
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlatinum clusters Ptn+/0/– (n = 3–21): from triangles to remarkable tubular architectures†
Bao-Ngan Nguyen-Ha ab,
My-Phuong Pham-Hocd,
Nguyen Minh Tam*e and
Minh Tho Nguyenab
ab,
My-Phuong Pham-Hocd,
Nguyen Minh Tam*e and
Minh Tho Nguyenab
aLaboratory for Chemical Computation and Modeling, Institute for Computational Science and Artificial Intelligence, Van Lang University, Ho Chi Minh City, Vietnam. E-mail: ngan.nguyenhabao@vlu.edu.vn
bFaculty of Applied Technology, School of Technology, Van Lang University, Ho Chi Minh City, Vietnam
cFaculty of Chemical Engineering, Ho Chi Minh City University of Technology (HCMUT), 268 Ly Thuong Kiet Street, District 10, Ho Chi Minh City, Vietnam
dVietnam National University Ho Chi Minh City, Linh Trung Ward, Thu Duc City, Ho Chi Minh City, Vietnam
eFaculty of Basic Sciences, University of Phan Thiet, 225 Nguyen Thong, Phan Thiet City, Binh Thuan, Vietnam. E-mail: nmtam@upt.edu.vn
First published on 6th June 2025
Abstract
This paper reports on a theoretical investigation of Ptn+/0/– clusters with n = 3–21 in three-charge states using density functional theory with the B3PW91 functional in conjunction with the aug-cc-pVTZ-PP and Def2-TZVP basis sets. Geometric structures of Ptn+/0/– in the small-to-medium size range are primarily derived from Pt6+/0/–; whereas Pt10+/0/–, Pt14+/0/– and Pt18+/0/– also emerge as building blocks. The sizes Pt4, Pt6, Pt10 and Pt18 emerge as magic clusters, in part in agreement with experimental mass spectrometry. Tubular Pt structures are identified at the sizes of Pt12, Pt18 and Pt24 clusters that can be constructed by either stacking planar Pt6 or assembling prismatic Pt6 units. These tubular configurations are energetically favorable and present an effective pathway for the design of more complex Pt-based nanostructures. The Pt6 triangle features six σ bonds, six conjugated π-bonds of (2c–1e) delocalization and six (6c–1e) bonds reflecting a fully SP2 hybridization. The Pt6 prism contains three (6c–1e) bonds and nine σ-bonds from (2c–1e) delocalization, spanning its edges and faces. These delocalized bonds facilitate the structural integrity and connectivity of tubular Pt12, Pt18 and Pt24 isomers. In these tubular clusters, increased bond coordination and redistribution of electron contributions among s, p and d orbitals enhance bonding interactions and promote stabilized structural assemblies.
1. Introduction
Pure and doped platinum clusters as well as their integration into nanomaterials have attracted considerable attention, in part due to their extensive applicability as efficient catalysts for diverse chemical reactions.1–4 These nanomaterials possess remarkable catalytic power, rendering them valuable across diverse fields of science and technology.5 They are variously utilized in, among others, fuel cells,6,7 gas reforming,8,9 automotive conversion,10 as well as various oxidation,11–13 and redox14,15 reactions. Furthermore, they hold application promise in medicine, particularly in anticarcinogenic treatments.16–18Nevertheless, particularly for heavy elements like the Pt atom, inherent relativistic effects can wield importance, influencing both the structure of their compounds and their chemical behavior. The notable relativistic impact of the Pt atom and its distinct metal bonding characteristics often present a significant obstacle to achieve a comprehensive understanding solely through the knowledge derived from individual Pt atoms and complexes. In this context, the understanding of platinum clusters' structures remains a big challenge due to, beside relativistic effects, the intricate interplay between spin exchange stabilization and electron correlation effects.19 The presence of a largely open d shell within these clusters gives rise to multiple electronic states that encompass a large range of spin multiplicities, thus introducing intricacy to their structural and spectroscopic attributes. Several studies were carried out to investigate the structure and magnetism of Pt clusters, but a number of contentions persist even at their small sizes.20–28
In previous studies, stable structures of Ptn clusters were found with intriguing shapes at high spin states. It has been observed that small size Ptn+/0/– clusters in the negative, neutral and positive charge states typically prefer planar shapes.27,29 In the negatively charged clusters, the Ptn− anions continue to favor planar structure up to size n = 7.28 However, it becomes more complicated in the cationic and neutral counterparts. It is particularly intriguing that in a 2014 investigation by Chaves et al.,24 the Pt6+ cations were reported to adopt a D3h planar triangular structure, despite the fact that three-dimensional (3D) configurations already emerge at smaller sizes n = 4 and 5.27 This raises a legitimate question as to whether the planar structure is truly favored for cations at size n = 6. A similar question arises for neutral Ptn clusters where planar structures reappear at sizes n = 8 and 9 after a shift to 3D configurations at size n = 7.27 The situation becomes even more complex when considering a subsequent report from Singh et al.,23 which found that the most stable structures for both neutral Pt8 and Pt9 clusters are, in fact, 3D structures.26 Both studies, remarkedly, used rather low-level computational methods that lack sufficient precision. While the Chaves et al. study24 employed the PBE functional with a light-tier 2 NAO basis set, the Singh et al. study23 used both B3LYP and B3PW91 functionals in conjunction with the small LAN2DZ and LAN2MB basis sets. More recently, Rodríguez-Kessler et al. used the PBE with a plane-wave basis set30 and found that the Pt7 structure aligns with Chaves et al.’s findings, whereas the Pt9 structure matches that of Singh et al. (cf. Scheme 1).
 | ||
| Scheme 1 Previously reported structures of neutral Ptn clusters with n = 7–9. | ||
When it comes to larger sizes, the problems become even more elusive when the number of itinerant d electrons are significantly pronounced. Apart from the stable tetrahedral structure observed across different charge states at size n = 10, the anionic Pt10−, neutral Pt10, cationic Pt10+ and di-cationic Pt102+, each of these displays high magnetic moment of approximately 7,28,29 8,23,25,26,29,31 9 (ref. 29) and 10 (ref. 32) μB, respectively. A considerable inconsistency still appears regarding the structures and spin states of other Ptn clusters. Previous findings reported by different groups produced varying results and no clear consensus emerges on either their preferred configurations or spin states.13,23,25,28,29,31
For neutral clusters, although numerous studies have focused on the n = 10–20 size range,13,23,30,33–42 the spin states corresponding to their ground state configurations remain largely uncertain (cf. Scheme 2), with significant discrepancies among these studies.33,42
 | ||
| Scheme 2 Previously reported structures and magnetism of neutral Ptn with n = 11–20. | ||
Concerning anionic Ptn− clusters, the stable Pt6− anion was identified as having a planar triangular structure in a doublet state.28 However, only the triangular Pt6− structure aligns well (profile factor Rw = 1.7%) with experimental data measured from trapped ion electron diffraction (TIED), while larger anions require the inclusion of higher-energy isomers to achieve reasonable agreement with experiment.28 Furthermore, beyond the Chaves et al.'s study mentioned above, there has been no other investigation into the positively charged counterparts, underscoring the need for more definitive studies in this area. A main issue is that the ground electronic states identified for the stable structures in these charge states also remain largely elusive.
