DOI:
10.1039/D4RA08691K
(Paper)
RSC Adv., 2025,
15, 2825-2839
Synthesis, cytotoxicity, apoptosis-inducing activity and molecular docking studies of novel isatin–podophyllotoxin hybrids†
Received
10th December 2024
, Accepted 13th January 2025
First published on 28th January 2025
Abstract
Podophyllotoxin, along with its numerous derivatives and related compounds, is well known for its broad-spectrum pharmacological activity, especially for anticancer potential. In this study, several isatin–podophyllotoxin hybrid compounds were successfully synthesized with good yields through microwave-prompted three-component reactions of 2-amino-1,4-naphthoquinone, various substituted isatins, and tetronic acid. Their cytotoxicity was assessed against four types of human cancer cell lines, HepG2 (hepatoma carcinoma), MCF7 (breast cancer), A549 (non-small lung cancer), and KB (epidermoid carcinoma), alongside nontumorigenic HEK-293 human embryonic kidney cells. Among 14 compounds screened, 7f possessed the strongest cytotoxicity to KB and A549 cell lines, with IC50 values of 1.99 ± 0.22 and 0.90 ± 0.09 μM, respectively. Further studies revealed that product 7f could arrest the cell cycle of A549 cells at S phase and induce apoptosis of A549 cells. This compound was examined for its binding ability against cyclin-dependent kinases (CDKs) and procaspase/caspase systems. The results indicated that 7f exhibited significant interactions with the residues of the ATP binding sites of CDK2/cyclin A and CDK5/p25 and also activated procaspase 6 through stable zinc chelation. Additionally, physicochemical and pharmacokinetic properties related to drug-likeness, in parallel with toxicity, were computationally assessed to identify the main issues that need to be addressed in structural optimization. Taken together, compound 7f was identified as a potent cytotoxic agent that could be considered for anticancer drug discovery and development.
Introduction
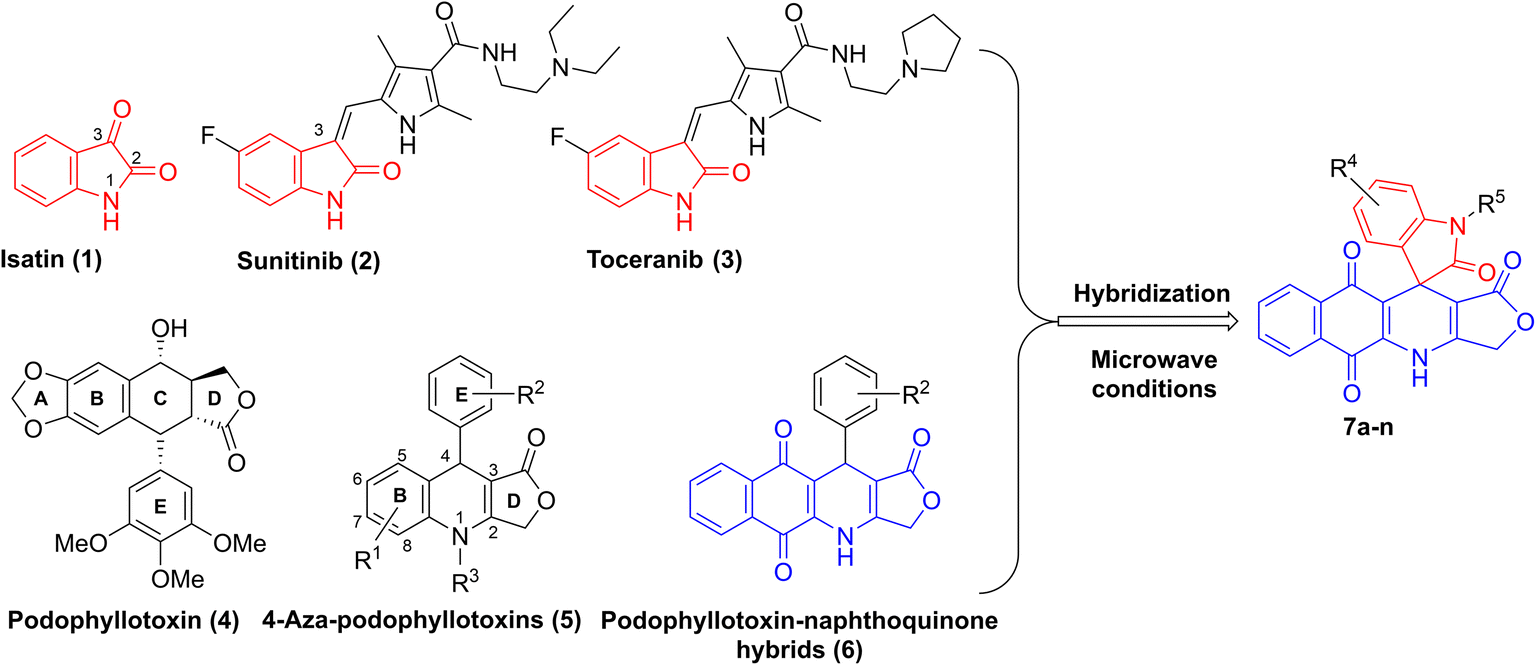
Isatin (1H-indol-2,3-dione, 1) is a natural alkaloid isolated from a diversity of plants such as Boronia koniamboensis, Couroupita guianensis Aubl, Isatis tinctoria, and Melochia tomentosa (Fig. 1).1 In humans, it is found in the central nervous system (CNS), peripheral tissues, as well as body fluids.2–4 As a versatile molecule with unique size and privileged electronic properties, isatin and its derivatives have presented vivid biological and pharmacological properties, including anti-microbial,5 anticancer,6 anti-convulsant,7 anti-inflammatory,8 anti-oxidant,9 and anti-viral activities.10 Isatin derivatives can act on a large number of biological targets, including tyrosine kinase, tubulin, phosphodiesterase 4B, histone deacetylase, and β-carbonic anhydrase, thereby triggering apoptosis.11,12 Several studies have highlighted the potential of developing isatin-based compounds as promising anticancer agents.13–15 Moreover, introducing various types of substituents at N-1, C-3 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and C in the benzene ring of isatin can modulate its biological properties.16 Notably, a huge number of bioactive 3-substituted isatins as potent anticancer agents have been reported in the literature.17–19 For example, sunitinib (2) and toceranib (3), known as isatin derivatives, are multi-receptor tyrosine kinase inhibitors with profound antitumor and antiangiogenic activities in clinical trials (Fig. 1).20
O) and C in the benzene ring of isatin can modulate its biological properties.16 Notably, a huge number of bioactive 3-substituted isatins as potent anticancer agents have been reported in the literature.17–19 For example, sunitinib (2) and toceranib (3), known as isatin derivatives, are multi-receptor tyrosine kinase inhibitors with profound antitumor and antiangiogenic activities in clinical trials (Fig. 1).20
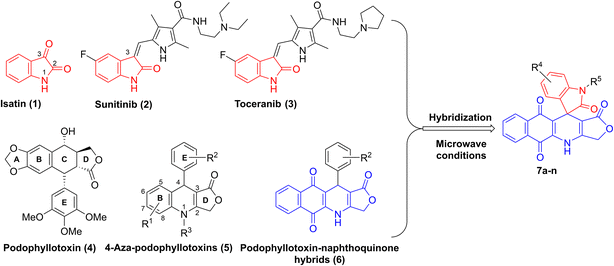 |
| | Fig. 1 Design of compounds 7a–n. | |
On the other hand, podophyllotoxin (4, Fig. 1), a naturally occurring cyclolignan, has been widely used as an anticancer agent due to its disruption of cell mitosis through the inhibition of tubulin polymerization; however, it failed clinical trials because of severe side effects and toxicity.21,22 Recently, 4-aza-podophyllotoxins (5), known as podophyllotoxin derivatives, have attracted much interest owing to their outstanding biological activities, such as their antiproliferative effect on cancer cells, induction of apoptosis and cell cycle arrest in the G2/M phase, destabilization of microtubules, and vascular disruption effects.23–26 Various aza-podophyllotoxins have exhibited activities comparable to podophyllotoxin (4).27 By using multicomponent synthesis, which is a powerful and efficient method to prepare diverse and complex products in a single synthetic step with high selectivity, a wide variety of 4-aza-podophyllotoxin analogs with structural modifications in the B, E and D rings of the scaffold have been synthesized.28–30 For instance, introducing 1,4-naphthoquinone to the B ring and different substituents in the E ring of 4-aza-podophyllotoxin, as well as keeping the intact γ-butylrolactone ring D, have furnished novel naphthoquinone–podophyllotoxin compounds 6 with strong cytotoxic effect on KB, HepG2 and SK-Lu-1 cancer cell lines (Fig. 1).31,32
The approach of merging two pharmacophores to enhance the biological and pharmacological potency of the resulting molecules is commonly employed in pharmaceutical and medicinal chemistry.33 Therefore, we propose that incorporating isatins into a podophyllotoxin–naphthoquinone framework could synergize the molecular targets of both components, leading to the development of novel and promising anticancer agents. To the best of our knowledge, only one study has reported the synthesis of five isatin–podophyllotoxin hybrids via ultrasound-assisted four-component reactions,34 and no research has been conducted on the biological activities of these hybrids. In light of the aforementioned findings and in a continuation of our endeavor with respect to the development of bioactive heterocyclic compounds,35–38 we herein present a new convenient, efficient pathway to synthesize new isatin–podophyllotoxin hybrids 7a–n (Fig. 1) as well as assessment of their cytotoxic activity against different cancer cell lines, namely hepatoma carcinoma (HepG2), breast (MCF7), non-small lung (A549) and epidermoid carcinoma (KB) cancer cell lines, using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Moreover, the most active molecule was further evaluated for its potential to induce apoptosis in the human A549 cancer cell line by conducting cell cycle analysis, apoptosis assay, and molecular docking analysis.
Results and discussion
Chemistry
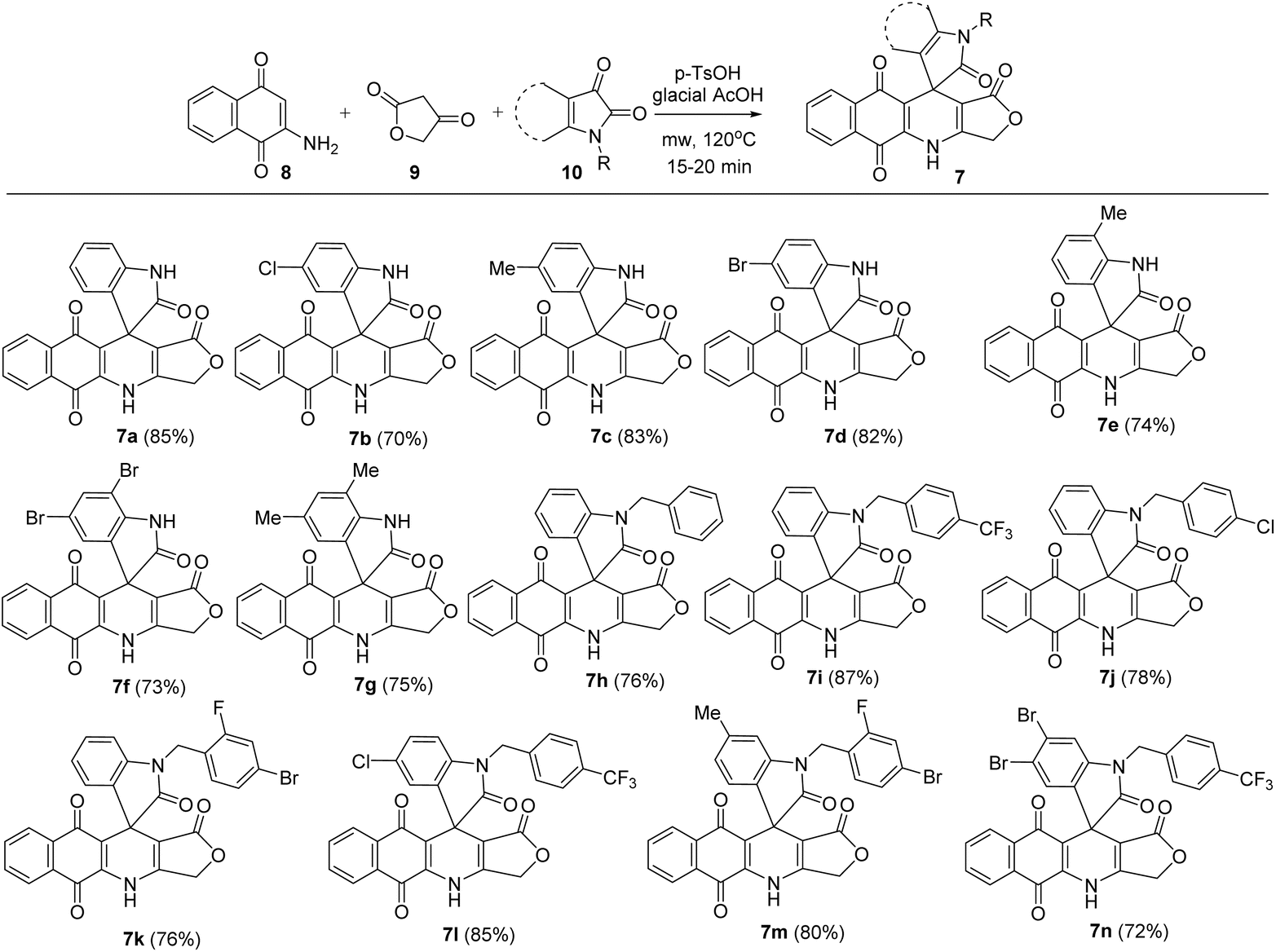
In this study, novel substituted 3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone derivatives (7) have been synthesized via microwave-assisted three-component reactions, in which various substituted isatins 10 (1 mmol) were applied to the mixture of 2-amino-1,4-naphthoquinone 8 (1 mmol) and tetronic acid 9 (1 mmol) in glacial acetic acid as solvent and in the presence of 20 mol% of p-toluenesulfonic acid (p-TsOH) as catalyst. The reactions were carried out under microwave irradiation (MW) at 120 °C for 20 min, resulting in the formation of substituted 3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone derivatives in 70–85% yield (Scheme 1). Substituents attached to the isatin moiety were found to have no noticeable effect on the reaction's outcome. All obtained compounds were analysed using 1H NMR, 13C NMR, MS and IR techniques to confirm their molecular structure. Structure of product 7m was reconfirmed using HSQC and HMBC.
