
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSeparator-free Li–S thin-film battery with spin-coated S/CNT/SP cathode and PEO/PVDF/LTFSI/LLZO composite electrolyte
Aiym Mashekova†
 abc,
Arman Umirzakov†ac,
Mukagali Yegamkulov†ac,
Marzhan Aliyakbarovaabc,
Berik Uzakbaiulycd,
Arailym Nurpeissovaabc,
Zhumabay Bakenov*abc and
Aliya Mukanova*ace
abc,
Arman Umirzakov†ac,
Mukagali Yegamkulov†ac,
Marzhan Aliyakbarovaabc,
Berik Uzakbaiulycd,
Arailym Nurpeissovaabc,
Zhumabay Bakenov*abc and
Aliya Mukanova*ace
aNational Laboratory Astana, Nazarbayev University, 53 Kabanbay Batyr Avenue, Astana 010000, Kazakhstan. E-mail: zbakenov@nu.edu.kz; aliya.mukanova@nu.edu.kz
bDepartment of Chemical and Materials Engineering, Nazarbayev University, 53 Kabanbay Batyr Avenue, Astana 010000, Kazakhstan
cInstitute of Batteries, 53 Kabanbay Batyr Avenue, Astana 010000, Kazakhstan
dFraunhofer IKTS, Ausere Nurnberger Str 62, Forchheim 91301, Germany
eInstitute of Petrochemical Engineering and Ecology. N.K. Nadirova, Atyrau Oil and Gas University, Baimukhanov 45, 060027 Atyrau, Kazakhstan
First published on 14th April 2025
Abstract
The advancement of miniaturized energy storage systems is essential for the next generation of electronics. Lithium–sulfur (Li–S) microbatteries are able to offer exceptional theoretical capacity and energy density for microdevices. However, their practical implementation is hindered by challenges in material stability and electrode design. In this study, we introduced a spin-coated sulfur–carbon nanotube–Super P (S/CNT/SP) cathode integrated with a spin-coated polyethylene oxide (PEO)/polyvinylidene fluoride (PVDF)/lithium lanthanum zirconium oxide (LLZO) composite electrolyte. The spin-coating technique ensured the formation of uniform electrode and electrolyte thin films, which could work without a separator. The polymer-ceramic composite electrolyte with nanopores effectively suppressed polysulfide dissolution, improved ionic conductivity, and stabilized the electrode–electrolyte interface. Electrochemical evaluation revealed that the quasi-solid-state Li–S battery achieved near-theoretical capacity with enhanced cycling stability, retaining approximately 1000 mA h g−1 (60% of its initial capacity) after 150 cycles across various C-rates. In a pouch-cell configuration, the cell retained 64% of its initial capacity over 60 cycles. These findings underscore the potential of spin-coating and composite quasi-solid electrolytes in enabling high-performance, safe, and compact Li–S battery technologies for next-generation energy storage applications.
1. Introduction
The necessity for advanced energy storage solutions at the microscale has become increasingly evident with the rapid development of miniaturized electronic devices. Among various battery technologies, lithium–sulfur (Li–S) batteries stand out due to their high theoretical specific capacity, elevated energy density, and cost-effectiveness.1 However, their integration into microdevices remains in an early stage, largely due to challenges related to material stability, electrode design, and electrolyte compatibility at the microscale.Sulfur, with its exceptional theoretical capacity of 1672 mA h g−1 and theoretical energy density of 2600 W h kg−1, emerges as a promising alternative to conventional cathode materials such as layered transition metal oxides.2 Nevertheless, its practical application is hindered by several intrinsic limitations, including poor electrical conductivity, dissolution of intermediate polysulfides, and significant volume expansion during cycling, leading to capacity fade and shortened cycle life.3–7 The well-known ‘shuttle effect’ caused by the migration of dissolved lithium polysulfides within the electrolyte exacerbates these challenges.4,8 Additionally, structural instabilities at the solid electrolyte interface (SEI) further degrade battery performance.5,9,10
State-of-the-art strategies addressing these issues include modifications in cathode composition, anode protection layers, electrolyte and separator engineering, and the incorporation of catalysts or dopants.11,12 However, adapting these solutions to microbattery applications introduces new challenges, primarily due to the limited research on techniques suitable for microscale sulfur-based cathodes. Magnetron sputtering has been extensively employed for depositing cathode, electrolyte, and anode materials in microbattery architectures, with efforts focusing on sulfur-based thin films using physical and chemical vapor deposition methods.13–15 TiOxSy,16,17 TiS2,18,19 FeS2,20 Li2S,21 and MoS2 (ref. 22) are among the thin-film S-based cathodes explored for microbattery applications. Additionally, ALD,23 electrodeposition,24,25 ink-based printing,26,27 and slurry casting28 have been investigated to fabricate thin-film S-cathodes.
Spin-coating has emerged as a promising technique in the fabrication of thin-film components for Li–S microbatteries. This method enables precise control over film thickness, uniformity, and morphology, making it highly suitable for microscale applications. Compared to other deposition techniques, spin-coating offers scalability, cost efficiency, and the ability to produce defect-free films with tunable properties.11
The nanostructured sulfur with carbon nanotubes (CNT) and Super P carbon additive form a conductive matrix that significantly enhances electron transport, mitigates sulfur dissolution, and stabilizes the electrode structure during cycling.29,30 CNTs, with their high electrical conductivity and large surface area, provide efficient charge pathways and mechanical reinforcement, reducing structural degradation caused by volumetric expansion. Super P, a high-surface-area conductive carbon, further improves electronic conductivity and helps in uniformly distributing sulfur within the cathode matrix. Additionally, nanostructured sulfur enhances electrochemical reactivity and shortens lithium-ion diffusion paths, ensuring improved sulfur utilization.11
Addressing the critical challenges of sulfur batteries – capacity loss during initial cycles and rapid degradation – has led to increasing interest in polymer-based solid-state electrolytes. Gel-polymer electrolytes exhibit enhanced ionic conductivity, mechanical robustness, and electrochemical stability.31 A polyethylene oxide (PEO)/polyvinylidene fluoride (PVDF)/lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) matrix is particularly advantageous, as it not only increases efficiency of a Li–S cell by reducing the sulfur shuttle but also improves interfacial stability.31 PEO is valued for its ability to solvate lithium salts, while PVDF helps maintain mechanical integrity and suppresses lithium dendrite formation. LiTFSI, as a lithium salt, further enhances ionic conductivity and electrochemical performance.11 However, achieving optimal performance requires careful engineering of the interlayer composition with ceramic fillers to increase conductivity and stabilize the interfaces between the anode, cathode, and electrolyte. A highly effective strategy is to cross-link the polymer with oxide electrolyte fillers, such as the well-known lithium lanthanum zirconium oxide (Li7La3Zr2O12, LLZO).32–34 Garnet LLZO as a filler in the electrolyte membrane has demonstrated its remarkable effectiveness, significantly enhancing ionic conductivity at room temperature and mitigating lithium dendrite growth. The combination of PEO, PVDF, LiTFSI, and LLZO in a composite electrolyte ensures both high ionic conductivity and structural stability, making it a promising choice of electrolyte for solid-state lithium–sulfur batteries.
Leveraging a combination of advanced materials and processing techniques, our study introduces an alternative paradigm for Li–S microbatteries. We prepared an S/CNT/Super P composite cathode using spin-coating techniques and encapsulated it with a PEO/PVDF–LTFSI–LLZO composite electrolyte. This electrolyte holds a dual role, acting as a protective coating in liquid cells and as an electrolyte in quasi-solid-state cells with a small addition of liquid electrolyte. The liquid electrolyte was intentionally introduced to enable stable cycling at room temperature. The integration of composite electrolyte structure not only enhanced the overall electrochemical stability but also improved sulfur utilization and suppressed the polysulfide shuttle effect. These findings contribute to the advancement of next-generation miniaturized energy storage systems, highlighting the crucial role of innovative material engineering in developing compact and high-performance battery technologies.