In this context, to gain a better understanding of the structural growth mechanisms of Ptn+/0/– clusters, especially in the small to medium size range, we set out to conduct a thorough re-evaluation and expanded investigation. With such an objective, the present study aims to re-evaluate and extend by employing higher accuracy quantum chemical methods to determine low-energy structures, exploring a more comprehensive range of structural configurations and examining a larger set of sizes of Ptn with n = 3–21, crossing the anionic, neutral and cationic charge states. This approach facilitates a detailed analysis of the preferred configurations and structural evolution of Ptn+/0/− clusters, offering deeper insights into the factors driving their formation across different sizes and charges.
2. Computational methods
All standard electronic structure calculations in this study are performed using the Gaussian 16 set of programs.43 We employ two basis sets where the effective core potentials include relativistic effects for heavy atoms, namely the Dunning correlation consistent aug-cc-pVTZ-PP44,45 and the Ahlrichs Def2-TZVP46 basis sets. The B3PW91 (ref.47 and 48) is used on the basis of its previously shown effectiveness in modeling Ptn cluster structures, offering a favorable balance between computational efficiency and accuracy compared with experimental data.4,32,36,42 This choice of functional ensures consistency with earlier theoretical studies allowing meaningful comparisons across studies.To identify low-energy geometric isomers, an extensive exploration of the potential energy surface of each size is conducted, involving a combination of both manual and automated structure generation methods. Initially, trial structures are constructed manually by considering previously reported structures (as discussed in Introduction) and by systematically expanding smaller clusters through the addition of atoms to vertices, edges, or faces to create larger ones. These guess structures are then optimized using the B3PW91 functional in conjunction with the small LANL2DZ basis set.49 To further enhance structural diversity for clusters from Pt10+/0/– onward, a stochastic generation approach50 is employed, utilizing an algorithm improved from the random kick method.51 In this process, the lowest-energy isomers within 2 eV obtained from the initial optimizations are used as seeds in both genetic algorithm and modified random kick routines. The two methods are used iteratively, feeding new structures from one into the other, until no additional unique structures are identified, thus allowing for a thorough sampling of the structural configuration.
All guess structures generated in such a way are optimized across multiple spin states using the B3PW91/LANL2DZ method. If a novel structure emerged in one spin state, it is then re-optimized under other multiplicities. For closely related structures with nearly identical energies, symmetry-constrained re-optimization is performed to confirm their point group. This systematic workflow allows a comprehensive set of stable configurations to be explored across different charge states and multiplicities.
For each size and charge state, local minima with relative energies lying within 5 eV to the lowest-energy minimum are re-optimized using the same B3PW91 but with the larger aug-cc-pVTZ-PP basis set for the sizes from n = 3–18 and the Def2-TZVP basis set for the sizes of n = 3–21. Harmonic vibrational frequency calculations at these levels are carried out to confirm equilibrium geometries and provide zero-point energy (ZPE) corrections.
Electronic properties, including natural electron configurations (NEC) and spin densities, are analyzed using the NBO 5.0 program52 to detail chemical bonding and charge transfer patterns. Additionally, the adaptive natural density partitioning (AdNDP)53 is performed using the Multiwfn program54 to further emphasize bonding characteristics. These methodologies allow us to obtain insights into the electronic and bonding properties of the lowest-energy isomers.
3. Results and discussion
3.1. Geometric structures
As for a convention, the notation x.y.z is used to specify the Ptn+/0/– isomers considered. Accordingly, x represents the charge state with c for cationic, a for anionic and n for neutral isomers. While y indicates the number of Pt atoms, z denotes the relative energy ranking, with z = 1, 2, 3, etc., corresponding to isomers of increasing relative energy. For instance, c.3.1 refers to the most stable isomer of the Pt3+ cation. Relative energies given in the following sections are determined from B3PW91/Def2-TZVP + ZPE computations. Isomers having an energy difference within 2.3 kcal mol−1 or 0.1 eV are considered as nearly degenerate. This threshold is smaller than the expected error margin of ±7 kcal mol−1 (∼0.3 eV) commonly associated with energetic parameters obtained from DFT computations.55,56For the Ptn+/0/– clusters with sizes ranging from n = 3 to 21, the potential energy surface for each size exhibits a large number of local minima and a variety of nearly degenerate isomers across different geometric configurations and spin states. Therefore, we focus exclusively only on the low-lying isomers, highlighting those with relative energies lying within ∼12 kcal mol−1 (∼0.5 eV) from the most stable configuration. The structures, spin states, and relative energies of the neutral Ptn clusters, obtained from B3PW91/Def2-TZVP computations, are presented in Fig. 1–8. Additional details, including data across all three charge states and results from both B3PW91/Def2-TZVP and B3PW91/aug-cc-pVTZ-PP calculations, are provided in Fig. S1–S8 of the (ESI)† file.
In general, results obtained from both sets of B3PW91/Def2-TZVP and B3PW91/aug-cc-pVTZ-PP calculations show no significant differences. However, the B3PW91/aug-cc-pVTZ-PP calculations require considerably more computational time and are more challenging to converge for both SCF and geometry optimization as the system size increases. Consequently, for a heavy transition metal cluster system like Ptn, the Def2-TZVP basis set offers a more practical and effective balance between computational cost and accuracy as compared to the augmented correlation consistent aug-cc-pVTZ-PP basis set.
 | ||
| Fig. 1 Structures, multiplicities (M, in bracket) and relative energies (rE, kcal mol−1) of the low-lying neutral Ptn with n = 3–6 calculated using the B3PW91/D-ef2TZVP + ZPE method. | ||
For neutral Pt5, unlike the structures previously reported by both Chaves et al. and Singh et al.,26,27 Pt5 exhibits an energy degeneracy between an edge-added tetrahedron and a pyramid (cf. Fig. 1), with an energy difference of only 0.7 kcal mol−1. Similarly to its neutral counterpart, the lowest-lying structure of cationic Pt5+ is not the shape of two congruent regular tetrahedra glued together at their bases as reported previously,27 but instead it adopts a pyramidal skeleton.