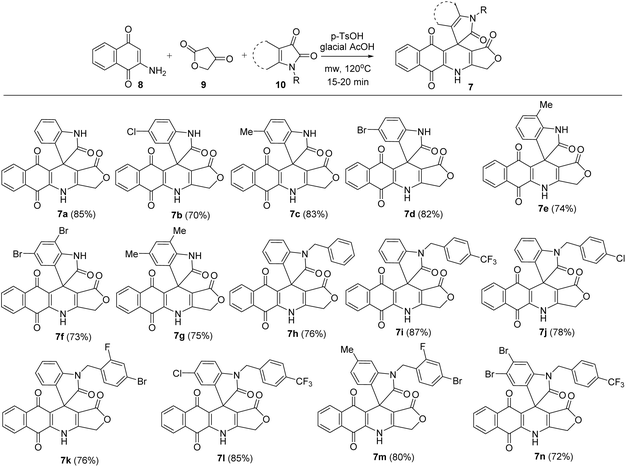 |
| | Scheme 1 Synthesis of isatin–podophyllotoxin hybrids 7a–n. | |
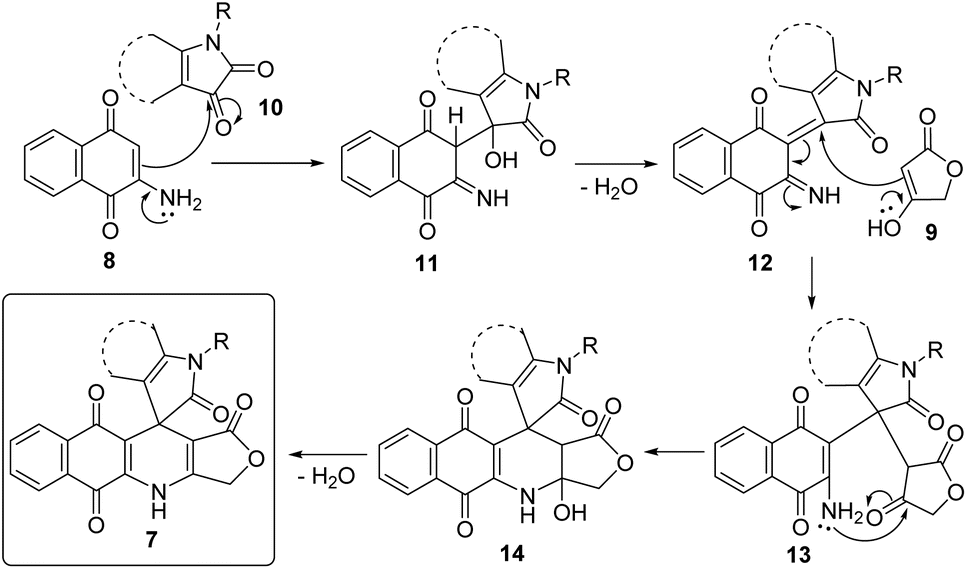
A possible mechanistic explanation for the formation of products 7 is depicted in Scheme 2. Initially, intermediates 11 were built upon the Knoevenagel condensation between 2-amino-1,4-naphthoquinone (8) and substituted isatin 10, followed by dehydration to afford intermediates 12. Subsequent Michael addition of tetronic acid (9) to Michael acceptors 12 furnished intermediates 13. An intramolecular cyclization followed by dehydration of intermediates 13 generated final products 7.
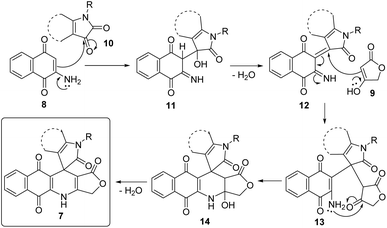 |
| | Scheme 2 The proposed reaction mechanism. | |
Cytotoxicity of isatin–podophyllotoxin hybrids
The next objective of this study was to measure the in vitro cytotoxic activity of obtained products 7a–n toward four types of human cancer cell lines (KB, HepG2, A549 and MCF7). For this objective, products 7 at different concentrations were firstly used to treat human cancer cells. Cytotoxic effects were measured using the MTT colorimetric assay, with ellipticine used as a ref. 39 and 40. The results revealed that all products 7a–n exhibited moderate to strong inhibitory activity against the four tested cancer cell lines, with IC50 values ranging from 0.90 to 51.68 μM. Generally, products 7a–n displayed lower cytotoxicity against HepG2 cells compared with that of ellipticine. Products 7a–d, g–h exhibited potent growth inhibition values against A549 in the micromolar range (IC50 = 1.41–1.98 μM), close to that of the reference ellipticine (IC50 = 1.34 ± 0.08 μM). The inhibitory activities against A549 cells of products 7f (IC50 = 0.90 ± 0.09 μM) and 7n (IC50 = 1.03 ± 0.13 μM) were better than that of ellipticine. In the case of MCF7 cells, products 7a (IC50 = 1.95 ± 0.21 μM), 7d (IC50 = 2.07 ± 0.26 μM), 7f (IC50 = 1.84 ± 0.17 μM), and 7n (IC50 = 2.11 ± 0.24 μM) showed similar or enhanced activity relative to ellipticine (IC50 = 2.07 ± 0.16 μM). Interestingly, product 7f possessed the strongest cytotoxicity to KB cells, with an IC50 value of 1.99 ± 0.22 μM. Moreover, the investigation on the toxicity of products 7a–n on immortalized human embryonic kidney cells (Hek-293) revealed that products 7a–n showed toxicity to Hek-293 (Table 1). Products 7f and 7n exhibited about 7-fold and 22-fold lower cytotoxicity against Hek-293 cells than to A549 cancer cell lines. Product 7n exhibited about 11-fold lower cytotoxicity against Hek-293 cells than to MCF7 cancer cell lines. Based on the cytotoxicity data, it was found that the presence of bromo substituents in the benzene ring of isatin improved the inhibitory activity, as exemplified by products 7d, 7f and 7n. Thus, the introduction of 4-bromo-2-fluorobenzyl substituent at N-1 of isatin produced products 7k and 7m with lower cytotoxic activity.
Table 1 Cytotoxicity of the products against KB, HepG2, MCF7, A549, and Hek-293 cell lines
| Entry |
Compound |
IC50, μM |
| KB |
A549 |
HepG2 |
MCF7 |
Hek-293 |
| 1 |
7a |
5.33 ± 0.62 |
1.98 ± 0.16 |
5.33 ± 0.62 |
1.95 ± 0.21 |
4.89 ± 0.44 |
| 2 |
7b |
3.15 ± 0.21 |
1.41 ± 0.12 |
2.32 ± 0.31 |
4.20 ± 0.50 |
3.32 ± 0.33 |
| 3 |
7c |
3.46 ± 0.33 |
1.91 ± 0.20 |
4.02 ± 0.45 |
4.17 ± 0.48 |
3.89 ± 0.40 |
| 4 |
7d |
4.75 ± 0.41 |
1.62 ± 0.17 |
4.34 ± 0.41 |
2.07 ± 0.26 |
3.52 ± 0.37 |
| 5 |
7e |
10.04 ± 0.93 |
2.28 ± 0.20 |
6.80 ± 0.63 |
18.83 ± 1.71 |
10.04 ± 1.08 |
| 6 |
7f |
1.99 ± 0.22 |
0.90 ± 0.09 |
3.28 ± 0.39 |
1.84 ± 0.17 |
6.70 ± 0.57 |
| 7 |
7g |
4.85 ± 0.41 |
1.67 ± 0.17 |
6.74 ± 0.68 |
2.62 ± 0.22 |
7.27 ± 0.78 |
| 8 |
7h |
3.96 ± 0.34 |
1.71 ± 0.19 |
4.24 ± 0.38 |
3.73 ± 0.38 |
2.72 ± 0.19 |
| 9 |
7i |
4.65 ± 0.44 |
7.37 ± 0.83 |
4.50 ± 0.48 |
3.72 ± 0.35 |
4.04 ± 0.42 |
| 10 |
7j |
5.60 ± 0.49 |
6.54 ± 0.69 |
5.78 ± 0.61 |
4.09 ± 0.41 |
10.34 ± 1.06 |
| 11 |
7k |
27.30 ± 2.15 |
14.00 ± 1.59 |
20.90 ± 2.36 |
15.09 ± 1.59 |
>28.00 |
| 12 |
7l |
17.75 ± 1.68 |
5.93 ± 0.64 |
15.05 ± 1.70 |
13.40 ± 1.23 |
>27.73 |
| 13 |
7m |
51.68 ± 1.78 |
13.28 ± 0.70 |
47.50 ± 2.15 |
19.85 ± 0.89 |
>64.96 |
| 14 |
7n |
4.36 ± 0.40 |
1.03 ± 0.13 |
6.74 ± 0.73 |
2.11 ± 0.24 |
22.85 ± 2.20 |
| 15 |
Ellipticine |
1.99 ± 0.16 |
1.34 ± 0.08 |
2.07 ± 0.12 |
2.07 ± 0.16 |
5.64 ± 0.32 |
Among all products, product 7f displayed the most potent activity against A549 cancer cell line, making it the selected compound for subsequent cell cycle analysis, apoptosis assay, and molecular docking studies.
Product 7f induced cell cycle S phase arrest in A549 cells
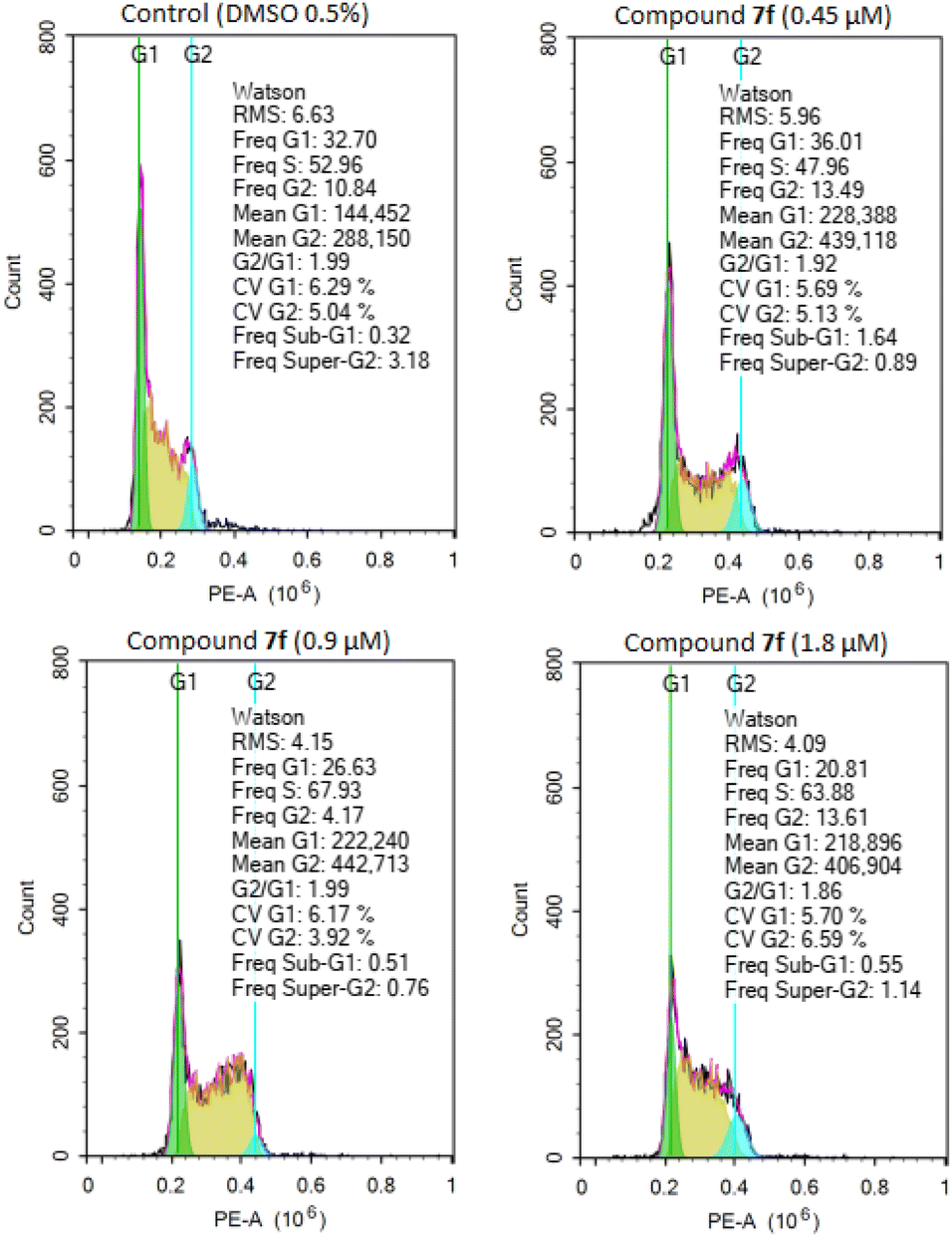
To determine whether the inhibition of cancer cell growth by product 7f was associated with cell cycle arrest, A549 cells were treated with different concentrations of product 7f (0.45 μM, 0.9 μM and 1.8 μM) for 24 hours. The distributions of cells in populations based on distinct phases including G0/G1 phase, S phase (synthesis), and G2/M phase (gap 2/mitosis) were analyzed by flow cytometry in cells stained with propidium iodide.32,41 As shown in Fig. 2 and Table 2, treatment with product 7f caused a significant rise of cell accumulation in S phase, accompanied by a decrease of cells in G0/G1 phase. These results suggested that the tested compound could dose-dependently arrest the cell cycle of A549 cells at S phase, thereby inhibiting cancer cell growth.
