in the natural charge on the M3. Also the complexes with electron-donating substituents exhibit greater amounts of charge transfer compared to those with electron-withdrawing substituents. Specifically, the highest and lowest charge transfer values correspond to [L2(C2H5)6→Au3]3+ and [L2(F)6→Cu3]3+ complexes which have charge transfers about −2.39e and −1.75e, respectively.
Wiberg index
Using the Wiberg bond index (WBI) method, the chemical bond orders of C(tris-NHC)→M3 in [L2(R)6→M3]3+ complexes; M = Cu(I), Ag(I), Au(I) and R = C2H5, CH3, H, F, Cl, Br, Ph and SiH3 are investigated. The results are summarized in Table S2 in the ESI.† Due to the symmetrical nature of the studied complexes, the bond order for each of the six C(tris-NHC)→M3 bonds are same. Therefore, only one bond order for the C(tris-NHC)→M3 bond is reported. By changing the M3 from Cu(I) to Au(I) while keeping the same R substituents, a well-known V-shaped trend emerges for the Wiberg Bond Index (WBI) values of the C(tris-NHC)→M3 bonds. This trend follows the order: Ag(I) < Cu(I) < Au(I), which correlates well with the interaction energies. The results indicate that varying the substituents (R) does not affect the Wiberg index for the corresponding complexes.
Donor–acceptor and natural hybrid orbital (NHO) analysis
The results of the natural hybrid orbital (NHO) analysis for the M and C atoms in the C(tris-NHC)→M3 bond within the [L2(R)6→M3]3+ complexes; M = Cu(I), Ag(I), Au(I); R = C2H5, CH3, H, F, Cl, Br, Ph and SiH3 are reported in Table 5. Apart from that, the occupancy of C atom in the latter bonds was ≃73% in the presence of Au(I). For silver and copper, no occupancy of the C atoms in the latter bonds was observed. The modification of the R substituent does not significantly affect the occupancy values of carbon atoms in the C(tris-NHC)→M3 bond. The calculated donor–acceptor interactions for the investigated complexes are given in Table S3.† The results of indicate that the σ* and Lp* orbitals of M metal ion in the C(tris-NHC)→M3 bonds are filled with the lone-pair electrons from the carbon atoms. The most significant and strong donor–acceptor interactions for Cu(I) and Ag(I) are observed as CNHC → Cu(I) and CNHC → Ag(I) transitions, which occur from the lone pair (LP) to the lone pair antibonding orbital (LP*). In the case of Au(I), the strongest interaction is from CNHC → Au(I)–CNHC, transitioning from the lone pair (LP) to the sigma antibonding orbital (σ*). These interactions highlight the varying nature of bonding in the metal complexes depending on the metal involved.
Table 5 Natural hybrid orbital (NHO) analysis of [L2(R)6→M3]3+ nano-sized complexes
| Occupancy |
C2H5 |
CH3 |
H |
F |
Cl |
Br |
SiH3 |
Ph |
| Au |
C |
Au |
C |
Au |
C |
Au |
C |
Au |
C |
Au |
C |
Au |
C |
Au |
C |
| Occupancy Au–C |
1.96140 |
1.96240 |
1.96259 |
1.95893 |
1.96252 |
1.96212 |
1.95799 |
1.96057 |
| % |
26.98 |
73.02 |
27.17 |
72.83 |
27.03 |
72.97 |
26.16 |
73.84 |
26.42 |
73.58 |
26.51 |
73.49 |
27.08 |
72.92 |
26.54 |
73.46 |
| % S |
75.37 |
38.64 |
75.14 |
38.62 |
75.02 |
38.38 |
75.80 |
39.41 |
75.56 |
39.28 |
75.43 |
39.06 |
75.39 |
38.53 |
75.54 |
39.61 |
| % p |
4.37 |
61.33 |
4.34 |
61.35 |
3.80 |
61.59 |
3.90 |
60.57 |
4.13 |
60.69 |
4.24 |
60.91 |
4.32 |
61.44 |
4.62 |
60.36 |
| % d |
20.22 |
0.01 |
20.48 |
0.01 |
21.14 |
0.01 |
20.26 |
0.01 |
20.28 |
0.01 |
20.28 |
0.01 |
20.24 |
0.02 |
19.79 |
0.01 |
| % f |
0.04 |
0.01 |
0.03 |
0.02 |
0.04 |
0.02 |
0.04 |
0.02 |
0.04 |
0.02 |
0.04 |
0.02 |
0.05 |
0.02 |
0.05 |
0.01 |
Energy decomposition analysis (EDA)
