
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-valent oxovanadium metallosupramolecular species as catalysts for the oxidation of benzyl alcohol derivatives†
Edi Topić a,
Josipa Sarjanovića,
Danijela Musijab,
Mirna Mandarića,
Tomica Hrenara,
Jana Pisk*a and
Višnja Vrdoljak*a
a,
Josipa Sarjanovića,
Danijela Musijab,
Mirna Mandarića,
Tomica Hrenara,
Jana Pisk*a and
Višnja Vrdoljak*a
aUniversity of Zagreb, Faculty of Science, Department of Chemistry, Horvatovac 102a, 10000 Zagreb, Croatia. E-mail: jana.pisk@chem.pmf.hr; visnja.vrdoljak@chem.pmf.hr
bUniversity of Zagreb, School of Medicine, Department of Chemistry and Biochemistry, Šalata 3, 10000 Zagreb, Croatia
First published on 4th February 2025
Abstract
Coordination-driven synthesis has been successfully utilized to prepare vanadium-based metallosupramolecular species. By systematically varying synthesis methods and reaction conditions, two series of isostructural oxovanadium(V) coordination polymers [VO(SIH)(OR)]n (where SIH2− represents salicylaldehyde isonicotinoylhydrazonate, and R corresponds to CH3 (1β·0.25CH3OH), C2H5 (2α and 2β·0.25C2H5OH), C3H7 (3α), C4H9 (4α and 4β), and C5H11 (5β)), were synthesized. Additionally, a phase-pure tetranuclear compound [VO(SIH)(OCH3)]4·4CH3OH (1t·4CH3OH) was also prepared. In these compounds the [VO(SIH)] units, featuring the isonicotinoylhydrazonate heterocyclic moiety, are interconnected through V–Nisonicotinoyl coordination bonds. This linkage enables formation of the one-dimensional (1D) zig–zag chain compounds and zero-dimensional (0D) metallocycle, as determined by single crystal X-ray crystallography. This study further explored the formation and transformation of the assemblies, thereby emphasizing the influence of the ancillary OR− ligand. The compounds were also characterized by powder X-ray diffraction, chemical analysis, thermogravimetric (TGA) measurements, and spectroscopic methods (IR-ATR, UV-Vis, and NMR). The catalytic activity of vanadium coordination entities 1t and 1β was tested for the oxidation of benzyl alcohol and its derivatives, including 2-nitrobenzyl, 2-chlorobenzyl, and 2-methylbenzyl alcohol. Several oxidizing agents were used, including tert-butyl hydroperoxide (TBHP) in aqueous and decane solutions, as well as hydrogen peroxide (H2O2). The study also assessed the impact of different solvents, such as toluene, acetonitrile, and methanol, thereby enhancing the understanding of these systems.
Introduction
High-valent metal–organic species are widely recognized as active oxidants in the catalytic oxidation of organic compounds, serving as a cornerstone in both academic research and industrial applications.1–4 Their utilization requires careful consideration of several factors to optimize performance and efficiency. These factors include not only the selection of the central metal but also the choice and variation of ligands. Ligand substituents significantly influence the solubility, stability, electronic structure, and steric properties of the complex system, all of which ultimately affect catalyst performance.Among these systems, oxovanadium complexes are particularly notable due to the advantageous characteristics of the vanadium metal center.5–7 Vanadium exhibits a variety of oxidation states, different coordination numbers, enhanced oxophilicity, and a good Lewis acidity.8,9 These properties make vanadium-based catalysts highly effective in promoting the oxidation of organic substrates, including saturated, aromatic, and olefinic hydrocarbons.10–12 Despite these advantages, the development and application of high-valent oxovanadium metallosupramolecular compounds as catalysts remain relatively underexplored. High-valent vanadium complexes tend to form oxo- or alkoxo-bridged compounds, limiting their use in creating well-defined supramolecular structures.13–15 To the best of our knowledge, there are only rare examples of VO coordination polymers or cyclic assemblies [VO(L)(L′)]x (L, L′ = ligands) comprising nitrogen and oxygen donor ligands.16–20
In contrast, significant progress in this area has been made for high-valent metallosupramolecular species, such as molybdenum(VI) and tungsten(VI) assemblies bearing terminal oxo co-ligands.21–25 These species are synthesized using carefully designed organic ligands and strict control of the reaction conditions, preventing oligomerization of the highly reactive metal-oxo units through the oxygen atom.21–24 The size, shape of these assemblies, and the functional groups of all building units are also essential and determine their properties and potential application.25,26
From a structural perspective, one could anticipate observations of multiple crystal forms for the same metallosupramolecular assembly, however, polymorphism in metal–organic chemistry is seldom reported on; less than 0.5% of metal–organic structures in the Cambridge Structural Database have corresponding polymorphs. Moreover, the occurrence of polymorphism is unevenly distributed, particularly among first-row transition metals. Among these, iron and copper compounds exhibit the highest frequency of polymorph occurrence, aligning with their abundant entries in the database. Vanadium compounds, however, stand out as an exception compared to their periodic table neighbours, displaying a relatively high number of polymorphs compared to the total number of published crystal structures (Fig. 1).
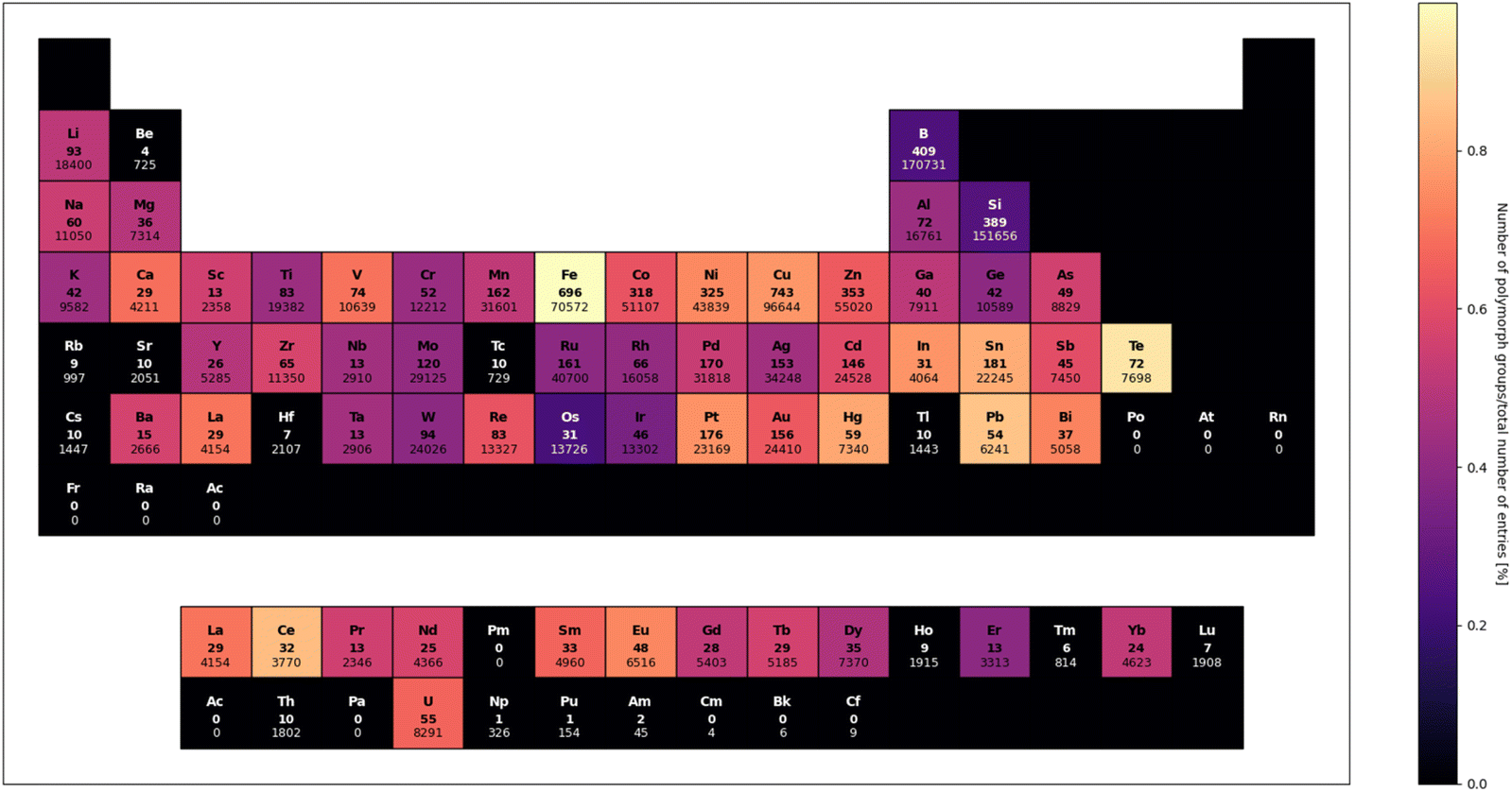 | ||
| Fig. 1 Periodic table of polymorph occurrence among crystal structures containing metal or semi-metal elements in Cambridge Structural Database. Polymorph group was defined as set of structures with the same refcode root and distances between reduced unit cell vectors of at least 0.5. Number in bold font represents number of polymorph groups (i.e. groups of crystal structures with same composition and molecular structure, but different unit cell or packing of molecules). Number in regular font represents total number of entries containing a particular element. Colour of element box (1) maps normalized polymorph occurrence or (2) is black if number of found polymorph groups is less or equal to 10. | ||
Focusing on neutral vanadium complexes, a few studies have reported polymorphs of vanadium(III), oxovanadium(IV), (alkoxo)oxovanadium(V), and dioxovanadium(V) species. These polymorphs are associated with a variety of ligands, including pyridine derivatives,27,28 Schiff bases,29–31 hydrazones, and related compounds.32–36 Additionally, polymorphism has been observed in heteropolymetallic coordination polymers containing dioxovanadium(V) ions37,38 and vanadium complexes with porphyrin derivatives.39–41 Interestingly, polymorphism is predominantly found in compounds lacking strong directional intermolecular interactions, such as hydrogen or halogen bonding. This suggests that vanadium compounds are prone to polymorphism in the absence of significant supramolecular synthons. However, this observation also implies that incorporating well-defined supramolecular synthons in the ligand design might hinder exploration of the polymorphic landscape.
Despite these findings, the structural chemistry of vanadium remains relatively underexplored compared to other transition metal complexes. Consequently, these insights should be regarded as preliminary rather than definitive, until more structural data is collected.
Although constructing metallosupramolecular architectures with oxovanadium(V) building blocks is more challenging, the potential of these types of compounds is highly promising and worth exploring.42 In this study, we therefore explored their synthesis by employing a coordination-driven self-assembly methodology as an effective tool for constructing discrete or polynuclear architectures.43–47 The [VO(SIH)] structural unit, incorporating the salicylaldehyde isonicotinoylhydrazonate ligand (SIH2−) has been selected since the ligand features a heterocyclic moiety that provides a coordinating platform with a nitrogen donor atom. The choice of this ligand without additional substituents ensures that neither electron donating nor electron withdrawing groups interfere with the formation of the desired superstructures. In the presence of primary alcohols ROH (CnH2n+1OH, n = 1–5) building units can combine and yield metallocyclic [VO(SIH)(OR)]4 or polymeric assemblies [VO(SIH)(OR)]n. We also explored transformation of the assemblies by alkoxo OR− ligand exchange.
By changing synthesis method and reaction conditions, two series of isostructural polymorphs were obtained (where R = CH3 (1β·0.25CH3OH), C2H5 (2α and 2β·0.25C2H5OH), C3H7 (3α), C4H9 (4α and 4β), and C5H11 (5β)). The structures of these oxovanadium(V) polymers and metallocyclic compound [VO(SIH)(OCH3)]4·4CH3OH (1t·4CH3OH), were elucidated using single-crystal X-ray diffraction (SCXRD) and powder X-ray diffraction (PXRD) analysis. They were further characterized using elemental analysis, infrared (IR-ATR), UV-Vis, and NMR spectroscopic methods. The thermal stability of the assembled structures was also studied using thermogravimetric analysis (TGA).
These above mentioned findings suggest that similar strategies could be employed to advance the field of high-valent oxovanadium catalysts, opening new avenues for the development of efficient and selective oxidation processes. Due to it, we chose to investigate the oxidation of benzyl alcohol as it is extensively used as a precursor in the synthesis of various products.48,49 Common oxidizing agents for the oxidation processes, potassium permanganate and sodium dichromate, are causing environmental issues.50 While nitric acid is another commonly used and cost-effective oxidant its use leads to the formation of side products that are both toxic and environmentally polluting. Environmentally acceptable oxidants would be hydrogen peroxide or molecular oxygen which were tested for benzaldehyde oxidation assisted by transition metals such as Co, Fe, Sn–W, V and Au.51–56
Our research was focused on the catalytic performance of V-coordination entities 1t and 1β whose activity was compared to the compounds [VO2(HSIH)] (6), V2O5, and [MoO2(SIH)]n.57 They were assessed in oxidizing benzyl alcohol and its derivatives: 2-nitrobenzyl, 2-chlorobenzyl, and 2-methylbenzyl alcohol. The oxidation of benzyl alcohol derivatives to the corresponding carbonyl compounds was explored using various oxidizing agents, including tert-butyl hydroperoxide (TBHP) in aqueous and decane solutions, as well as hydrogen peroxide (H2O2), while also evaluating the influence of different solvents such as toluene, acetonitrile, and methanol.
Results and discussion
Synthesis and characterization of metallocycle and two series of coordination polymers
The hydrazone derived from salicylaldehyde and isonicotinoylhydrazide was synthesized as described in the literature.58 Supramolecular coordination complexes were afforded through the reaction of NH4VO3 or [VO(acac)2], hydrazone (H2SIH), and the corresponding primary alcohol (CnH2n+1OH, n = 1–5) (Table 1, Scheme 1, Procedures I and II). The solvent was evaporated leaving a dark red coloured solution from which a crystalline precipitate formed. All compounds were characterized using various analytical and spectroscopic methods and were identified as tetranuclear metallocyclic [VO(SIH)(OCH3)]4·4CH3OH (1t·4CH3OH), and infinite zig–zag chain products [VO(SIH)(OR)]n (where R = CH3 (1β·0.25CH3OH), C2H5 (2α and 2β·0.25C2H5OH), C3H7 (3α), C4H9 (4α and 4β), and C5H11 (5β), Table 1). Their formation was driven by the denticity of the ligand and donor atom positions within the linking unit. The crystalline powders were analysed using PXRD and the results were then compared to the simulated powder patterns obtained from the SCXRD data (Fig. S1–S3, see ESI†). In the case of polynuclear assemblies, two series of polymorphic forms were characterized. The structures of 2α, 3α, and 4α belong to the first arrangement (Fig. S2†), while the structures of 1β·0.25CH3OH, 2β·0.25CH3OH, 4β, and 5β belong to the second one (Fig. S3†). The observed similarities in the powder diffractograms within each series facilitate the determination of the polymorphic type associated for each compound. Despite our attempts, we didn't obtain 1α, 3β, and 5α forms by the mentioned procedures. Following Procedure I, 4β polymorph can be isolated upon the evaporation of the reaction solution on the same day. In contrast, the 4α form is generated when the solution remains undisturbed for an extended period.| [VO(SIH)(OR)]x | OR | x | Procedure |
|---|---|---|---|
| 1t·4CH3OH | OCH3 | 4 | I |
| 1β·0.25CH3OH | OCH3 | n | II |
| 2α | OC2H5 | n | I or III |
| 2β·0.25C2H5OH | OC2H5 | n | II |
| 3α | OC3H7 | n | I or III |
| 4α | OC4H9 | n | I or II or III |
| 4β | OC4H9 | n | I |
| 5β | OC5H11 | n | I or III |
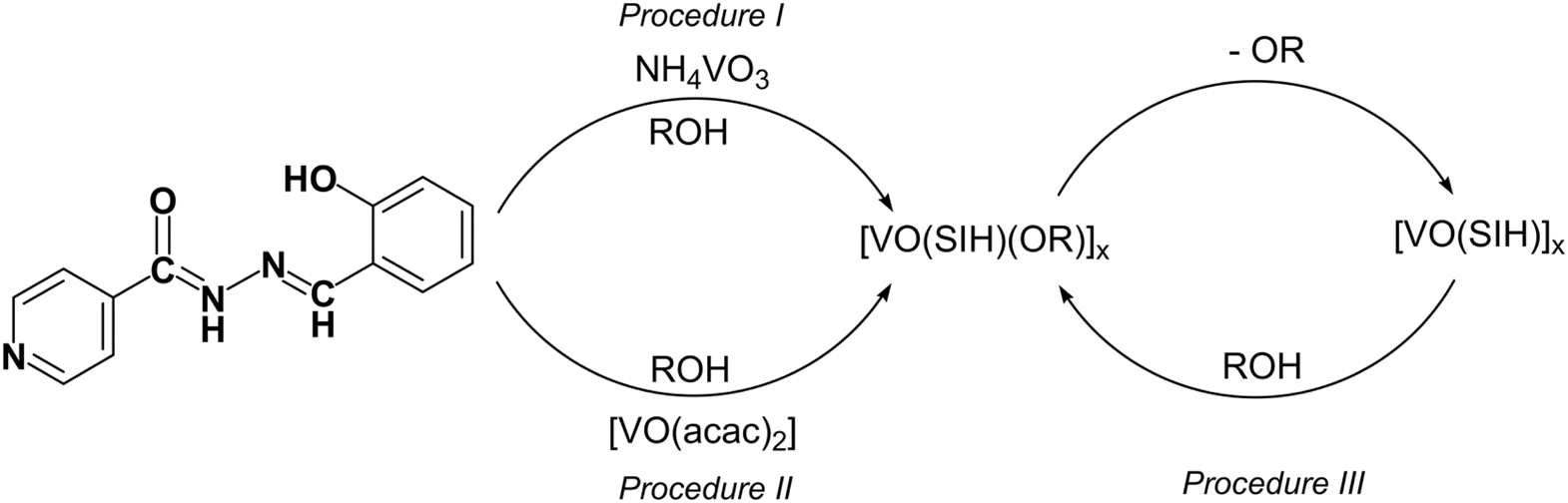 | ||
| Scheme 1 Structure of the H2SIH ligand and synthetic pathways to oxovanadium(V) supramolecular species. | ||
Furthermore, by immersing crystals of the tetramer [VO(SIH)(OCH3)]4·4CH3OH (1t·4CH3OH) in an appropriate coordinating solvents ROH (R = CnH2n+1, n = 2–5), the ancillary ligand was replaced with a different one, OR. However, in the case of ethanol and butanol, it resulted in a mixture of polymorphs 2α and 2β·0.25CH3OH, 4α and 4β, respectively. A similar result was obtained by higher alkoxo ligand substitution when submersing crystals in methanol. Polymer and tetramer with coordinated methoxo ligand were achieved as a mixture.
We were therefore interested in studying the thermally assisted structural transformation of [VO(SIH)(OCH3)]4·4CH3OH (1t·4CH3OH) in solvent-free conditions. Consistent with findings on its thermal stability (vide infra), a crystalline sample of 1t·4CH3OH was gradually heated from the ambient temperature up to 165 °C with a heating rate of 10 °C min−1. After being held at that temperature for one hour, the amorphous residue of the thermally activated complex [VO(SIH)]x was immersed in the corresponding ROH for one week (Procedure III). This procedure resulted in the formation of 1t, 2α, 3α, 4α, and 5β which were identified by IR-ATR and X-ray powder diffraction method (Fig. S4, see ESI†). The formation of polymers can be explained by the cleavage of the V–Nisonicotinoyl bonds in the metallocycle and formation of new V–Nisonicotinoyl bonds in solution. It is worth noting that the formation of a supramolecular architecture indirectly through the solid-state transformation of vanadium supramolecular complex has never been reported.
In the reaction between the NH4VO3 and hydrazone ligand in methanol, followed by the evaporation in an airstream, the [VO2(HSIH)] (6) compound crystallized. A single-crystal X-ray study of 6 confirmed the structure to be mononuclear. However, it was previously reported (CSD refcode LAMLES).59
Description of molecular and crystal structures
High-quality single crystals suitable for single crystal X-ray diffraction were obtained for total of 7 compounds, namely 1t·4CH3OH, 2α, 3α, 4α, 1β·0.25CH3OH, 2β·0.25C2H5OH and 5β. General and crystallographic data can be found in ESI, Tables S1–S2 and Fig. S5–S7.† Determined crystal structures represent a diverse set of metallosupramolecular assemblies based on the same building block – {VO(OR)(L)} (Fig. 2, top left). The geometry of the monomeric building unit is practically identical across the observed structures: vanadium center is situated in an expected ONO coordination environment provided by a doubly deprotonated hydrazonato ligand in its enolato-imino tautomeric form (Fig. S5†). In each compound, oligomerization or polymerization is facilitated through coordination of the nitrogen atom from an ancillary pyridyl functional group on the sixth coordination site of the oxovanadium(V) centre. | ||
| Fig. 2 [VO(OR)(L)] monomer (L = SIH) can oligomerize into porous tetramer 1t, or polymerize into polymorphs α (densely packed 1D chains) or β (porous assembly of 1D chains). Structures of α and β are shown as structural overlays of complexes with 2 and 3 different alkoxy chain lengths, respectively, emphasizing the isostructurality of the assemblies. | ||
Tetranuclear complex 1t crystallizes in the tetragonal space group I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) . Although the complex has a fourth order rotoinversion symmetry (Fig. 2, top right), the complex conforms to a shape of a diagonally folded square, with vanadium centers significantly shifted from the tetramer equatorial plane (Fig. S7†). Moreover, the complex molecules are stacked along the ring axis, forming large channels comprising up to 20.7% of unit cell volume. This arrangement of molecules is certainly unexpected and could represent a new moment in supramolecular metal–organic frameworks (sMOF), and the detailed investigations are underway. The contents of the void volume could not be modelled from the difference Fourier map, but according to the TGA analysis the voids are natively filled with residual solvent molecules with ratio V
. Although the complex has a fourth order rotoinversion symmetry (Fig. 2, top right), the complex conforms to a shape of a diagonally folded square, with vanadium centers significantly shifted from the tetramer equatorial plane (Fig. S7†). Moreover, the complex molecules are stacked along the ring axis, forming large channels comprising up to 20.7% of unit cell volume. This arrangement of molecules is certainly unexpected and could represent a new moment in supramolecular metal–organic frameworks (sMOF), and the detailed investigations are underway. The contents of the void volume could not be modelled from the difference Fourier map, but according to the TGA analysis the voids are natively filled with residual solvent molecules with ratio V![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CH3OH equal to unity.
:CH3OH equal to unity.
Besides this tetranuclear complex, two major groups of compounds representing (pseudo)polymorphic pairs were identified, nα and nβ, although a complete structural determination of both polymorphs was only achieved for compound 2, namely 2α and 2β·0.25C2H5OH.
Crystallographic analysis showed that compounds in the α form crystallize as densely packed 1D polymeric chains in space group Pbca, whereas those in the β form crystallize in space group Ibca with a different arrangement of 1D polymeric chains, but with the same 2C1 underlying topology (Fig. 2, bottom, Fig. S6†). As evidenced by the structural data, as well as PXRD data, the isolated α and β solid forms are isostructural within each group, up to a difference in the alkoxy chain. The coordination environment of vanadium ion remains consistent throughout the structures. The arrangement of chains in β polymorphs allows for significant void volume in the structures. For instance, 1β contains approximately 9.7% void volume, 2β contains about 12.7%, and 5β also has an appreciable void volume, but its determination is obstructed by disordered pentoxy chains, which partially occupy the voids. It is nevertheless inferred that β-type polymorphs generally permit the formation of channels enclosed by partially disordered alkoxy groups. As in the case of 1t, the difference Fourier map in the channels is featureless, so the final composition is inferred from the void volume, solvent molar volume and TGA analysis.
All three types of assemblies are characterized by the lack of strong or directed supramolecular interactions, as there are no suitable hydrogen bond donors. In line with the introductory findings, the lack of supramolecular interactions favours the existence of polymorphs. However, to the best of our knowledge, the α–β polymorphic system could be the first one where such a stark difference in porosity is observed. This fact, along with no significant stability differences between the polymorphs, could serve as motivation for investigation of this polymorphic system (and related systems lacking strong supramolecular interactions) as solid-state porosity-on-demand materials.
Principal component analysis
We focused on gaining a more profound comprehension of this reaction system. The reaction between NH4VO3 and H2SIH (with initial reactant concentrations of 4.0 × 10−5 mol dm−3) was studied in methanol at ambient temperature using time-dependent UV-Vis spectroscopy (Fig. 3). The time-dependent spectra were analyzed by employing principal component analysis, a 2nd-order tensor decomposition tool. The results indicated that two principal components could accurately represent the initial dataset (comprising 221 spectra). These components accounted for more than 98% of the total variance in the original data. | ||
| Fig. 3 Time-dependent UV-Vis spectra of a reaction mixture of NH4VO3 and H2SIH in methanol at room temperature. | ||
The first principal component accounted for 80.79% of the total variance highlighting a significant reaction involving the coordination of the hydrazonato ligand to vanadium. The second component accounted for 17.35% of the total variance, reflecting a secondary reaction occurring within the system (Fig. 4). Furthermore, the analysis of the UV spectra from the V-hydrazonato complex showed a complete conversion of the initially formed hydrazonato complex into the related dioxovanadium(V) species in very dilute methanolic solutions.
 | ||
| Fig. 4 Principal component scores for the reaction of NH4VO3 and H2SIH in methanol at room temperature monitored in time by UV-Vis spectrophotometry. | ||
Spectroscopic characterization
In methanolic solution, H2SIH exhibits distinct absorption maxima at wavelengths of 214 nm, 288 nm, and 333 nm, along with several shoulder peaks (see ESI†). The absorption band at 214 nm is associated with π → π* transitions occurring in the aromatic rings. The bands observed at 288 nm and 333 nm correspond to π → π* and n → π* transitions, respectively, which are associated with the azomethine and carbonyl functionalities.60,61 In the spectra of methanolic solutions of complexes 1t and 1–6, these intraligand transitions are exhibited at around 230 nm and 320 nm alongside the band at about 404 nm that is assignable to charge transfer from the coordinated hydrazonato ligand to vanadium (LMCT band).60
The 1H NMR spectra of 1–5 are similar, except for resonances attributed to free ROH. The 13C NMR spectra revealed deshielding of the chemical shifts for C1, C4, and C12 carbons, with maximum shifts of 3.00 ppm, 6.81 ppm, and 5.99 ppm, respectively, because of metal complexation. On the other hand, in highly diluted deuterated methanol solutions, it appears that the coordination of the pyridine ring to the coordinatively unsaturated vanadium center is not maintained. The isonicotinoyl carbons C5–C10 exhibited minimal changes in chemical shifts compared to the free ligand, with a maximum of Δδ = 0.98 ppm.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NH, and CO bands (at 3200 cm−1, 3180 cm−1, and 1683 cm−1, respectively),58 which are characteristic of free hydrazone ligand H2SIH, disappeared upon vanadium–ligand complexation (Fig. S11 and S12 in the ESI†). The presence of bands belonging to CNimine (at about 1620 cm−1), C–Ohydrazonato (at about 1345 cm−1), and C–Ophenolic (at about 1275 cm−1) in the spectra of 1t and 1–5 indicated SIH2− coordination to the metal centre via oxygen and nitrogen donor atoms.21–23 This is further confirmed by the weaker intensity bands for V–Nimine at 750 cm−1, V–Ohydrazonato at 580 cm−1, and V–Ophenolato at 560 cm−1. The spectra also exhibited a medium-intensity stretching band at 1600 cm−1 associated with CN of the isonicotinoyl pyridine ring moiety. This band is shifted to a lower wavenumber, when compared to free hydrazone, indicating coordination of the isonicotinic-nitrogen atom to the {VO}3+ core. A medium intensity band observed at 970 cm−1 was assigned to N–N stretching vibrations.62
N–NH, and CO bands (at 3200 cm−1, 3180 cm−1, and 1683 cm−1, respectively),58 which are characteristic of free hydrazone ligand H2SIH, disappeared upon vanadium–ligand complexation (Fig. S11 and S12 in the ESI†). The presence of bands belonging to CNimine (at about 1620 cm−1), C–Ohydrazonato (at about 1345 cm−1), and C–Ophenolic (at about 1275 cm−1) in the spectra of 1t and 1–5 indicated SIH2− coordination to the metal centre via oxygen and nitrogen donor atoms.21–23 This is further confirmed by the weaker intensity bands for V–Nimine at 750 cm−1, V–Ohydrazonato at 580 cm−1, and V–Ophenolato at 560 cm−1. The spectra also exhibited a medium-intensity stretching band at 1600 cm−1 associated with CN of the isonicotinoyl pyridine ring moiety. This band is shifted to a lower wavenumber, when compared to free hydrazone, indicating coordination of the isonicotinic-nitrogen atom to the {VO}3+ core. A medium intensity band observed at 970 cm−1 was assigned to N–N stretching vibrations.62Bands at about 710 cm−1 characteristic for V–N bonds corroborated the metallosupramolecular complex formation. The coordination of RO− in complexes 1t and 1–5 was supported by a new absorption band typical of C–OOR at approximately 1040 cm−1.63 This band was missing in the spectra of the thermally activated complex [VO(SIH)]x. It should be noted that C–OOR stretching frequencies in metallosupramolecular species slightly differ from those in the corresponding free alcohol.
The spectra of complexes 1t and 1–5 displayed intense bands in the region of 970–956 cm−1 (Fig. S11 and S12†), which were characteristic of the {VO}3+ core.64 It should be noted that there is a certain similarity of spectra belonging to the same series of polymorphs.
It is worth noting that the absence of coordinated isonicotinoyl moiety and alkoxide co-ligand in the case of the mononuclear complex 6 resulted in a slightly lower intensity stretching bands at 953 cm−1 and 942 cm−1 belonging to the {OVO}+ core. The bands characteristic for C–Ophenolic, CN, and C–Ohydrazone were accompanied by the broad band in the range of 2400–2600 cm−1 assigned to the N–H stretching vibration of the protonated HSIH− ligand.65
The thermal stabilities of all compounds were studied using thermogravimetric analysis (Fig. S13–S17 in the ESI†) by heating the crystalline sample in the temperature range of 25–600 °C under an oxygen atmosphere. The TGA analysis of solvates showed a steady decline at the beginning of the thermogram due to the loss of the lattice solvent molecules followed by the escape of oxidized alkoxide products in the temperature ranges of 29–153 °C and 154–237 °C (for 1t·2CH3OH), 25–77 °C and 148–256 °C (for 1β·0.25CH3OH), and 40–144 °C and 148–259 °C (for 2β·0.25C2H5OH). The onset temperatures of the first step in TGA are very low, signifying the presence of weak noncovalent interactions in the solid state.
All desolvated polymeric compounds exhibited a comparable pattern of thermal decomposition steps. The mass loss process, due to the oxidation of one alkoxide for each metal–organic unit, started above 100 °C (at 148 °C (for 1β), 165 °C (for 2α), 148 °C (for 2β), 185 °C (for 3α), 135 °C (for 4α), 114 °C (for 4β), 149 °C (for 5β)) with the butoxide derivative being the most unstable and propoxide the most stable. This step was followed by the final hydrazone decomposition of the corresponding complex.
 | ||
| Fig. 5 Kinetic profile of benzyl alcohol conversion with catalyst 1t. Light blue presents the reaction with TBHP in decane, purple with TBHP in water, orange with TBHP in water and toluene, yellow with TBHP in water and methanol, grey with TBHP in water and acetonitrile and green with hydrogen peroxide and acetonitrile. | ||
| Catalyst | 1t | 1β | 6 | [MoO2(SIH)]n | V2O5 | [VO(SIH)]x | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Time/min | 120 | 20 | 120 | 20 | 120 | 20 | 120 | 20 | 120 | 20 | 120 | 20 | |
| a Substrate consumed at the end of the reaction.b Formed aldehyde per converted alcohol at the end of the reaction.c n(substrate) transformed/n(catalyst)/time(h) at 20 min.d n(substrate) transformed/n(catalyst) at the end of reaction. | |||||||||||||
| TBHP in water | |||||||||||||
| No solvent | Conversiona | 85 | 23 | 90 | 70 | 75 | 16 | 14 | 9 | 78 | 3 | 79 | 36 |
| Selectivityb | 18 | 74 | 12 | 33 | 29 | 77 | 50 | 31 | 27 | 2 | 26 | 60 | |
| TOF20 minc | 140 | 391 | 92 | 59 | 42 | 223 | |||||||
| TONd | 174 | 168 | 149 | 30 | 323 | 163 | |||||||
| Toluene | Conversion | 68 | 19 | 63 | 16 | 50 | 3 | 5 | 1 | 30 | 7 | — | |
| Selectivity | 3 | 70 | 43 | 70 | 56 | 89 | 47 | 63 | 79 | 72 | |||
| TOF20 min | 114 | 93 | 21 | 3 | 35 | ||||||||
| TON | 136 | 123 | 100 | 10 | 52 | ||||||||
| MeCN | Conversion | 47 | 9 | 54 | 11 | 39 | 12 | 6 | 3 | 43 | 8 | ||
| Selectivity | 66 | 90 | 58 | 85 | 61 | 35 | 53 | 39 | 65 | 63 | |||
| TOF20 min | 46 | 61 | 70 | 17 | 49 | ||||||||
| TON | 83 | 102 | 75 | 12 | 82 | ||||||||
| MeOH | Conversion | 53 | 12 | 52 | 4 | 47 | 3 | 10 | 2 | 45 | 8 | ||
| Selectivity | 49 | 68 | 49 | 90 | 54 | 88 | 30 | 48 | 55 | 63 | |||
| TOF20 min | 63 | 24 | 17 | 15 | 45 | ||||||||
| TON | 93 | 102 | 91 | 21 | 82 | ||||||||
|
|||||||||||||
| TBHP in decane | |||||||||||||
| No solvent | Conversion | 85 | 60 | 87 | 67 | 84 | 40 | 14 | 6 | 82 | 46 | 82 | 49 |
| Selectivity | 16 | 38 | 13 | 31 | 16 | 48 | 68 | 46 | 20 | 53 | 19 | 39 | |
| TOF20 min | 346 | 402 | 226 | 34 | 282 | 284 | |||||||
| TON | 163 | 175 | 161 | 26 | 169 | 160 | |||||||
|
|||||||||||||
| H2O2 | |||||||||||||
| MeCN | Conversion | 33 | 25 | 38 | 30 | 77 | 28 | 11 | 4 | 33 | 28 | 42 | 11 |
| Selectivity | 40 | 42 | 36 | 38 | 26 | 49 | 90 | 99 | 39 | 48 | 30 | 76 | |
| TOF20 min | 165 | 197 | 181 | 23 | 172 | 67 | |||||||
| TON | 72 | 81 | 168 | 22 | 68 | 85 | |||||||
Catalysts 1t and 1β show consistently high conversions across various conditions, particularly with TBHP in decane (85%) and TBHP in water (>85%). These values are comparable to 6 [VO2(HSIH)], suggesting its effectiveness. TOF20 min values are among the highest (346 for 1t and 402 for 1β with TBHP in decane), indicating rapid catalytic activity. Selectivity remains moderate to low across most conditions. For instance, aldehyde selectivity for 1t is only 16% with TBHP in decane and 18% with TBHP in water. This indicates the catalyst prioritizes reactivity over specificity. Catalyst 1β performs slightly better than 1t, with TBHP in decane, it achieves a conversion of 60% and a selectivity of 38%, striking a better balance between reactivity and selectivity. TOF20 min values for 1β (402 with TBHP in decane) are larger than 1t, making it a faster-acting catalyst in benzyl alcohol oxidation. The selectivity parameter is similar for 1β than 1t in aqueous TBHP-driven systems. Fig. 5 illustrates the performance comparison of the tested conditions for catalyst 1t. The data reveal that 1t exhibits the highest activity when TBHP is utilized in decane or water as the oxidant, whereas its activity is significantly reduced when H2O2 and MeCN are employed. Furthermore, the choice of solvent added to TBHP in water substantially affects the conversion rate, with the activity following the order: toluene > methanol > acetonitrile. However, aldehyde selectivity is the highest when TBHP in water and MeCN or MeOH were applied as oxidants. Catalyst [VO2(HSIH)] (6) showed good conversion rates in oxidant-rich conditions, especially with TBHP in water and decane, 75 and 84%, respectively. Aldehyde selectivity in the same oxidants is moderate (13 and 29%, respectively), highlighting its preference for reactivity over product-desired aldehyde.
[MoO2(SIH)]n has low conversion parameters under all the tested conditions, although the aldehyde selectivity is quite high, spanning to 90% when hydrogen peroxide with the addition of acetonitrile is used as an oxidant. It can be noted that the Mo catalyst is not very active for the benzyl alcohol oxidations, no matter the oxidant chosen or solvent added to the reaction mixture.
V2O5 exhibits high conversion rates with TBHP in water (78%) and decane (82%). Conversion decreases in aqueous TBHP-driven systems when the solvent is added. Aldehyde selectivity with TBHP in water is 27%, while in decane is 20%. In contrast, the addition of solvent to TBHP in water increases the selectivity (79% in toluene, 65% in MeCN, 55% in MeOH). It has to be noted that the aldehyde selectivity remains the same through all the reactions and does not decrease with time, highlighting that the overoxidation of aldehyde to the corresponding carboxylic acid is not present.
[VO(SIH)]x has been tested with TBHP in water, decane and H2O2. It can be observed that in TBHP benzyl alcohol oxidation is quite high, but the aldehyde selectivity remains low, while with H2O2 as an oxidant, the conversion of benzyl alcohol and aldehyde selectivity achieve similar values (30% of selectivity and 42% of conversion).
 | ||
| Fig. 6 Kinetic profile of benzyl alcohol conversion with catalyst 1t. Blue circles presents the reaction with 2-nitrobenzyl alcohol, green circles with 2-chlorobenzyl alcohol and red circles with 2-methylbenzyl alcohol. Full circles are reaction with TBHP in water and empty circles with TBHP in decane. | ||
| Catalyst | 1t | 1β | 6 | V2O5 | [VO(SIH)]x | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Time/min | 120 | 20 | 120 | 20 | 120 | 20 | 120 | 20 | 120 | 20 |
| a Substrate consumed at the end of the reaction.b Formed aldehyde per converted alcohol at the end of the reaction.c n(substrate) transformed/n(catalyst)/time(h) at 20 min.d n(substrate) transformed/n(catalyst) at the end of reaction. | ||||||||||
| 2-Chloro benzyl alcohol | ||||||||||
| TBHP in water | ||||||||||
| Conversiona | 87 | 42 | 89 | 55 | 84 | 44 | 58 | 7 | 90 | 45 |
| Selectivityb | 32 | 73 | 26 | 58 | 37 | 96 | 62 | 96 | 27 | 73 |
| TOF20 minc | 251 | 395 | 28 | 44 | 275 | |||||
| TONd | 176 | 213 | 173 | 126 | 181 | |||||
| TBHP in decane | ||||||||||
| Conversion | 88 | 74 | 87 | 61 | 88 | 51 | 86 | 68 | 89 | 74 |
| Selectivity | 30 | 42 | 31 | 40 | 26 | 58 | 29 | 39 | 24 | 43 |
| TOF20 min | 460 | 382 | 334 | 439 | 422 | |||||
| TON | 182 | 180 | 192 | 184 | 171 | |||||
|
||||||||||
| 2-Nitro benzyl alcohol | ||||||||||
| TBHP in water | ||||||||||
| Conversion | 54 | 10 | 71 | 24 | 48 | 11 | 22 | 3 | 53 | 17 |
| Selectivity | 65 | 98 | 41 | 87 | 70 | 70 | 93 | 51 | 64 | 60 |
| TOF20 min | 55 | 132 | 66 | 19 | 93 | |||||
| TON | 103 | 130 | 93 | 39 | 96 | |||||
| TBHP in decane | ||||||||||
| Conversion | 76 | 56 | 78 | 50 | 78 | 24 | 71 | 39 | 79 | 51 |
| Selectivity | 32 | 48 | 28 | 42 | 29 | 57 | 36 | 69 | 28 | 49 |
| TOF20 min | 298 | 240 | 142 | 201 | 273 | |||||
| TON | 133 | 125 | 157 | 122 | 142 | |||||
|
||||||||||
| 2-Methyl benzyl alcohol | ||||||||||
| TBHP in water | ||||||||||
| Conversion | 83 | 21 | 93 | 30 | 90 | 6 | 78 | 3 | 93 | 32 |
| Selectivity | 8 | 73 | 3 | 60 | 5 | 97 | 12 | 87 | 33 | 53 |
| TOF20 min | 117 | 170 | 27 | 15 | 185 | |||||
| TON | 156 | 178 | 144 | 144 | 184 | |||||
| TBHP in decane | ||||||||||
| Conversion | 93 | 65 | 95 | 41 | 86 | 67 | 93 | 67 | 93 | 72 |
| Selectivity | 3 | 7 | 2 | 22 | 6 | 11 | 3 | 15 | 7 | 8 |
| TOF20 min | 429 | 258 | 479 | 441 | 463 | |||||
| TON | 205 | 200 | 207 | 205 | 199 | |||||
For 2-chlorobenzyl alcohol, the catalysts 1t, 1β, [VO2(HSIH)], and [VO(SIH)]x achieved high conversion rates (>84%) with both oxidants. Selectivity toward aldehyde at the reaction's conclusion was moderate regardless of the oxidant. However, after 20 minutes, aldehyde selectivity was notably higher with TBHP in water, peaking at 96% with [VO2(HSIH)]. V2O5 exhibited a higher conversion rate with TBHP in decane compared to water (86% vs. 58%), while aldehyde selectivity was superior with TBHP in water (62%) compared to decane (29%). TON values for the catalysts 1t, 1β, [VO2(HSIH)], and [VO(SIH)]x are similar, no matter the oxidant used, achieving values 171-213, while V2O5 shows the lowest value of 126 with TBHP in water. TOF values for all the catalysts with TBHP in decane are almost two times higher than with TBHP in water.
For 2-nitrobenzyl alcohol, the catalytic activity in water is notably lower compared to decane, whereas selectivity toward the aldehyde shows the opposite trend. The oxidative conversion of aldehyde to carboxylic acid, evident from the comparison of selectivity parameters at 20 and 120 minutes, is less pronounced than in the case of 2-chlorobenzyl alcohol. This difference is likely due to the electron-withdrawing nitro group stabilizing the aldehyde, thereby reducing its susceptibility to overoxidation. Catalysts 1t, [VO2(HSIH)], and [VO(SIH)]x demonstrate similar performance trends. Alcohol conversions in water hover around 50%, with aldehyde selectivity at the reaction's conclusion reaching approximately 65%. In decane, alcohol conversions increase to ∼58%, while aldehyde selectivity decreases significantly to ∼30%. Catalyst 1β displays consistent activity irrespective of the oxidant, achieving alcohol conversions exceeding 70%. However, aldehyde selectivity is higher in water, reaching 42% at the reaction's end. Notably, at 20 minutes, aldehyde selectivity in water is nearly maximal, peaking at 98%. The TON values for all catalysts are lower for 2-nitrobenzyl alcohol than for 2-chlorobenzyl alcohol, with slightly higher values observed in decane. The TOF20 min values, however, are significantly higher with TBHP in decane, reaching a maximum of 298 for 1t. These findings highlight the delicate interplay between catalyst, oxidant, and substrate electronic properties in determining reaction efficiency and selectivity.
A comparison of the oxidation reactions of 2-chlorobenzyl alcohol and 2-nitrobenzyl alcohol reveals that 2-nitrobenzyl alcohol is a superior substrate for selective oxidation to aldehydes. This superiority arises from the strong electron-withdrawing properties of the nitro group, which significantly increase the electrophilicity of the benzyl carbon, thereby facilitating the oxidation process under milder conditions. Furthermore, the aldehyde product derived from 2-nitrobenzyl alcohol is stabilized by the resonance and inductive effects of the nitro group, reducing its susceptibility to overoxidation into the corresponding carboxylic acid. In contrast, the weaker electron-withdrawing inductive effect of the chloro substituent in 2-chlorobenzyl alcohol renders the benzyl carbon less electrophilic, necessitating severe oxidation conditions and resulting in a less stable aldehyde product.
The conversion of 2-methylbenzyl alcohol is high, whether TBHP is used in water or decane. However, the aldehyde selectivity showed very low values. The obtained results seem to not favour 2-methylbenzyl alcohol as a substrate. The methyl group is an electron-donating group due to its inductive effect that could reduce electrophilicity at the benzyl position and destabilization of the intermediates compared to the 2-chloro- and 2-nitro-benzyl alcohol. Furthermore, the methyl group introduces significant steric hindrance near the benzyl alcohol group. As expected, the desired aldehyde is less stabilized and more prone to overoxidation to the carboxylic acid under strong oxidative conditions present in the reaction.
Experimental
General details
[VO(acac)2], [MoO2(SIH)]n, and salicylaldehyde isonicotinoyl hydrazone (H2SIH), were synthesized according to the procedures described in the literature.57,58,66 Ammonium metavanadate, salicylaldehyde, and isonicotinoyl hydrazide were commercially available. All chemicals and solvents were used without further purification. They were purchased from Alfa Aesar or Aldrich. Details about the instruments and characterization methods (elemental and thermal analyses, ATR-IR, UV, NMR spectroscopies, PXRD, and SCXRD methods) and obtained data can be found in the ESI.† The crystals obtained from the reaction were used directly for the IR-ATR and X-ray crystallographic studies. The samples for elemental analysis were completely desolvated until constant weight and were analysed. The samples for TG measurements were transferred into a desiccator and placed in a freezer (at −15 °C).Synthesis of the metallosupramolecular compounds
[VO(SIH)(OCH3)]4·4CH3OH (1t·4CH3OH). The complex was prepared by Procedure I in methanol. The solution was concentrated after two days to about 5 mL. Yield: 0.040 g; 54%.
Anal. calcd for 1t, C56H48N12O16V4 (1348.817): C, 49.87; H, 3.59; N, 12.46. Found: C, 49.68; H, 3.27; N, 12.33%. TG for 1t·2CH3OH: CH3OH, 4.18 (calcd 4.54%), CH3O, 8.53 (calcd 8.79%); V2O5, 25.88% (calcd 25.75%). Selected IR data (cm−1): 1622, 1601 (CN), 1349 (C–Ohydrazone), 1274 (C–Ophenolate), 1041 (C–Oalkoxo), 964 (N–N), 956 (VO), 759 (V–Nimine), 713 (V–Nisonicotinoyl), 627 (V–O)alkoxo, 582 (V–O)hydrazonato, 563 (V–O)phenolato.
[VO(SIH)(OCH3)]n·0.25CH3OH (1β·0.25CH3OH). The complex was prepared by Procedure II in methanol. Yield: 0.056 g; 81%. Anal. calcd for 1β, C14H12N3O4V (337.204): C, 49.87; H, 3.59; N, 12.46. Found: C, 49.68; H, 3.25; N, 12.12%. TG for 1β·0.25CH3OH: CH3OH, 1.97 (calcd 2.32%), CH3O, 8.58 (calcd 8.99%); V2O5, 25.99% (calcd 26.34%). Selected IR data (cm−1): 1616, 1600 (C
N), 1345 (C–Ohydrazone), 1279 (C–Ophenolate), 1051 (C–Oalkoxo), 973 (N–N), 959 (VO), 750 (V–Nimine), 709 (V–Nisonicotinoyl), 623 (V–O)alkoxo, 570 (V–O)hydrazonato, 559 (V–O)phenolato.
[VO(SIH)(OC2H5)]n (2α). The complex was prepared by Procedure I in ethanol. The solution was concentrated after one day to about 20 mL and a small amount of precipitated solid filtered the next day. The product 2α was obtained from the filtrate. Yield: 0.027 g; 39%. Anal. calcd for 2α, C15H14N3O4V (351.230): C, 51.29; H, 4.02; N, 11.96. Found: C, 51.03; H, 3.86; N, 11.78%. TG for 2α: C2H5O, 12.56 (calcd 12.83%); V2O5, 25.99% (calcd 25.89%). Selected IR data (cm−1): 1620, 1599 (C
N), 1349 (C–Ohydrazone), 1278 (C–Ophenolate), 1046 (C–Oalkoxo), 969 (N–N, VO), 756 (V–Nimine), 710 (V–Nisonicotinoyl), 633 (V–O)alkoxo, 587 (V–O)hydrazonato, 562 (V–O)phenolato.
[VO(SIH)(OC2H5)]n·0.25C2H5OH (2β·0.25C2H5OH). The complex was prepared by Procedure II in ethanol. Yield: 0.063 g; 87%.
Anal. calcd for 2β, C15H14N3O4V (351.230): C, 51.29; H, 4.02; N, 11.96. Found: C, 50.98; H, 3.79; N, 11.75%. TG for 2β·0.25C2H5OH: C2H5OH, 3.12 (calcd 3.18%); C2H5O, 12.56 (calcd. 12.42%); V2O5, 24.82% (calcd 25.07%). Selected IR data (cm−1): 1615, 1598 (CN), 1344 (C–Ohydrazone), 1277 (C–Ophenolate), 1038 (C–Oalkoxo), 974 (N–N), 964 (VO), 750 (V–Nimine), 709 (V–Nisonicotinoyl), 630 (V–O)alkoxo, 574 (V–O)hydrazonato, 560 (V–O)phenolato.
[VO(SIH)(OC3H7)]n (3α). The complex was prepared by Procedure I in 1-propanol. The solution was concentrated after one day to about 20 mL. Yield: 0.062 g; 85%. Anal. calcd for 3α, C16H16N3O4V (365.257): C, 52.61; H, 4.42; N, 11.50. Found: C, 52.45; H, 4.32; N, 11.36%. TG for 3α: C3H7O, 16.44 (calcd 16.18%); V2O5, 24.62% (calcd 24.90%). Selected IR data (cm−1): 1617, 1596 (C
N), 1348 (C–Ohydrazone), 1277 (C–Ophenolate), 1049 (C–Oalkoxo), 9727 (N–N), 970 (VO), 756 (V–Nimine), 706 (V–Nisonicotinoyl), 629 (V–O)alkoxo, 590 (V–O)hydrazonato, 562 (V–O)phenolato.
[VO(SIH)(OC4H9)]n (4α). (a) The complex was prepared by Procedure I in 1-butanol. After the reaction, the solution was left at room temperature for 5 days and evaporated to 10 mL. The resulting crystalline product was filtered the next day. Yield: 0.054 g; 71%. (b) The complex was also prepared by Procedure II in 1-butanol. Yield: 0.053 g; 70%. Anal. calcd for 4α, C17H18N3O4V (379.284): C, 53.83; H, 4.78; N, 11.08. Found: C, 52.62; H, 4.56; N, 10.84%. TG for 4α: C4H9O, 18.85 (Calcd. 19.28%); V2O5, 24.24% (Calcd. 23.98%). Selected IR data (cm−1): 1614, 1598 (C
N), 1348 (C–Ohydrazone), 1277 (C–Ophenolate), 1042 (C–Oalkoxo), 972 (N–N), 969 (VO), 754 (V–Nimine), 699 (V–Nisonicotinoyl), 630 (V–O)alkoxo, 585 (V–O)hydrazonato, 561 (V–O)phenolato.
[VO(SIH)(OC4H9)]n (4β). The complex was prepared by Procedure I in 1-butanol. After completion of the reaction, the solution was evaporated to about 10 mL the same day. The resulting crystalline product was filtered the next day. Yield: 0.023 g; 30%. Anal. calcd for 4β, C17H18N3O4V (379.284): C, 53.83; H, 4.78; N, 11.08. Found: C, 53.66; H, 4.43; N, 10.88%.
TG for 4β: C4H9O, 19.12 (calcd 19.28%); V2O5, 24.32% (calcd 23.98%). Selected IR data (cm−1): 1621, 1598 (CN), 1343 (C–Ohydrazone), 1276 (C–Ophenolate), 1060 (C–Oalkoxo), 968 (N–N), 957 (VO), 750 (V–Nimine), 694 (V–Nisonicotinoyl), 629 (V–O)alkoxo, 577 (V–O)hydrazonato, 560 (V–O)phenolato.
[VO(SIH)(OC5H11)]n (5β). The complex was prepared by Procedure I in 1-pentanol. The solution was concentrated after one day to about 10 mL. Yield: 0.053 g; 67%. Anal. calcd for 5β, C18H20N3O4V (393.310): C, 54.97; H, 5.13; N, 10.68. Found: C, 54.77; H, 4.96; N, 10.48%. TG for 5β: C5H11O, 21.85 (calcd 22.16%); V2O5, 23.28% (calcd 23.12%). Selected IR data (cm−1): 1621, 1598 (C
N), 1344 (C–Ohydrazone), 1275 (C–Ophenolate), 1040 (C–Oalkoxo), 968 (N–N), 958 (VO), 749 (V–Nimine), 694 (V–Nisonicotinoyl), 629 (V–O)alkoxo, 578 (V–O)hydrazonato, 560 (V–O)phenolato.
Synthesis of the mononuclear compound
N), 1351 (C–Ohydrazone), 1258 (C–Ophenolate), 969 (N–N), 953, 942 (VO), 739 (V–Nimine), 571 (V–Ohydrazonato), 555 (V–Ophenolato).Catalytic investigation
Benzyl alcohol. A reaction mixture was prepared with benzyl alcohol (2.16 g, 20 mmol), biphenyl (0.1152 g, 0.75 mmol) as the internal standard, and catalyst (0.1 mmol). The mixture was heated to 80 °C prior to the addition of aqueous tert-butyl hydroperoxide (TBHP, 70% w/w, 5.54 mL, 40 mmol) or heated to 70 °C before the addition of hydrogen peroxide (H2O2) (4.12 mL, 40 mmol) in the presence of 3 mL of acetonitrile.
2-Nitrobenzyl alcohol. A reaction mixture was prepared with 2-nitrobenzyl alcohol (0.51 g, 3.3 mmol), acetophenone (0.412 g, 3.43 mmol) as the internal standard, and catalyst (0.0165 mmol). The mixture was heated to 80 °C before the addition of aqueous TBHP (70% w/w, 0.91 mL, 6.6 mmol).
2-Chlorobenzyl alcohol. A reaction mixture was prepared with 2-chlorobenzyl alcohol (0.47 g, 3.3 mmol), acetophenone (0.412 g, 3.43 mmol) as the internal standard, and catalyst (0.0165 mmol). The mixture was heated to 80 °C prior to the addition of aqueous TBHP (70% w/w, 0.91 mL, 6.6 mmol).
2-Methylbenzyl alcohol. A reaction mixture was prepared with 2-methylbenzyl alcohol (0.40 g, 3.3 mmol), dodecane (0.225 g, 1.34 mmol) as the internal standard, and catalyst (0.0165 mmol). The mixture was heated to 80 °C prior to the addition of aqueous TBHP (70% w/w, 0.91 mL, 6.6 mmol).
The reaction progress was monitored over a two-hour period by collecting 0.1 mL aliquots of the organic phase at predefined time intervals (0, 20, 40, 60, and 120 minutes). Each aliquot was diluted with 1.4 mL of diethyl ether and subjected to gas chromatography (GC) analysis to assess the conversion and formation of reaction products.
Principal component analysis
Data acquired through time-dependent UV-Vis spectrophotometry during the monitored chemical reaction were exported in ASCII format and arranged into a matrix of numerical entries. This process resulted in a 2nd-order data tensor characterized by dimensions corresponding to the number-of-spectra × number-of-wavelengths. We then applied principal component analysis (PCA), a second-order tensor reduction technique, to decompose the dataset. PCA facilitates the identification of the best linear projections for a high-dimensional data set based on the least-squares approach. The resulting scores represent the projections of the original sample points on the principal component (PC) direction, thereby enabling a comprehensive description of the reactions in a reduced space. Consequently, each point in the score plots corresponds to one sample UV-Vis spectra. Data were mean-centered and PCA of the covariance matrix was performed utilizing the NIPALS algorithm, which was implemented within our software application, moonee.67Conclusions
This research contributes to a better understanding of the vanadium-based metallosupramolecular polymorphic assemblies, particularly regarding their formation and properties. Namely, two series of isostructural oxovanadium(V) coordination 1D zig–zag chain polymers were synthesized. These include [VO(SIH)(OR)]n, with R representing different alkyl groups (R = CH3 (1β·0.25CH3OH), C2H5 (2α and 2β·0.25C2H5OH), C3H7 (3α), C4H9 (4α and 4β), and C5H11 (5β)). The synthesis involved combining salicylaldehyde isonicotinoylhydrazonate (SIH2−) and the corresponding alkoxide (OR−) with {VO}3+ units. The similarity observed in the powder diffractograms across the series allowed the identification of each compound's polymorphic type. Although detailed studies are still needed, initial results suggest that the formation of the pure phase may be controlled by the suitable reaction conditions – solution concentrations, crystallization temperatures, evaporation rates, solvent selection, and the chosen synthesis method. The occurrence of concomitant polymorphs likely arises from the capacity of the {VO}3+ ion and SIH2− ligand to form two crystalline phases with only slight differences in formation energy. Additionally, a phase-pure metallocyclic supramolecular isomer [VO(SIH)(OCH3)]4·4CH3OH (1t·4CH3OH), was identified. This compound features the interconnection of the mononuclear units into 0D squares through V–Nisonicotinoyl coordination bonds, which adds to the diversity of the compounds studied. The research also emphasizes the potential of vanadium-based metallosupramolecular architectures in catalytic applications, further contributing to a better understanding of the structure–activity relationships in these complex systems.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for 1t·4CH3OH, 2α, 3α, 4α, 1β·0.25CH3OH, 2β·0.25CH3CH2OH and 5β have been deposited at the Cambridge Structural Data base under with deposition numbers CCDC 2367532–2367538.Author contributions
Edi Topić: investigation, writing – original draft preparation, review and editing. Josipa Sarjanović: investigation and formal analysis. Danijela Musija: investigation and formal analysis, and writing – original draft preparation. Mirna Mandarić: investigation and formal analysis. Tomica Hrenar: chemometric analysis, writing – original draft preparation, visualization. Jana Pisk: investigation, supervision, formal analysis, writing – original draft preparation. Višnja Vrdoljak: conceptualization, supervision, funding acquisition, investigation, writing – original draft preparation, visualization, review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Croatian Science Foundation under the project number HRZZ-IP-2022-10-7368. We acknowledge the support of project CIuK co-financed by the Croatian Government and the European Union through the European Regional Development Fund-Competitiveness and Cohesion Operational Programme (Grant KK.01.1.1.02.0016).Notes and references
- Y. Duan, W. Wei, F. Xiao, Y. Xi, S.-L. Chen, J.-L. Wang, Y. Xu and C. Hua, High-valent cationic metal–organic macrocycles as novel supports for immobilization and enhancement of activity of polyoxometalate catalysts, Catal. Sci. Technol., 2016, 6, 8540 RSC.
- S. C. A. Sousa, I. Cabrita and A. C. Fernandes, High-valent oxo-molybdenum and oxo-rhenium complexes as efficient catalysts for X–H (X = Si, B, P and H) bond activation and for organic reductions, Chem. Soc. Rev., 2012, 41, 5641 RSC.
- W. R. Thiel, On the Way to a New Class of Catalysts—High-Valent Transition-Metal Complexes That Catalyze Reductions, Angew. Chem., Int. Ed., 2003, 42, 5390 CrossRef CAS PubMed.
- S. Kim, K.-B. Cho, Y.-M. Lee, J. Chen, S. Fukuzumi and W. Nam, Factors Controlling the Chemoselectivity in the Oxidation of Olefins by Nonheme Manganese(IV)-Oxo Complexes, J. Am. Chem. Soc., 2016, 138, 10654 CrossRef CAS PubMed.
- J. A. L. da Silva, J. J. R. F. da Silva and A. J. L. Pombeiro, Oxovanadium complexes in catalytic oxidations, Coord. Chem. Rev., 2011, 255, 2232 CrossRef CAS.
- R. R. Langeslay, D. M. Kaphan, C. L. Marshall, P. C. Stair, A. P. Sattelberger and M. Delferro, Catalytic Applications of Vanadium: A Mechanistic Perspective, Chem. Rev., 2019, 119, 2128 Search PubMed.
- J. C. Pessoa and M. R. Maurya, Vanadium Complexes Supported on Organic Polymers as Sustainable Systems for Catalytic Oxidations, Inorg. Chim. Acta, 2017, 455, 415 CrossRef.
- M. Sutradhar, M. V. Kirillova, M. F. C. G. da Silva, L. M. D. R. S. Martins and A. J. L. Pombeiro, A Hexanuclear Mixed-Valence Oxovanadium(IV,V) Complex as a Highly Efficient Alkane Oxidation Catalyst, Inorg. Chem., 2012, 51, 11229 Search PubMed.
- X. Xu, H. Wang, C.-H. Tan and X. Ye, Applications of Vanadium, Niobium, and Tantalum Complexes in Organic and Inorganic Synthesis, ACS Org. Inorg. Au, 2023, 3, 74 Search PubMed.
- I. Gryca, K. Czerwińska, B. Machura, A. Chrobok, L. S. Shul’pina, M. L. Kuznetsov, D. S. Nesterov, Y. N. Kozlov, A. L. Pombeiro, I. A. Varyan and G. B. Shul’pin, High Catalytic Activity of Vanadium Complexes in Alkane Oxidations with Hydrogen Peroxide: An Effect of 8-Hydroxyquinoline Derivatives as Noninnocent Ligands, Inorg. Chem., 2018, 57, 1824 CrossRef CAS PubMed.
- D. Dragancea, N. Talmaci, S. Shova, G. Novitchi, D. Darvasiová, P. Rapta, M. Breza, M. Galanski, J. Kožíšek, N. M. R. Martins, L. M. D. R. S. Martins, A. J. L. Pombeiro and V. B. Arion, Vanadium(V) Complexes with Substituted 1,5-bis(2-hydroxybenzaldehyde)carbohydrazones and their Use As Catalyst Precursors in Oxidation of Cyclohexane, Inorg. Chem., 2016, 55, 9187 CrossRef CAS PubMed.
- G. B. Shul’pin and Y. N. Kozlov, Kinetics and mechanism of alkane hydroperoxidation with tert-butyl hydroperoxide catalysed by a vanadate anion, Org. Biomol. Chem., 2003, 1, 2303 RSC.
- M. Sutradhar, L. M. D. R. S. Martins, T. Roy Barman, M. L. Kuznetsov, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Vanadium complexes of different nuclearities in the catalytic oxidation of cyclohexane and cyclohexanol – an experimental and theoretical investigation, New J. Chem., 2019, 43, 17557 RSC.
- M. R. Maurya, Probing the synthetic protocols and coordination chemistry of oxido-, dioxido-, oxidoperoxido-vanadium and related complexes of higher nuclearity, Coord. Chem. Rev., 2019, 383, 43 CrossRef CAS.
- R. Dinda, P. Sengupta, M. Sutradhar, T. C. W. Mak and S. Ghosh, Solution Study of a Structurally Characterized Monoalkoxo-Bound Monooxo-Vanadium(V) Complex: Spontaneous Generation of the Corresponding Oxobridged Divanadium(V,V) Complex and its Electroreduction to a Mixed-Valence Species in Solution, Inorg. Chem., 2008, 47, 5634 CrossRef CAS PubMed.
- M. Y. Enbo, W. Ying, L. Shutao, W. Yangguang, L. Wang and C. Hu, A novel chain-like binuclear vanadium(V) coordination polymer containing mixed ligands: hydrothermal synthesis and crystal structure of [{VO2(2,2′-bipy)}2(tp)]∞ (tp=terephthalate), Inorg. Chim. Acta, 2003, 344, 257 CrossRef.
- E. Kiss, A. Benyei and T. Kiss, VO(IV) complexes of 3-hydroxypicolinic acid: a solution study and the structure of a supramolecular assembly in the solid state, Polyhedron, 2003, 22, 27 Search PubMed.
- X.-M. Zhang, M. Liang Tong, H. Kay Lee and X.-M. Chen, The First Noncluster Vanadium(IV) Coordination Polymers: Solvothermal Syntheses, Crystal Structure, and Ion Exchange, J. Solid State Chem., 2001, 160, 118 CrossRef CAS.
- K. Su, M. Wu, D. Yuan and M. Hong, Interconvertible vanadium-seamed hexameric pyrogallol[4]arene nanocapsules, Nat. Commun., 2018, 9, 4941 CrossRef PubMed.
- S. P. Dash, A. K. Panda, S. Pasayat, R. Dinda, A. Biswas, E. R. T. Tiekink, S. Mukhopadhyay, S. K. Bhutia, W. Kaminsky and E. Sinn, RSC Adv., 2015, 5, 51852 RSC.
- V. Vrdoljak, M. Mandarić, T. Hrenar, I. Đilović, J. Pisk, G. Pavlović, M. Cindrić and D. Agustin, Geometrically constrained molybdenum(VI) metallosupramolecular architectures: conventional Synthesis versus vapor and thermally induced solid-state structural transformations, Cryst. Growth Des., 2019, 19, 3000 Search PubMed.
- D. Cvijanović, J. Pisk, G. Pavlović, D. Šišak-Jung, D. Matković-Čalogović, M. Cindrić, D. Agustin and V. Vrdoljak, Discrete mononuclear and dinuclear compounds containing a MoO22+ core and 4-aminobenzhydrazone ligands: synthesis, structure and organic-solvent-free epoxidation activity, New J. Chem., 2019, 43, 1791 RSC.
- J. Pisk, M. Rubčić, D. Kuzman, M. Cindrić, D. Agustin and V. Vrdoljak, Molybdenum(VI) complexes of hemilabile aroylhydrazone ligands as efficient catalysts for greener cyclooctene epoxidation: an experimental and theoretical approach, New J. Chem., 2019, 43, 5531 RSC.
- V. Vrdoljak, B. Prugovečki, D. Matković-Čalogović, R. Dreos, P. Siega and C. Tavagnacco, Zigzag Chain, Square Tetranuclear, and Polyoxometalate-Based Inorganic–Organic Hybrid Compounds - Molybdenum vs. Tungsten, Cryst. Growth Des., 2010, 10(3), 1373 CrossRef CAS.
- J. Pisk, D. Agustin and V. Vrdoljak, Tetranuclear molybdenum(VI) hydrazonato epoxidation (pre)catalysts: Is water always the best choice?, Catal. Commun., 2020, 142, 106027 Search PubMed.
- M. Mandarić, E. Topić, D. Agustin, J. Pisk and V. Vrdoljak, Preparative and Catalytic Properties of MoVI Mononuclear and Metallosupramolecular Coordination Assemblies Bearing Hydrazonato Ligands, Int. J. Mol. Sci., 2024, 25, 1503 CrossRef PubMed.
- M. A. Fik, A. Gorczyński, M. Kubicki, Z. Hnatejko, A. Wadas, P. J. Kulesza, A. Lewińska, M. Giel-Pietraszuk, E. Wyszko and V. Patroniak, New vanadium complexes with 6,6′′-dimethyl-2,2′:6′,2′′-terpyridine in terms of structure and biological properties, Polyhedron, 2015, 97, 83 CrossRef CAS.
- M. Motevalli, D. Shah, S. A. A. Shah and A. C. Sullivan, A novel route to oxovanadium halides. Synthesis and X-ray structures of VOCl2·3Py and VO2Cl·2Py, Polyhedron, 1996, 15, 2387 CrossRef CAS.
- E. Salojärvi, A. Peuronen, M. Lahtinen, H. Huhtinen, L. S. Vlasenko, M. Lastusaari and A. Lehtonen, Series of Near-IR-Absorbing Transition Metal Complexes with Redox Active Ligands, Molecules, 2020, 25, 2531 CrossRef PubMed.
- L. M. Mokry and C. J. Carrano, Steric control of vanadium(V) coordination geometry: a mononuclear structural model for transition-state-analog RNase inhibitors, Inorg. Chem., 1993, 32, 6119 CrossRef CAS.
- B. Baruah, S. P. Rath and A. Chakravorty, A Novel Pentacoordinated Dioxovanadium(V) Salicylaldiminate: Solvent Specific Crystallization of Dimorphs with Contrasting Coordination Geometries, Ligand Conformations and Supramolecular Architectures, Eur. J. Inorg. Chem., 2004, 2004, 1873 CrossRef.
- P. I. d. S. Maia, V. M. Deflon, E. J. d. Souza, E. Garcia, G. F. d. Sousa, A. A. Batista, A. T. d. Figueiredo and E. Niquet, Biomimetic oxovanadium(IV) and (V) complexes with a tridentate (N,N,O)-donor hydrazonic ligand. Two X-ray crystal structure modifications of (2-acetylpyridine-benzoylhydrazonato)dioxovanadium(V), Transition Met. Chem., 2005, 30, 404 CrossRef CAS.
- M. R. P. Kurup, E. B. Seena and M. Kuriakose, Synthesis, spectral and structural studies of oxovanadium(IV/V) complexes derived from 2-hydroxyacetophenone-3-hydroxy-2-naphthoylhydrazone: polymorphs of oxovanadium(V) complex [VOL(OCH3)], Struct. Chem., 2010, 21, 599 CrossRef CAS.
- Y. Jin and M. S. Lah, Polymorphism Driven by π–π Stacking and van der Waals Interactions: Preparation and Characterization of Polymorphic Vanadium Crystals of [VVO(Hacshz)(OEt)] and [VIV(Hacshz)2], Eur. J. Inorg. Chem., 2005, 2005, 4944 CrossRef.
- N. Heydari, R. Bikas, M. Shaterian, M. S. Krawczyk and T. Lis, Investigation of the substituent effects on the oxidation of styrene derivatives by silica-supported heterogeneous oxidovanadium(V) coordination compound, Appl. Organomet. Chem., 2023, 37, e6976 CrossRef CAS.
- M. Rubčić, D. Milić, G. Pavlović and M. Cindrić, Conformational Diversity of Thiosemicarbazonatovanadium(V) Complexes in the Solid State: From Polymorphism to Isostructurality, Cryst. Growth Des., 2011, 11, 5227 CrossRef.
- T. Hu, Q. Wang, W. You, D. Song, C. Huang, Y. Xu and Z. Sun, Two new layered complexes supported by helical chains: Hydrothermal syntheses and crystal structures of [M(dpa)V2O6] (M = Zn(II) and Cu(II); dpa = 2,2′-dipyridylamine), Inorg. Chem. Commun., 2008, 11, 470 CrossRef CAS.
- J.-S. Qin, D.-Y. Du, S.-L. Li, Y.-Q. Lan, K.-Z. Shao and Z.-M. Su, pH-Tuned self-assembly of organic–inorganic hybrids based on different vanadate chains, Zn(II) ions and flexible ligands: crystallizing in polar and centrosymmetric space group, CrystEngComm, 2011, 13, 779 RSC.
- A. Sukhikh, D. Bonegardt, D. Klyamer and T. Basova, Effect of non-peripheral fluorosubstitution on the structure of metal phthalocyanines and their films, Dyes Pigm., 2021, 192, 109442 CrossRef CAS.
- D. Klyamer, A. Sukhikh, D. Bonegardt, P. Krasnov, P. Popovetskiy and T. Basova, Thin Films of Chlorinated Vanadyl Phthalocyanines as Active Layers of Chemiresistive Sensors for the Detection of Ammonia, Micromachines, 2023, 14, 1773 CrossRef PubMed.
- Y. Horii, M. Makino, T. Yamamoto, S. Tatsumi, H. Suzuki, M. Noguchi, T. Yoshida, T. Kajiwara, Z.-Y. Li and M. Yamashita, Solid polymorphism and dynamic magnetic properties of a dodecylated vanadyl–porphyrinato complex: spin–lattice relaxations modulated by phase stabilisation, Inorg. Chem. Front., 2022, 9, 6271 RSC.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Supramolecular Catalysis in Metal–Ligand Cluster Hosts, Chem. Rev., 2015, 115, 3012 CrossRef CAS PubMed.
- C. Lescop, Coordination-Driven Syntheses of Compact Supramolecular Metallacycles toward Extended Metallo-organic Stacked Supramolecular Assemblies, Acc. Chem. Res., 2017, 50, 885 CrossRef CAS PubMed.
- B. H. Northrop, H.-B. Yang and P. J. Stang, Coordination-driven self-assembly of functionalized supramolecular metallacycles, Chem. Commun., 2008, 5896 Search PubMed.
- T. R. Cook and P. J. Stang, Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages via Coordination, Chem. Rev., 2015, 115, 7001 Search PubMed.
- S. Leininger, B. Olenyuk and P. J. Stang, Self-assembly of discrete cyclic nanostructures mediated by transition metals, Chem. Rev., 2000, 100, 853 CrossRef CAS PubMed.
- B. H. Northrop, Y.-R. Zheng, K.-W. Chi and P. J. Stang, Self-Organization in Coordination-Driven Self-Assembly, Acc. Chem. Res., 2009, 42, 1554 Search PubMed.
- R. J. J. Jachuck, D. K. Selvaraj and R. S. Varma, Process intensification: oxidation of benzyl alcohol using a continuous isothermal reactor under microwave irradiation, Green Chem., 2006, 8, 29 RSC.
- C. Ragupathi, J. J. Vijaya, S. Narayanan, S. K. Jesudoss and L. J. Kennedy, Highly selective oxidation of benzyl alcohol to benzaldehyde with hydrogen peroxide by cobalt aluminate catalysis: a comparison of conventional and microwave methods, Ceram. Int., 2015, 41, 2069 Search PubMed.
- A. Mahmood, G. E. Robinson and L. Powell, An improved oxidation of an alcohol using aqueous permanganate and phase-transfer catalyst, Org. Process Res. Dev., 1999, 3, 363 CrossRef CAS.
- A. L. Cánepa, V. R. Elías, V. M. Vaschetti, E. V. Sabre, G. A. Eimer and S. G. Casuscelli, Selective oxidation of benzyl alcohol through ecofriendly processes using mesoporous V-MCM-41, Fe-MCM-41 and Co-MCM41 materials, Appl. Catal., A, 2017, 545, 72 Search PubMed.
- S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott, A. D. Sutton and D. L. Thorn, Aerobic oxidation of pinacol by vanadium (V) dipicolinate complexes: evidence for reduction to vanadium (III), J. Am. Chem. Soc., 2008, 131, 428 CrossRef PubMed.
- V. R. Choudhary, A. Dhar, P. Jana, R. Jha and B. S. Uphade, A green process for chlorine-free benzaldehyde from the solvent-free oxidation of benzyl alcohol with molecular oxygen over a supported nano-size gold catalyst, Green Chem., 2005, 7, 768 RSC.
- C. Xu, L. Zhang, Y. An, X. Wang, G. Xu, Y. Chen and L. Dai, Promotional synergistic effect of Sn doping into a novel bimetallic Sn-W oxides/graphene catalyst for selective oxidation of alcohols using aqueous H2O2 without additives, Appl. Catal., A, 2018, 558, 26 Search PubMed.
- C. Kamonsatikul, T. Khamnaen, P. Phiriyawirut, S. Charoenchaidet and E. Somsook, Synergistic activities of magnetic iron-oxide nanoparticles and stabilizing ligands containing ferrocene moieties in selective oxidation of benzyl alcohol, Catal. Commun., 2012, 26, 1 CrossRef CAS.
- F. Shi, M. K. Tse, M.-M. Pohl, A. Brückner, S. Zhang and M. Beller, Tuning catalytic activity between homogeneous and heterogeneous catalysis: Improved activity and selectivity of free nano-Fe2O3 in selective oxidations, Angew. Chem., Int. Ed., 2007, 46, 8866 CrossRef CAS PubMed.
- V. Vrdoljak, B. Prugovečki, D. Matković-Čalogović, J. Pisk, R. Dreos and P. Siega, Supramolecular Hexagon and Chain Coordination Polymer Containing the MoO22+ Core: Structural Transformation in the Solid State, Cryst. Growth Des., 2011, 11, 1244 CrossRef CAS.
- J. Pisk, T. Hrenar, M. Rubčić, G. Pavlović, V. Damjanović, J. Lovrić, M. Cindrić and V. Vrdoljak, Comparative studies on conventional and solvent-free synthesis toward hydrazones: application of PXRD and chemometric data analysis in mechanochemical reaction monitoring, CrystEngComm, 2018, 20, 1804 RSC.
- G. H. Shahverdizadeh, S. W. Ng, E. R. T. Tiekink and B. Mirtamizdoust, cis-Dioxido[N′-(2-oxidobenzylidene)pyridinium-4-carbohydrazidato-κ3O,N′,O′]vanadium(V), Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m236 Search PubMed.
- S. Y. Ebrahimipour, M. Mohamadi, I. Sheikhshoaie, S. Suárez, R. Baggio and M. Khaleghi, A novel oxido-vanadium(V) Schiff base complex: synthesis, spectral characterization, crystal structure, electrochemical evaluation, and biological activity, Res. Chem. Intermed., 2016, 42, 611 Search PubMed.
- Y.-H. Lu, Y.-W. Lu, C.-L. Wu, Q. Shao, X.-L. Chen and R. N. B. Bimbong, UV-visible spectroscopic study of the salicylaldehyde benzoylhydrazone and its cobalt complexes, Spectrochim. Acta, Part A, 2006, 65, 695 CrossRef PubMed.
- M. R. Maurya, S. Khurana, C. Schulzke and D. Rehder, Dioxo- and Oxovanadium(V) Complexes of Biomimetic Hydrazone ONO Donor Ligands: Synthesis, Characterisation, and Reactivity, Eur. J. Inorg. Chem., 2001, 2001, 779 CrossRef.
- L. M. Martínez-Prieto, P. Palma, E. Álvarez and J. Cámpora, Nickel Pincer Complexes with Frequent Aliphatic Alkoxo Ligands [(iPrPCP)Ni-OR] (R = Et, nBu, iPr, 2-hydroxyethyl). An Assessment of the Hydrolytic Stability of Nickel and Palladium Alkoxides, Inorg. Chem., 2017, 56(21), 13086 Search PubMed.
- S. R. Patra, S. Mondal, D. Sinha and K. K. Rajak, Mono Versus Dinuclear Vanadium(V) Complexes: Solvent Dependent Structural Versatility and Electro Syntheses of Mixed-Valence Oxovanadium(IV/V) Entities in Solution, ACS Omega, 2022, 7, 11710 CrossRef PubMed.
- M. Mandarić, B. Prugovečki, D. Cvijanović, J. Parlov Vuković, J. Lovrić, M. Skočibušić, R. Odžak, M. Cindrić and V. Vrdoljak, Vapour- and solvent-mediated crystalline transformations in Mo(VI) hydrazone complexes controlled by noncovalent interactions, CrystEngComm, 2019, 21, 6281 Search PubMed.
- B. E. Bryant, W. C. Fernelius, D. H. Busch, R. C. Stoufer and W. Stratton, Vanadium(IV) Oxy(acetylacetonate), in Inorganic Syntheses, ed. T. Moeller, John Wiley & Sons, Inc, 1957, vol. 5, pp. 113–116 Search PubMed.
- O. Jović, T. Smolić, I. Primožič and T. Hrenar, Spectroscopic and Chemometric Analysis of Binary and Ternary Edible Oil Mixtures: Qualitative and Quantitative Study, Anal. Chem., 2016, 88, 4516 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: (1) Powder diffraction patterns, (2) additional figures for compounds, (3) tables of selected bond distances and angles and of hydrogen bonds parameter, (4) UV-Vis spectra, (5) NMR spectra ATR-IR spectra, (6) TGA curves, (7) crystallographic data sets for the structures 1t·4CH3OH, 2α, 3α, 4α, 1β·0.25CH3OH, 2β·0.25C2H5OH and 5β. CCDC 2367532–2367538. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra09073j |
| This journal is © The Royal Society of Chemistry 2025 |