
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the activity origin and mechanisms of the oxygen reduction reaction on the tetramethyl metalloporphyrin/MoS2 electrocatalyst†
Tran Phuong Dungab,
Pham Tran Nguyen Nguyena,
Viorel Chihaia c and
Do Ngoc Son*de
c and
Do Ngoc Son*de
aDepartment of Chemistry, University of Science, Vietnam National University, Ho Chi Minh City, Vietnam
bDepartment of Chemistry, Ho Chi Minh City University of Education, Ho Chi Minh City, Vietnam
cInstitute of Physical Chemistry “Ilie Murgulescu” of the Romanian Academy, Splaiul Independentei 202, Sector 6, Bucharest, 060021, Romania
dHo Chi Minh City University of Technology (HCMUT), 268 Ly Thuong Kiet Street, District 10, Ho Chi Minh City, Vietnam. E-mail: dnson@hcmut.edu.vn
eVietnam National University Ho Chi Minh City, Linh Trung Ward, Ho Chi Minh City, Vietnam
First published on 26th March 2025
Abstract
The efficiency of the oxygen reduction reaction (ORR) on the cathode plays a crucial role in determining the performance of proton exchange membrane fuel cells. Porphyrin, distinguished by its cost-effectiveness, eco-friendly nature, and efficient utilization of its metal, stands out as a promising candidate for a metal single-atom catalyst in fuel cell cathodes. The metal and support modifications significantly impact the porphyrin's ORR activity. Nevertheless, the effects of Ni, Co, and Fe metals in tetramethyl metalloporphyrin/MoS2, named MeTMP/MoS2, catalyst on the mechanisms and activity of the ORR remain unknown. This study elucidates the topic using van der Waals dispersion-corrected density functional theory (DFT) calculations and thermodynamic model. Results showed that the rate-limiting step is located at the first and second hydrogenation steps in the associative mechanisms for Ni and Co (Fe) substitutions, respectively. For the dissociative mechanisms, the dissociation of molecular oxygen to two oxygen atoms is the rate-determining step on all the NiTMP/MoS2, CoTMP/MoS2, and FeTMP/MoS2 catalysts. The presence of the MoS2 support significantly reduces the thermodynamic activation barrier of the ORR, and hence improves the ORR activity in the dissociative mechanisms. This activation barrier is 3.45, 0.92, and 1.82 eV for NiTMP/MoS2, CoTMP/MoS2, and FeTMP/MoS2, which is much better compared to 4.85, 3.34, and 2.19 eV for NiTMP, CoTMP, and FeTMP, respectively. CoTMP/MoS2 is the best candidate among the considered catalysts for the ORR. Furthermore, we provide a detailed explanation of the physical insights into the interaction between the ORR intermediates and the catalysts.
1. Introduction
The ORR is a critical process in energy technologies, such as proton exchange membrane fuel cells and metal-air batteries.1,2 However, the sluggish kinetics of the ORR on the cathodes hinder the efficiency of these devices. Therefore, finding efficient and sustainable ORR catalysts is crucial for advancing energy technologies. Metalloporphyrins, incorporating a non-precious metal atom in the centre of the porphyrin ring, have emerged as promising alternatives to traditional noble metal catalysts.3–11 The metal centre, often transition metals such as iron, cobalt, nickel, and manganese, serves as an active site for catalysing the ORR.10,11 A unique advantage of metal porphyrins lies in their ability to host a single metal atom as the catalytic site, minimizing metal usage and maximizing the catalytic efficiency. Besides, the reactivity of the metal porphyrins toward ORR is highly dependent on the substituents of the porphyrin ring functional groups, as evidenced by experimental12–14 and theoretical investigations.15–17 Our previous study showed that substituting tetramethyl (–CH3), amino (–NH2), and carboxyl (–COOH) functional groups at the meso-positions of the iron porphyrin can alter the electron density distribution within the ring, affecting the ORR mechanisms and activity of the iron porphyrin.15 Among the functional groups studied, the amino and methyl groups exhibited high oxygen reduction activity due to lowering the activation energy of the rate-determining steps.15Because of the low stability of pure metalloporphyrins, they are typically coated onto electrodes made of various materials, such as glassy carbon and graphite for the ORR.7–10,17–19 Porphyrins supported on carbon substrates were found to significantly enhance the ORR catalytic activity.18,19 Schilling et al. also showed that metalloporphyrin membranes integrated into carbon nanotubes offered superior electrical conductivity, and significantly improved the ORR activity.18 Electrode materials with highly porous structures, such as metal–organic frameworks integrated with metal porphyrin linkers, also exhibited selective catalytic activity for the ORR via the 4-electron mechanism.19 Although carbon materials are commonly used as supports for the ORR catalysts, they are corroded under electrochemical operating conditions of the ORR. Therefore, they weaken the long-term stability of the catalysts.20,21
Recently, transition metal dichalcogenides such as MoS2, a class of carbon-free materials, have attracted much attention as support materials for electrocatalysts due to the high flexibility and stability of their structure.22–26 For example, under appropriate conditions, MoS2 offered high stability and a large surface area for dispersing catalysts.27 The literature revealed that the layered structure of MoS2 provides well-defined active sites for anchoring precious metal catalysts, such as platinum for electro-catalytic water splitting.28 Additionally, the MoS2-based materials have been demonstrated as alternative catalysts for the hydrogen evolution reaction (HER) and the ORR.22,23,29 The N-doped MoS2 supported on carbon catalysts also showed a dramatic improvement in their ORR activity and stability for microbial fuel cells.30 Recently, iron porphyrins reinforced on the MoS2 support showed good oxygen reduction activity.24 However, no research is available on the ORR mechanisms and activity on the metalloporphyrin/MoS2 electro-catalysts, particularly, for the MeTMP/MoS2 catalyst with Me = Ni, Co, Fe; and TMP = tetramethyl porphyrin.
This study clarifies the effects of metal substitutions in the metalloporphyrin/MoS2 catalyst on the mechanisms and activity of the ORR using the van der Waals DFT calculations and the thermodynamic model. The research focuses on iron-series elements (Fe, Co, Ni) because they are earth-abundant, and their compounds with porphyrins have shown good ORR activities, as reported in previous studies.3,7–13 The results of this work will be useful for designing a rational electrocatalyst for ORR.
2. Computational details
This study utilizes the metallic phase of MoS2 with the distorted octahedral coordinate structure, denoted as 1T′-MoS2, because of the following reasons: (1) the semiconducting phase 2H-MoS2 undergoes a phase transition naturally or by heat treatments, and hydrothermal synthesis into the metallic phase 1T′-MoS2.29,31 (2) The metallic phase has a higher electrical conductivity; therefore, it is beneficial for making an electrode with a better electrical conductivity and therefore enhances catalytic performance.32 (3) the recent study showed that the metallic phase 1T′-MoS2 exhibited significantly higher activity for the electrocatalytic HER in an acidic medium than that of 2H-MoS2.33 Based on the primitive unit cell of the monolayer structure of the 1T′-MoS2 phase,34 we built a supercell in the slab model by repeating 3 and 5 times along the a and b unit vectors or the x and y axes, as shown in Fig. S1† (top view) of ESI, respectively. The monolayer has one atomic layer of Mo atoms, sandwiched between two atomic layers of S atoms. Each atomic layer has thirty atoms. The optimized structure of the supercell by our DFT calculations has the Mo–Mo distance of 3.17, 3.81, and 2.77 Å along the y-axis and two diagonal axes (Fig. S1†), respectively. The free space of the supercell along the c direction (see the side view in Fig. S1†) is 17 Å, which is large enough for the adsorption of the metalloporphyrin molecule and the ORR intermediates on the 1T′-MoS2 substrate without the crossing interaction between the supercell images.Our previous study also showed that the meso-tetra-methyl (–CH3) functional group is the best substitution among the considered substituents.15 The metal centre (Me) of the metalloporphyrin (MeTMP) is replaced sequentially with Ni, Co, and Fe. Here, we used the oxidation state +2 for Fe, Co, and Ni, which agrees with the literature.3,7–9,35,36 It is worth noting that the ORR study for the FeTMP molecule has been done in our previous work.15 Therefore, we can use our earlier results for the FeTMP substrate to make comparisons, from which we can gauge the new outcomes for the presence of the MoS2 support on the FeTMP/MoS2 catalyst.