Energy decomposition analysis (EDA) is considered fundamental tool for obtaining information about the driving forces in molecular structure and for quantitatively interpreting chemical bonds. EDA calculations were performed based on the DFT method using the BP86-D3 functional and the TZ2P basis set on the relevant nano-sized complexes, utilizing ADF 2013 software. Four parameters are examined in the EDA calculations. The first parameter, ΔEpauli, corresponds to the repulsive interactions between fragments, based on the fact that two electrons with similar spins cannot occupy the same region in space, which typically results in positive values. The second parameter, ΔEelstat, determines the electrostatic interaction energy between the fragments, calculated using the frozen electron density distribution of the fragments in the molecular structure. The third parameter, ΔEorb, includes covalent attraction in the M-L bonds. The fourth parameter, ΔEdis, represents the change in energy due to dispersion interactions in a molecular system, arising from the attractive forces between temporary dipoles that form in molecules. These calculations were conducted to analyze the nature of the C(tris-NHC)→M3 bond in the complexes [L2(R)6→M3]3+; (M = Cu(I), Ag(I), Au(I) and R = C2H5, CH3, H, F, Cl, Br, Ph and SiH3). The total interaction energy was calculated using the following equation eqn (6)| | |
ΔEint = ΔEelstat + ΔEorb + ΔEPauli +ΔEdisp
| (6) |
Three fragments A, A′ and B have been previously defined for the complexes [L2(R)6→M3]3+. In this study, the interaction energies for the fragments A–B and A–BA′ in the complexes, [L2(R)6→M3]3+; M = Cu(I), Ag(I), Au(I) and R = C2H5, CH3, H, F, Cl, Br, Ph and SiH3, were calculated using the ADF 2013 software at the BP86-D3, TZ2P theoretical level. Optimized output files were used to create the input files for ADF 2013. The results of the energy decomposition analysis (EDA) for the mentioned complexes are presented in Table 6. Assuming that the M3 remains unchanged, the interaction energy increases as the substituents change from electron-withdrawing to electron-donating. For example, the interaction energy of the [L2(F)6→Au3]3+ and [L2(Ph)6→Au3]3+ complexes, for the fragments A–B and A–BA′ were (−461.36, −387.17) and (−371.51, −291.15) in kcal mol−1, respectively. However, if the substituent R remains unchanged, the interaction energy exhibits a V-shaped trend, following the order Ag(I) < Cu(I) < Au(I). For instance, the interaction energy of, the [L2(C2H5)6→Au3]3+, [L2(C2H5)6→Ag3]3+ and [L2(C2H5)6→Cu3]3+ complexes for the fragments A–B and A–BA′ were (−460.90, −313.57), (−317.88, −248.38) and (−394.96, −290.59) in kcal mol−1, respectively. On the other hand, the highest and lowest interaction energies for A–B fragments in the complexes investigated here correspond to the [L2(Ph)6→Au3]3+ and [L2(F)6→Ag3]3+ complexes and those for A–BA′ fragments are (−461.26, −317.51) and (−267.55, −225.16) in kcal mol−1, respectively. The computational results align with the interaction energies calculated using Gaussian, at the PBE-D3/def2-TZVP theoretical level. The EDA results indicated that among the three energy decomposition terms in the mentioned complexes. ΔEelstat is the most significant energy term. The values of ΔEelstat for the A–B fragment in the respective complexes of Cu(I), Ag(I) and Au(I) were observed in the ranges of (61.23–65.08) %, (63.78–67.72) %, and (61.53–64.33) %, respectively and for the fragment A–BA′ in ranges of (64.92–69.25) %, (67.71–71.93) % and (67.03–70.17) %, respectively. On the other hand the value of ΔEorb in [L2(R)6→Cu3]3+, [L2(R)6→Ag3]3+ and [L2(R)6→Au3]3+ complexes for fragment A–B were (32.87–36.48)%, (30.05–33.14)% and (33.88–36.45)%, respectively, and for the fragment A–BA′ were (27.25–30.53)%, (23.81–28.38)% and (27.23–30.06)% respectively. According to the obtained results, the highest and lowest values of the ΔEelstat in the studied complexes for fragment A–B correspond to complexes [L2(H)6→Ag3]3+, (67.72%) and [L2(Ph)6→Cu3]3+ (61.23%), respectively and for fragment A–BA and those correspond to complexes [L2(Ph)6→Ag3]3+, (71.93%) and [L2(Ph)6→Cu3]3+, (64.92%), respectively. The results indicated that the highest and lowest values of ΔEorb for the A–B fragment in the aforesaid complexes correspond to complex [L2(Br)6→Cu3]3+ with)36.48% (and complex [L2(H)6→Ag3]3 with) 30.05% (, respectively. For the A–BA′ fragment, the highest and lowest values of ΔEorb correspond to complex [L2(F)6→Cu3]3+ with (30.53%) and complex [L2(Ph)6→Ag3]3+ with (23.81%), respectively. The results of the energy decomposition analysis showed that in all studied complexes, the ΔEelstat accounted for the largest share, indicating that the interaction between the fragments is predominantly electrostatic.
