
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-pronged molecular insights into flavonoid-mediated inhibition of squalene epoxidase: a pathway to novel therapeutics
Emadeldin M. Kamel *a,
Sarah I. Othmanb,
Hassan A. Rudaynic,
Ahmed A. Allamcd and
Al Mokhtar Lamsabhief
*a,
Sarah I. Othmanb,
Hassan A. Rudaynic,
Ahmed A. Allamcd and
Al Mokhtar Lamsabhief
aChemistry Department, Faculty of Science, Beni-Suef University, Beni-Suef 62514, Egypt. E-mail: emad.abdelhameed@science.bsu.edu.eg
bDepartment of Biology, College of Science, Princess Nourah bint Abdulrahman University, P. O. BOX 84428, Riyadh 11671, Saudi Arabia
cDepartment of Biology, College of Science, Imam Mohammad Ibn Saud Islamic University, Riyadh 11623, Saudi Arabia
dDepartment of Zoology, Faculty of Science, Beni-suef University, Beni-suef 65211, Egypt
eDepartamento de Química, Módulo 13, Universidad Autónoma de Madrid, Campus de Excelencia UAM-CSIC Cantoblanco, 28049 Madrid, Spain
fInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 6th February 2025
Abstract
Squalene epoxidase (SQLE) is a crucial enzyme in the sterol biosynthesis pathway and a promising target for therapeutic intervention in hypercholesterolemia and fungal infections. This study evaluates the inhibitory potential of six flavonoids namely silibinin, baicalin, naringenin, chrysin, apigenin-7-O-glucoside, and isorhamnetin against SQLE using an integrative approach combining in silico and experimental methods. Molecular docking revealed that apigenin-7-O-glucoside, silibinin, and baicalin displayed the highest binding affinities (−10.7, −10.2, and −10.0 kcal mol−1, respectively) and robust interactions with the SQLE binding site. These findings were corroborated by 200 ns molecular dynamics (MD) simulations, which demonstrated stable binding trajectories, minimal structural fluctuations, a thermodynamically favored potential energy landscape (PEL) and favorable MM/PBSA binding free energies for three flavonoids. Experimental validation via in vitro inhibition assays confirmed the computational predictions, with apigenin-7-O-glucoside emerging as the most potent inhibitor (IC50 = 1.74 ± 0.05 μM), followed by silibinin (IC50 = 1.88 ± 0.28 μM) and baicalin (IC50 = 2.50 ± 0.46 μM). Enzyme kinetics studies revealed distinct mechanisms of action: apigenin-7-O-glucoside exhibited competitive inhibition, while silibinin and baicalin showed mixed inhibition. Furthermore, in silico ADMET analysis indicated favorable pharmacokinetic and pharmacodynamic profiles for these flavonoids, with silibinin demonstrating particularly high bioavailability and lipophilicity. This study highlights apigenin-7-O-glucoside, silibinin, and baicalin as potent SQLE inhibitors with promising therapeutic potential. The congruence between in silico predictions and experimental results underscores the reliability of computational approaches in drug discovery, paving the way for future preclinical development of these compounds as novel SQLE-targeted therapeutics.
1. Introduction
Squalene epoxidase (SQLE) is a pivotal enzyme in the cholesterol biosynthesis pathway, catalyzing the epoxidation of squalene to 2,3-oxidosqualene, a critical step in the production of sterols, including cholesterol.1 As a rate-limiting enzyme, SQLE represents a crucial control point in the regulation of cholesterol levels within the body.2 Elevated cholesterol levels, particularly low-density lipoprotein (LDL) cholesterol, are directly associated with the development of atherosclerosis and cardiovascular diseases, making cholesterol regulation an important therapeutic target.3 Additionally, aberrant SQLE activity has been implicated in cancer, where disruptions in lipid metabolism can fuel tumor progression.4 As a result, inhibiting SQLE is emerging as a promising therapeutic approach for managing both hypercholesterolemia and cancer.1 Targeting this enzyme can not only reduce cholesterol synthesis but also limit the accumulation of toxic sterol intermediates, positioning SQLE inhibition as a valuable strategy for combating disorders associated with abnormal lipid metabolism.5Flavonoids, a diverse group of polyphenolic compounds found in various fruits, vegetables, and medicinal plants, have garnered attention for their wide range of pharmacological activities, including antioxidant, anti-inflammatory, and cholesterol-lowering effects.6–11 Their structural versatility and capacity to modulate key biological pathways make flavonoids promising candidates for SQLE inhibition. In the search for novel inhibitors of SQLE, we selected six flavonoid compounds namely, silibinin, baicalin, naringenin, chrysin, apigenin-7-O-glucoside, and isorhamnetin, based on their structural uniqueness, biological activities, and commercial availability (Fig. 1). Structurally, these compounds offer a diverse set of functional groups (e.g., glycosylation, methylation) that enhance their bioavailability and potential interactions with the enzyme's active site. Their ability to modulate lipid-metabolizing enzymes makes them suitable candidates for SQLE inhibition.12 Furthermore, these flavonoids are commercially available and have established safety profiles, supporting their further investigation in both in vitro and in silico studies. By targeting SQLE, these flavonoids could contribute to the development of potential treatments for hypercholesterolemia and associated cardiovascular diseases.
 | ||
| Fig. 1 Structures of investigated flavonoids. | ||
Silibinin, derived from Silybum marianum (milk thistle), has well-documented hepatoprotective properties and its role in regulating cholesterol metabolism makes it a compelling candidate for SQLE inhibition.13 Baicalin, from Scutellaria baicalensis (Chinese skullcap), exhibits strong anti-inflammatory and lipid-lowering effects, providing a rationale for its inclusion in this study.14 Naringenin, a flavonoid present in citrus fruits, has demonstrated hypocholesterolemic activity, making it a promising natural inhibitor of cholesterol biosynthesis enzymes like SQLE.5 Chrysin, found in honey and propolis, is known for its antioxidant and lipid-regulating properties, while apigenin-7-O-glucoside, a glycosylated form of apigenin, has been shown to exhibit cholesterol-lowering effects.15,16 Lastly, isorhamnetin, a methylated flavonol present in various plants like Hippophae rhamnoides (sea buckthorn), has demonstrated anti-inflammatory and cholesterol-lowering activities, further supporting its potential as an SQLE inhibitor.17
Combining in vitro experiments with in silico tools offers a comprehensive and integrated approach to studying enzyme inhibition by providing both experimental validation and detailed mechanistic insights.18–20 In vitro assays are essential for directly measuring the inhibitory efficacy of compounds, allowing for the quantification of activity in controlled environments.7,20,21 However, these experiments can be resource-intensive and time-consuming, making in silico methods a valuable complement.22,23 Molecular docking predicts how compounds interact with enzymes at the molecular level, identifying critical binding interactions and affinities at the active site.24–26 Molecular dynamics (MD) simulations build on this by providing dynamic insights into the stability and behavior of enzyme–ligand complexes over time.27–29 ADMET predictions evaluate the absorption, distribution, metabolism, excretion, and toxicity properties of potential inhibitors, ensuring they have favorable pharmacokinetic profiles.30 MM/PBSA calculations offer precise estimates of binding free energies, providing insights into the strength of enzyme-inhibitor interactions.7,9 Principal component analysis (PCA) simplifies complex data, highlighting key conformational changes that contribute to inhibition.20 Together, these methods create a powerful framework for discovering and optimizing enzyme inhibitors while significantly enhancing the efficiency and predictive capabilities of the drug discovery process.
The aim of this study is to explore the inhibitory potential of these selected flavonoids against SQLE through both in vitro and in silico approaches. By combining molecular docking, MD simulations, in silico ADMET profiling, and in vitro enzyme inhibition assays, we seek to provide a comprehensive evaluation of their efficacy in inhibiting SQLE. This approach will not only allow us to identify novel natural inhibitors of SQLE but also to assess their potential for therapeutic application in the management of cholesterol-related diseases, offering a new perspective on flavonoid-based strategies for modulating cholesterol biosynthesis.
2. Materials and methods
2.1. In silico experiments
To validate the reliability of our docking protocol, we performed a redocking experiment using the original co-crystallized ligand from the PDB structure of the target. First, the protein was prepared by removing water molecules and adding missing hydrogen atoms. The co-crystallized ligand was then extracted and subjected to the same docking procedure employed for the test compounds. After docking, we superimposed the best-ranked pose on the experimentally determined bound conformation to calculate the root-mean-square deviation (RMSD). An RMSD value below 2.0 Å was considered indicative of a successful redocking, confirming that the chosen docking parameters and scoring functions could accurately recapitulate the native binding mode.