We designed the MeTMP/MoS2 substrates (Me = Fe, Co, and Ni). We then optimized the MeTMP/MoS2 systems with several initial positions of the MeTMP molecule on the optimized MoS2 surface to search for the most stable configuration of the MeTMP molecule on the MoS2 support via the binding energy calculation:
| Eb = [EPor + EMoS2] − EPor/MoS2. | (1) |
We employed the Vienna Ab Initio Simulation Package (VASP) to perform DFT calculations with the van der Waals corrections (vdW-DF), the revised version of the generalized gradient approximation proposed by Perdew–Burke–Ernzerhof (GGA-revPBE) for the exchange-correlation energy.37 The vdw-DF method was chosen due to its rigorous theoretical foundation, which self-consistently incorporates nonlocal correlation effects.38,39 Moreover, this method has demonstrated high accuracy in adsorption energy calculations for similar systems.40 The projector-augmented wave technique for pseudopotentials was employed with a plane-wave cutoff energy of 450 eV.41 The Monkhorst and Pack method was utilized for sampling the special k-points with a 3 × 3 × 1 k-point mesh grid.42 We employed the Gaussian smearing method with a smearing width of 0.1 eV for structural optimization and total energy calculations, and 0.01 eV for calculating the electronic density of states. The criteria for the convergence of the force for atomic position optimization and the energy difference for self-consistent electronic loops are 0.001 eV Å−1 and 10−5 eV, respectively. Spin-polarized calculations were performed with a dipole correction applied to the c direction.
The formula used to calculate the adsorption energy of each intermediate (A) on the surface of substrate B is as follows:
| Ea = EAB − [EA + EB]. | (2) |
The interaction between the substrate and the adsorbed intermediates is often visualized by charge density difference plots,43,44 which can be calculated by
| Δρ = ρABC − [ρA + ρB + ρC]. | (3) |
The Gibbs free energy (ΔG) for each intermediate step was calculated using the proton and electron exchange model,45–48 where the Gibbs free energy involved in the proton and electron combination process is equivalent to that of ½H2 in the gas phase at the reversible hydrogen electrode potential, H+ + e− = ½H2:
| ΔG(U) = ΔE + ΔZPE − TΔS + neU. | (4) |
The Gibbs free energy for the transition state (TS) of the dissociation from  to 2O* has been calculated by:
to 2O* has been calculated by:
| ΔGTS = ΔEbr + ΔZPE − TΔS. | (5) |
 to (TS). Like the ORR, ΔZPE and ΔS are the change of the zero-point energy and the entropy change between (TS) and
to (TS). Like the ORR, ΔZPE and ΔS are the change of the zero-point energy and the entropy change between (TS) and 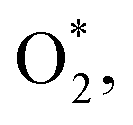 respectively. However, the electrode potential does not influence the Gibbs free energy for the transition state because there is no electron reduction in the
respectively. However, the electrode potential does not influence the Gibbs free energy for the transition state because there is no electron reduction in the  dissociation process. The asterisk denotes the adsorbed state of the intermediate on the MeTMP/MoS2 catalyst.
dissociation process. The asterisk denotes the adsorbed state of the intermediate on the MeTMP/MoS2 catalyst.
To consider the possibility that the HER can compete with the ORR on the MeTMP/MoS2 substrates, we are going to investigate the HER, which is
| H+ + e− = 1/2H2. | (R1) |
This reaction on the MeTMP/MoS2 substrate (Sub) is described as:
| Sub* + H+ + e− = Sub–H*, | (R2) |
| Sub–H* = Sub* + 1/2H2. | (R3) |
For the HER, we calculated the adsorption energy of the H* intermediate by the equation:
 | (6) |
| ΔGH* = ΔEH* + ΔZPE − TΔS + meU. | (7) |
Furthermore, we studied the dissolution potential for the Fe, Co, and Ni metals in the MeTMP/MoS2 systems to estimate the electrochemical stability of the substrates. The potential dissolution reaction is:
| MeTMP/MoS2 ⇌ TMP/MoS2 + MeZ+ + Ze−. | (R4) |
The dissolution potential for the metals was calculated by Greeley and Norskov's method,49 as follows:
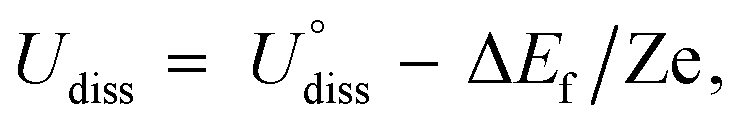 | (8) |
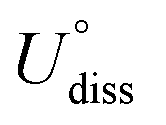 is the standard dissolution potential of bulk metals from the NIST database,50 and Z is the number of electrons released when the metal dissolution occurs. The formation energy ΔEf of the MeTMP/MoS2 system was calculated by:
is the standard dissolution potential of bulk metals from the NIST database,50 and Z is the number of electrons released when the metal dissolution occurs. The formation energy ΔEf of the MeTMP/MoS2 system was calculated by:| ΔEf = EMeTMP/MoS2 − (ETMP/MoS2 + EMe). | (9) |
3. Results and discussion
3.1 Designing the MeTMP/MoS2 substrates
Because the metalloporphyrin has a planar structure, we initially set metalloporphyrin at various locations on MoS2 in the following configurations: (1) inclined, and (2) parallel to the surface of MoS2. However, we did not consider the vertical configuration of metalloporphyrin because the vacuum space in our system is limited. After optimizing the stacking configurations of MeTMP/MoS2, we calculated the binding energy of the MeTMP molecule, using expression (1), and its optimized average distance to the MoS2 surface. We found that the parallel configuration is much more stable than the inclined one for each MeTMP/MoS2 system.Fig. 1a (top and side views) shows that the porphyrin molecule in the parallel configuration is most stable as its metal atom (green colour) is located on the hollow site of three S atoms in the upper atomic layer of the MoS2 support. Fig. 1b shows that the binding energy of the porphyrin molecule with the MoS2 support varies with its average distance (d) to the support surface, where the most stable position of the porphyrin molecule corresponds to the highest binding energy versus the average distance. We found that the optimized average distance is approximately 3.15, 3.08, and 3.07 Å with the highest binding energy of 1.50, 1.92, and 2.17 eV for the tetra methyl nickel, cobalt, and iron porphyrins, respectively. Moreover, the S atoms on each surface layer are not in the same plane. Several S atoms are shifted upwards and downwards compared to those of the ideal structure (see the side view in Fig. 1a). These most stable MeTMP/MoS2 systems (Me = Ni, Co, and Fe) were chosen for studying the adsorption of the ORR intermediates and the ORR mechanisms.
 | ||
| Fig. 1 Structure of the MeTMP/MoS2 substrate: (a) side view and top view. (b) The binding energy (Eb) of the metal porphyrin molecule (MeTMP) on MoS2 varies with its average distance to the MoS2 surface, where Me = Fe, Co, and Ni. Light blue (Mo), orange (S), green (Me), dark blue (C), violet (N), and yellow (H). | ||
3.2 Proposed reaction scenarios and mechanisms
The total ORR is as follows:| O2 + 4(H+ + e−) ⇄ 2H2O. | (R5) |
This reaction can proceed through many steps. Scheme 1 shows our proposed possible reaction intermediates and pathways, i.e., step 1: the adsorption of molecular oxygen on the MeTMP/MoS2 surface transforming O2 to  According to the hydrogen standard electrode model,45 we replaced H+ + e− by one hydrogen atom (H). Therefore, the proton and electron transfer processes can be modelled via the successive hydrogenation steps of the ORR intermediates by sequentially adding a hydrogen atom to an oxygen atom of the previously formed intermediate, starting from transforming
According to the hydrogen standard electrode model,45 we replaced H+ + e− by one hydrogen atom (H). Therefore, the proton and electron transfer processes can be modelled via the successive hydrogenation steps of the ORR intermediates by sequentially adding a hydrogen atom to an oxygen atom of the previously formed intermediate, starting from transforming  to HOO* (step 2). There are three possibilities for the subsequent step in converting HOO*, i.e., (1) by loading an H atom onto the available O* atom of HOO* to form HOOH* (step 3), and then HO* + H2O* (step 4); (2) by loading a H atom onto the O atom near the H atom of HOO* to form O* + H2O* (step 5), and then transform to HO* + H2O* (step 6); (3) HOO* can also dissociate into O* + HO* (step 7). Besides the molecular oxygen adsorption state,
to HOO* (step 2). There are three possibilities for the subsequent step in converting HOO*, i.e., (1) by loading an H atom onto the available O* atom of HOO* to form HOOH* (step 3), and then HO* + H2O* (step 4); (2) by loading a H atom onto the O atom near the H atom of HOO* to form O* + H2O* (step 5), and then transform to HO* + H2O* (step 6); (3) HOO* can also dissociate into O* + HO* (step 7). Besides the molecular oxygen adsorption state, 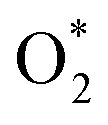 can also dissociate into two separate oxygen atoms, O* + O* or 2O* (step 8). The hydrogenation of O* + O* can transform it to O* + HO* (step 9), then HO* + HO* (step 10), and HO* + H2O (step 11). The final product of the ORR is 2H2O due to transforming HO* + H2O* (step 12).