Table 6 Energy decomposition analysis (EDA), (kcal mol−1) of [L2(R)6→M3]3+ nano-sized complexes
| M |
R |
Fragment |
ΔEPauli |
ΔEelstat |
ΔEorb |
ΔEDis |
ΔEint |
| |
C2H5 |
A–B |
332.9 |
−462.6(63.6)% |
−246.9(33.9)% |
−18.4(2.6)% |
−395.0 |
| |
C2H5 |
A–BA′ |
319.3 |
−413.7(67.8)% |
−167.6(27.5)% |
−28.6(4.7)% |
−290.6 |
| |
CH3 |
A–B |
328.3 |
−458.7(64.9)% |
−239.1(33.9)% |
−8.3(1.1)% |
−378.1 |
| |
CH3 |
A–BA′ |
315.4 |
−412.0(68.3)% |
−165.8(27.5)% |
−25.8(4.2)% |
−288.2 |
| |
H |
A–B |
326.6 |
−454.7(65.1)% |
−229.6(32.9)% |
−14.4(2.0)% |
−372.1 |
| |
H |
A–BA′ |
305.9 |
−408.2(69.3)% |
−160.6(27.2)% |
−20.6(3.5)% |
−283.6 |
| Cu |
F |
A–B |
300.6 |
−396.2(61.8)% |
−231.5(36.1)% |
−13.7(2.1)% |
−340.7 |
| |
F |
A–BA′ |
275.2 |
−357.5(65.9)% |
−165.5(30.5)% |
−19.1(3.6)% |
−266.9 |
| |
Cl |
A–B |
312.8 |
−416.8(61.9)% |
−241.8(35.9)% |
−14.9(2.2)% |
−360.8 |
| |
Cl |
A–BA′ |
290.3 |
−375.2(66.3)% |
−168.5(29.8)% |
−22.4(3.9)% |
−275.6 |
| |
Br |
A–B |
314.4 |
−419.3(61.3)% |
−249.2(36.5)% |
−15.4(2.2)% |
−369.5 |
| |
Br |
A–BA′ |
294.0 |
−377.2(65.9)% |
−171.3(29.9)% |
−24.00(4.2)% |
−278.5 |
| |
SiH3 |
A–B |
334.3 |
−459.4(63.1)% |
−249.0(34.2)% |
−19.7(2.7)% |
−393.7 |
| |
SiH3 |
A–BA′ |
322.9 |
−413.4(67.7)% |
−169.7(27.8)% |
−27.5(4.5)% |
−287.8 |
| |
Ph |
A–B |
330.2 |
−451.9(61.2)% |
−266.0(36.1)% |
−20.1(2.7)% |
−407.8 |
| |
Ph |
A–BA′ |
326.1 |
−404.6(64.9)% |
−177.1(28.4)% |
−41.5(6.7)% |
−297.2 |
| |
C2H5 |
A–B |
366.8 |
−454.30(66.4)% |
−212.6(31.0)% |
−17.8(2.6)% |
−317.9 |
| |
C2H5 |
A–BA′ |
349.2 |
−419.6(70.2)% |
−153.6(25.7)% |
−24.4(4.1)% |
−248.4 |
| |
CH3 |
A–B |
363.4 |
−449.0(66.8)% |
−206.8(30.8)% |
−16.4(2.4)% |
−308.7 |
| |
CH3 |
A–BA′ |
345.4 |
−416.6(70.6)% |
−151.7(25.7)% |
−21.8(3.7)% |
−244.8 |
| |
H |
A–B |
353.3 |
−439.4(67.7)% |
−195.0(30.1)% |
−14.4(2.2)% |
−295.6 |
| |
H |
A–BA′ |
332.4 |
−408.1(71.2)% |
−146.9(25.6)% |
−18.3(3.2)% |
−240.9 |
| Ag |
F |
A–B |
328.1 |
−385.4(64.7)% |
−195.3(32.8)% |
−115.0(2.5)% |
−267.6 |
| |
F |
A–BA′ |
304.6 |
−360.8(68.1)% |
−150.4(28.4)% |
−18.6(3.5)% |
−225.2 |
| |
Cl |
A–B |
344.7 |
−413.8 (65.1)% |
−203.6(32.0)% |
−18.40(2.9)% |
−291.1 |
| |
Cl |
A–BA′ |
320.7 |
−379.2(68.2)% |
−153.5(27.6)% |
−23.2(4.2)% |
−235.4 |
| |
Br |
A–B |
344.4 |
−408.9(63.8)% |
−212.5(33.1)% |
−19.7(3.1)% |
−296.8 |
| |
Br |
A–BA′ |
324.1 |
−381.2(67.7)% |
−156.4(27.8)% |
−25.5(4.5)% |
−238.9 |
| |
SiH3 |
A–B |
368.7 |
−450.2(66.0)% |
−212.9(31.2)% |
−19.2(2.8)% |
−313.5 |
| |
SiH3 |
A–BA′ |
351.8 |
−418.4(69.7)% |
−154.9(25.8)% |
−26.7(4.5)% |
−248.3 |
| |
Ph |
A–B |
360.2 |
−442.8(64.1)% |
−228.2(33.0)% |
−20.1(2.9)% |
−330.8 |
| |
Ph |
A–BA′ |
655.5 |