 | ||
| Fig. 2 Structures, multiplicities (M, in bracket) and relative energies (rE, kcal mol−1) of the low-lying neutral Ptn with n = 7–10 calculated at the B3PW91/Def2-TZVP + ZPE method. | ||
 | ||
| Fig. 3 Structures, multiplicities (M, in bracket) and relative energies (rE, kcal mol−1) of the low-lying neutral Ptn with n = 11–14 calculated at the B3PW91/Def2-TZVP + ZPE method. | ||
 | ||
| Fig. 4 Structures, multiplicities (M, in bracket) and relative energies (rE, kcal mol−1) of the low-lying neutral Ptn with n = 15–16 calculated at the B3PW91/Def2-TZVP + ZPE method. (*) denotes structures reported in ref. 36. | ||
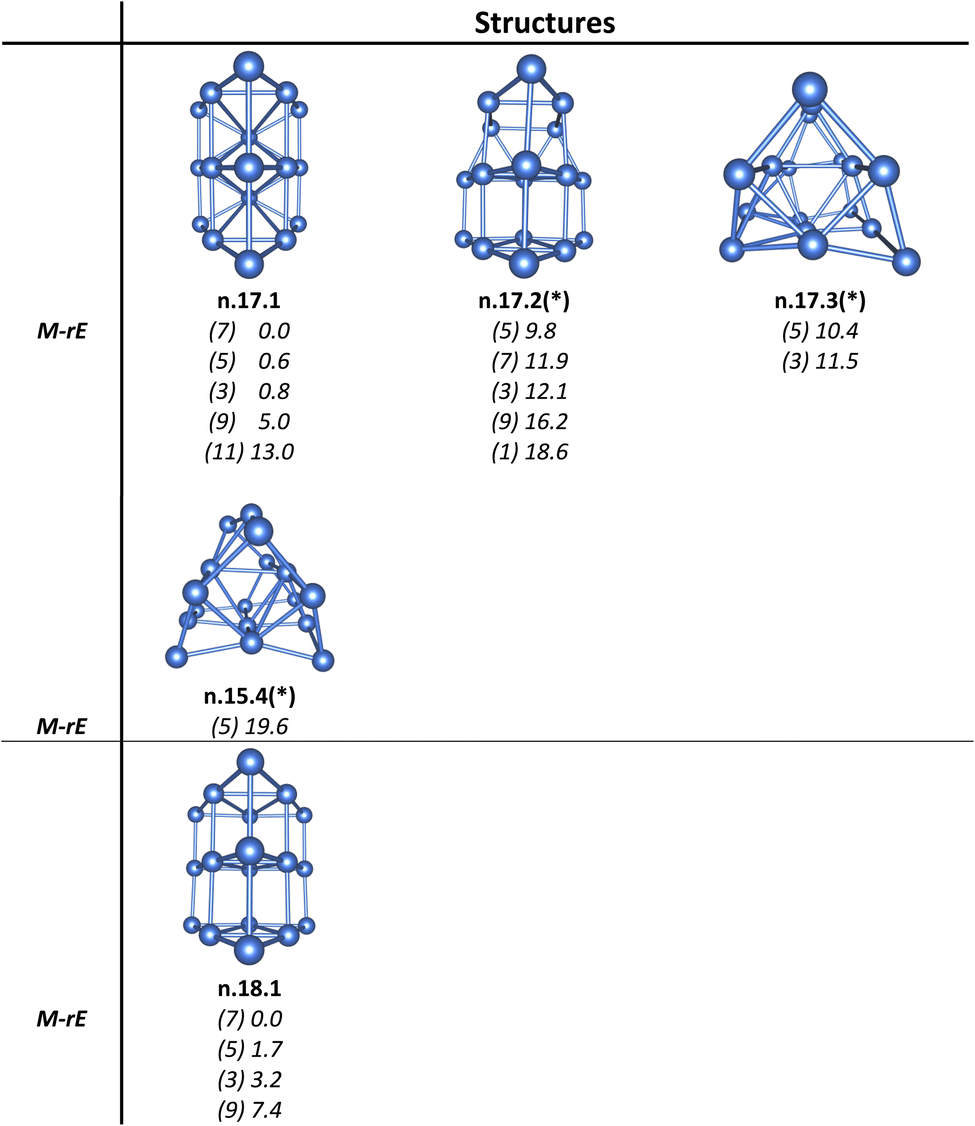 | ||
| Fig. 5 Structures, multiplicities (M, in bracket), and relative energies (rE, kcal mol−1) of the low-lying neutral Ptn with n = 17–18 calculated at the B3PW91/Def2-TZVP + ZPE method. (*) denotes structures reported from ref. 36. | ||
 | ||
| Fig. 6 Structures, multiplicities (M, in bracket), and relative energies (rE, kcal mol−1) of the low-lying neutral Pt19 calculated at the B3PW91/Def2-TZVP + ZPE method. (*) denotes structures reported from ref. 42. | ||
 | ||
| Fig. 7 Structures, multiplicities (M, in bracket), and relative energies (rE, kcal mol−1) of the low-lying neutral Pt20 calculated at the B3PW91/Def2-TZVP + ZPE method. (*) denotes structures reported from ref. 42. | ||
 | ||
| Fig. 8 Structures, multiplicities (M, in bracket), and relative energies (rE, kcal mol−1) of the low-lying neutral Pt21 calculated at the B3PW91/Def2-TZVP + ZPE method. | ||
Accordingly, Pt6+/0/–, Pt10+/0/–, Pt14+/0/– and Pt18+/0/– serve as key structural building blocks in the assembly of various Ptn+/0/– clusters in the medium-to-large size range. These units appear repeatedly as core frameworks that guide the growth and structural evolution of larger clusters (see Fig. 9 for neutral clusters and Fig. S9† for anionic and cationic counterparts).