Energy minimization was initially performed using the steepest descent algorithm over 10 picoseconds (ps) to eliminate steric clashes or unfavorable atomic interactions.43 This was followed by a two-stage equilibration protocol: first, an NVT ensemble equilibration to stabilize temperature and volume, followed by an NPT ensemble equilibration to stabilize pressure, with both phases carried out for 100 ps at a target temperature of 300 K.44 A 200 ns production phase ensued under stable conditions of 300 K and 1 bar pressure, enabling comprehensive analysis of the interactions between SQLE and each inhibitor across the simulation period. This approach allowed us to assess the dynamic stability and binding interactions within the enzyme-inhibitor complexes.
F = −kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(P) ln(P) |
The resulting energy landscape was visualized using a contour plot or heatmap in the PC1–PC2 space, where color gradients illustrated the free energy levels, with lower-energy regions marking stable conformational states and higher-energy areas representing less probable conformations. Key conformational states, including global minima and any transition states, were annotated where identifiable. Through the analysis of the PEL, we identified stable low-energy basins, representing favored conformational states of the enzyme, and examined pathways between these basins to infer potential transitions. The overall landscape enabled an assessment of the enzyme's structural stability and provided insights into conformational states that may influence its biological activity or ligand interactions. This PEL analysis yielded a comprehensive view of the protein's conformational variability and stability throughout the simulation, highlighting primary conformational states and possible transition pathways that contribute to its functional dynamics.
P), gastrointestinal (GI) absorption, blood–brain barrier (BBB) permeability, and potential interactions with P-glycoprotein and cytochrome P450 enzymes. Additionally, the analysis provided insights into drug-likeness criteria based on established rules such as Lipinski's rule of five, Veber, and Ghose filters. The results enabled an assessment of each compound's potential as a drug candidate by highlighting favorable pharmacokinetic attributes and identifying any potential liabilities related to bioavailability or metabolism, thus guiding the selection of promising compounds for further study.2.2. In vitro inhibitory activity assays
To determine the inhibition mechanism, a Lineweaver–Burk plot was constructed by plotting the inverse of substrate concentration (1/[S]) against the inverse of reaction velocity (1/V). The inhibition constant (Ki) was derived from the plot to assess the type and intensity of inhibition. Ki and IC50 values were calculated through nonlinear regression analysis using GraphPad Prism 9.0 software.
3. Results and discussion
3.1. Molecular docking analysis
The docking study of six flavonoids namely, apigenin-7-O-glucoside, baicalin, chrysin, isorhamnetin, naringenin, and silibinin against SQLE yielded binding affinities of −10.7, −10.0, −9.9, −8.5, −9.4, and −10.2 kcal mol−1, respectively. The binding energies indicate that all tested flavonoids have strong interactions with SQLE, suggesting potential for effective inhibition. Among the compounds, apigenin-7-O-glucoside demonstrated the highest binding affinity at −10.7 kcal mol−1, followed closely by silibinin at −10.2 kcal mol−1, indicating stable binding within the enzyme's active site.In Fig. 2, the binding poses of apigenin-7-O-glucoside and baicalin reveal their interactions with several key residues within the SQLE binding pocket. Apigenin-7-O-glucoside forms hydrogen bonds and hydrophobic interactions with residues such as His435, Tyr236, and Phe479, stabilizing its deeply embedded position in the main binding site. These interactions likely enhance its binding affinity and potential inhibitory effect. Baicalin similarly interacts with His435 and Tyr236, along with additional contacts with Trp388, which collectively contribute to its favorable binding affinity of −10.0 kcal mol−1. On the other hand, Fig. 3 illustrates the docking poses of chrysin and isorhamnetin. Chrysin, with a binding affinity of −9.9 kcal mol−1, establishes hydrogen bonding and hydrophobic contacts with key residues, including Tyr236, His435, and Ala236. These interactions stabilize chrysin within the binding site, supporting its potential as an SQLE inhibitor. Isorhamnetin, which showed a binding affinity of −8.5 kcal mol−1, is positioned in a similar binding orientation and interacts with His435, Phe479, and Trp388. However, its slightly lower binding affinity may be attributed to fewer or less robust interactions with the active site residues compared to the other compounds.
 | ||
| Fig. 2 Structural docking configurations of apigenin-7-O-glucoside and baicalin within the active site of squalene epoxidase (SQLE). (A) Display of the binding orientations of each ligand nestled within the enzyme's active site, (B) surface model illustrating the positioning of both ligands inside the enzyme's binding pocket, and (C) identification of residues involved in both polar and hydrophobic interactions with the inhibitors. | ||
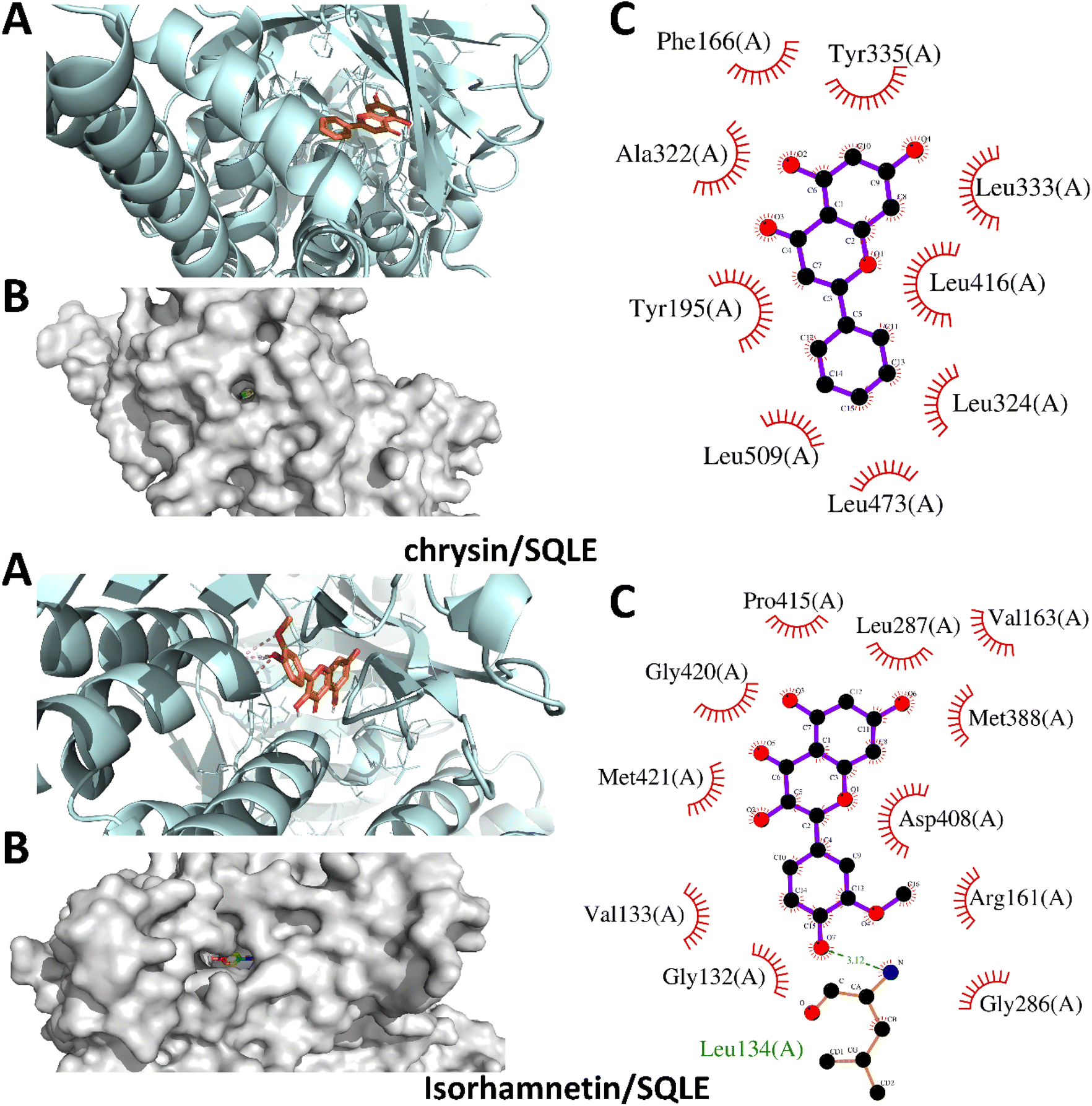 | ||
| Fig. 3 Structural docking configurations of chrysin and isorhamnetin within the active site of squalene epoxidase (SQLE). (A) Display of the binding orientations of each ligand nestled within the enzyme's active site, (B) surface model illustrating the positioning of both ligands inside the enzyme's binding pocket, and (C) identification of residues involved in both polar and hydrophobic interactions with the inhibitors. | ||
The docking poses for naringenin and silibinin show both compounds to be deeply submerged within the primary active site of SQLE, as represented in Fig. 4. Naringenin interacts with critical residues such as His435, Tyr236, and Ala236, which helps stabilize its position within the pocket. Silibinin, with a binding affinity of −10.2 kcal mol−1, forms multiple interactions with His435, Tyr236, and Phe479, aligning with its high binding affinity and stable orientation within the enzyme. These interactions suggest that silibinin may serve as an effective inhibitor of SQLE by blocking access to key catalytic regions within the active site.
 | ||
| Fig. 4 Structural docking configurations of naringenin and silibinin within the active site of squalene epoxidase (SQLE). (A) Display of the binding orientations of each ligand nestled within the enzyme's active site, (B) surface model illustrating the positioning of both ligands inside the enzyme's binding pocket, and (C) identification of residues involved in both polar and hydrophobic interactions with the inhibitors. | ||
The strong binding affinities and deeply submerged binding positions of all six flavonoids highlight their potential as SQLE inhibitors. Apigenin-7-O-glucoside and silibinin, which exhibit the highest binding affinities, are stabilized within the active site by multiple interactions with essential residues such as His435, Tyr236, and Phe479. These key interactions suggest these flavonoids could effectively disrupt SQLE activity, as they are optimally positioned to interfere with squalene conversion within cholesterol biosynthesis pathways. Baicalin, chrysin, isorhamnetin, and naringenin also demonstrate favorable binding interactions with essential residues, though with slightly lower binding affinities. The close interactions of these compounds with His435 and Tyr236, residues critical for SQLE's catalytic activity, reinforce the likelihood of effective inhibition. Furthermore, the ability of these structurally varied flavonoids to fit into the binding site suggests flexibility within SQLE's active site, opening possibilities for the design of related inhibitors with improved potency.