2. Experimental
2.1 Materials
The following materials were used in this study: polyethylene oxide (PEO) with an average molecular weight of approximately 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, acetonitrile (ACN, anhydrous, 99.8%), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, ≥99.0%), polyvinylidene fluoride (PVDF), 1,3-dioxolane (DOL), dimethoxyethane (DME), lithium nitrate (LiNO3), and sulfur (S), all purchased from Sigma-Aldrich (Merck). Multi-walled carbon nanotubes (CNT) and Super P (SP) were sourced from US Research Nanomaterials and MTI Corp., respectively. Lithium lanthanum zirconium oxide (Li7La3Zr2O12, LLZO), a commercial powder with a purity of ≥99.99%, and carboxymethyl cellulose (CMC, ≥99.5%) were obtained from MTI Corp. All chemicals were of analytical grade and were used as received without further purification.
000, acetonitrile (ACN, anhydrous, 99.8%), lithium bis(trifluoromethanesulfonyl)imide (LiTFSI, ≥99.0%), polyvinylidene fluoride (PVDF), 1,3-dioxolane (DOL), dimethoxyethane (DME), lithium nitrate (LiNO3), and sulfur (S), all purchased from Sigma-Aldrich (Merck). Multi-walled carbon nanotubes (CNT) and Super P (SP) were sourced from US Research Nanomaterials and MTI Corp., respectively. Lithium lanthanum zirconium oxide (Li7La3Zr2O12, LLZO), a commercial powder with a purity of ≥99.99%, and carboxymethyl cellulose (CMC, ≥99.5%) were obtained from MTI Corp. All chemicals were of analytical grade and were used as received without further purification.
2.2 Electrode preparation
A schematic illustration of the cathode preparation process is presented in Fig. 1a and b. The synthesis of the S/CNT/SP composite precursor was carried out by grinding commercial sulfur powder in ethanol using zirconium balls in a planetary ball mill (Pulverisette 7, Germany) at 400 rpm for 6 hours. Subsequently, CNT and Super P were mixed and milled in a 1:2 weight ratio under identical conditions for 3 to 6 hours to obtain a fine mixture of a conductive agent. This agent was mixed with the resulting sulfur nanopowder in ethanol and further milled under the same conditions to ensure homogeneity of the mixture. The final composite maintained a sulfur-to-carbon additive weight ratio of 7:3.
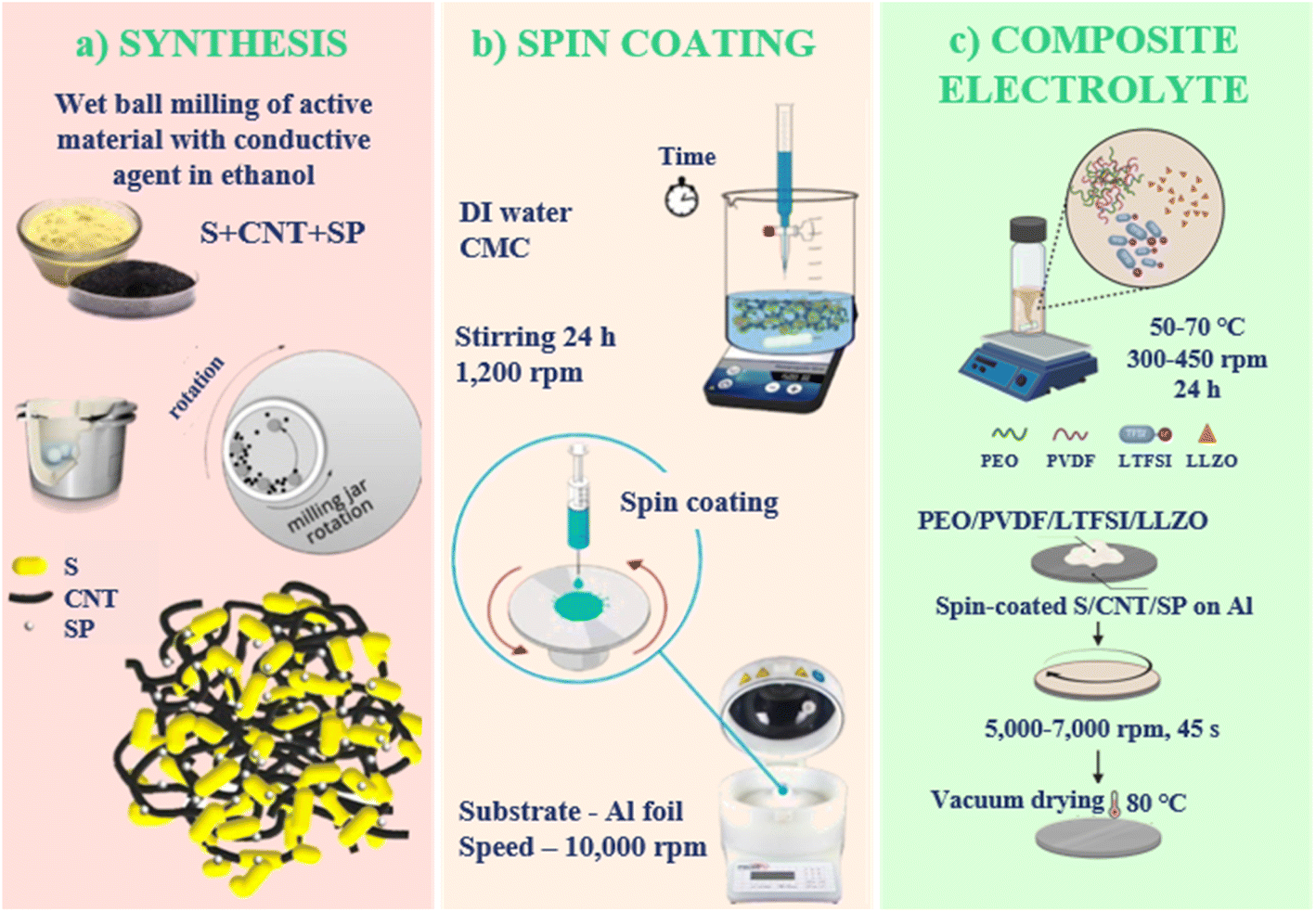 | ||
| Fig. 1 Fabrication process of spin-coated S/CNT/SP cathode: (a) wet ball milling of sulfur, CNT, and Super P to achieve a well-dispersed active material mixture; (b) spin-coating of cathode slurry onto Al foil substrate; (c) schematic illustration of preparation of PEO/PVDF/LiTFSI/LLZO composite electrolyte layer by stirring polymer and ceramic components to achieve a homogeneous electrolyte mixture and its spin-coating onto Al foil with a precoated S/CNT/SP cathode, followed by vacuum drying at 80 °C. | ||
The sulfur cathode slurries were carefully prepared by sequentially mixing the components using a magnetic stirring bar in a controlled environment (Fig. 1a). Carboxymethyl cellulose (CMC) served as the binder and was dissolved in 4 mL of deionized water by mixing for 6 hours. The process began by blending the S/CNT mixture into the CMC solution for 1 hour using a Planetary Vacuum Mixer (Model: ARV-310CE, Thinky, Japan) at 1200 rpm to obtain a homogeneous slurry.
The sulfur cathode slurry was then spin-coated (Fig. 1b) onto ultra-high vacuum (UHV) aluminum foil (from All-foils, Inc.). The spin-coating process was optimized with a rotation speed of 10000 rpm, acceleration of 1000–2000 rpm per second, and a duration of 1 minute to prepare a uniform and dense coating with a minimal thickness. The UHV Al foil was chosen for its rough surface, which improves slurry adhesion. After spin-coating, the coated slurries were dried in an oven at 60 °C for 16 hours to stabilize the cathode structure. Following this, an additional 16 hours of vacuum drying removed any residual solvent, ensuring a solvent-free cathode that enhances both battery performance and stability. Finally, the prepared cathodes were roll-pressed to achieve uniformity and density of the film. The mass loading of the composite material in the electrode was measured using a ultra-precise microbalance (MSE2.7S-000-DM, Sartorius) and was around 0.5 mg cm−2.
