
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The “factory in a lab”: telescoping the Matteson and Matteson–Hoppe–Aggarwal boronate chemistry under flow conditions†‡
Florian
Fricke
a,
Gerald
Dräger
 a and
Andreas
Kirschning
*ab
a and
Andreas
Kirschning
*ab
aInstitute of Organic Chemistry, Leibniz University Hannover, Schneiderberg 1B, 30167 Hannover, Germany. E-mail: andreas.kirschning@oci.uni-hannover.de
bUppsala Biomedical Center (BMC), Uppsala University, Husargatan 3, 752 37 Uppsala, Sweden
First published on 12th December 2024
Abstract
The Matteson reaction and the related Matteson–Hoppe–Aggarwal variant were combined in a compartmentalized flow system and the doubly homologated resulting boronate was transformed into the corresponding alcohols after terminal oxidation. Terpenoids were included allowing the generation of new terpene alcohols in excellent yields which were evaluated for their olfactory properties.
One concept of why Nature appears to be such a “formidable chemist” is associated with the fact that it does rely on the concepts of modularity and iteration as manifested in polypeptides, oligonucleotides and oligosaccharides. Also, the biosyntheses of terpenes and polyketides are based on these synthetic concepts.1 Strategically, such iterations can be combined very well with flow chemistry2 by creating assembly line systems using modular flow elements specially designed for this purpose. Such assembly line systems are easier to handle in terms of rate control than corresponding single-pot batch processes and often lead to higher yields in much shorter process times.3
The study also revealed for the first time that transfer from batch to flow provides accurate data on reaction times and ideal temperatures for the individual steps (lithiation 1 → 2, I, boronate formation 2 → 4, II, and migration 4 → 5, III) of the Matteson reaction. These three steps can be conducted in less than 10 s in total.4