−634.5(71.9)% |
−210.1(23.8)% |
−37.5(4.3)% |
−226.6 |
| |
C2H5 |
A–B |
553.3 |
−633.4(62.2)% |
−347.9(34.2)% |
−36.8(3.6)% |
−460.9 |
| |
C2H5 |
A–BA′ |
526.0 |
−580.3(69.1)% |
−228.8(27.3)% |
−30.5(3.6)% |
−313.6 |
| |
CH3 |
A–B |
547.8 |
−626.5(63.5)% |
−340.4(34.5)% |
−20.0(2.0)% |
−439.1 |
| |
CH3 |
A–BA′ |
519.5 |
−576.9(69.6)% |
−226.7(27.3)% |
−25.6(3.1)% |
−309.8 |
| |
H |
A–B |
546.1 |
−623.8(64.3)% |
−328.5(33.9)% |
−17.4(1.8)% |
−423.6 |
| |
H |
A–BA′ |
509.3 |
−572.7(70.2)% |
−222.3(27.2)% |
−21.3(2.6)% |
−306.9 |
| |
F |
A–B |
507.7 |
−551.4(61.5)% |
−326.5(36.5)% |
−17.9(2.0)% |
−388.2 |
| Au |
F |
A–BA′ |
463.5 |
−506.2(67.0)% |
−226.9(30.1)% |
−21.6(2.9)% |
−291.2 |
| |
Cl |
A–B |
523.4 |
−575.8(61.5)% |
−339.1(36.3)% |
−20.1(2.2)% |
−412.4 |
| |
Cl |
A–BA′ |
488.4 |
−531.7(67.4)% |
−231.1(29.3)% |
−26.1(3.3)% |
−300.5 |
| |
Br |
A–B |
549.6 |
−591.8(61.5)% |
−344.7(35.0)% |
−24.4(2.5)% |
−411.3 |
| |
Br |
A–BA′ |
491.2 |
−533.3(67.1)% |
−233.0(29.2)% |
−29.4(3.7)% |
−304.5 |
| |
SiH3 |
A–B |
555.1 |
−630.4(63.0)% |
−347.9(34.8)% |
−22.6(2.2)% |
−445.9 |
| |
SiH3 |
A–BA′ |
531.4 |
−583.1(69.1)% |
−230.9(27.3)% |
−30.7(3.6)% |
−313.3 |
| |
Ph |
A–B |
545.0 |
−619.2(61.5)% |
−362.6(36.1)% |
−24.4(2.4)% |
−461.3 |
| |
Ph |
A–BA′ |
523.0 |
−564.6(67.2)% |
−234.4(27.9)% |
−41.5(4.9)% |
−317.5 |
EDA-NOCV analysis
NOCV (Natural Orbital for Chemical Valence) provides insights into orbital interactions for asymmetric molecules, as the density of shape changes decomposes the chemical bond into various components (σ, π, δ). The NOCV method indicates the interactions between fragments, which, through energy decomposition calculations, relate to significant orbital interactions between the fragments. Moreover the ΔEorb term can be further analyzed using the NOCV (Natural Orbital for Chemical Valence) extension of the EDA method. The EDA-NOCV approach offers pairwise energy contributions from each set of interacting orbitals to the total bond energy. Δρ refers to the change in electron density between the fragments L2(R)6 and M3+. The eigenvalues (ν) give the size of the charge migration. The NOCV analysis for the two fragments, A–B and A–BA′, was performed, and the results for the [L2(C2H5)6→Au3]3+ complex are presented in Fig. 5. The density of shape between the fragments M3+ and (L2(R)6), along with the significant energy results of the remaining complexes [L2(R)6→M3]3+; M = Cu(I), Ag(I), Au(I) and R = C2H5, CH3, H, F, Cl, Br, Ph and SiH3 for the fragments A–B and A–BA′, is summarized in Fig. S3–S5.† NOCV analysis showed that the types of interactions Δρ1, Δρ2 and Δρ3 present in the C(tris-NHC)→M3 bond in both fragments A–B and A–BA′ were of the σ type, arising from the stable non-bonding electron pairs on the carbon atom. It may be referred to donation from the non-bonding electron pair of carbon towards the empty d orbital of the metal ions.