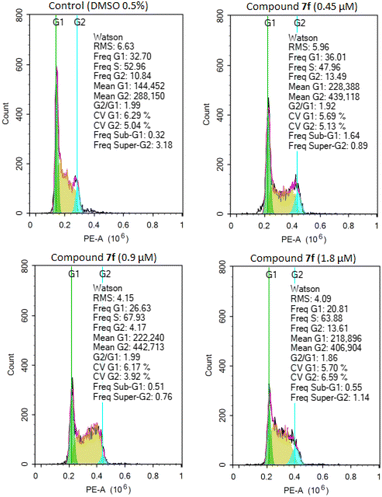 |
| | Fig. 2 Effect of compound 7f on cell cycle distribution in A549 cells. Cells were treated with different concentrations of product 7f (0.45 μM, 0.9 μM and 1.8 μM), DMSO 0.5% for 24 hours, stained with propidium iodide (PI) and analyzed for cell cycle using flow cytometry. | |
Table 2 Percentage of cells by phases of product 7f in A549 cell line
| Sample |
Percentage of cells by phases (%) |
| % G0/G1 |
% S |
% G2/M |
| Control (DMSO 0.5%) |
32.70 |
52.96 |
10.84 |
| 7f (0.45 μM) |
36.01 |
47.96 |
13.49 |
| 7f (0.9 μM) |
26.63 |
67.93 |
4.17 |
| 7f (1.8 μM) |
20.81 |
63.88 |
13.61 |
Product 7f induced apoptosis in A549 cells
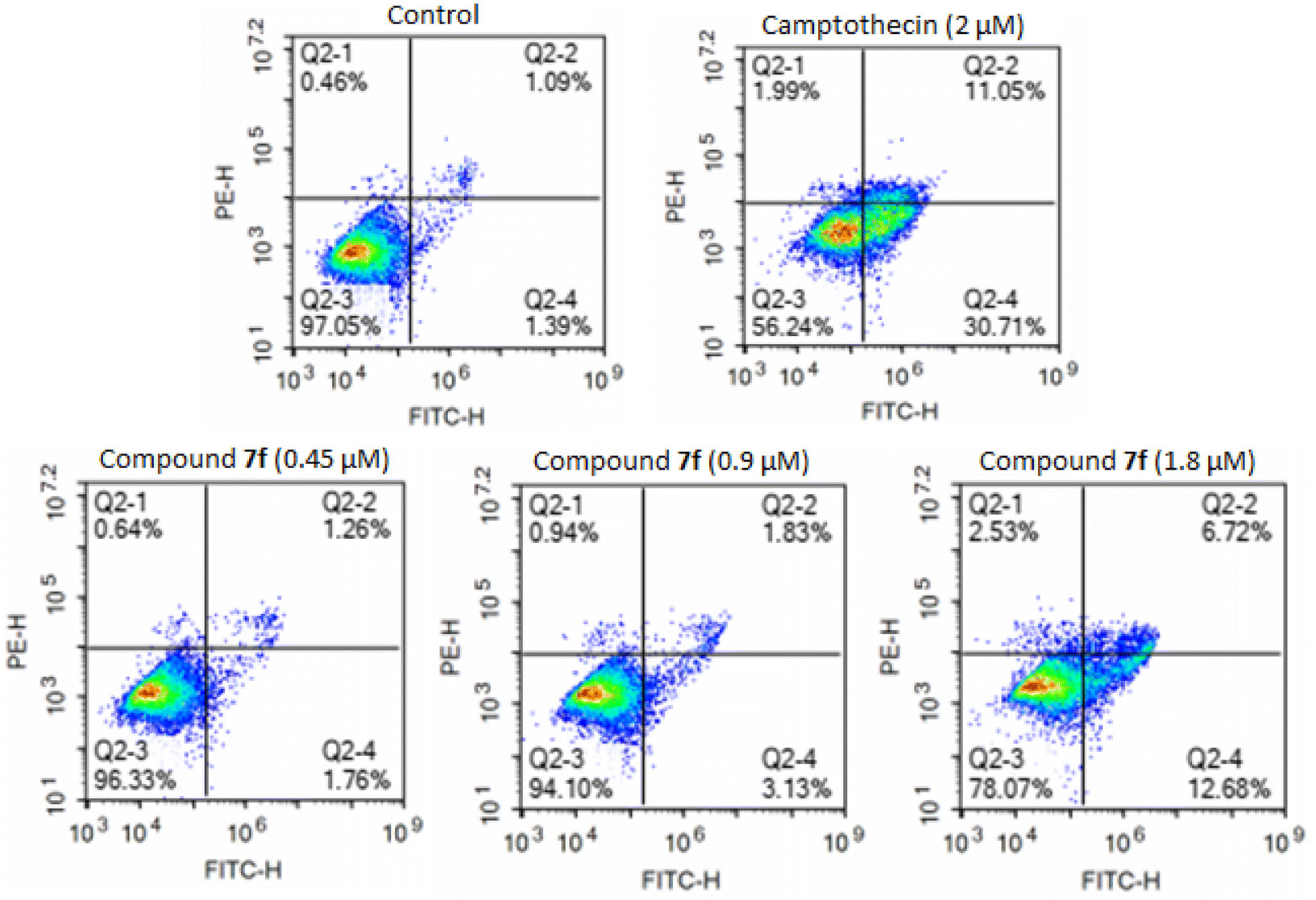
Annexin-V FITC and PI flow cytometry assay for apoptosis. Fluorescent conjugates of annexin V can identify apoptotic cells by binding to phosphatidylserine (PS) exposed on the outer leaflet of their plasma membrane.42,43 Thus, the apoptotic effects of product 7f were further assessed in the A549 cell line upon treatment by product 7f for 48 hours at concentrations of 0.45 μM, 0.9 μM, and 1.8 μM using annexin V.32,41 As illustrated in Fig. 3 and Table 3, treatment of cells with product 7f at concentrations of 0.45 μM, 0.9 μM, and 1.8 μM resulted in a dose-dependent increase in apoptotic cell accumulation, from 2.48% (control) to 3.02%, 8.09%, and 19.4%, respectively. Therefore, the results indicated that product 7f inhibited A549 cancer cell growth through inducing cellular apoptosis of A549 cells in a dose-dependent manner, particularly in the early apoptotic stage.
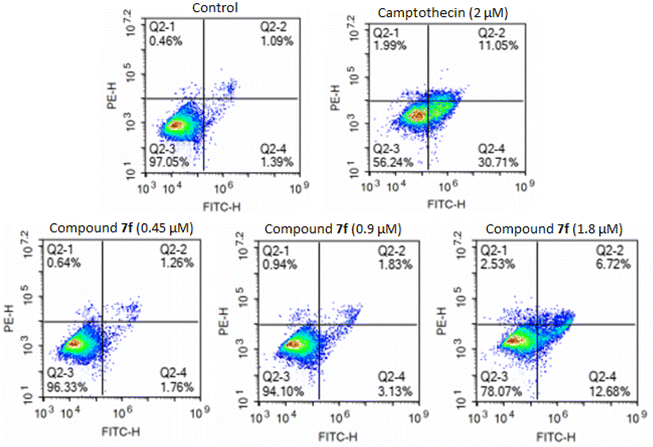 |
| | Fig. 3 Apoptosis population distribution of A549 cells treated with product 7f using annexin-V/PI staining assay. Cells were treated with different concentrations of compound 7f (0.45 μM, 0.9 μM and 1.8 μM), control, and camptothecin (2 μM) for 48 hours, stained with annexin-V/PI and analyzed for apoptosis using flow cytometry. The diverse cell stages were given as live (Q2-3), early apoptotic (Q2-4), late apoptotic (Q2-2), and necrotic cells (Q2-1). | |
Table 3 Percentage of apoptosis cells
| Sample |
% necrotic cells |
% early apoptosis cells |
% late apoptosis cells |
% total apoptosis cells |
| Control |
0.46 |
1.39 |
1.09 |
2.48 |
| Camptothecin (2 μM) |
1.99 |
30.71 |
11.05 |
41.76 |
| 7f (0.45 μM) |
0.64 |
1.76 |
1.26 |
3.02 |
| 7f (0.9 μM) |
0.94 |
3.13 |
1.83 |
8.09 |
| 7f (1.8 μM) |
2.53 |
12.68 |
6.72 |
19.4 |
Molecular docking study
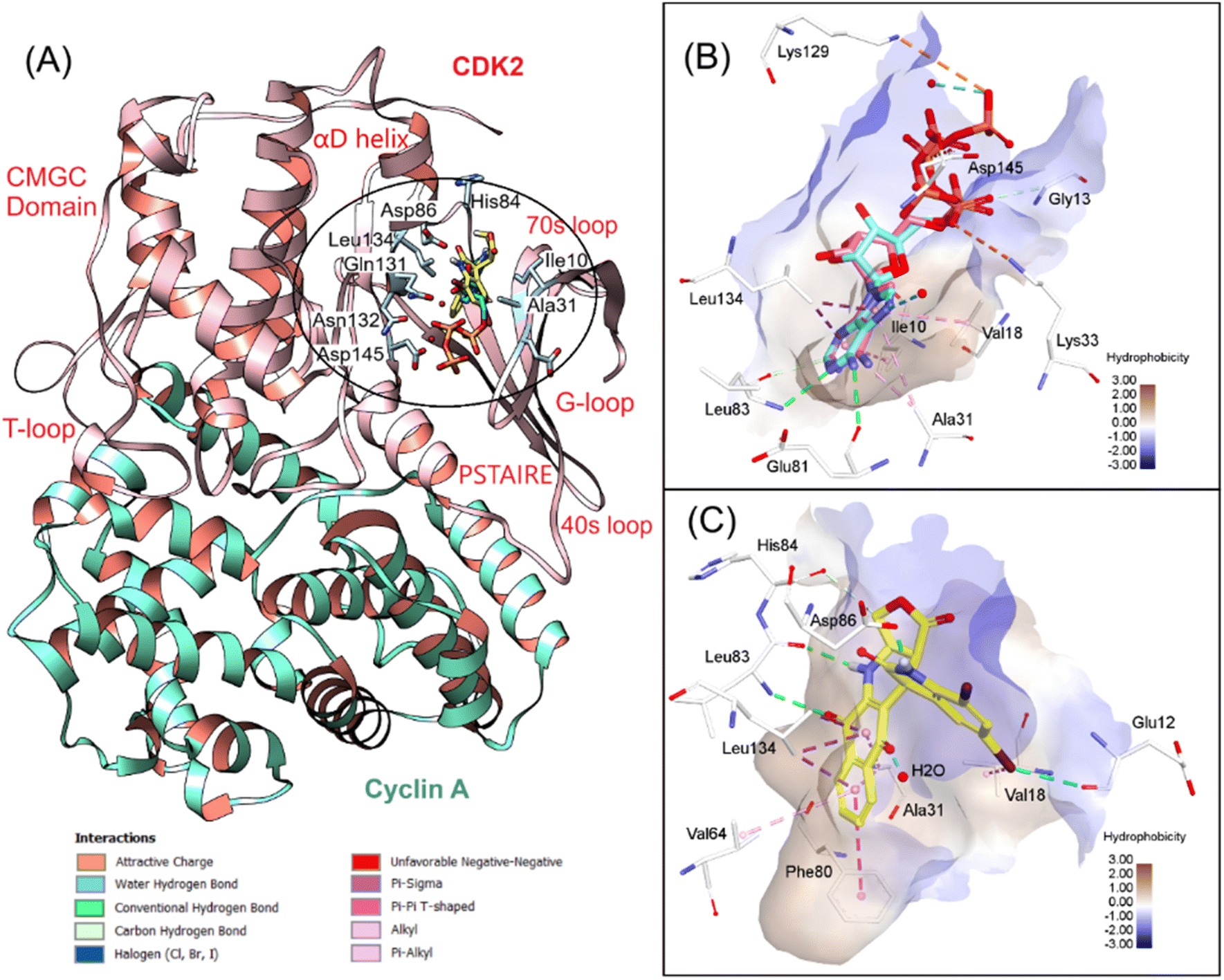
According to the design strategy and experimental assays, we found 7f as a potential hit compound that could induce G1/S cell cycle arrest of cancer cells targeting apoptotic pathways, e.g., procaspase/caspase activation and cyclin-dependent kinases (CDKs). It is worth noting that isatin–podophyllotoxin derivatives have not been explored for their binding interactions with CDKs. We therefore decided to perform docking simulations for 7f and two CDK complexes, including CDK2/cyclin A and CDK5/p25, and one apoptosis effector, procaspase/caspase 6. CDK2 and CDK5 are the main regulators of the cell division cycle, in which CDK2 plays a significant role in the progression from G1 to S phase.44 The association of CDK2 with cyclin A (or cyclin E) and subsequent phosphorylation of the active loop are pivotal to make CDK2 fully active. CDK5 in turn exhibits regulatory function in a different manner. The binding with the activator p25 (or p35/p39) is the only step to activate this CDK without a phosphorylation process. The dysregulation of CDKs has been linked to various diseases, including cancer, severe neurodegenerative disorders, etc.44
As typical kinases, CDK2 and CDK5 have two globular domains, the N-terminal domain composed mostly of β-sheet and the C-terminal domain which is predominantly α-helix. According to the 3D structures published by Jeffrey et al.,45 we identified that the catalytic cleft of CDK2/cyclin A includes a Gly rich loop (G-loop) which is located above the ATP binding pocket. The other α-helix, referred to as the PSTAIRE (Pro–Ser–Thr–Ala–Isoleu–Arg–Glu) helix, undergoes rearrangement and assists in repositioning the key residues within the active site (Fig. 4A). The T-loop, located at the interface of CDK2 and cyclin A, is more flexible than the G-loop and plays a role in phosphorylation. Therefore, we focused solely on the ATP binding site, as the compound is believed to disrupt the interaction between CDK2 and cyclin A. We initially redocked an ATP molecule and compared it with the co-crystal ligand to validate the protocol. The redocking result was suitable, as the redocked and co-crystal conformers showed a RMSD of 1.02 Å with a docking score of −35.84 kcal mol−1 and conserved important interactions in the active site. We then applied the validated docking method to estimate the binding modes of 7f with CDK2/cyclin A. This compound bound to the hydrophobic secondary pocket, as Gleevec-like (type-II) kinase inhibitors normally act. As can be seen in Fig. 4C, isatin–podophyllotoxin derivative was found to be involved in numerous stacking interactions with Ala31, Val64, Leu134, and especially gatekeeper Phe80. In addition, H-bonds from NH and CO groups, as well as the 5,7-dibromoisatin moiety towards backbone O of Glu12, Leu83, and side chain O of Asp86, significantly stabilize the accommodation of 7f at the ATP binding site of CDK2/cyclin A. A docking score of −27.35 kcal mol−1 was calculated for 7f, which is slightly lower than that of the native ligand ATP.