can also dissociate into two separate oxygen atoms, O* + O* or 2O* (step 8). The hydrogenation of O* + O* can transform it to O* + HO* (step 9), then HO* + HO* (step 10), and HO* + H2O (step 11). The final product of the ORR is 2H2O due to transforming HO* + H2O* (step 12).
 | ||
| Scheme 1 Possible pathways for the ORR on the substrates. | ||
From the proposed intermediates and pathways, we can formulate the ORR process explicitly by the following reaction equations:
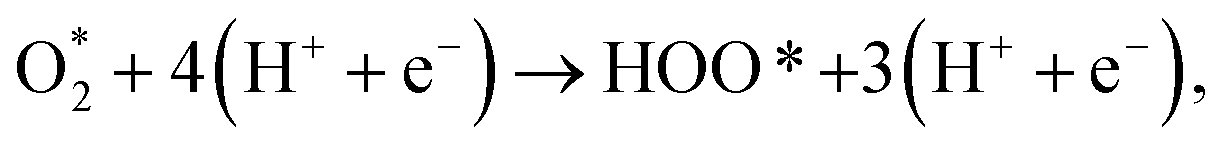 | (R6) |
| HOO* + 3(H+ + e−) → HOOH* + 2(H+ + e−), | (R7) |
| HOOH* + 2(H+ + e−) → HO* + H2O* + (H+ + e−), | (R8) |
| HOO* + 3(H+ + e−) → O* + H2O* + 2(H+ + e−), | (R9) |
| O* + H2O* + 2(H+ + e−) → HO* + H2O* + (H+ + e−), | (R10) |
| HOO* + 3(H+ + e−) → O* + HO* + 3(H+ + e−), | (R11) |
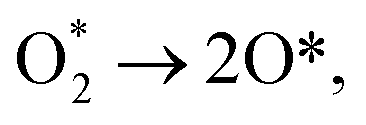 | (R12) |
| 2O* + 4(H+ + e−) → O* + HO* + 3(H+ + e−), | (R13) |
| O* + HO* + 3(H+ + e−) → HO* + HO* + 2(H+ + e−), | (R14) |
| HO* + HO* + 2(H+ + e−) → HO* + H2O* + (H+ + e−), | (R15) |
| HO* + H2O* + (H+ + e−) → 2H2O. | (R16) |
3.3 Explore the adsorption of ORR intermediates on MeTMP/MoS2
As shown in Scheme 1, the possible intermediates of the ORR include HOO*, HOOH*, O* + H2O*, HO* + H2O*, 2O*, O* + HO*, and HO* + HO*. Each reactive intermediate undergoes structural optimization, with the vdW-DF calculations, at various adsorption sites on the MeTMP/MoS2 surface. The adsorption energy of each configuration is determined via formula (2) to assess its adsorption strength. It should be noted that we optimized the structure of O2, HOO, HOOH, and HO + H2O to obtain the total energy of these isolated intermediates as a reference for calculating the adsorption energy of
HOO*, HOOH*, O* + H2O*, HO* + H2O*, 2O*, O* + HO*, and HO* + HO*. Each reactive intermediate undergoes structural optimization, with the vdW-DF calculations, at various adsorption sites on the MeTMP/MoS2 surface. The adsorption energy of each configuration is determined via formula (2) to assess its adsorption strength. It should be noted that we optimized the structure of O2, HOO, HOOH, and HO + H2O to obtain the total energy of these isolated intermediates as a reference for calculating the adsorption energy of  HOO*, HOOH*, and HO* + H2O*, respectively. However, the isolated O + O, O + HO, and HO + HO (O + H2O) intermediates do not exist naturally in the gas phase, so they have no physical meaning to use as references. Therefore, we used the total energy of the isolated O2, HOO, and H2O2 for calculating the adsorption energy of the O* + O*, O* + HO*, and HO* + HO* (O* + H2O*) intermediates, respectively.
HOO*, HOOH*, and HO* + H2O*, respectively. However, the isolated O + O, O + HO, and HO + HO (O + H2O) intermediates do not exist naturally in the gas phase, so they have no physical meaning to use as references. Therefore, we used the total energy of the isolated O2, HOO, and H2O2 for calculating the adsorption energy of the O* + O*, O* + HO*, and HO* + HO* (O* + H2O*) intermediates, respectively.
According to the definition of eqn (2), the more negative the adsorption energy, the more stable the adsorption is. We found that the most favourable adsorption site for the ORR intermediates is around the metal site (Me) of the MeTMP molecules of the cobalt, nickel, and iron tetramethyl porphyrins/MoS2 substrates (Fig. 2). We found that the molecular-oxygen-containing intermediates, i.e.,  HOO*, and HOOH* adsorbed with the end-on configurations, where one oxygen atom is on the top of the metal atom Me and another one is over the hollow site of the Me–N–C ring of the MeTMP molecules (Fig. 2a–c). Meanwhile, the atomic-oxygen-containing intermediates such as O* + H2O*, HO* + H2O*, 2O*, O* + HO*, and HO* + HO* adsorbed most stably with one oxygen atom always on the top of the metal atom (Me), and the other oxygen atom can be over the hollow site or another place of the porphyrin ring (Fig. 2d–h).
HOO*, and HOOH* adsorbed with the end-on configurations, where one oxygen atom is on the top of the metal atom Me and another one is over the hollow site of the Me–N–C ring of the MeTMP molecules (Fig. 2a–c). Meanwhile, the atomic-oxygen-containing intermediates such as O* + H2O*, HO* + H2O*, 2O*, O* + HO*, and HO* + HO* adsorbed most stably with one oxygen atom always on the top of the metal atom (Me), and the other oxygen atom can be over the hollow site or another place of the porphyrin ring (Fig. 2d–h).
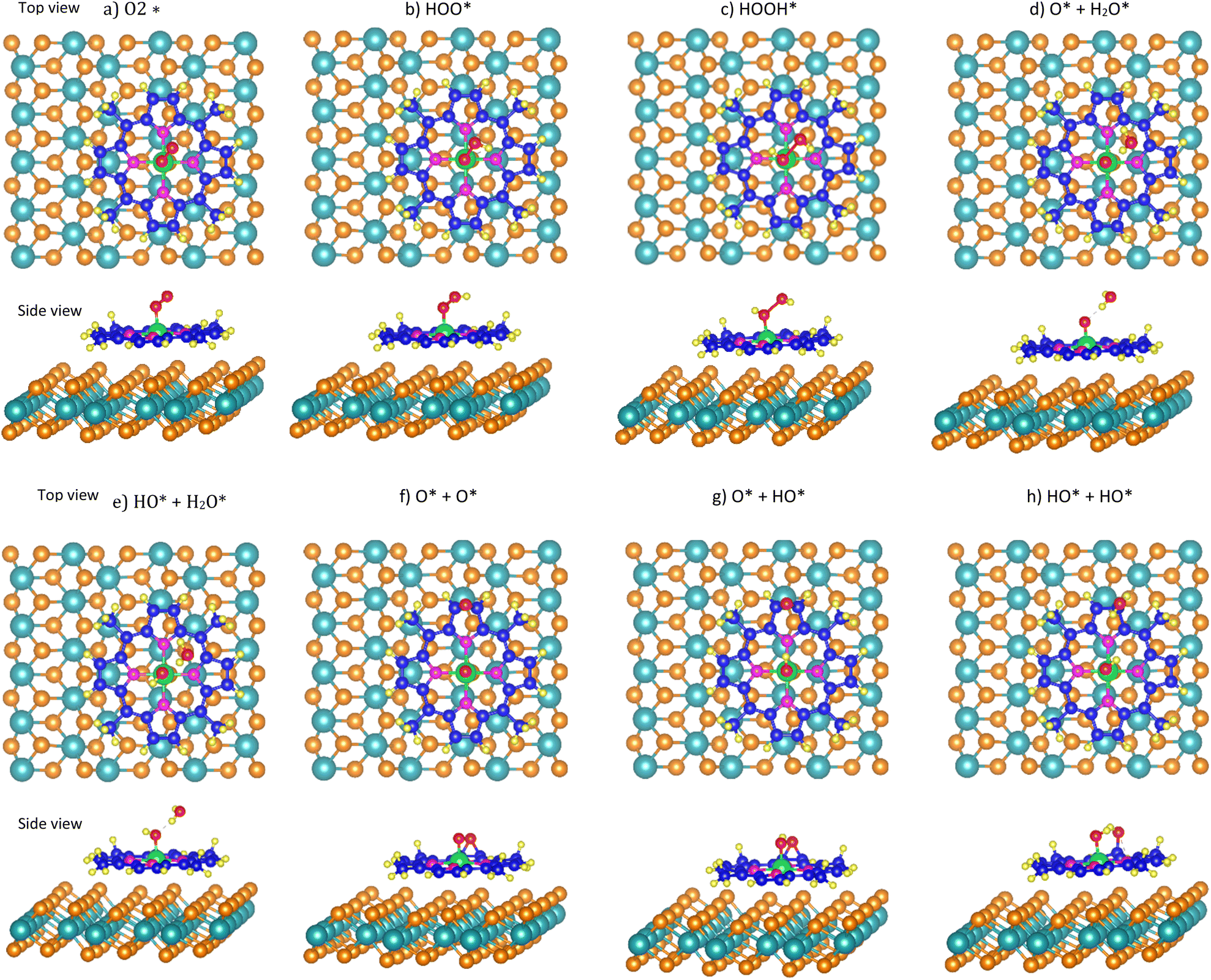 | ||
| Fig. 2 Most favourable adsorption configuration of the ORR intermediates on the MeTMP/MoS2 substrates from the top and side views. Light blue (Mo), orange (S), green (Me metal), dark blue (C), violet (N), yellow (H), and red (O). | ||
Table S1† shows the bond distances from the nearest oxygen atom to the metal atom Me of the MeTMP molecule, dO–M, and between two oxygen atoms of the intermediates, dO–O. We also found the values of these bond distances for the FeTMP substrate in our previous publication.15 For all MeTMP/MoS2 substrates, we found that the dO–M value of  (row 1) is always significantly larger than that of 2O* (row 6). Meanwhile, dO–M of HOO* (row 2, column 2) > dO–M of O* + HO* (row 7, column 2) and dO–M of HOOH* (row 3, column 2) > dO–M of 2HO* (row 8, column 2) for the NiTMP/MoS2 substrate, and dO–M of HOO* ≈ dO–M of O* + HO* and dO–M of HOOH* ≈ dO–M of 2HO* for the CoTMP/MoS2 and FeTMP/MoS2 substrates. Remarkably, the dO–O value of the molecular-oxygen-containing intermediates is always much shorter than that of the atomic-oxygen-containing intermediates with the same number of hydrogen atoms, i.e., dO–O of
(row 1) is always significantly larger than that of 2O* (row 6). Meanwhile, dO–M of HOO* (row 2, column 2) > dO–M of O* + HO* (row 7, column 2) and dO–M of HOOH* (row 3, column 2) > dO–M of 2HO* (row 8, column 2) for the NiTMP/MoS2 substrate, and dO–M of HOO* ≈ dO–M of O* + HO* and dO–M of HOOH* ≈ dO–M of 2HO* for the CoTMP/MoS2 and FeTMP/MoS2 substrates. Remarkably, the dO–O value of the molecular-oxygen-containing intermediates is always much shorter than that of the atomic-oxygen-containing intermediates with the same number of hydrogen atoms, i.e., dO–O of  of 2O*, dO–O of HOO* ≪ dO–O of O* + HO*, and dO–O of HOOH* ≪ dO–O of 2HO* for all the MeTMP/MoS2 substrates. It should be noted that the O* + H2O* and HO* + H2O* intermediates belong to both molecular and atomic oxygen adsorption scenarios, which have the dO–M and dO–O bond distances in the middle range between that of the molecular-oxygen-containing intermediates and that of the atomic-oxygen-containing ones for all the MeTMP/MoS2 substrates.