2.3 Composite electrolyte preparation
The ionically conductive protection layer was prepared by dissolving PEO, PVDF, LiTFSI, and LLZO in acetonitrile, as shown in Fig. 1c. The components PEO:PVDF:LiTFSI (7:3:2) were thoroughly mixed with controlled heating (50–70 °C) and rotation speed (300–450 rpm) for 24 hours. The ratio was chosen to balance the benefits of PEO for ionic conduction, PVDF for mechanical stability, and LiTFSI for high ionic dissociation. The effects of increasing the amount of PVDF or LiTFSI were considered to avoid compromising the electrolyte's conductivity and stability.31 The LLZO was incorporated in varying amounts (0–25 w%) due to its ability to drastically change the ionic conductivity. The resulting slurry was then spin-coated onto the prepared S/CNT cathode using a rotation speed of 5000–7000 rpm, acceleration of 1000–2000 rps, and a duration of 45 seconds.
The prepared samples were dried in a vacuum oven for 16 hours at 80 °C. To optimize the electrochemical properties of the composite electrolyte, various compositions with different LLZO contents (ranging from 0 to 25 wt% of the total composite mass) were prepared and tested electrochemically. Through this process, the optimal LLZO content was identified. Once the optimal composition was determined, the mixture was thoroughly stirred and applied to the cathode-coated substrate (UHV Al foil) via spin-coating using the same parameters as described for the electrode. This ensured a uniform distribution and effective integration of the solid electrolyte within the polymer matrix.
2.4 Material characterization
Material characterization was performed using a variety of techniques to assess the structure and morphology of the samples. Scanning electron microscopy (SEM) (ZEISS Crossbeam 540) and transmission electron microscopy (TEM, JEOL JEM-1400 Plus) were used for morphology analysis, operating at 80 kV. X-ray diffraction (XRD) (SmartLab, Rigaku) with Cu Kα radiation (λCuKα1 = 1.54056 Å, λCuKα2 = 1.54439 Å) in Bragg–Brentano reflection geometry was employed for crystallographic analysis.2.5 Cell assembly and electrochemical testing
The electrochemical performance of the cells was evaluated with the thin-film S-cathodes prepared in this work, Celgard 2400 separator or the prepared composite electrolyte, and lithium foil. The assembly of CR2032 coin cells was carried out in a glove box (MBRAUN, LABMaster Pro Glovebox, Germany) in a high-purity Ar atmosphere with <0.1 ppm O2 and <0.1 ppm H2O to avoid moisture and oxygen contamination. A composite electrolyte, consisting of a PEO/PVDF/LiTFSI/LLZO and a drop (15 μL) of 1 M LiTFSI in DOL:DME (1:1) with 0.1% LiNO3, was used without separator.
Electrochemical testing was performed using cyclic voltammetry (CV, VMP3, BioLogic, France) and galvanostatic charge–discharge cycling with a multichannel battery testing system (Neware Co., Shenzhen, China). The ionic conductivity of the electrolyte was measured via electrochemical impedance spectroscopy (EIS) in a frequency range from 1 Hz to 1 MHz with a potential amplitude of 10 mV at room temperature using a potentiostat/galvanostat (Autolab PGSTAT204, Metrohm, Switzerland). The data presented obtained for dry composite electrolyte without liquid electrolyte.
The system long-term cycling performance was evaluated at a 0.1C rate within a potential range of 1.6 to 2.8 V vs. Li+/Li. Rate performance tests were conducted to assess the electrochemical stability of the electrode under high current between 0.1 and 2C rate. The specific capacities of the cells were calculated based on the sulfur loading, ensuring that the reported capacities reflected the actual performance of the active material. Consistent sulfur loading (0.5 mg cm−2) across all cells was maintained to facilitate a fair comparison of their electrochemical behaviour. All tests were conducted at room temperature under controlled environmental conditions for reliable and reproducible results.
For the post-mortem analysis, the electrodes were carefully retrieved from the cycled coin cells in Ar-filled glovebox with minimized exposure to ambient conditions to prevent unwanted side reactions. The cathodes were gently rinsed with anhydrous dimethyl ether (DME) to remove any residual electrolyte and loosely bound lithium polysulfides, dried and transferred to SEM in an inert atmosphere to prevent surface change and contamination.
2.6 Pouch-cell microbattery assembly
A pouch-cell microbattery was assembled using spin-coated S/CNT/SP cathode and composite PEO/PVDF/LiTFSI/LLZO electrolyte. The assembly included a 1.5 × 1.5 cm electrode, a 1.3 × 1.3 cm nickel mesh, and rolled 1.3 × 1.3 cm Li foil (0.35 mm thick, from MTI Corp.), with designated contacts for each cell component. Notably, the electrolyte layer eliminated the need for a conventional separator. A minimal amount (15 μL) of 1 M LiTFSI DOL:DME (1:1) with 0.1% LiNO3 liquid electrolyte was introduced to facilitate the sulfur–lithium electrochemical kinetics. The entire assembly process was performed in an Ar-filled glove box, followed by evacuation and sealing of the pouch-type microbattery.
3. Results and discussion
As it was reported before,11 the PEO matrix facilitates primary Li+ transport through segmental motion, while the LLZO particles provide secondary conduction pathways via their bulk and grain boundaries. Additionally, the interfaces between the polymer matrix and LLZO act as high-conductivity regions for Li+ hopping. Although PVDF mainly contributes to mechanical stability, its integration with PEO ensures the structural integrity of the composite without significantly hindering ion mobility, as illustrated in Fig. 2a. This multi-pathway Li+ transport mechanism, enabled by the optimized preparation process, results in a composite electrolyte with high ionic conductivity and robust electrochemical performance. | ||
| Fig. 2 (a) Schematic ion transport pathways diagram within composite electrolyte; (b) EIS results for spin-coated composite thin-film electrolytes PEO/PVDF/LiTFSI/LLZO at room temperature; (c) ionic conductivity dependence of composite thin-film solid electrolyte on LLZO concentration in polymer matrix. | ||
Building on this, the incorporation of LLZO solid electrolyte plays a crucial role in further enhancing the ionic conductivity of the composite electrolyte.32–37 By careful adjusting the LLZO concentration, it is possible to tailor the ionic transport properties and strike the balance between conductivity and mechanical strength of the electrolyte. The presence of LLZO not only provides additional percolation pathways for lithium-ion movement but also helps to suppress the polymer crystallinity, thereby increasing segmental motion of the PEO chains for better Li-ion mobility.34
To evaluate the influence of LLZO content on ionic conductivity, a series of composite electrolytes with the LLZO concentration ranging from 0 to 25% were prepared and investigated using electrochemical impedance spectroscopy. As shown in Fig. 2b, the Nyquist impedance plots reveal a clear trend for the studied systems. The results indicate that increasing the LLZO content up to an optimal level significantly reduces the cell impedance, confirming the contribution of LLZO to enhanced Li+ conduction. However, beyond certain concentrations, excessive LLZO loading may lead to particle agglomeration and disruption of the polymer matrix continuity, which negatively impact the overall ionic mobility. This highlights the importance of compositional tuning in achieving high-performance solid-state electrolytes.
To better understand the electrochemical processes, the impedance spectra were fitted using an equivalent circuit model shown in the inset of the Nyquist plot in Fig. 2b. This model consists of several components that represent different resistive and capacitive contributions: bulk resistance (R1), which accounts for the intrinsic resistance of the polymer electrolyte, influenced by both the polymer matrix and LLZO filler; capacitance (C), which reflects charge storage at the interfaces; interfacial resistance (R2), associated with charge transfer at the electrode–electrolyte interface; and constant phase element (CPE), which models non-ideal capacitive behaviour due to surface roughness, heterogeneity, or inhomogeneous particle distribution.