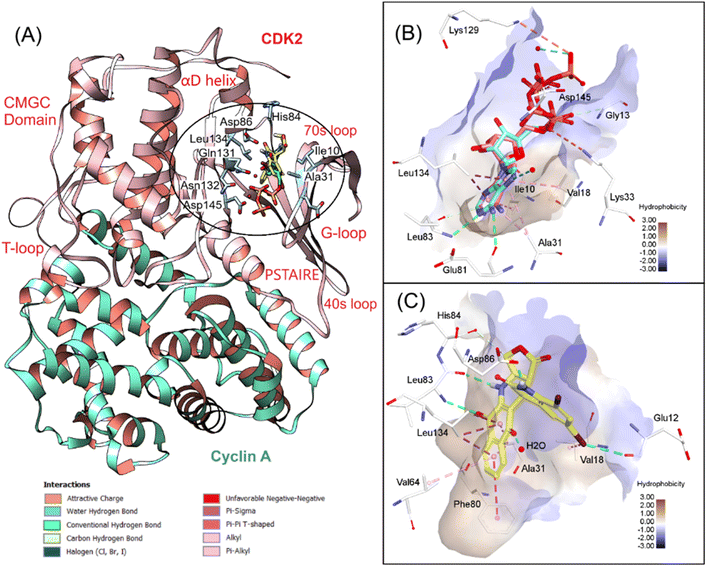 |
| | Fig. 4 3D visualization of (A) superposition of 7f and ATP in CDK2/cyclin A (PDB ID 1FIN), (B) redocking results, and (C) 7f interactions with the key residues at the ATP binding site. | |
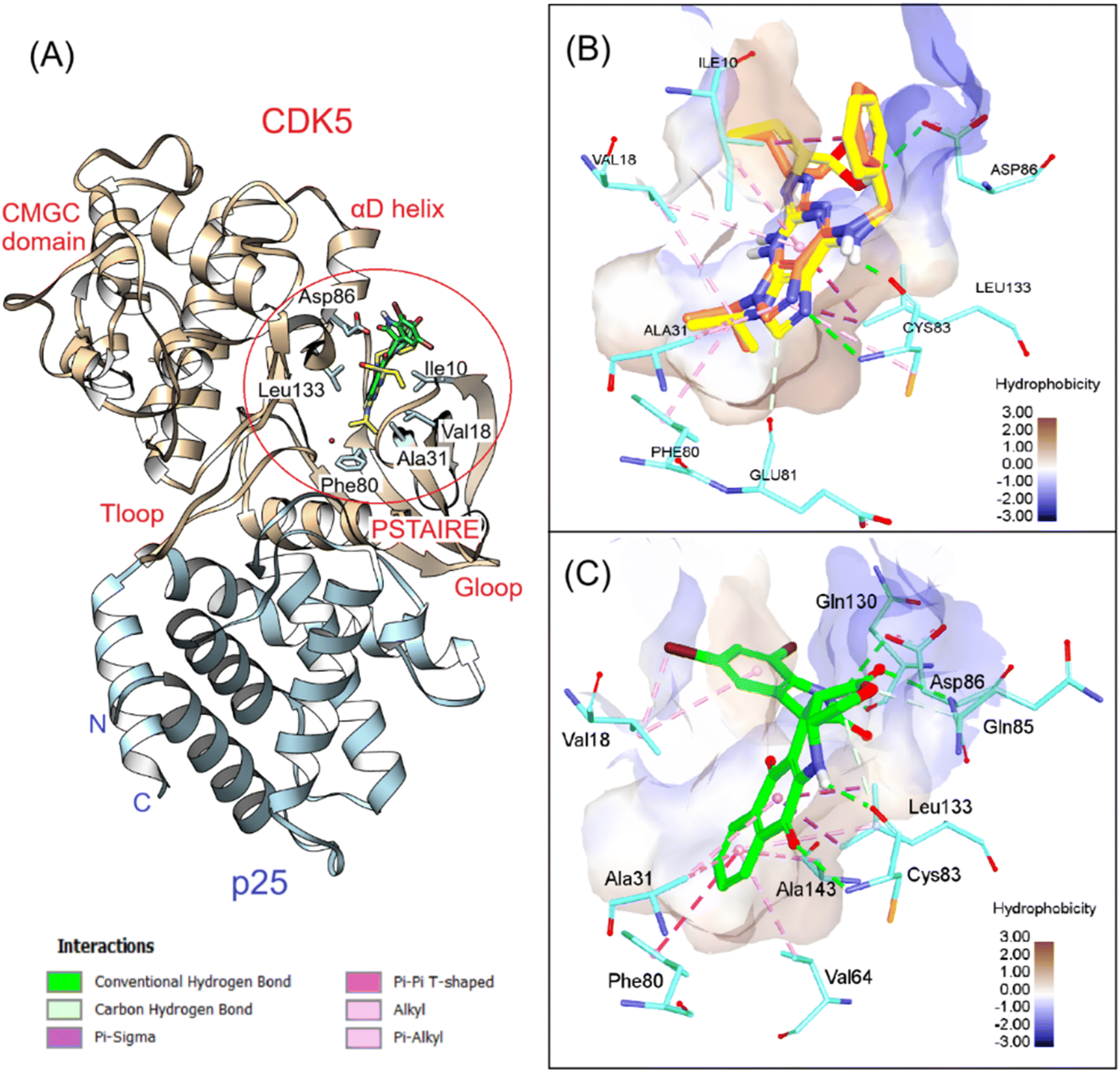
We continued analyzing the structure of CDK5/p25 complex and performed docking simulations with the crystallographic structure reported by Mapelli et al.46 Several studies have been conducted to understand the mechanism of activation of this complex.47 In general, the structures of CDK5/p25 and CDK2/cyclin A are quite similar. Some dissimilarity was noted at the activation T-loop, but it does not affect the binding of roscovitine, a cell proliferation inhibitor, toward CDK5 N-terminal domain.47 After redocking, roscovitine was found to be highly overlapped with the co-crystal ligand (RMSD = 0.791 Å), with a docking score of −23.14 kcal mol−1. The key interactions, including H-bonds from the purine moiety with Glu81, Cys83, Asp86, as well as hydrophobic interactions between pyrazole and isopropyl rings with Leu10, Val18, Ala31 and Phe80, were conserved, suggesting the validity of the docking protocol for this system. We continued docking 7f into the ATP binding site of CDK5/p25 and found that this compound even bound better to this pocket in comparison to roscovitine. Numerous H-bonds were observed between this compound and the hinge region residues such as Cys83, Gln85, Cys86, and Gln130. The extra stacking interactions formed between 7f and the hydrophobic binding pocket were favorable for the binding of this derivative in the G-loop. The binding energy estimated by GBSA/MM-type scoring function was −26.02 kcal mol−1, suggesting good affinity of 7f to CDK5 (Fig. 5).
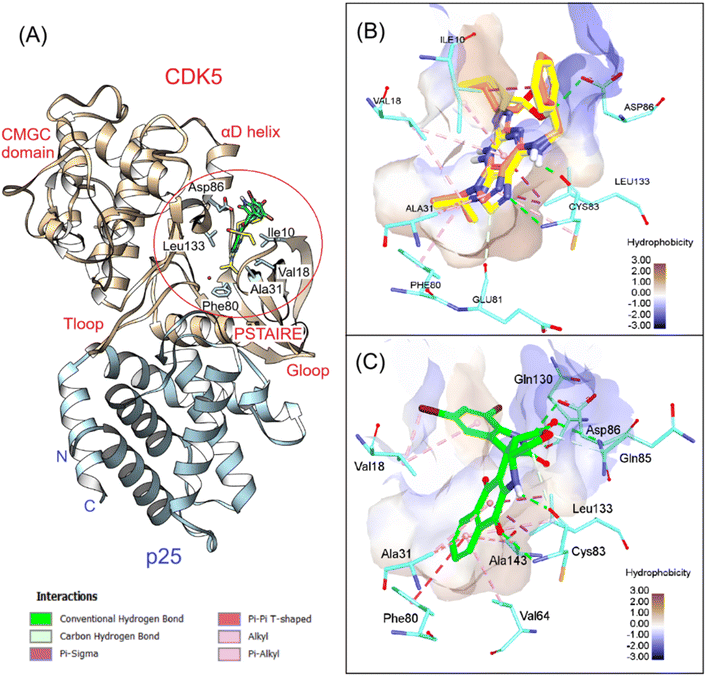 |
| | Fig. 5 3D visualization of (A) superposition of 7f and roscovitine in CDK5/p25 (PDB ID 1UNL), (B) redocking results, and (C) 7f interactions with the key residues at the ATP binding site. | |
In the next step, the apoptosis induction activity of 7f was examined by using docking simulations with a procaspase 6/caspase 6 in complex with PAC-1 (PDB ID 4FXO).48 We have demonstrated previously that podophyllotoxin–naphthoquinone compounds can form stable bidentate chelation with inhibitory Zn ion, which is associated with their anticancer activity.30,31 By running multiple sequence alignment (MSA) and phylogenetic analysis with seven caspase data using Clustal Omega (https://www.ebi.ac.uk/jdispatcher/msa/clustalo), we classified caspases 3, 6 and 7 as effectors of apoptosis (Fig. 6A). The catalytic activities of these effectors are blocked by Zn, which is coordinated with Lys36, Glu244, His287 and a water molecule.31,48 By docking PAC-1 to the allosteric site of procaspase 6, we identified that this caspase activator could form stable chelation with inhibitory Zn ion and allow sequestering of Zn bound to procaspase.49,50 We continued docking 7f into the same pocket and revealed that this derivative also formed bidentate chelation with Zn ion via the two CO groups of 5,7-dibromoisatin and trans lactone fragments (Fig. 6B and C). These results indicated that the synthesized compound could activate procaspase in the same manner that PAC-1 did. However, there is a difference between the coordination geometries of 7f and PAC-1 with Zn, and 7f displayed lower docking scores (−19.10 kcal mol−1) compared to PAC-1 (−23.81 kcal mol−1).
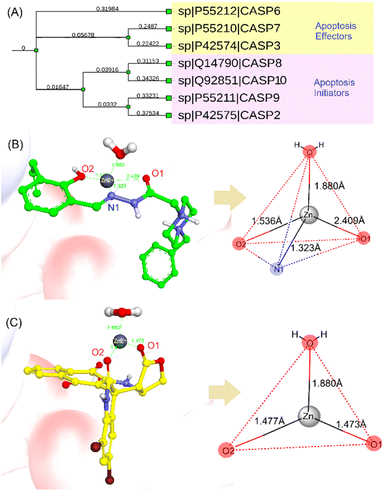 |
| | Fig. 6 (A) A dendrogram of selected caspases showing their role in apoptosis. Metal complex and geometries of PAC-1 of PAC-1 (B) and 7f (C) against Zn ion extracted from procaspase-6 (PDB ID 4FXO). | |
Prediction of physicochemical and ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties
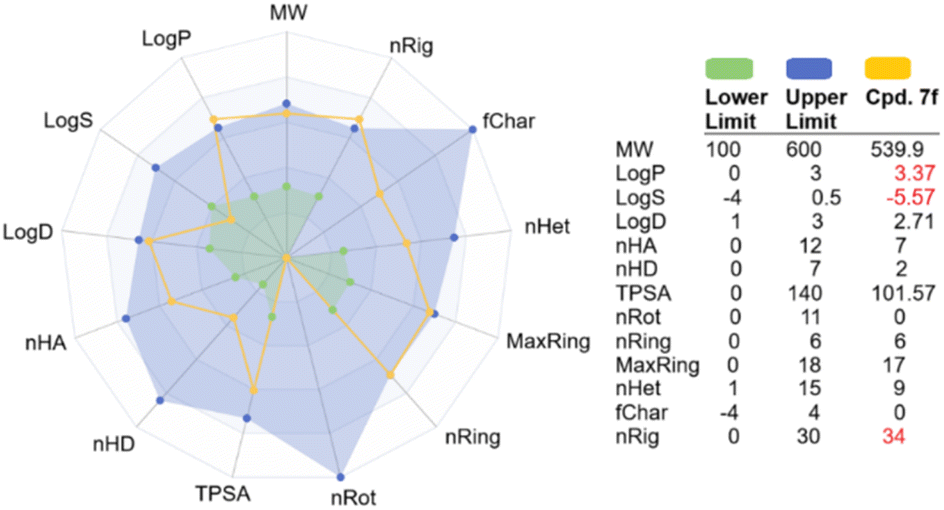
It is desirable to examine the pharmacokinetic properties in parallel with biological activity of hit compound 7f. We therefore applied several cheminformatics tools for the calculation of the physicochemical and ADMET properties of this compound.51–56 The results are summarized in Fig. 7 and Table 4. Based on several drug-like rules, we identified that this compound accomplished the Lipinski and Pfizer rules but did not obey the GSK rule. Among the physicochemical properties calculated, lipophilicity (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P), solubility (logS), and number of rings (nRig) are out of drug-like ranges. In addition, it is provisionally classified as BCS class II, which may need some modifications to improve aqueous solubility.52,55 Subsequently, the ADMET properties were evaluated, revealing that the primary concern would be the extensive metabolism by cytochrome P450. Compound 7f was predicted to be a substrate for several first-pass metabolizing enzymes, including CYP3A4, CYP1A2, CYP2C9, and others (Table 4). It is therefore estimated that this compound would have low oral bioavailability. In terms of toxicity, 7f did not show interaction with the human ether-à-go-go-related gene (hERG) channel, which is considered the main cause of cardiotoxicity. In addition, this compound showed low toxicity against skin, respiratory system and AMES except for the liver. Its oral acute toxicity in rats was determined to be low, with a lethal dose > 500 mg kg−1.51
P), solubility (logS), and number of rings (nRig) are out of drug-like ranges. In addition, it is provisionally classified as BCS class II, which may need some modifications to improve aqueous solubility.52,55 Subsequently, the ADMET properties were evaluated, revealing that the primary concern would be the extensive metabolism by cytochrome P450. Compound 7f was predicted to be a substrate for several first-pass metabolizing enzymes, including CYP3A4, CYP1A2, CYP2C9, and others (Table 4). It is therefore estimated that this compound would have low oral bioavailability. In terms of toxicity, 7f did not show interaction with the human ether-à-go-go-related gene (hERG) channel, which is considered the main cause of cardiotoxicity. In addition, this compound showed low toxicity against skin, respiratory system and AMES except for the liver. Its oral acute toxicity in rats was determined to be low, with a lethal dose > 500 mg kg−1.51
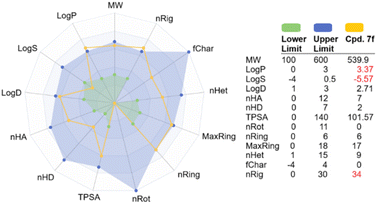 |
| | Fig. 7 Predicted physicochemical properties of 7f compared to optimal drug ranges. Abbreviations can be found in ADMETlab 3.0 web server (https://admetlab3.scbdd.com/). | |
Table 4 Summary of ADMET properties of 7f
| Property |
Predicted value |
Unit |
| Absorption |
| Caco2 permeability |
−4.788 |
logPapp in 10−6 cm s−1 |
| P-glycoprotein substrate |
Yes |
Categorical (yes/no) |
| Human intestinal absorption (HIA) |
>30 |
% absorbed |
| Bioavailability (F) |
<20 |
% oral bioavailable |
|
| Distribution |
| Human distribution volume (VDss) |
4.236 |
log L kg−1 |
| Fraction unbound |
2.1 |
Fu% |
| Blood–brain barrier (BBB) permeability |
Yes |
Categorical (yes/no) |
|
| Metabolism |
| CYP2D6 substrate |
No |
Categorical (yes/no) |
| CYP3A4 substrate |
Yes |
Categorical (yes/no) |
| CYP1A2 substrate |
Yes |
Categorical (yes/no) |
| CYP2C19 substrate |
Yes |
Categorical (yes/no) |
| CYP2C9 substrate |
Yes |
Categorical (yes/no) |
|
| Excretion |
| Total clearance |
1.935 |
CLplasma, mL min−1 kg−1 |
| T1/2 |
1.105 |
Hour |
|
| Toxicity |
| AMES toxicity |
No |
Categorical (yes/no) |
| hERG inhibitor |
No |
Categorical (yes/no) |
| Hepatotoxicity |
Yes |
Categorical (yes/no) |
| Skin sensitization |
No |
Categorical (yes/no) |
| Rat oral acute toxicity |
0.832 |
Low-toxicity, >500 mg kg−1 |
Experimental
General considerations
All chemicals and solvents were purchased from Aldrich or Merck unless otherwise specified. Reactions were conducted in an Anton Paar Microwave Synthetic Reactor Monowave 400. To monitor reaction progress, thin layer chromatography (TLC) was performed using TLC silica gel 60 F254 visualized under UV light at 254 nm. Purification of the compounds was achieved through open silica gel column flash chromatography, employing Merck silica gel 60 (240–400 mesh) as the stationary phase. Melting points were determined using a Buchi Melting Point B-545 and were uncorrected. Infrared (IR) analysis was conducted with a PerkinElmer Spectrum Two spectrometer using KBr pellets. NMR spectra (1H, 13C) were recorded on a Bruker Avance III spectrometer (600 and 150 MHz) with tetramethylsilane used as the internal standard. HRMS was performed on an SCIEX X500 QTOF mass spectrometer.