of 2O*, dO–O of HOO* ≪ dO–O of O* + HO*, and dO–O of HOOH* ≪ dO–O of 2HO* for all the MeTMP/MoS2 substrates. It should be noted that the O* + H2O* and HO* + H2O* intermediates belong to both molecular and atomic oxygen adsorption scenarios, which have the dO–M and dO–O bond distances in the middle range between that of the molecular-oxygen-containing intermediates and that of the atomic-oxygen-containing ones for all the MeTMP/MoS2 substrates.
Table 1 presents the adsorption energy of the ORR intermediates in their most favourable configuration on the substrates. The negative and positive adsorption energies indicate the thermodynamically favourable and unfavourable adsorption of the intermediates, respectively. Therefore, we found in Table 1 that the O* + H2O* and 2O* intermediates on NiTMP/MoS2 and 2O* on NiTMP are unfavourable. For each substrate with and without the MoS2 support, HO* + H2O* has the greatest negative adsorption energy among all the ORR intermediates. We also compare the adsorption energy calculated by the same vdW-DF scheme for the FeTMP molecule in ref. 15. Notably, the substitution of Ni, Co, and Fe metals into the tetramethyl porphyrin molecule causes a modification in the adsorption energy in increasingly negative order: Ni < Co < Fe for all the intermediates except for 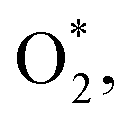 which follows the order: Ni < Fe < Co on all the MeTMP/MoS2 and MeTMP substrates. This order has a similar trend as the previous study,54 where the authors investigated the ORR on phenyl metalloporphyrins and metal phthalocyanines without supports.
which follows the order: Ni < Fe < Co on all the MeTMP/MoS2 and MeTMP substrates. This order has a similar trend as the previous study,54 where the authors investigated the ORR on phenyl metalloporphyrins and metal phthalocyanines without supports.
| Intermediates | NiTMP/MoS2 | CoTMP/MoS2 | FeTMP/MoS2 | NiTMP | CoTMP |
|---|---|---|---|---|---|
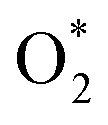 |
−0.15 | −0.68 | −0.48 | −0.20 | −0.75 |
| HOO* | −0.61 | −1.29 | −1.30 | −0.44 | −1.29 |
| HOOH* | −0.05 | −0.37 | −0.44 | −0.29 | −0.46 |
| O* + H2O* | 0.13 | −1.48 | −2.06 | −0.06 | −1.40 |
| HO* + H2O* | −1.84 | −2.61 | −2.79 | −1.73 | −2.65 |
| 2O* | 1.14 | −0.41 | −0.96 | 1.03 | −0.01 |
| O* + HO* | −1.13 | −1.99 | −2.13 | −0.92 | −1.89 |
| 2HO* | −0.38 | −1.44 | −1.45 | −0.19 | −1.13 |
In the presence of the MoS2 support, the adsorption energy of all the ORR intermediates on FeTMP/MoS2 uniquely becomes less negative than that on the FeTMP (ref. 15). However, the MoS2 support affects the adsorption of the intermediates differently for the Ni and Co substitutions. By comparing the second with the fifth and third with the sixth columns of Table 1, we found that the adsorption strength of  HOOH*, O* + H2O*, and 2O* on NiTMP/MoS2,
HOOH*, O* + H2O*, and 2O* on NiTMP/MoS2,  HOO*, HOOH*, and HO* + H2O* on CoTMP/MoS2 decreases and otherwise increases, compared to that on NiTMP and CoTMP, respectively.
HOO*, HOOH*, and HO* + H2O* on CoTMP/MoS2 decreases and otherwise increases, compared to that on NiTMP and CoTMP, respectively.
3.4 Electronic properties
Analysing the electronic structural characteristics, such as the Bader charge and the charge density difference, can expose the physical insights into the interaction between the ORR intermediates and the MeTMP/MoS2 substrates. The Bader charge in Table S2† indicates that the metal Me and C atoms of the MeTMP molecule consistently donate the negative charge (e−). Meanwhile, the N and H atoms of the MeTMP molecule always accumulate the charge for every case, including having the adsorption of the ORR intermediates. However, in the presence of the MoS2 support, the H atoms of the MeTMP molecule can donate or accumulate the charge for different intermediates. As a result, the clean MeTMP molecules (without MoS2 support) retain their neutral charge (0.000 e−). However, they become positively charged in the presence of the MoS2 support or they donate the negative charge of about 0.5 e− to the MoS2 support, thereby strengthening the bond between the MeTMP molecule and MoS2. The charge gain of MoS2 can be arranged in the following order of magnitude: NiTMP/MoS2 < CoTMP/MoS2 < FeTMP/MoS2, which is consistent with the trend of the maximum binding energy of the MeTMP molecule on the MoS2 support, as analysed in Section 3.1. In the presence of the ORR intermediates, the MeTMP molecules with and without the MoS2 support always donate the charge to the intermediates for all cases. Moreover, the MeTMP molecule always transfers a significant amount of its charge to the MoS2 support.For each case of intermediate adsorption, by comparing MeTMP/MoS2 to MeTMP (the Bader charge for the FeTMP substrate found in Table 2, column 4 of ref. 15), we found that the MoS2 support causes the Me and C atoms to donate more and less charge, except for Me![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Fe in the HOOH*@FeTMP/MoS2 system and C atoms in the [O* + HO*]@NiTMP/MoS2 system, respectively. The MoS2 support causes the N and H atoms to gain less charge for every case of the ORR intermediate adsorption when compared to the MeTMP molecule without the MoS2 support. Furthermore, the charge accumulation of the dissociative intermediates is significantly higher than that of the associative intermediates, i.e.,
Fe in the HOOH*@FeTMP/MoS2 system and C atoms in the [O* + HO*]@NiTMP/MoS2 system, respectively. The MoS2 support causes the N and H atoms to gain less charge for every case of the ORR intermediate adsorption when compared to the MeTMP molecule without the MoS2 support. Furthermore, the charge accumulation of the dissociative intermediates is significantly higher than that of the associative intermediates, i.e.,  O* + HO* > HOO*, and HO* + HO* > HOOH*. The charge gains of O* + H2O* and HO* + H2O* lie within the middle range of that of the dissociative and associative intermediates. This agrees with the trend of their dO–M and dO–O bond distances, as analysed previously. Upon adding the MoS2 support, we observed a reduction in the charge accumulation of the ORR intermediates, particularly in the case of O* + HO* and HO* + HO*. Most importantly, we found that the charge exchange between the MeTMP and MeTMP/MoS2 substrates with the ORR intermediates is significant. Therefore, we can conclude that the nature of the interaction between the intermediates and the substrates is due to the charge exchange.