The redocking procedure for the co-crystallized ligand (PDB: 6C6N) produced four docking modes, with the best-ranked pose (Mode 1) showing a binding affinity of −11.9 kcal mol−1 and an RMSD of 0.0 Å relative to the native conformation. This indicates that the original binding orientation was reproduced with high fidelity, thereby validating the robustness of the docking protocol. Additional modes also exhibited acceptable binding affinities (−9.0 to −10.8 kcal mol−1) with RMSD values ranging from 1.27 to 2.13 Å, further supporting the consistency of the docking parameters and scoring function for this system.
3.2. Molecular dynamics simulation
To gain deeper insights into the interactions between our selected phytochemicals and SQLE, we conducted a comprehensive series of molecular dynamics (MD) simulations. These simulations aimed to reveal the stability of each complex over time, as well as to elucidate the specific binding modes and potential inhibitory mechanisms at play. By closely analyzing the resulting trajectories, we focused on how the structural behaviors of these inhibitors might impact their effectiveness in inhibiting SQLE activity. Such insights are essential for advancing the design of new, highly effective SQLE inhibitors with therapeutic potential. We prioritized complexes identified as having the strongest binding affinities in our initial docking studies and extended the simulations to 200 ns to ensure robust results. This extensive analysis involved assessing a range of critical parameters: interaction energies, molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) calculations, potential energy landscapes (PEL), hydrogen-bonding patterns, root mean square deviations (RMSD), solvent-accessible surface area (SASA), radius of gyration (Rg), and root mean square fluctuations (RMSF). By evaluating these diverse metrics, we aimed to build a comprehensive understanding of the structural dynamics and binding stability of these phytochemical inhibitors. | ||
| Fig. 5 Analysis results of 200 ns MD simulation, (A) the RMSD of studied flavonoids–SQLE complexes, and (B) the SQLE backbone RMSD of unbound enzyme and various flavonoid complexes. | ||
The RMSD of the SQLE backbone further supports the stability of the enzyme in complex with different flavonoids (Fig. 5B). The enzyme maintained relatively low RMSD values across all complexes, staying within a narrow range throughout the 200 ns simulation. This low RMSD for the backbone indicates that the binding of the flavonoids did not induce significant structural perturbations in the enzyme, suggesting that SQLE remained stable regardless of the bound ligand. Notably, the black line representing the unbound SQLE backbone also demonstrated low RMSD, affirming that ligand binding did not destabilize the enzyme structure.
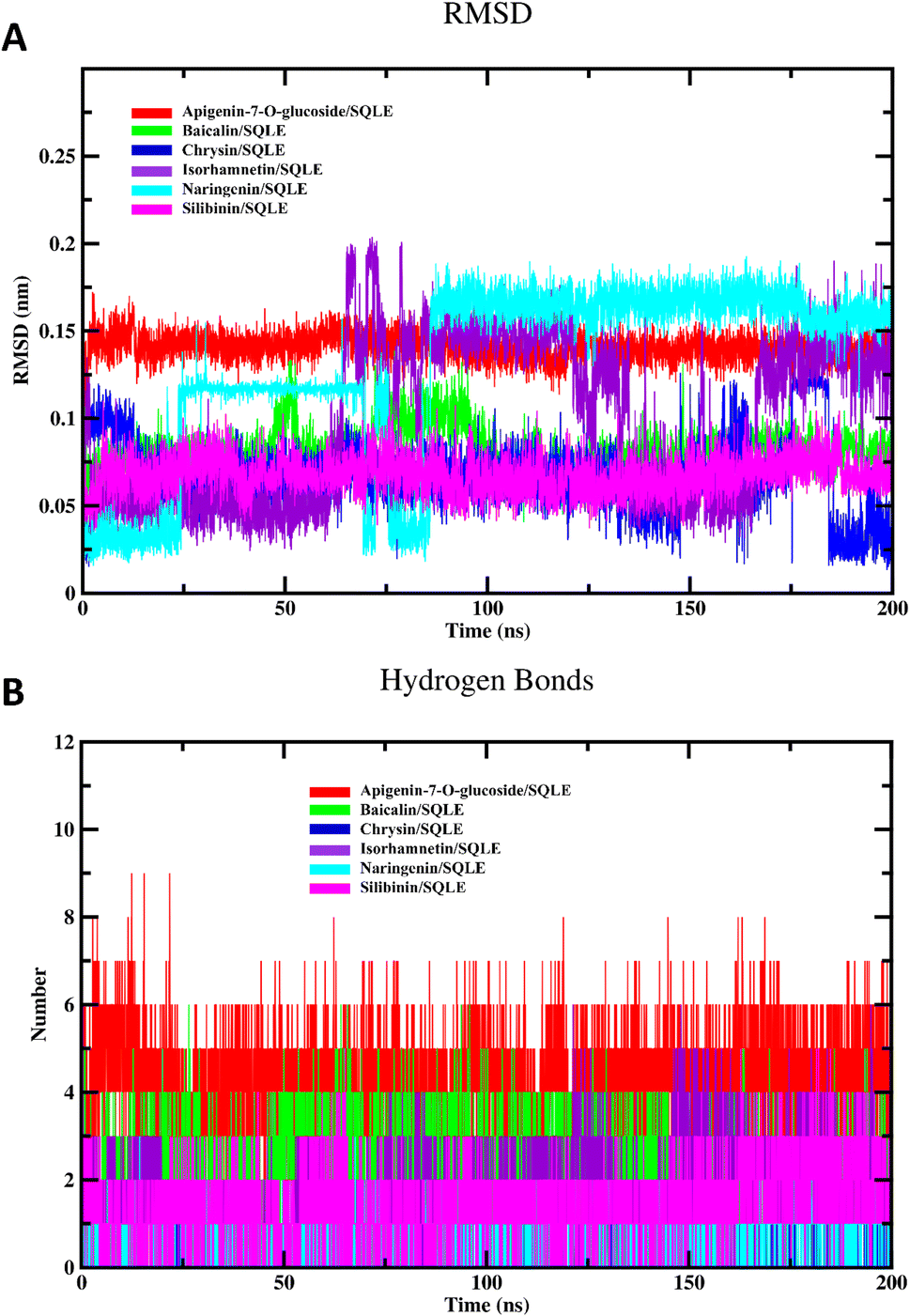
The RMSD values in Fig. 6A reflect the stability of each flavonoid within the SQLE binding pocket over the course of the simulation. Apigenin-7-O-glucoside, silibinin and baicalin showed consistently stable RMSD profile, suggesting that these flavonoids remained relatively stable in their binding conformations with minimal fluctuations, potentially indicating a stronger binding affinity. In contrast, chrysin and isorhamnetin displayed higher RMSD fluctuations, particularly after 100 ns, which could suggest either greater conformational flexibility or weaker binding stability. Naringenin also showed moderate stability, although they experienced occasional spikes in RMSD values, indicative of minor adjustments in binding orientation.
 | ||
| Fig. 6 (A) The RMSD of investigated flavonoids relative to their initial structures, and (B) the hydrogen bonding profiles of investigated complexes. | ||
Fig. 6B displays the hydrogen bond interactions between each flavonoid and SQLE throughout the simulation. Apigenin-7-O-glucoside consistently maintained a higher number of hydrogen bonds, often forming 6–8 hydrogen bonds during the simulation. This extensive hydrogen bonding network may contribute to its stability in the binding pocket, as observed in its RMSD profile. Baicalin also showed a relatively high number of hydrogen bonds, typically around 3–5, which could explain its stable binding conformation. In contrast, chrysin, isorhamnetin, and naringenin exhibited fewer hydrogen bonds, with numbers generally ranging from 1 to 3. Silibinin displayed similar behavior, with limited hydrogen bonding interactions. The reduced hydrogen bond formation for these flavonoids may correlate with their observed RMSD fluctuations, suggesting that a lack of stable hydrogen bonds might lead to less consistent binding within the active site.