General procedure for the synthesis of products 7a–n
A vial containing a mixture of 2-amino-1,4-naphthoquinone 8 (1 mmol), tetronic acid 9 (1 mmol), isatin derivative 10 (1 mmol), and p-TsOH (0.02 mmol) in glacial acetic acid (3 mL) was subjected to microwave irradiation (120 °C and 150 W). Following a reaction time of 15 to 20 minutes, the vial was cooled to room temperature, and 20 mL of water was added to the mixture. The product was extracted with dichloromethane (3 × 20 mL), washed with brine (3 × 10 mL), and dried using Na2SO4. The solvent was evaporated to yield crude product which was purified by column chromatography with a dichloromethane–ethyl acetate eluent (25:4, 20:1, v/v) to afford product 7.
3H-Spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7a). Yield 327 mg (85%), red-brown solid, mp 356–357 °C. IR (KBr) νmax/cm−1 3133, 3081, 3032, 2840, 1756, 1708, 1663, 1617, 1592, 1498, 1473, 1394, 1335, 1302, 1265, 1202, 1180, 1143, 1082, 1020, 970, 789, 753, 723, 621. 1H NMR (DMSO-d6, 600 MHz) δ 10.90 (1H, s, NH), 10.51 (1H, s, NH), 8.08 (1H, dd, J = 6.6, 1.8 Hz, H-6), 7.86–7.80 (3H, m, H-7, H-8, H-9), 7.15 (1H, td, J = 7.2, 1.2 Hz, H-4′), 7.10 (1H, d, J = 7.2 Hz, H-5′), 6.83 (1H, td, J = 7.2, 1.2 Hz, H-6′), 6.81 (1H, d, J = 7.2 Hz, H-7′), 4.99 (1H, d, J = 16.8 Hz, H-3), 4.97 (1H, d, J = 16.8 Hz, H-3). 13C NMR (DMSO-d6, 150 MHz) δ 181.1 (C-10), 179.1 (C-5), 177.9 (C-2′), 169.2 (C-1), 156.1, 141.9, 140.7, 135.5, 135.1, 133.6, 131.5, 129.9, 128.4, 126.1, 125.9, 124.1, 121.3, 117.5, 108.8, 101.0, 66.1 (C-3), 48.0 (C-11). HR-ESI-MS: found m/z 385.0804 [M + H]+ (calcd for [C22H13N2O5]+: 385.0819) and m/z 407.0631 [M + Na]+ (calcd for [C22H12N2NaO5]+: 407.0639).
5′-Chloro-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7b). Yield 293 mg (70%), red-brown solid, mp 365–366 °C. IR (KBr) νmax/cm−1 3153, 3115, 3048, 2859, 1757, 1713, 1686, 1665, 1638, 1619, 1590, 1496, 1475, 1395, 1337, 1302, 1266, 1198, 1085, 1041, 1021, 1007, 973, 812, 725, 642, 554. 1H NMR (DMSO-d6, 600 MHz) δ 10.96 (1H, s, NH), 10.67 (1H, s, NH), 8.10–8.07 (1H, m, H-6), 7.86–7.81 (3H, m, H-7, H-8, H-9), 7.24 (1H, d, J = 2.4 Hz, H-4′), 7.20 (1H, dd, J = 8.4, 2.4 Hz, H-6′), 6.83 (1H, d, J = 7.8 Hz, H-7′), 5.00 (2H, s, H-3). 13C NMR (DMSO-d6, 125 MHz) δ 181.4 (C-10), 179.1 (C-5), 177.8 (C-2′), 169.3 (C-1), 156.6, 141.0, 140.9, 137.1, 135.2, 133.7, 131.5, 130.0, 128.3, 126.2, 126.0, 125.4, 124.5, 116.9, 110.3, 100.5, 66.3 (C-3), 48.3 (C-11). HR-ESI-MS: found m/z 419.0434 [M + H]+ (calcd for [C22H12ClN2O5]+: 419.0429).
5′-Methyl-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7c). Yield 331 mg (83%), red solid, mp 361–362 °C. IR (KBr) νmax/cm−1 3328, 3031, 2915, 2862, 2796, 2735, 1753, 1730, 1669, 1631, 1588, 1494, 1396, 1336, 1310, 1266, 1203, 1072, 1042, 1020, 999, 820, 722, 691, 601. 1H NMR (DMSO-d6, 600 MHz) δ 10.89 (1H, s, NH), 10.41 (1H, s, NH), 8.07 (1H, dd, J = 7.8, 1.8 Hz, H-6), 7.85–7.80 (3H, m, H-7, H-8, H-9), 6.95 (2H, d, J = 10.8 Hz, H-4′, H-6′), 6.70 (1H, d, J = 7.8 Hz, H-7′), 4.99 (2H, s, H-3), 2.14 (3H, s, CH3). 13C NMR (DMSO-d6, 150 MHz) δ 181.1 (C-10), 179.1 (C-5), 177.9 (C-2′), 169.2 (C-1), 156.1, 140.6, 139.4, 135.6, 135.1, 133.6, 131.5, 130.2, 129.9, 128.6, 126.1, 125.9, 124.8, 117.6, 108.6, 101.1, 66.0 (C-3), 48.1 (C-11), 20.5 (CH3). HR-ESI-MS: found m/z 399.0964 [M + H]+ (calcd for [C23H15N2O5]+: 399.0976) and m/z 421.0788 [M + Na]+ (calcd for [C23H14N2NaO5]+: 421.0795).
5′-Bromo-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7d). Yield 379 mg (82%), red solid, mp (decomp.) 310–311 °C. IR (KBr) νmax/cm−1 3112, 3045, 2976, 2856, 2255, 1757, 1713, 1663, 1637, 1616, 1590, 1495, 1475, 1395, 1337, 1303, 1265, 1197, 1085, 1022, 1004, 809, 724, 640, 535. 1H NMR (DMSO-d6, 600 MHz) δ 10.96 (1H, s, NH), 10.68 (1H, s, NH), 8.07 (1H, dd, J = 7.8, 1.8 Hz, H-6), 7.87–7.81 (3H, m, H-7, H-8, H-9), 7.36 (1H, d, J = 1.8 Hz, H-4′), 7.33 (1H, dd, J = 8.4, 1.8 Hz, H-6′), 6.79 (1H, d, J = 8.4 Hz, H-7′), 5.00 (2H, s, H-3). 13C NMR (DMSO-d6, 150 MHz) δ 181.3 (C-10), 179.0 (C-5), 177.6 (C-2′), 169.2 (C-3), 156.5, 141.3, 141.0, 137.5, 135.1, 133.6, 131.5, 131.1, 130.0, 127.1, 126.1, 125.9, 116.8, 113.0, 110.7, 100.5, 66.2 (C-3), 48.2 (C-11). HR-ESI-MS: found m/z 462.9936 and 464.9912 [M + H]+ (calcd for [C22H12BrN2O5]+: 462.9924 and 464.9904) and m/z 484.9749 and 486.9726 [M + Na]+ (calcd for [C22H11BrN2NaO5]+: 484.9744 and 486.9724).
7′-Methyl-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7e). Yield 295 mg (74%), brown-orange solid, mp (decomp.) 367–368 °C. IR (KBr) νmax/cm−1 3153, 2854, 1721, 1667, 1625, 1595, 1490, 1396, 1331, 1296, 1269, 1214, 1084, 1041, 1028, 839, 767, 721, 569. 1H NMR (DMSO-d6, 600 MHz) δ 10.89 (1H, s, NH), 10.55 (1H, s, NH), 8.08–8.06 (1H, m, H-6), 7.86–7.80 (3H, m, H-7, H-8, H-9), 6.97 (1H, d, J = 7.2 Hz, H-4′), 6.92 (1H, d, J = 7.2 Hz, H-6′), 6.75 (1H, t, J = 7.2 Hz, H-5′), 4.99 (1H, dd, J = 16.8 Hz, H-3), 4.97 (1H, d, J = 16.8 Hz, H-3), 2.27 (3H, s, CH3). 13C NMR (DMSO-d6, 125 MHz) δ 181.1 (C-10), 179.1 (C-5), 178.4 (C-2′), 169.2 (C-1), 156.0, 140.5, 140.4, 135.2, 135.1, 133.6, 131.5, 129.9, 129.8, 126.1, 125.9, 121.6, 121.3, 117.8, 117.6, 101.1, 66.0 (C-3), 48.3 (C-11), 16.4 (CH3). HR-ESI-MS: found m/z 399.0967 [M + H]+ (calcd for [C23H15N2O5]+: 399.0976) and m/z 421.0780 [M + Na]+ (calcd for [C23H14N2NaO5]+: 421.0795).
5′,7′-Dibromo-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7f). Yield 396 mg (73%), orange solid, mp (decomp.) 364–365 °C. IR (KBr) νmax/cm−1 3164, 3084, 2860, 1724, 1667, 1645, 1593, 1501, 1463, 1400, 1336, 1299, 1183, 1078, 1043, 1022, 1003, 979, 803, 725, 560. 1H NMR (DMSO-d6, 600 MHz) δ 11.05 (1H, s, NH), 11.03 (1H, s, NH), 8.09 (1H, dd, J = 6.6, 1.8 Hz, H-6), 7.86–7.82 (3H, m, H-7, H-8, H-9), 7.59 (1H, d, J = 1.8 Hz, H-6′), 7.42 (1H, d, J = 1.8 Hz, H-4′), 5.04 (1H, dd, J = 16.8 Hz, H-3), 5.02 (1H, d, J = 16.8 Hz, H-3). 13C NMR (DMSO-d6, 125 MHz) δ 181.4 (C-10), 179.0 (C-5), 177.6 (C-2′), 169.3 (C-1), 156.8, 141.2, 141.1, 138.2, 135.2, 133.8, 133.1, 131.4, 130.1, 126.5, 126.2, 126.1, 116.6, 113.6, 102.1, 100.3, 66.4 (C-3), 49.4 (C-11). HR-ESI-MS: found m/z 542.9003 and 540.9027 and 544.9012 [M + H]+ (calcd for [C22H11Br2N2O5]+: 542.9009 and 540.9030 and 544.8989) and m/z 564.8824 and 562.8872 and 566.8793 [M + Na]+ (calcd for [C22H10Br2N2NaO5]+: 564.8829 and 562.8849 and 566.8809).