The trends in the Nyquist plot confirmed that moderate LLZO content (up to 15%) significantly lowers both bulk and interfacial resistance, thereby improving ionic conductivity. However, beyond 15% LLZO, the impedance begins to rise, indicating the negative effect of further increase of the LLZO content. This effect is likely due to particle agglomeration or disrupting the polymer–ion interactions at higher LLZO concentrations, which hinders lithium-ion mobility.
The ionic conductivity (σ) was determined using the equation:
| σ = d/RtS, |
The conductivity plot in Fig. 2c further supports these findings, showing a steady increase in total ionic conductivity as the LLZO concentration increases from 0% to 15%. At this composition, the electrolyte achieves an optimal balance between polymer matrix flexibility and ion transport efficiency, maximizing lithium-ion mobility. However, above 15% LLZO, the conductivity declines sharply, reinforcing the idea that an excess of ceramic filler obstructs ion conduction rather than facilitating it.
Overall, these results suggest that incorporating LLZO into the polymer electrolyte enhances its ionic conductivity up to an optimal concentration of approximately 15%. Beyond this point, the negative effects of excessive filler content outweigh the benefits, leading to increased resistance and reduced conductivity. This finding is particularly relevant for solid-state lithium–sulfur batteries, where optimizing the ceramic-polymer composition is crucial for achieving high-performance electrolyte systems.
All the studies samples were thoroughly analyzed using a range of characterization techniques to assess their structural, and morphological properties. The samples' thickness and morphology were observed using scanning electron microscopy. Fig. 3a and b display the sulfur powder before and after ball milling, respectively, revealing a significant reduction in particle size from approximately 20 μm to nanoscale. Fig. 3c shows the SEM image of the S/CNT/SP mixture, clearly illustrating the uniform dispersion of the composite components. Energy dispersive X-ray spectroscopy (EDS) analysis, shown in Fig. 3d, confirms the presence and even distribution of sulfur and carbon elements within the composite. Fig. 3e presents a transmission electron microscopy (TEM) image of the CNT after interaction with sulfur, highlighting the substantial presence of sulfur and indicating the size of sulfur particles to be up to 50 nm. Such a small particle size is critical for ensuring efficient electrical conductivity throughout the composite material. Fig. 3f and g show cross sectional SEM images of the cathode before and after coating by the ionically conductive PEO/PVDF/LiTFSI/LLZO layer. Fig. 3f reveals the cross-section of the cathode (after roll-press) with the thickness of around 3 μm. In Fig. 3g, a cross-sectional SEM image clearly demonstrates a uniform and well-adhered PEO/PVDF/LiTFSI/LLZO coating, which made the total thickness of the cathode approximately 8 μm. These results confirm that the thin-film PEO/PVDF/LiTFSI/LLZO-electrolyte-coated S/CNT/SP cathode has successfully been prepared. Finally, Fig. 3h presents the morphology of the coated cathode, which appears smooth and free of significant microcracks or defects. However, in the magnified image, tiny pores are visible within the coating, which may facilitate Li-ion penetration through the polymeric scaffold during cycling, enhancing the cell performance.
 | ||
| Fig. 3 SEM images of (a) fresh commercial sulfur and (b) sulfur after ball-milling; (c) SEM images of S/CNT/SP; (d) EDS of S/CNT/SP, (e) TEM image of S/CNT; cross-sectional SEM of S/CNT/SP cathode (f) without and (g) with coating; (h and i) top SEM view of PEO/PVDF/LiTFSI/LLZO on cathode surface. | ||
The CV curves of the Li–S cells with S/CNT/SP cathode in Fig. 4a and b reveal distinct electrochemical behaviour difference of the cathode without the PEO/PVDF/LiTFSI/LLZO coating and with it. In the uncoated configuration (Fig. 4a), where a conventional separator and electrolyte are used, the redox peaks are broader and less defined across the cycles. The first cathodic peak, associated with the reduction of sulfur to higher-order polysulfides, appears around 2.3 V, followed by the second peak around 2.0 V related to the further reduction to Li2S2/Li2S. The corresponding anodic peak, which reflects the reverse oxidation process, is centered broadly around 2.4 V. A relatively large separation between the anodic and cathodic peaks indicates significant polarization and sluggish kinetics. This peak separation, along with evolving current responses over the first few cycles, suggests limited reversibility and increasing cell resistance, possibly due to poor ionic transport and polysulfide shuttle effects. Meanwhile, the coated configuration (Fig. 4b), where no separator is used and ionic conduction is provided by the porous solid-state layer, shows sharper and more symmetric redox peaks. These features indicate reduced polarization, reflecting improved charge transfer kinetics and more efficient redox reactions, suggesting that the PEO/PVDF/LiTFSI/LLZO coating supports effective lithium-ion transport along with mitigating the diffusion of polysulfides. The porous structure likely enables better electrolyte infiltration and contact with the active materials, leading to more stable electrochemical operation over repeated cycling.
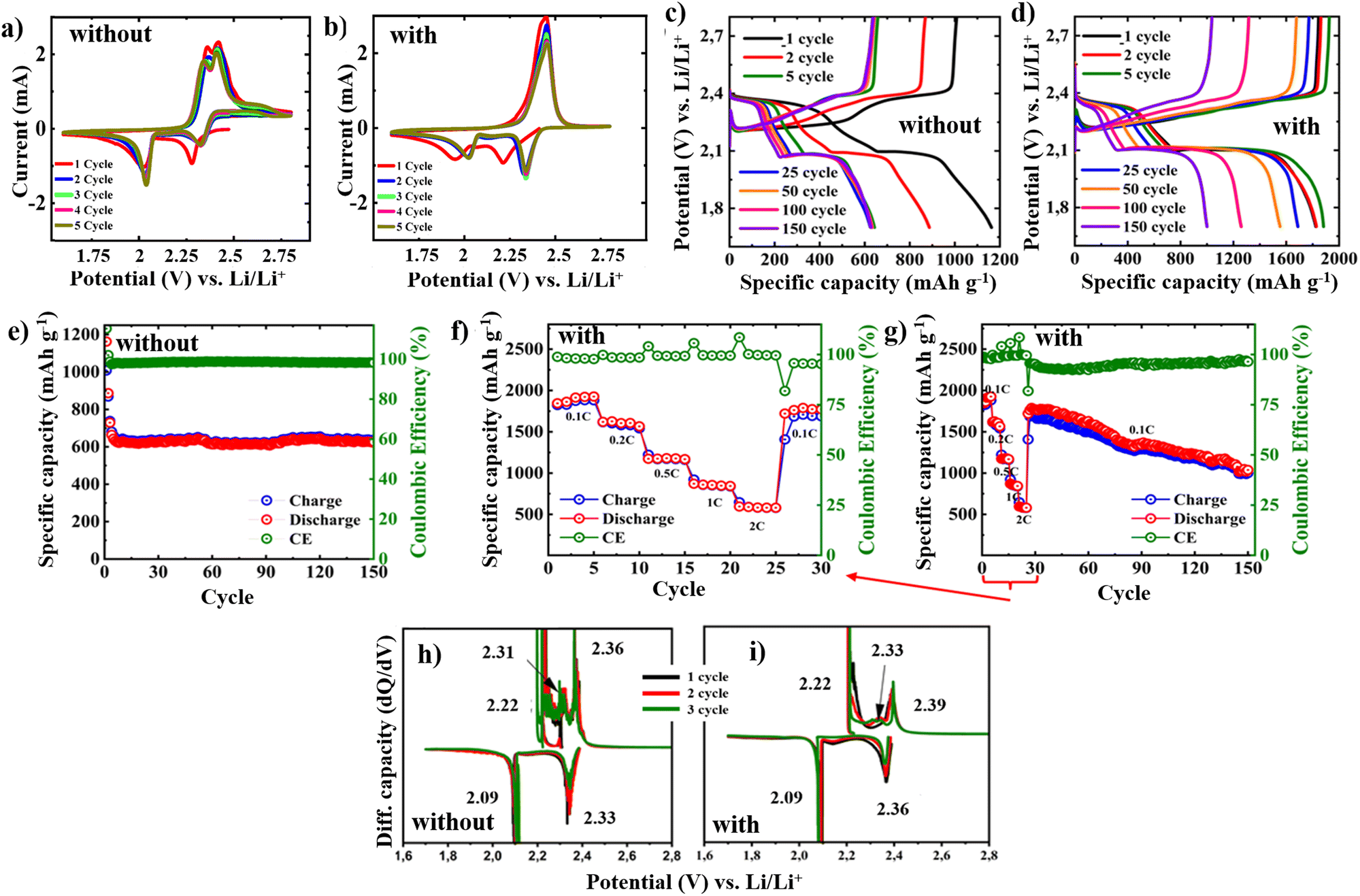 | ||
| Fig. 4 Results of electrochemical tests: CV of S/CNT/SP electrode (a) without and (b) with PEO/PVDF/LiTFSI/LLZO; galvanostatic cycling in potential range 1.7–2.8 V: potential profiles of (c) bare and (d) coated S/CNT/SP electrode at 0.1C; (e) cycling performance of bare S/CNT/SP; (f) C-rate capability and (g) cycling performance of S/CNT/SP with PEO/PVDF/LiTFSI/LLZO at 0.1–2C; comparative differential capacity plots of (h) bare and (i) coated S/CNT/SP. All tests were performed at RT. | ||
Galvanostatic charge–discharge profiles offer crucial understanding of electrochemical behaviour of the investigated systems (Fig. 4c–i). The charge–discharge profiles of bare electrode in Fig. 4c and that of coated counterpart in Fig. 4d highlight distinct differences in their electrochemical behaviour. The potential plateaus correspond to the multi-step conversion of sulfur species during lithiation and delithiation and matches with the trends in the CV scans. The bare electrode shows well-defined discharge plateaus at ∼2.3 V and ∼2.1 V, indicating the stepwise reduction of sulfur to lithium polysulfides Li2Sn and ultimately to Li2S. The electrode initially delivers a discharge capacity of 1162 mA h g−1 and a charge capacity of 1008 mA h g−1. However, a rapid decline in capacity is observed during the initial cycles. As cycling progresses, the potential plateaus during both discharge and charge exhibit notable shifts, with the discharge plateau (initially at ∼2.3 V) gradually shifting to the lower potentials and the charge plateau moving to the higher potential values. This increasing potential gap between charge and discharge plateaus indicates rising polarization, which can be attributed to side reactions, loss of active material, and reduced electrolyte conductivity due to the polysulfide shuttle effect. Moreover, upon extended cycling, these plateaus become shorter, likely due to the polysulfide dissolution and electrode degradation.