 | ||
| Fig. 9 Structural evolution of the Ptn clusters with n = 3–21. | ||
3.2. Thermodynamic stability
To evaluate the thermodynamic stability of the clusters considered as well as the influence of sizes and charge states on their stability, the average binding energies (Eb), adiabatic ionization energies (IEa), adiabatic electron affinities (EAa) and second-order energy differences (Δ2E) are employed. In this study, the Eb, IEa, EAa and Δ2E values of the lowest-lying Ptn+/0/– (n = 3–21) structures in all three charge states are calculated using the following formulas (eqn (1)–(8)):❖ Average binding energy (eV)
| For neutral Ptn clusters: Eb(Ptn) = [nE(Pt) – E(Ptn)]/n | (1) |
| For cationic Ptn+ clusters: Eb(Ptn+) = [(n – 1)E(Pt) + E(Pt+) – E(Ptn+)]/n | (2) |
| For anionic Ptn− clusters: Eb(Ptn−) = [(n – 1)E(Pt) + E(Pt−) – E(Ptn−)]/n | (3) |
❖ Adiabatic ionization energy:
| IEa(Ptn) = E(Ptn+) – E(Ptn) | (4) |
❖ Adiabatic electron affinity:
| EAa(Ptn) = E(Ptn) – E(Ptn−) | (5) |
❖ Second-order energy differences:
| For neutral Ptn clusters: Δ2E (Ptn) = E(Ptn+1) + E(Ptn−1) −2E(Ptn) | (6) |
| For cationic Ptn+ clusters: Δ2E (Ptn+) = E(Ptn+1+) + E(Ptn−1+) − 2E(Ptn+) | (7) |
| For anionic Ptn− clusters: Δ2E (Ptn−) = E(Ptn+1−) + E(Ptn−1−) − 2E(Ptn−) | (8) |
The results shown in Fig. 10 highlight the critical role of charge in determining overall stability (see Table S1 in the ESI† file for detailed data). Fig. 10A clearly reveals a stability trend across clusters studied, following this order: Ptn+ > Ptn− > Ptn. Specifically, charged species demonstrate greater stability as compared to their neutral counterparts, with cationic states being the most stable.
 | ||
| Fig. 10 (A): Average binding energies of Ptn+/0/– clusters considered. (B): Adiabatic ionization energies of Ptn clusters, (C): Adiabatic electron affinities of Ptn clusters and (D): Second-order energy difference of Ptn+/0/– clusters with n = 3–21. Values are obtained from B3PW91/Def2-TZVP + ZPE computations. | ||
The adiabatic ionization energies IEa and the adiabatic electron affinities EAa illustrated in Fig. 10B and C further highlight the size- and charge-dependent trends. While the IEa decreases with increasing cluster size, indicating that larger clusters facilitate easier electron detachment, the EAa shows an increasing trend with respect to cluster size, signifying that electron acceptance becomes progressively more favorable as the clusters grow. The electron affinities of the sizes n = 20 and 21 amount to ∼3.4 eV making them quasi-super halogen species.
This stability's preference of charged clusters over the neutral counterpart is also shown via the dissociation energies De(x) in Table 1, where x represents the fragmentation channels De(1) to De(5). The formulae for these channels are outlined below:
| De(1) = E(Ptn−1) + E(Pt) − E(Ptn); |
| De(2) = E(Ptn−1) + E(Pt−) − E(Ptn−); De(3) = E(Ptn−1−) + E(Pt) − E(Ptn−); |
| De(4) = E(Ptn−1) + E(Pt+) − E(Ptn+); De(5) = E(Ptn−1+) + E(Pt) − E(Ptn+) |
| n | De(1) | De(2) | De(3) | De(4) | De(5) |
|---|---|---|---|---|---|
| 2 | 2.76 | 2.87 | 2.87 | 2.88 | 2.88 |
| 3 | 3.01 | 3.24 | 3.13 | 4.43 | 4.31 |
| 4 | 3.02 | 3.52 | 3.28 | 5.62 | 4.19 |
| 5 | 2.97 | 3.79 | 3.30 | 5.19 | 2.60 |
| 6 | 3.30 | 4.51 | 3.69 | 5.62 | 3.41 |
| 7 | 3.35 | 4.54 | 3.33 | 5.58 | 3.26 |
| 8 | 3.52 | 4.81 | 3.63 | 5.51 | 3.28 |
| 9 | 3.69 | 5.04 | 3.75 | 5.87 | 3.89 |
| 10 | 4.13 | 5.23 | 3.89 | 6.17 | 4.00 |
| 11 | 3.20 | 4.49 | 3.38 | 5.47 | 3.43 |
| 12 | 3.67 | 4.84 | 3.55 | 6.21 | 3.93 |
| 13 | 3.65 | 4.94 | 3.77 | 6.25 | 3.72 |
| 14 | 3.55 | 4.94 | 3.66 | 6.15 | 3.55 |
| 15 | 3.60 | 5.00 | 3.61 | 6.26 | 3.66 |
| 16 | 3.90 | 5.35 | 3.94 | 6.56 | 3.89 |
| 17 | 4.01 | 5.49 | 4.05 | 6.69 | 4.03 |
| 18 | 3.91 | 5.29 | 3.82 | 6.74 | 4.06 |
| 19 | 3.24 | 4.67 | 3.29 | 6.12 | 3.29 |
| 20 | 3.72 | 5.23 | 3.81 | 6.55 | 3.67 |
| 21 | 3.85 | 5.41 | 3.90 | 6.77 | 3.94 |
Ptn+/0/– clusters tend to favor a detachment of a neutral Pt atom over a charged one, indicating a preference for charge retention during dissociation. This behavior aligns with the above discussion on the greater stability of charged clusters compared to their neutral counterparts.
In addition, relative stabilities of clusters are investigated using second-order energy differences (cf. Fig. 10D). As shown in both Fig. 10A and D, the cationic Pt4+ cluster exhibits unusually high stability, which is consistent with previous studies that combined experimental mass spectrometry with calculated second-order energy differences.57 Likewise, Pt10+/– clusters were also reported as particularly stable and even chemically inert.29 In the present study, in addition to the confirmed stability of Pt clusters at the sizes n = 4 and 10, clusters with enhanced stability are also identified at the sizes n = 6 and 18 across all three charge states.
Results presented in Fig. 10D indicate that clusters at sizes n = 4, 6, 10 and 18 exhibit significantly higher thermodynamic stability. Addition or removal of a Pt atom from these magic clusters tend to lead to a marked decrease in stability of the resulting derivatives. Consequently, clusters at sizes n = 5, 7, 9, 11, 15 and 19, with odd numbers of atoms, become considerably less stable (cf. Fig. 10D). Overall, cationic, neutral and anionic Ptn+/0/– clusters are consistently more stable at even-numbered sizes, in part in agreement with previous experiment.29,57
3.3. Formation of multi-layer structures from the planar Pt6 triangle
Although the platinum clusters studied here exhibit relatively uniform charge distributions and low overall polarity (cf. Tables S2–S7†), and thus lack distinct Lewis acidic sites, their metallic bonding and delocalized electronic structures remain effective in facilitating strong interactions and maintaining high electron density on the cluster surfaces.The planar triangular structure of neutral Pt6 at the 7A1 electronic state possesses 60 valence electrons distributed in a set of 42 localized d-electrons and a set of 18 delocalized electrons. The localized electrons reside on individual atoms, with 24 paired and 18 unpaired (cf. Fig. S10†). The 18 delocalized electrons form twelve (2c–1e) bonds and six (6c–1e) bonds, collectively strengthening the interactions among the six Pt atoms and stabilizing the triangular configuration (cf. Fig. 11 and S10†).