4′,6′-Dimethyl-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7g). Yield 309 mg (75%), orange-red solid, mp 372–373 °C. IR (KBr) νmax/cm−1 3353, 3183, 1749, 1725, 1666, 1592, 1488, 1396, 1333, 1298, 1269, 1183, 1075, 1040, 1020, 752, 725. 1H NMR (DMSO-d6, 600 MHz) δ 10.87 (1H, s, NH), 10.45 (1H, s, NH), 8.08 (1H, m, H-6), 7.84–7.80 (3H, m, H-7, H-8, H-9), 6.76 (2H, d, J = 17.4 Hz, H-4′, H-6′), 4.98 (2H, s, H-3), 2.23 (3H, s, CH3-5′), 2.11 (3H, s, CH3-7′). 13C NMR (DMSO-d6, 150 MHz) δ 181.1 (C-10), 179.1 (C-5), 178.3 (C-2′), 169.2 (C-1), 155.9, 140.5, 138.0, 135.3, 135.1, 133.5, 131.5, 130.3, 130.1, 129.9, 126.1, 125.9, 122.2, 117.8, 117.5, 101.2, 66.0 (C-3), 48.3 (C-11), 20.4 (CH3-5′), 16.3 (CH3-7′). HR-ESI-MS: found m/z 413.1131 [M + H]+ (calcd for [C24H17N2O5]+: 413.1132) and m/z 435.0948 [M + Na]+ (calcd for [C24H16N2NaO5]+: 435.0952).
1′-Benzyl-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7h). Yield 361 mg (76%), red-brown solid, mp (decomp.) 320–321 °C. IR (KBr) νmax/cm−1 3431, 3061, 1752, 1712, 1667, 1608, 1497, 1333, 1300, 1267, 1181, 1075, 1041, 1018, 961, 747, 721. 1H NMR (DMSO-d6, 600 MHz) δ 11.03 (1H, s, NH), 8.11–8.08 (1H, m, H-6), 7.85–7.81 (3H, m, H-7, H-8, H-9), 7.63 (2H, d, J = 7.2 Hz), 7.36 (2H, t, J = 7.2 Hz), 7.29 (1H, t, J = 7.2 Hz), 7.21 (1H, dd, J = 7.8, 0.6 Hz), 7.14 (1H, td, J = 7.8, 1.2 Hz), 6.91 (1H, d, J = 7.8, 1.2 Hz), 6.69 (1H, d, J = 7.8 Hz), 5.05 (1H, d, J = 16.2 Hz), 5.04 (2H, d, J = 3.6 Hz), 4.94 (1H, d, J = 16.2 Hz). 13C NMR (DMSO-d6, 125 MHz) δ 181.2 (C-10), 179.0 (C-5), 176.7 (C-2′), 169.2 (C-1), 156.5, 142.5, 140.8, 136.3, 135.1, 134.5, 133.6, 131.4, 129.9, 128.5, 128.3 (2C), 127.2 (2C), 127.0, 126.2, 126.0, 124.1, 122.2, 117.2, 108.4, 100.8, 66.2 (C-3), 47.7 (C-11), 43.7 (C-7′′). HR-ESI-MS: found m/z 475.1281 [M + H]+ (calcd for [C29H19N2O5]+: 475.1289) and m/z 497.1094 [M + Na]+ (calcd for [C29H18N2NaO5]+: 497.1108).
1′-(4-(Trifluoromethyl)benzyl)-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7i). Yield 472 mg (87%), orange-red solid, mp (decomp.) 335–336 °C. IR (KBr) νmax/cm−1 3314, 3066, 1763, 1708, 1671, 1611, 1505, 1421, 1371, 1323, 1300, 1268, 1163, 1124, 1067, 1015, 907, 803, 751, 726. 1H NMR (DMSO-d6, 600 MHz) δ 11.05 (1H, s, NH), 8.11–8.08 (1H, m, H-6), 7.86 (2H, d, J = 7.8 Hz), 7.85–7.82 (3H, m), 7.73 (2H, d, J = 7.8 Hz), 7.23 (1H, dd, J = 7.8, 1.2 Hz), 7.16 (1H, td, J = 7.8, 1.2 Hz), 6.93 (1H, td, J = 7.8, 1.2 Hz), 6.75 (1H, d, J = 7.8 Hz), 5.15 (1H, d, J = 16.8 Hz), 5.07 (1H, d, J = 16.8 Hz), 5.05 (2H, d, J = 3.6 Hz). 13C NMR (DMSO-d6, 125 MHz) δ 181.3 (C-10), 179.0 (C-5), 176.7 (C-2′), 169.4 (C-1), 156.6, 142.2, 141.4, 140.9, 135.2, 134.5, 133.7, 131.4, 130.0, 128.6, 128.0 (3C), 126.2, 126.1, 125.2 (2C, d, J = 3.8 Hz), 124.3 (1C, q, J = 271.3 Hz, CF3), 124.3, 122.5, 117.1, 108.3, 100.8, 66.3 (C-3), 47.7 (C-11), 43.2 (C-7′′). HR-ESI-MS: found m/z 543.1137 [M + H]+ (calcd for [C30H18F3N2O5]+: 543.1162) and m/z 565.0952 [M + Na]+ (calcd for [C30H17F3N2NaO5]+: 565.0982).
1′-(4-Chlorobenzyl)-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7j). Yield 397 mg (78%), orange-brown solid, mp (decomp.) 292–293 °C. IR (KBr) νmax/cm−1 3164, 3061, 1760, 1708, 1671, 1645, 1595, 1504, 1490, 1400, 1370, 1331, 1299, 1268, 1188, 1176, 1070, 1042, 1010, 907, 800, 748, 726, 572, 476. 1H NMR (DMSO-d6, 600 MHz) δ 11.04 (1H, s, NH), 8.11–8.07 (1H, m, H-6), 7.85–7.81 (3H, m, H-7, H-8, H-9), 7.65 (2H, d, J = 8.4 Hz), 7.42 (2H, dt, J = 8.4, 2.4 Hz), 7.21 (1H, d, J = 7.8, 1.2 Hz), 7.15 (1H, td, J = 7.8, 1.2 Hz), 6.91 (1H, td, J = 7.8, 1.2 Hz), 6.73 (1H, d, J = 7.8 Hz), 5.04 (1H, d, J = 16.2 Hz), 5.03 (2H, d, J = 3.6 Hz), 4.95 (1H, d, J = 16.2 Hz, H-7′′). 13C NMR (DMSO-d6, 125 MHz) δ 181.3 (C-10), 179.0 (C-5), 176.7 (C-2′), 169.3 (C-1), 156.5, 142.3, 140.9, 135.4, 135.2, 134.5, 133.7, 131.8, 131.4, 130.0, 129.3 (2C), 128.6, 128.3 (2C), 126.2, 126.1, 124.2, 122.4, 117.1, 108.4, 100.8, 66.3 (C-3), 47.7 (C-11), 43.0 (C-7′′). HR-ESI-MS: found m/z 509.0895 [M + H]+ (calcd for [C29H18ClN2O5]+: 509.0899) and m/z 531.0694 [M + Na]+ (calcd for [C29H17ClN2NaO5]+: 531.0719).
1′-(4-Bromo-2-fluorobenzyl)-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7k). Yield 434 mg (76%), orange solid, mp (decomp.) 350–351 °C. IR (KBr) νmax/cm−1 3440, 1760, 1708, 1673, 1648, 1611, 1505, 1488, 1370, 1332, 1299, 1269, 1176, 1070, 1042, 1012, 873, 747, 727. 1H NMR (DMSO-d6, 600 MHz) δ 11.05 (1H, s, NH), 8.10–8.08 (1H, m), 7.85–7.82 (3H, m, H-7, H-8, H-9), 7.68 (1H, t, J = 8.4 Hz), 7.61 (1H, dd, J = 9.6, 1.8 Hz), 7.40 (1H, dd, J = 8.4, 1.8 Hz), 7.24 (1H, dd, J = 7.8, 0.6 Hz), 7.20 (1H, td, J = 7.8, 1.2 Hz), 6.94 (1H, td, J = 7.8, 1.2 Hz), 6.83 (1H, d, J = 7.8 Hz), 5.04 (2H, d, J = 3.0 Hz), 5.02 (1H, dd, J = 16.2, 2.4 Hz), 4.93 (1H, d, J = 16.2 Hz). 13C NMR (DMSO-d6, 125 MHz) δ 181.3 (C-10), 179.0 (C-5), 176.7 (C-2′), 169.4 (C-1), 159.9 (1C, d, J = 248.8 Hz, C–F), 156.6, 142.0, 140.9, 135.2, 134.5, 133.7, 131.4, 130.9, 130.0, 128.8, 127.5, 126.2 (1C, d, J = 18,75 Hz, C-1′′), 124.4, 122.8 (1C, d, J = 13.8 Hz), 122.6, 120.6 (1C, d, J = 8.8 Hz, C-4′′), 118.6 (1C, d, J = 25 Hz, C-3′′), 117.0, 108.0, 100.7, 66.4 (C-3), 47.6 (C-11), 39.83 (C-7′′). HR-ESI-MS: found m/z 571.0284 and 573.0271 [M + H]+ (calcd for [C29H17BrFN2O5]+: 571.0300 and 573.0279) and m/z 593.0107 and 595.0093 [M + Na]+ (calcd for [C29H16BrFN2NaO5]+: 593.0119 and 595.0099).
5′-Chloro-1′-(4-(trifluoromethyl)benzyl)-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7l). Yield 490 mg (85%), red-brown solid, mp 354–355 °C. IR (KBr) νmax/cm−1 3442, 3303, 1762, 1705, 1670, 1595, 1503, 1485, 1431, 1323, 1301, 1268, 1160, 1121, 1067, 1042, 1014, 824, 808, 729, 695, 553. 1H NMR (DMSO-d6, 600 MHz) δ 11.10 (1H, s, NH), 8.12–8.08 (1H, m, H-6), 7.88–7.84 (3H, m, H-7, H-8, H-9), 7.84 (2H, d, J = 8.4 Hz), 7.74 (2H, d, J = 8.4 Hz), 7.38 (1H, d, J = 2.4 Hz), 7.23 (1H, dd, J = 8.4, 2.4 Hz), 6.78 (1H, d, J = 8.4 Hz), 5.14 (1H, d, J = 16.8 Hz), 5.07 (1H, d, J = 16.8 Hz), 5.06 (2H, s). 13C NMR (DMSO-d6, 125 MHz) δ 181.4 (C-10), 179.0 (C-5), 176.5 (C-2′), 169.4 (C-1), 156.9, 141.2, 141.2, 141.0, 136.1, 135.1, 133.7, 131.4, 130.1, 128.5, 128.0 (3C), 126.6, 126.2, 126.1, 125.2 (2C, d, J = 3.8 Hz), 124.7, 124.3 (1C, q, J = 270.0 Hz, CF3), 116.4, 109.7, 100.2, 66.5 (C-3), 47.9 (C-11), 43.3 (C-7′′). HR-ESI-MS: found m/z 577.0772 [M + H]+ (calcd for [C30H17ClF3N2O5]+: 577.0773) and m/z 599.0593 [M + Na]+ (calcd for [C30H17ClF3N2NaO5]+: 599.0593).
1′-(4-Bromo-2-fluorobenzyl)-6′-methyl-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7m). Yield 468 mg (80%), red-brown solid, mp (decomp.) 337–338 °C. IR (KBr) νmax/cm−1 3314, 2970, 1761, 1725, 1685, 1655, 1595, 1496, 1451, 1396, 1341, 1301, 1267, 1242, 1174, 1077, 1027, 875, 800, 743, 726. 1H NMR (DMSO-d6, 600 MHz) δ 11.06 (1H, s, NH), 8.09 (1H, m, H-6), 7.89–7.88 (1H, m, H-9), 7.86–7.83 (2H, m, H7, H8), 7.81 (1H, t, J = 8.4 Hz, H-6′′), 7.64 (1H, dd, J = 9.6 Hz, J = 1.8 Hz, H-3′′), 7.44 (1H, dd, J = 8.4 Hz, J = 1.8 Hz, H-5′′), 7.09 (1H, d, J = 7.2 Hz, H-8′), 6.99 (1H, d, J = 7.2 Hz, H-5′), 6.87 (1H, t, J = 7.2 Hz, H-7′), 5.18 (1H, d, J = 17.4 Hz, H-7′′), 5.13 (1H, d, J = 17.4 Hz, H-7′′), 5.05 (2H, d, J = 16.8 Hz, H-3), 2.23 (3H, s, H-9). 13C NMR (DMSO-d6, 125 MHz) δ 181.3 (C-10), 178.9 (C-5), 177.8 (C-2′), 169.4 (C-1), 158.9 (1C, d, J = 247.5 Hz, C–F), 156.4 (C-3a), 140.8 (C-4a), 139.98 (C-4′), 135.5 (C-6′), 135.1 (C-8), 133.7 (C-7), 132.5 (C-5′), 131.4 (C-9a), 129.98 (C-5a), 129.8 (1C, d, J = 4.5 Hz, C-6′′), 127.8 (C-5′′), 126.2 (C-6), 126.1 (C-9), 125.2 (1C, d, J = 15.0 Hz, C-1′′), 122.6 (C-7′, C-8′), 120.4 (1C, d, J = 9.0 Hz, C-4′′), 118.6 (1C, d, J = 24 Hz, C-3′′), 118.3 (C-3′), 117.3 (C-10a), 100.9 (C-11a), 66.4 (C-3), 47.1 (C-11), 39.92 (C-7′′), 17.3 (C-9′). HR-ESI-MS: found m/z 585.04564 and 587.0436 [M + H]+ (calcd for [C30H19BrFN2O5]+: 585.0456 and 587.0436) and m/z 607.0273 and 609.0256 [M + Na]+ (calcd for [C30H18BrFN2NaO5]+: 607.0276 and 609.0255).