O* + HO* > HOO*, and HO* + HO* > HOOH*. The charge gains of O* + H2O* and HO* + H2O* lie within the middle range of that of the dissociative and associative intermediates. This agrees with the trend of their dO–M and dO–O bond distances, as analysed previously. Upon adding the MoS2 support, we observed a reduction in the charge accumulation of the ORR intermediates, particularly in the case of O* + HO* and HO* + HO*. Most importantly, we found that the charge exchange between the MeTMP and MeTMP/MoS2 substrates with the ORR intermediates is significant. Therefore, we can conclude that the nature of the interaction between the intermediates and the substrates is due to the charge exchange.
| ORR intermediates | NiTMP/MoS2 | CoTMP/MoS2 | FeTMP/MoS2 | NiTMP | CoTMP |
|---|---|---|---|---|---|
 |
1.20 | 1.05 | 0.84 | 1.15 | 1.10 |
| HOO* + 3/2H2 → HOOH* + H2 | 0.59 | 1.08 | 1.03 | 0.60 | 1.01 |
| HOO* + 3/2H2 → O* + H2O* + H2 | 0.99 | 0.06 | 0.00 | 0.89 | 0.15 |
| O* + H2O* + H2 → HO* + H2O* + ½H2 | 0.00 | 0.06 | 0.46 | 0.00 | 0.00 |
| HO* + H2O* + ½H2 → 2H2O | 0.00 | 0.00 | 0.01 | 0.00 | 0.00 |
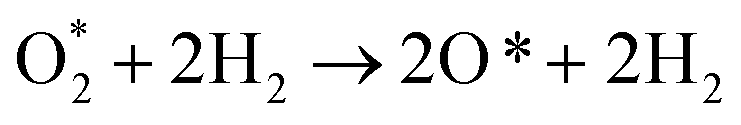 |
3.45 | 0.92 | 1.82 | 4.85 | 3.34 |
| 2O* + 2H2 → O* + HO* + 3/2H2 | 0.00 | 0.07 | 0.49 | 0.95 | 0.92 |
| O* + HO* + 3/2H2 → HO* + HO* + H2 | 0.00 | 0.00 | 0.85 | 0.00 | 0.00 |
| O* + HO* + 3/2H2 → O* + H2O* + H2 | 1.48 | 0.74 | 0.29 | 0.10 | 0.00 |
The charge density difference of the MeTMP/MoS2 substrates with the adsorbed ORR intermediates, calculated by the formula (3) (Fig. S2–S4 in ESI†), shows that the centre of the charge gain is always the oxygen atoms of the ORR intermediates. While the metal and C atoms of MeTMP porphyrins donate, the upper atomic layer of the S atoms gains the charge. These results agree with the Bader charge analysis, as shown above.
3.5 Mechanism of oxygen reduction reaction on MeTMP/MoS2
The Gibbs free energy for each side of the reactions (R6) to (R16) was calculated relative to that of two water molecules, which are the final product of the ORR. The Gibbs free energy was studied at the standard condition. The values are listed in Table S3.† Dissociating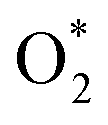 into 2O* is possible if overcoming an energy barrier at the transition state (TS), ETS, which was identified using the nudged elastic bands method, as shown in Fig. 3 for MeTMP/MoS2 and Fig. S5† for MeTMP. The calculation involved the initial and final states (denoted as (IS) and (FS)) corresponding to the most stable molecular adsorption configuration of the
into 2O* is possible if overcoming an energy barrier at the transition state (TS), ETS, which was identified using the nudged elastic bands method, as shown in Fig. 3 for MeTMP/MoS2 and Fig. S5† for MeTMP. The calculation involved the initial and final states (denoted as (IS) and (FS)) corresponding to the most stable molecular adsorption configuration of the 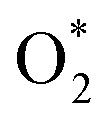 molecule and the most stable atomic adsorption configuration of 2O*. Once the transition state structure was determined, the total energy and the Gibbs free energy of (TS) + 2H2 were calculated following eqn (5).
molecule and the most stable atomic adsorption configuration of 2O*. Once the transition state structure was determined, the total energy and the Gibbs free energy of (TS) + 2H2 were calculated following eqn (5).
 | ||
Fig. 3 Dissociating  to 2O*, from the initial state (IS) overcoming the transition state (TS) to the final state (FS), on NiTMP/MoS2 (a), CoTMP/MoS2 (b), and FeTMP/MoS2 (c). Light blue (Mo), orange (S), gray (Ni), green (Co), brown (Fe), dark blue (C), violet (N), yellow (H), and red (O). to 2O*, from the initial state (IS) overcoming the transition state (TS) to the final state (FS), on NiTMP/MoS2 (a), CoTMP/MoS2 (b), and FeTMP/MoS2 (c). Light blue (Mo), orange (S), gray (Ni), green (Co), brown (Fe), dark blue (C), violet (N), yellow (H), and red (O). | ||
We can write the reaction series as,  The energy barrier for the dissociation process of
The energy barrier for the dissociation process of 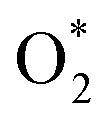 into 2O*, which is the energy of the (TS) state relative to that of the (IS) state, is 3.69, 1.43, 1.94, 5.89, and 4.79 eV on NiTMP/MoS2, CoTMP/MoS2, FeTMP/MoS2, NiTMP, and CoTMP, respectively. The energy barrier of 2.67 eV for this process on FeTMP was given in our previous publication.15 We found that the MoS2 support could significantly reduce the energy barrier for the
into 2O*, which is the energy of the (TS) state relative to that of the (IS) state, is 3.69, 1.43, 1.94, 5.89, and 4.79 eV on NiTMP/MoS2, CoTMP/MoS2, FeTMP/MoS2, NiTMP, and CoTMP, respectively. The energy barrier of 2.67 eV for this process on FeTMP was given in our previous publication.15 We found that the MoS2 support could significantly reduce the energy barrier for the  dissociation. Therefore, it can facilitate the dissociative pathway of the ORR.
dissociation. Therefore, it can facilitate the dissociative pathway of the ORR.