Conclusion
In this study, the effect of substituents including: C2H5, CH3, H, F, Cl, Br, Ph and SiH3 on the M–C bond in the nano-sized [L2(R)6→M3]3+; [M = Cu(I), Ag(I), Au(I)] complexes was investigated at the PBE-D3/def2-TZVP level of theory. The total interaction energies for all mentioned complexes for the fragments AB and A–BA′ were also calculated. The interaction energy values for both AB and A–BA′ fragments in group 11 metals followed a V-shaped pattern, the trend in energy change observed as (IE Au(I) > IE Cu(I) > IE Ag(I)). The cooperativity energy values for the complexes [L2(R)6→M3]3+; M = Cu(I), Ag(I), Au(I) and R = C2H5, CH3, H, F, Cl, Br, Ph and SiH3 were calculated. The results indicate that all complexes have positive cooperativity values, suggesting anti-cooperativity. The cooperativity trend for Cu(I), Ag(I), Au(I) metals showed a V-shaped pattern, with the order Ag(I) < Cu(I) < Au(I), similar to the trend observed in interaction energies. The natural charge and charge transfer amounts for the complexes [L2(R)6→M3]3+ with the mentioned substituents were calculated, showing that the charge transfer for the corresponding gold complexes is greater than that for silver, and silver is slightly greater than copper. It seems that in most cases, the complexes with electron-donating substituents have slightly more charge transfer compared to those with electron-withdrawing substituents. Energy decomposition analysis was performed for the two fragments AB and A–BA′ in the corresponding complexes. The results showed that in all studied complexes, the ΔEelstat accounted for the largest share, indicating that the interaction between the fragments is predominantly electrostatic. The values of ΔEelstat for fragment A–B in the respective complexes Cu(I), Ag(I) and Au(I) were observed in ranges (61.23–65.08)%, (63.78–67.72)% and (61.53–64.33)% respectively, and for the fragment A–BA′ in ranges (64.92–69.25)%, (67.71–71.93)% and (67.03–70.17)%, respectively.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†
Conflicts of interest
The authors have no conficts of interest to declare.
References
- S. P. Nolan, N-heterocyclic Carbenes in Synthesis, John Wiley & Sons, 2006 Search PubMed.
- D. Bourissou, O. Guerret, F. P. Gabbai and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- F. Lazreg, F. Nahra and C. S. J. Cazin, Coord. Chem. Rev., 2015, 293–294, 48–79 CrossRef CAS.
- F. Wang, L.-j. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804–853 CrossRef CAS.
- T. Zou, C. N. Lok, P. K. Wan, Z. F. Zhang, S. K. Fung and C. M. Che, Curr. Opin. Chem. Biol., 2018, 43, 30–36 CrossRef CAS PubMed.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC.
- L. Oehninger, R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269–3284 RSC.
- W. A. Herrmann, Angew Chem. Int. Ed. Engl., 2002, 41, 1290–1309 CrossRef CAS PubMed.
- L. Boselli, M. Carraz, S. Mazères, L. Paloque, G. González, F. Benoit-Vical, A. Valentin, C. Hemmert and H. Gornitzka, Organometallics, 2015, 34, 1046–1055 CrossRef CAS.
- S. Nayak and S. L. Gaonkar, ChemMedChem, 2021, 16, 1360–1390 CrossRef CAS PubMed.
- M. E. Garner, W. Niu, X. Chen, I. Ghiviriga, K. A. Abboud, W. Tan and A. S. Veige, Dalton Trans., 2015, 44, 1914–1923 RSC.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anticancer Agents Med. Chem., 2009, 9, 185–211 CrossRef CAS PubMed.
- W. R. Hill and D. M. Pillsbury, Argyria : the Pharmacology of Silver, Williams & Wilkins, 1939 Search PubMed.
- S. Medici, M. Peana, G. Crisponi, V. M. Nurchi, J. I. Lachowicz, M. Remelli and M. A. Zoroddu, Coord. Chem. Rev., 2016, 327–328, 349–359 CrossRef CAS.
- P. Kumar and I. Cisarova, J. Organomet. Chem., 2013, 735, 32–37 CrossRef CAS.
- A. Melaiye, R. S. Simons, A. Milsted, F. Pingitore, C. Wesdemiotis, C. A. Tessier and W. J. Youngs, J. Med. Chem., 2004, 47, 973–977 CrossRef CAS PubMed.
- F. Hackenberg, G. Lally, H. Müller-Bunz, F. Paradisi, D. Quaglia, W. Streciwilk and M. Tacke, Inorganica Chim. Acta, 2013, 395, 135–144 CrossRef CAS.
- S. Patil, K. Dietrich, A. Deally, B. Gleeson, H. Müller-Bunz, F. Paradisi and M. Tacke, Helv. Chim. Acta, 2010, 93, 2347–2364 CrossRef CAS.
- V. Gandin, M. Pellei, M. Marinelli, C. Marzano, A. Dolmella, M. Giorgetti and C. Santini, J. Inorg. Biochem., 2013, 129, 135–144 CrossRef CAS PubMed.
- G. J. Higby, Gold Bull., 1982, 15, 130–140 CrossRef CAS PubMed.
- I. C. Shaw, Chem. Rev., 1999, 99, 2589–2600 CrossRef PubMed.
- P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, J. Inorg. Biochem., 2004, 98, 1642–1647 CrossRef CAS PubMed.
- O. Sanchez, S. González, M. Fernández, A. R. Higuera-Padilla, Y. Leon, D. Coll, A. Vidal, P. Taylor, I. Urdanibia, M. C. Goite and W. Castro, Inorganica Chim. Acta, 2015, 437, 143–151 CrossRef CAS.
- R. W. Sun, M. Zhang, D. Li, Z. F. Zhang, H. Cai, M. Li, Y. J. Xian, S. W. Ng and A. S. Wong, Chemistry, 2015, 21, 18534–18538 CrossRef CAS PubMed.
- J. Weaver, S. Gaillard, C. Toye, S. Macpherson, S. P. Nolan and A. Riches, Chemistry, 2011, 17, 6620–6624 CrossRef CAS PubMed.
- P. de Frémont, E. D. Stevens, M. D. Eelman, D. E. Fogg and S. P. Nolan, Organometallics, 2006, 25, 5824–5828 CrossRef.
- C. H. G. Jakob, A. W. Muñoz, J. F. Schlagintweit, V. Weiß, R. M. Reich, S. A. Sieber, J. D. G. Correia and F. E. Kühn, J. Organomet. Chem., 2021, 932 Search PubMed.
- J. Rieb, B. Dominelli, D. Mayer, C. Jandl, J. Drechsel, W. Heydenreuter, S. A. Sieber and F. E. Kuhn, Dalton Trans., 2017, 46, 2722–2735 RSC.
- M. Rezaeivala, M. Ahmadi, B. Captain, M. Bayat, M. Saeidirad, S. Şahin-Bölükbaşı, B. Yıldız and R. W. Gable, Inorganica Chim. Acta, 2020, 513, 119935 CrossRef CAS.
- B. Naderizadeh and M. Bayat, ACS Omega, 2020, 5, 26999–27015 CrossRef CAS PubMed.
- B. Naderizadeh and M. Bayat, Mendeleev Commun., 2021, 31, 179–181 CrossRef CAS.
- B. Naderizadeh, M. Bayat, M. Ranjbaran and S. Salehzadeh, J. Mol. Liq., 2021, 341, 117310 CrossRef CAS.
- B. Naderizadeh and M. Bayat, J. Iran. Chem. Soc., 2024, 1–24 Search PubMed.
- A. Rit, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2010, 132, 4572–4573 CrossRef CAS PubMed.
- A. Rit, T. Pape, A. Hepp and F. E. Hahn, Organometallics, 2010, 30, 334–347 CrossRef.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K.
N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. Bayat, F. Amraie and S. Salehzadeh, J. Theor. Comput. Chem., 2016, 15(4), 1650032 CrossRef.
- S. Salehzadeh and F. Maleki, J. Comput. Chem., 2016, 37, 2799–2807 CrossRef CAS PubMed.
- H. S. Frank and W.-Y. Wen, Faraday Discuss. R. Soc. Chem., 1957, 24, 133–140 RSC.
- I. Alkorta, F. Blanco, P. M. Deyà, J. Elguero, C. Estarellas, A. Frontera and D. Quiñonero, Theor. Chem. Acc., 2009, 126, 1–14 Search PubMed.
- D. Hankins, J. W. Moskowitz and F. H. Stillinger, Chem. Phys. Lett., 1970, 4, 527–530 CrossRef CAS.
- Q. Li, H. Liu, X. An, B. Gong and J. Cheng, J. Mol. Struct. THEOCHEM, 2008, 861, 14–17 CrossRef CAS.
- C. Estarellas, A. Frontera, D. Quinonero, I. Alkorta, P. M. Deya and J. Elguero, J. Phys. Chem. A, 2009, 113, 3266–3273 CrossRef CAS PubMed.
- D. Escudero, C. Estarellas, A. Frontera, D. Quiñonero and P. M. Deyà, Chem. Phys. Lett., 2010, 485, 221–225 CrossRef CAS.
- C. Estarellas, A. Frontera, D. Quinonero and P. M. Deya, ChemPhysChem, 2011, 12, 2742–2750 CrossRef CAS PubMed.
- M. Solimannejad, ChemPhysChem, 2012, 13, 3158–3162 CrossRef CAS PubMed.
- M. Solimannejad, Z. Rezaei and M. D. Esrafili, J. Mol. Model, 2013, 19, 5031–5035 CrossRef CAS PubMed.
- M. D. Esrafili, P. Esmailpour, F. Mohammadian-Sabet and M. Solimannejad, Chem. Phys. Lett., 2013, 588, 47–50 CrossRef CAS.
- B. Gong, B. Jing, Q. Li, Z. Liu, W. Li, J. Cheng, Q. Zheng and J. Sun, Theor. Chem. Acc., 2009, 127, 303–309 Search PubMed.
- X. Guo, Y.-W. Liu, Q.-Z. Li, W.-Z. Li and J.-B. Cheng, Chem. Phys. Lett., 2015, 620, 7–12 CrossRef CAS.
- Q. Li, R. Li, Z. Zhou, W. Li and J. Cheng, J. Chem. Phys., 2012, 136, 014302 CrossRef PubMed.
- W. Wang and Q. Li, Comput. Theor. Chem., 2011, 977, 128–133 CrossRef CAS.
- A. Whitty, Nat. Chem. Biol., 2008, 4, 435–439 CrossRef CAS PubMed.
- M. F. Perutz, Q. Rev. Biophys., 1989, 22, 139–237 CrossRef CAS PubMed.
- S. S. Movafagh and S. Salehzadeh, Phys. Chem. Chem. Phys., 2024, 26, 15005–15017 RSC.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b and
Ehsan Alavi Poura
*b and
Ehsan Alavi Poura