Fig. 7A illustrates the RMSF for the free SQLE enzyme and the enzyme in complex with various flavonoids. RMSF analysis highlights the flexibility of residues within the protein structure, revealing the impact of ligand binding on protein dynamics. In this study, all complexes exhibited similar RMSF patterns, suggesting consistent flexibility trends across the different ligands. However, some specific residues displayed elevated fluctuations in certain complexes, indicating regions with increased mobility in the presence of particular flavonoids. Notably, apigenin-7-O-glucoside and silibinin complexes showed slightly higher fluctuations in the regions around residues 200–300 and 450–500, which might be associated with their binding interactions. Overall, these RMSF patterns imply that the binding of flavonoids generally stabilizes the enzyme, with minor localized fluctuations likely due to specific ligand interactions.
 | ||
| Fig. 7 (A) The RMSF profile of free SQLE and various flavonoids–SQLE complexes, (B) the Rg values of different systems, and (C) the SASA values of investigated complexes and the free enzyme. | ||
The Rg values, shown in Fig. 7B, provide insights into the overall compactness and structural stability of the SQLE enzyme and its complexes. Throughout the 200 ns simulation, the Rg values for all systems, including the free enzyme and each flavonoid complex, were stable, fluctuating between 2.3 and 2.5 nm. This indicates that the binding of flavonoids does not significantly affect the compactness or overall structural integrity of SQLE. Among the flavonoid complexes, naringenin and chrysin exhibited slightly lower Rg values, suggesting these complexes may enhance structural stability, leading to a more compact enzyme structure. However, the differences in Rg across all systems were minimal, indicating that flavonoid binding generally maintains the enzyme's native structural compactness.
Fig. 7C presents the SASA for the free enzyme and its flavonoid complexes. SASA provides information on the surface exposure of the protein, reflecting conformational changes and potential stabilization effects. Throughout the simulation, the SASA values for all systems were relatively consistent, ranging from 190 to 230 nm2. The observed SASA values indicate minimal conformational alterations upon flavonoid binding, as there were no drastic changes in surface exposure. However, the complexes with apigenin-7-O-glucoside and naringenin displayed slightly higher SASA values, suggesting these ligands may induce subtle conformational changes, exposing more of the protein surface. The stable SASA patterns across all systems suggest that flavonoid binding does not significantly disrupt the overall conformation of SQLE.
 | ||
| Fig. 8 (A) The Coul-SR interaction energies of various flavonoids–SQLE complexes, and (B) the LJ-SR interaction energies of different systems. | ||
The Lennard-Jones short-range (LJ-SR) interaction energies for the SQLE-flavonoid complexes, reflecting the van der Waals interactions (Fig. 8B). Similar to Coul-SR, more negative LJ-SR values indicate stronger van der Waals interactions. Throughout the simulation, the LJ-SR energies for all flavonoid complexes remained consistently negative, with naringenin/SQLE and silibinin/SQLE showing the most negative values, often reaching below −200 kJ mol−1. This suggests that van der Waals interactions are particularly favorable for these complexes, potentially enhancing the stability of the flavonoid–SQLE binding. Baicalin, chrysin, and isorhamnetin complexes, while also displaying negative LJ-SR energies, had slightly higher values, indicating comparatively weaker van der Waals interactions.
 | ||
| Fig. 9 Potential Energy Landscape (PEL) analysis of SQLE and its complexes with apigenin-7-O-glucoside, baicalin, and chrysin. (A) 2D heat maps of the PEL estimated from principal components PC1 and PC2. (B) Structural representation of SQLE and the lowest-energy conformations. (C) 3D PEL plots, visualizing the conformational energy wells of each complex. | ||
 | ||
| Fig. 10 Potential Energy Landscape (PEL) analysis of SQLE and its complexes with isorhamnetin, naringenin, and silibinin. (A) 2D heat maps of the PEL estimated from principal components PC1 and PC2. (B) Structural depiction of SQLE with the lowest-energy conformations. (C) 3D PEL plots based on PC1 and PC2, showing the conformational energy wells for each complex. | ||
Fig. 9B and 10B depict the lowest-energy conformation of each flavonoid–SQLE complex and highlights the key residues involved in the binding interactions. The interactions include hydrogen bonding, hydrophobic contacts, and π–π stacking, which are essential for stabilizing the complex. Notably, apigenin-7-O-glucoside form multiple hydrogen bonds with active site residues, contributing to their stability in the SQLE binding pocket. Other flavonoids, such as chrysin and silibinin, also exhibit hydrophobic interactions that enhance binding stability. These residue-level interactions provide insight into the specific binding modes, which could influence each flavonoid's potential inhibitory effect on SQLE. Panel C presents the 3D representations of the PEL for each flavonoid–SQLE complex. The 3D landscape visualizations show well-defined energy wells corresponding to stable conformational states. The depth and shape of these wells indicate the binding strength and stability of each flavonoid. For example, apigenin-7-O-glucoside and baicalin have deeper and more pronounced energy minima, suggesting stronger and more stable binding interactions with SQLE. These findings support the 2D contour data, reinforcing that certain flavonoids may achieve greater binding stability due to their favorable conformational positioning within the SQLE active site.
| Complex | ΔEvdw | ΔEele | ΔGsolv | ΔGgas | ΔGtotal |
|---|---|---|---|---|---|
| Apigenin-7-O-glucoside/SQLE | −54.84 ± 1.16 | −46.05 ± 2.19 | 56.36 ± 1.12 | −100.89 ± 2.54 | −44.53 ± 2.77 |
| Baicalin/SQLE | −49.40 ± 2.54 | −25.50 ± 4.77 | 42.58 ± 3.48 | −74.90 ± 3.97 | −32.31 ± 3.01 |
| Chrysin/SQLE | −35.73 ± 0.47 | −7.86 ± 0.99 | 18.48 ± 0.70 | −43.59 ± 1.16 | −25.11 ± 1.35 |
| Isorhamnetin/SQLE | −30.46 ± 0.76 | −22.84 ± 4.60 | 33.65 ± 1.66 | −53.30 ± 4.68 | −19.65 ± 4.96 |
| Naringenin/SQLE | −36.39 ± 2.01 | −5.97 ± 3.17 | 17.16 ± 2.51 | −42.35 ± 3.40 | −25.19 ± 2.41 |
| Silibinin/SQLE | −59.00 ± 0.81 | −22.76 ± 3.10 | 43.75 ± 1.43 | −81.76 ± 3.27 | −38.01 ± 3.57 |
Electrostatic interactions also varied among the complexes. Apigenin-7-O-glucoside displayed the most favorable electrostatic energy, indicating strong polar interactions, which could contribute to its stability within the binding site. In comparison, naringenin and chrysin showed weaker electrostatic interactions, suggesting a lesser degree of polar interactions with SQLE. The solvation free energy values were positive for all complexes, indicating that desolvation is energetically unfavorable upon binding, as expected due to the reduction in surface area exposed to the solvent. Apigenin-7-O-glucoside had the highest ΔGsolv, followed by silibinin, implying that these complexes require more energy for desolvation, possibly due to their polar or exposed functional groups. In terms of gas-phase energy, apigenin-7-O-glucoside and silibinin again showed the most favorable values. This suggests that their combined van der Waals and electrostatic interactions create a stable environment within the SQLE binding site, making them strong candidates for effective binding.
The overall binding free energy (ΔGtotal) reflects the net binding affinity, with more negative values indicating stronger binding. Apigenin-7-O-glucoside showed the most favorable ΔGtotal, followed by silibinin and baicalin. These values suggest that apigenin-7-O-glucoside and silibinin have the highest potential as SQLE inhibitors, as they exhibit the strongest binding affinities. In contrast, isorhamnetin had the least favorable ΔGtotal, indicating a weaker overall binding interaction with SQLE. Thus, the MM/PBSA results highlight that investigated flavonoids demonstrated favorable binding affinities toward the target enzyme.