In contrast, the separator-free cell utilizing the electrolyte-coated electrode exhibits more stable and well-defined plateaus (Fig. 4d), indicating enhanced sulfur redox kinetics and more complete conversion reactions. A gradual delithiation observed in this system results from diffusion-limited lithium-ion transport through the porous PEO/PVDF/LiTFSI/LLZO layer, which effectively moderates the redox kinetics and suppresses the sharp phase transitions inherent to Li–S batteries. As a result, smoother CV curves (Fig. 4b) and sloped potential profiles (Fig. 4d) without distinct plateaus emerge. Moreover, the coating likely mitigates polysulfide shuttling, promoting more uniform and continuous electrochemical reactions. While some plateau shifting is still noticeable during prolonged cycling, it is significantly less severe than in the case of bare electrode (Fig. 4c). The relatively modest increase in polarization over cycles points to improved reaction kinetics and efficient charge transport, which is particularly notable given the reduced liquid electrolyte content. As a result, the initial discharge and charge capacities reach 1829 mA h g−1 and 1845 mA h g−1, respectively, corresponding to an initial coulombic efficiency exceeding 98%. This marked enhancement in cycling stability can be attributed to the improved interfacial contact, suppressed polysulfide diffusion, and the composite electrolyte's dual role in offering mechanical stability and facilitating ion conduction—further aided by the presence of nanopores that maintain ion accessibility throughout the film.
Fig. 4e illustrates the cycling performance of the bare spin-coated S/CNT/SP electrode without use of the composite electrolyte coating. Despite the initial rapid fade, the capacity stabilizes at approximately 600 mA h g−1 and remains relatively unchanged up to 150 cycles. These results suggest that the spin-coated thin-film electrode itself exhibits stable long-term performance, maintaining capacity retention beyond the initial decline phase. Meanwhile, the S/CNT/SP electrode coated with the PEO/PVDF/LiTFSI/LLZO electrolyte in Fig. 4f and g demonstrates significantly enhanced electrochemical performance. To assess the impact of the composite electrolyte, the rate capability tests were performed across a range of current densities from 0.1 to 2C (Fig. 4f). The discharge capacities at increasing rates were recorded as 1810, 1616, 1221, 921, and 647 mA h g−1 at 0.1, 0.2, 0.5, 1, and 2C, respectively. A notable feature in the rate-dependent cycling behaviour is the fluctuation in CE when transitioning between different C-rates. This behaviour can be attributed to lithium stripping/plating effects, which become less controlled at higher currents, potentially leading to lithium loss and side reactions. Conversely, when the rate is lowered back to 0.1C, the CE temporarily dips as the system readjusts before stabilizing in the 92–99% range. Importantly, after the rate capability tests, the capacity successfully recovers to 1640 mA h g−1 upon returning to 0.1C, demonstrating the robustness of the composite electrolyte in preserving electrode integrity and maintaining lithium-ion transport. Further long-term cycling at 0.1C revealed a gradual capacity decline (Fig. 4g), with a final capacity of approximately 1000 mA h g−1 after 150 cycles.
A particularly intriguing aspect of the cycling behaviour of the cells is the presence of an excess capacity upon the initial few cycles, exceeding the theoretical limit. Looking at the descending discharge profile in the initial five cycles at 0.1C (Fig. 4d), one can notice that the plateau of sulfur lithiation ends at around its theoretical capacity value, after which an additional small plateau appears at around 1.95 V. Moreover, it becomes more pronounced at the 5th cycle, indicating that some redox reactions take place. At the charge–discharge curves of the bare S/CNT/SP cathode (Fig. 4c), we can also notice the same plateau, indicating that such a reaction happens in both cathodes, suggesting that it is unlikely to be primarily attributed to the solid electrolyte. From the literature, a similar phenomenon has been observed for LiNO3 additives in Li–S batteries and assigned to the reduction of LiNO3, resulting in slowing down the electrode reaction kinetics and a permanent loss in the reversibility of the Li–S cell. It was even suggested that deep discharge must be avoided for a long cycle life Li–S battery when LiNO3 is used as an additive or a co-salt in the electrolyte solution.35–37 It is worth noting that the uncoated cell contained more liquid electrolyte and exhibited a longer plateau compared to the coated cell. This suggests that the excess capacity of the cell may be attributed to LiNO3, which was present in the liquid electrolyte. However, as shown in Fig. 4d, this plateau gradually diminishes over subsequent cycles, and the excess theoretical capacity is no longer observed.
Further insights into the electrode stability and reaction kinetics are obtained from the differential capacity (dQ/dV) plots of the S/CNT/SP electrodes with and without the composite electrolyte (Fig. 4h and i). The CV behaviour of both electrodes in Fig. 4a and b are mirrored in their dQ/dV plots in Fig. 4h and i. Both electrodes exhibit similar peak positions, indicating comparable reaction pathways. The bare cathode in Fig. 4h demonstrates irregular and somewhat noisy peaks, especially around 2.2–2.4 V, indicative of side reactions or interface instability. For the cell with the PEO/PVDF/LiTFSI/LLZO-coated cathode, the peaks in the dQ/dV plot are more distinct and stable, showing consistent lithium insertion/extraction processes. The coated electrode also displays slightly higher redox potentials, suggesting better ionic conductivity and reduced resistance. Overall, the coating significantly improved the electrochemical performance by enhancing stability, reducing polarization, and ensuring more reversible lithium–sulfur reactions.
The SEM analysis was conducted to evaluate the morphological evolution of the thin-film cathodes after 150 charge/discharge cycles (Fig. 5a–d). Fig. 5a presents the morphology of the bare S/CNT/SP cathode, whereas Fig. 5b shows the same cathode but coated with the PEO/PVDF/LiTFSI/LLZO layer. In the bare cathode, the surface reveals significant agglomeration of micron-sized particles and formation of microcracks, which can contribute to structural degradation, delamination and loss of electrical connectivity of the electrode. These observations are consistent with the galvanostatic cycling results, where the bare cathode exhibited significant polarization growth and capacity decay over cycling. The development of microcracks in the bare cathode could lead to excessive electrolyte penetration, exacerbating side reactions and irreversible loss of the active material.
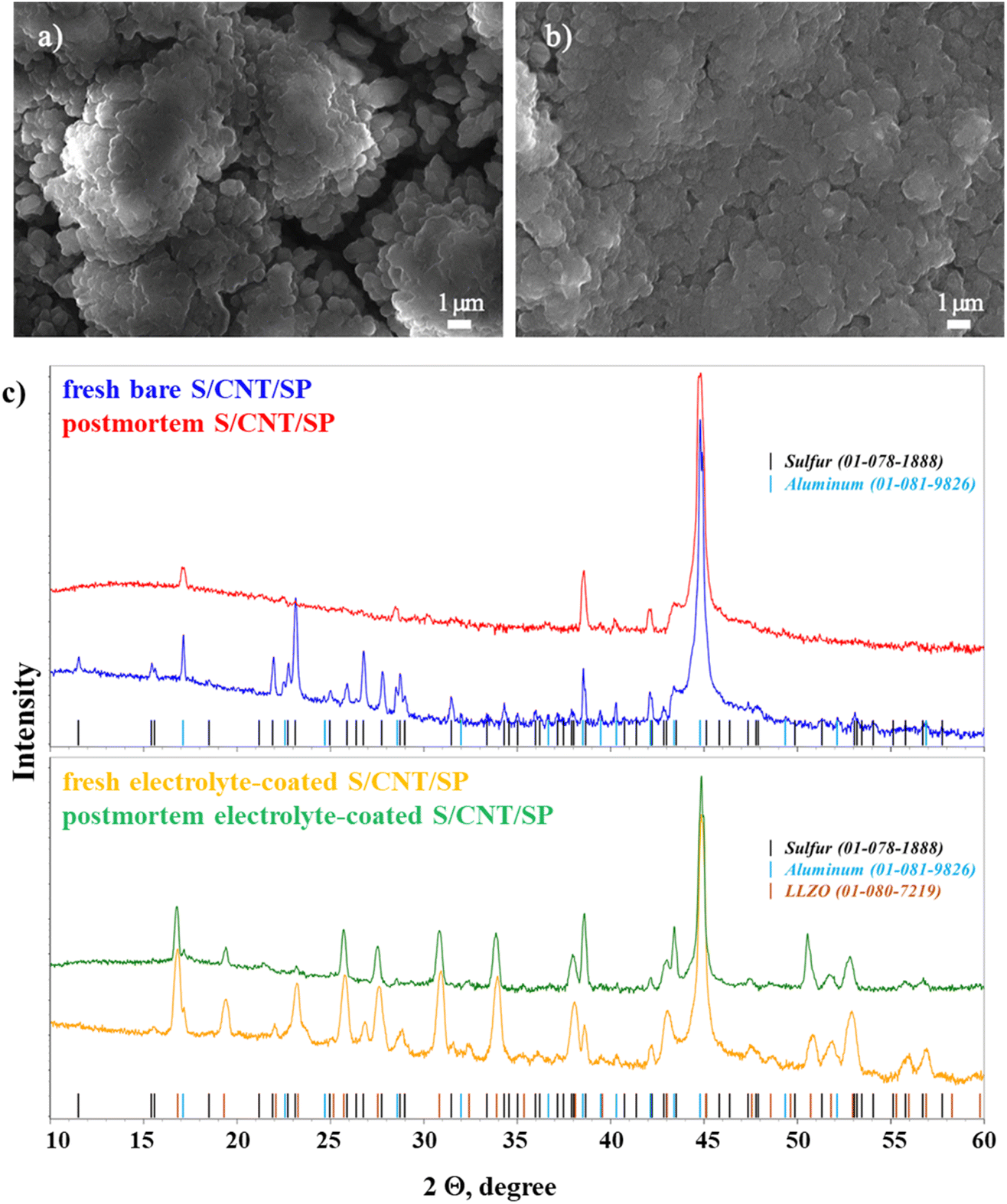 | ||
| Fig. 5 Results of post-mortem analysis of electrodes after 150 cycles: (a) bare S/CNT/SP electrode; (b) S electrode with PEO/PVDF/LiTFSI/LLZO; (c) XRD spectra of bare and electrolyte-coated S/CNT/SP electrodes before and after cycling. | ||
The second image (Fig. 5b) presents the electrolyte-coated electrode after 150 cycles. Compared to the fresh electrode (Fig. 4f), initially uniform polymer surface has roughened, and distinct agglomerations were formed. This suggests that prolonged cycling leads to structural changes, potentially caused by polymer degradation, redistribution of sulfur species, or partial loss of contact between the components. However, compared to the uncoated electrodes, the electrolyte-coated cathode retains its mechanical stability, exhibiting no delamination. This improved structural integrity likely contributed to better cycling stability by suppressing polysulfide dissolution and maintaining electrode cohesion.
To further understand the structural evolution of the cathodes, XRD analysis of both electrodes was conducted before and after cycling (Fig. 5c). In the uncoated electrode, the fresh sample displays sharp and well-defined peaks corresponding to crystalline S and Al (current collector), indicating the presence of highly crystalline active material. However, after cycling, the postmortem spectrum shows a noticeable reduction in sulfur peak intensity and sharpness. This suggests a loss of crystallinity, likely due to polysulfide dissolution and structural degradation, which are common issues in Li–S batteries. The overall intensity decrease indicates a partial loss of active material and/or electrode degradation.
In contrast, the coated electrode maintains a significantly better structural stability. The fresh sample exhibits strong S and Al peaks, along with additional peaks corresponding to LLZO, confirming the presence of the lithium-conducting electrolyte. After cycling, the postmortem spectrum shows that the sulfur peaks remain more pronounced and well-defined compared to the uncoated counterpart. This indicates better retention of the active material and reduced structural degradation. The coating effectively suppresses polysulfide dissolution and enhances the stability of the cathode, leading to improved electrochemical performance and longevity.
Overall, these findings demonstrate that incorporation of the PEO/PVDF/LiTFSI/LLZO composite layer significantly improved the electrochemical and structural stability of the cathode. The reduction in microcracks, suppression of polysulfide dissolution, and preservation of interfacial stability collectively contribute to the observed enhancement in cycling performance, evidenced by the lower polarization and higher capacity retention.
3.1 Pouch cell-assembly
To move close toward miniaturized and wearable applications, following successful evaluation of the electrochemical performance of the designed sulfur cathode, the next step was the fabrication of a microbattery prototype. Designing an efficient pouch-cell configuration for a Li–S microbattery presents both a technical challenge and an opportunity to advance energy storage technology. Utilizing the developed S/CNT/SP cathode in combination with a quasi-solid PEO/PVDF/LiTFSI/LLZO electrolyte, a pouch-type Li–S microbattery with sandwich structure was successfully assembled without separator (Fig. 6a). The initial open-circuit voltage (OCV) was measured at approximately 2.67 V, as depicted in Fig. 6b. Notably, the pouch-cell structure exhibited flexibility, a feature that could be highly advantageous for future applications in wearable and portable electronic devices. | ||
| Fig. 6 S|PEO/PVDF/LiTFSI/LLZO|Li pouch cell: (a) assembly scheme, (b) OCV measurement, (c) cycling performance at a current density of 20 μA cm−2. | ||
To evaluate the electrochemical performance of the Li–S microbattery, the pouch cell was subjected to charge–discharge cycling within a potential range of 1.6 to 2.8 V at a current density of 20 μA cm−2. The cycling data, presented in Fig. 6c, reveal the evolution of specific capacity over cycling at room temperature. The microbattery initially exhibited an areal capacity of approximately 110 μA h cm−2 (577.6 mA h g−1). Over the course of 60 cycles, the capacity gradually declined, stabilizing at around 70 μA h cm−2 (367.5 mA h g−1), effectively retaining 64% of its original capacity.
Despite a lower performance compared to a traditional coin-cell configuration, likely due to a less compact packaging structure, the pouch-cell format maintained a stable CE of approximately 97%, indicating a highly efficient charge–discharge process. The assembly of a pouch-type Li–S microbattery with stable electrochemical performance marks a first step toward practical miniaturization of Li–S energy storage devices. Although further optimization is necessary to increase mass loading and enhance capacity retention, the demonstrated flexibility and efficiency position this technology as a viable candidate for next-generation portable and wearable electronics.
4. Conclusion
In this study, we successfully developed and characterized an advanced spin-coated thin-film S/CNT/SP cathode and quasi-solid PEO/PVDF/LiTFSI/LLZO electrolyte for Li–S microbatteries. The incorporation of LLZO into the PEO/PVDF/LiTFSI composite electrolyte significantly enhanced the ionic conductivity and electrochemical performance of the solid-state electrolyte system. The results demonstrated that moderate LLZO loading (up to 15%) optimizes the balance between ionic transport and mechanical stability by creating additional conduction pathways and reducing polymer crystallinity. EIS and conductivity measurements confirmed that these compositional tuning lowers bulk and interfacial resistance, facilitating more efficient lithium-ion transport. Beyond the optimal LLZO concentration, however, agglomeration effects and polymer matrix disruption hinder conductivity, highlighting the importance of precise ceramic filler content control. SEM images confirmed the presence of nanopores in the PEO/PVDF/LiTFSI/LLZO layer, which facilitated lithium-ion penetration through the polymeric scaffold during cycling, enhancing performance.The electrochemical tests further validated the superior performance of the PEO/PVDF/LiTFSI/LLZO-coated S/CNT/SP cathode in Li–S batteries. The solid-state electrolyte layer improved charge transfer kinetics, reduced polarization, and suppressed polysulfide shuttling, leading to enhanced cycling stability and higher capacity retention. Galvanostatic cycling and CV tests revealed more distinct and stable redox peaks, lower potential hysteresis, and reduced side reactions in the coated system compared to the bare cathode. This enabled the cell to maintain a capacity of approximately 1000 mA h g−1 even after 150 cycles across various C-rates. Furthermore, post-cycling analysis revealed that the coated cathode preserved its morphological integrity and structural stability.
Furthermore, we demonstrated the Li–S pouch-type microbattery with a sandwich structure, which was able to maintain an areal capacity of approximately 70 μA h cm−2 (367.5 mA h g−1) after 60 cycles at 20 μA cm−2. Indeed, further optimization is needed to achieve a higher energy density, but the proved electrochemical performance of the developed cathode and electrolyte make it promising for next-generation portable and wearable electronics.
Overall, this research highlights the critical role of advanced composite cathode materials and quasi-solid composite electrolytes in enhancing the performance of Li–S microbatteries. The promising results of this work provide a strong foundation for further optimization and development of safe, high-energy-density, long-lasting microbattery solutions. These findings pave the way for next-generation miniaturized energy storage systems, emphasizing the importance of innovative material engineering in the pursuit of compact, high-performance battery technologies.
Data availability
The data supporting the findings of this study were obtained from standard sources such as the research articles; for crystallographic information the PDF4+ database was utilized. No new datasets were generated or analyzed specifically for this study. All relevant data are presented in the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The work was funded under the research targeted programs BR21882402 and BR24992766 from the Ministry of Science and Higher Education of the Republic of Kazakhstan and research grant 0122022FD4136 from Nazarbayev University.References
- A. Uali, A. Kazymbetova, A. Belgibayeva, A. Nurpeissova, Z. Bakenov and A. Mukanova, Industrial sulfur separation and purification: Paving the way to energy applications, Chem. Eng. J., 2025, 510, 161574, DOI:10.1016/j.cej.2025.161574.
- L. Ren, J. Liu, A. H. Pato, Y. Wang, X. Lu, I. A. Chandio, M. Zhou, W. Liu, H. Xu and X. Sun, Rational design of nanoarray structures for lithium-sulfur batteries: recent advances and future prospects, Mater. Futures, 2023, 2, 042103, DOI:10.1088/2752-5724/ace7e4.
- Q. Shao, S. Zhu and J. Chen, A review on lithium-sulfur batteries: challenge, development, and perspective, Nano Res., 2023, 16, 8097–8138, DOI:10.1007/s12274-022-5227-0.
- J. Du, X. Zhou, X. Cheng and G. Jiang, Theoretical evaluation of monolayer MA2Z4 (M = Ti, Zr, or Hf; A = Si or Ge; and Z = P or As) family as promising candidates for lithium–sulfur batteries, J. Colloid Interface Sci., 2024, 678, 150–158, DOI:10.1016/j.jcis.2024.09.106.
- Y. Bao, B. Yue, L. Li, H. Shao, Y. Xie, Q. Ma, W. Yu, J. Wang and X. Dong, LaF3@SiO2 yolk–shell heterostructure nanofiber-modified separator enhances the long-cycling performance of lithium–sulfur batteries, J. Colloid Interface Sci., 2025, 683, 358–374, DOI:10.1016/j.jcis.2024.12.093.
- M. Liu, L. Wan, P. Su, T. Guo, R. Yin, H. Jin, H. Jia and F. Tang, Co/Co3O4@NC-CNTs modified separator of Li-S battery achieving the synergistic effect of adsorption-directional migration-catalysis via built-in electric field, J. Colloid Interface Sci., 2024, 682, 436–445, DOI:10.1016/j.jcis.2024.11.211.
- Z. Liu, L. Sun, X. Liu and Q. Lu, Stabilization Strategies of Lithium Metal Anode toward Dendrite-Free Lithium-Sulfur Batteries, Chemistry, 2024, 30(60), e202402032, DOI:10.1002/chem.202402032.
- W. Ren, W. Ma, S. Zhang and B. Tang, Recent advances in shuttle effect inhibition for lithium sulfur batteries, Energy Storage Mater., 2019, 23, 707–732, DOI:10.1016/j.ensm.2019.02.022.
- K. G. Naik, D. Chatterjee and P. P. Mukherjee, Solid Electrolyte–Cathode Interface Dictates Reaction Heterogeneity and Anode Stability, ACS Appl. Mater. Interfaces, 2022, 14, 45308–45319, DOI:10.1021/acsami.2c11339.
- W. Yu, H. Xiang, J. Yue, X. Feng, W. Duan, Y. Feng, B. Cheng, N. Deng and W. Kang, The multi-scale dissipation mechanism of composite solid electrolyte based on nanofiber elastomer for all-solid-state lithium metal batteries, J. Colloid Interface Sci., 2024, 682, 1073–1084, DOI:10.1016/j.jcis.2024.12.042.
- A. Jetybayeva, A. Umirzakov, B. Uzakbaiuly, Z. Bakenov and A. Mukanova, Towards Li–S microbatteries: a perspective review, J. Power Sources, 2023, 573, 233158, DOI:10.1016/j.jpowsour.2023.233158.
- Y. Feng, H. Liu and X. Liu, et al., Enlarged interlayer of separator coating enabling high-performance lithium–sulfur batteries, Front. Chem. Sci. Eng., 2024, 18(2), 20, DOI:10.1007/s11705-024-2385-5.
- A. Jetybayeva, B. Uzakbaiuly, A. Mukanova, S. T. Myung and Z. Bakenov, Recent advancements in solid electrolytes integrated into all-solid-state 2D and 3D lithium-ion microbatteries, J. Mater. Chem. A, 2021, 9, 15140–15178, 10.1039/d1ta02652f.
- B. Uzakbaiuly, A. Mukanova, Y. Zhang and Z. Bakenov, Physical Vapor Deposition of Cathode Materials for All Solid-State Li Ion Batteries: A Review, Frontiers in Energy Research, 2021, 9, 1–16, DOI:10.3389/fenrg.2021.625123.
- A. Mukanova, A. Jetybayeva, S. T. Myung, S. S. Kim and Z. Bakenov, A mini-review on the development of Si-based thin film anodes for Li-ion batteries, Mater. Today Energy, 2018, 9, 49–66, DOI:10.1016/j.mtener.2018.05.004.
- M. H. Lindic, H. Martinez, A. Benayad, B. Pecquenard, P. Vinatier, A. Levasseur and D. Gonbeau, XPS investigations of TiOySz amorphous thin films used as positive electrode in lithium microbatteries, Solid State Ionics, 2005, 176, 1529–1537, DOI:10.1016/j.ssi.2005.04.007.
- M. H. Lindic, B. Pecquenard, P. Vinatier, A. Levasseur, H. Martinez, D. Gonbeau, P. E. Petit and G. Ouvrard, Electrochemical Mechanisms during Lithium Insertion into TiO0.6S2.8 Thin Film Positive Electrode in Lithium Microbatteries, J. Electrochem. Soc., 2005, 152, A141, DOI:10.1149/1.1834893.
- S. R. V. Bugga, S. C. Jones, J. Pasalic, C. S. Seu, J.-P. Jones and L. Torres, Metal sulphide blended sulfur cathodes in high energy lithium-sulfur cells, J. Electrochem. Soc., 2017, 164, A265–A276, DOI:10.1149/2.0941702jes.
- S. D. Jones, J. R. Akridge and F. K. Shokoohi, Thin film rechargeable Li batteries, Solid State Ionics, 1994, 69, 357–368, DOI:10.1016/0167-2738(94)90423-5.
- V. Pelé, F. Flamary, L. Bourgeois, B. Pecquenard and F. Le Cras, Perfect reversibility of the lithium insertion in FeS2: the combined effects of all-solid-state and thin film cell configurations, Electrochem. Commun., 2015, 51, 81–84, DOI:10.1016/j.elecom.2014.12.009.
- M. J. Klein, G. M. Veith and A. Manthiram, Chemistry of Sputter-Deposited Lithium Sulfide Films, J. Am. Chem. Soc., 2017, 139, 9229–9237, DOI:10.1021/jacs.7b03379.
- V. Shokhen, Y. Miroshnikov, G. Gershinsky, N. Gotlib, C. Stern, D. Naveh and D. Zitoun, On the Impact of Vertical Alignment of MoS2 for Efficient Lithium Storage, Sci. Rep., 2017, 7, 1–11, DOI:10.1038/s41598-017-03453-x.
- X. Meng, D. J. Comstock, T. T. Fister and J. W. Elam, Vapor-Phase Atomic-Controllable Growth of Amorphous Li2S for High-Performance Lithium–Sulfur Batteries, ACS Nano, 2014, 8, 10963–10972, DOI:10.1021/nn505480w.
- M. Nathan, D. Golodnitsky, V. Yufit, E. Strauss, T. Ripenbein, I. Shechtman, S. Menkin and E. Peled, Three-dimensional thin-film Li-ion microbatteries for autonomous MEMS, J. Microelectromech. Syst., 2005, 14, 879–885, DOI:10.1109/JMEMS.2005.851860.
- D. Golodnitsky, M. Nathan, V. Yufit, E. Strauss, K. Freedman, L. Burstein, A. Gladkich and E. Peled, Progress in three-dimensional (3D) Li-ion microbatteries, Solid State Ionics, 2006, 177, 2811–2819, DOI:10.1016/j.ssi.2006.02.048.
- C. Milroy and A. Manthiram, Printed microelectrodes for scalable, high-areal capacity lithium-sulfur batteries, Chem. Commun., 2016, 52, 4282–4285, 10.1039/c5cc10503j.
- C. A. Milroy, S. Jang, T. Fujimori, A. Dodabalapur and A. Manthiram, Inkjet-printed lithium–sulfur microcathodes for all-printed, integrated nanomanufacturing, Small, 2017, 13, 1603786, DOI:10.1002/smll.201603786.
- Y. Mao, G. Li, Y. Guo, Z. Li, C. Liang, X. Peng and Z. Lin, Foldable interpenetrated metal-organic frameworks/carbon nanotubes thin film for lithium-sulfur batteries, Nat. Commun., 2017, 8, 1–8, DOI:10.1038/ncomms14628.
- Y. Fu, Y. S. Su and A. Manthiram, Highly reversible lithium/dissolved polysulfide batteries with carbon nanotube electrodes, Angew. Chem., Int. Ed., 2013, 52, 6930–6935, DOI:10.1002/anie.201301250.
- F. Sultanov, N. Zhumasheva, A. Dangaliyeva, A. Zhaisanova, N. Baikalov, B. Tatykayev, M. Yeleuov, Z. Bakenov and A. Mentbayeva, Enhancing lithium-sulfur battery performance with biomass-derived graphene-like porous carbon and NiO nanoparticles composites, J. Power Sources, 2023, 593, 233959, DOI:10.1016/j.jpowsour.2023.233959.
- J. Li, K. Zhu and Z. Yao, et al., A promising composite solid electrolyte incorporating LLZO into PEO/PVDF matrix for all-solid-state lithium-ion batteries, Ionics, 2020, 26, 1101–1108, DOI:10.1007/s11581-019-03320-x.
- M. Wu, D. Liu, D. Qu, Z. Xie, J. Li, J. Lei and H. Tang, 3D Coral-like LLZO/PVDF Composite Electrolytes with Enhanced Ionic Conductivity and Mechanical Flexibility for Solid-State Lithium Batteries, ACS Appl. Mater. Interfaces, 2020, 12, 52652–52659, DOI:10.1021/acsami.0c15004.
- Y. Zhang, Y. Xiao, Z. Tang, L. Zou, Q. Liu, X. Fu, X. Xiang and J. Deng, Poly(ethylene oxide)-based composite solid electrolyte modified by Li7La3Zr2O12 inorganic filler with grafted imidazolium-based ionic liquids functional groups, Chem. Eng. J., 2024, 500, 157191, DOI:10.1016/j.cej.2024.157191.
- M. J. Counihan, J. Lee, P. Mirmira, P. Barai, M. E. Burns, C. V. Amanchukwu, V. Srinivasan, Y. Zhang and S. Tepavcevic, Improved interfacial Li-ion transport in composite polymer electrolytes via surface modification of LLZO, Energy Mater., 2025, 5, 500032, DOI:10.20517/energymater.2024.195.
- Y. Mao, T. Li, S. Abuelgasim, X. Hao, Y. Xiao, C. Li, W. Wang, Y. Li and E. Bao, Systematic insight of the behavior of LiNO3 additive in LiS batteries with gradient S loading, J. Energy Storage, 2024, 79, 110215, DOI:10.1016/j.est.2023.110215.
- S. S. Zhang, A review on lithium-sulfur battery systems, J. Electrochem. Soc., 2012, 159, A920, DOI:10.1149/2.002207jes.
- A. Rosenman, E. Markevich, G. Salitra, D. Aurbach, A. Garsuch and F. F. Chesneau, Review on Li-Sulfur Battery Systems: An Integral Perspective, Adv. Energy Mater., 2015, 5, 1500212, DOI:10.1002/aenm.201500212.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2025 |