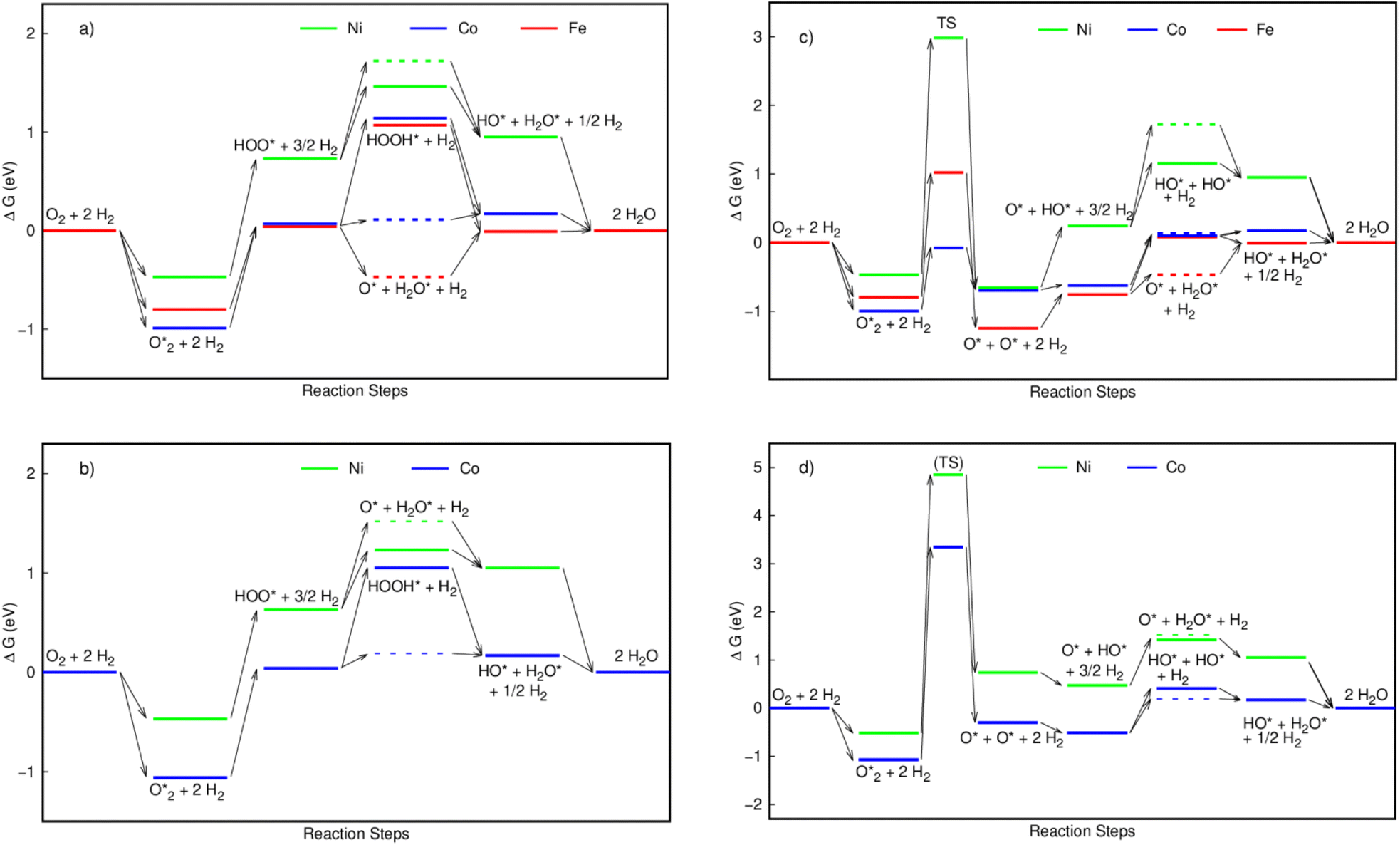
Fig. 4 displays the Gibbs free energy diagrams of ORR for the associative (left panel) and the dissociative (right panel) mechanisms on MeTMP/MoS2 (upper panel) and MeTMP (lower panel). As shown in Fig. 4a–d, forming  on all of the substrates is automatic and does not require any activation energy. Fig. 4a shows that the first two hydrogenation steps go uphill, except for the second hydrogenation converting HOO* + 3/2H2 to O* + H2O* + H2 (the red dashed line) on the FeTMP/MoS2 substrate, indicating the need for an activation energy. The third hydrogenation step converting HOOH* + H2 and O* + H2O* + H2 to HO* + H2O* + ½H2 goes downhill, except for transforming O* + H2O* + H2 to HO* + H2O* + ½H2 on the CoTMP/MoS2 and FeTMP/MoS2 substrates. The fourth hydrogenation goes downhill on NiTMP/MoS2 and CoTMP/MoS2, and slightly uphill on FeTMP/MoS2. We also found the same characteristics for the Gibbs free energy diagrams of the associative pathway on MeTMP without MoS2 support (Fig. 4b) with a slight increase of the energy level of O* + H2O* + H2 on CoTMP, so that it is almost the same height as that of HO* + H2O* + ½H2 on CoTMP. The downhill steps do not require activation energy, and can proceed automatically.
on all of the substrates is automatic and does not require any activation energy. Fig. 4a shows that the first two hydrogenation steps go uphill, except for the second hydrogenation converting HOO* + 3/2H2 to O* + H2O* + H2 (the red dashed line) on the FeTMP/MoS2 substrate, indicating the need for an activation energy. The third hydrogenation step converting HOOH* + H2 and O* + H2O* + H2 to HO* + H2O* + ½H2 goes downhill, except for transforming O* + H2O* + H2 to HO* + H2O* + ½H2 on the CoTMP/MoS2 and FeTMP/MoS2 substrates. The fourth hydrogenation goes downhill on NiTMP/MoS2 and CoTMP/MoS2, and slightly uphill on FeTMP/MoS2. We also found the same characteristics for the Gibbs free energy diagrams of the associative pathway on MeTMP without MoS2 support (Fig. 4b) with a slight increase of the energy level of O* + H2O* + H2 on CoTMP, so that it is almost the same height as that of HO* + H2O* + ½H2 on CoTMP. The downhill steps do not require activation energy, and can proceed automatically.
 | ||
| Fig. 4 Relative Gibbs energy diagrams for the associative pathways on MeTMP/MoS2 (a) and MeTMP (b), and the dissociative pathways on MeTMP/MoS2 (c) and MeTMP (d) at the reversible electrode potential of U = 1.23 V. The dashed lines depict the Gibbs free energy level of O* + H2O* + H2, while the solid lines depict the remaining ones. | ||
For dissociation mechanisms on MeTMP/MoS2 (Fig. 4c), to proceed in the forward direction (from left to right of the diagram), the ORR must overcome an activation barrier at the dissociation step converting  to 2O*. Afterward, the first, second, third, and fourth hydrogenation steps go uphill except for the third hydrogenation that converts HO* + HO* + H2 and O* + H2O* + H2 to HO* + H2O* + ½H2 on NiTMP/MoS2, and the fourth hydrogenation that transforms HO* + H2O* + ½H2 to 2H2O on the NiTMP/MoS2 and CoTMP/MoS2 substrate. For the dissociation mechanisms on MeTMP without MoS2 support (Fig. 4d), the relative position of the Gibbs free energy levels is more complicated. However, the most striking feature is that the activation barrier for the
to 2O*. Afterward, the first, second, third, and fourth hydrogenation steps go uphill except for the third hydrogenation that converts HO* + HO* + H2 and O* + H2O* + H2 to HO* + H2O* + ½H2 on NiTMP/MoS2, and the fourth hydrogenation that transforms HO* + H2O* + ½H2 to 2H2O on the NiTMP/MoS2 and CoTMP/MoS2 substrate. For the dissociation mechanisms on MeTMP without MoS2 support (Fig. 4d), the relative position of the Gibbs free energy levels is more complicated. However, the most striking feature is that the activation barrier for the  dissociation is much higher than that on MeTMP with the MoS2 support.
dissociation is much higher than that on MeTMP with the MoS2 support.
For the backward direction (from right to left of each diagram), the Gibbs free energy levels of HO* + H2O* + ½H2 and O* + H2O* + H2 in the associative mechanisms (Fig. 4a and b) and HO* + H2O* + ½H2, O* + H2O* + H2, O* + HO* + 3/2H2, and O* + O* + 2H2 in the dissociative mechanisms on the FeTMP/MoS2 substrates are lower than that of 2H2O. These results indicate that the backward processes of the ORR may occur, and therefore hinder the catalytic efficiency of the FeTMP/MoS2 substrate. The activation barrier for the associative and dissociative mechanisms on various substrates is listed in Table 2. The highest activation barrier determines the rate-limiting step for each reaction pathway. For the associative pathways, we found that the rate-limiting step occurs at the first hydrogenation step for NiTMP/MoS2 and NiTMP, and the second hydrogenation step for FeTMP/MoS2. However, the presence of the MoS2 support modified the rate-limiting step of the ORR from the first hydrogenation on CoTMP to the second hydrogenation on CoTMP/MoS2.
For the dissociative mechanisms, we found that the rate-limiting step always occurs at the dissociating step, forming 2O* + 2H2 from 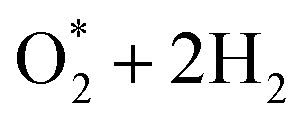 for all substrates. The highest thermodynamic barrier for the dissociative mechanisms on NiTMP/MoS2, CoTMP/MoS2, and FeTMP/MoS2 is 3.45, 0.92, and 1.82 eV, which is much lower than 4.85, 3.34, and 2.19 eV on NiTMP, CoTMP, and FeTMP,15 respectively. We also found that the MoS2 support does not lower the energy barrier for the associative mechanisms. However, it does significantly lower the energy barrier for the dissociation ones. In particular, it made the CoTMP/MoS2 and FeTMP/MoS2 systems the best and second-best catalysts among the considered ones for the dissociative mechanisms, respectively. Notably, nickel does not favour the ORR for both mechanisms. Considering all forward and backward directions, we can conclude that CoTMP/MoS2 is the superior catalyst.
for all substrates. The highest thermodynamic barrier for the dissociative mechanisms on NiTMP/MoS2, CoTMP/MoS2, and FeTMP/MoS2 is 3.45, 0.92, and 1.82 eV, which is much lower than 4.85, 3.34, and 2.19 eV on NiTMP, CoTMP, and FeTMP,15 respectively. We also found that the MoS2 support does not lower the energy barrier for the associative mechanisms. However, it does significantly lower the energy barrier for the dissociation ones. In particular, it made the CoTMP/MoS2 and FeTMP/MoS2 systems the best and second-best catalysts among the considered ones for the dissociative mechanisms, respectively. Notably, nickel does not favour the ORR for both mechanisms. Considering all forward and backward directions, we can conclude that CoTMP/MoS2 is the superior catalyst.
The superior catalytic activity of CoTMP/MoS2 is because of its most significant reduction in the thermodynamic energy barrier, as seen in Fig. 4c, which stems from its lowest energy level of the transition state for the  to 2O* conversion. Also, the electronic configuration of the Fe, Co, and Ni atoms is [Ar] 3d64s2, [Ar] 3d74s2, and [Ar] 3d84s2, respectively. We find that the number of electrons existing in the 3d orbital of the valence state of the Fe, Co, and Ni atoms is 6, 7, and 8, respectively. We guess that the odd number of electrons in the 3d orbital of the Co atom is responsible for the superior catalytic activity of CoTMP/MoS2 over the FeTMP/MoS2 and NiTMP/MoS2 substrates.
to 2O* conversion. Also, the electronic configuration of the Fe, Co, and Ni atoms is [Ar] 3d64s2, [Ar] 3d74s2, and [Ar] 3d84s2, respectively. We find that the number of electrons existing in the 3d orbital of the valence state of the Fe, Co, and Ni atoms is 6, 7, and 8, respectively. We guess that the odd number of electrons in the 3d orbital of the Co atom is responsible for the superior catalytic activity of CoTMP/MoS2 over the FeTMP/MoS2 and NiTMP/MoS2 substrates.
For the HER, we optimized the position of H* at all possible sites, such as on the metal (Fe, Co, and Ni), S atom, Mo atom, S–S bridge, and hollows. We found the optimized configuration, as shown in Fig. S6.† The positive adsorption energy of H* (Table 3) indicates that the adsorption of H* is unfavourable on the MeTMP/MoS2 substrates, which differs from the negative adsorption energy of the ORR intermediates. We then calculated the Gibbs free energy of reaction steps (R2) and (R3) under standard thermodynamic conditions (pH = 0, 298 K, and 1.0 bar). According to reactions (R1) and (R2), we have m = 1 and ΔEH* = Eads.
| Adsorption site | CoTMP/MoS2 | FeTMP/MoS2 | NiTMP/MoS2 |
|---|---|---|---|
| Metal (Fe, Co, Ni) atom | 0.17 | 0.54 | 0.89 |
| S atom | 0.12 | 0.12 | 0.17 |
| Mo atom | 1.05 | 1.03 | 1.03 |
| S–S bridge | 0.68 | 0.57 | 0.78 |
Fig. 5 shows that HER is thermodynamically unfavourable when it requires an activation energy. This energy is significantly lower than or comparable to the activation barrier of about 1.0 eV for the ORR at the electrode potential of U = 1.23 V. However, the reverse process of HER occurs spontaneously for all of the substrates. Therefore, there are always protons on the surface of the substrates, indicating that the HER does not interfere with ORR.
 | ||
| Fig. 5 Gibbs free diagram of the HER for the most favourable configurations of H* on MeTMP/MoS2 at zero potential (a) and the equilibrium potential (b). The line for H-on-S/FeTMP/MoS2 is identical to that for H-on-S/CoTMP/MoS2. | ||
The dissolution potential, presented in Table 4, shows that the Co, Fe, and Ni metals are stabilized in the MeTMP/MoS2 system because their dissolution potential is much higher than the equilibrium potential of 1.23 V for the ORR.
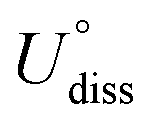
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
4. Conclusions
Using vdW-DF with the revised PBE version of the generalized gradient approximation and thermodynamic model, this study elucidated the impact of metal substitution in MeTMP/MoS2 on the overall catalytic performance of the ORR. The Co atom is the best substitution in the MeTMP/MoS2 catalysts. In the association mechanisms, the first and second hydrogenation steps emerge as the rate-determining steps for NiTMP/MoS2 (NiTMP) and FeTMP/MoS2, respectively. However, the rate-limiting step is the first hydrogenation for CoTMP and the second hydrogenation for CoTMP/MoS2. Notably, the dissociation mechanisms have the rate-determining step occurring at the dissociation on all substrates. The metals in the MeTMP/MoS2 catalyst are stable due to their high dissolution potentials. Moreover, the HER does not interfere with the ORR. The charge exchange dominates the interaction between the ORR intermediates and the substrates.
dissociation on all substrates. The metals in the MeTMP/MoS2 catalyst are stable due to their high dissolution potentials. Moreover, the HER does not interfere with the ORR. The charge exchange dominates the interaction between the ORR intermediates and the substrates.
Data availability
The data supporting this article have been included in the ESI.†Author contributions
Conceptualization (DNS), investigation (TPD), formal analysis (DNS), resources (DNS and VC), supervision (DNS, PTNN, and VC), validation (DNS), visualization (TPD), writing manuscript (DNS and TPD), and reviewing and editing (DNS, TPD, PTNN and VC).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research is funded by the Vietnam National University Ho Chi Minh City (VNU-HCM) under grant number VL2022-20-01.References
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Activity benchmarks and requirements for Pt, Pt-alloy, and non-Pt oxygen reduction catalysts for PEMFCs, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- F. Cheng and J. Chen, Metal–air batteries: from oxygen reduction electrochemistry to cathode catalysts, Chem. Soc. Rev., 2012, 41, 2172–2192 Search PubMed.
- L. Jiang, M. Li, L. Lin, Y. Li, X. He and L. Cui, Electrocatalytic activity of metalloporphyrins grown in situ on graphene sheets toward oxygen reduction reaction in an alkaline medium, RSC Adv., 2014, 4, 26653–26661 Search PubMed.
- D. N. Son, H. Nakanishi, M. Y. David and H. Kasai, Oxygen Reduction on Pt(111) Cathode of Fuel Cells, J. Phys. Soc. Jpn., 2009, 78, 114601 CrossRef.
- D. N. Son, O. K. Le, V. Chihaia and K. Takahashi, Effects of Co Content in Pd-Skin/PdCo Alloys for Oxygen Reduction Reaction: Density Functional Theory Predictions, J. Phys. Chem. C, 2015, 119, 24364–24372 CrossRef CAS.
- D. N. Son, P. N. Thanh, N. D. Quang, K. Takahashi and M. P. Pham-Ho, First-principles study of Pd-skin/Pd3Fe(111) electrocatalyst for oxygen reduction reaction, J. Appl. Electrochem., 2017, 47, 747–754 CrossRef CAS.
- C. J. Kaminsky, J. Wright and Y. Surendranath, Graphite-conjugation enhances porphyrin electrocatalysis, ACS Catal., 2019, 9, 3667–3671 CrossRef CAS.
- A. N. Marianov, A. S. Kochubei, S. Gu and Y. Jiang, Charge-Transfer Mechanism in Oxygen Reduction over Co Porphyrins: Single-Site Molecular Electrocatalysts to Macromolecular Frameworks, ACS Catal., 2022, 12, 8610–8622 CrossRef CAS.
- Y. Liu, G. Zhou, Z. Zhang, H. Lei, Z. Yao, J. Li, J. Lin and R. Cao, Significantly improved electrocatalytic oxygen reduction by an asymmetrical Pacman dinuclear cobalt (II) porphyrin–porphyrin dyad, Chem. Sci., 2020, 11, 87–96 RSC.
- G. Passard, D. K. Dogutan, M. Qiu and C. Costentin, et al., Oxygen Reduction Reaction Promoted by Manganese Porphyrins, ACS Catal., 2018, 8671–8679 CrossRef CAS.
- D.-H. Kim, D.-H. Kwak and S.-B. Han, et al., Electrochemical catalytic contribution of transition metals at the center of porphyrin macrocycle structures as catalysts for oxygen reduction reaction, J. Ind. Eng. Chem., 2017, 54, 200–204 CrossRef CAS.
- L. Ye, Y. Fang, Z. Ou, S. Xue and K.-M. Kadish, Cobalt tetrabutano-and tetrabenzotetraarylporphyrin complexes: effect of substituents on the electrochemical properties and catalytic activity of oxygen reduction reactions, Inorg. Chem., 2017, 56, 13613–13626 CAS.
- B. Sun, Z. Ou and D. Meng, et al., Electrochemistry and catalytic properties for dioxygen reduction using ferrocene-substituted cobalt porphyrins, Inorg. Chem., 2014, 53, 8600–8609 CAS.
- S. Amanullah, P. Saha and A. Dey, O2 reduction by iron porphyrins with electron withdrawing groups: to scale or not to scale, Faraday Discuss., 2022, 234, 143–158 CAS.
- T. P. Dung, V. Chihaia and D. N. Son, Effects of functional groups in iron porphyrin on the mechanism and activity of oxygen reduction reaction, RSC Adv., 2023, 13, 8523–8534 CAS.