3.3. In silico ADMET analysis
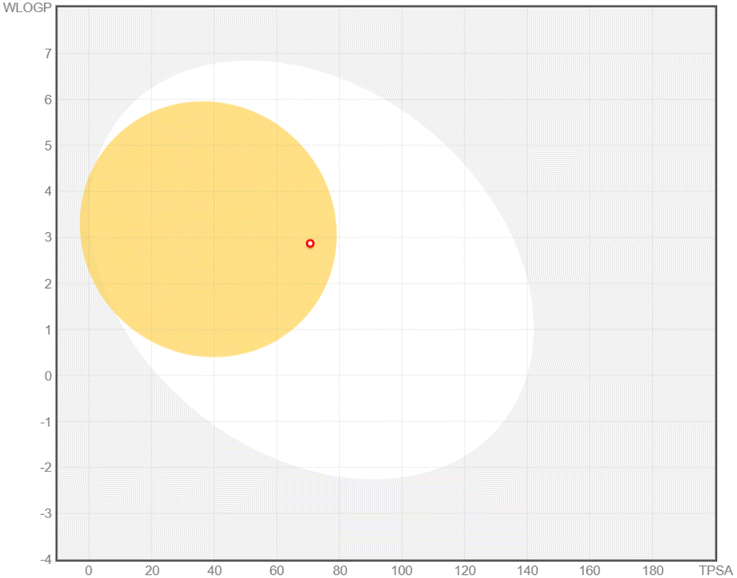
The ADMET analysis provided valuable insights into the drug-likeness properties and potential pharmacokinetic behavior of six flavonoid compounds targeting SQLE (Table 2). This assessment, in conjunction with the MM/PBSA results and MD simulations, supports the evaluation of their inhibitory efficacy and therapeutic potential against SQLE. Wherever possible, the predicted ADMET profiles were correlated with existing experimental findings from the literature to bolster confidence in these in silico outcomes.| Bioavailability radar | BOILED-egg model | ADMET properties | |
|---|---|---|---|
| Apigenin-7-O-glucoside | 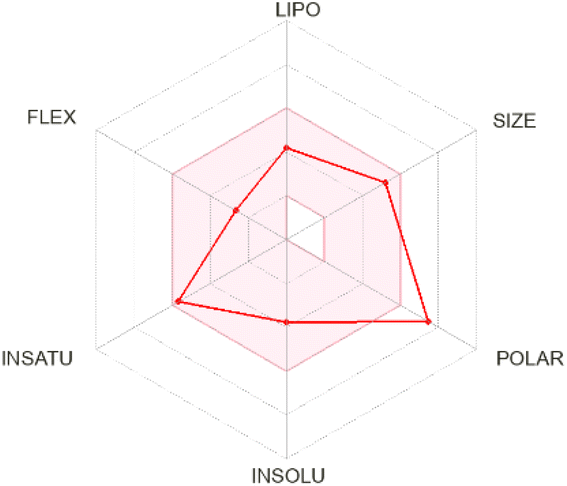 |
 |
Size: MW = 432.38 g mol−1 |
| Num. H-bond donors: 6 | |||
| Num. H-bond acceptors: 10 | |||
| Flexibility: number of rotatable bonds = 4 | |||
| Molar refractivity: 106.11 | |||
| Saturation: fraction Csp3 = 0.29 | |||
| TPSA (polarity): 170.05 Å2 | |||
| LogPo/w = 0.52 |
|||
| Lipophilicity: XlogP3 = 1.81 |
|||
| Solubility: logS (ESOL) = −3.78 |
|||
| Bioavailability score = 0.55 | |||
| LogKp (skin permeation) = −7.65 cm s−1 |
|||
| P-gp substrate, yes | |||
| CYP1A2 (No), CYP2C19 (No), CYP2C9 (No), CYP2D6 (No), and CYP3A4 (No) | |||
| BBB permeant: no | |||
| Baicalin |  |
 |
Size: MW = 446.36 g mol−1 |
| Num. H-bond donors: 6 | |||
| Num. H-bond acceptors: 11 | |||
| Flexibility: number of rotatable bonds = 4 | |||
| Molar refractivity: 106.72 | |||
| Saturation: fraction Csp3 = 0.24 | |||
| TPSA (polarity): 187.12 Å2 | |||
| LogPo/w = 0.25 |
|||
| Lipophilicity: XlogP3 = 1.11 |
|||
| Solubility: logS (ESOL) = −3.41 |
|||
| Bioavailability score = 0.55 | |||
| LogKp (skin permeation) = −8.23 cm s−1 |
|||
| P-gp substrate, yes | |||
| CYP1A2 (No), CYP2C19 (No), CYP2C9 (No), CYP2D6 (No), and CYP3A4 (No) | |||
| BBB permeant: no | |||
| Chrysin |  |
 |
Size: MW = 254.24 g mol−1 |
| Num. H-bond donors: 2 | |||
| Num. H-bond acceptors: 4 | |||
| Flexibility: number of rotatable bonds = 1 | |||
| Molar refractivity: 71.97 | |||
| Saturation: fraction Csp3 = 0.00 | |||
| Lipophilicity: XlogP3 = 3.52 |
|||
| TPSA (polarity): 70.67 Å2 | |||
| Solubility: logS (ESOL) = −4.19 |
|||
| LogPo/w = 2.55 |
|||
| P-gp substrate, no | |||
| LogKp (skin permeation) = −5.35 cm s−1 |
|||
| Bioavailability score = 0.55 | |||
| CYP1A2 (Yes), CYP2C19 (No), CYP2C9 (No), CYP2D6 (Yes), and CYP3A4 (Yes) | |||
| BBB permeant: yes | |||
| Isorhamnetin | 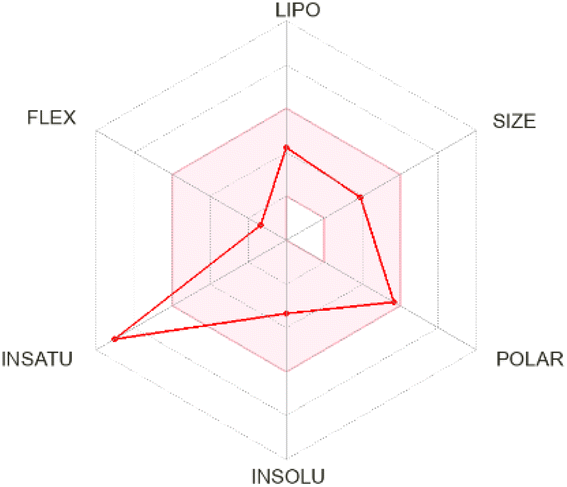 |
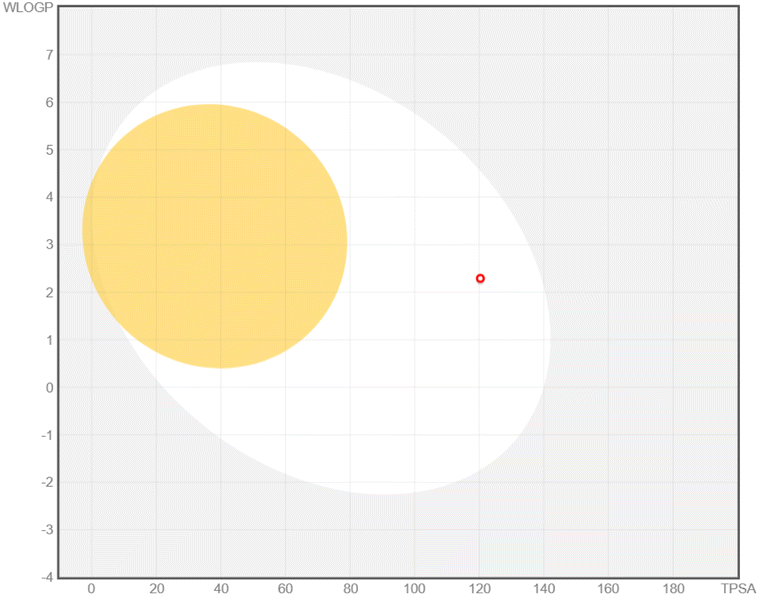 |
Size: MW = 316.26 g mol−1 |
| Num. H-bond donors: 4 | |||
| Num. H-bond acceptors: 7 | |||
| Molar refractivity: 82.50 | |||
| Flexibility: number of rotatable bonds = 2 | |||
| Saturation: fraction Csp3 = 0.06 | |||
| LogPo/w = 1.65 |
|||
| Lipophilicity: XlogP3 = 1.87 |
|||
| TPSA (polarity): 120.36 Å2 | |||
| Solubility: logS (ESOL) = −3.36 |
|||
| LogKp (skin permeation) = −6.90 cm s−1 |
|||
| P-gp substrate, no | |||
| Bioavailability score = 0.55 | |||
| CYP1A2 (Yes), CYP2C19 (No), CYP2C9 (No), CYP2D6 (Yes), and CYP3A4 (Yes) | |||
| BBB permeant: no | |||
| Naringenin |  |
 |
Size: MW = 272.25 g mol−1 |
| Num. H-bond donors: 3 | |||
| Num. H-bond acceptors: 5 | |||
| Flexibility: number of rotatable bonds = 1 | |||
| Molar refractivity: 71.57 | |||
| Saturation: fraction Csp3 = 0.13 | |||
| Lipophilicity: XlogP3 = 2.52 |
|||
| TPSA (polarity): 86.99 Å2 | |||
| LogPo/w = 1.84 |
|||
| LogKp (skin permeation) = −6.17 cm s−1 |
|||
| Solubility: logS (ESOL) = −3.49 |
|||
| P-gp substrate, yes | |||
| Bioavailability score = 0.55 | |||
| CYP1A2 (Yes), CYP2C19 (No), CYP2C9 (No), CYP2D6 (No), and CYP3A4 (Yes) | |||
| BBB permeant: no | |||
| Silibinin |  |
 |
Size: MW = 482.44 g mol−1 |
| Num. H-bond acceptors: 10 | |||
| Num. H-bond donors 5 | |||
| Saturation: fraction Csp3 = 0.24 | |||
| Molar refractivity: 120.55 | |||
| TPSA (polarity): 155.14 Å2 | |||
| Lipophilicity: XlogP3 = 1.90 |
|||
| LogPo/w = 1.51 |
|||
| Solubility: logS (ESOL) = −4.14 |
|||
| BBB permeant: no | |||
| Flexibility: Number of rotatable bonds = 4 | |||
| LogKp (skin permeation) = −7.89 cm s−1 |
|||
| Bioavailability score = 0.55 | |||
| P-gp substrate, no | |||
| CYP1A2 (No), CYP2C19 (No), CYP2C9 (No), CYP2D6 (No), and CYP3A4 (Yes) |
Apigenin-7-O-glucoside demonstrated moderate drug-likeness attributes, including a bioavailability score of 0.55. It was identified as a P-glycoprotein (P-gp) substrate and did not inhibit cytochrome P450 enzymes, aligning with previous in vitro studies indicating that apigenin derivatives can undergo active efflux by P-gp transporters.52,53 Although this compound lacks blood–brain barrier (BBB) permeability according to our predictions, pharmacokinetic data for structurally similar apigenin glycosides also suggest limited CNS uptake.54 Its high polarity and favorable aqueous solubility are supportive of systemic distribution, consistent with experimental reports on apigenin-7-O-glucoside's moderate to high solubility profile.53 Baicalin presented a slightly higher molecular weight and displayed drug-likeness features comparable to apigenin-7-O-glucoside, including P-gp substrate characteristics and no inhibition of major cytochrome P450 enzymes. Previous studies have shown that baicalin undergoes limited gastrointestinal absorption but retains notable systemic bioavailability when administered in higher doses or via certain formulations.55 Its high polar surface area and substantial solubility predicted here are consistent with reports of baicalin's hydrophilic nature, which supports adequate circulation and potential distribution in vivo.55
Chrysin showed distinct pharmacokinetic properties, including a smaller molecular size, lower polarity, and favorable lipophilicity, leading to a predicted BBB permeability—unique among the compounds studied. This aligns with animal model data that have demonstrated chrysin's ability to penetrate the CNS to exert neuroprotective and anxiolytic effects.56 However, chrysin's lack of P-gp substrate activity may influence its distribution and clearance, echoing in vitro observations indicating limited interaction with efflux transporters.57 Isorhamnetin exhibited a balanced pharmacokinetic profile, with moderate polarity and a logP of 1.87, suggesting systemic availability without BBB penetration. Its predicted P-gp non-substrate properties and partial inhibition of cytochrome P450 enzymes are supported by experimental data showing that certain methylated flavonoids can moderately modulate CYP activity.58,59 Although the MM/PBSA results indicated a relatively low ΔGtotal, the combination of systemic availability and possible enzyme inhibition warrants further exploration for optimizing its structure–activity relationship.