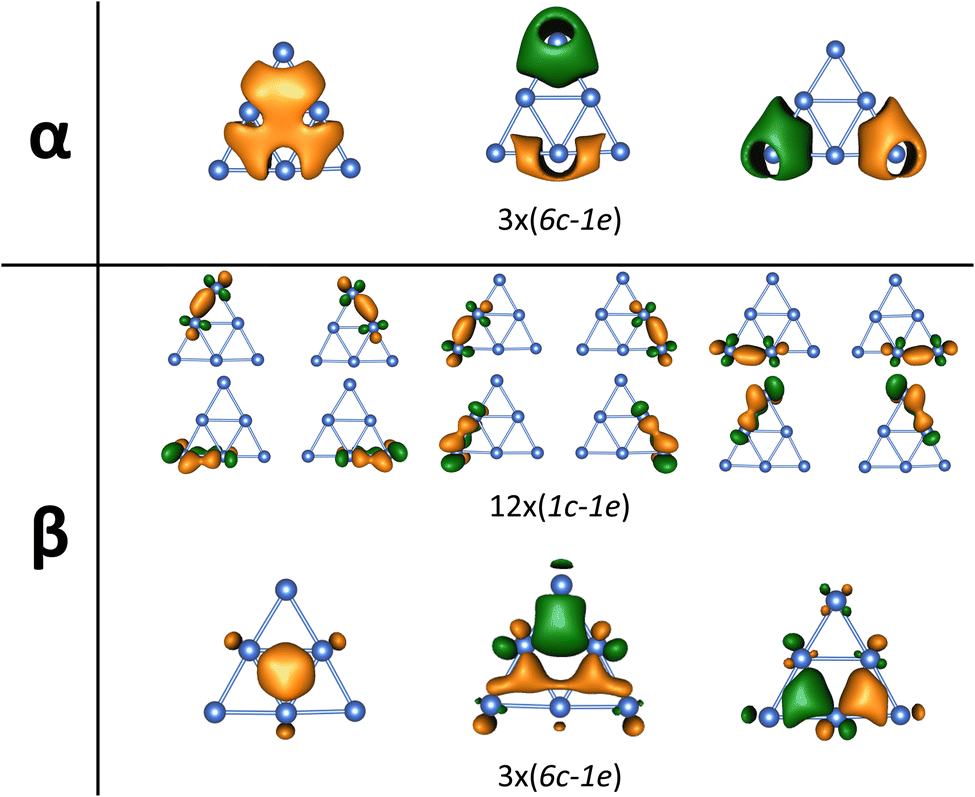 | ||
| Fig. 11 The metallic aromaticity arising from (2c–1e) and (6c–1e) bonds of the triangular Pt6 cluster via AdNDP analysis (B3PW91/Def2-TZVP). | ||
Specifically, the twelve (2c–1e) bonds include six σ bonds and six conjugated π bonds arising from d-orbital overlaps, which tend to strengthen the outer triangular framework. In addition to these two-center bonds, the six (6c–1e) bonds are delocalized across one S-type molecular orbital (S-MO) and two P-type molecular orbitals (P-MOs) in both alpha and beta sides. This distribution reflects a SP2 hybridization which accounts for the planar triangular geometry of Pt6, with three central Pt atoms acting as a unified center of hybridization.
Evidence of a SP2 hybridization is further supported by electron populations that show a decrease of ∼0.2 e on 6S orbitals and an increase of ∼0.4 e on 6P orbitals of the three central Pt atoms (cf. Table S2 for neutral isomers and Tables S3 and S4† for charged isomers). Together, these delocalized bonds lead to an aromatic character in the Pt6 triangle.
A structural motif can be constructed by stacking multiple layers of the planar Pt6 triangle (cf. Scheme 3). Following such an assembling, the triangular Pt6 units can evolve into tubular structures forming larger clusters including Pt12, Pt18 and Pt24. These structures are energetically favorable, representing local minima on the potential energy surface. The repeated stacking along a longitudinal axis suggests a viable pathway for constructing one-dimensional, tube-like architectures. This approach offers an effective strategy for designing larger, more complex Pt-based nanostructures, facilitating the creation of Pt tubes with promising electronic and catalytic properties.
 | ||
| Scheme 3 Growth into tubular structures through the stacking of multiple planar Pt6 triangles or connecting prismatic Pt6 by sharing their base and side faces. | ||
The larger clusters of Pt12, Pt18, and Pt24 can also be regarded as being assembled from prismatic Pt6 units, interconnected by sharing their base and side faces (Scheme 3). Each prismatic Pt6 unit features twelve delocalized bonds, comprising nine (2c–1e) bonds and three (6c–1e) bonds that span across the edges and faces of the prism (cf. Fig. 12 and S11†). These bonds not only secure the six atoms within each Pt6 unit but also facilitate robust connections between adjacent Pt6 units, further contributing to the structural integrity and thermodynamic stability of the larger Pt12, Pt18, and Pt24 tubular clusters.
 | ||
| Fig. 12 The metal aromaticity arising from (2c–1e) and (6c–1e) bonds of the prismatic Pt6 cluster via AdNDP analysis (B3PW91/Def2-TZVP). | ||
The coordination numbers of Pt atoms in the larger Pt12, Pt18, and Pt24 clusters are higher than the ones in a single triangular Pt6. Correspondingly, the Wiberg bond indices for the vertex Pt atoms in the Pt6 layers of the larger clusters amount to ∼2.5, while those for the central Pt atoms are ∼3 (cf. Table S5–S7†). In comparison, Wiberg bond indices for the vertex and central Pt atoms in the single Pt6 triangular cluster amount to ∼2 and ∼2.5, respectively (cf. Table S2†).
Furthermore, the density of states (DOS) maps shown in Fig. 13–15 reveal notable differences in the DOS profiles and the contributions from s, p, and d orbitals between the single Pt6 triangular cluster and the Pt6 layers within the Pt12, Pt18 and Pt24 clusters, irrespective of whether the layers are positioned at the outer or middle region. Specifically, in the single Pt6 triangle, the d-orbital contributions are more sharply defined and localized within specific regions. Conversely, in the Pt6 layers of the larger Pt12, Pt18 and Pt24, the d-orbital contributions exhibit a more balanced distribution. Additionally, the s and p orbital contributions are enhanced in the Pt6 layers of the larger clusters as compared to the single Pt6 cluster.
 | ||
| Fig. 13 Calculated density of states (DOS) for (A) n.6.3 isomer at 7A state and (B) the Pt6 layer (highlighted in pink) within n.12.3 isomer at 3A state. Positive and negative DOS represent spin-up (alpha) and spin-down (beta) electrons, respectively. | ||
 | ||
| Fig. 14 Calculated density of states (DOS) for (A) the outer Pt6 layer (highlighted in pink) and (B) the middle Pt6 layer (highlighted in pink) within the n.18.1 isomer at 7A state. Positive and negative DOS represent spin-up (alpha) and spin-down (beta) electrons, respectively. | ||
 | ||
| Fig. 15 Calculated density of states (DOS) for (A) the outer Pt6 layer (highlighted in pink) and (B) the middle Pt6 layer (highlighted in pink) within the n.24.1 isomer at 13A state. Positive and negative DOS represent spin-up (alpha) and spin-down (beta) electrons, respectively. | ||
The latter change in electron contribution is further evidenced in the natural electron configurations, which reveal a significant increase in the 6S and 6P electrons and a decrease in the 5d electrons for the Pt atoms in the larger clusters compared to those in the single Pt6 triangular cluster (cf. Tables S2 and S5–S7†). Such an electron redistribution indicates an increased hybridization between the s, p, and d orbitals, contributing to stronger bonding interactions and greater electron delocalization.
As a result, the combined effect of increased bond coordination and altered electron contribution leads to stronger bonding and effective structural construction in larger Pt12, Pt18, and Pt24 clusters.
4. Concluding remarks
In this theoretical study, the pure platinum clusters Ptn+/0/– with n = 3–21 are reinvestigated using DFT calculations with the B3PW91 functional in conjunction with both aug-cc-pVTZ-PP and Def2-TZVP basis sets. Computed findings yield several key conclusions:❖ A comprehensive and updated depiction of the structures of cationic, neutral and anionic Ptn+/0/– clusters is presented, along with the identification of newly discovered lowest-lying isomers across a broad range of sizes from n = 3 to n = 21. In general, the structures of Ptn+/0/– clusters in the small-to-medium size range are primarily derived from key building blocks, including Pt6+/0/–, Pt10+/0/–, Pt14+/0/–, and Pt18+/0/– clusters.
❖ Clusters with even number of atoms tend to be thermodynamically more stable than their odd number counterparts. The sizes Pt4, Pt6, Pt10 and Pt18 emerge as magic clusters, in part in agreement with experimental mass spectrometry.
❖ Charged clusters exhibit greater stability than their neutral counterparts with respect to atom detachment, with the cationic state being the most stable.
❖ Tubular structures of Pt12, Pt18, and Pt24 clusters are constructed by either stacking planar Pt6 triangles or assembling prismatic Pt6 units interconnected through shared base and side faces. These tubular configurations are energetically favorable and offer an appropriate strategy for designing complex Pt-based structures such as nanowires.
❖ Both triangular and prismatic Pt6 clusters exhibit a metallic aromaticity characterized by effective electron delocalization, facilitating bonding interactions:
• Pt6 triangle: this form features six σ and six conjugated π bonds arising from (2c–1e) delocalization and six (6c–1e) bonds reflecting a fully SP2 hybridization and accounting for its geometry.
• Pt6 prism: this form possesses three (6c–1e) bonds and nine σ bonds from (2c–1e) delocalization, spanning across edges and faces of the prism.
❖ In larger Pt12, Pt18 and Pt24 tubes, the combined effect of increased bond coordination on Pt atoms and redistributed electron contribution among s, p and d orbitals tend to strengthen bonding interactions and contribute to effective structural assembly.
❖ Last but not least, concerning the methods, the Def2-TZVP basis set offers a more practical and efficient balance between computational cost and accuracy as compared to the correlation consistent aug-cc-pVTZ-PP basis set.
Data availability
The quantum chemical program Gaussian 16 has been used. Reference of this program is given in the list of references. The additional datasets supporting this article are given as part of the ESI† file which includes the Cartesian coordinates of the optimized geometries, information on the electronic structures, density of states maps of the clusters considered.Conflicts of interest
There are no conflicts to declare.Acknowledgements
BNNH and MTN are grateful to Van Lang University. PHMP acknowledges Ho Chi Minh City University of Technology (HCMUT), VNU-HCM for supporting this study.References
- T. Imaoka, Y. Akanuma, N. Haruta, S. Tsuchiya, K. Ishihara, T. Okayasu, W.-J. Chun, M. Takahashi and K. Yamamoto, Platinum clusters with precise numbers of atoms for preparative-scale catalysis, Nat. Commun., 2017, 8(1), 688 CrossRef.
- P. L. Rodríguez-Kessler, A. Muñoz-Castro, P. A. Alonso-Dávila, F. Aguilera-Granja and A. R. Rodríguez-Domínguez, Structural, electronic and catalytic properties of bimetallic PtnAgn (n = 1–7) clusters, J. Alloys Compd., 2020, 845, 155897 CrossRef.
- P. L. Rodríguez-Kessler, A. R. Rodríguez-Domínguez, P. Alonso-Dávila, P. Navarro-Santos and A. Muñoz-Castro, Structural and electronic properties for Be-doped Ptn (n = 1–12) clusters obtained by DFT calculations, Phys. Chem. Chem. Phys., 2022, 24(13), 7856–7861 RSC.
- P. Ferrari, J. Vanbuel, N. M. Tam, M. T. Nguyen, S. Gewinner, W. Schöllkopf, A. Fielicke and E. Janssens, Effects of Charge Transfer on the Adsorption of CO on Small Molybdenum-Doped Platinum Clusters, Chem. A Eur. J., 2017, 23(17), 4120–4127 CrossRef CAS.
- C. Schmitt, N. Da Roit, M. Neumaier, C. B. Maliakkal, D. Wang, T. Henrich, C. Kübel, M. Kappes and S. Behrens, Continuous flow synthesis of atom-precise platinum clusters, Nanoscale Adv., 2024, 6(9), 2459–2468 RSC.
- S. Zhu, X. Wang, E. Luo, L. Yang, Y. Chu, L. Gao, Z. Jin, C. Liu, J. Ge and W. Xing, Stabilized Pt Cluster-Based Catalysts Used as Low-Loading Cathode in Proton-Exchange Membrane Fuel Cells, ACS Energy Lett., 2020, 5(9), 3021–3028 CrossRef CAS.
- R. Wang, D. Li, S. Maurya, Y. S. Kim, Y. Wu, Y. Liu, D. Strmcnik, N. M. Markovic and V. R. Stamenkovic, Ultrafine Pt cluster and RuO2 heterojunction anode catalysts designed for ultra-low Pt-loading anion exchange membrane fuel cells, Nanoscale Horiz., 2020, 5(2), 316–324 RSC.
- J. Niu, Y. Wang, E. S. Liland, K. S. Regli, J. Yang, K. R. Rout, J. Luo, M. Rønning, J. Ran and D. Chen, Unraveling Enhanced Activity, Selectivity, and Coke Resistance of Pt–Ni Bimetallic Clusters in Dry Reforming, ACS Catal., 2021, 11(4), 2398–2411 CrossRef CAS.
- A. Gamal, K. Eid and A. M. Abdullah, Engineering of Pt-based nanostructures for efficient dry (CO2) reforming: Strategy and mechanism for rich-hydrogen production, Int. J. Hydrogen Energy, 2022, 47(9), 5901–5928 CrossRef CAS.
- Y. Li, K.-B. Low, A. Sundermann, H. Zhu, L. E. Betancourt, C. Kang, S. Johnson, S. Xie and F. Liu, Understanding the nature of Pt-Rh synergy for three-way conversion catalysis, Appl. Catal., B, 2023, 334, 122821 CrossRef CAS.
- J. Kim, H. Kim, S. Kim, J.-H. Jang, H. Sohn, S. J. Hong, J. Kim, G. H. Han, S. Choe, Y.-E. Sung, S. Y. Kim, H. W. Jang, T. H. Jo, H.-K. Lim, S. J. Yoo and S. H. Ahn, Atomic Pt clusters on Au dendrite for formic acid oxidation, Chem. Eng. J., 2023, 451, 138664 CrossRef CAS.
- M. Taleblou, M. F. Camellone, S. Fabris and S. Piccinin, Oxidation of Gas-Phase and Supported Pt Nanoclusters: An Ab Initio Investigation, J. Phys. Chem. C, 2022, 126(26), 10880–10888 CrossRef CAS.
- T. Imaoka, H. Kitazawa, W.-J. Chun, S. Omura, K. Albrecht and K. Yamamoto, Magic Number Pt13 and Misshapen Pt12 Clusters: Which One is the Better Catalyst?, J. Am. Chem. Soc., 2013, 135(35), 13089–13095 CrossRef CAS.
- G. Shu, Y. Lin, S. Wang, S. Zhang, L. Fan, C. Wang, C. Zhou, L. Song, L. Zheng, J. Zhang, K. Ma and H. Yue, Dynamic Metal–Support Interaction-Activated Sub-Nanometer Pt Clusters on FeOx Supports for Aqueous Phase Reforming and Hydrogenolysis of Glycerol, ACS Catal., 2023, 13(13), 8423–8436 CrossRef CAS.
- C. Especel, G. Lafaye and F. Epron, Bimetallic Catalysts for Sustainable Chemistry: Surface Redox Reactions For Tuning The Catalytic Surface Composition, ChemCatChem, 2023, 15(3), e202201478 CrossRef CAS.
- J. Depciuch, M. Stec, B. Klebowski, A. Maximenko, E. Drzymała, J. Baran and M. Parlinska-Wojtan, Size effect of platinum nanoparticles in simulated anticancer photothermal therapy, Photodiagn. Photodyn. Ther., 2020, 29, 101594 CrossRef CAS PubMed.
- A. Abed, M. Derakhshan, M. Karimi, M. Shirazinia, M. Mahjoubin-Tehran, M. Homayonfal, M. R. Hamblin, S. A. Mirzaei, H. Soleimanpour, S. Dehghani, F. F. Dehkordi and H. Mirzaei, Platinum Nanoparticles in Biomedicine: Preparation, Anti-Cancer Activity, and Drug Delivery Vehicles, Front. pharmacol., 2022, 13, 797804 CrossRef CAS.
- C. Zhang, C. Xu, X. Gao and Q. Yao, Platinum-based drugs for cancer therapy and anti-tumor strategies, Theranostics, 2022, 12(5), 2115–2132 CrossRef CAS.
- D. Dai and K. Balasubramanian, Potential energy surfaces for platinum (Pt3)+ hydrogen atom and palladium (Pd3)+ hydrogen atom interactions, J. Phys. Chem., 1992, 96, 3279–3282 CrossRef CAS.
- V. Kumar and Y. Kawazoe, Evolution of atomic and electronic structure of Pt clusters: Planar, layered, pyramidal, cage, cubic, and octahedral growth, Phys. Rev. B, 2008, 77(20), 205418 CrossRef.
- A. Nie, J. Wu, C. Zhou, S. Yao, C. Luo, R. C. Forrey and H. Cheng, Structural evolution of subnano platinum clusters, Int. J. Quantum Chem., 2007, 107(1), 219–224 CrossRef CAS.
- L. Xiao and L. Wang, Structures of Platinum Clusters: Planar or Spherical?, J. Phys. Chem. A, 2004, 108(41), 8605–8614 CrossRef CAS.
- V. Fung and D.-e. Jiang, Exploring Structural Diversity and Fluxionality of Ptn (n = 10–13) Clusters from First-Principles, J. Phys. Chem. C, 2017, 121(20), 10796–10802 CrossRef CAS.
- R. Li, M. Odunlami and P. Carbonnière, Low-lying Ptn cluster structures (n = 6–10) from global optimizations based on DFT potential energy surfaces: sensitivity of the chemical ordering with the functional, Comput. Theor. Chem., 2017, 1107, 136–141 CrossRef CAS.
- A. S. Chaves, M. J. Piotrowski and J. L. F. Da Silva, Evolution of the structural, energetic, and electronic properties of the 3d, 4d, and 5d transition-metal clusters (30 TMn systems for n = 2–15): a density functional theory investigation, Phys. Chem. Chem. Phys., 2017, 19(23), 15484–15502 RSC.
- N. B. Singh and U. Sarkar, Structure, vibrational, and optical properties of platinum cluster: a density functional theory approach, J. Mol. Model., 2014, 20(12), 2537 CrossRef PubMed.
- A. S. Chaves, G. G. Rondina, M. J. Piotrowski, P. Tereshchuk and J. L. F. Da Silva, The Role of Charge States in the Atomic Structure of Cun and Ptn (n = 2–14 atoms) Clusters: A DFT Investigation, J. Phys. Chem. A, 2014, 118(45), 10813–10821 CrossRef CAS.
- D. Bumüller, A. G. Yohannes, S. Kohaut, I. Kondov, M. M. Kappes, K. Fink and D. Schooss, Structures of Small Platinum Cluster Anions Ptn–: Experiment and Theory, J. Phys. Chem. A, 2022, 126(22), 3502–3510 CrossRef PubMed.
- Y. Jia, X. Yu, H. Zhang, L. Cheng and Z. Luo, Tetrahedral Pt10– Cluster with Unique Beta Aromaticity and Superatomic Feature in Mimicking Methane, J. Phys. Chem. Lett., 2021, 12(21), 5115–5122 CrossRef CAS PubMed.
- P. L. Rodríguez-Kessler, A. R. Rodríguez-Domínguez and A. Muñoz-Castro, Systematic cluster growth: a structure search method for transition metal clusters, Phys. Chem. Chem. Phys., 2021, 23(8), 4935–4943 RSC.
- K. Bhattacharyya and C. Majumder, Growth pattern and bonding trends in Ptn (n = 2–13) clusters: Theoretical investigation based on first principle calculations, Chem. Phys. Lett., 2007, 446(4), 374–379 CrossRef CAS.
- B.-N. Nguyen-Ha, C.-T. Phan Dang, L. Van Duong, M. P. Pham-Ho, M. T. Nguyen and N. M. Tam, Formation of pyramidal structures through mixing gold and platinum atoms: the AuxPty2+ clusters with x + y = 10, RSC Adv., 2023, 13(47), 32893–32903 RSC.
- G.-F. Wei and Z.-P. Liu, Subnano Pt Particles from a First-Principles Stochastic Surface Walking Global Search, J. Chem. Theory Comput., 2016, 12(9), 4698–4706 CrossRef CAS PubMed.
- X. Wang and D. Tian, Structures and structural evolution of Ptn (n = 15–24) clusters with combined density functional and genetic algorithm methods, Comput. Mater. Sci., 2009, 46(1), 239–244 CrossRef CAS.
- P. L. Rodríguez-Kessler, A. Muñoz-Castro, A. R. Rodríguez-Domínguez and J. L. Cabellos, Structure effects of Pt15 clusters for the oxygen reduction reaction: first-principles calculations, Phys. Chem. Chem. Phys., 2023, 25(6), 4764–4772 RSC.
- J. M. Guevara-Vela, T. Rocha-Rinza, P. L. Rodríguez-Kessler and A. Muñoz-Castro, On the structure and electronic properties of Ptn clusters: new most stable structures for n = 16–17, Phys. Chem. Chem. Phys., 2023, 25(42), 28835–28840 RSC.
- S. K. Ignatov, A. G. Razuvaev, A. S. Loginova and A. E. Masunov, Global Structure Optimization of Pt Clusters Based on the Modified Empirical Potentials, Calibrated using Density Functional Theory, J. Phys. Chem. C, 2019, 123(47), 29024–29036 CrossRef CAS.
- P. L. Rodríguez-Kessler and A. R. Rodríguez-Domínguez, Size and structure effects of PtN (N = 12−13) clusters for the oxygen reduction reaction: first-principles calculations, J. Chem. Phys., 2015, 143(18), 184312 CrossRef PubMed.
- N. Watari and S. Ohnishi, Atomic and electronic structures of Pd13 and Pt13 clusters, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58(3), 1665–1677 CrossRef CAS.
- E. Aprà and A. Fortunelli, Density-Functional Calculations on Platinum Nanoclusters: Pt13, Pt38, and Pt55, J. Phys. Chem. A, 2003, 107(16), 2934–2942 CrossRef.
- C. M. Chang and M. Y. Chou, Alternative Low-Symmetry Structure for 13-Atom Metal Clusters, Phys. Rev. Lett., 2004, 93(13), 133401 CrossRef CAS.
- J. M. Guevara-Vela, M. Gallegos, T. Rocha-Rinza, Á. Muñoz-Castro, P. L. R. Kessler and Á. Martín Pendás, New global minimum conformers for the Pt19 and Pt20 clusters: low symmetric species featuring different active sites, J. Mol. Model., 2024, 30(9), 310 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda; J. Hasegawa; M. Ishida; T. Nakajima; Y. Honda; O. Kitao; H. Nakai; T. Vreven; K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers; K. N. Kudin, V. N. Staroverov, T. A. Keith; R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell; J. C. Burant, S. S. Iyengar, J. Tomasi; M. Cossi, J. M. Millam, M. Klene, C. Adamo; R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- D. Figgen, K. A. Peterson, M. Dolg and H. Stoll, Energy-consistent pseudopotentials and correlation consistent basis sets for the 5d elements Hf–Pt, J. Chem. Phys., 2009, 130(16), 164108 CrossRef PubMed.
- T. H. Dunning and P. J. Hay, Gaussian Basis Sets for Molecular Calculations, in Methods of Electronic Structure Theory, ed. Schaefer, H. F., Springer US, Boston, MA, 1977; pp pp 1–27 Search PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7(18), 3297–3305 RSC.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- J. P. Perdew, P. Ziesche and H. Eschrig, Electronic Structure of Solids' 91, Akademie Verlag, Berlin, 1991 Search PubMed.
- P. J. Hay and W. R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals, J. Chem. Phys., 1985, 82(1), 299–310 CrossRef CAS.
- H. T. Pham, L. V. Duong, B. Q. Pham and M. T. Nguyen, The 2D-to-3D geometry hopping in small boron clusters: The charge effect, Chem. Phys. Lett., 2013, 577, 32–37 CrossRef CAS.
- M. Saunders, Stochastic search for isomers on a quantum mechanical surface, J. Comput. Chem., 2004, 25(5), 621–626 CrossRef CAS.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO, version 5.0, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2001 Search PubMed.
- D. Y. Zubarev and A. I. Boldyrev, Developing paradigms of chemical bonding: adaptive natural density partitioning, Phys. Chem. Chem. Phys., 2008, 10(34), 5207–5217 RSC.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS.
- E. J. Baerends, V. Branchadell and M. Sodupe, Atomic reference energies for density functional calculations, Chem. Phys. Lett., 1997, 265(3), 481–489 CrossRef CAS.
- S. Pittalis, S. Kurth and E. K. U. Gross, On the degeneracy of atomic states within exact-exchange (spin-) density functional theory, J. Chem. Phys., 2006, 125(8), 084105 CrossRef CAS PubMed.
- P. Ferrari, K. Hansen, P. Lievens and E. Janssens, Stability of small cationic platinum clusters, Phys. Chem. Chem. Phys., 2018, 20(46), 29085–29090 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: (i) The structures, multiplicities and relative energies (rE, kcal mol−1) of the of the low-lying Ptn+/0/− (n = 3–21) calculated at the B3PW91/ aug-cc-pVTZ-PP + ZPE and B3PW91/ Def2-TZVP + ZPE methods; (ii) structural evolution of anionic and cationic Ptn−/+ clusters (n = 3–21); (iii) AdNDP analysis showing muti-center bonds in the n.6.1 and n.6.3 isomers; (iv) detailed values of the average binding energy of Ptn+/0/− clusters (n = 1–21), as well as adiabatic ionization energy and adiabatic electron affinity of Ptn clusters (n = 1–21); (v) natural electron configuration (NEC), natural charge and Wiberg bond index of low-lying isomers of Pt6+/0/−, Pt12, Pt18 and Pt24, (vi) Coordinates of the lowest-lying Ptn+/0/− clusters (n = 3–21). See DOI: https://doi.org/10.1039/d5ra02668g |
| This journal is © The Royal Society of Chemistry 2025 |