Flow chemistry is commonly associated with the term “lab on a chip”, which in our view does not fully reflect reality. Industrial reality would probably rather describe the possibilities and potential of flow chemistry with the term “factory in a lab”. Since the pioneering work of Yoshida and co-workers, it is well known that organolithium chemistry in combination with “flow” technology is perfectly suited to perform carbanion chemistry in high yield under highly optimized conditions.5 The easy handling of highly reactive organolithium species in flow devices6 was demonstrated in the synthesis of the tricyclic antidepressant amitriptyline (Elavil).7 Besides carbanions, other reactive intermediates such as carbenes and radicals can be well controlled in flow reactors.8
Also, the Matteson homologation reaction9 was found to be ideally suited for iterative operation. At each stage, a boronate ester is homologated by one carbon atom yielding a new boronate ester (Scheme 1, top).10 Recently, we disclosed a Matteson flow protocol that allowed (per-) homologation of terpenes in a controlled manner to yield homo-, bishomo- and trishomo-terpenols after oxidative workup.4
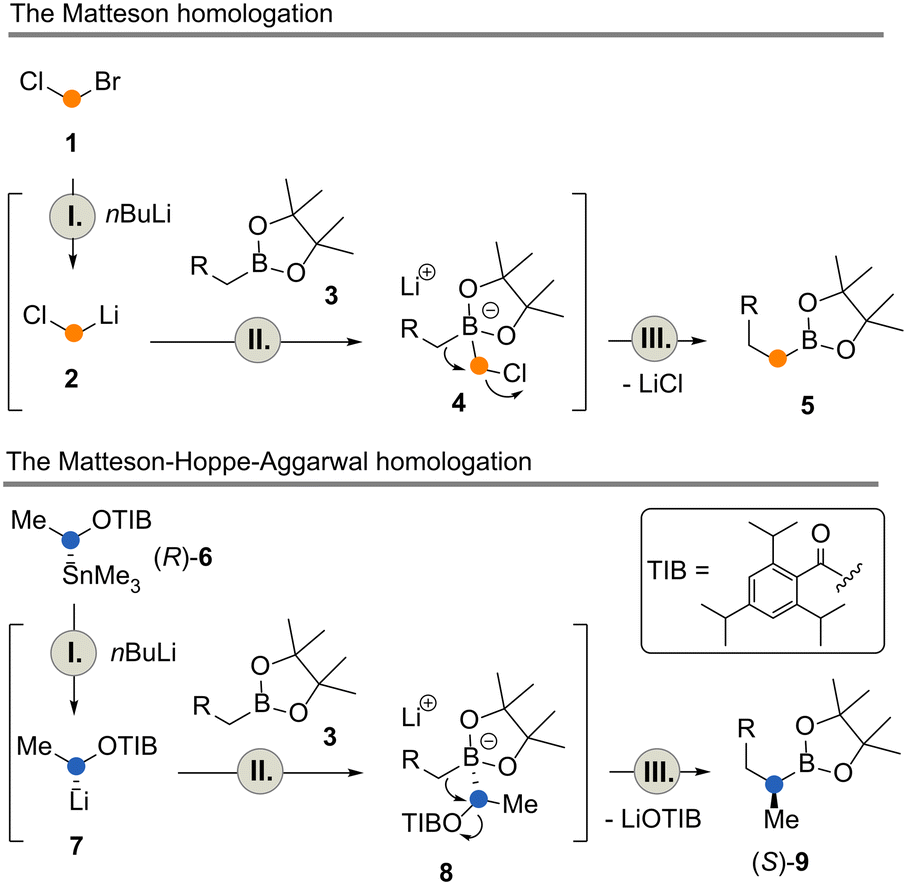 | ||
| Scheme 1 The Matteson and the Matteson–Hoppe–Aggarwal homologations. | ||
An extension of the Matteson reaction has recently been popularized by the group of Aggarwal.11 Here, Matteson's boronate chemistry is combined with the controlled formation of stable chiral organolithium intermediates developed by Hoppe and co-workers (Scheme 1, bottom).12 Mechanistically, this reaction is very similar to the Matteson homologation (lithiation 6 → 7, I, boronate formation 7 → 8, II, and migration 8 → 9, III) but it allows the introduction of side chains, mainly methyl groups, in a highly enantiocontrolled manner. Aggarwal and coworkers also devoted their studies to iterative applications13 and, more recently, to iterative automation under batch-type conditions in which they were able to prepare the core fragment of the natural product (+)-kalkitoxin.14
In the current work, we describe the combination of the classical Matteson reaction with the Matteson–Hoppe–Aggarwal protocol under flow conditions. Again, we included terpenes as substrates in our studies because organo-lithium flow chemistry offers great potential for “factory in the lab” production, and the flavour and fragrance industries could very well benefit from this enabling technology.15,16 Continuous flow processes can be controlled through flow rates and residence times and efficient mixing is a key to success, especially when the concentration of reactants or the flow rates in two streams to be mixed differ considerably.
We began our investigations with the conversion of the organotin compound 6 to the lithiated intermediate 7 by altering reaction parameters and in-line quenching with EtOH (Scheme 2). We kept the reactor system as compact as possible and decided to use moderate flow rates with residence times compatible with the second step. For optimal mixing, we employed a 3D-printed static mixer17 that was designed during the investigation of the Matteson reaction to prevent side reactions such as per-homologation18 and Wurtz coupling. It is important that the lithiation step has to be conducted below −42 °C (see the ESI†) as otherwise formation of substantial amounts of by-products was observed. Likely the lithiated species underwent degradation e.g. α-elimination. We then went on to introduce the boronic ester via a third stream (Scheme 3).
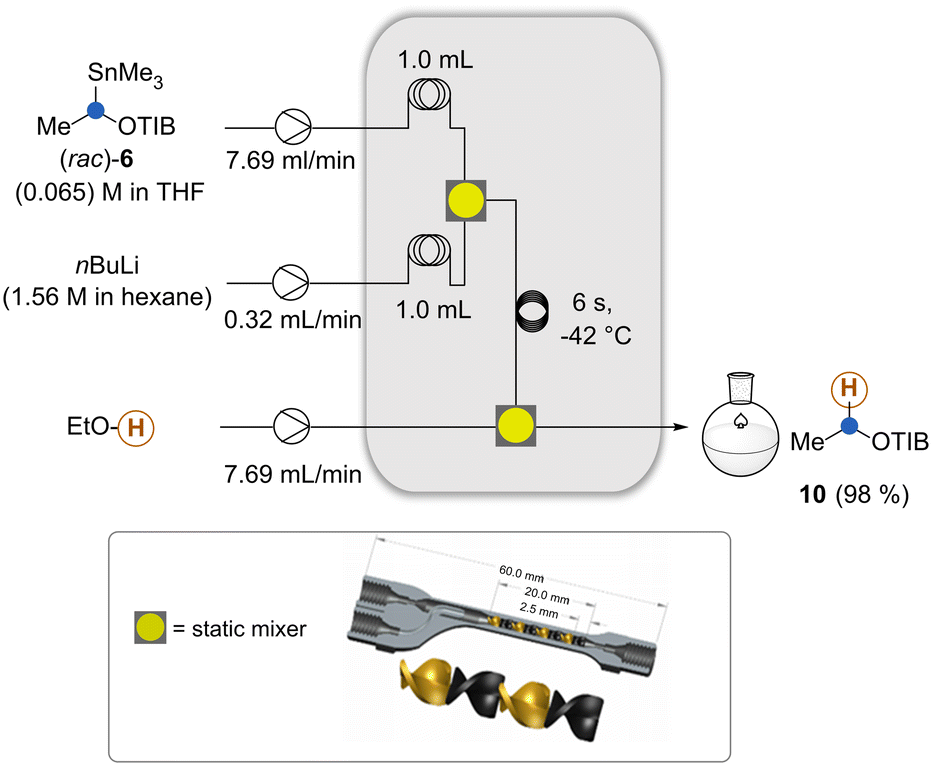 | ||
| Scheme 2 Intercepting the in situ generated organolithium species 7 and formation of ester 10 to determine the residence time for the lithiation of 6. Graphic presentation of the static mixer used here (see also ref. 4). | ||
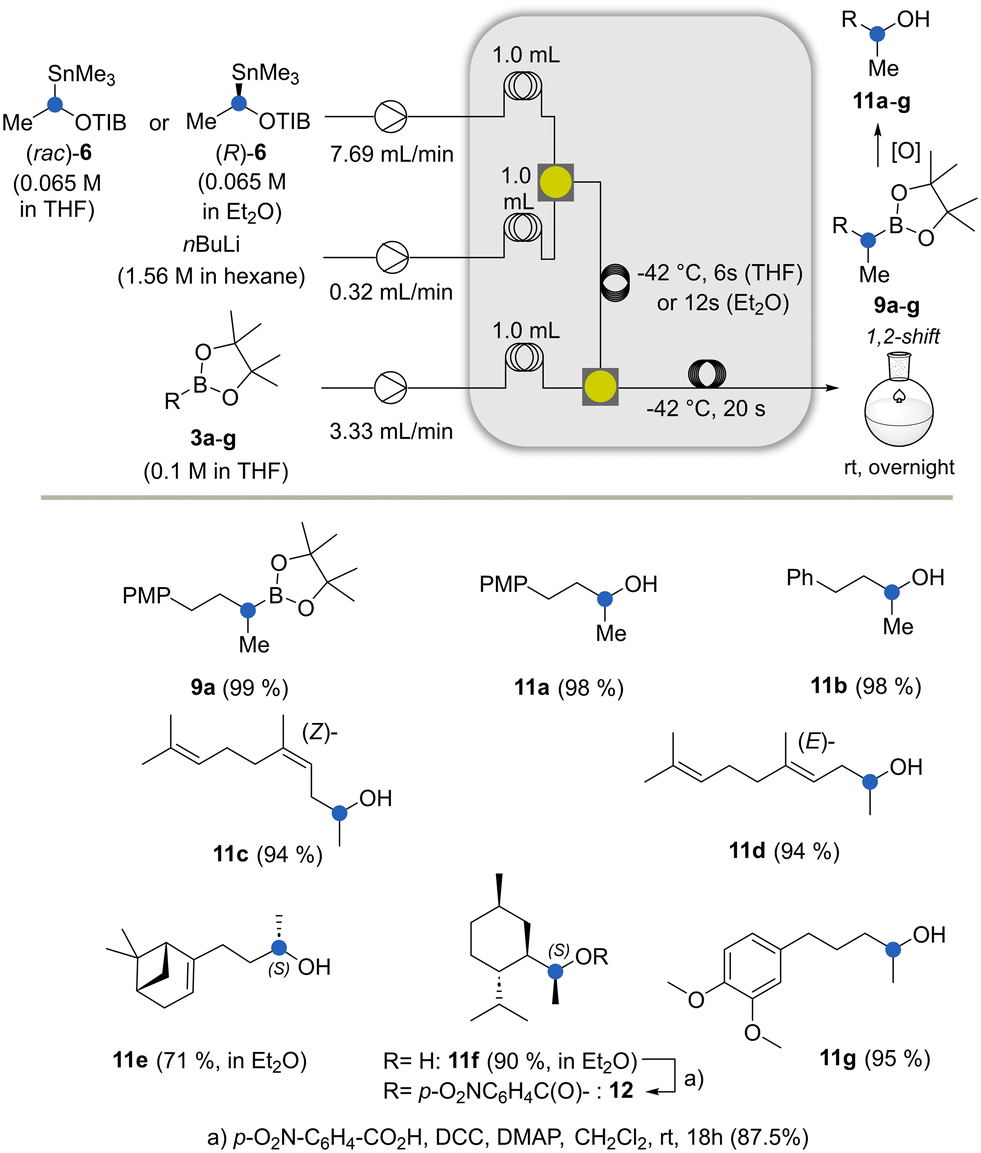 | ||
| Scheme 3 The Matteson–Hoppe–Aggarwal homologation under flow conditions and oxidation of boronic esters (PMP = p-methoxybenzyl, DCC= dicyclohexylcarbodiimide, DMAP = 4-dimethylaminopyridine). | ||
After the optimisation of the reaction parameters that included temperature, concentration, residence time and equivalents of reactants (for details, see the ESI†), we found the flow conditions that yielded the homologated boronate 9a in excellent yields (Scheme 3). The spatial compartmentalisation of the lithiation step and the formation of the boronate complex led to an efficient suppression of side reactions. Except for the conversion of boronate 3a to the homologous product 9a, which served as a model reaction for the development of the Matteson–Hoppe–Aggarwal flow protocol, we generally terminated the process with the oxidation of the final boronic esters (aq. H2O2, (35%), 0 °C → rt). As such, the corresponding alcohols 11a–11g were obtained from the corresponding intermediate boronates 9a–9g.
Experiments using enantiopure 6 in THF with the boronic esters 3e and 3f led to the partial erosion of the integrity of the stereogenic center and the formation of a diastereomeric mixture of about 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. This problem was circumvented by generating the carbanion from (R)-configuration stannane 6 in Et2O instead of THF. Afterwards, the lithiated species were mixed with the boronic ester dissolved in THF, leading to a single diastereomer. Clearly, a THF solution can be added to the carbenoid dissolved in a second solvent like Et2O, which drastically enhances the rate of borylation while achieving high ee values. In contrast, the exclusive use of THF leads to decreased ee values.19
:1. This problem was circumvented by generating the carbanion from (R)-configuration stannane 6 in Et2O instead of THF. Afterwards, the lithiated species were mixed with the boronic ester dissolved in THF, leading to a single diastereomer. Clearly, a THF solution can be added to the carbenoid dissolved in a second solvent like Et2O, which drastically enhances the rate of borylation while achieving high ee values. In contrast, the exclusive use of THF leads to decreased ee values.19
Under these conditions, the residence time was found to be 12 s to achieve full conversion. The absolute configuration of the resulting alcohol 11f was confirmed by X-ray crystallography of the p-nitrobenzoate 12 which was obtained under standard esterification conditions (Fig. 1).
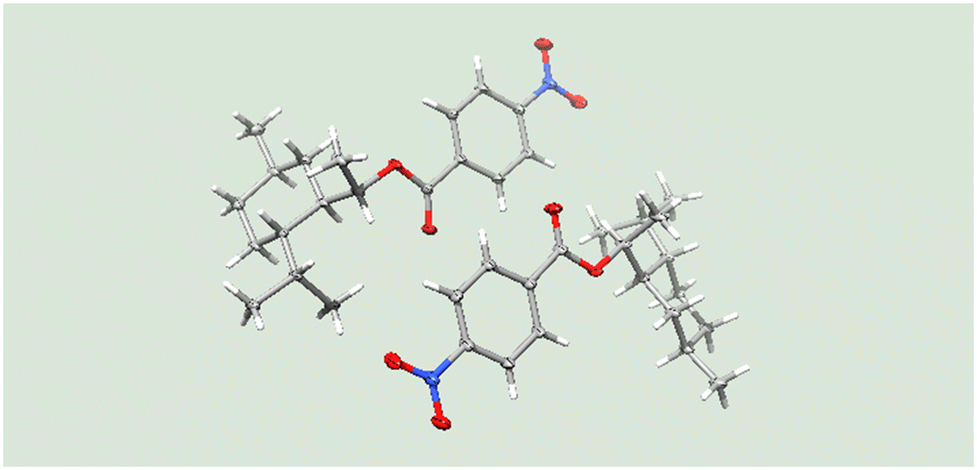 | ||
| Fig. 1 Crystallographic analysis of p-nitrobenzoic ester 12. | ||
To fit the reaction parameters for coupling the aforementioned method with our previously reported Matteson homologation,4 we modified the original flow protocol (Scheme 4). By adjusting the substrate concentrations, equivalents and the individual flow rates in conjunction with the reactor length and the reactor temperature (see the ESI†), a flow module was created that delivered excellent yields of new boronates (Scheme 4). The module consists of two pumps, two cooling loops to cool the two input streams before they are efficiently mixed, and a reactor element where lithiation and boronate formation take place.
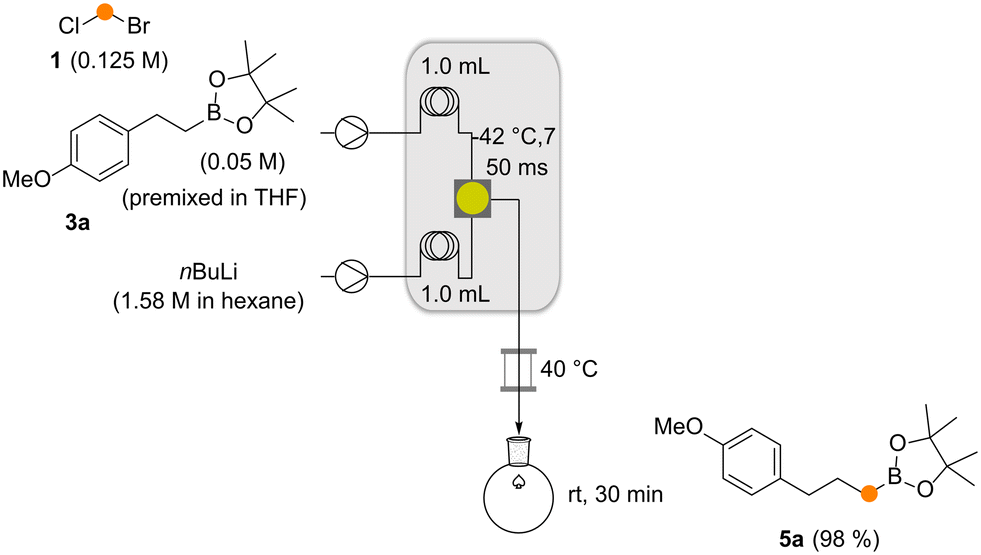 | ||
| Scheme 4 The Matteson homologation under flow (3a → 5a). Conditions had to be optimized with respect to the protocol reported in ref. 4 for effective telescoping of this reaction with the Matteson–Hoppe–Aggarwal flow protocol. | ||
The two flow processes reveal differences between the Matteson homologation and the Matteson–Hoppe–Aggarwal variant. The most striking and important one is the ease of formation of the boronate complex 4 compared to the corresponding ate-complex 8 and the ease with which the rearrangement 4 → 5versus the one from 8 to 9 occurs (Scheme 1). In the classical Matteson reaction, the lithium intermediate bears chloride, a very good leaving group and usually it is not branched, so both steps can proceed very rapidly (within 250 ms at −40 °C for the first step and 9 s at 40 °C for the second step, according to ref. 4). In contrast, organolithium species 7 is branched with a methyl group creating a certain degree of steric hindrance but foremost the 2,4,6-triisopropylbenzoate group has poorer leaving group properties compared to chloride typically used in the Matteson homologation protocol. These factors explain the significant difference of 1,2-migration reaction rates observed for the two homologation protocols. While boronate formation is completed within 20 s at −40 °C, the rearrangement requires reaction times that are beyond the regime of seconds. The 1,2 migration can be monitored by 11B-NMR spectroscopy and was reported to take place within 1–2 h at temperatures between 20 °C and 35 °C for selected boronate complexes containing the benzoate leaving group.13,20 With the optimized conditions for both reactions in hand, we set out to telescope the Matteson homologation flow protocol with the Matteson–Hoppe–Aggarwal homologation flow protocol developed here (Scheme 5) by connecting homologation module 1 with module 2. In module 1, the Matteson reaction was carried out by first forming the lithiated intermediate 2 by halogen lithium exchange.
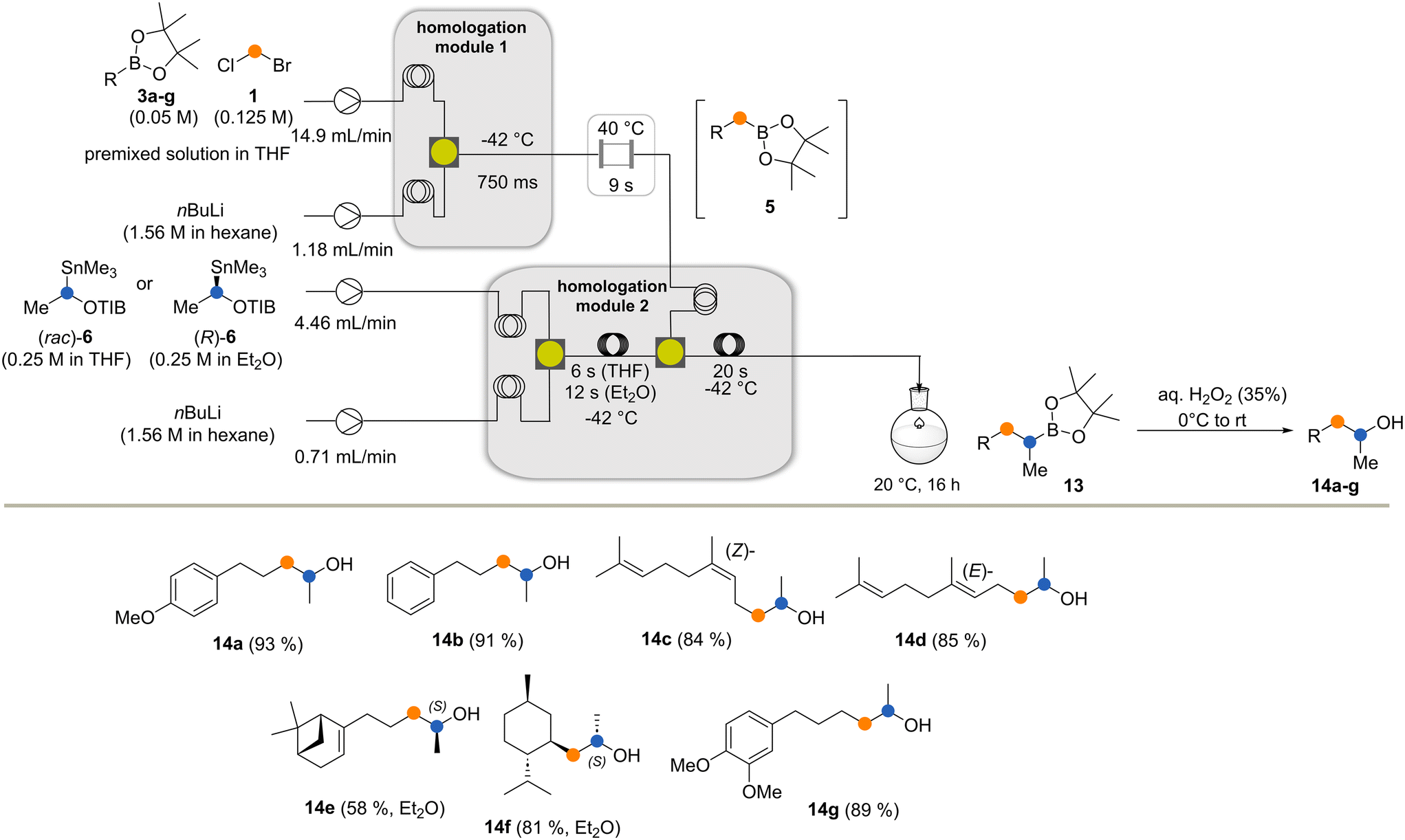 | ||
| Scheme 5 Telescoping the Matteson homologation with the Matteson–Hoppe–Aggarwal homologation under flow conditions starting from boronates 3. The process is terminated by the oxidation of the final boronates 13a–g (yields refer to isolated yields; the solvent is mentioned for the enantioselective protocol). | ||
After boronation and rearrangement, the new boronates 5 were pumped into homologation module 2, where they reacted with lithiated species 7. This was generated in a second compartment by tin lithium exchange prior to boronate formation. The resulting ate complexes that leave homologation module 2 were kept for up to 16 h at rt to guarantee complete rearrangement and formation of boronates 13a–g. Finally, the boronic esters were directly oxidized with H2O2 to furnish the corresponding alcohols 14a–g in overall very good yields.
Finally, the new alcohols 11a–g and 14a–g were examined for their olfactory profiles using GC-O (gas chromatography-olfactometry) (Table 1).21 Except for alcohol 14e, all products showed interesting olfactory properties. Alcohols 11c and 11d exhibited a floral, rosy scent, with the (Z)-isomer outperforming the (E)-isomer. The –CH2– homologated alcohols 14c and 14d showed similar profiles, with 14d exhibiting an additional note of apple and pear. Alcohol 11b exhibited a profile that can be described as flowery, herbal and green with additional notes of lavender and banana. The corresponding methylene elongated alcohol 14b also revealed a strong scent, specifically fruity and green with a metallic note. Alcohol 11a was judged to be woody and mossy with a hint of mold while the corresponding homologated alcohol 14a exhibited a balsamic, powdery profile with notes of vanilla and maltol. Alcohol 11e exhibited a weak scent with notes of lavender and muguet, whereas alcohol 14e has an unpleasant odor profile. Alcohols 14f and 11f were found to have a scent linked to menthol and peppermint with a cooling touch, with 11f showing an additional element of chamomile. Finally, the monohomologated alcohol 11g exhibited a weak floral scent, and the doubly homologated analog 14g showed a strong scent reminiscent of vanilla, dry fruits and rum.
| Alcohol | Olfactory analysis | Alcohol | Olfactory analysis |
|---|---|---|---|
| 11a | Woody, mossy with a hint of mold | 14a | Balsamic, powdery profile with notes of vanilla and maltol |
| 11b | Flowery, herbal and green; additional notes of lavender and banana | 14b | Fruity and green with a metallic note |
| 11c | Floral, rosy | 14c | Floral, rosy |
| 11d | Floral, rosy | 14d | Floral, rosy with a note of apple and pear |
| 11e | Weak scent with notes of lavender and muguet | 14e | Strong, sulphurous, technical |
| 11f | Menthol, peppermint with a cooling touch and a note of chamomile | 14f | Menthol, peppermint with a cooling touch |
| 11g | Weak, floral, salicyl-like | 14g | Strong, vanilla, dry fruits, rum |
Conclusions
In conclusion, we report on the development of a flow module for performing the Matteson–Hoppe–Aggarwal homologation protocol under continuous mode conditions. The use of a specially designed static mixer allowed us to carry out the homologation in a highly efficient manner with high yields. This sets the stage for combining this Matteson variant with the classical Matteson homologation by optimizing individual flow rates, residence times and reactor temperature and then linking two homologation flow modules. Double homologation and oxidation (1. Matteson, 2. Matteson–Hoppe–Aggarwal and 3. oxidation) also proceeded in very good yields. This work represents another example that organolithium chemistry and flow technology are a perfect match being superior to the corresponding batch chemistry. This opens doors for expanding the synthetic opportunities for industrial applications which includes the fragrance and flavour industries as demonstrated in this work.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
F. F. conducted the research and performed the experiments. G. D. carried out the X-ray analysis. A.K. formulated the overarching research goals and aims and supervised the research programme. The manuscript was written by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Symrise AG, Holzminden, Germany and particularly Dr. J. Panten for financial support and Lara-Joy Kleine-Benne for the olfactory evaluation of new products.Notes and references
- (a) G. Jürjens, A. Kirschning and D. Candito, Lessons from the Synthetic Chemist Nature, Nat. Prod. Rep., 2015, 32, 723 RSC; (b) J. Hartwig and A. Kirschning, Iterative Syntheses-The Gateway to New Automation Protocols, Angew. Chem., Int. Ed., 2015, 54, 10412 CrossRef CAS PubMed.
- Selected reviews on flow chemistry: (a) A. Kirschning, L. Kupracz and J. Hartwig, New Synthetic Opportunities in Miniaturized Flow Reactors with Inductive Heating, Chem. Lett., 2012, 41, 562 CrossRef CAS; (b) J. Wegner, S. Ceylan and A. Kirschning, Flow Chemistry - A Key Enabling Technology for (Multistep) Organic Synthesis, Adv. Synth. Catal., 2012, 354, 17 CrossRef CAS; (c) D. T. McQuade and P. H. Seeberger, Applying Flow Chemistry: Methods, Materials, and Multistep Synthesis, J. Org. Chem., 2013, 78, 6384 CrossRef CAS PubMed; (d) J. C. Pastre, D. L. Browne and S. V. Ley, Flow chemistry syntheses of natural products, Chem. Soc. Rev., 2013, 42, 8849 RSC; (e) I. R. Baxendale, The integration of flow reactors into synthetic organic chemistry, J. Chem. Technol. Biotechnol., 2013, 88, 519 CrossRef CAS; (f) T. Fukuyama, T. Totoki and I. Ryu, Carbonylation in microflow: close encounters of CO and reactive species, Green Chem., 2014, 16, 2042 RSC; (g) H. P. L. Gemoets, Y. Su, M. Shang, V. Hessel, R. Luque and T. Noel, Liquid phase oxidation chemistry in continuous-flow microreactors, Chem. Soc. Rev., 2016, 45, 83 RSC; (h) D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel and T. Noel, Applications of Continuous-Flow Photochemistry in Organic Synthesis, Material Science, and Water Treatment, Chem. Rev., 2016, 116, 10276 CrossRef PubMed; (i) M. B. Plutschack, B. Pieber, K. Gilmore and P. H. Seeberger, The Hitchhiker's Guide to Flow Chemistry, Chem. Rev., 2017, 117, 11796 CrossRef CAS PubMed.
- (a) C. P. Gordon, The renascence of continuous-flow peptide synthesis – an abridged account of solid and solution-based approaches, Org. Biomol. Chem., 2018, 16, 180 RSC , and references cited therein; (b) C. Battilocchio, F. Feist, A. Hafner, M. Simon, D. N. Tran, D. M. Allwood, D. C. Blakemore and S. V. Ley, Iterative reactions of transient boronic acids enable sequential C–C bond formation, Nat. Chem., 2016, 8, 360 CrossRef CAS PubMed.
- C. Kuhwald and A. Kirschning, The Matteson reaction under flow conditions – iterative flash homologations of terpenes, Org. Lett., 2021, 23, 4300–4304 CrossRef CAS PubMed.
- (a) H. Usutani, Y. Tomida, A. Nagaki, H. Okamoto, T. Nokami and J. Yoshida, Generation and Reactions of o-Bromophenyllithium without Benzyne Formation Using a Microreactor, J. Am. Chem. Soc., 2007, 129, 11692 CrossRef PubMed; (b) A. Nagaki, E. Takizawa and J. Yoshida, Oxiranyl Anion Methodology Using Microflow Systems, J. Am. Chem. Soc., 2009, 131, 1654 CrossRef CAS PubMed; (c) A. Nagaki, A. Kenmoku, Y. Moriwaki, A. Hayashi and J. Yoshida, Cross-coupling in a flow microreactor: space integration of lithiation and Murahashi coupling, Angew. Chem., Int. Ed., 2010, 49, 7543 ( Angew. Chem. , 2010 , 122 , 7705 ) CrossRef CAS PubMed; (d) Y. Tomida, A. Nagaki and J. Yoshida, Asymmetric Carbolithiation of Conjugated Enynes: A Flow Microreactor Enables the Use of Configurationally Unstable Intermediates before They Epimerize, J. Am. Chem. Soc., 2011, 133, 3744 CrossRef CAS PubMed; (e) H. Kim, A. Nagaki and J. Yoshida, A flow-microreactor approach to protecting-group-free synthesis using organolithium compounds, Nat. Commun., 2011, 2, 264 CrossRef PubMed; (f) A. Nagaki, C. Matsuo, S. Kim, K. Saito, A. Miyazaki and J. Yoshida, Lithiation of 1,2-dichloroethene in flow microreactors: versatile synthesis of alkenes and alkynes by precise residence-time control, Angew. Chem., Int. Ed., 2012, 51, 3245 ( Angew. Chem. , 2012 , 124 , 3299 ) CrossRef CAS PubMed; (g) A. Nagaki, Y. Takahashi and J. Yoshida, Extremely Fast Gas/Liquid Reactions in Flow Microreactors: Carboxylation of Short-Lived Organolithiums, Chem. – Eur. J., 2014, 20, 7931 CrossRef CAS PubMed; (h) A. Nagaki, Y. Takahashi and J. Yoshida, Generation and Reaction of Carbamoyl Anions in Flow: Applications in the Three-Component Synthesis of Functionalized α-Ketoamides, Angew. Chem., Int. Ed., 2016, 55, 5327 ( Angew. Chem. , 2016 , 128 , 5413 ) CrossRef CAS PubMed; (i) A. Nagaki, H. Yamashita, K. Hirose, Y. Tsuchihashi and J. Yoshida, Alkyllithiums Bearing Electrophilic Functional Groups: A Flash Chemistry Approach, Angew. Chem., Int. Ed., 2019, 58, 4027 ( Angew. Chem. , 2019 , 131 , 4067 ) CrossRef CAS PubMed.
- A. Nagaki, Recent topics of functionalized organolithiums using flow microreactor chemistry, Tetrahedron Lett., 2019, 60, 150923 CrossRef CAS.
- L. Kupracz and A. Kirschning, Multiple Organolithium Generation in the Continuous Flow Synthesis of Amitriptyline, Adv. Synth. Catal., 2013, 35, 3369 Search PubMed.
- Reviews on “flash” chemistry: (a) J. Yoshida, A. Nagaki and T. Yamada, Flash Chemistry: Fast Chemical Synthesis by Using Microreactors, Chem. – Eur. J., 2008, 14, 7450 CrossRef CAS PubMed; (b) J. Yoshida, Y. Takahashi and A. Nagaki, Flash chemistry: flow chemistry that cannot be done in batch, Chem. Commun., 2013, 49, 9896 RSC.
- D. S. Matteson and R. W. H. Mah, Neighboring Boron in Nucleophilic Displacement, J. Am. Chem. Soc., 1963, 85, 2599 CrossRef CAS.
- (a) T. von Kreutz, D. Cantillo and C. O. Kappe, Flow Microreactor Technology for Taming Highly Reactive Chloroiodomethyllithium Carbenoid: Direct and Chemoselective Synthesis of α-Chloroaldehydes, Org. Lett., 2020, 22, 7537 CrossRef PubMed; (b) P. Musci, M. Colella, A. Sivo, G. Romanazzi, R. Luisi and L. Degennaro, Organomagnesium Based Flash Chemistry: Continuous Flow Generation and Utilization of Halomethylmagnesium Intermediates, Org. Lett., 2020, 22, 3623 CrossRef CAS PubMed; (c) C. Stueckler, P. Hermsen, B. Ritzen, M. Vasiloiu, P. Poechlauer, S. Steinhofer, A. Pelz, C. Zinganell, U. Felfer, S. Boyer, M. Goldbach, A. de Vries, T. Pabst, G. Winkler, V. LaVopa, S. Hecker and C. Schuster, Development of a continuous flow process for a Matteson reaction: from lab scale to full-scale production of a pharmaceutical intermediate, Org. Process Res. Dev., 2019, 23, 1069 CrossRef CAS.
- M. Burns, S. Essafi, J. R. Bame, S. P. Bull, M. P. Webster, S. Balieu, J. W. Dale, C. P. Butts, J. N. Harvey and V. K. Aggarwal, Assembly-line synthesis of organic molecules with tailored shapes, Nature, 2014, 513, 183 CrossRef CAS PubMed.
- D. Hoppe and T. Hense, Enantioselective synthesis with lithium/(−)-sparteine carbanion pairs, Angew. Chem., Int. Ed. Engl., 1997, 36, 2282 CrossRef CAS.
- S. Balieu, G. E. Hallett, M. Burns, T. Bootwicha, J. Studley and V. K. Aggarwal, Toward Ideality: The Synthesis of (+)-Kalkitoxin and (+)-Hydroxyphthioceranic Acid by Assembly-Line Synthesis, J. Am. Chem. Soc., 2015, 137, 4398–4403 CrossRef CAS PubMed.
- V. Fasano, R. C. Mykura, J. M. Fordham, J. J. Rogers, B. Banecki, A. Noble and V. K. Aggarwal, Automated stereocontrolled assembly-line synthesis of organic molecules, Nat. Synth., 2022, 1, 902 CrossRef CAS.
- A. Kirschning, W. Solodenko and K. Mennecke, Combining enabling techniques in organic synthesis: Continuous flow processes with heterogenized catalysts, Chem. – Eur. J., 2006, 12, 5972 CrossRef CAS PubMed.
- (a) M. Kleoff, P. Kiler and P. Heretsch, Synthesis of odorants in flow and their applications in perfumery, Beilstein J. Org. Chem., 2022, 18, 754 CrossRef CAS PubMed; (b) G. Gambacorta, J. S. Sharley and I. R. Baxendale, A comprehensive review of flow chemistry techniques tailored to the flavours and fragrances industries, Beilstein J. Org. Chem., 2021, 17, 1181 CrossRef CAS PubMed.
- (a) O. S. Galaktionov, P. D. Anderson, G. W. M. Peters and H. E. H. Meijer, Analysis and optimization of Kenics static mixers, Int. Polym. Process., 2003, 18, 138–150 CrossRef CAS; (b) D. S. Kim, I. H. Lee, T. H. Kwon and D.-W. Cho, A barrier embedded Kenics micromixer, J. Micromech. Microeng., 2004, 14, 1294 CrossRef.
- The perhomologation can occur when the first homologation product 5a reacts with a second equivalent of (chloromethyl)lithium (2), followed by rearrangement.
- R. C. Mykura, S. Veth, A. Varela, L. Dewis, J. J. Farndon, E. L. Myers and V. K. Aggarwal, Investigation of the Deprotonative Generation and Borylation of Diamine-Ligated α-Lithiated Carbamates and Benzoates by in Situ IR Spectroscopy, J. Am. Chem. Soc., 2018, 140, 14677 CrossRef CAS PubMed.
- R. Larouche-Gauthier, C. J. Fletcher, I. Couto and V. K. Aggarwal, Use of alkyl 2,4,6-triisopropylbenzoates in the asymmetric homologation of challenging boronic esters, Chem. Commun., 2011, 47, 12592 RSC.
- Evaluation of the olfactoric profile was conducted at Symrise AG (Holzminden, Germany).
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4re00394b |
| ‡ Dedicated to Dieter Hoppe († 20 March 2024). |
| This journal is © The Royal Society of Chemistry 2025 |