Naringenin demonstrated moderate lipophilicity, no BBB permeability, and an adequate bioavailability score. Its profile as a P-gp substrate and lack of CYP inhibition align with literature findings on naringenin metabolism, which indicate that it is actively effluxed and does not substantially interfere with key CYP isoforms.60,61 These favorable in silico ADMET properties, especially its documented oral bioavailability and moderate clearance, highlight naringenin as a promising systemic agent against SQLE. On the other hand, silibinin displayed the highest molecular weight and moderate lipophilicity, alongside a high TPSA and favorable solubility. These in silico observations are consistent with experimental data showing that silibinin, a major active component of silymarin, is relatively well absorbed under certain formulations and exhibits good systemic circulation (Kidd & Head, 2005). Its potential as a potent and stable SQLE-targeting compound is further underscored by literature describing silibinin's efficacy in inhibiting cancer-related targets and regulating lipid metabolism.62
In summary, apigenin-7-O-glucoside and silibinin emerged as the most promising SQLE inhibitors based on their robust MM/PBSA binding energies and stable MD profiles, supported by favorable ADMET characteristics—including high bioavailability scores and, in the case of apigenin-7-O-glucoside, experimentally corroborated P-gp substrate activity. Similarly, silibinin's predicted stability and solubility align with literature reports of its therapeutic efficacy and manageable pharmacokinetic profile in vivo. Taken together, these findings indicate that the in silico ADMET predictions correlate well with available experimental data for the examined flavonoids, reinforcing their potential for therapeutic development against SQLE.
3.4. In vitro inhibitory activity assay
The inhibitory activities of six flavonoids against SQLE were evaluated, with TNSCPA serving as the positive control (IC50: 2.68 ± 0.18 μM). The concentration–response curve of these flavonoids against SQLE is shown in Fig. 11A. Among these, apigenin-7-O-glucoside exhibited the most potent inhibition with an IC50 of 1.74 ± 0.05 μM, followed by silibinin (1.88 ± 0.28 μM) and baicalin (2.50 ± 0.46 μM). Chrysin (4.47 ± 0.19 μM), naringenin (6.52 ± 0.9 μM), and isorhamnetin (11.50 ± 1.47 μM) showed comparatively lower inhibitory activities. These results, shown in Fig. 11A and 11B, identify apigenin-7-O-glucoside, baicalin, and silibinin as lead compounds for further kinetic analysis. | ||
| Fig. 11 In vitro inhibition of SQLE by selected flavonoids and enzyme kinetics analysis. (A) Concentration-response curves showing the percentage relative activity of SQLE at varying concentrations of TNSCPA (positive control) and the proposed flavonoids. (B) IC50 values of TNSCPA and the six flavonoids. (C–E) Lineweaver–Burk plots for apigenin-7-O-glucoside (C), baicalin (D), and silibinin (E). (F–H) Michaelis–Menten plots for apigenin-7-O-glucoside (F), baicalin (G), and silibinin (H). | ||
The Lineweaver–Burk plots (Fig. 11C–E) and Michaelis–Menten plots (Fig. 11F–H) were used to characterize the modes of inhibition for the three selected compounds. Apigenin-7-O-glucoside exhibited competitive inhibition, as indicated by increasing slopes and unchanged y-intercepts in its Lineweaver–Burk plot (Fig. 11C). The competitive mode suggests that apigenin-7-O-glucoside binds to the enzyme's active site, preventing substrate access. Conversely, baicalin and silibinin displayed mixed inhibition (Fig. 11D and E), where both the binding to the active site and allosteric interactions are likely involved. The changes in both slopes and y-intercepts in their Lineweaver–Burk plots support this conclusion. The Ki values for apigenin-7-O-glucoside (0.66 μM), baicalin (1.75 μM), and silibinin (1.42 μM) further underscore their strong binding affinities to SQLE.
The experimental results align with in silico docking and MD simulation studies, which predicted high binding energies and stable trajectories for apigenin-7-O-glucoside, baicalin, and silibinin to SQLE. This congruence highlights the reliability of computational approaches in guiding experimental validation. The in silico ADMET properties of the three flavonoids provide insights into their interaction mechanisms. Apigenin-7-O-glucoside, with its moderate lipophilicity (XlogP3 = 1.81) and favorable TPSA (170.05 Å2), supports its competitive inhibition by facilitating active site binding without extensive steric hindrance. Its limited bioavailability (logS = −3.78) and poor blood–brain barrier permeability suggest its effect is likely localized, enhancing specificity for SQLE. Baicalin and silibinin, exhibiting mixed inhibition, possess higher molecular weights (446.36 g mol−1 and 482.44 g mol−1, respectively) and greater polarity (TPSA = 187.12 Å2 and 155.14 Å2, respectively). These properties align with their dual interaction modes, potentially involving both active site and allosteric binding. Notably, silibinin's higher lipophilicity (XlogP3 = 1.90) compared to baicalin (XlogP3 = 1.11) may contribute to its slightly stronger binding affinity (Ki = 1.61 μM).
Although our study focused on flavonoid-based inhibitors, contextualizing these in vitro findings with respect to well-established SQLE inhibitors can provide deeper insights into structure–activity relationships (SAR) and binding mechanisms. Known clinically used compounds such as terbinafine, naftifine, butenafine, and NB-598 typically act through high-affinity interactions within the enzyme's catalytic region, often displaying IC50 values in the low micromolar to submicromolar range.63 For instance, terbinafine, an allylamine derivative, is reported to bind predominantly in a hydrophobic pocket of SQLE, forming crucial hydrogen bonds with active-site residues that stabilize its binding mode.64 In this regard, our top-performing flavonoids—apigenin-7-O-glucoside, silibinin, and baicalin—exhibited IC50 values (1.74–2.50 μM) that fall into a similar potency range for early-stage hit compounds.