5′,6′-Dibromo-1′-(4-(trifluoromethyl)benzyl)-3H-spiro[benzo[g]furo[3,4-b]quinoline-11,3′-indoline]-1,2′,5,10(4H)-tetraone (7n). Yield 504 mg (72%), orange-red solid, mp 278–279 °C. IR (KBr) νmax/cm−1 3431, 3067, 2957, 2926, 1751, 1728, 1668, 1645, 1596, 1501, 1456, 1397, 1324, 1267, 1161, 1127, 1067, 1039, 1023, 1010, 974, 804, 739, 727, 551. 1H NMR (DMSO-d6, 600 MHz) δ 11.19 (1H, s, NH), 8.11 (1H, dd, J = 6.6, 2.4 Hz, H-6), 7.93 (1H, dd, J = 6.6, 2.4 Hz, H-9), 7.89–7.85 (2H, m, H-7, H-8), 7.79 (2H, d, J = 8.4 Hz, H-3′′, H-5′′), 7.73 (2H, d, J = 8.4 Hz, H-2′′, H-6′′), 7.63 (1H, d, J = 2.4 Hz, H-8′), 7.59 (1H, d, J = 2.4 Hz, H-5′), 5.42 (2H, s), 5.08 (2H, s). 13C NMR (DMSO-d6, 125 MHz) δ 181.6 (C-10), 178.8 (C-5), 177.4 (C-2′), 169.5 (C-1), 157.1, 143.0, 141.4, 139.3, 139.2, 135.5, 135.2, 133.9, 131.3, 130.2, 127.2, 127.0 (2C), 126.3, 126.2, 125.2 (2C, d, J = 3.8 Hz), 124.3 (1C, q, J = 270.0 Hz, CF3), 116.1, 115.1, 101.6, 100.0, 66.7 (C-3), 47.7 (C-11), 44.9 (C-7′′). HR-ESI-MS: found m/z 700.9359 and 698.9366 and 702.9299 [M + H]+ (calcd for [C30H16Br2F3N2O5]+: 700.9353 and 698.9373 and 702.9332).
Cell culture and cell viability assay
Epidermoid carcinoma (KB, ATCC number CCL-17), hepatocellular carcinoma (HepG2, ATCC number HB-8065), lung (A549) cancer, breast (MCF7, HTB-22™) cancer cell lines, and human embryonic kidney (HEK-293, ATCC number CRL-1573) cell line were sourced from the American Type Culture Collection (ATCC, USA). The cells were cultured in DMEM (Dulbecco's Modified Eagle Medium) medium, supplemented with 10% fetal bovine serum, 100 U per mL penicillin, and 100 μg per mL streptomycin, under a humidified atmosphere (95% air and 5% CO2) at 37 °C. The exponentially growing cells were used throughout the experiments. Cytotoxic effects of products 7a–n on the growth of human cancer cells were evaluated using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. Human cancer cell lines (3 × 104 cells per mL) were treated with solutions of products 7 in DMSO. After incubating for 3 days, 10 μL solution of 5 mg mL−1 MTT in sodium phosphate buffer (PBS, 0.1 M, pH 7.4) was added to each well, and the cells were incubated at 37 °C for 4 hours. The medium was then carefully removed, and DMSO (150 μL) was added to each well to dissolve the obtained formazan crystals. Absorbance of the solutions was measured at a 540 nm wavelength with a Biotek Epoch 2 microplate reader. Percentage of growth inhibition was determined by comparing the absorbance of treated samples to that of control samples without the compounds. A dose–response curve was generated, and the IC50 values for each compound against both cell lines were determined.32,41
Cell cycle analysis
A549 cancer cells were seeded in 25 cm2 flasks (Corning, USA) at a density of 3 × 105 cells and treated with different concentrations of product 7f at 0.45 μM, 0.9 μM and 1.8 μM or DMSO 0.5% (negative reference) for 24 hours. After treatment, the cells were harvested and washed twice with phosphate-buffered saline (PBS). They were then fixed with ice-cold ethanol 70% at −20 °C for 2 hours, followed by low-speed centrifugation at 2000 rpm for 5 minutes to collect the cells. After washing, the cells were resuspended in 0.45 mL PBS and incubated with RNase A (2.5 mg mL−1) at 37 °C for 15 minutes. Next, the cells were stained with 2.5 μL propidium iodide (PI) (1 mg mL−1) for 30 minutes and diluted with 500 μL PBS. The samples were analyzed by a flow cytometry (Novocyte, ACEA Biosciences, San Diego, CA, USA) and data were analyzed and reported by ACEA's NovoExpress software.32,41
Annexin-V FITC and PI flow cytometry assay for apoptosis
The extent of apoptosis was quantitatively measured using the kit annexin V and PI/dead cell apoptosis® from Invitrogen. Briefly, A549 cancer cells were seeded in 25 cm2 flasks (Corning, USA) at a density of 5 × 105 cells and treated with different concentrations of product 7f (0.45 μM, 0.9 μM and 1.8 μM) or DMSO 1% and camptothecin (2 μM) for 48 h. Then, the cells were collected into falcon tubes, centrifuged, and washed with cold PBS. The washed cells were re-centrifuged, and the supernatant was discarded. Then, the cells were resuspended in 100 μL of binding buffer and stained with 5 μL of annexin V and 1 μL of propidium iodide (PI) (1 mg mL−1). The cells were incubated for 15 minutes at 37 °C, after which 400 μL of binding buffer was added. The samples were analyzed by a Novocyte flow cytometry system, and data were analyzed and reported by NoVo Express software (ACEA Bioscience Inc.).32,41
Molecular docking study
Molecular docking simulations were performed to evaluate the binding ability of compound 7f against common cytotoxic targets according to our chemical design. ICM-pro 3.8.3 (Molsoft, L.L.C.) was employed for this purpose, taking into consideration the previous configurations.30,31,57,58 Three proteins with crystalized three-dimensional (3D) structures in the Protein Data Bank were selected, including two human cyclin-dependent kinase (CDK) complexes, CDK2/cyclin A in complex with ATP (PDB ID 1FIN) and CDK5/p25 in complex with (R)-roscovitine (PDB ID 1UNL), and procaspase locked by Zn ion (PDB ID 4FXO).45,46,48 The 3D structure of compound 7f was created by ChemAxon Marvin Suite version 22.5 and optimized using the force field MMFF94s through the steepest-descent method with a convergence criterion of 1 × 10−5. Receptor preparation was carried out following the previous reports which include consecutive steps such as correction of protonation states, active site identification using ICM Pocket Finder, occupancy and b-factor check, optimization of polar hydrogen atoms, setup of rotameric states for His–Pro–Gly–Cys residues, and ICMFF force field for global system. The docking simulations were then performed, generating 103 conformations. The top-scoring poses were ranked and analyzed based on the GBSA/MM-type scoring function, whose features include the conventional interactions and internal force field energy, e.g., hydrophobic van der Waals, electrostatic interaction, solvation/desolvation, conformational loss energy and hydrogen bonding.
Prediction of physicochemical and ADMET (absorption, distribution, metabolism, excretion, and toxicity) properties
Significant physicochemical properties related to ADMET processes were computed using ADMETlab 3.0 web server (https://admetlab3.scbdd.com/) and other tools.51–54 The properties related to the Biopharmaceutics Classification System (BCS) of drugs, including aqueous solubility, epithelial permeability, human intestinal absorption, and metabolism extent, were computed.52 In addition, drug-like indices were calculated based on rule-of-thumb methods,51,54,55 including Lipinski, Prizer, SGK, 3PRule, etc. In total, 120 pharmacokinetic and toxicity properties were computed and discussed in accordance with the current standard for preclinical screening of drug candidates.
Conclusions
In summary, the effective synthesis of isatin–podophyllotoxin derivatives 7a–n through a microwave-facilitated three-component reaction of 2-amino-1,4-naphthoquinone, tetronic acid, and substituted isatin derivatives in glacial acetic acid in the presence of p-toluenesulfonic acid as a catalyst has been described. Cytotoxic effects of products were evaluated using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. All products 7a–n displayed moderate to strong inhibitory activity against four cancer cell lines, with IC50 values ranging from 0.90 to 51.68 μM, indicating the potential of these compounds for the treatment of cancer. Among the products, product 7f displayed the most potent cytotoxicity, with IC50 values of 1.99 ± 0.22 and 0.90 ± 0.09 μM against KB and A549 cell lines, respectively. In addition, product 7f could arrest the cell cycle of A549 cells at S phase and trigger apoptosis in a dose-dependent manner. Molecular docking simulation of product 7f revealed the expected binding mode which involves the accommodation of product 7f at the ATP binding sites of CDK2/cyclin A and CDK5/p25 as well as stable bidentate chelation of 7f with the inhibitory Zn ion. Based on the docking results, this compound was found to have antiproliferative activity against cancer cells by inhibiting activation of CDK2/cyclin A complex as well as anti-tumorigenesis by targeting CDK5/p25 complex. In addition, 7f was estimated to induce the apoptosis process by sequestering Zn bound to procaspase in the same manner as PAC-1, a procaspase activator that induces G1/S cell cycle arrest and blocks DNA synthesis in cancer cells. These findings are in agreement with the previous reports relating to the anticancer mechanism of podophyllotoxin derivatives. Physicochemical and ADMET calculations for 7f revealed that first-pass metabolism and liver toxicity would be significant issues to be addressed in the next hit-to-lead optimization. Otherwise, it accomplished almost all the drug-like rule and preclinical safety requirements in the anticancer drug discovery process.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†
Author contributions
Ha Thanh Nguyen: investigation: synthesis and characterizations of compounds; writing – original draft. Ket Tran Van: investigation: synthesis and characterizations of compounds. Hai Pham-The and Bao Le-Quang: investigation: molecular docking and computational studies. Giang Le-Nhat-Thuy and Tuyet Anh Dang Thi: investigation: biological activity evaluation of potent compounds. Phuong Hoang Thi and Quynh Giang Nguyen Thi: investigation: characterizations of compounds. Tuan Anh Nguyen and Doan Vu Ngoc: investigation: revised draft. Tuyen Van Nguyen: project administration; supervision: conceptual design; writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by National Foundation for Science and Technology Development (NAFOSTED) [grant number: 104.01-2023.19].
Notes and references
- M. B. Parmar, M. K. Vara and J. H. Pandya, A brief review on imidazole, triazine, and isatin derivatives: synthesis approaches and their applications, Discover Chemistry, 2024, 1, 56 CrossRef.
- A. E. Medvedev, A. Clow, M. Sandler and V. Glover, Isatin: a link between natriuretic peptides and monoamines?, Biochem. Pharmacol., 1996, 52, 385–391 CrossRef PubMed.
- A. E. Medvedev and V. Glover, Tribulin and endogenous mao-inhibitory regulation in vivo, Neurotoxicology, 2004, 25, 185–192 CrossRef PubMed.
- A. Medvedev, N. Igosheva, M. Crumeyrolle-Arias and V. Glover, Isatin: role in stress and anxiety, Stress, 2005, 8, 175–183 CrossRef PubMed.
- T. E. Malah, H. Farag, B. A. Hemdan, R. E. A. Mageid, M. T. Abdelrahman, M. A. El-Manawaty and H. F. Nour, Synthesis, in vitro antimicrobial evaluation, and molecular docking studies of new isatin-1,2,3-triazole hybrids, J. Mol. Struct., 2022, 1250, 131855 CrossRef.
- C. S. Marques, A. González-Bakker, J. M. Padrón and A. J. Burke, Easy access to Ugi-derived isatin-peptoids and their potential as small-molecule anticancer agents, New J. Chem., 2023, 47, 743–750 RSC.
- R. S. Cheke, V. M. Patil, S. D. Firke, J. P. Ambhore, I. A. Ansari, H. M. Patel, S. D. Shinde, V. R. Pasupuleti, M. I. Hassan, M. Adnan, A. Kadri and M. Snoussi, Therapeutic outcomes of isatin and its derivatives against multiple diseases: recent developments in drug discovery, Pharmaceuticals, 2022, 15, 272 CrossRef CAS PubMed.
- K. Lahari and R. Sundararajan, Design and synthesis of novel isatin derivatives as potent analgesic, anti-inflammatory and antimicrobial agents, J. Chem. Sci., 2020, 132, 94 CrossRef CAS.
- T. K. Bakır, M. S. Çavuş, H. Muğlu and H. Yakan, Synthesis, structure elucidation, antioxidant properties, and theoretical calculations of new Schiff bases–isatin derivatives, Res. Chem. Intermed., 2024, 50, 3937–3962 CrossRef.
- V. Varun, S. Sonam and R. Kakkar, Isatin and its derivatives: a survey of recent syntheses, reactions, and applications, Med. Chem. Commun., 2019, 10, 351–368 RSC.
- H. M. Alkahtani, M. M. Alanazi, F. S. Aleanizy, F. Y. Alqahtani, A. Alhoshani, F. E. Alanazi, A. A. Almehizia, A. N. Abdalla, M. G. Alanazi, A. S. El-Azab and A. A.-M. Abdel-Aziz, Synthesis, anticancer, apoptosis-inducing activities and EGFR and VEGFR2 assay mechanistic studies of 5,5-diphenylimidazolidine-2,4-dione derivatives: molecular docking studies, Saudi Pharm. J., 2019, 27, 682–693 CrossRef.
- A. S. Alanazi, T. O. Mirgany, A. A. Alsfouk, N. A. Alsaif and M. M. Alanazi, Antiproliferative activity, multikinase inhibition, apoptosis inducing effects and molecular docking of novel isatin–purine hybrids, Medicina, 2023, 59, 610 CrossRef PubMed.
- Z. Li, J. Ma, M. Tian, P. Xia, X. Lv, R. Hou, Y. Jiang, X. Xu, Z. Jia, J. Wang and Z. Li, Synthesis, biological activity evaluation and mechanism of action of novel bis-isatin derivatives as potential anti-liver cancer agents, Bioorg. Med. Chem. Lett., 2024, 99, 129613 CrossRef CAS PubMed.
- M. Altamimi, S. A. Syed, B. Tuzun, M. R. Alhazani, O. Alnemer and A. Bari, Synthesis, biological evaluation and molecular docking of isatin hybrids as anti-cancer and anti-microbial agents, J. Enzyme Inhib. Med. Chem., 2024, 39, 2288548 CrossRef PubMed.
- A. Mushtaq, R. Asif, W. A. Humayun and M. M. Naseer, Novel isatin–triazole based thiosemicarbazones as potential anticancer agents: synthesis, DFT and molecular docking studies, RSC Adv., 2024, 14, 14051–14067 RSC.
- G. Kumar, N. P. Singh and K. Kumar, Recent advancement of synthesis of isatins as a versatile
pharmacophore: a review, Drug Res., 2021, 71, 115–121 CrossRef CAS.
- Preeti, A. Raza, A. Anand, N. Henry, A. K. Sharma, P. Roussel and V. Kumar, Stereo/regio-selective access to substituted 3-hydroxy-oxindoles with anti-proliferative assessment and in silico validation, RSC Adv., 2023, 13, 28434–28443 RSC.