- H. Wan, T. M. Østergaard, L. Arnarson and J. Rossmeisl, Climbing the 3D volcano for the oxygen reduction reaction using porphyrin motifs, ACS Sustainable Chem. Eng., 2018, 7, 611–617 Search PubMed.
- G. Tei, T. Tamaki and T. Hayashi, et al., Oxygen Reduction Reaction (ORR) Activity of a Phenol-Substituted Linear FeIII–Porphyrin Dimer, Eur. J. Inorg. Chem., 2017, 26, 3229–3232 CrossRef.
- T. Schilling, A. Okunola, J. Masa, W. Schuhmann and M. Bron, Carbon nanotubes modified with electrodeposited metal porphyrins and phenanthrolines for electrocatalytic applications, Electrochim. Acta, 2010, 55, 7597–7602 CrossRef CAS.
- M. Jahan, Q. Bao and K. P. Loh, Electrocatalytically Active Graphene−Porphyrin MOF Composite for Oxygen Reduction Reaction, J. Am. Chem. Soc., 2012, 134, 6707–6713 CrossRef CAS PubMed.
- L. Wei and Y. Chen, Degradation of carbon materials in electrocatalysis, Curr. Opin. Electrochem., 2022, 36, 101159 CAS.
- J. Kwon, P. Choi, S. Jo, H. Oh, K. Y. Cho and Y. K. Lee, et al., Identification of electrode degradation by carbon corrosion in polymer electrolyte membrane fuel cells using the distribution of relaxation time analysis, Electrochim. Acta, 2022, 414, 140219 CAS.
- S. Jayabal, G. Saranya, J. Wu and Y. Liu, et al., Understanding the high-electrocatalytic performance of two-dimensional MoS2 nanosheets and their composite materials, J. Mater. Chem. A, 2017, 5, 24540–24563 CAS.
- I. S. Amiinu, Z. Pu and X. Liu, et al., Multifunctional Mo-N/C@MoS2 Electrocatalysts for HER, OER, ORR, and Zn-Air Batteries, Adv. Funct. Mater., 2017, 27, 1702300 Search PubMed.
- Q. Hua, K. E. Madsen and A. M. Esposito, et al., Effect of support on oxygen reduction reaction activity of supported iron porphyrins, ACS Catal., 2022, 12, 1139–1149 CAS.
- C. Suresh, S. Mutyala and J. Mathiyarasu, Support interactive synthesis of nanostructured MoS2 electrocatalyst for oxygen reduction reaction, Mater. Lett., 2016, 164, 417–420 CAS.
- M. T. Anwar, X. Yan and M. R. Asghar, et al., MoS2-rGO hybrid architecture as durable support for cathode catalyst in proton exchange membrane fuel cells, Chin. J. Catal., 2019, 40, 1160–1167 CrossRef CAS.
- D. Sun, D. Huang and H. Wang, et al., 1T MoS2 nanosheets with extraordinary sodium storage properties via thermal-driven ion intercalation assisted exfoliation of bulky MoS2, Nano Energy, 2019, 61, 361–369 CrossRef CAS.
- D. Hou, W. Zhou and X. Liu, et al., Pt nanoparticles/MoS2 nanosheets/carbon fibers as efficient catalyst for the hydrogen evolution reaction, Electrochim. Acta, 2015, 166, 26–31 CrossRef CAS.
- I. S Kwon, I. H. Kwak and J. Kim, et al., Two-Dimensional MoS2/Fe-Phthalocyanine Hybrid Nanostructures as Excellent Electrocatalysts for Hydrogen Evolution and Oxygen Reduction Reactions, Nanoscale, 2019, 11, 14266–14275 RSC.
- L. Hao, J. Yu and X. Xu, et al., Nitrogen-doped MoS2/carbon as highly oxygen-permeable and stable catalysts for oxygen reduction reaction in microbial fuel cells, J. Power Sources, 2017, 339, 68–79 CrossRef CAS.
- G. Gao, Y. Jiao and F. Ma, et al., Charge Mediated Semiconducting-to-Metallic Phase Transition in Molybdenum Disulfide Monolayer and Hydrogen Evolution Reaction in New 1T′ Phase, J. Phys. Chem. C, 2015, 119, 13124–13128 CrossRef CAS.
- Y. Yu, et al., High phase-purity 1T′-MoS2- and 1T′-MoSe2- layered crystals, Nat. Chem., 2018, 10, 638–643 CrossRef CAS.
- A. Ejigu, I. A. Kinloch and E. Prestat, et al., A simple electrochemical route to metallic phase trilayer MoS2: evaluation as electrocatalysts and supercapacitors, J. Mater. Chem. A, 2017, 5, 11316–11330 RSC.
- X. Qian, J. Liu, L. Fu and J. Li, Quantum spin Hall effect in two-dimensional transition metal dichalcogenides, Science, 2014, 346, 1344–1347 CrossRef CAS PubMed.
- M. S. Liao and S. Scheiner, Electronic structure and bonding in metal porphyrins, metal=Fe, Co, Ni, Cu, Zn, J. Chem. Phys., 2002, 117(1), 205–219 CrossRef CAS.
- K. Sengupta, S. Chatterjee and A. Dey, In situ mechanistic investigation of O2 reduction by iron porphyrin electrocatalysts using surface-enhanced resonance Raman spectroscopy coupled to rotating disk electrode (SERRS-RDE) setup, ACS Catal., 2016, 6(10), 6838–6852 CrossRef CAS.
- J. Hafner, Ab-initio simulations of materials using VASP: Density-functional theory and beyond, J. Comput. Chem., 2008, 29, 2044–2078 CrossRef CAS.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Van der Waals Density Functional for General Geometries, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- J. Hermann, R. A. DiStasio and A. Tkatchenko, First-Principles Models for van der Waals Interactions in Molecules and Materials: Concepts, Theory, and Applications, Chem. Rev., 2017, 117, 4714–4758 CrossRef CAS PubMed.
- P. G. Moses, J. J. Mortensen, B. I. Lundqvist and J. K. Nørskov, Density functional study of the adsorption and van der Waals binding of aromatic and conjugated compounds on the basal plane of MoS2, J. Chem. Phys., 2009, 130, 104709 Search PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- A. J. Hensley, R. Zhang, Y. Wang and J. S. McEwen, Tailoring the adsorption of benzene on PdFe surfaces: a density functional theory study, J. Phys. Chem. C, 2013, 117(46), 24317–24328 CrossRef CAS.
- D. Tozini, M. Forti, P. Gargano, P. R. Alonso and G. H. Rubiolo, Charge difference calculation in Fe/Fe3O4 interfaces from DFT results, Procedia Mater. Sci., 2015, 9, 612–618 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
- O. My Na, N. T. Xuan Huynh, P. T. Thi, V. Chihaia and D. N. Son, Mechanism and activity of the oxygen reduction reaction on WTe2 transition metal dichalcogenide with Te vacancy, RSC Adv., 2020, 10, 8460–8469 CAS.
- H. T. Thanh, O. K. Le, V. Chihaia and D. N. Son, Carbon dioxide conversion to methanol on a PdCo bimetallic catalyst, Phys. Chem. Chem. Phys., 2024, 26, 3963–3973 RSC.
- D. L. Quan, V. Chihaia and D. N. Son, Mechanisms and selectivity of methanol oxidation on PtRuM3/C-MWCNT (M = Fe and Co) electrocatalysts, RSC Adv., 2024, 14, 25918–25931 RSC.
- J. Greeley and J. K. Nørskov, Electrochemical dissolution of surface alloys in acids: Thermodynamic trends from first-principles calculations, Electrochim. Acta, 2007, 52(19), 5829–5836 CrossRef CAS.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, CRC Press, New York, 95th edn, 2014, pp. 5–80 Search PubMed.
- W. Pepperhoff and M. Acet, The structure of iron, in Constitution and Magnetism of Iron and its Alloys, Engineering Materials, Springer, Berlin, Heidelberg, 2001, pp. 1–13 Search PubMed.
- C. S. Yoo, P. Söderlind and H. Cynn, The phase diagram of cobalt at high pressure and temperature: the stability of-cobalt and new-cobalt, J. Phys.: Condens. Matter, 1988, 10(20), L311–L318 CrossRef.
- N. N. Lathiotakis, A. N. Andriotis, M. Menon and J. Connolly, Tight binding molecular dynamics study of Ni clusters, J. Chem. Phys., 1996, 104, 992–1003 CrossRef CAS.
- G. Di Liberto, L. Giordano and G. Pacchioni, Predicting the Stability of Single-Atom Catalysts in Electrochemical Reactions, ACS Catal., 2024, 14(1), 45–55 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Understanding activity origin and mechanisms of oxygen reduction reaction on the tetra-methyl metalloporphyrin/MoS2 electrocatalyst. See DOI: https://doi.org/10.1039/d5ra00814j |
| This journal is © The Royal Society of Chemistry 2025 |