From an SAR perspective, both allylamine-based inhibitors (e.g., terbinafine) and our flavonoids appear to exploit the enzyme's substrate-binding region, yet they do so with distinct chemical scaffolds. Flavonoids include polar functional groups (e.g., hydroxyl and glycosyl moieties) capable of forming hydrogen bonds with key residues, while allylamine inhibitors use more lipophilic anchors to occupy the predominantly non-polar cavity of SQLE.65 In apigenin-7-O-glucoside, for example, the glycosidic substituent likely contributes specific hydrogen-bonding interactions that enable its competitive inhibition mechanism. This contrasts with the mixed inhibition observed for silibinin and baicalin, suggesting these molecules may also engage allosteric residues beyond the core catalytic site—a possibility also observed in certain azole derivatives or second-generation SQLE inhibitors such as NB-598.66
In terms of binding modes, our computational data indicate stable interactions between flavonoid ligands and residues that are commonly implicated in the binding of allylamine- and benzylamine-class inhibitors, underscoring the importance of conserved hot spots within the enzyme's active site.67 Moreover, key hydrophobic contacts seen in known inhibitors appear partially mirrored by the flavonoids' planar aromatic rings, whereas polar moieties in the flavonoids create new hydrogen bonds and electrostatic interactions not seen in purely lipophilic scaffolds. These additional contacts may open avenues for further structural modifications to enhance specificity and potency.
Overall, while the flavonoid derivatives and conventional SQLE inhibitors originate from distinct chemical families, they converge on similar critical binding residues in SQLE, illustrating both the enzyme's versatility in accommodating different scaffolds and the potential for novel SAR insights. Future efforts to optimize flavonoid structures—leveraging the hydrogen-bonding capability and mixed binding modes—may lead to more potent next-generation SQLE inhibitors that rival or complement existing agents like terbinafine and butenafine.
4. Conclusion
In this study, we employed an integrative approach combining in silico predictions and experimental validation to evaluate the inhibitory potential of six flavonoids—silibinin, baicalin, naringenin, chrysin, apigenin-7-O-glucoside, and isorhamnetin—against squalene epoxidase (SQLE). The selected flavonoids were chosen based on their structural diversity, reported bioactivity, and relevance to SQLE inhibition. Comprehensive computational studies, including molecular docking, MD simulations, potential energy landscape (PEL) analysis, MM/PBSA calculations, and ADMET profiling, identified apigenin-7-O-glucoside, baicalin, and silibinin as the most promising inhibitors. These compounds displayed the highest binding affinities, robust interaction profiles, and stable dynamic trajectories, coupled with favorable pharmacokinetic and pharmacodynamic properties. Experimental validation confirmed the computational predictions, with apigenin-7-O-glucoside exhibiting the most potent inhibition of SQLE (IC50 = 1.74 ± 0.05 μM), followed by silibinin (IC50 = 1.88 ± 0.28 μM) and baicalin (IC50 = 2.50 ± 0.46 μM). The Lineweaver–Burk and Michaelis–Menten analyses further revealed that apigenin-7-O-glucoside acts as a competitive inhibitor, while silibinin and baicalin exhibit mixed inhibition, suggesting distinct mechanisms of action.From a stereostructural perspective, each of these top inhibitors shares a core flavonoid scaffold that supports key hydrogen-bonding and π–π stacking interactions with SQLE. However, their varied substitutions and glycosylation patterns profoundly influence pharmacokinetic behavior. Silibinin features a more complex stereochemistry and multiple hydroxyl groups, which enhance its binding surface area and facilitate robust lipophilic and hydrogen-bond interactions. Experimentally and in silico, silibinin's favorable lipophilicity, moderate molecular weight, and P-glycoprotein substrate profile collectively suggest enhanced bioavailability and systemic distribution. Apigenin-7-O-glucoside contains a glycosidic moiety that increases polarity and water solubility, translating to strong systemic bioavailability but reduced blood–brain barrier permeability. The glycoside portion also participates in additional hydrogen bonding, contributing to potent binding despite its higher polarity. Baicalin, likewise a glycosylated flavonoid, shares structural parallels with apigenin-7-O-glucoside in its ring system and glycosylation, displaying robust but mixed binding interactions. Its polar surface area and solubility support broad distribution, making it a promising prototype for further structural refinements.
By comparing these flavonoids' stereogeometric analogies and their distinct ADMET profiles, we highlight silibinin's particularly strong drug-likeness, apigenin-7-O-glucoside's potent inhibitory action with high solubility, and baicalin's balanced profile bridging polarity and efficacy. These findings underscore the potential of these flavonoids as versatile SQLE inhibitors and illustrate how structural modifications—especially glycosylation and hydroxylation patterns—modulate both affinity and pharmacokinetics. Overall, this study provides a comprehensive framework for identifying and validating potential SQLE inhibitors, paving the way for future preclinical and clinical studies. Apigenin-7-O-glucoside, silibinin, and baicalin emerge as strong candidates for further development, with silibinin in particular showing promising stereogeometric and pharmacokinetic attributes that warrant continued optimization for therapeutic applications in treating SQLE-related diseases, including hypercholesterolemia and fungal infections.
Data availability
The data supporting the findings of this study are available within the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out with the support from the project PID2023-150717NB-I00 from Ministerio de Ciencia, Innovacion y Universidades in Spain and the PRIES-CM project Ref:Y2020/EMT-6290 from the Comunidad de Madrid. Lastly, the authors express their gratitude to the Centro de Computación Científica of the UAM (CCC-UAM) for providing the computing time. The authors acknowledge Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2025R5), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
References
- N. K. Chua, H. W. Coates and A. J. Brown, Prog. Lipid Res., 2020, 79, 101033 CrossRef CAS PubMed
.
- H. Xu, S. Zhou, Q. Tang, H. Xia and F. Bi, Biochim. Biophys. Acta, Rev. Cancer, 2020, 1874, 188394 CrossRef CAS PubMed
- N. K. Kapur, D. Ashen and R. S. Blumenthal, Vasc. Health Risk Manage., 2008, 4, 39–57 CrossRef CAS
- Y.-C. Zhao, Y.-F. Li, L. Qiu, S.-Z. Jin, Y.-N. Shen, C.-H. Zhang, J. Cui and T.-J. Wang, BMC Cancer, 2024, 24, 1133 CrossRef CAS PubMed
- A. Chugh, A. Ray and J. B. Gupta, Prog. Lipid Res., 2003, 42, 37–50 CrossRef CAS PubMed
- D. Tungmunnithum, A. Thongboonyou, A. Pholboon and A. Yangsabai, Medicines, 2018, 5, 93 CrossRef CAS PubMed
- M. A. Alwaili, F. F. Aba Alkhayl, H. A. Rudayni, A. A. Allam, N. G. Altoom, A. M. Lamsabhi and E. M. Kamel, New J. Chem., 2024, 48, 14236–14252 RSC