- A. Andreani, M. Granaiola, A. Leoni, A. Locatelli, R. Morigi, M. Rambaldi, V. Garaliene, W. Welsh, S. Arora, G. Farruggia and L. Masotti, antitumor activity of new substituted 3-(5-imidazo[2,1-b]thiazolylmethylene)-2-indolinones and study of their effect on the cell cycle, J. Med. Chem., 2005, 48, 5604–5607 CrossRef PubMed.
- R. E. Ferraz De Paiva, E. G. Vieira, D. Rodrigues Da Silva, C. A. Wegermann and A. M. C. Ferreira, Anticancer compounds based on isatin-derivatives: strategies to ameliorate selectivity and efficiency, Front. Mol. Biosci., 2021, 7, 627272 CrossRef PubMed.
- A. K. Gupta, S. Tulsyan, M. Bharadwaj and R. Mehrotra, systematic review on cytotoxic and anticancer potential of N-substituted isatins as novel class of compounds useful in multidrug-resistant cancer therapy: in silico and in vitro analysis, Top. Curr. Chem., 2019, 377, 15 CrossRef.
- M. Gordaliza, P. A. García, J. M. Miguel del Corral, M. A. Castro and M. A. Gómez-Zurita, Podophyllotoxin: distribution, sources, applications and new cytotoxic derivatives, Toxicon, 2004, 44, 441–459 CrossRef.
- H. Ardalani, A. Avan and M. Ghayour-Mobarhan, Podophyllotoxin: a novel potential natural anticancer agent, Avicenna J. Phytomed., 2017, 7, 10 Search PubMed.
- N. H. Thanh, L. Q. Bao, H. Pham-The, D. T. T. Anh and P. V. Kiem, Synthesis, molecular docking, and cytotoxic evaluation of fluorinated podophyllotoxin derivatives, Nat. Prod. Commun., 2023, 18(2), 1–7 CrossRef.
- N. Jeedimalla, M. Flint, L. Smith, A. Haces, D. Minond and S. P. Roche, Multicomponent assembly of 4-aza-podophyllotoxins: a fast entry to highly selective and potent anti-leukemic agents, Eur. J. Med. Chem., 2015, 106, 167–179 CrossRef PubMed.
- N. H. Thanh, H. T. Phuong, L. T. T. Anh, L. N. T. Giang, N. T. Q. Giang, N. T. Anh, D. T. T. Anh and P. V. Kiem, Synthesis and cytotoxic evaluation of fluoro and trifluoromethyl substituents containing novel naphthoquinone-fused podophyllotoxins, Nat. Prod. Commun., 2022, 17(10), 1–6 CrossRef.
- H. T. Nguyen, G. Le-Nhat-Thuy, P. H. Thi, Q. G. N. Thi, T. A. Nguyen, T. H. N. Thi, T. A. D. Thi and T. V. Nguyen, Microwave-assisted three-component synthesis of novel n-arylated-dihydrobenzo[g]quinoline-5,10-diones and their potential cytotoxic activity, Chem. Biodiversity, 2022, 19, e202200359 CrossRef.
- Y. Hitotsuyanagi, M. Fukuyo, K. Tsuda, M. Kobayashi, A. Ozeki, H. Itokawa and K. Takeya, 4-Aza-2,3-dehydro-4-deoxypodophyllotoxins: simple aza-podophyllotoxin analogues possessing potent cytotoxicity, Bioorg. Med. Chem. Lett., 2000, 10, 315–317 CrossRef.
- H. Naeimi, Z. Rashid, A. H. Zarnani and R. Ghahremanzadeh, An Efficient one-pot multicomponent synthesis of 4-aza-podophyllotoxin derivatives in ionic liquid, J. Chem., 2013, 2013, 1–9 Search PubMed.
- X. Yang, C. Zhang and L. Wu, l -Proline catalyzed three-component synthesis of para-naphthoquinone–4-aza-podophyllotoxin hybrids as potent antitumor agents, RSC Adv., 2015, 5, 18945–18951 RSC.
- H. T. Nguyen, K. T. Van, H. Pham-The, J. Braire, P. H. Thi, T. A. Nguyen, Q. G. N. Thi, T. A. D. Thi, G. Le-Nhat-Thuy, T. A. L. Thi, D. V. Ngoc and T. N. Van, Synthesis, molecular docking analysis and in vitro evaluation of new heterocyclic hybrids of 4-aza-podophyllotoxin
as potent cytotoxic agents, RSC Adv., 2024, 14, 1838–1853 RSC.
- H. T. Nguyen, Q. G. N. Thi, T. H. N. Thi, P. H. Thi, G. Le-Nhat-Thuy, T. A. D. Thi, B. Le-Quang, H. Pham-The and T. V. Nguyen, Synthesis and biological activity, and molecular modelling studies of potent cytotoxic podophyllotoxin-naphthoquinone compounds, RSC Adv., 2022, 12, 22004–22019 RSC.
- Q. G. N. Thi, G. Le-Nhat-Thuy, T. A. D. Thi, P. H. Thi, A. N. Tuan, T. H. N. Thi, T. T. Nguyen, T. N. Ha, H. H. Mai and T. V. Nguyen, Synthesis of novel potent cytotoxicy podophyllotoxin-naphthoquinone compounds via microwave-assisted multicomponent domino reactions, Bioorg. Med. Chem. Lett., 2021, 37, 127841 CrossRef.
- T. S. Morais, Recent advances in the development of hybrid drugs, Pharmaceutics, 2024, 16(7), 889 CrossRef PubMed.
- M. Dabiri, Z. N. Tisseh, M. Bahramnejad and A. Bazgir, Sonochemical multi-component synthesis of spirooxindoles, Ultrason. Sonochem., 2011, 18, 1153–1159 CrossRef CAS.
- H. T. Nguyen, H. Pham-The, A. N. Tuan, H. N. T. Thu, T. A. D. Thi, G. Le-Nhat-Thuy, P. H. Thi, Q. G. N. Thi and T. V. Nguyen, Improved synthesis, molecular modeling and anti-inflammatory activity of new fluorinated dihydrofurano-naphthoquinone compounds, Bioorg. Med. Chem. Lett., 2024, 104, 129714 CrossRef CAS.
- G. Le-Nhat-Thuy, T. A. D. Thi, P. H. Thi, Q. G. N. Thi, H.-T. Nguyen, D. V. Ngoc, T.-A. Nguyen and T. V. Nguyen, Multicomponent synthesis of novel 3-benzoyl-4h-benzo[g]chromene-5,10-dione derivatives, Tetrahedron Lett., 2021, 75, 153215 CrossRef CAS.
- H. T. Nguyen, T. H. N. Thi, J. Braire, T. A. D. Thi and T. N. Van, Microwave-assisted multicomponent synthesis of new 6-arylated 5-hydroxy-benzo[ a ]phenazine derivatives and their potential anti-inflammatory activity, ChemistrySelect, 2023, 8, e202204376 CrossRef CAS.
- H. T. Nguyen, A. N. Tuan, T. A. D. Thi, K. T. Van, G. Le-Nhat-Thuy, P. H. Thi, Q. G. N. Thi, C. B. Thi, H. T. Quang and T. V. Nguyen, Synthesis, in vitro A-glucosidase, and acetylcholinesterase inhibitory activities of novel indol-fused pyrano[2,3-D]pyrimidine compounds, Bioorg. Med. Chem. Lett., 2024, 98, 129566 CrossRef CAS PubMed.
- H. T. Nguyen, H. N. Van, P. H. Thi, T. A. D. Thi, G. Le-Nhat-Thuy, Q. G. N. Thi, A. N. Tuan, C. B. Thi, H. T. Quang and T. V. Nguyen, Synthesis and cytotoxic evaluation of new fluoro and trifluoromethyl substituents containing chromeno[2,3- d ]pyrimidines, ChemistrySelect, 2023, 8, e202300227 CrossRef CAS.
- N. H. Thanh, N. T. Q. Giang, N. V. Ha, H. T. Phuong, L. N. T. Giang, N. T. Anh, B. T. Cham, L. D. Huy, D. T. T. Anh, P. V. Kiem and N. V. Tuyen, Synthesis and evaluation of acetylcholinesterase inhibitory and cytotoxic activities of pyrano[2,3- d ]pyrimidines, Nat. Prod. Commun., 2023, 18(9), 1–10 CrossRef.
- N. H. Thanh, N. T. T. Ha, N. T. Tra, L. T. T. Anh, N. T. Son, N. V. Tuyen and P. V. Kiem, Bannaxanthone E induced cell-cycle arrest and apoptosis in human lung cancer cell line, Nat. Prod. Commun., 2021, 16(11), 1–6 Search PubMed.
- V. A. Fadok, D. L. Bratton, S. C. Frasch, M. L. Warner and P. M. Henson, The role of phosphatidylserine in recognition of apoptotic cells by phagocytes, Cell Death Differ., 1998, 5, 551–562 CrossRef.
- H. A. Andree, C. P. Reutelingsperger, R. Hauptmann, H. C. Hemker, W. T. Hermens and G. M. Willems, Binding of vascular anticoagulant alpha (VAC alpha) to planar phospholipid
bilayers, J. Biol. Chem., 1990, 265, 4923–4928 CrossRef PubMed.
- S. L. Rath and S. Senapati, Molecular basis of differential selectivity of cyclobutyl-substituted imidazole inhibitors against cdks: insights for rational drug design, PLoS One, 2013, 8, e73836 CrossRef PubMed.
- P. D. Jeffrey, A. A. Russo, K. Polyak, E. Gibbs, J. Hurwitz, J. Massagué and N. P. Pavletich, Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex, Nature, 1995, 376, 313–320 CrossRef PubMed.
- M. Mapelli, L. Massimiliano, C. Crovace, M. A. Seeliger, L.-H. Tsai, L. Meijer and A. Musacchio, Mechanism of cdk5/p25 binding by cdk inhibitors, J. Med. Chem., 2005, 48, 671–679 CrossRef PubMed.
- M. D. Vivo, G. Bottegoni, A. Berteotti, M. Recanatini, F. L. Gervasio and A. Cavalli, Cyclin-dependent kinases: bridging their structure and function through computations, Future Med. Chem., 2011, 3, 1551–1559 CrossRef.
- E. M. Velázquez-Delgado and J. A. Hardy, Zinc-mediated allosteric inhibition of caspase-6, J. Biol. Chem., 2012, 287, 36000–36011 CrossRef.
- D. T. M. Dung, E. J. Park, D. T. Anh, P.-T. Hai, L. Q. Bao, A. Y. Ji, J. S. Kang, T. T. Tung, S.-B. Han and N.-H. Nam, Design, synthesis and evaluation of novel (E)-N'-((1-(4-chlorobenzyl)-1H-indol-3- yl)methylene)-2-(4-oxoquinazolin-3(4H)-yl)acetohydrazides as antitumor agents, Anti-Cancer Agents Med. Chem., 2022, 22, 2586–2598 CrossRef PubMed.
- L. C. Huan, D. T. Anh, B. X. Truong, P. H. Duc, P.-T. Hai, L. Duc-Anh, L.-T.-T. Huong, E. J. Park, H. J. Lee, J. S. Kang, P.-T. Tran, D. T. T. Hai, D. T. K. Oanh, S.-B. Han and N.-H. Nam, New acetohydrazides incorporating 2-oxoindoline and 4-oxoquinazoline: synthesis and evaluation of cytotoxicity and caspase activation activity, Chem. Biodiversity, 2020, 17, e1900670 CrossRef.
- L. Fu, S. Shi, J. Yi, N. Wang, Y. He, Z. Wu, J. Peng, Y. Deng, W. Wang, C. Wu, A. Lyu, X. Zeng, W. Zhao, T. Hou and D. Cao, ADMETlab 3.0: an updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support, Nucleic Acids Res., 2024, 52, W422–W431 CrossRef.
- H. Pham-The, T. Garrigues, M. Bermejo, I. González-Álvarez, M. C. Monteagudo and M. Á. Cabrera-Pérez, Provisional classification and in silico study of biopharmaceutical system based on caco-2 cell permeability and dose number, Mol. Pharmaceutics, 2013, 10, 2445–2461 CrossRef.
- J. A. Castillo-Garit, G. M. Casanola-Martin, H. Le-Thi-Thu, H. Pham-The and S. J. Barigye, A simple method to predict blood-brain barrier permeability of drug- like compounds using classification trees, Med. Chem., 2017, 13, 664–669 Search PubMed.
- H. Pham-The, I. González-Álvarez, M. Bermejo, T. Garrigues, H. Le-Thi-Thu and M. Á. Cabrera-Pérez, The use of rule-based and qspr approaches in AMDE profiling: a case study on caco-2 permeability, Mol. Inf., 2013, 32, 459–479 CrossRef CAS PubMed.
- H. Pham-The, M. Á. Cabrera-Pérez, N.-H. Nam, J. A. Castillo-Garit, B. Rasulev, H. Le-Thi-Thu and G. M. Casañola-Martin, In silico assessment of ADME properties: advances in caco-2 cell monolayer permeability modeling, Curr. Top. Med. Chem., 2019, 18, 2209–2229 CrossRef.
- Y. Perez-Castillo, A. Sánchez-Rodríguez, E. Tejera, M. Cruz-Monteagudo, F. Borges, M. N. D. S. Cordeiro, H. Le-Thi-Thu and H. Pham-The, A desirability-based multi objective approach for the virtual screening discovery of broad-spectrum anti-gastric cancer agents, PLoS One, 2018, 13, e0192176 CrossRef PubMed.
- L. C. Huan, C. V. Phuong, L. C. Truc, V. N. Thanh, H. Pham-The, L.-T.-T. Huong, N. T. Thuan, E. J. Park, A. Y. Ji, J. S. Kang, S.-B. Han, P.-T. Tran and N.-H. Nam, (E)- N’ -Arylidene-2-(4-oxoquinazolin-4(3 H)-yl) acetohydrazides: Synthesis and evaluation of antitumor cytotoxicity and caspase activation activity, J. Enzyme Inhib. Med. Chem., 2019, 34, 465–478 CrossRef CAS PubMed.
- M. A. C. Neves, M. Totrov and R. Abagyan, Docking and scoring with ICM: the benchmarking results and strategies for improvement, J. Comput.-Aided Mol. Des., 2012, 26, 675–686 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Ket Tran Van*bd,
Hai Pham-The
ab,
Ket Tran Van*bd,
Hai Pham-The