
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A mini review on aromatization of n-alkanes
Hongqi
Wang
 and
N. Raveendran
Shiju
*
and
N. Raveendran
Shiju
*
Catalysis Engineering Group, Van't Hoff Institute for Molecular Sciences, University of Amsterdam, 1090 GD Amsterdam, The Netherlands. E-mail: h.wang4@uva.nl; n.r.shiju@uva.nl
First published on 24th October 2024
Abstract
The catalytic aromatization of n-alkanes is an important process in the chemical industry, especially for the production of value-added aromatics from the abundant and unreactive small alkanes. This mini review summarizes the recent progress on the development of catalysts for the aromatization of n-alkanes and the mechanistic studies. The effects of various catalysts (e.g. shape selective zeolites and noble metals) and reactant compositions on the aromatization performance are discussed to shed light on the rational design of novel heterogeneous catalysts.
1. Introduction
The catalytic aromatization of n-alkanes is an important process to upgrade the low-cost feedstock into value-added aromatic chemicals such as benzene, toluene and xylene (BTX) and to improve the octane number of gasoline.1–3 The abundant alkanes could be acquired from the traditional fossil fuels (e.g. crude oil and natural gas, especially shale gas), as well as from recycled sources (e.g. chemical degradation of plastic waste). Catalytic aromatization is one of the most promising methods to valorize these alkanes.4 The produced aromatic compounds, especially BTX, are crucial building blocks in the chemical industry, and currently BTX is mainly produced from naphtha reforming.5 Another advantage of alkane aromatization, especially for utilizing methane and ethane, is the easier transportation of the liquid-form aromatic compounds than the feedstock gases.Alkane aromatization is generally composed of complicated multi-step reactions in parallel or series over a catalyst, including cracking, dehydrogenation, isomerization, oligomerization and cyclization reactions.6 Various parameters, including the reactant composition, catalyst and operating parameters (e.g. temperature and pressure), will affect the reaction routes and thus the product distribution and yield. Among these factors, the reactant and catalyst are of paramount importance and would be discussed in this review. Among the reactants, the aromatization of CH4 and C2H6 is still in the laboratory developmental stage. However, for C3+ alkanes, some aromatization processes are commercial, such as Cyclar (UOP and BP) and M2-reforming (Mobil). The Zeolite Socony Mobil-5 (ZSM-5) zeolite and its various modifications (e.g. gallium modified) are effectively utilized in these processes due to their shape selectivity and ability to inhibit coke formation.7 However, almost all the aromatization processes are limited by their low yield because of side reactions and catalyst deactivation due to coke formation.8 Various new catalysts are developed to overcome these bottleneck problems by improving the activity, selectivity and long-term stability.
While there are some reviews focusing on certain alkanes (e.g. naphtha3) or catalysts (e.g. ZSM-5 (ref. 9)), a generalized mechanism is necessary to understand the interaction between alkanes and catalysts. This review summarizes the recent advances of the catalyst development and discusses the effects of the reactant composition during the catalytic aromatization of linear alkanes, aiming to provide readers a comprehensive insight into the generalized mechanism of alkane aromatization (especially the importance of an unsaturated intermediate pool) and to give a perspective about future direction. Firstly, zeolite catalysts together with their modifications, noble metals and other types of catalysts are summarized. Then the effects of reactants including single alkanes, co-aromatization and process coupling with oxidation are also discussed.
2. Catalysts
2.1. Zeolites
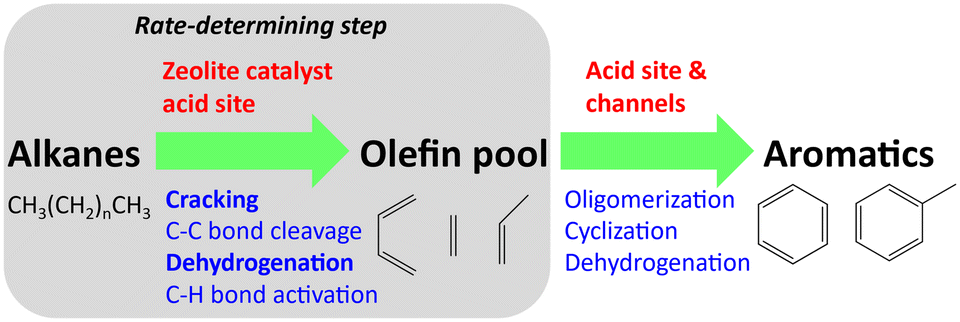 | ||
| Fig. 1 Generalized steps for the catalytic aromatization of alkanes on zeolite catalysts. | ||
 | ||
| Fig. 2 Cracking and dehydrogenation rates at different Ga/Al ratios (this figure is reproduced from ref. 19 with permission). | ||
The nature and dispersion of the metal modifier have an impact on the activity, which could be tuned by preparation and pretreatment methods. For example, it is reported that Zn2+ on H-BEA zeolite prepared by a reaction with zinc vapor has higher activity than ZnO/H-BEA synthesized by hydrolysis of zinc–organic species during propane aromatization.23 However, in another study, the opposite conclusion was reached indicating that subnanometric ZnO clusters on H-ZSM-5 might have higher selectivity than Zn2+ ions.24 This contradictory result might be caused by a support effect. For example, a Zn/SiO2@ZSM-5 core–shell catalyst synthesized by a hydrothermal coating method would lead to higher alkane conversion and aromatic selectivity than the conventional Zn/ZSM-5 during the co-aromatization of methane and propane.25 Moreover, H2-chemisorbed Zn species could significantly improve the aromatic selectivity to 30% by hydrogen treatment compared to 11% of H-ZSM-5 during pentane aromatization.26 Additionally, it is found that a Zn/ZSM-5 microsphere catalyst with a hierarchical structure exhibited improved stability compared to the conventional microporous Zn/ZSM-5 because of the presence of intergranular mesopores which could enhance the molecular transport and resistance to coke formation.27
Furthermore, Zn and Pt are commonly used as promoters for alkane reforming to achieve high BTX yields due to the incorporation of Zn into the framework of ZSM-5 and the enhanced metal dispersion by Pt.1,5,28 It is reported that Zn, as a promoter, can increase the aromatic selectivity.29 Similarly, Pt could enhance the catalytic conversion of ethane because of the strengthened reducibility of Ga species on ZSM-5 (ref. 30) and the synergistic interaction.31 One disadvantage of Zn/ZSM is the agglomeration and loss of Zn after a certain time on stream even though it could be re-dispersed evenly after regeneration.29,32 Zn/ZSM-5 should be modified with higher thermal stability and activity, such as introducing other metal elements.
Similarly, the Lewis acid sites of Ga modified zeolite promoted the aromatization, and Ga introduced by direct (or in situ) synthesis seemed to have better performance.31,33 The GaH2+ species was highly active for hexane dehydroaromatization over Ga modified H-ZSM-5 and could be acquired by pretreatment using hydrogen.34 The Ga species incorporated in the framework favored the BTX formation than the extra framework ones because of the insignificant shielding of acid sites for the framework ones.35 As for the effects of dispersion, the highly dispersed Ga on hierarchical zeolite nanosheets also contributed to the activation of n-pentane.36 However, the mesopores of desilicated hierarchical Ga/H-ZSM-5 via alkaline treatment decreased the activity and selectivity for propane aromatization apparently because the mesopores lowered the residence time of olefins and inhibited the dehydrogenation which is the rate-determining step.37 The investigation of structure–activity relationships by correlating the catalyst structure and performance could give some insights into the reaction pathway, which also emphasizes the importance of the olefin pool shown in Fig. 1.38 In addition, operando characterization is required to analyse the active sites even though the coke formation could make the analysis difficult.39
Mo-modified zeolite can be used to aromatize small alkanes (e.g. C1–C3).18 The induction period is consistently present when using Mo/ZSM-5.40 This is because Mo carbide as the active species will be formed by carburization at the beginning.41 Mg as a promoter can enhance the catalytic activity and stability of Mo/ZSM-5.42
High Cu loading decreases the acidic strength of the zeolite catalyst, which can maintain the alkane conversion and selectivity to aromatics.13 It is reported that mononuclear Cu is the dominant species at low Cu loading (0.1 wt%), while multinuclear Cu with a Cu–O–Cu bridge is the main species at higher Cu loading (1.4 wt%).43
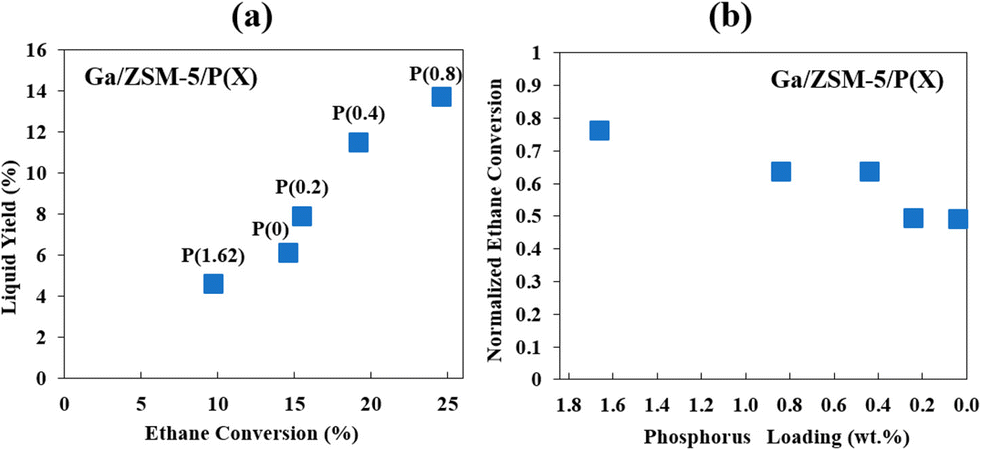 | ||
| Fig. 3 Effects of phosphorus (wt%) on the yield of liquid aromatics (a) and ethane conversion (b) (this figure is reproduced from ref. 45 with permission). | ||
Similar to phosphorus, boron, being near to Al and Si in the periodic table, could also reduce the deactivation rate in a Zn–B/ZSM-5 catalyst during hexane aromatization even though it decreased the catalyst activity initially.46 Boron was incorporated into the framework and reduced the acidity of the Brønsted acid sites, which could further limit the conversion of coke precursors on these sites.
2.2. Noble metals
Noble metal catalysts such as platinum and palladium47 are generally featured with high activity and are extensively applied on the scale of single atoms, clusters and nanoparticles loaded on various supports such as carbon, zeolite and alumina. Table 1 shows various catalyst combinations and their performance. A single site platinum catalyst on a CeO2 support exhibited high selectivity for dehydrocyclization and aromatization reactions compared to Pt cluster and nanoparticle catalysts during hexane reforming even though its thermal stability degraded after 450 °C.48 Besides the noble metal sizes, the location of the metal atoms on the support could also affect the catalyst performance and therefore the product distribution. For example, the Pt nanoparticles inside the zeolite channels led to higher selectivity to aromatics than those on the external surface because the Pt sites inside the pores would favor the cyclization.49 However, the Pt particles inside the channels might relocate to the outside during the reaction and thus lead to deactivation.50 Besides the relocation of Pt particles, the higher cost of noble metals than Ga and Zn should also be taken into consideration. The development of single atom noble metal catalysts may help to decrease the cost due to the maximum utilisation of noble metals.48| Noble metal | Support | Reactant | Conditions | Conversion/selectivity | Key findings | Ref. |
|---|---|---|---|---|---|---|
| Pd | C | n-Octane | 500 °C, WHSV = 2 h−1, hydrogen to hydrocarbon molar ratio = 2 | Conversion = 54.4 wt%; aromatic selectivity = 58 wt% | Pd/C has higher selectivity to xylenes | 47 |
| Pt | CeO2 | n-Hexane | 500 °C | Conversion = 5%; aromatization selectivity = 40% | The single-site catalyst Pt/CeO2 shows higher selectivity to dehydrocyclization and aromatization than CeO2-supported Pt clusters and nanoparticles | 48 |
| Pt | KL zeolite | n-Heptane | 420 °C, hydrogen/n-heptane molar ratio = 6, WHSV = 0.68 h−1 | Conversion = 88 mol%; aromatic selectivity = 84.3 mol% | The location of Pt nanoparticles in KL zeolites could be controlled by changing the exposure time of the Pt precursor | 49 |
| Pt | KL zeolite | n-Heptane | 500 °C, 1 atm, WHSV = 1 h−1, H2/n-heptane = 6 mol mol−1, Pt loading = 0.6 wt% | Conversion = 63.89 wt%; aromatic selectivity = 58.84 wt% | High dispersion of Pt in KL zeolite could improve the stability | 50 |
| Pt | Desilicated ZSM-5 | Ethane | 550 °C, 2 barg, GHSV = 0.3 h−1 | Conversion = 25%; selectivity = 42% | Pt nanoparticles in desilicated ZSM-5 are near the acid sites, which could increase the activity and selectivity to BTX | 51 |
The catalytic performance of noble metal catalysts is also support-dependent. The hierarchical support structure (e.g. micro-/mesoporous Pt/KL) can enhance the aromatization by a combined effect of the high activity of Pt in micropores and the inhibited side reactions due to the mesoporous catalyst.52 For example, the mesopores in Pt/desilicated ZSM-5 contributed to the higher ethane conversion by 60% and BTX selectivity by 75% compared to the Pt/conventional ZSM-5.51 Some macroporous oxide (e.g. Ta2O5) supports can improve the desired selectivity (up to 97% towards isomerization) by allowing charge transfer at the metal–oxide interface and high electron density of Pt.53
Furthermore, metal promoters such as Zn54,55 and Fe56 could be introduced to improve the dispersion of noble metal atoms on the support. The formed bimetallic catalyst enhanced the alkane conversion and BTX selectivity by accelerating the initial dehydrogenation to form olefins and the subsequent dehydrocyclization to produce aromatics. Another study showed that the introduction of Zn to Pt over H-ZSM-5 could reduce the coke yield from 8.3 to 1.7 μg gcat−1 h−1 and thus alleviate catalyst deactivation.57 Moreover, for bimetallic catalysts, the interaction between both metals (e.g. Pt–Zn nanoparticles) and the support (e.g. uniform compact cylindrical ZSM-5) also improved the selectivity over 90% towards aromatics compared to the selectivity of less than 40% from Pt–Zn over the conventional ZSM-5.58
2.3. Other catalysts
Besides the widely-utilized zeolites and noble metals, other catalysts were also used to aromatize alkanes. GaN as a nitride semiconductor could catalyze the aromatization of light alkanes to benzene with high selectivity.59,60 However, the small surface area (e.g. 8 m2 g−1) of GaN required high temperature (>650 °C) to initiate benzene formation because the activity is limited by the number of active sites which are dependent on the surface area.3 Activated carbon doped with phosphorus is also reported to aromatize n-hexane into benzene with the –P(O)(OH) functional group as the active site.61,62 The active sites of the P@AC catalyst are weak/medium-strength acidic centers in nature, which could selectively activate the C–H bond instead of the C–C bond.2.4. Catalyst deactivation
During the catalytic aromatization of n-alkanes, coke formation is almost inevitable and is the major reason for the catalyst deactivation because the formed coke will block the active sites and zeolite channels.63,64 However, measures could be taken to suppress the coke formation, such as developing bimetallic catalysts. Incorporation of Co into Mo/ZSM-5 could enhance the resistance to deactivation by reducing the coke formation during the aromatization of methane.65 Similarly, introduction of Pt into Ga/ZSM-5 could facilitate the hydrogenolysis of coke precursors.66 Besides modifying the catalyst, conducting aromatization under a reduction environment (e.g. with H2 in the feed) could also suppress coke formation.21,673. Reactants
The aromatization of methane,64 ethane,7 and propane68 has been separately reviewed in the literature. The purpose of discussion here is to provide a big picture about the influence of the alkane structure and a generalized reaction mechanism of alkane aromatization.3.1. Single alkanes
CH4 is abundant in natural gas and is produced as a by-product in many chemical processes. The strong C–H bond in CH4 makes it the most unreactive hydrocarbon species. The most widely investigated catalyst for CH4 aromatization is Mo/H-ZSM-5.64 As shown in Fig. 1, the aromatization of CH4 over Mo/H-ZSM-5 at 700 °C involves the formation of a hydrocarbon pool. An induction period was also observed due to the accumulation of adsorbed hydrocarbons on the Mo sites.69 It is believed that the molybdenum carbide species are only responsible for the activation of CH4 molecules and Mo-carbide/H-ZSM-5 is more active and stable than Mo-oxide/H-ZSM-5 during CH4 aromatization.70,71 This could explain the presence of an induction period when the Mo-oxide is converted into Mo-carbide. The formation of carbides is facilitated by the reduction and carburization pretreatment and thus improved the catalytic properties.72 As shown in Fig. 4, the catalysts with pre-carburization demonstrated a higher benzene yield and a slower deactivation rate. Various Mo-carbide species such as MoC,39 Mo2C,73 Mo2C6,74 and MoCxOy,75 each with distinct structural and catalytic properties, may be the active sites to catalyze the aromatization. There is still debate about the evolution of the species during the reaction and their effects on the catalytic performance. For instance, it is reported that the isolated Mo-oxide evolves into Mo-carbide nanoparticles during the reaction.76 It is also proved that even though MoCxOy could activate CH4, MoCx is essential for the formation of aromatics.75 To resolve the debate, advanced operando techniques are required to determine the reaction pathways. In addition, the doping of metal elements such as Co and Pd to the Mo/ZSM-5 catalyst enhanced the stability and improved the resistance to deactivation.65,73 It is also reported that the physical mixing of NiO with Mo/H-ZSM-5 enhanced the aromatic yield and catalytic stability because NiO could increase the dispersion of molybdenum carbide and metal Ni can decrease the coke formation by converting the coke precursors to carbon nanotubes.41 Besides zeolite, sulfated zirconia supported Mo with various promoters could also promote the aromatization with little coke formation.77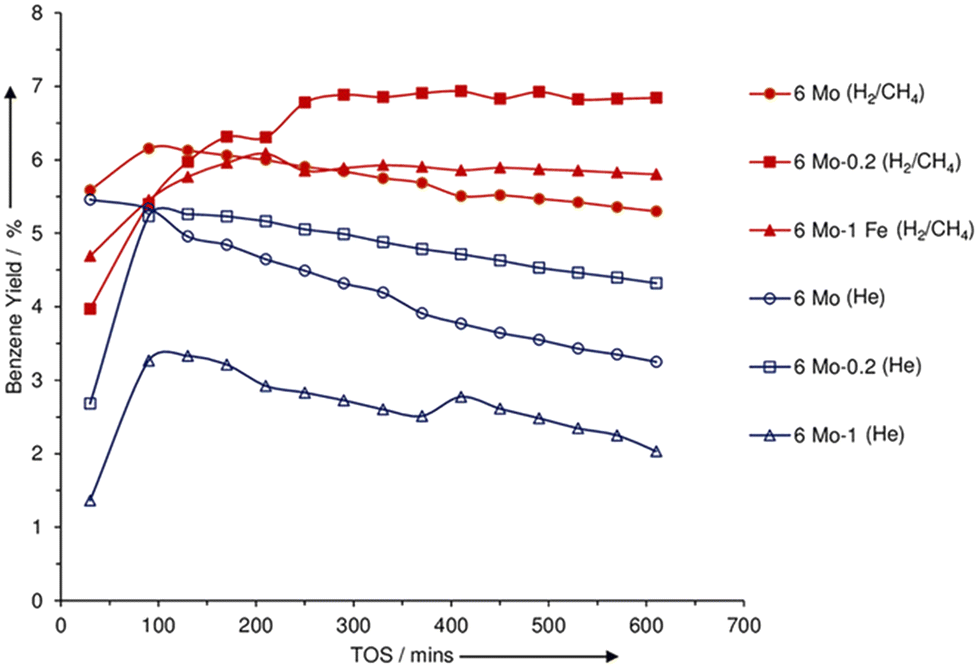 | ||
| Fig. 4 Benzene yield for 6Mo/ZSM-5, 6Mo-0.2Fe/ZSM-5, and 6Mo-1Fe/ZSM-5 catalysts under He treatment (open symbols) and pre-carburization (closed symbols) (this figure is reproduced from ref. 72 with permission). | ||
Similar to the unreactive methane, ethane aromatization has not been commercialized until now as well. Besides Mo/ZSM-5 and its modification (e.g. Fe or Zn as the promoter),29 other active metals such as Pt,78 Zn,21 Ga,40 Re,79 GaPt30,66 and NiGa80 could be supported on ZSM-5 to catalyze the ethane aromatization reaction. After ethane activation (i.e. dehydrogenation), oligomerization would also take place to form new C–C bonds over both Brønsted and Lewis acid sites.81 The newly formed unsaturated intermediate pool of a certain size (e.g. C6–C8) would further undergo cyclization and dehydrogenation to produce aromatics.
The aromatization of C3+ alkanes, especially the C3–C6 species and their mixture, has been commercialized using a Ga or Zn modified H-ZSM-5 catalyst or supported noble metals, such as Cyclar and M2-reforming processes. Typically, the yield of aromatic compounds could reach up to 58–60 wt% when C3 and C4 are utilized as the feedstocks.7 Lower temperature is generally required for the aromatization of C3+ alkanes compared to methane or ethane.82
The aromatization of C6+ alkanes is also called “reforming” in the industry. The main aim of reforming is to convert alkanes and cycloalkanes into aromatics with platinum catalysts such as Pt/γ-Al2O3 and Pt/KL zeolite.83–86 Besides the supported noble metal catalysts, other metals such as Mo,63 Cu,13 and Ga34 could also be utilized to achieve the aromatization. Even though it is thought that the straight carbon chain of C6–C8 alkanes could be converted into aromatics without cracking or oligomerization reactions,58 BTX will be present in the products when a single alkane acts as the reactant,87 which could prove the existence of the unsaturated intermediate pool.
The reports of the aromatization of C9+ alkanes are fewer than the lighter alkanes. The cracking reaction of C9+ alkanes would be more desired compared to the light alkanes,12 because it would be easier for the C2–C8 species in the unsaturated intermediate pool to form the aromatics. After cracking, the species in the unsaturated intermediate pool would undergo a similar aromatization route.88
3.2. Co-aromatization
Co-aromatization is generally conducted for CH4 with other alkanes such as propane89,90 and pentane91 because it is supposed that the presence of a co-reactant could reduce the thermodynamic limitation. Conversely, CH4 could also function as a co-reactant to promote BTX production during the aromatization of light alkanes over Zn-Ga/H-ZSM-5 (ref. 92) or the aromatization of complex naphtha over Zn–Pt/H-ZSM-5.28 As shown in Fig. 5, the presence of methane could increase the liquid and BTX yields compared to N2. The Hx and CHx intermediates formed from CH4 are anticipated to participate in the formation of light aromatics and suppress the formation of heavy aromatics compared to N2.67 The incorporation of fragments from CH4 in the products during the co-aromatization with propane was supported by the detection of 13C of CH4 in the aromatic rings of the products.93,94 The catalyst properties might affect the CH4 reaction pathway, such as the acidity influenced by the Al sites95 and the location of Zn species over the Zn/H-ZSM-5 zeolite.96 The aromatization of naphtha as a paraffin mixture could also be considered as co-aromatization of similar alkanes.97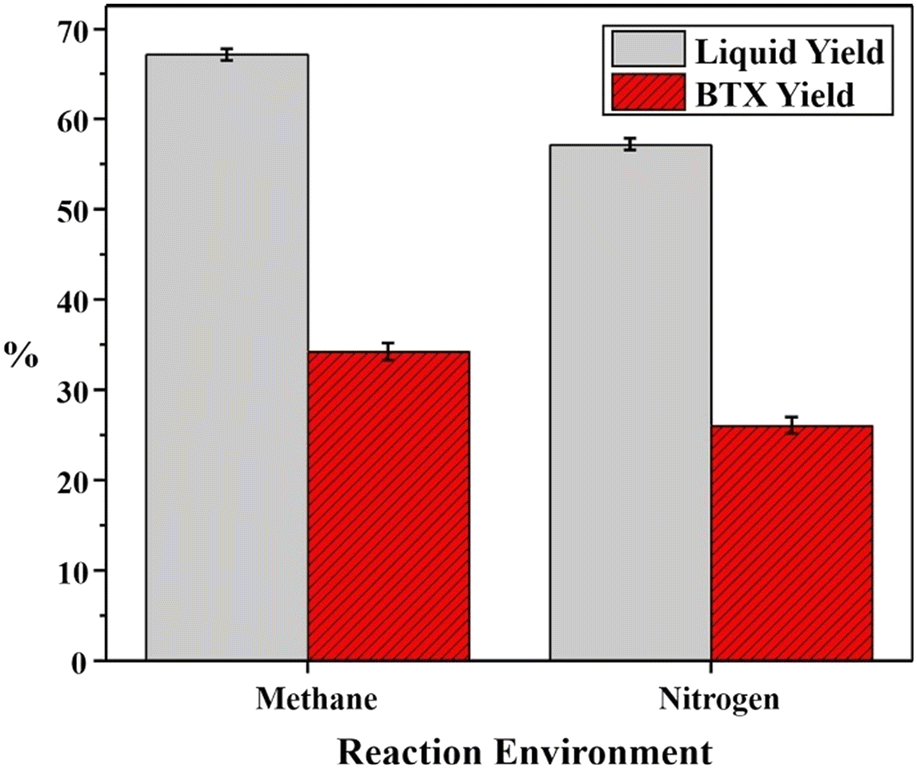 | ||
| Fig. 5 Effects of environments on liquid and BTX yields during the reforming of naphtha utilizing a Pt–Zn/ZSM-5 catalyst (this figure is reproduced from ref. 28 with permission). | ||
3.3. Coupling with the oxidation process
Besides the direct aromatization discussed above, oxidative dehydrogenation could be coupled with aromatization. CO2 has been used already to produce valuable olefins or synthesis gas from alkanes via CO2-oxidative dehydrogenation (CO2-ODH) or dry reforming.98–100 Chen et al. proposed a tandem CO2 oxidative dehydrogenation–aromatization reaction, which could promote the dehydrogenation and enhance the subsequent aromatization by consuming H2.45Fig. 6 demonstrates that CO2-oxidative dehydrogenation and aromatization could achieve a higher equilibrium ethane conversion than direct dehydrogenation and aromatization. It is proposed that CO2 consumes the coke via the reverse Boudouard reaction (CO2 + C → 2CO), thus enhancing stability.101 Moreover, the reverse water gas shift reaction (RWGS) is promoted by CO2, consuming H2 and thereby enhancing the dehydrogenation activity.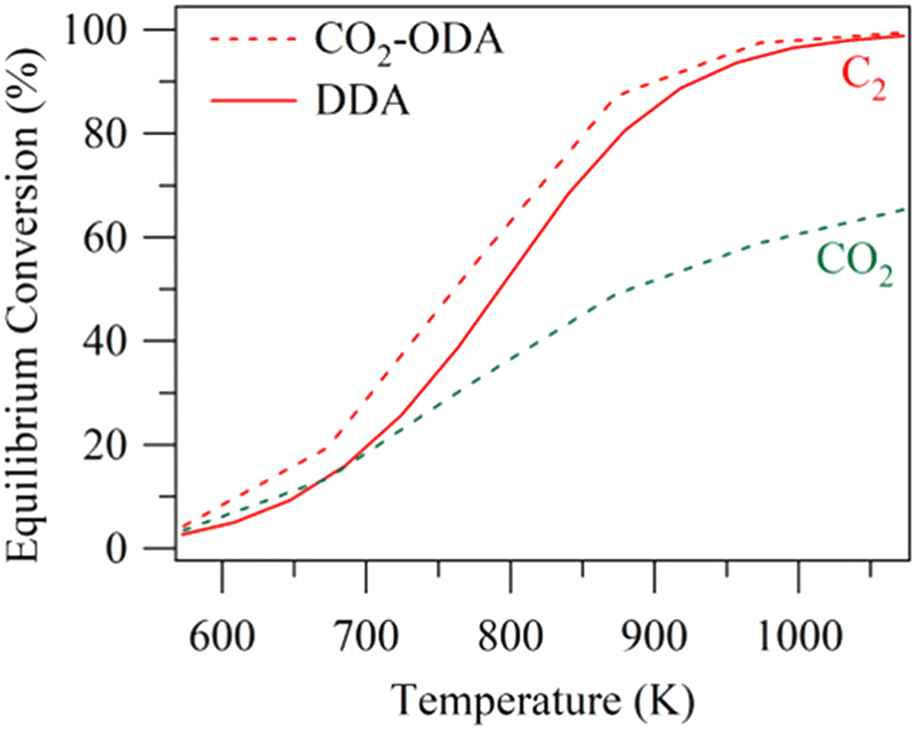 | ||
| Fig. 6 Effects of temperature on the equilibrium ethane conversion (DDA: direct dehydrogenation and aromatization; CO2-ODA: CO2-oxidative dehydrogenation and aromatization) (this figure is reproduced from ref. 45 with permission). | ||
Besides the direct coupling of chemical reactions, the methane aromatization process can also be integrated with the chemical looping for selective H2 oxidation and H2O removal so that a high combined aromatic yield could be acquired for the whole integrated process.102
4. Challenges and outlook
During the catalytic aromatization, low activity/selectivity especially for methane conversion and catalyst deactivation are the main challenges.29 As discussed in section 3.1, methane is very difficult to activate, which requires high temperatures. On the other hand, elevated temperatures could cause side reactions that lower the selectivity and increased coke formation accelerating the deactivation. Promising catalysts are reported in the literature; however, improvements are still needed in activity and stability. Moreover, the scale-up should be carefully considered, as most of the studies are on a lab-scale. Alternate reactor configurations also need to be studied, considering the heat effects associated with the reaction.Future efforts should be focusing on using small alkanes such as methane from abundant natural gas7 and carbonous waste such as biomass and plastic waste. New catalysts and process technologies need to be developed to address the challenges, as mentioned above.23
Conclusions
This paper reviews the recent advances about the development of catalysts and the mechanism investigations of alkane aromatization. The most important step in the alkane aromatization is the activation of alkanes to form an unsaturated intermediate pool with the help of a catalyst. The C–H bond activation is essential for the formation of an unsaturated intermediate pool from the alkanes, whereas the C–C bond breakage would occur via cracking. For zeolite catalysts, Brønsted acid sites favor the C–C bond breakage while Lewis acid sites promote C–H bond cleavage during the activation of the reactant alkane. Oligomerization might also occur depending on the exact carbon number of the alkane feedstock, such as methane and ethane, which are more difficult to activate compared to higher alkanes. Besides zeolites and their modified materials, noble metals and other catalysts such as GaN could also be utilized to aromatize alkanes. Based on the summary of the past studies, we believe that there is still a large scope for future research in different aspects of this reaction. There is potential in either improving the existing catalysts or designing new catalysts which could enhance the formation of the unsaturated intermediate pool. The use of predictive modeling or a rapidly developing machine learning approach may help to identify novel potential compositions that were not tested so far in this reaction.103 Materials such as niobia, whose Lewis/Brønsted acidity is tunable, could also be examined as part of such compositions.104–106 It would also be interesting to try non-conventional synthesis methods such as hydrothermal methods and microwave-assisted methods to accelerate the catalyst synthesis or to control the compositions more accurately.107 Doping with suitable components will also help to modulate the activity and stability of the new catalyst compositions.108 The use of tandem reactions, as described above, could be another option to overcome the issues in the direct aromatization such as coke deposition and consequent deactivation. It would also help in having a deeper insight into the mechanism through in situ spectroscopy and computational calculations. Simultaneously, studies in the reaction engineering will help to address the probable challenges in the potential scale-up possibilities of the aromatization of alkanes.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
Hongqi Wang: conceptualization, investigation, data curation, visualization, writing – original draft, writing – review & editing. N. Raveendran Shiju: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.References
- Q.-M. Zhou, S. Wang, Z.-F. Qin, M. Dong, J.-G. Wang and W.-B. Fan, J. Fuel Chem. Technol., 2023, 51, 1529–1539 CrossRef CAS.
- K. Li, M. Yan, H. Wang, L. Cai, P. Wang and H. Chen, Fuel Process. Technol., 2023, 252, 107982 CrossRef CAS.
- M. N. Akhtar, A. M. Aitani, A. C. Ummer and H. S. Alasiri, Energy Fuels, 2023, 37, 2586–2607 CrossRef CAS.
- F. Yu, S. Liu and B. Liu, Molecules, 2024, 29, 1288 CrossRef CAS PubMed.
- D. Xu, L. Wei, M. Yan, F. Yi, G. Zhao, A. Jia, D. Zhu, S. Wang and Y. Li, Appl. Catal., A, 2023, 663, 119308 CrossRef CAS.
- G. He and D. Mei, J. Catal., 2024, 436, 115623 CrossRef CAS.
- Y. Xiang, H. Wang, J. Cheng and J. Matsubu, Catal. Sci. Technol., 2018, 8, 1500–1516 RSC.
- V. I. Bogdan, A. E. Koklin, I. I. Mishanin, T. V. Bogdan, N. V. Mashchenko and L. M. Kustov, Mendeleev Commun., 2021, 31, 230–232 CrossRef CAS.
- D. Liu, L. Cao, G. Zhang, L. Zhao, J. Gao and C. Xu, Fuel Process. Technol., 2021, 216, 106770 CrossRef CAS.
- A. A. Rownaghi, F. Rezaei and J. Hedlund, Chem. Eng. J., 2012, 191, 528–533 CrossRef CAS.
- T. F. Narbeshuber, H. Vinek and J. A. Lercher, J. Catal., 1995, 157, 388–395 CrossRef CAS.
- S. Pradhan, R. Lloyd, J. K. Bartley, D. Bethell, S. Golunski, R. L. Jenkins and G. J. Hutchings, Chem. Sci., 2012, 3, 2958–2964 RSC.
- O. Tursunov, L. Kustov and Z. Tilyabaev, J. Pet. Sci. Eng., 2019, 180, 773–778 CrossRef CAS.
- C. M. Lok, J. Van Doorn and G. Aranda Almansa, Renewable Sustainable Energy Rev., 2019, 113, 109248 CrossRef.
- F. Goodarzi, R. P. Thumbayil, K. Enemark-Rasmussen, J. Mielby, T. T. M. Nguyen, P. Beato, F. Joensen and S. Kegnæs, ChemCatChem, 2020, 12, 1519–1526 CrossRef CAS.
- B. Xu, M. Tan, X. Wu, H. Geng, F. Song, Q. Ma, C. Luan, G. Yang and Y. Tan, Fuel, 2021, 283, 118889 CrossRef CAS.
- Z. Hou, X. Mi, X. Li and H. Liu, Ind. Eng. Chem. Res., 2021, 60, 12100–12108 CrossRef CAS.
- Y. Hyun Lim, H. Won Ryu, K. Nam, Y. Hwang, J. Roh and D. Heui Kim, Fuel, 2023, 342, 127906 CrossRef CAS.
- M. W. Schreiber, C. P. Plaisance, M. Baumgärtl, K. Reuter, A. Jentys, R. Bermejo-Deval and J. A. Lercher, J. Am. Chem. Soc., 2018, 140, 4849–4859 CrossRef CAS PubMed.
- N. M. Phadke, E. Mansoor, M. Head-Gordon and A. T. Bell, ACS Catal., 2021, 11, 2062–2075 CrossRef CAS.
- T. Liang, S. Fadaeerayeni, J. Shan, T. Li, H. Wang, J. Cheng, H. Toghiani and Y. Xiang, Ind. Eng. Chem. Res., 2019, 58, 17699–17708 CrossRef CAS.
- A. K. Panda, A. Alotaibi, I. V. Kozhevnikov and N. R. Shiju, Waste Biomass Valoriz., 2020, 11, 6337–6345 CrossRef CAS.
- S. S. Arzumanov, A. A. Gabrienko, A. V. Toktarev, Z. N. Lashchinskaya, D. Freude, J. Haase and A. G. Stepanov, J. Phys. Chem. C, 2019, 123, 30473–30485 CrossRef CAS.
- L. Lin, J. Liu, X. Zhang, J. Wang, C. Liu, G. Xiong and H. Guo, Ind. Eng. Chem. Res., 2020, 59, 16146–16160 CrossRef CAS.
- G. Xu and X. Zhu, Appl. Catal., B, 2021, 293, 120241 CrossRef CAS.
- S. Tamiyakul, T. Sooknoi, L. L. Lobban and S. Jongpatiwut, Appl. Catal., A, 2016, 525, 190–196 CrossRef CAS.
- J. Wang, C. Liu, P. Zhu, H. Liu, X. Zhang, Y. Zhang, J. Liu, L. Zhang and W. Zhang, New J. Chem., 2021, 45, 18659–18668 RSC.
- J. Jarvis, A. Wong, P. He, Q. Li and H. Song, Fuel, 2018, 223, 211–221 CrossRef CAS.
- B. Robinson, X. Bai, A. Samanta, V. Abdelsayed, D. Shekhawat and J. Hu, Energy Fuels, 2018, 32, 7810–7819 CrossRef CAS.
- A. Caiola, B. Robinson, X. Bai, D. Shekhawat and J. Hu, Ind. Eng. Chem. Res., 2021, 60, 11421–11431 CrossRef CAS.
- Y. Wang, A. Caiola, B. Robinson, Q. Li and J. Hu, Energy Fuels, 2020, 34, 3100–3109 CrossRef CAS.
- R. Geng, Y. Liu, Y. Guo, P. Wang, M. Dong, S. Wang, J. Wang, Z. Qin and W. Fan, ACS Catal., 2022, 12, 14735–14747 CrossRef CAS.
- Y. Zhou, H. Thirumalai, S. K. Smith, K. H. Whitmire, J. Liu, A. I. Frenkel, L. C. Grabow and J. D. Rimer, Angew. Chem., Int. Ed., 2020, 59, 19592–19601 CrossRef CAS PubMed.
- A. Thivasasith, T. Maihom, S. Pengpanich, J. Limtrakul and C. Wattanakit, Phys. Chem. Chem. Phys., 2019, 21, 5359–5367 RSC.
- B. J. Lee, J. H. Lee, D. H. Kim, Y. G. Hur and K.-Y. Lee, Microporous Mesoporous Mater., 2021, 323, 111243 CrossRef CAS.
- W. Wannapakdee, D. Suttipat, P. Dugkhuntod, T. Yutthalekha, A. Thivasasith, P. Kidkhunthod, S. Nokbin, S. Pengpanich, J. Limtrakul and C. Wattanakit, Fuel, 2019, 236, 1243–1253 CrossRef CAS.
- M. Raad, S. Hamieh, J. Toufaily, T. Hamieh and L. Pinard, J. Catal., 2018, 366, 223–236 CrossRef CAS.
- Y. Wu, Y. Lv, R. Wang, L. Bao, Z. Zhang, D. Shi, A. Zhang, Y. Zhang, Q. Liu, Q. Wu, D. Shi, K. Chen, G. Jiang and H. Li, Langmuir, 2024, 40, 11998–12008 CrossRef CAS PubMed.
- I. Vollmer, I. Yarulina, F. Kapteijn and J. Gascon, ChemCatChem, 2019, 11, 39–52 CrossRef CAS.
- E. A. Uslamin, H. Saito, Y. Sekine, E. J. M. Hensen and N. Kosinov, Catal. Today, 2021, 369, 184–192 CrossRef CAS.
- K. Nam, H. W. Ryu, M. Y. Gim and D. H. Kim, Appl. Catal., B, 2021, 296, 120377 CrossRef CAS.
- H. W. Ryu, Y. H. Lim, W. Jung, K. Nam, Y. Hwang, J. E. Roh and D. H. Kim, Chem. Eng. J., 2023, 467, 143404 CrossRef CAS.
- A. A. Kolganov, A. A. Gabrienko, S. A. Yashnik, E. A. Pidko and A. G. Stepanov, J. Phys. Chem. C, 2020, 124, 6242–6252 CrossRef CAS.
- W. Lin, Y. Song, L. Han, X. Yang, J. Liu and B. Peng, Fuel, 2021, 299, 120890 CrossRef CAS.
- E. Gomez, X. Nie, J. H. Lee, Z. Xie and J. G. Chen, J. Am. Chem. Soc., 2019, 141, 17771–17782 CrossRef CAS PubMed.
- P. T. Huyen, V. D. Trinh, M. Teresa Portilla and C. Martínez, Catal. Today, 2021, 366, 97–102 CrossRef CAS.
- M. Yin, R. H. Natelson, A. A. Campos, P. Kolar and W. L. Roberts, Fuel, 2013, 103, 408–413 CrossRef CAS.
- S. Zhang, L. Chen, Z. Qi, L. Zhuo, J.-L. Chen, C.-W. Pao, J. Su and G. A. Somorjai, J. Am. Chem. Soc., 2020, 142, 16533–16537 CrossRef CAS PubMed.
- D. Xu, S. Wang, B. Wu, C. Huo, Y. Qin, B. Zhang, J. Yin, L. Huang, X. Wen, Y. Yang and Y. Li, J. Catal., 2018, 365, 163–173 CrossRef CAS.
- K. Li, Q. Chang, J. Yin, C. Zhao, L. Huang, Z. Tao, Y. Yun, C. Zhang, H. Xiang, Y. Yang and Y. Li, J. Catal., 2018, 361, 193–203 CrossRef CAS.
- F. Goodarzi, D. B. Christensen, F. Joensen, S. Kegnæs and J. Mielby, Appl. Catal., A, 2020, 592, 117383 CrossRef CAS.
- K. Lee and M. Choi, J. Catal., 2016, 340, 66–75 CrossRef CAS.
- K. An, S. Alayoglu, N. Musselwhite, K. Na and G. A. Somorjai, J. Am. Chem. Soc., 2014, 136, 6830–6833 CrossRef CAS PubMed.
- P. He, Y. Chen, J. Jarvis, S. Meng, L. Liu, X.-D. Wen and H. Song, ACS Appl. Mater. Interfaces, 2020, 12, 28273–28287 CrossRef CAS PubMed.
- G. Liu, J. Liu, N. He, S. Sheng, G. Wang and H. Guo, J. Energy Chem., 2019, 34, 96–103 CrossRef.
- D. Xu, S. Wang, B. Wu, B. Zhang, Y. Qin, C. Huo, L. Huang, X. Wen, Y. Yang and Y. Li, ACS Appl. Mater. Interfaces, 2019, 11, 29858–29867 CrossRef CAS PubMed.
- J. Jarvis, P. He, A. Wang and H. Song, Fuel, 2019, 236, 1301–1310 CrossRef CAS.
- J. S. Jarvis, J. H. Harrhy, P. He, A. Wang, L. Liu and H. Song, Chem. Commun., 2019, 55, 3355–3358 RSC.
- L. Li, X. Mu, W. Liu, X. Kong, S. Fan, Z. Mi and C.-J. Li, Angew. Chem., Int. Ed., 2014, 53, 14106–14109 CrossRef CAS PubMed.
- K. Dutta, L. Li, P. Gupta, D. P. Gutierrez and J. Kopyscinski, Catal. Commun., 2018, 106, 16–19 CrossRef CAS.
- Y. Li, H. Zhao, S. Chen, S. Bao, F. Xing and B. Jiang, Catal. Commun., 2021, 156, 106318 CrossRef CAS.
- Y. Li, S. Bao, H. Zhao, B. Feng, S. Chen, T. Gu, B. Yang and B. Jiang, Chem. Commun., 2021, 57, 4166–4169 RSC.
- R. Barthos and F. Solymosi, J. Catal., 2005, 235, 60–68 CrossRef CAS.
- J. J. Spivey and G. Hutchings, Chem. Soc. Rev., 2014, 43, 792–803 RSC.
- V. Fila, M. Bernauer, B. Bernauer and Z. Sobalik, Catal. Today, 2015, 256, 269–275 CrossRef CAS.
- A. Samanta, X. Bai, B. Robinson, H. Chen and J. Hu, Ind. Eng. Chem. Res., 2017, 56, 11006–11012 CrossRef CAS.
- Q. Li, P. He, J. Jarvis, A. Bhattacharya, X. Mao, A. Wang, G. M. Bernard, V. K. Michaelis, H. Zeng, L. Liu and H. Song, Appl. Catal., B, 2018, 236, 13–24 CrossRef CAS.
- A. Bhan and W. Nicholas Delgass, Catal. Rev.: Sci. Eng., 2008, 50, 19–151 CrossRef CAS.
- I. Vollmer, E. Abou-Hamad, J. Gascon and F. Kapteijn, ChemCatChem, 2020, 12, 544–549 CrossRef CAS.
- M. Rahman, A. Sridhar and S. J. Khatib, Appl. Catal., A, 2018, 558, 67–80 CrossRef CAS.
- V. Ramasubramanian, H. Ramsurn and G. L. Price, J. Energy Chem., 2019, 34, 20–32 CrossRef.
- A. Sridhar, M. Rahman and S. J. Khatib, ChemCatChem, 2018, 10, 2571–2583 CrossRef CAS.
- N. Pasupulety, A. A. Al-Zahrani, M. A. Daous, H. Driss and L. A. Petrov, J. Mater. Res. Technol., 2021, 14, 363–373 CrossRef CAS.
- T. S. Khan, S. Balyan, S. Mishra, K. K. Pant and M. A. Haider, J. Phys. Chem. C, 2018, 122, 11754–11764 CrossRef CAS.
- I. Lezcano-González, R. Oord, M. Rovezzi, P. Glatzel, S. W. Botchway, B. M. Weckhuysen and A. M. Beale, Angew. Chem., Int. Ed., 2016, 55, 5215–5219 CrossRef PubMed.
- J. Gao, Y. Zheng, J.-M. Jehng, Y. Tang, I. E. Wachs and S. G. Podkolzin, Science, 2015, 348, 686–690 CrossRef CAS PubMed.
- M. A. Abedin, S. Kanitkar, S. Bhattar and J. J. Spivey, Catal. Today, 2021, 365, 71–79 CrossRef CAS.
- H. Liu, H. Wang, A.-H. Xing and J.-H. Cheng, J. Phys. Chem. C, 2019, 123, 15637–15647 CrossRef CAS.
- L. Ma and X. Zou, Appl. Catal., B, 2019, 243, 703–710 CrossRef CAS.
- S. Fadaeerayeni, J. Shan, E. Sarnello, H. Xu, H. Wang, J. Cheng, T. Li, H. Toghiani and Y. Xiang, Appl. Catal., B, 2020, 601, 117629 CrossRef CAS.
- A. Mehdad and R. F. Lobo, Catal. Sci. Technol., 2017, 7, 3562–3572 RSC.
- D. Zeng, G. Zhu and C. Xia, Fuel Process. Technol., 2022, 226, 107087 CrossRef CAS.
- E. A. Belopukhov, D. I. Kir'yanov, M. D. Smolikov, V. A. Shkurenok, A. S. Belyi, A. V. Lavrenov, A. V. Kleimenov and D. O. Kondrashev, Catal. Today, 2021, 378, 113–118 CrossRef CAS.
- V. Y. Tregubenko, K. V. Veretelnikov, N. V. Vinichenko, T. I. Gulyaeva, I. V. Muromtsev and A. S. Belyi, Catal. Today, 2019, 329, 102–107 CrossRef CAS.
- D. Xu, B. Wu, P. Ren, S. Wang, C. Huo, B. Zhang, W. Guo, L. Huang, X. Wen, Y. Qin, Y. Yang and Y. Li, Catal. Sci. Technol., 2017, 7, 1342–1350 RSC.
- W. D. Shafer, G. Jacobs, U. M. Graham, V. R. R. Pendyala, M. Martinelli, G. A. Thomas, T. Jermwongratanachai, A. MacLennan, Y. Hu and B. H. Davis, Catal. Lett., 2018, 148, 97–107 CrossRef CAS.
- T. E. Tshabalala and M. S. Scurrell, Catal. Commun., 2015, 72, 49–52 CrossRef CAS.
- A. Zaker, G. A. Tompsett, S. Wang, J. Q. Bond and M. T. Timko, Fuel, 2022, 310, 122360 CrossRef CAS.
- C. Song, M. Y. Gim, Y. H. Lim and D. H. Kim, Fuel, 2019, 251, 404–412 CrossRef CAS.
- G. Xu, P. Zhang, J. Cheng, L. Xu, J. Fan, X. Zhu and F. Yang, Microporous Mesoporous Mater., 2022, 330, 111494 CrossRef CAS.
- P. He, J. Jarvis, L. Liu and H. Song, Fuel, 2019, 239, 946–954 CrossRef CAS.
- Q. Li, F. Zhang, J. Jarvis, P. He, M. M. Yung, A. Wang, K. Zhao and H. Song, Fuel, 2018, 219, 331–339 CrossRef CAS.
- M. V. Luzgin, V. A. Rogov, S. S. Arzumanov, A. V. Toktarev, A. G. Stepanov and V. N. Parmon, Catal. Today, 2009, 144, 265–272 CrossRef CAS.
- M. V. Luzgin, A. A. Gabrienko, V. A. Rogov, A. V. Toktarev, V. N. Parmon and A. G. Stepanov, J. Phys. Chem. C, 2010, 114, 21555–21561 CrossRef CAS.
- P. He, A. Wang, S. Meng, G. M. Bernard, L. Liu, V. K. Michaelis and H. Song, Catal. Today, 2019, 323, 94–104 CrossRef CAS.
- P. He, J. S. Jarvis, S. Meng, Q. Li, G. M. Bernard, L. Liu, X. Mao, Z. Jiang, H. Zeng, V. K. Michaelis and H. Song, Appl. Catal., B, 2019, 250, 99–111 CrossRef CAS.
- M. Ellouh, Z. S. Qureshi, A. Aitani, M. N. Akhtar, Y. Jin, O. Koseoglu and H. Alasiri, ChemistrySelect, 2020, 5, 13807–13813 CrossRef CAS.
- M. Ronda-Lloret, T. K. Slot, N. P. van Leest, B. de Bruin, W. G. Sloof, E. Batyrev, A. Sepúlveda-Escribano, E. V. Ramos-Fernandez, G. Rothenberg and N. R. Shiju, ChemCatChem, 2022, 14, e202200446 CrossRef CAS.
- M. Ronda-Lloret, L. Yang, M. Hammerton, V. S. Marakatti, M. Tromp, Z. Sofer, A. Sepúlveda-Escribano, E. V. Ramos-Fernandez, J. J. Delgado, G. Rothenberg, T. Ramirez Reina and N. R. Shiju, ACS Sustainable Chem. Eng., 2021, 9, 4957–4966 CrossRef CAS PubMed.
- M. Ronda-Lloret, V. S. Marakatti, W. G. Sloof, J. J. Delgado, A. Sepúlveda-Escribano, E. V. Ramos-Fernandez, G. Rothenberg and N. R. Shiju, ChemSusChem, 2020, 13, 6401–6408 CrossRef CAS PubMed.
- E. Devid, M. Ronda-Lloret, Q. Huang, G. Rothenberg, N. R. Shiju and A. Kleyn, Chin. J. Chem. Phys., 2020, 33, 243–251 CrossRef CAS.
- C. Brady, Q. Debruyne, A. Majumder, B. Goodfellow, R. Lobo, T. Calverley and B. Xu, Chem. Eng. J., 2021, 406, 127168 CrossRef CAS.
- N. Madaan, N. R. Shiju and G. Rothenberg, Catal. Sci. Technol., 2016, 6, 125–133 RSC.
- S.-H. Chung, V. Lachman, T. K. Slot and N. R. Shiju, Catal. Commun., 2023, 182, 106754 CrossRef CAS.
- S. Shirazimoghaddam, I. Amin, J. A. Faria Albanese and N. R. Shiju, ACS Eng. Au, 2023, 3, 37–44 CrossRef CAS PubMed.
- E. S. Gnanakumar, N. Chandran, I. V. Kozhevnikov, A. Grau-Atienza, E. V. Ramos Fernández, A. Sepulveda-Escribano and N. R. Shiju, Chem. Eng. Sci., 2019, 194, 2–9 CrossRef CAS.
- N. R. Shiju and V. V. Guliants, ChemPhysChem, 2007, 8, 1615–1617 CrossRef CAS PubMed.
- E. V. Ramos-Fernandez, N. R. Shiju and G. Rothenberg, RSC Adv., 2014, 4, 16456–16463 RSC.
| This journal is © The Royal Society of Chemistry 2025 |