- M. A. Alwaili, F. F. A. Alkhayl, H. A. Rudayni, A. A. Allam, A. M. Lamsabhi and E. M. Kamel, J. Mol. Struct., 2024, 140354, DOI:10.1016/j.molstruc.2024.140354
- E. M. Kamel, A. Bin-Ammar, A. A. El-Bassuony, M. M. Alanazi, A. Altharawi, A. F. Ahmeda, A. S. Alanazi, A. M. Lamsabhi and A. M. Mahmoud, RSC Adv., 2023, 13, 12361–12374 RSC
- S. A. Ramadan, E. M. Kamel, M. A. Ewais, A. A. Khowailed, E. H. M. Hassanein and A. M. Mahmoud, Environ. Sci. Pollut. Res., 2023, 30, 49197–49214 CrossRef CAS PubMed
- G. M.-H. R. S. Alruhaimi, M. Siddiq Abduh, A. Bin-Ammar, E. H. M. Hassanein, E. M. Kamel and A. M. Mahmoud, Front. Pharmacol, 2023, 14, 1204641 CrossRef PubMed
- B. M. ALRashdi, H. A. Elgebaly, M. O. Germoush, M. M. Qarmush, M. S. Azab, R. S. Alruhaimi, A. F. Ahmeda, M. H. Abukhalil, E. M. Kamel and H. H. Arab, Environ. Sci. Pollut. Res., 2022, 29, 63520–63532 CrossRef CAS PubMed
- N. P. E. Kadoglou, C. Panayiotou, M. Vardas, N. Balaskas, N. G. Kostomitsopoulos, A. K. Tsaroucha and G. Valsami, Pharmaceuticals, 2022, 15, 538 CrossRef CAS PubMed
- T. Wang, H. Jiang, S. Cao, Q. Chen, M. Cui, Z. Wang, D. Li, J. Zhou, T. Wang, F. Qiu and N. Kang, Eur. J. Med. Chem., 2017, 141, 92–100 CrossRef CAS PubMed
- V. P. Chavda, S. Vuppu, P. C. Balar, T. Mishra, R. Bezbaruah, D. Teli, N. Sharma and S. Alom, Int. J. Biol. Macromol., 2024, 266, 131219 CrossRef CAS PubMed
- H. H. Orak, M. Karamać, R. Amarowicz, A. Orak and K. Penkacik, Molecules, 2019, 24, 1130 CrossRef CAS PubMed
- M. Mihal, S. Roychoudhury, A. V. Sirotkin and A. Kolesarova, Front. Endocrinol., 2023, 14 DOI:10.3389/fendo.2023.1244300
- E. M. Kamel, H. A. Alqhtani, M. Bin-Jumah, H. A. Rudayni, A. A. El-Bassuony and A. Mokhtar Lamsabhi, Bioorg. Chem., 2024, 150, 107609 CrossRef CAS PubMed
- E. M. Kamel, F. F. Aba Alkhayl, H. A. Alqhtani, M. Bin-Jumah, H. A. Rudayni and A. M. Lamsabhi, Biophys. Chem., 2024, 313, 107304 CrossRef CAS PubMed
- E. M. Kamel, F. F. A. Alkhayl, H. A. Alqhtani, M. Bin-Jumah, H. A. Rudayni and A. M. Lamsabhi, Comput. Biol. Med., 2024, 180, 108969 CrossRef CAS PubMed
- R. S. Alruhaimi, A. M. Mahmoud, I. Elbagory, A. F. Ahmeda, A. A. El-Bassuony, A. M. Lamsabhi and E. M. Kamel, Bioorg. Chem., 2024, 147, 107397 CrossRef CAS PubMed
- E. M. Kamel and A. M. Lamsabhi, Org. Biomol. Chem., 2020, 18, 3334–3345 RSC
- E. M. Kamel, F. F. Aba Alkhayl, H. A. Alqhtani, M. Bin-Jumah and A. M. Lamsabhi, Int. J. Biol. Macromol., 2024, 282, 136982 CrossRef CAS PubMed
- E. M. Kamel, M. A. Alwaili, H. A. Rudayni, A. A. Allam and A. M. Lamsabhi, Molecules, 2024, 29, 1433 CrossRef CAS PubMed
- E. M. Kamel, S. Maodaa, E. M. Al-Shaebi and A. M. Lamsabhi, Org. Biomol. Chem., 2024, 22, 6561–6574 RSC
- E. M. Kamel and A. M. Lamsabhi, Org. Biomol. Chem., 2021, 19, 9031–9042 RSC
- E. M. Kamel, N. A. Ahmed, A. A. El-Bassuony, O. E. Hussein, B. Alrashdi, S. A. Ahmed, A. M. Lamsabhi, H. H. Arab and A. M. Mahmoud, Comb. Chem. High Throughput Screening, 2022, 25, 1336–1344 CrossRef CAS PubMed
- E. M. Kamel, A. M. Tawfeek, A. A. El-Bassuony and A. M. Lamsabhi, Org. Biomol. Chem., 2023, 21, 7158–7172 RSC
- E. M. Kamel, A. M. Tawfeek, A. A. El-Bassuony and A. M. Lamsabhi, New J. Chem., 2023, 47, 16429–16443 RSC
- U. Norinder and C. A. S. Bergström, ChemMedChem, 2006, 1, 920–937 CrossRef CAS PubMed
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed
- A. D. Becke, Phys. Rev. A:At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed
- W. J. Hehre, L. Radom, P. V. R. Schleyer and J. A. Pople, Ab Initio Molecular Orbital Theory, Wiley New York, 1986 Search PubMed
- N. Guex and M. C. Peitsch, Electrophoresis, 1997, 18, 2714–2723 CrossRef CAS PubMed
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CrossRef CAS PubMed
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed
- H. A. Alqhtani, S. I. Othman, F. F. A. Alkhayl, N. G. Altoom, A. M. Lamsabhi and E. M. Kamel, ChemistrySelect, 2025, 10, e202402984 CrossRef CAS
- A. K. Padyana, S. Gross, L. Jin, G. Cianchetta, R. Narayanaswamy, F. Wang, R. Wang, C. Fang, X. Lv, S. A. Biller, L. Dang, C. E. Mahoney, N. Nagaraja, D. Pirman, Z. Sui, J. Popovici-Muller and G. A. Smolen, Nat. Commun., 2019, 10, 97 CrossRef PubMed
- P. Bauer, B. Hess and E. Lindahl, GROMACS 2022.4 Manual, 2022 Search PubMed
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef
- A. D. MacKerell, Jr., D. Bashford, M. Bellott, R. L. Dunbrack, Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef PubMed
- B. Hess, C. Kutzner, D. Van Der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed
- M. Parrinello and A. Rahman, J. Appl. Phys., 1981, 52, 7182–7190 CrossRef CAS
- M. S. Valdés-Tresanco, M. E. Valdés-Tresanco, P. A. Valiente and E. Moreno, J. Chem. Theory Comput., 2021, 17, 6281–6291 CrossRef PubMed
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed
- N. Philippe, L. Rivron, B. De Bruin, J. Schofield and S. Roy, J. Labelled Compd. Radiopharm., 2018, 61, 878–884 CrossRef CAS PubMed
- I. Abe, T. Seki and H. Noguchi, Biochem. Biophys. Res. Commun., 2000, 270, 137–140 CrossRef CAS PubMed
- Y. S. Ha, W. Lee, J.-M. Jung, N. Soni, D. N. Pandya, G. I. An, S. Sarkar, W. K. Lee and J. Yoo, Anal. Chem., 2018, 90, 8927–8935 CrossRef CAS PubMed
- T. S. Laferriere-Holloway, A. Rios, Y. Lu, C. C. Okoro and R. M. van Dam, J. Chromatogr. A, 2023, 1687, 463656 CrossRef CAS PubMed
- H. Y. Kim and N. Salem, J. Lipid Res., 1990, 31, 2285–2289 CrossRef CAS
- A. I. Alvarez, R. Real, M. Pérez, G. Mendoza, J. G. Prieto and G. Merino, J. Pharm. Sci., 2010, 99, 598–617 CrossRef CAS PubMed
- M. Smiljkovic, D. Stanisavljevic, D. Stojkovic, I. Petrovic, J. Marjanovic Vicentic, J. Popovic, S. Golic Grdadolnik, D. Markovic, S. Sankovic-Babice, J. Glamoclija, M. Stevanovic and M. Sokovic, EXCLI J., 2017, 16, 795–807 Search PubMed
- A. Waheed, S. Zameer, K. Ashrafi, A. Ali, N. Sultana, M. Aqil, Y. Sultana and Z. Iqbal, Curr. Pharm. Des., 2023, 29, 1326–1340 CrossRef CAS PubMed
- Y.-F. Yang, Z. Li, W.-F. Xin, Y.-Y. Wang and W.-S. Zhang, Chin. J. Nat. Med., 2014, 12, 632–640 CAS
- A. Mishra, P. S. Mishra, R. Bandopadhyay, N. Khurana, E. Angelopoulou, Y. N. Paudel and C. Piperi, Molecules, 2021, 26, 6456 CrossRef CAS PubMed
- W. Li, H. Sun, X. Zhang, H. Wang and B. Wu, Biochem. Pharmacol., 2015, 98, 203–214 CrossRef CAS PubMed
- T. K. H. Chang, J. Chen and E. Y. H. Yeung, Toxicol. Appl. Pharmacol., 2006, 213, 18–26 CrossRef CAS PubMed
- S. Zhou, Y. Gao, W. Jiang, M. Huang, A. Xu and J. W. Paxton, Drug Metab. Rev., 2003, 35, 35–98 CrossRef CAS PubMed
- J. M. Assini, E. E. Mulvihill and M. W. Huff, Curr. Opin. Lipidol., 2013, 24, 34–40 CrossRef CAS PubMed
- A. Miron, A. C. Aprotosoaie, A. Trifan and J. Xiao, Ann. N. Y. Acad. Sci., 2017, 1398, 152–167 CrossRef CAS PubMed
- P. Kidd and K. Head, Altern. Med. Rev, 2005, 10, 193–203 Search PubMed
- A. A. Sagatova, J. Fungi, 2021, 7, 49 CrossRef CAS PubMed
- G. Scheiffer, K. Z. A. Domingues, D. Gorski, A. F. Cobre, R. E. L. Lazo, H. H. L. Borba, L. M. Ferreira and R. Pontarolo, EXCLI J., 2024, 23, 1117–1169 Search PubMed
- J. E. Birnbaum, J. Am. Acad. Dermatol., 1990, 23, 782–785 CrossRef CAS PubMed
- L. Zhang, Z. Cao, Y. Hong, H. He, L. Chen, Z. Yu and Y. Gao, Int. J. Mol. Sci., 2024, 25, 3874 CrossRef CAS PubMed
- D. Hammoudi Halat, S. Younes, N. Mourad and M. Rahal, Membranes, 2022, 12, 1171 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |